

Abstract
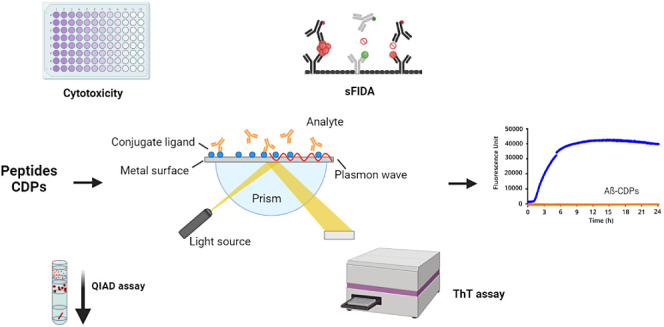
Over a century has passed since Alois Alzheimer first described Alzheimer’s disease (AD), and since then, researchers have made significant strides in understanding its pathology. One key feature of AD is the presence of amyloid-β (Aβ) peptides, which form amyloid plaques, and therefore, it is a primary target for treatment studies. Naturally occurring peptides have garnered attention for their potential pharmacological benefits, particularly in the central nervous system. In this study, nine peptide derivatives of Crotamine, a polypeptide from Crotalus durissus terrificus Rattlesnake venom, as well as one d-enantiomer, were evaluated for their ability to modulate Aβ42 aggregation through various assays such as ThT, QIAD, SPR, and sFIDA. All tested peptides were able to decrease Aβ42 aggregation and eliminate Aβ42 aggregates. Additionally, all of the peptides showed an affinity for Aβ42. This study is the first to describe the potential of crotamine derivative peptides against Aβ42 aggregation and to identify a promising d-peptide that could be used as an effective pharmacological tool against AD in the future.
Keywords: peptide, snake venom, Aβ42, aggregation, deaggregation, d-peptide
Introduction
The increase in life expectancy today can be associated with a higher incidence of age-related diseases, such as Alzheimer’s disease (AD).1−3 AD is known to affect elderly by inducing cognitive deficits as well as operational impairment.4 Consequently, AD patients initially experience difficulty with daily tasks, which progressively leads to a complete dependence on caretakers. Currently, only one drug, called lecanemab, is approved by the Food and Drug Administration (FDA) as a curative treatment of AD.5
The hallmarks of AD are the presence of neuritic plaques, neurofibrillary tangles, and neurodegeneration.6−9 The first above-mentioned structures are composed of protein aggregates, which then induce the observed neurodegeneration. Neuritic plaques are primarily composed of amyloid-β (Aβ) misfolded peptides. These peptides assemble into oligomers, described as the most toxic conformation, and eventually form into fibrils.10−14 Aβ is produced by the sequential cleavage of Amyloid Precursor Protein (APP) by different secretases.15 First, APP is cleaved at the C-terminal part of the protein by β-secretase, then Aβ is formed from APP cleavage by γ-secretase.16−18 This cleavage produces different Aβ isoforms, among which Aβ (1–42) (Aβ42) is one of the most toxic and prone to aggregation.19,20
Considering that Aβ aggregation seems to be the initial downstream event in AD and the fact that only one drug, lecanemab, has been approved that directly interacts with Aβ, the development of new drugs targeting this protein remains essential. Since the beginning of civilization, natural compounds have played a significant role in treating various diseases, including neurological disorders. Therefore, the naturally occurring peptides may hold an important place in the drug development against AD.21 In this context, snake venom has been studied for its potential compounds with antimicrobial and anticancer properties. Beyond these properties, some snake venom compounds have already been approved for treating high blood pressure (Captopril) and as antiplatelet (Tirofiban and Eptifibatide).22 In the central nervous system, snake venom compounds are known to interact with distinct receptors, reducing pain, neuroinflammation, anxiety, and depression.23
Crotamine, which is a protein isolated from Crotalus durissus terrificus, has different positive biological effects; when injected in the hippocampus, crotamine has been shown to improve cognition in rats.24 Additionally, this polypeptide possesses cell-penetrating properties, which can play a role in drug delivery.25,26 For years, many cell-penetrating peptides have been studied for AD treatment due to their nontoxic and high activity properties. Crotamine has two specific regions that enable it to translocate quickly and efficiently into actively proliferating cells.26 These regions are classified as nucleolar targeting peptides (NrTPs). Based on the literature, we selected the amino acids regions from Lys27 to Lys39,26,27 which retain some properties of crotamine and it is smaller in size.
In this study, we evaluated peptides derived from crotamine (Lys27–Lys39). Subsequently, some of those amino acids were replaced to improve the peptide performance. CDPs and one of its d-enantiomer were investigated for their ability to (1) prevent Aβ42 aggregation; (2) eliminate Aβ42, and (3) exhibit affinity to Aβ42. The results we are describing here suggested that CDPs could serve as potential lead peptides targeting Aβ42 aggregation.
In the context of advances in biotechnology, d-enantiomeric peptides present a solution to the challenges associated with peptides in clinical applications. They are resistant to proteases and exhibit lower immunogenicity.28 Several distinct d-peptides have been investigated for Alzheimer’s disease treatment, showing promising results.
Results and Discussion
Naturally Occurring Peptides Prevent Aβ42 Aggregation
Thioflavin T-based assays are used for the in vivo and in vitro detection of amyloid aggregates. First, the potential of the eight CD peptides (CDPs) to reduce Aβ42 aggregation was evaluated at a concentration of 28 μM. Four CDPs showed a significant decrease of the ThT signal compared to Aβ42 alone (Figures 1A and S3). The strongest effect could be observed for CDP-1, where no Aβ42 aggregation was determined over the experimental time (Figure 1C; two-way ANOVA, F (1440, 4800) = 5,349; p < 0.0001). CDP-2, -6, and -8, were able to reduce the aggregation, however, not in the same proportion indicated in the negative control. The signal was four times smaller than in the control (Figure 1D–F; two-way ANOVA, F (1440, 4800) = 5,349; p < 0.0001). The CDP-1 peptide had the strongest reduction potential over the incubation time, where no Aβ42 aggregation was detected (Figure 1C).
Figure 1.

The effect of CDPs on Aβ42 aggregation using Thioflavin T assays. The ThT fluorescence signal with only Aβ42 is shown in blue. In orange is the action of CDP-1, -2, -6, and -8 in the signal of Thioflavin T, which decreased over time. (A) Provides an overview of the effect of the tested peptides against Aβ42 aggregation. (B) Demonstrates the effect of different doses of CDP-1 and CDP-2 (0.5, 1, 3, 6, 13, and 28 μM) on Aβ42 aggregation. The end point is shown for the relative fluorescence during a ThT assay. The Aβ42 aggregation serves as the control. (C) The effect of CDP-1 against Aβ42 aggregation. (D) The effect of CDP-2 against Aβ42 aggregation. (E) The effect of CDP-6 against Aβ42 aggregation. (F) The effect of CDP-8 against Aβ42 aggregation. Data shown are the mean ± SEM from three independent measurements (n = 3). Asterisks mean that the data differ from the Aβ42 control significantly at *p < 0.05, **p < 0.01, and ***p < 0.001 levels according to analyses by two-way ANOVA.
Additional ThT experiments were performed to evaluate the dose dependency of the CDP-1 and CDP-2 peptides, as they revealed the most substantial effect against the Aβ42 aggregation. The results of those experiments demonstrated the dose response relationship of the peptides. The graph in Figure 1B effectively communicates the percentage reduction in the Aβ42 aggregation after 24 h, showing that CDP-1 was much more effective compared to CDP-2 (Figures 1B and S4).
Effect of CDPs on Aβ42 Oligomer Size Distribution Using QIAD
ThT analysis indicated that four crotamine-derived peptides, CDPs (CDP-1, -2, -6, and -8), eliminated or efficiently decreased Aβ42 aggregation. To quantify the effect of the CDPs on the Aβ42 oligomer and aggregate size distribution, we performed QIAD assays. For this assay, RP-HPLC was performed to disassemble all of the different Aβ42 assemblies. The oligomer elimination efficiency is defined as the reduction of Aβ42 contents in fractions 4 to 6 in the presence of the CDPs. Fractions 4–6 containing the Aβ42 oligomers were explicitly sensitive to the studied peptides (Figure 2). The CDPs proved to be significantly efficient.
Figure 2.

CDP derivatives eliminated the Aβ42 oligomers. In the QIAD assay, the Aβ42 solution was separated into different fractions according to the particle size. All peptides were able to reduce the toxic Aß oligomers. When Aβ42 was incubated with CDP-1, CDP-2, CDP-6, and CDP-8, there was a reduction in the peak area corresponding to Aβ42 in the HPLC chromatogram. The data presented the mean ± SEM obtained from three independent measurements (n = 3). Asterisks denote significant differences from the control group at varying levels of significance. Specifically, * represents p < 0.05, ** represents p < 0.01, *** represents p < 0.001, and **** represents p < 0.0001, as determined by analyses conducted through two-way ANOVA.
As observed in the ThT assay, Aβ42 was not detected in fractions 4–5 when incubated with CDP-1, CDP-2, CDP-6, and CDP-8 (two-way ANOVA, p < 0.001; F (12, 39) = 2,276) compared to the control (Figure 2). In fraction 6, however, Aβ42 was detectable even after incubation with CDP-6 (two-way ANOVA; p = 0.7985).
Surface-Based Fluorescence Intensity Distribution Analysis Assay to Follow Aβ42 Oligomer Elimination
Surface-based fluorescence intensity distribution (sFIDA) employs a biochemical setup similar to that of ELISA-like techniques. However, sFIDA uses the same epitope for capturing and detecting antibodies, leading to only recognizing oligomers and aggregates without detecting monomers.29 The microscopy-based readout ensures single-particle sensitivity.29 sFIDA was performed to demonstrate the ability of selected CDPs to eliminate Aβ42 aggregates through a different methodology. Initially, analysis of Aβ42 aggregates at different concentrations was carried out (Figure S5). To follow the elimination of Aβ42 aggregates by the peptides, 50 nM of CDP-1, CDP-2, CDP-6, and CDP-8 were incubated, separately, with 1 nM Aβ42 aggregates (Figure 3). It could be observed that all samples containing the studied peptides have a reduction of the Aβ aggregates, with the most substantial effect for CDP-2 with a reduction of 95.2%, followed by CDP-6 with a 91.2% reduction in the aggregate (Figure 3A–C).
Figure 3.

sFIDA experiments to follow Aβ42 aggregates elimination by CDPs. Antibody Nab228 was captured on the plate surface. After incubation of the samples, the Aβ targets were detected with IC-16 labeled with CF633. The assay surface was then imaged with the TIRFM. (A) TIRFM images of 1 nM Aβ42 aggregates (1 nM) were treated with 50 nM CDPs or without CDPs (control). (B) Pixel count analysis of the TIRFM images from A. One nM Aβ42 aggregates treated with 50 nM CDPs or without CDPs (control). (C) Each peptide’s signal reduction compared with the control (1 nM Aβ42 aggregates). The data presented represent the mean ± SEM from three independent measurements (n = 3). Asterisks denote significant differences from the control group, where ** indicates p < 0.01 and *** indicates p < 0.001 levels of significance, as determined by analyses conducted through a two-sample t test.
sFIDA experiments demonstrated that CDP-1, -1D, -2, -6, and -8 eliminate Aβ42 aggregates. However, the effect of CDP-1 and its d-enantiomer was not pronounced as observed in the ThT and QIAD assay experiments. Further adjustment and optimization of the sFIDA assay conditions are in progress.
Determination of CDP-Binding Affinities with Aβ42
The interaction kinetics of CDP-1, -2, -6, and -8 with Aβ42 was determined using surface plasmon resonance (SPR) experiments. Equilibrium dissociation constant (KD) of the peptides CDP-1, -2, -6, and -8 was determined under the assay described in Material and Methods. Aβ42 was immobilized via covalent primary amino group coupling, and CDP-2, -6, or -8 peptides were injected as analytes. In the case of CDP-1, the peptide was immobilized, and Aβ42 were the analyte. Figure S6 shows the SPR sensorgrams and saturation curves for the tested peptides. The affinity interaction was determined using steady-state model. All peptides were able to interact with Aβ42, although with varying affinity (Figure S6 and Table 1). CDP-1 reveled the lowest KD and therefore, the highest affinity for Aβ42 with a KD value of 406.8 nM; our findings unveiled a better affinity compared to a widely studied peptide, D3D3, a head-to-tail tandem version of D3, a fully d-enantiomeric peptide targeting Aβ42 (N, O), followed by CDP-2, which exhibited a KD value of 3.25 μM, falling within a comparable range of affinities observed with D3 and RD2 (derived from D3) d-enantiomer peptides. Interestingly, CDP-6 (KD 26.38 μM) and CDP-8 (KD 569.6 μM) exhibited a very lower affinity to Aβ42 than CDP-1, suggesting that the Cys residue plays a crucial role in shaping the secondary structure of the peptides.
Table 1. KD Values Were Determined by SPR Experiments.
| steady-state
fitting | |
|---|---|
| peptide | KD ± STD (μM) |
| CDP-1 | 0.41 ± |
| CDP-2 | 3.25 ± 0.2 |
| CDP-6 | 26.28 ± 5.7 |
| CDP-8 | 569.6 ± |
SPR results revealed substantial differences between CDP-1 and its derivatives CDP-2, -6, and -8, which may be related to the change in the secondary structure of the derivatives compared to the original CDP-1 peptide. CD experiments conducted on the peptides demonstrated differences in the secondary structure of CDP-1 compared to CDP-2, -6, and -8. These differences are likely to impact the binding behavior of the peptides with Aβ42 (Figure S9). The secondary structure analysis based on the CD results, using the BeStSel online tool,30,31 revealed a notable decline in α helix content among the peptide variants. For instance, while CDP-1 exhibited a significant α helix content of 25%, this characteristic diminished in subsequent derivatives such as CDP-2, -6, and -8 (<7%) (Table S1). The loss of structural composition suggests a nuance alteration in the peptide’s conformational landscape, potentially influencing its interaction dynamics, as already described for CDPs targeting the SARS-CoV-2 protease.27 Further, Jiang and collaborators (2019) described α-helical peptide inhibitors against Aβ oligomer formation. Their findings underscore the correlation between the loss of secondary structure and the functionality of these inhibitors.32
Effect of CDP-1 d-Enantiomer against Aβ42 Aggregation and Toxic Aβ42 Aggregates
Peptides are attractive drug candidates and have increasingly become the leading molecules in drug development. However, their application is limited due to the susceptibility of l-peptides to endogenous enzymes. On the contrary, peptides composed of d-amino acids are rarely accessible to these enzymes. d-Peptides, when compared with their l-enantiomeric counterparts, possess several therapeutic advantages. As shown previously, the proteolytic stability of d-peptides is superior to l-peptides, which can significantly extend the serum half-life,33,34 resulting in reduced immunogenicity and increased bioavailability of the d-peptides.35 CDP-1 exhibited the most promising results in inhibiting Aβ42 aggregation. Therefore, building upon the aforementioned findings, CDP-1 was synthesized in its d-enantiomeric form, designated as CDP-1D, and subsequently assessed for its potential in mitigating Aβ42 aggregation and eliminating toxic aggregates using ThT, QIAD, and sFIDA assays (Figure 4).
Figure 4.

The effect of CDP-1D against Aß42 aggregation and toxic Aß42 aggregates. (A) D-CDP-1 inhibits Aβ42 aggregation in the Thioflavin T assay (orange). The ThT fluorescence signal with only Aβ42 increased over time (blue). (B) Eliminated Aβ42 oligomers in the QIAD assay. The results are shown in comparison with the mother l-peptide CDP-1. (C) sFIDA assay results for CDP-1 and CDP-1D. TIRFM images of Aβ42 aggregates treated with CDP-1 and CDP-1D. (D) Pixel count analysis of the TIRFM images from C. The data presented represent the mean ± SEM from three independent measurements (n = 3). Asterisks denote significant differences from the control group, where ** indicates p < 0.01 levels of significance, as determined by analyses conducted through a two-sample t test.
Like CDP-1, the d-enantiomeric form of the peptide showed the potential to inhibit Aβ42 aggregation by 100% at the tested concentration of 28 μM in the ThT assay (Figure 4A). To demonstrate the efficacy of the d-enantiomeric peptide, we also applied QIAD assay’s. We demonstrate that both peptides showed efficiency in eliminating the Aβ42 aggregates. However, contrary to the results reported for CDP-1, Aβ42 aggregates were detected after CDP-1D treatment (Figure 4B), and we suspect that CDP-1D agent yielded significant reduction of Aβ42 oligomers in fraction 6; however, it was not able to eliminate it by 100%. The sFIDA experiment revealed a decrease in aggregates after CDP-1D treatment, detected in the same quantity described for the l-enantiomeric counterparts (Figure 4C).
PRI-002, D3, RD2D3, and their cyclic forms demonstrated the ability to reduce Aβ aggregation in the ThT assay and eliminate Aβ oligomers in the QIAD assay.36−38 In a similar vein, CDP-1D demonstrated the capacity to reduce Aβ42 aggregation by 0% and to eliminate Aβ42 oligomers. Therefore, the chiral modification did not affect the efficacy of CDP-1D in vitro, suggesting its potential as a reliable candidate for in vivo treatment studies. Currently, the majority of treatment studies prioritize the disruption of Aβ42 aggregation due to its significant role in Alzheimer’s disease pathology, toxicity in AD.19 Aβ42 is known to be a toxic species and, therefore, was chosen in this study. In the preclinical stages of AD, where clinical symptom are absent, Aβ42 is already present in the brain, transitioning from monomeric form to oligomers, and ultimately fibrils.11,13,14 The different stages can be evaluated in both QIAD and ThT assay.39,40
Cytotoxicity Assay of CDPs against SH-SY5Y and HEK293 Cells
Different concentrations of CDP-1, -2, -6, -8, and -1D were evaluated regarding a cytotoxic effect (Figures S7 and S8). A cytotoxicity assay was performed aiming the safety of the peptides using two different cells: SH-SY5Y (Human neuroblastoma) and HEK293 (Human embryonic kidney) cells. Figure 5A,B displays the viability of SH-SY5Y and HEK293 cells treated with 20 and 40 μM of each peptide, respectively. The peptide concentration determination represents the same and 1.5- to 2-fold concentrations used in the ThT (28 μM) and QIAD (20 μM) assays.
Figure 5.

MTT assay of CDPs in SH-SY5Y and HEK293 cells. MTT assay evaluated the cytotoxicity of four l-peptides and one d-peptide. The effects of 20 and 40 μM peptides on the viability of both cell lines are shown. The complete MTT assay (concentrations tested between 0 and 100 μM) for each peptide is shown in Figures S7 and S8. The control shows the cell viability without peptide, and 0.1% Triton X-100 was used as the negative control. (A) SH-SY5Y cell line and (B) HEK293 cell line. The data displayed represent the mean ± SEM from three independent measurements (n = 3). Asterisks indicate significant differences from the control group, where * represents p < 0.05 and ** represents p < 0.01 levels of significance, as determined by analyses conducted through two-way ANOVA.
The MTT assay revealed that CDP-1, CDP-2, and CDP-8 were nontoxic to both tested human cell lines at 20 and 40 μM concentrations (Figure 5); even at high test dose (100 μM), the SH-SY5Y cell viability was higher than 90% for the tested peptides: CDP-1: 98%; CDP-2: 94.4%, and CDP-8: 91.4% (Figure S7). In comparison, at the same peptide concentration, the viability of HEK293 cells was significantly reduced to 66% (CDP-1), 53% (CDP-2), and 64% (CDP-8), which demonstrated dose-dependent toxicity, with significant reductions in HEK293 cell viability at 100 μM (Figure S8). The cell viability of CDP-6 and CDP-1D (20 and 40 μM final concentrations) tested in SH-SY5Y cells was >85% and for HEK293 cells >70%. At higher concentration (100 μM), the cell viability of SH-SY5Y was reduced, >75% (CDP-6: 86% and CDP-1D: 78%). The peptides’ toxicity was assessed at an elevated concentration (100 μM) in HEK293 cells, resulting in an anticipated substantial decrease in cell viability: CDP-6 exhibited 48% viability, while CDP-1D showed 47%. Our results clearly illustrate that the chosen peptides exhibit different effects depending on both the concentration and cell type, which can be explained due to differences between cell types, tissue origin, and biological function. The literature extensively discusses how the choice of tissue or cell type used in a study can alter the performance and results of cytotoxicity and/or cell viability assays.41 Cell viability and cytotoxicity assays rely on diverse cellular functions, such as cell membrane permeability, enzyme activity, cell adherence, ATP production, coenzyme production, and nucleotide uptake activity.42 The latter may contribute to the heightened cytotoxicity of CDP-1D compared to its l-enantiomeric counterparts (CDP-1) at high doses. d-Peptides exhibit lower enzyme sensitivity relative to l-peptides and may lead to more pronounced adverse effects in vitro.43−45
In this study, we report a novel capability to mitigate Aβ42 aggregation and facilitate the dissolution of Aβ42 aggregates in vitro. Crotamine, a polypeptide isolated from Crotalus durissus terrificus, has been the subject of extensive study for many years. Crotamine can cross membranes and have anticancer properties.26 Besides, crotamine improved memory in rats when infused intrahippocampally.24 The crotamine derivative peptides (CDPs) used in this study also demonstrated inhibitory potential against both SARS-CoV-2 cell culture and the virus’s main protease in vitro.27 CDP-1 was initially conceived as a segment of the crotamine polypeptide, whereas CDP-2 and CDP-8 were derivatives with cysteine/serine substitution. Additionally, based on the original CDP-1 peptide, a d-enantiomer peptide named CDP-1D was synthesized. d-Enantiomeric peptides are considered useful tool in the drug development since those peptides are resistant to proteases degradation and less immunogenic.46,47 Several publications have previously demonstrated the potential of peptides to eliminate Aβ aggregation and, to a lesser extent, to disassemble Aβ aggregates (Table 2). Among these peptides are synthetically developed d-enantiomers.
Table 2. Peptide Inhibitors Targeting Aβ.
| peptidesa | prevent Aβ42 aggregation | eliminate Aβ42 aggregates | references |
|---|---|---|---|
| RYYAAFFARR | yes | - | (48) |
| pgklvya and kklvffarrrra | yes | - | (49) |
| FDYKAEFMPWDT | yes | - | (50) |
| Ac-LPFFN-NH2 | yes | - | (51) |
| KLVFF and variants | yes | - | (52,53) |
| MLRTKDLIWTLFFLGTAVS | yes | - | (54) |
| KFFEAAAKKFFE and variants | yes | - | (55) |
| TLWYK, EHWYH, HYFKY, HYYIK, and KYYEI | yes | - | (56) |
| AFRADVRAERAE and variants | yes | - | (32) |
| γ-AApeptides | yes | yes | (57) |
| lLwHsK and sHwHsK | yes | yes | (58) |
| rprtrlhthrnr, rprtrlhthrnrrprtrlhthrnr, ptlhthnrrrrrrprtrlhthrnr, and ptlhthnrrrrr | yes | yes | (36,39,59−61) |
| CDPs | yes | yes | Described in this manuscript |
Capital letters corresponds to l-amino acids and small letters to d-amino acids in the peptide sequences.
Further experiments are required to unravel the precise mechanism by which CDPs inhibit Aβ aggregation and facilitate the elimination of Aβ aggregates. However, depending on the charge distribution on the surface of the molecules, we suggest that the cationic CDPs (with a net charge of +5) could potentially interact with negatively charged regions present on the surface of the Aβ monomer, oligomer, or fibrils. The N-terminal region of the Aβ monomer (Asp1-Lys16) contains four negatively charged residues (Asp1, Glu3, Asp7, and Glu11). Along with Glu23 and Asp23, they form a negatively charged surface that might be attractable to interact with the positively charged CDP residues (Figure S10). Several studies demonstrated a particular contribution of the Aβ N-terminus to its aggregation behavior.62 Building on this knowledge, we hypothesize that CDPs may interact with the Aβ N-terminus, thereby impeding the aggregation process. This interaction potentially disrupts key steps in the aggregation pathway, representing a promising avenue for therapeutic intervention against Alzheimer’s disease.
Further studies have revealed that in large Aβ aggregates and mature fibrils, the N-terminus becomes exposed while C-terminus remains concealed.63,64 The N-terminal domain plays a crucial role in shaping the structures of aggregates and fibrils. Residues of the N-terminus form essential salt bridges during fibril assembly, such as the interaction between Asp1-Lys28 and Asp7 with Arg5.64 These salt bridges contribute significantly to the stability and architecture of the fibrils. Besides, the N-terminal portion of Aβ(1–10) forms a β-sheet structure by binding with Aβ (12–22) within fibrils.65 The interaction of CDPs with the N-terminus not only serves to prevent aggregation but also has the potential to destabilize oligomers or aggregates, leading to their elimination. This effect was explored by Mallesh et al. 2023, who investigated how peptides interact with Aβ42 and their antiaggregation effects, which were characterized by a reduction in β-sheet formation.52
CDP-1 exhibited an nM affinity for interacting with Aβ42, efficiently eliminated toxic Aβ42 oligomers in the QIAD assay, and completely inhibited ThT-positive Aβ42 fibrils in de novo ThT aggregation assays. Based on this performance, CDP-1D, the d-enantiomer of CDP-1, displayed promising effects in preventing Aβ42 aggregation and eliminating toxic Aβ42 aggregates. Additionally, MTT assays revealed either no or minimal cytotoxicity of the peptides against SH-SY5Y and HEK293 cells at concentrations used in the ThT and QIAD assays (ranging from 20 to 28 μM). However, at a concentration exceeding 60 μM, the CDPs exhibited significant cytotoxic effects against HEK293 cells compared to SH-SY5Y cells, as demonstrated in this study. Similar cytotoxic effects were observed in Vero cells27 and NIH-3T3 cells.66 It is anticipated that the sensitivity of different cell lines to cytotoxic effects of the same compound will vary.67−69 Despite this, the properties of CDP-1 and CDP-1D make them promising lead peptides, as they advance to the next stage of the development process.
Material and Methods
Peptides
Synthetic crotamine derivative peptides (CDP) were synthesized by Genscript (Leiden, NL) with a purity of >95%. HPLC chromatograms demonstrate the purity of each peptide (Figures S1 and S2). The peptides were acetylated at the N-terminus and methylated at the C-terminus. Essential information about the CDPs used in this study is summarized in Table 3. All peptides were diluted in water in a stock solution of 500 mM and placed at 4 °C until further use. Aβ42 (Bachem, Bubendorf, Switzerland) was suspended in HFIP (1 mg/mL) overnight, lyophilized, and stored at room temperature.
Table 3. Basic Information about the Tested Peptides.
| name | sequence | conformation | solvent |
|---|---|---|---|
| CDP-1 | KMDCRWRWKCCKK | L | H2O |
| CDP-2 | KMDSRWRWKSSKK | L | H2O |
| CDP-3 | KMDCRWRWKSSKK | L | H2O |
| CDP-4 | KMDSRWRWKCCKK | L | H2O |
| CDP-5 | KMDSRWRWKSCKK | L | H2O |
| CDP-6 | KMDSRWRWKCSKK | L | H2O |
| CDP-7 | KMDCRWRWKSCKK | L | H2O |
| CDP-8 | KMDCRWRWKCSKK | L | H2O |
| CDP-1D | kkcckwrwrcdmk | D | H2O |
Circular Dichroism Spectroscopy of CDPs
Circular dichroism measurements were carried out with a Jasco J-1100 Spectropolarimeter (Jasco, Germany). Far-UV spectra were measured at 190 to 260 nm using a peptide concentration of 30 μM in ddH2O. The secondary structures of CDPs (1 to 8) and CDP-1D were checked. A 1-mm path length cell was used for the measurements; 10 repeat scans were obtained for each sample, and five scans were conducted to establish the respective baselines. The average baseline spectrum was subtracted from the average sample spectrum. The results are presented as molar ellipticity [θ], according to eq 1:
| 1 |
where θ is the ellipticity measured at the wavelength λ (deg), c is the peptide concentration (mol/L), 0.001 is the cell path length (cm), and n is the number of amino acids. The secondary structure determination was performed using the BeStSel online tool (A,B).
Thioflavin T Assay
To evaluate the ability of the peptides to prevent Aβ42 aggregation, a ThT assay was performed. Aβ42 (10 μM), ThT (5 μM), and the peptides (28 μM) were incubated in 96-well plates for 24 h at room temperature in a plate reader (Clariostar, BMG Labtech, Ortenberg, Germany). During this time, the ThT fluorescence was measured every 6 minutes with an excitation of 440 nm and emission of 490 nm. The data were corrected considering the blank wells (without Aβ42 and the peptides). None of the peptides interact with ThT alone or has autofluorescence properties. All measurements were performed in triplicate (n = 3), and data are presented as mean ± SM.
Investigation of CDP Doses Dependency on the Aβ42 Aggregation Process
Different concentrations were titrated to investigate CDP-1 and -2 dose dependency on the Aβ42 aggregate formation, and a ThT assay was performed as described before. The effect of 0, 0.5, 1, 3, 6, 13, and 28 μM peptides was tested against 10 μM Aβ42 over 24 h. All experiments were performed in triplicate (n = 3), and data are presented as mean ± SM.
Quantitative Determination of Interference with Aβ42 Aggregate Size Distribution (QIAD)
In order to evaluate the efficacy of the peptides in eliminating Aβ42 oligomers, QIAD assays were performed.39 Briefly, lyophilized Aβ42 (80 μM) was incubated for 2 h in sodium phosphate buffer for Aβ42 oligomerization. Then, each peptide (20 μM) was added to the Aβ42 solution and incubated for 30 min. Finally, samples were added on top of an iodixanol density gradient (5–50% (w/v) (OptiPrep, Sigma-Aldrich, Darmstadt, Germany) and centrifuged for 3 h at 4 °C and 259,000 x g (Optima TL-100, Beckman Coulter, Brea, CA, USA). For the sample analysis, 14 fractions (140 μL each) were collected from top to bottom. The top fractions, named fractions 1–2, contained Aβ42-monomers; fractions 4–6 contained the Aβ42-oligomers, which are of special interest; and the bottom fractions 11–14 contained high molecular weight of aggregated Aβ42. Each density gradient fraction was analyzed by Reversed Phase Liquid Chromatography. For this an Agilent 1260 Infinity II (Santa Clara, California, USA) system was equipped with an Agilent Zorbax SB-300 C-8 5 μm, 4.6 × 250 mm Column (Santa Clara, California, USA) and a multi-wavelength detector set to acquire the UV absorbance at 214 nm. H2O + 0.1% trifluoroacetic acid (AppliChem, Darmstadt, Germany) and acetonitrile (Roth, Karlsruhe, Germany) + 0.1% trifluoroacetic acid were used as eluent A and B, respectively. The acquisition method consisted of an initial isocratic step at 15% B, followed by a gradient from 15% B to 45% B in 10 min and another isocratic step at 45% B. The column temperature was set to 80 °C for the entire analysis. This method ensured full separation of the iodixanol density gradient medium and Aβ42 in all fractions. Aβ42 peaks were analyzed and integrated by the Agilent OpenLab 2.5 software. All measurements were performed in triplicate (n = 3), and data are presented as mean ± SM.
Surface Plasmon Resonance
The dissociation constant (KD) of CDP-2, CDP-6, and CDP-8 binding to Aβ42 was determined by SPR spectroscopy using a Biacore T200 instrument (Cytiva, formerly GE Healthcare, Uppsala, Sweden). Aβ42 was immobilized on a series S CM-5 sensor chip (Cytiva, Uppsala, Sweden) by amino coupling. For this, two flow cells of the chip were activated with a freshly prepared solution containing 50 mM N-hydroxysuccinimide (NHS) and 16.1 mM N-ethyl-N′-(dimethylaminopropyl)carbodiimid (EDC) (XanTec, Düsseldorf, Germany) for 7 min. Aβ42 which was stored as a 1 mg/mL solution in HFIP was lyophilized and resolved in 10 mM sodium acetate pH 5.0 buffer (Merck, Darmstadt, Germany) to a final concentration of 50 μg/mL. The Aβ42 solution was injected over one of the two activated flow cells until a signal of 900 RU was reached. After immobilization of Aβ42, both flow cells were deactivated by a 7 min injection of 1 M ethanol at pH 8.5 (XanTec, Düsseldorf, Germany). The activated and deactivated flow cells without Aβ42 served as a reference.
Multicycle kinetic experiments were performed with 10 mM HEPES + 50 mM NaCl + 0.05% Tween 20 (AppliChem, Darmstadt, Germany) as the running buffer at 25 °C at a flow rate of 30 μL/min flow rate. The peptides were diluted in running buffer to final concentrations ranging from 50 to 0.02 μM with 1:2 dilution steps. Each sample was injected for 360 s, followed by a dissociation time of 600 s with running buffer. After each sample, the chip was regenerated with a 45 s injection of 2 M guanidinium hydrochloride (AppliChem, Darmstadt, Germany). The chip was allowed to equilibrate with a running buffer before the next sample injection. The reference flow cell and buffer injections (c = 0 nM) were used to double reference the sensorgrams. Data were evaluated and fitted to a steady-state affinity model with the Biacore T200 Evaluation Software 3.2.
The experimental setup was slightly changed to determine the KD for binding of CDP-1 with Aβ42. Here, CDP-1 was immobilized by the procedure as mentioned above to a signal of 1916.5 RU. For multicycle experiments, Aβ42 was used as the analyte in concentrations ranging from 1.85 μM to 0.02 μM with 1:2 dilution steps with running buffer. All buffers and injection times were the same for the measurement of CDP-2, CDP-6, and CDP-8, except that the dissociation time was extended from 600 to 900 s. The data were fitted to a 1:1 kinetic fit implemented in the Biacore T200 Evaluation Software, with a global Rmax value.
Surface-Based Fluorescence Intensity Distribution Analysis Assay
To determine the influence of peptides on Aβ42 aggregates, an sFIDA assay was performed. The principle was previously described.29 Therefore, we used pretreated 384 glass bottom microtiterplates (Sensoplate plus, Greiner Bio-One GmbH, Frickenhausen, Germany) to immobilize the capture-antibody Nab228 (Sigma-Aldrich, St. Louis, Missouri, USA) in a concentration of 2.5 μg/mL in 0.1 M carbonate buffer (Carl Roth, Karlsruhe, Germany). After an overnight incubation at 4 °C, the plate was washed five times with TBS and blocked with 80 μL of a 0.5% BSA solution in TBS with 0.03% ProClin 300 (Sigma-Aldrich, St. Louis, Mo, USA) for 1.5 h at room temperature.
The influence of 50 nM CDPs on 1 nM Aβ42 aggregates (prepared according to Ref (70)) in PBS was determined by incubating them overnight at room temperature and 600 rpm. As a control, the same Aβ42 aggregates concentration was incubated without additions of peptides; instead, the buffer was added to contain the same end concentrations. The plate was washed as previously described, and 20 μL each of the peptide solution and a serial dilution of Aβ42 aggregates standards in PBS were added and incubated for 2 h at room temperature.
Afterward, 20 μL of 0.625 μg/mL IC-16 CF633 (Heinrich Heine Universität Düsseldorf, Germany) in TBS with 0.1% BSA was added to each well and incubated for 1 h at room temperature. This red fluorescently labeled detection antibody was labeled with CF633 succinimidyl ester (Sigma-Aldrich, St. Louis, Missouri, USA) after the manufacturer’s protocol. After a washing step to remove the redundant antibodies, 80 μL of TBS containing 0.03% Proclin were added.
By using a total internal reflection microscopy (TIRFM) (Leica Camera AG, Wetzlar, Germany), measurement (excitation: 635 nm, emission filter: 705/22 nm) was carried out with an oil immersion objective in 100× magnifications as previously described by Kass et al. 2022, and for each well, 25 of 14-bit grayscale images were measured with a size of 1000 × 1000 pixel each. These data were analyzed with our developed sFIDAta software tool.29 All measurements were performed in triplicate (n = 3), and data are presented as mean ± SM.
Cell Viability Assay
The cell viability assay was performed using the reduction of [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide-MTT] (Merck, Darmstadt, Germany) to investigate the cytotoxicity of the CDPs against SH-SY5Y (human neuroblastoma) and HEK293 (human embryonic kidney) cells. Both cell lines were purchased from the Leibniz Institute DSMZ-German collection of microorganisms and cell cultures GmbH. HEK293 cells were cultivated in Dulbecco’s modified Eagle’s medium high glucose (Merck, Darmstadt, Germany) containing 2% antibiotic solution of 10 000 units Penicillin, 10 mg streptomycin/ml (Merck, Darmstadt, Germany), and 10% fetal bovine serum (Merck, Darmstadt, Germany) at 37 °C with 5% CO2. SH-SY5Y cells were also cultivated in Dulbecco’s modified Eagle’s medium high glucose containing 1% antibiotic solution of 10 000 unit Penicillin,10 mg streptomycin/ml, and 20% fetal bovine serum (Sigma-Aldrich) at 37 °C with 5% CO2.
According to the manufacturer’s instructions, cell viability was measured using the Cell Proliferation Kit I (Roche, Basel, Switzerland). The absorbance of the formazan product was determined by measuring the absorption at 570 nm and subtracted from the absorbance at 660 nm.
The SH-SY5Y cells with a density of 10 000 cells (for HEK293 5000 cells) per well (total volume of 100 μL) were added to each well of a 96-well tissue culture plate (VWR North American) and incubated overnight at 37 °C with 5% CO2. Following seeding, the cells were subjected to treatment by incubation with varying peptide concentrations ranging from 0 to 100 μM. As a negative control, Triton X-100 was added to five wells, resulting in a final concentration of 0.1%. Subsequently, the plates were placed in a humidified 5% CO2 incubator at 37 °C overnight. The next day, 10 μL of MTT labeling reagent from the cell viability kit (Roche, Basel, Switzerland) was added to each well. After 4 h of incubation with the MTT labeling reagent, 100 μL of solubilization buffer was added to each well. The plates were then incubated overnight to ensure complete solubilization of the formazan crystals. Finally, the absorbance was measured at 570 and 660 nm using a CLARIO star plate reader (BMG labtech, Ortenberg, Germany), and the cell viability was calculated using eq 2:
| 2 |
A represents the absorbance readings taken from the wells, likely measured using a spectrophotometer or a microplate reader. These absorbance readings are indicative of the metabolic activity of the cells and can be used to assess the cell viability or proliferation. All measurements were performed in triplicate (n = 3), and data are presented as mean ± SM.
Statistical Analysis
For statistical analysis, GraphPad Prism 8.1 was used. For the ThT assay and cell viability assay, two-way ANOVA and Tukey’s test were performed between the groups at different times. For the QIAD assay, two-way ANOVA and Tukey’s test was performed between the groups in fractions 4–6. For the sFIDA assay, a two sample t test was used.
Acknowledgments
We thank Carsten Corte, who provided us with the IC-16 antibody.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.4c00089.
HPLC chromatograms of CDP-1 to -4; HPLC chromatograms of CDP-5 to -8 and CDP-1D; effect of CDP-3, -4, -5, and -7 on Aβ42 aggregation using Thioflavin T assays; dose dependency of CDP-1 and CDP-2 against the Aβ42 aggregation; sFIDA Aβ aggregate control; Biacore SPR kinetic analyses of peptides to Aβ42; MTT assay of CDPs on SH-SY5Y cells; MTT assay of CDPs on HEK293 cells; CD spectra of CDPs; negatively charged residues in the Aβ structure and surface; Secondary structure content of CDPs, based on CD-experiments (PDF)
Author Contributions
Conceptualization was contributed by L.C.C., M.A.C., and R.J.E; methodology was contributed by L.C.C., I.G., M.M., V.K.S., T.B., and M.A.C.; validation was contributed by L.C.C., I.G., M.M., V.K.S., T.B., M.A.C, and R.J.E. formal analysis was contributed by L.C.C., I.G., M.M., V.K.S., T.B., and M.A.C.; investigation was performed by L.C.C., M.A.C., and R.J.E; resources were by D.W.; writing—original draft preparation was by L.C.C., M.A.C., and R.J.E; writing—review and editing was by L.C.C., I.G., M.M., V.K.S., T.B., D.W., M.A.C, and R.J.E. All authors have read and agreed to the published version of the manuscript.
V.K.S. and T.B. received funding from the Deutsche Forschungsgemeinschaft (INST 208/616–1 FUGG, INST 208/794–1 FUGG) and the Helmholtz Association (HVF0079, DB001822).
The authors declare no competing financial interest.
Supplementary Material
References
- Brookmeyer R.; Johnson E.; Ziegler-Graham K.; Arrighi H. M. Forecasting the Global Burden of Alzheimer’s Disease. Alzheimers Dement. 2007, 3 (3), 186–191. 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- Prince M.; Comas-Herrera A.; Knapp M.; Guerchet M.; Karagiannidou M.. World Alzheimer Report 2016: Improving Healthcare for People Living with Dementia: Coverage, Quality and Costs Now and in the Future, Doctoral Dissertation, Alzheimer’s Disease International, 2016. [Google Scholar]
- Nichols E.; Szoeke C. E. I.; Vollset S. E.; Abbasi N.; Abd-Allah F.; Abdela J.; Aichour M. T. E.; Akinyemi R. O.; Alahdab F.; Asgedom S. W.; et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18 (1), 88–106. 10.1016/S1474-4422(18)30403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheltens P.; Blennow K.; Breteler M. M. B.; De Strooper B.; Frisoni G. B.; Salloway S.; Van der Flier W. M. Alzheimer’s Disease. Lancet 2016, 388 (10043), 505–517. 10.1016/S0140-6736(15)01124-1. [DOI] [PubMed] [Google Scholar]
- Larkin H. D. Lecanemab Gains FDA Approval for Early Alzheimer Disease. JAMA 2023, 329 (5), 363–364. 10.1001/jama.2022.24494. [DOI] [PubMed] [Google Scholar]
- Alzheimer A. Uber Eigenartige Krankheitsfalle Des Spateren Alters. Psychiatr. Nervenkr. Z Gesamte Neurol. Psychiatr. 1911, 4, 356–385. 10.1007/BF02866241. [DOI] [Google Scholar]
- DeTure M. A.; Dickson D. W. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol. Neurodegener. 2019, 14 (1), 32. 10.1186/s13024-019-0333-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KoSIK K. S.; Joachim C. L.; Selkoe D. J. Microtubule-Associated Protein Tau (Tau) Is a Major Antigenic Component of Paired Helical Filaments in Alzheimer Disease. Proc. Natl. Acad. Sci. U. S. A. 1986, 83 (11), 4044–4048. 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi R. E.; Gusella J. F.; Watkins P. C.; Bruns G. A. P.; St George-Hyslop P.; Van Keuren M. L.; Patterson D.; Pagan S.; Kurnit D. M.; Neve R. L. Amyloid β Protein Gene: CDNA, MRNA Distribution, and Genetic Linkage near the Alzheimer Locus. Science 1987, 235 (4791), 880–884. 10.1126/science.2949367. [DOI] [PubMed] [Google Scholar]
- Willbold D.; Strodel B.; Schröder G. F.; Hoyer W.; Heise H. Amyloid-Type Protein Aggregation and Prion-like Properties of Amyloids. Chem. Rev. 2021, 121 (13), 8285–8307. 10.1021/acs.chemrev.1c00196. [DOI] [PubMed] [Google Scholar]
- Chen G.; Xu T.; Yan Y.; Zhou Y.; Jiang Y.; Melcher K.; Xu H. E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38 (9), 1205–1235. 10.1038/aps.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C.; Selkoe D. J. Soluble Protein Oligomers in Neurodegeneration: Lessons from the Alzheimer’s Amyloid β-Peptide. Nat. Rev. Mol. Cell Biol. 2007, 8 (2), 101–112. 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- McLean C. A.; Cherny R. A.; Fraser F. W.; Fuller S. J.; Smith M. J.; Vbeyreuther K.; Bush A. I.; Masters C. L. Soluble Pool of Aβ Amyloid as a Determinant of Severity of Neurodegeneration in Alzheimer’s Disease. Ann. Neurol. 1999, 46 (6), 860–866. . [DOI] [PubMed] [Google Scholar]
- Stroud J. C.; Liu C.; Teng P. K.; Eisenberg D. Toxic Fibrillar Oligomers of Amyloid-β Have Cross-β Structure. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (20), 7717–7722. 10.1073/pnas.1203193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer T. A.; Cappai R.; Masters C. L.; Beyreuther K.; Multhaup G. It All Sticks Together-the APP-Related Family of Proteins and Alzheimer’s Disease. Mol. Psychiatry 1999, 4 (6), 524–528. 10.1038/sj.mp.4000552. [DOI] [PubMed] [Google Scholar]
- Bolduc D. M.; Montagna D. R.; Seghers M. C.; Wolfe M. S.; Selkoe D. J. The Amyloid-Beta Forming Tripeptide Cleavage Mechanism of γ-Secretase. Elife 2016, 5, e17578 10.7554/eLife.17578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H.; Wang Y.; McCarthy D.; Wen H.; Borchelt D. R.; Price D. L.; Wong P. C. BACE1 Is the Major β-Secretase for Generation of Aβ Peptides by Neurons. Nat. Neurosci. 2001, 4 (3), 233–234. 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- Fernandez M. A.; Klutkowski J. A.; Freret T.; Wolfe M. S. Alzheimer Presenilin-1 Mutations Dramatically Reduce Trimming of Long Amyloid β-Peptides (Aβ) by γ-Secretase to Increase 42-to-40-Residue Aβ. J. Biol. Chem. 2014, 289 (45), 31043–31052. 10.1074/jbc.M114.581165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings B. J.; Satou T.; Head E.; Milgram N. W.; Cole G. M.; Savage M. J.; Podlisny M. B.; Selkoe D. J.; Siman R.; Greenberg B. D. Diffuse Plaques Contain C-Terminal Aβ42 and Not Aβ40: Evidence from Cats and Dogs. Neurobiol. Aging 1996, 17 (4), 653–659. 10.1016/S0197-4580(96)00062-0. [DOI] [PubMed] [Google Scholar]
- McGowan E.; Pickford F.; Kim J.; Onstead L.; Eriksen J.; Yu C.; Skipper L.; Murphy M. P.; Beard J.; Das P.; et al. Aβ42 Is Essential for Parenchymal and Vascular Amyloid Deposition in Mice. Neuron 2005, 47 (2), 191–199. 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargo L. C.; Campos G. A. A.; Galante P.; Biolchi A. M.; Goncalves J. C.; Lopes K. S.; Mortari M. R. Peptides Isolated from Animal Venom as a Platform for New Therapeutics for the Treatment of Alzheimer’s Disease. Neuropeptides 2018, 67, 79–86. 10.1016/j.npep.2017.11.010. [DOI] [PubMed] [Google Scholar]
- Diniz-Sousa R.; Caldeira C. A. D. S.; Pereira S. S.; Da Silva S. L.; Fernandes P. A.; Teixeira L. M. C.; Zuliani J. P.; Soares A. M. Therapeutic Applications of Snake Venoms: An Invaluable Potential of New Drug Candidates. Int. J. Biol. Macromol. 2023, 238, 124357. 10.1016/j.ijbiomac.2023.124357. [DOI] [PubMed] [Google Scholar]
- Talukdar A.; Maddhesiya P.; Namsa N. D.; Doley R. Snake Venom Toxins Targeting the Central Nervous System. Toxin Rev. 2023, 42 (1), 382–406. 10.1080/15569543.2022.2084418. [DOI] [Google Scholar]
- Vargas L. S.; Lara M. V. S.; Gonçalves R.; Mandredini V.; Ponce-Soto L. A.; Marangoni S.; Dal Belo C. A.; Mello-Carpes P. B. The Intrahippocampal Infusion of Crotamine from Crotalus Durissus Terrificus Venom Enhances Memory Persistence in Rats. Toxicon 2014, 85, 52–58. 10.1016/j.toxicon.2014.04.017. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Zhang Y.; Zhang C.; Yu H.; Ma Y.; Li Z.; Shi N. Recent Advances of Cell-Penetrating Peptides and Their Application as Vectors for Delivery of Peptide and Protein-Based Cargo Molecules. Pharmaceutics 2023, 15 (8), 2093. 10.3390/pharmaceutics15082093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkis I.; Hayashi M. A. F.; Prieto da Silva A. R. B.; Pereira A.; De Sa Junior P. L.; Zaharenko A. J.; Rádis-Baptista G.; Kerkis A.; Yamane T. State of the Art in the Studies on Crotamine, a Cell Penetrating Peptide from South American Rattlesnake. BioMed. Res. Int. 2014, 2014, 675985. 10.1155/2014/675985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle R. J.; Gering I.; Tusche M.; Ostermann P. N.; Müller L.; Adams O.; Schaal H.; Olivier D. S.; Amaral M. S.; Arni R. K. Design of D-Amino Acids SARS-CoV-2 Main Protease Inhibitors Using the Cationic Peptide from Rattlesnake Venom as a Scaffold. Pharmaceuticals 2022, 15 (5), 540. 10.3390/ph15050540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesehan K.; Buder K.; Linke R. P.; Patt S.; Stoldt M.; Unger E.; Schmitt B.; Bucci E.; Willbold D. Selection of D-amino-acid Peptides That Bind to Alzheimer’s Disease Amyloid Peptide Aβ1–42 by Mirror Image Phage Display. ChemBiochem 2003, 4 (8), 748–753. 10.1002/cbic.200300631. [DOI] [PubMed] [Google Scholar]
- Blömeke L.; Pils M.; Kraemer-Schulien V.; Dybala A.; Schaffrath A.; Kulawik A.; Rehn F.; Cousin A.; Nischwitz V.; Willbold J. Quantitative Detection of α-Synuclein and Tau Oligomers and Other Aggregates by Digital Single Particle Counting. NPJ. Parkinsons Dis. 2022, 8 (1), 68. 10.1038/s41531-022-00330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micsonai A.; Wien F.; Kernya L.; Lee Y.-H.; Goto Y.; Réfrégiers M.; Kardos J. Accurate Secondary Structure Prediction and Fold Recognition for Circular Dichroism Spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (24), E3095–E3103 10.1073/pnas.1500851112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micsonai A.; Wien F.; Bulyáki É.; Kun J.; Moussong É.; Lee Y.-H.; Goto Y.; Réfrégiers M.; Kardos J. BeStSel: A Web Server for Accurate Protein Secondary Structure Prediction and Fold Recognition from the Circular Dichroism Spectra. Nucleic Acids Res. 2018, 46 (W1), W315–W322. 10.1093/nar/gky497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y.; Jiang X.; Shi X.; Yang F.; Cao Y.; Qin X.; Hou Z.; Xie M.; Liu N.; Fang Q.; et al. α-Helical Motif as Inhibitors of Toxic Amyloid-β Oligomer Generation via Highly Specific Recognition of Amyloid Surface. iScience 2019, 17, 87–100. 10.1016/j.isci.2019.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Regenmortel M. H. V.; Muller S. D-Peptides as Immunogens and Diagnostic Reagents. Curr. Opin. Biotechnol. 1998, 9 (4), 377–382. 10.1016/S0958-1669(98)80011-6. [DOI] [PubMed] [Google Scholar]
- Sadowski M.; Pankiewicz J.; Scholtzova H.; Ripellino J. A.; Li Y.; Schmidt S. D.; Mathews P. M.; Fryer J. D.; Holtzman D. M.; Sigurdsson E. M.; et al. A Synthetic Peptide Blocking the Apolipoprotein E/β-Amyloid Binding Mitigates β-Amyloid Toxicity and Fibril Formation in Vitro and Reduces β-Amyloid Plaques in Transgenic Mice. Am. J. Pathol. 2004, 165 (3), 937–948. 10.1016/S0002-9440(10)63355-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dintzis H. M.; Symer D. E.; Dintzis R. Z.; Zawadzke L. E.; Berg J. M. A Comparison of the Immunogenicity of a Pair of Enantiomeric Proteins. Proteins: struct., Funct., Bioinf. 1993, 16 (3), 306–308. 10.1002/prot.340160309. [DOI] [PubMed] [Google Scholar]
- Kutzsche J.; Schemmert S.; Tusche M.; Neddens J.; Rabl R.; Jürgens D.; Brener O.; Willuweit A.; Hutter-Paier B.; Willbold D. Large-Scale Oral Treatment Study with the Four Most Promising D3-Derivatives for the Treatment of Alzheimer’s Disease. Molecules 2017, 22 (10), 1693. 10.3390/molecules22101693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schemmert S.; Camargo L. C.; Honold D.; Gering I.; Kutzsche J.; Willuweit A.; Willbold D. In Vitro and in Vivo Efficacies of the Linear and the Cyclic Version of an All-d-Enantiomeric Peptide Developed for the Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22 (12), 6553. 10.3390/ijms22126553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Groen T.; Schemmert S.; Brener O.; Gremer L.; Ziehm T.; Tusche M.; Nagel-Steger L.; Kadish I.; Schartmann E.; Elfgen A. The Aβ Oligomer Eliminating D-Enantiomeric Peptide RD2 Improves Cognition without Changing Plaque Pathology. Sci. Rep. 2017, 7 (1), 16275. 10.1038/s41598-017-16565-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brener O.; Dunkelmann T.; Gremer L.; Van Groen T.; Mirecka E. A.; Kadish I.; Willuweit A.; Kutzsche J.; Jürgens D.; Rudolph S. QIAD Assay for Quantitating a Compound’s Efficacy in Elimination of Toxic Aβ Oligomers. Sci. Rep. 2015, 5 (1), 13222. 10.1038/srep13222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gade Malmos K.; Blancas-Mejia L. M.; Weber B.; Buchner J.; Ramirez-Alvarado M.; Naiki H.; Otzen D. ThT 101: A Primer on the Use of Thioflavin T to Investigate Amyloid Formation. Amyloid 2017, 24 (1), 1–16. 10.1080/13506129.2017.1304905. [DOI] [PubMed] [Google Scholar]
- Khalef L.; Lydia R.; Filicia K.; Moussa B. Cell Viability and Cytotoxicity Assays: Biochemical Elements and Cellular Compartments. Cell Biochem. Funct. 2024, 42 (3), e4007 10.1002/cbf.4007. [DOI] [PubMed] [Google Scholar]
- Ishiyama M.; Tominaga H.; Shiga M.; Sasamoto K.; Ohkura Y.; Ueno K. A Combined Assay of Cell Vability and in Vitro Cytotoxicity with a Highly Water-Soluble Tetrazolium Salt, Neutral Red and Crystal Violet. Biol. Pharm. Bull. 1996, 19 (11), 1518–1520. 10.1248/bpb.19.1518. [DOI] [PubMed] [Google Scholar]
- Melchionna M.; E Styan K.; Marchesan S. The Unexpected Advantages of Using D-Amino Acids for Peptide Self-Assembly into Nanostructured Hydrogels for Medicine. Curr. Top. Med. Chem. 2016, 16 (18), 2009–2018. 10.2174/1568026616999160212120302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander A. J.; Jin Y.; Luk L. Y. P. D-Peptide and D-Protein Technology: Recent Advances, Challenges, and Opportunities**. ChemBiochem 2023, 24 (4), e202200537 10.1002/cbic.202200537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Xu H.; Xia J.; Ma J.; Xu J.; Li Y.; Feng J. D- and Unnatural Amino Acid Substituted Antimicrobial Peptides With Improved Proteolytic Resistance and Their Proteolytic Degradation Characteristics. Front. Microbiol. 2020, 11, 563030. 10.3389/fmicb.2020.563030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien S.; Lowman H. B. Therapeutic Peptides. Trends Biotechnol. 2003, 21 (12), 556–562. 10.1016/j.tibtech.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Schumacher T. N. M.; Mayr L. M.; Minor D. L. Jr.; Milhollen M. A.; Burgess M. W.; Kim P. S. Identification of D-Peptide Ligands through Mirror-Image Phage Display. Science 1996, 271 (5257), 1854–1857. 10.1126/science.271.5257.1854. [DOI] [PubMed] [Google Scholar]
- Liu J.; Wang W.; Zhang Q.; Zhang S.; Yuan Z. Study on the Efficiency and Interaction Mechanism of a Decapeptide Inhibitor of β-Amyloid Aggregation. Biomacromolecules 2014, 15 (3), 931–939. 10.1021/bm401795e. [DOI] [PubMed] [Google Scholar]
- Jagota S.; Rajadas J. Synthesis of D-Amino Acid Peptides and Their Effect on Beta-Amyloid Aggregation and Toxicity in Transgenic Caenorhabditis Elegans. Med. Chem. Res. 2013, 22 (8), 3991–4000. 10.1007/s00044-012-0386-2. [DOI] [Google Scholar]
- Zhang Y.; Wang S.; Lu S.; Zhang L.; Liu D.; Ji M.; Wang W.; Liu R. A Mimotope of Aβ Oligomers May Also Behave as a β-Sheet Inhibitor. FEBS Lett. 2017, 591 (21), 3615–3624. 10.1002/1873-3468.12871. [DOI] [PubMed] [Google Scholar]
- Minicozzi V.; Chiaraluce R.; Consalvi V.; Giordano C.; Narcisi C.; Punzi P.; Rossi G. C.; Morante S. Computational and Experimental Studies on β-Sheet Breakers Targeting Aβ1–40 Fibrils. J. Biol. Chem. 2014, 289 (16), 11242–11252. 10.1074/jbc.M113.537472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallesh R.; Khan J.; Gharai P. K.; Gupta V.; Roy R.; Ghosh S. Controlling Amyloid Beta Peptide Aggregation and Toxicity by Protease-Stable Ligands. ACS Bio Med. Chem. Au 2023, 3 (2), 158–173. 10.1021/acsbiomedchemau.2c00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsley J. R.; Jovcevski B.; Wegener K. L.; Yu J.; Pukala T. L.; Abell A. D. Rationally Designed Peptide-Based Inhibitor of Aβ42 Fibril Formation and Toxicity: A Potential Therapeutic Strategy for Alzheimer’s Disease. Biochem. J. 2020, 477 (11), 2039–2054. 10.1042/BCJ20200290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henning-Knechtel A.; Kumar S.; Wallin C.; Król S.; Wärmländer S. K. T. S.; Jarvet J.; Esposito G.; Kirmizialtin S.; Gräslund A.; Hamilton A. D.; Magzoub M. Designed Cell-Penetrating Peptide Inhibitors of Amyloid-Beta Aggregation and Cytotoxicity. Cell Rep. Phys. Sci. 2020, 1 (2), 100014. 10.1016/j.xcrp.2020.100014. [DOI] [Google Scholar]
- Kumar J.; Namsechi R.; Sim V. L. Structure-Based Peptide Design to Modulate Amyloid Beta Aggregation and Reduce Cytotoxicity. PLoS One 2015, 10 (6), e0129087– 10.1371/journal.pone.0129087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Cao Q.; Wang C.; Zheng J.; Luo F.; Xie J.; Li Y.; Ma X.; He L.; Eisenberg D.; et al. Structure-Based Peptide Inhibitor Design of Amyloid-β Aggregation. Front. Mol. Neurosci. 2019, 12, 54. 10.3389/fnmol.2019.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Li Y.; Bai G.; Niu Y.; Qiao Q.; Tipton J. D.; Cao C.; Cai J. γ-AApeptide-Based Small-Molecule Ligands That Inhibit Aβ Aggregation. Chem. Commun. 2014, 50 (40), 5206–5208. 10.1039/C3CC46685J. [DOI] [PubMed] [Google Scholar]
- Richman M.; Wilk S.; Chemerovski M.; Wärmländer S. K. T. S.; Wahlström A.; Gräslund A.; Rahimipour S. In Vitro and Mechanistic Studies of an Antiamyloidogenic Self-Assembled Cyclic d,l-α-Peptide Architecture. J. Am. Chem. Soc. 2013, 135 (9), 3474–3484. 10.1021/ja310064v. [DOI] [PubMed] [Google Scholar]
- Klein A. N.; Ziehm T.; van Groen T.; Kadish I.; Elfgen A.; Tusche M.; Thomaier M.; Reiss K.; Brener O.; Gremer L.; et al. Optimization of D-Peptides for Aβ Monomer Binding Specificity Enhances Their Potential to Eliminate Toxic Aβ Oligomers. ACS Chem. Neurosci. 2017, 8 (9), 1889–1900. 10.1021/acschemneuro.7b00045. [DOI] [PubMed] [Google Scholar]
- Van Groen T.; Schemmert S.; Brener O.; Gremer L.; Ziehm T.; Tusche M.; Nagel-Steger L.; Kadish I.; Schartmann E.; Elfgen A.; Jürgens D.; Willuweit A.; Kutzsche J.; Willbold D. The Aβ Oligomer Eliminating D-Enantiomeric Peptide RD2 Improves Cognition without Changing Plaque Pathology. Sci. Rep. 2017, 7 (1), 1–12. 10.1038/s41598-017-16565-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leithold L. H. E.; Jiang N.; Post J.; Niemietz N.; Schartmann E.; Ziehm T.; Kutzsche J.; Shah N. J.; Breitkreutz J.; Langen K. J.; Willuweit A.; Willbold D. Pharmacokinetic Properties of Tandem D-Peptides Designed for Treatment of Alzheimer’s Disease. Eur. J. Pharm. Sci. 2016, 89, 31–38. 10.1016/j.ejps.2016.04.016. [DOI] [PubMed] [Google Scholar]
- Söldner C. A.; Sticht H.; Horn A. H. C. Role of the N-Terminus for the Stability of an Amyloid-β Fibril with Three-Fold Symmetry. PLoS One 2017, 12 (10), e0186347 10.1371/journal.pone.0186347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrdenovic D.; Pieta I. S.; Nowakowski R.; Kutner W.; Lipkowski J.; Pieta P. Amyloid β Interaction with Model Cell Membranes–What Are the Toxicity-Defining Properties of Amyloid β?. Int. J. Biol. Macromol. 2022, 200, 520–531. 10.1016/j.ijbiomac.2022.01.117. [DOI] [PubMed] [Google Scholar]
- Gremer L.; Schölzel D.; Schenk C.; Reinartz E.; Labahn J.; Ravelli R. B. G.; Tusche M.; Lopez-Iglesias C.; Hoyer W.; Heise H. Fibril Structure of Amyloid-β (1–42) by Cryo–Electron Microscopy. Science 2017, 358 (6359), 116–119. 10.1126/science.aao2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollmer M.; Close W.; Funk L.; Rasmussen J.; Bsoul A.; Schierhorn A.; Schmidt M.; Sigurdson C. J.; Jucker M.; Fändrich M. Cryo-EM Structure and Polymorphism of Aβ Amyloid Fibrils Purified from Alzheimer’s Brain Tissue. Nat. Commun. 2019, 10 (1), 4760. 10.1038/s41467-019-12683-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha D.; Mishra R.; Gottschalk S.; Wiesmüller K.-H.; Ugurbil K.; Maier M. E.; Engelmann J. CyLoP-1: A Novel Cysteine-Rich Cell-Penetrating Peptide for Cytosolic Delivery of Cargoes. Bioconjugate Chem. 2011, 22 (3), 319–328. 10.1021/bc100045s. [DOI] [PubMed] [Google Scholar]
- Wojtowicz K.; Sterzyńska K.; Świerczewska M.; Nowicki M.; Zabel M.; Januchowski R. Piperine Targets Different Drug Resistance Mechanisms in Human Ovarian Cancer Cell Lines Leading to Increased Sensitivity to Cytotoxic Drugs. Int. J. Mol. Sci. 2021, 22 (8), 4243. 10.3390/ijms22084243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Femina T. A.; Barghavi V.; Archana K.; Swethaa N. G.; Maddaly R. Non-Uniformity in in Vitro Drug-Induced Cytotoxicity as Evidenced by Differences in IC50 Values–Implications and Way Forward. J. Pharmacol. Toxicol. Methods 2023, 119, 107238. 10.1016/j.vascn.2022.107238. [DOI] [PubMed] [Google Scholar]
- Sazonova E. V.; Chesnokov M. S.; Zhivotovsky B.; Kopeina G. S. Drug Toxicity Assessment: Cell Proliferation versus Cell Death. Cell Death Discovery 2022, 8 (1), 417. 10.1038/s41420-022-01207-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pils M.; Dybala A.; Rehn F.; Blömeke L.; Bujnicki T.; Kraemer-Schulien V.; Hoyer W.; Riesner D.; Willbold D.; Bannach O. Development and Implementation of an Internal Quality Control Sample to Standardize Oligomer-Based Diagnostics of Alzheimer’s Disease. Diagnostics 2023, 13 (10), 1702. 10.3390/diagnostics13101702. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.