

Abstract
Mitochondria carry their own genetic information encoding for a subset of protein-coding genes and translational machinery essential for cellular respiration and metabolism. Despite its small size, the mitochondrial genome, its natural genetic variation and molecular phenotypes have been challenging to study using bulk sequencing approaches, due to its variation in cellular copy number, non-Mendelian modes of inheritance and propensity for mutations. Here we highlight emerging strategies designed to capture mitochondrial genetic variation across individual cells for lineage tracing and studying mitochondrial genetics in primary human cells and clinical specimens. We review recent advances surrounding single-cell mitochondrial genome sequencing and its integration with functional genomic readouts, including leveraging somatic mitochondrial DNA mutations as clonal markers that can resolve cellular population dynamics in complex human tissues. Finally, we discuss how single-cell whole mitochondrial genome sequencing approaches can be utilized to investigate mitochondrial genetics and its contribution to cellular heterogeneity and disease.
The mitochondrial genome was discovered in 1963 and is present within the powerhouses of our cells known as mitochondria1. According to the endosymbiotic theory, mitochondria descended from formerly free-living prokaryotes and evolved into a highly specialized compartment within modern eukaryotic cells, housing key metabolic processes and forming a metabolic hub essential for diverse cellular functions2. As such, mitochondrial DNA (mtDNA) presents a special form of genetic information that is physically separated from nuclear DNA (nDNA) with distinct modes of copy number regulation, replication, transcription and translation (Fig. 1a). In humans, the circular and relatively small mtDNA encodes for 2 rRNAs and 22 tRNAs to support the translation of its 13 protein-coding genes (Fig. 1b). While the mitochondrial genome (approximately 16.5 kilobases) is substantially smaller than the haploid linear chromosomal DNA encoded in the nucleus (approximately 3 gigabases), different cell types may vary in their mitochondrial content as a function of their metabolic needs. Depending on cell type, mtDNA copy number can range between hundreds and thousands of copies per cell on average, with mature oocytes containing more than 100,000 copies3,4 and erythrocytes being devoid of mitochondria5 at the extreme ends of variation. Thus, genetic variants may be present at various allelic frequencies within a cell, but also within single mitochondria, a state referred to as heteroplasmy6,7 (Fig. 1c).
Fig. 1 |. Fundamental aspects of mitochondrial genetics.
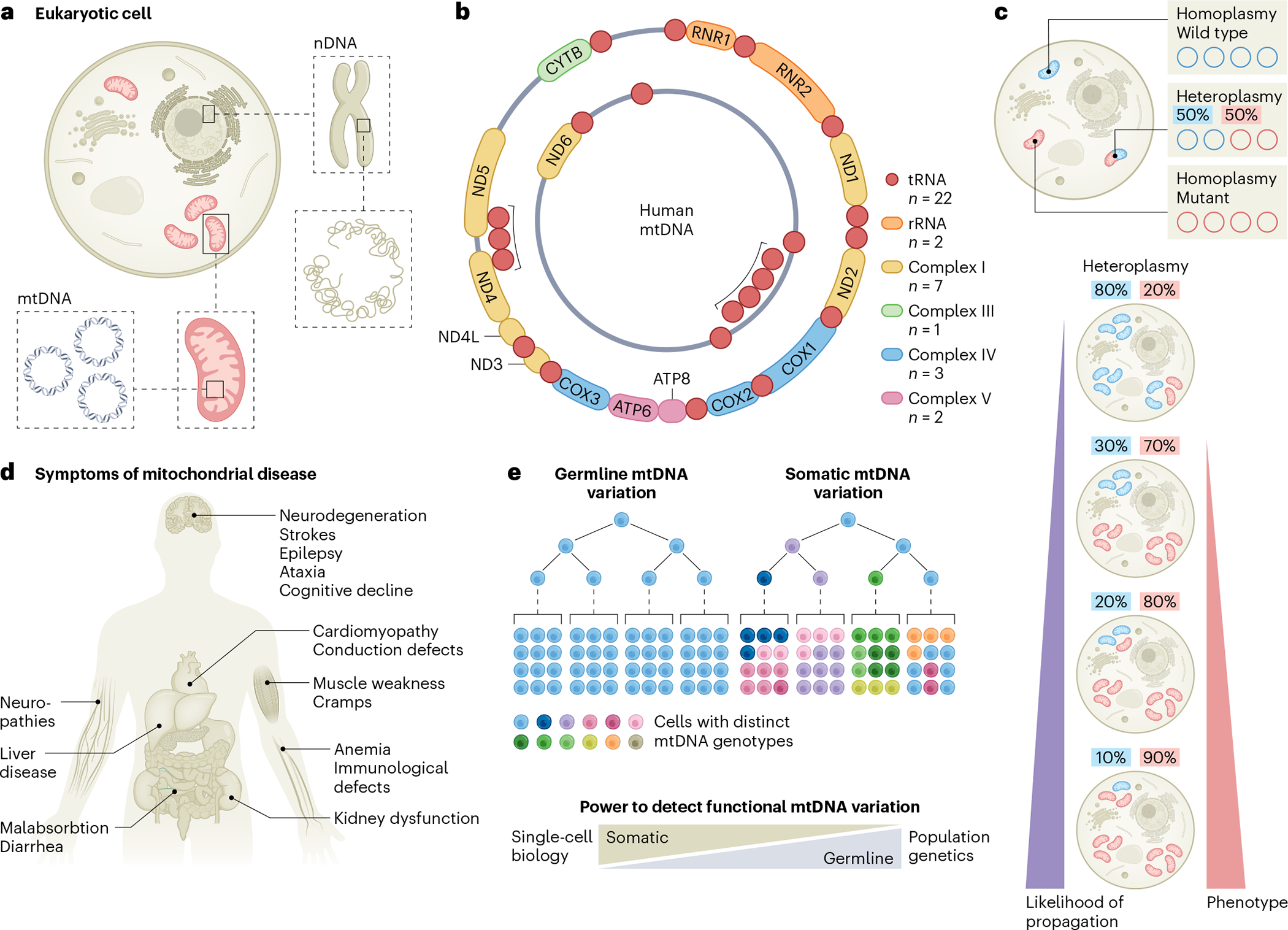
a, Schematic illustrating the physical separation of the set of linear chromosomes representing the nuclear genome and mtDNA located in mitochondria. b, Schematic of the double-stranded, circular mitochondrial genome encoding 22 tRNAs and 2 rRNAs alongside 13 protein-coding genes, which contribute to indicated complexes of the respiratory chain (I, III, IV and V; color-coded). c, Individual cells have multiple mitochondria with potentially multiple copies of mtDNA. Each copy may have unique mutations leading to a state of heteroplasmy. As such, individual mitochondria, as well as cells, may have different allele frequencies of any given mutation. While low-allele-frequency variants may be more dynamic, also given the natural turnover of mitochondria and mtDNA within, individual mtDNA mutations may reach high allele frequencies, even up to homoplasmy owing to, for example, genetic drift. With increasing heteroplasmy, the likelihood of their stable propagation across cell divisions is increased, serving as natural genetic barcodes of clonality. Pathogenic mtDNA mutations may show variable phenotypic effects as a function of heteroplasmy, with a high mutational burden leading to pronounced cellular pathology and human disease. d, Various organ systems may be affected in patients with mitochondrial disorders. e, Germline mtDNA variants, including mtDNA haplotypes, exhibit largely no variation among cells of a single individual. By contrast, a major fraction of cells may develop unique mitochondrial genomes due to somatic mutations that may arise with age. Emerging single-cell omics approaches now enable a more sensitive detection of rare and functionally relevant mtDNA mutations compared to widely used population-based genetic approaches. Panel b adapted from ref. 78, Springer Nature Limited.
Over the course of human history, common groups of germline mtDNA variations evolved, referred to as mtDNA haplotypes3,8, that arose from the gradual development of mutations over generations in the matrilineal lineage. Originally used to trace the migrations of human populations across the globe, mtDNA haplotypes continue to evolve and have been suggested to modulate disease risk and severity9–11. The functional impact of mitochondrial genetic variation is, however, best appreciated in the context of a range of congenital pathogenic mtDNA mutations and deletions associated with a heterogeneous group of mitochondrial disorders, collectively known as mitochondriopathies6,7. Given the ubiquity of mitochondria, these diseases tend to be multi-systemic, often affecting the central nervous and muscular systems. Notably, organ- and tissue-specific phenotypes are not uncommon and point to cell-type- or cell-state-specific metabolic requirements (Fig. 1d). Independent of germline variants, the distinct fidelity of the human mtDNA polymerase (PolG) results in a 10- to 270-fold higher mutational rate of the mitochondrial genome compared to the nuclear genome12, further implying that the large majority of our cells may present with a highly mosaic repertoire of somatically arising mtDNA genetic variation13,14 (Fig. 1e).
In this Review, we highlight recent advances in mitochondrial population genetics enabled by biobank-scale genetic databases. Further, we discuss the potential of emerging single-cell multi-omic technologies in providing new capabilities to investigate mitochondrial genetic variation and its functional analysis, and infer clonal relationships and cellular population dynamics. From these technical innovations, we envision a breadth of future studies to further elucidate functional consequences of mtDNA variation with cell-type specificity that will provide new fundamental insights into this ancient genome.
Mitochondrial genetics in population genetic studies
Human mitochondrial genetics studies have historically focused on mitochondriopathies or discerned the lineage relationships via mitochondrial haplogroups of modern humans toward a common ancestor15. However, advances in mtDNA genotype determination (also referred to as variant calling), and the refinement of quality control analyses, including in whole‐genome sequencing (WGS) data, have accelerated large‐population genetic studies (Fig. 2a). These studies have established links between genetic variations within the mitochondrial genome and complex human phenotypes. For example, by determining genotype–phenotype associations from biobank-scale genome-wide association studies in cohorts such as the UK Biobank, recent studies have linked the contribution of mtDNA variation to common disease and human traits, including haplotype-defining variants with large effect sizes, such as variants that affect human height and are mediated by variation in ATP synthesis16,17. In complementary studies, genome-wide association studies of nuclear variation have revealed the complex genetic architecture underlying measurable properties of the mitochondrial genome, including copy number and the accumulation of heteroplasmy in the form of somatic variants6. As the mitochondrial genome copy number and mutations can be readily determined by genotyping arrays and conventional WGS technologies, we expect similar analyses in other cohorts to become commonplace, by linking both mtDNA mutations to complex traits and nuclear architectures to mtDNA regulation.
Fig. 2 |. Emerging biomedical research avenues of mtDNA.
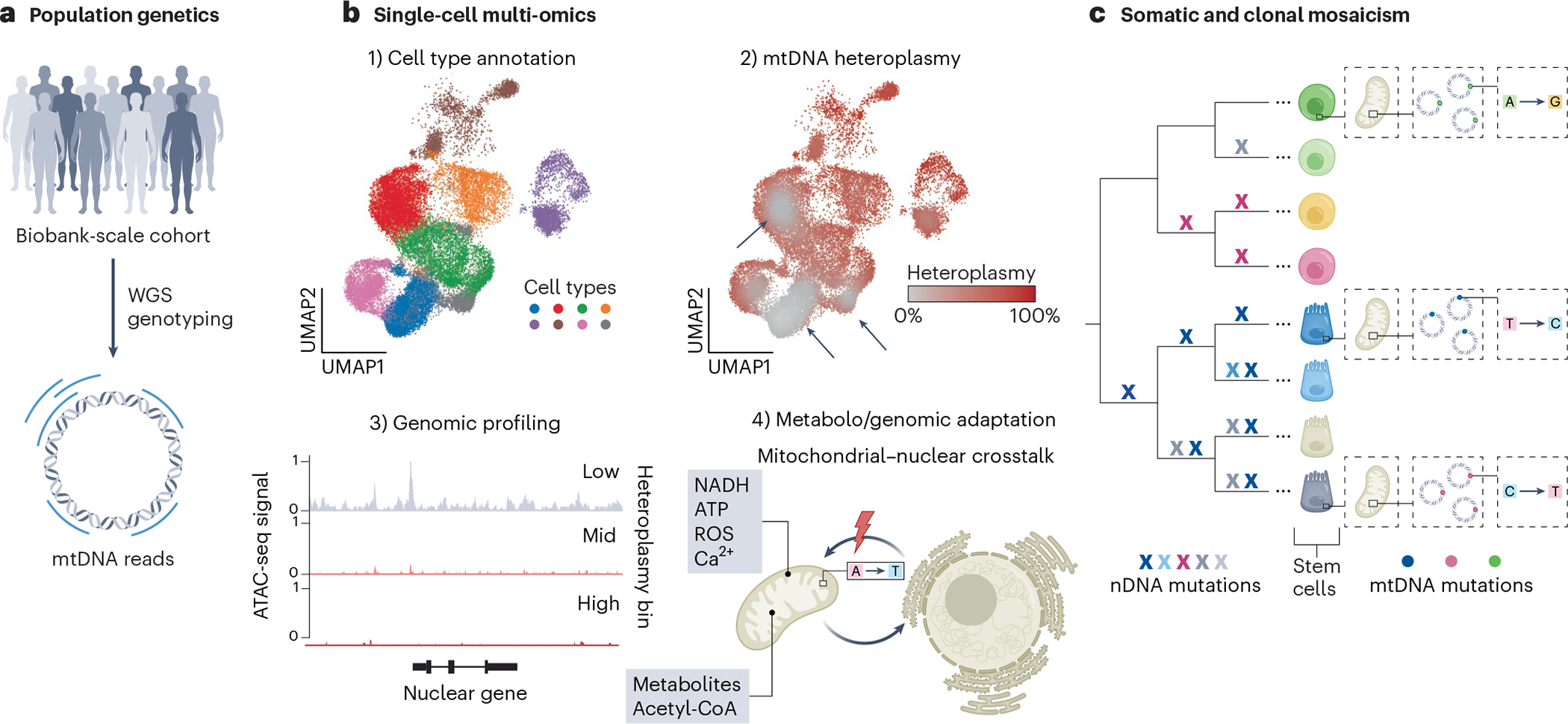
a, Large-scale population genetic studies using genotyping and WGS have revealed a multitude of complex mitochondrial genetic associations surrounding human disease and traits. b, Single-cell multi-omics approaches enable the study of mtDNA genetic variation across (1) cell types and states as identified by transcriptome or chromatin profiling and can reveal (2) selection dynamics of pathogenic mtDNA variants as evident by their depletion in specific cell types (arrows). (3) They further enable the profiling of genomic states as a function of heteroplasmy (that is, low, intermediate or high levels of a pathogenic variant) and may (4) aid in further investigating how alterations in mitochondrial genetic integrity lead to nuclear genomic responses and more generally affect mitochondrial–nuclear crosstalk, including by small metabolites. ROS, reactive oxygen species; UMAP, uniform manifold approximation and projection. c, The constant acquisition of somatic mtDNA mutations contributes to a high degree of cellular genetic mosaicism, which may also be leveraged to trace cellular lineages and clonal population dynamics. Although nDNA mutations arise over individual cell divisions and so enable a retrospective inference of high-resolution phylogenies, their detection in WGS remains costly and is not yet readily scalable at the single-cell level. By contrast, somatically arising mtDNA mutations may mark individual adult stem cells and their clonal progeny.
In addition to analyzing germline variation in these biobank studies, the utility of mitochondrial genetics has emerged in other settings. Specifically, cancer genome sequencing has provided a rich resource of mtDNA genotype associations with patient phenotypes, including findings that have implications for patient stratification and therapeutic development18–22. For example, pathogenic mtDNA mutations are associated with increases in overall survival in colorectal cancer and broadly appear to modulate transcriptional programs18. Additionally, clinical and sequencing analyses of patients diagnosed with mitochondriopathies have revealed a diversity of human disease phenotypes caused by mtDNA variation, indicating that mtDNA variation can variably affect dozens of cell states throughout distinct tissues23. As an extension of these findings, recent technological advances in single-cell sequencing have and will further expedite the study of mitochondrial genetics from functional variant assessment in individual cell types to using these mutations as markers of clonal activity (Fig. 2b,c).
Single-cell methods for detecting mtDNA mutations
Single-cell sequencing technologies have substantially evolved to provide nuanced characterizations of heterogeneous cell states throughout complex tissues. However, efforts to capture genetic variation and its impact on a cell are relatively underdeveloped. Despite the ever-growing wealth of single-cell genomic or multi-omic data from the human cell atlas24 or related efforts25, systematic assessments of the diversity of pathogenic or functional germline and/or somatic mtDNA variants across organ and disease states along with their functional implications are essentially absent. Single-cell WGS (scWGS) to capture nuclear genetic variation has been successfully leveraged to identify somatic variants and devise phylogenetic trees to reconstruct developmental processes (Fig. 2c), for example, describing the polyclonal architecture of the human cortex26,27. However, scWGS remains cost-limiting due to the large size of the nuclear genome, technically challenging and susceptible to amplification-related artifacts when attempting to confidently detect heterozygous variants in single cells that are only present in a single copy of DNA.
By stark contrast, most single-cell transcriptomics (scRNA)- or chromatin accessibility (scATAC)-seq genomic techniques inherently cover large proportions of the mitochondrial genome as a ‘by-product’28,29 (Fig. 3a and Box 1). As >90% of mtDNA is transcribed, full-length RNA-seq techniques (for example, Smart-seq) that capture the entire sequence of a transcript are particularly attractive to capture genetic variants30–33 (Fig. 3b, left), although the high degree of inherent transcriptional error of the mitochondrial RNA polymerase may confound variant determination28. The more high-throughput and thus more popular 3′- or 5′-based scRNA-seq approaches sequence only part of the transcript and thus tend to suffer from limited coverage of the mitochondrial transcriptome for confident variant detection28,34. However, the primer-based tiling of mitochondrial transcripts and a modified computational toolkit (‘MAESTER’) has successfully demonstrated variant determination from cDNA libraries35 (Fig. 3b, right). One recent application of MAESTER resolved the malignant evolution in Barrett’s esophagus36, providing a unified view of the cell states and dynamics along a disease trajectory. Yet, RNA-based methods do have limitations. For example, not all mtDNA molecules may be equally transcribed or captured; for example, the 22 mitochondrial tRNAs are not reverse transcribed by poly(A)-based priming, which is most commonly utilized in scRNA-seq. Here, technologies that poly-adenylate all transcripts before poly(A)-based priming and amplification (for example, vast transcriptome analysis of single cells by dA-tailing (VASA)-seq37) may provide a full characterization of the mitochondrial transcriptome.
Fig. 3 |. Overview of single-cell multi-omic assays for genomic profiling and the co-detection of mtDNA mutations.
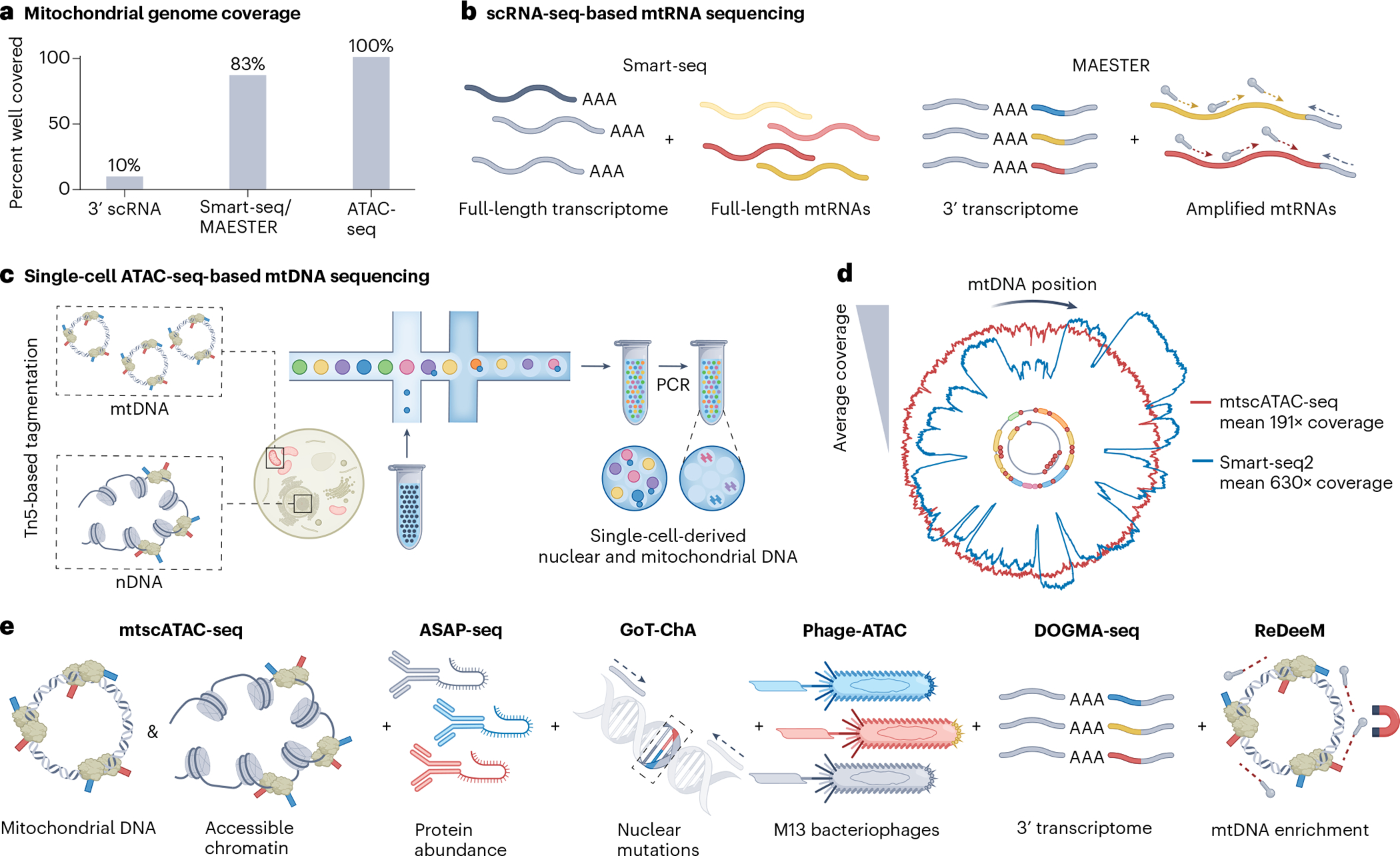
a, Several single-cell omics assays inherently capture mtDNA or may be modified to do so, but cover mtDNA sequences to different degrees, with ATAC-seq-based assays showing the highest mitochondrial genome coverage. b, Among the full-length RNA sequencing approaches, Smart-seq uses poly(A)-based priming to capture large fractions of the mitochondrial transcriptome, including rRNAs, but not tRNAs (left). scRNA-seq techniques can be augmented via the tiling of primers across mitochondrial transcripts to increase mtRNA variant coverage as done in MAESTER (right). c, Schematic illustration of droplet-based encapsulation of cells in mtscATAC-seq. As mtDNA is not densely packed, it is thus readily accessible to tagmentation, analogous to regions of accessible chromatin (left). Following bulk Tn5 tagmentation of cells, individual cells may be droplet-encapsulated (middle), after which PCR enables the amplification of accessible nuclear and mtDNA fragments from single cells (right) for genome-wide mtDNA variant identification in combination with profiling of accessible chromatin. d, Mitochondrial genome coverage is variable depending on technology and type of oligonucleotide (mtDNA for mtscATAC-seq and mtRNA for Smart-seq2) used, as exemplified here for the hematopoietic TF1 cell line. For example, dips in coverage for Smart-seq relate to the inability to capture mitochondrial tRNA, which is not polyadenylated. Note that coverages may differ substantially by cell type. e, Schematic illustration of approaches for single-cell mtDNA genotyping based on mtscATAC-seq (left) that can be variably combined with different genomic readouts (to the right). Antibody-mediated protein marker profiling is enabled by ASAP-seq. Phage-ATAC leverages nanobodies displayed on phagemids to quantify protein markers. The GoT-ChA assay co-detects nDNA mutations using targeted primers. DOGMA-seq enables additional transcriptome profiling, by combining mtscATAC-seq with scRNA-seq. However, mitochondrial genome coverage is less pronounced compared to the standalone mtscATAC-seq assay, leading to the development of ReDeeM, which experimentally enriches mtDNA during library preparation. See Box 1 for further details on the different methods. Panel c adapted from ref. 91, Springer Nature Limited, panel d adapted with permission from ref. 28, Elsevier.
Box 1. Single-cell methods for mtDNA and RNA sequencing.
In 3′-based scRNA-seq, transcripts are captured and reverse transcribed from the 3′ end via their polyadenylated (poly(A)) tails using barcoded oligo dT primers34,93. The resulting cDNA is fragmented and only sequenced from the 3′ end, resulting in limited coverage of the rest of the transcript to determine genetic variants.
5′-Based scRNA-seq is similar to 3′-based scRNA-seq, but the 5′ end of the transcript is sequenced. It is most often applied together with T cell receptor profiling94.
ASAP-seq is a combination of (mt)scATAC-seq with antibody-mediated protein marker profiling40. This proteogenomics approach leverages antibodies coupled to specific oligonucleotide sequences that can be co-detected with accessible chromatin profiles and optional mitochondrial genome sequencing.
DOGMA-seq presents an adaptation of the 10x Genomics Multiome assay40, which integrates principles of ASAP-seq and mtscATAC-seq. It thereby can concomitantly profile the accessible chromatin, the transcriptome, protein markers and whole mitochondrial genome sequencing from the same cell.
Full-length scRNA-seq approaches are typically plate-based and enable sequencing of the entire transcript (from the 5′ to its 3′ end). The increased coverage of the full transcript thereby facilitates the determination of splice isoforms as well as genetic variants, including fusion transcripts. Popular methods are based on the Smart-seq protocol that has been continuously refined (that is, Smart-seq2 (ref. 30), Smart-seq3 (ref. 31), Smart-seq3xpress32 and FLASH-seq33).
GoT-ChA presents a variation of the scATAC-seq assay to co-detect nDNA mutations using targeted primers flanking the genomic site of interest42. It can further be combined with proteogenomics profiling (via ASAP-seq) as well as mtDNA genotyping.
Mitochondrial alteration enrichment from single-cell transcriptomes to establish relatedness (MAESTER) tiles primers across mitochondrial cDNAs captured, for example, via 3′-based scRNA-seq methods to increase the coverage of mitochondrial transcripts for determining genetic variants35,36.
mtscATAC-seq presents an adaptation of the scATAC-seq protocol38,95. By fixing and permeabilizing cells (versus just nuclei), Tn5 also tagments the relatively much more accessible mitochondrial genome. Accessible chromatin profiling can thereby be readily combined with single-cell whole mitochondrial genome sequencing.
Multiomic assays integrate diferent types of genomic modalities93, for example, profiling of accessible chromatin (scATAC-seq) with the transcriptome (via 3′ scRNA-seq) of the same single nuclei or cell.
PHAGE-ATAC leverages nanobodies displayed on phagemids to detect and quantify protein markers41 analogous to ASAP-seq. The phagemid encodes the sequence information for the nanobody, which is detected via sequencing to enable the quantification of the target protein.
ReDeeM is based on the 10x Genomics Multiome assay and leverages oligonucleotides tiling the mitochondrial genome to enrich mtDNA from the ATAC-seq library part of the assay43. Enriched mtDNA sequence libraries are thereby separated for deep sequencing.
scATAC-seq leverages the hyperactive Tn5 transposase to insert (tagment) genomic adapters for PCR amplification of regions of accessible chromatin (for example, typically active promoter and enhancer regions)96. Typically, nuclei are profiled to investigate the epigenetic state of a cell.
scRNA-seq methodologies typically capture mRNA profiles (the transcriptome) of single cells. Experimentally, single cells are separated in wells of a microwell plate or encapsulated in droplets93, enabling the addition of cell-specific oligonucleotide barcodes (cell barcodes) during reverse transcription or cDNA amplification to uniquely label the transcriptome of individual cells. Most scRNA-seq workflows capture nuclear-derived as well as mitochondrial transcripts for gene expression studies and single-nucleotide variant determination28.
Tn5-based transposition of mtDNA as used in mitochondrial scATAC-seq (mtscATAC‐seq)38 currently provides the most scalable and uniform coverage across the mitochondrial genome in addition to cell-state measurements (that is, accessible chromatin profiling) (Fig. 3c–e and Box 1). Differential compaction of mtDNA nucleoid by, for example, the mitochondrial transcription factor A (TFAM)39, may enable the survey of only a variable fraction of mitochondrial genomes within a cell, though effects on variant determination remain to be investigated. Single-cell mtDNA genome coverages range from 40- to 60-fold in primary immune cells, but they can be substantially higher depending on the cell type or cell line (Fig. 3d). Multiple mtscATAC-seq variant assays have been developed, for example, ATAC with select antigen profiling by sequencing (ASAP-seq)40 and PHAGE-ATAC41, which in addition facilitate surface-marker profiling using oligonucleotide-conjugated antibodies or nanobodies displayed on phagemids, respectively (Fig. 3e and Box 1). Genotyping of targeted loci with single-cell chromatin accessibility (GoT-ChA) further leverages targeted primers to genotype nuclear variants in addition to optional mtDNA sequencing to characterize epigenetic landscapes in a clone-specific manner in conditions, such as clonal hematopoiesis or (blood) malignancies42. The 10x Genomics Multiome kit has further facilitated the integration of transcriptional profiles, yielding DOGMA-seq40; this enables the single-cell capture of up to four data modalities, including transcriptome, accessible chromatin, surface markers and mtDNA mutations. Ultimately, unique molecular biology requirements for each data type may limit mtDNA detection relative to the standalone mtscATAC-seq assay. With regulatory multi-omics with deep mitochondrial mutation profiling (ReDeeM), the addition of a probe-based mtDNA enrichment step as part of the 10x Multiome workflow has recently been demonstrated to increase the yield of mtDNA-derived sequences43. Together, these techniques effectively combine mtDNA genotyping with (multiple) phenotypic genomic readouts to annotate cell states in a more unbiased manner and have thus opened up a breadth of biological applications, including the retrospective tracing of clones and lineages (Fig. 2b,c).
History of mtDNA mutations for clonal tracing
Our cells constantly acquire somatic mutations in both the nuclear and mitochondrial genomes that accumulate linearly with age6,12. As these mutations may be propagated across cell divisions, they enable the retrospective inference of ancestral relationships among cells, an approach that has been systematically leveraged to study tumor evolution and progression44,45. The vast space of the nuclear genome and the frequent acquisition of somatic variants with each cell cycle, therefore, in principle, enable a WGS-based reconstruction of deeply resolved phylogenetic trees among cells; this has further been applied to study early human developmental46 or homeostatic processes such as blood production47. Although the high mutational rate of mtDNA make it similarly amenable to reconstructing phylogenies among different species and human populations across evolution, the multi-copy-number nature of mtDNA with its particular mode of inheritance may complicate such inferences at the cellular scale. Specifically, the vast array of constantly newly arising mutations at low allelic frequencies may also be quickly lost, given their asymmetric propagation across cell divisions and the natural turnover of mitochondria and mtDNA variants within (for example, via mitophagy48).
Among the first notions highlighting the utility of somatic mtDNA variants for clonal tracing came from immunohistochemical studies of the healthy human liver49 and colon50 (Fig. 4a). Staining for the activity of mtDNA-encoded subunit of cytochrome c oxidase (mtCCO) in combination with the nuclear-encoded succinate dehydrogenase (SDH) notably revealed the high prevalence of clonal patches of cells that are deficient in mtCCO activity. Targeted sequencing of these cells revealed somatic mtDNA variants at high allelic frequencies that led to altered mtCCO activity. When arising in intestinal stem cells, mtCCO-activity deficiency essentially marked all of their clonal progeny50, a feature that was further leveraged to capture clonal processes in the human lung51, prostate52, breast epithelium53, liver49 and endometrium54 (Fig. 4b), highlighting the potential of somatic mtDNA variation to reveal clonal cellular dynamics across the human body. However, while a powerful in situ readout, it only captures a fraction of mtDNA genetic variation that can be attained via sequencing28,29 (Fig. 3). The particular promise of mtDNA-based clonal tracking lies in the substantial scalability of mtDNA sequencing compared to nuclear WGS efforts. The confined but otherwise highly mutable barcoding space of mtDNA provides a readily targetable search space. Furthermore, the high copy number of mtDNA and resulting high mitochondrial genome coverage facilitates the confident detection of variants with higher allelic frequencies (for example, >10%), features that were first recognized in the study of hematological cells28.
Fig. 4 |. Revealing in situ clonal tracing by mtDNA genetic variation.
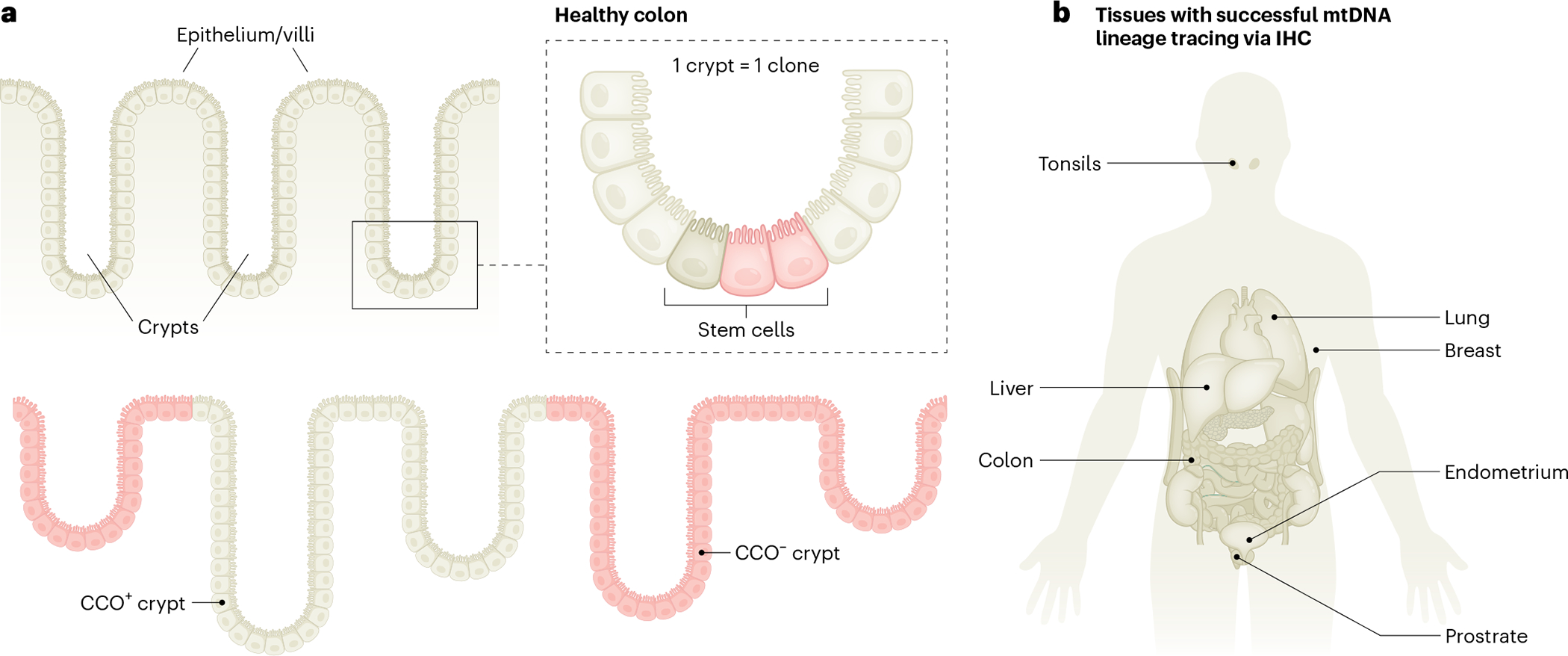
a, Schematic illustrating how stem cells in colonic crypts may acquire somatic mtDNA mutations in cytochrome c oxidase (CCO), leading to the loss of biochemical activity (pink) compared to wild-type cells (beige), as measured by immunohistochemistry (IHC). As mtDNA mutations may be propagated across cell divisions, all stem-cell-derived daughter cells are ‘visually’ marked as they all exhibit a lack of CCO activity, thereby facilitating the identification of clonally related cells. b, Overview of human organ systems surveyed with classical immunohistochemical or immunofluorescence approaches to identify clonal patches of cells on the basis of the loss of protein activity or expression of mtDNA-encoded genes.
Biological insights and prospects from sequencing mtDNA mutations
Clonal tracing and cell fate in human hematopoiesis
Hematopoietic stem cells (HSCs) self-renew and sustain the formation of all major blood cell lineages through division and differentiation (Fig. 5a). However, a quantitative understanding of their clonal activity and regulation in humans is only beginning to emerge. Classically, studies quantifying dynamics via genetic lineage tracing approaches were performed in model organisms through the introduction of heritable tags (for example, DNA barcoding, transposon tagging and CRISPR-based scarring)55,56. In the context of human gene therapy, pro-viral integration sites have been leveraged to trace the output of transplanted stem cells57, but this has not been performed at the single-cell level. Aside from these for now relatively rare cell therapies, the broad application of genetic barcoding is not readily applicable in humans. In this regard, the use of single-cell multi-omics and somatic mtDNA mutations as lineage and clonal barcodes provides a powerful approach to quantify clonal activities across peripheral blood and bone marrow compartments38 and investigate alterations of blood and immune cell population dynamics longitudinally and in relation to clinical events58. Independent of clonal measurements, two recent studies, one leveraging WGS of individual hematopoietic colonies (also co-detecting somatic nDNA variants)12 and the other using single-cell-derived somatic mtDNA mutational profiles43, notably provide conflicting evidence concerning the ability of somatic mtDNA mutations to infer high-resolution phylogenies between hematopoietic cells. Differences in mtDNA variant determination and thresholding of variants with variable heteroplasmy may account for these differences. Further computational innovation, also extending to defining clones and families, will be essential in this area of high-confidence variant calling and downstream biological interpretations.
Fig. 5 |. Examples of biological insights into hematopoiesis and the immune system afforded by single-cell mtDNA sequencing.
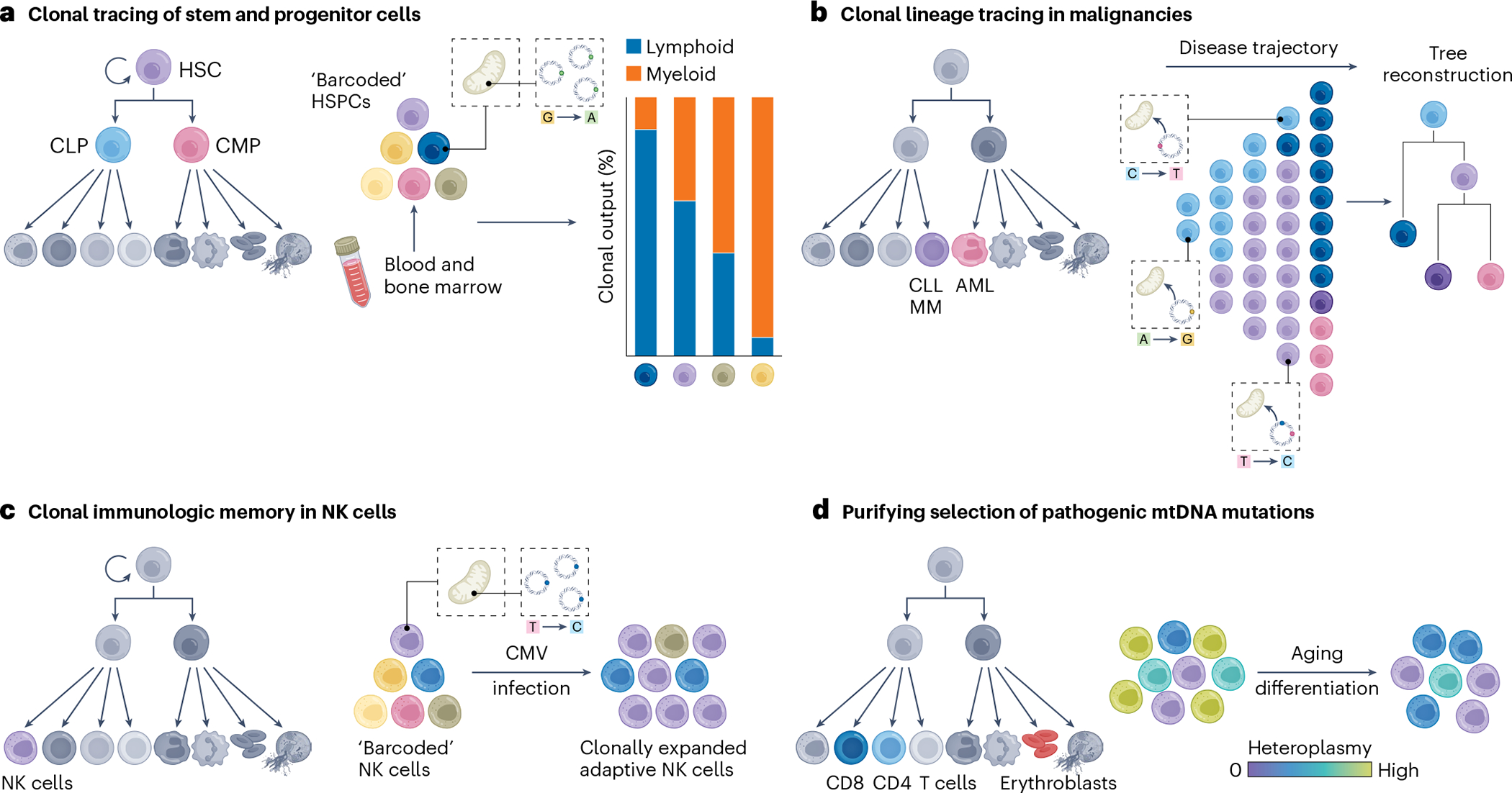
a, Somatic mtDNA mutations enable the inference of the clonal activity, dynamics and potential lineage biases of stem and progenitor cells, such as of HSCs. CMP, common myeloid progenitor; CLP, common lymphoid progenitor; HSPCs, hematopoietic stem and progenitor cells. b, Somatic mtDNA mutations have aided in discerning functional heterogeneity that allows reconstructing disease trajectories and subclonal population structures across different blood cancer entities, including chronic lymphocytic leukemia (CLL), multiple myeloma (MM) or acute myeloid leukemia (AML). c, Somatic mtDNA mutations can help to resolve clonal memory and expansion of immune cells, including of adaptive NK cells after CMV infection. d, Pathogenic mtDNA mutations are selected against in specific T cell populations, suggesting they give rise to distinct metabolic vulnerabilities as well as causing distinct pathologies in erythroblasts. Over the human lifetime, pathogenic mtDNA mutations appear to be largely purified away in the hematopoietic system, probably already at the HSC level. Adapted from ref. 92, Springer Nature Limited.
Recent literature further suggests the horizontal mitochondrial transfer between cells in distinct situations, including tumor progression or tissue repair59,60, which could potentially confound clonal tracing applications. However, simulations suggest such transfer would have to be substantial to confound any clonal relationship, in particular, when heteroplasmic variants of high allelic frequencies define such clones28. Moreover, mtDNA mutations that mark clonal populations may elicit phenotypic effects, impacting cellular and clonal fitness. The altered size of a given clonal family marked by an mtDNA variant may thus also reflect the biological impact of the variant. As such, naturally arising somatic mutations do not necessitate the otherwise required phenotypic neutrality of synthetic systems, where the genetic introduction of a DNA barcode may artificially confound cellular phenotype55,56. The prospective integration of evolving means to infer clonalities61, as well as advances in computational and experimental approaches, will be required to further evaluate discrepancies between clonal and phylogeny reconstruction and overcome (technical) challenges related to clonally dissecting highly polyclonal systems such as hematopoiesis, which may be sustained by 10,000–200,000s of HSC clones62. In a clinical context, the diagnostic assessment of the state of the HSC pool as a function of age or more broadly clonality and clonal evolution would thereby not only be restricted to investigate (pre-) malignant diseases but readily extend to monitoring of transplant and gene cellular therapies, or investigating cellular dynamics in, for example, (chronic) inflammatory processes. Moreover, single-cell efforts to resolve couplings between cell state and cell fate are emerging with the potential to further advance our molecular understanding of cell-fate decision-making43,63,64 and may now be more readily amenable to investigation in an in vivo human context.
Tracking clonal evolution and heterogeneity of human leukemias and tumors
One of the first genetic models for cancer described the occurrence of colorectal cancer through the mutational activation of oncogenes coupled with the mutational inactivation of tumor suppressor genes, whereafter the tumor continues to accumulate passenger and driver mutations to develop full malignancy65. Single-cell mtDNA sequencing of colorectal cancer demonstrated the co-segregation of individual mtDNA mutations with copy-number variants, thereby independently validating the utility of mtDNA as clonal markers, while further resolving subclonal structures within the tumor28,38 (Fig. 5b). Notably, mtDNA mutations also resolved clonalities in the tumor-resident immune cells, highlighting the potential of mtDNA mutations as a versatile tool to track clonal evolution and heterogeneity in clinical specimens, as well as in the immune system beyond T cell and B cell receptor profiling38 (see below).
mtDNA-based clonal tracing has further been applied across a range of hematologic cancers, which are often characterized by high clonality. In acute myeloid leukemia, this confirmed the previously appreciated lineage hierarchy of acute myeloid leukemia and aided in defining the genetic and epigenomic clonal evolution and heterogeneity of ‘pre-leukemic’ HSCs and their progenies29. In chronic lymphocytic leukemia, mtDNA mutations were found to be stably propagated over many years in the absence of strong selective pressures, whereas chemotherapy, disease transformation and relapse lead to dramatic changes in the mtDNA mutational landscape and associated clonal architecture (for example, clonal eradication after chemotherapy or clonal outgrowth)66. Furthermore, the concomitant alterations in chromatin states revealed nongenetic features of disease progression. Hence, mtDNA mutations not only mirrored disease history but also enabled patient-specific studies of subclonal evolution66. In a study addressing multiple myeloma, the further development of cross-modality data integration in the context of longitudinally profiled multiple myeloma specimens enabled resolving transcriptomic and epigenomic adaptations as a function of the subclonal architecture67. Here, defining multi-drug resistant subclones based on mtDNA mutations and copy number variants aided in identifying potentially actionable therapeutic targets67.
Together, somatic mtDNA mutations have been proven broadly valuable to aid in resolving the subclonal architecture across diverse (hematological) cancers. The integration with nuclear variant determination, including cancer-associated single-nucleotide variants42,68, is therefore crucial to further advance the study of cancer evolution at a single-cell resolution. Developing a deeper molecular understanding of the functional effects of somatic mtDNA variation on tumor heterogeneity18–22 will thus constitute an important challenge in the years to come.
Clonal tracing and dynamics of innate immune cells
Upon antigen exposure, immune cells activate, divide and/or clonally expand, recruit additional immune components and so may establish immune memory for a more effective response upon future pathogen exposure69,70. The substantial repertoire of B lymphocyte and T lymphocyte receptor sequences following genetic V(D)J recombination, the rearrangement of so-called V (variable), D (diversity) and J (joining) gene segments of immunoglobulins and T cell receptors71, has been long used as a measure to assess clonal dynamics in the adaptive immune system, be it upon infection or in the context of immunotherapies, including checkpoint inhibition72,73. However, clonal events preceding T cell and/or B cell receptor recombination or across the vast space of innate immune cells have been challenging to resolve in humans. Recently, Rückert et al.74 leveraged the identification of somatic mtDNA mutations to shed light on the oligoclonal origin, clonal expansion and persistence of human adaptive natural killer (NK) cells upon exposure to cytomegalovirus (CMV). Notably, distinct adaptive NK cell clones marked by individual mtDNA mutations showed unique and stably heritable chromatin accessibility profiles, providing novel insights into fundamental aspects of human adaptive NK cell memory74 (Fig. 5c). Hence, determining somatic mtDNA variation may be broadly applicable to investigate fundamental aspects of clonality and memory formation within the innate immune system, including within the myeloid lineage and microglia, across a variety of disease contexts, including tumor immunology or chronic inflammation.
Prospects for studies of mitochondrial genetics
Selection dynamics and functional effects of mtDNA mutations
While most single-cell multi-omic efforts so far have used mtDNA genotyping for purposes of clonal tracing, the substantial utility of these techniques for studies of mitochondrial genetics has only become evident more recently in the context of mitochondriopathies. Clinically, these heterogeneous diseases are well recognized, yet we lack a detailed molecular understanding of how alterations in mitochondrial genetic integrity elicit phenotypes in a cell-type- and cell-state-specific manner. Within this realm, mtscATAC-seq-based analysis of peripheral blood mononuclear cells from adult patients with mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS), who carry the pathogenic m.3243G > A mtDNA variant, revealed the surprising, almost complete absence of the mutation, specifically in T cells but not in other immune cells75 (Fig. 5d). Notably, T cells with low m.3243G > A allele frequencies were also only infrequently observed, suggesting that T cells with any heteroplasmy, rather than only those cells with higher pathogenic allele frequencies, may be metabolically impaired, resulting in their negative selection. While it is worth mentioning that mtDNA is subject to relaxed replication and random genetic drift, which may be differentially pronounced in dividing compared to postmitotic cells76, the question remains as to why the selection is so remarkably cell-type-specific. In light of mitochondrial threshold effects77 but also cell-type- and cell-state-specific effects, single-cell multi-omics, therefore, may enable a more sensitive and unbiased means to identify phenotypic consequences of pathogenic mtDNA compared to previously used (bulk) approaches.
With the use of additional single-cell approaches, the diversity of cell populations that are impacted by purifying selection can be further refined, as has been shown, for instance, in an alanine-tRNA-mutant murine model and in the context of Pearson syndrome78–80, which is characterized by large mtDNA deletions. Collectively, these studies revealed a purifying selection of pathogenic mtDNA in cell states that are probably reflective of specific metabolic requirements, in particular toward attaining and/or also potentially maintaining the mucosal-associated invariant T cell (MAIT) and CD8+ effector/memory T cell states. From these studies, single-cell analyses of cells harboring pathogenic mtDNA revealed several gene regulatory adaptations spanning thousands of differentially expressed genes, including upregulation of serine/glycine biosynthesis and compensatory nuclear oxidative phosphorylation-related gene expression programs78, but their contributions to pathogenesis remain to be investigated.
Moving forward
Technological advances have always been a catalyst for scientific discoveries, with recent innovations in single-cell multi-omics providing exciting prospects for the study of mitochondrial genetics. We foresee numerous applications in this area to provide a valuable tool to investigate, for example, mitochondrial genetic bottlenecks during oogenesis and transmission of varying proportions of mutant mtDNA to one’s offspring81,82. An important outstanding question in the field is how mutant alleles segregate or are selected against during mammalian organogenesis83. Here, model organisms provide an exciting avenue to study mutant mtDNA heteroplasmy organism-wide across tissues, as well as a function of age, in particular, as an elevated mutational mtDNA burden has been prominently linked to aging phenotypes84,85. Specifically, this raises the question of whether we can now quantify or estimate the full spectrum of mutations across the mitochondrial genome per cell and associate these with phenotypic effects. While the same pathogenic mtDNA mutation across a fraction of mitochondrial genomes may lead to pronounced defects, it is unclear whether different mutations in the same gene or within genes that encode for protein subunits of the same complex of the respiratory chain, similarly cooperate to diminish mitochondrial capacities. Another goal is to clarify the origin of mtDNA mutations, including whether they stem from oxidative damage or replication errors in a tissue-specific manner86. Conplastic mice enable the dissection of the consequences of different mtDNA haplogroups and variants within the same nuclear genomic background and have revealed peculiar phenotypes and effects on health longevity87,88. More broadly, and from a genome biology perspective, the molecular interplay between alterations in mitochondrial genetic integrity and associated mitochondrial (dys-)function as a function of cell-state-specific metabolic needs and programs that mediate gene regulatory responses at chromatin and transcription levels, remains to be investigated. Here, the full repertoire of single-cell multi-omics and evolving spatial approaches that capture mitochondrial genetic variation will reveal intriguing insights at multiple molecular layers into mitochondrial genetics and biology. Finally, these technologies may provide valuable avenues to investigate the efficacy of mitochondrial augmentation therapies89, illustrating their potential application across the entire spectrum ranging from basic science to translational medicine.
Outlook
The special aspects of the mitochondrial genome have motivated many groundbreaking studies over the past decades that have refined our understanding of (complex) human diseases. New molecular technologies have accelerated many of these insights, including single-cell sequencing that can contextualize the impact of mtDNA variation in otherwise isogenic settings. Moving forward, we expect that the synthesis of molecular and population genetics studies continues and anticipate further advances in our understanding of the functional role of mtDNA variation. As more biobank-scale datasets linking human (mitochondrial) genetic variation to deep phenotyping are collected, a more comprehensive catalog of the effects of mtDNA variants will emerge for an increasing number of human phenotypes. The inclusion of diverse ethnic backgrounds may further reveal the consequences of germline variants restricted to specific haplogroups for phenotypes that vary between ethnicities (for example, height). Similarly, WGS studies will uncover rare mitochondrial variants that are independent of haplogroups, which may have large effect sizes within individual populations.
While association studies can clarify the interplay between mtDNA, nDNA and complex human traits, molecular assays will be required to further mechanistically dissect the underlying biological pathways and cell types, through which mtDNA variants impact molecular and physiological traits. Specifically, the question remains of how genetically attributable mitochondrial dysfunction is sensed and translated into an adaptive gene regulatory and/or metabolic compensatory response. Furthermore, at what level does homeostatic compensation fail before pathology manifests itself? How can we measure metabolic traits as a function of mtDNA genotype within single cells? Answers to these questions can be derived from continuing to study naturally occurring mtDNA variation, as well as through engineered mtDNA base-editing approaches90 that generate heteroplasmic mutations in diverse cellular and organismal systems.
Beyond the resolution required to identify all relevant variations that affect cellular or organismal phenotypic changes, the utility of mtDNA mutations for clonal lineage tracing allows for functional inferences in diverse tissue contexts. Although the features of mtDNA that facilitate tracing in an in vivo human context have been established for decades, the recent emergence of scalable, single-cell mtDNA genotyping strategies has catalyzed additional biological insights. Rather than only revealing distinct aspects of mitochondrial biology, future applications of these single-cell multi-omics assays may uncover somatic changes important for disease progression and immune evolution. Though it is challenging to predict which contexts are most poised for new discoveries, the ubiquity of mitochondria in cells and the broad applicability of emerging technologies will probably open new dimensions for biological insights for decades to come.
Acknowledgements
We are grateful to the members of the Lareau and Ludwig labs for valuable discussions. This work was supported by NIH K99/R00 HG012579 (C.A.L.), UM1 HG012076 (C.A.L. and L.S.L.), the MDC-NYU exchange program (L.N.), the Hector Fellow Academy (L.N. and L.S.L.), a Longevity Impetus Grant (L.S.L.), the Paul Ehrlich Foundation (L.S.L.), the EMBO Young Investigator Programme (L.S.L.), an Emmy Noether fellowship (LU 2336/2–1) and grants by the German Research Foundation (DFG, LU 2336/3–1, LU 2336/6–1, STA 1586/5–1, TRR241, SFB1588, Heinz Maier-Leibnitz Award to L.S.L.). Individual figures and panels were created with BioRender.com. We apologize to all our colleagues whose work could not be specifically mentioned due to space restrictions.
Footnotes
Competing interests
The Broad Institute has filed for patents relating to the use of technologies described in this paper where C.A.L. and L.S.L., are named inventors (US patent applications 17/251,451 and 17/928,696). C.A.L. and L.S.L. are consultants to Cartography Biosciences. L.N. declares no competing interests.
Peer review information Nature Genetics thanks Aleksandra Trifunovic and the anonymous, reviewer(s), for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Nass S & Nass MMK Intramitochondrial fibers with DNA characteristics: II. Enzymatic and other hydrolytic treatments. J. Cell Biol. 19, 613 (1963). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wallace DC Why do we still have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu. Rev. Biochem. 76, 781–821 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Stewart JB & Chinnery PF The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet. 16, 530–542 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Wai T et al. The role of mitochondrial DNA copy number in mammalian fertility. Biol. Reprod. 83, 52–62 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palis J Primitive and definitive erythropoiesis in mammals. Front. Physiol. 5, 3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta R et al. Nuclear genetic control of mtDNA copy number and heteroplasmy in humans. Nature 620, 839–848 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stewart JB & Chinnery PF Extreme heterogeneity of human mitochondrial DNA from organelles to populations. Nat. Rev. Genet. 22, 106–118 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Emery LS, Magnaye KM, Bigham AW, Akey JM & Bamshad MJ Estimates of continental ancestry vary widely among individuals with the same mtDNA haplogroup. Am. J. Hum. Genet. 96, 183–193 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kandasamy J, Rezonzew G, Jilling T, Ballinger S & Ambalavanan N Mitochondrial DNA variation modulates alveolar development in newborn mice exposed to hyperoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 317, L740–L747 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farha S et al. Mitochondrial haplogroups and risk of pulmonary arterial hypertension. PLoS ONE 11, e0156042 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kenney MC et al. Molecular and bioenergetic differences between cells with African versus European inherited mitochondrial DNA haplogroups: implications for population susceptibility to diseases. Biochim. Biophys. Acta 1842, 208–219 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campbell P et al. Mitochondrial mutation, drift and selection during human development and ageing. Preprint at Research Square 10.21203/rs.3.rs-3083262/v1 (2023). [DOI] [Google Scholar]
- 13.Pesole G, Gissi C, De Chirico A & Saccone C Nucleotide substitution rate of mammalian mitochondrial genomes. J. Mol. Evol. 48, 427–434 (1999). [DOI] [PubMed] [Google Scholar]
- 14.Haag-Liautard C et al. Direct estimation of the mitochondrial DNA mutation rate in Drosophila melanogaster. PLoS Biol. 6, e204 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallace DC Mitochondrial genetic medicine. Nat. Genet. 50, 1642–1649 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Yonova-Doing E et al. An atlas of mitochondrial DNA genotype–phenotype associations in the UK Biobank. Nat. Genet. 53, 982–993 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cai N et al. Mitochondrial DNA variants modulate N-formylmethionine, proteostasis and risk of late-onset human diseases. Nat. Med. 27, 1564–1575 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Gorelick AN et al. Respiratory complex and tissue lineage drive recurrent mutations in tumour mtDNA. Nat. Metab. 3, 558–570 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Triska P et al. Landscape of germline and somatic mitochondrial DNA mutations in pediatric malignancies. Cancer Res. 79, 1318–1330 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopinski PK, Singh LN, Zhang S, Lott MT & Wallace DC Mitochondrial DNA variation and cancer. Nat. Rev. Cancer 21, 431–445 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Yuan Y et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet. 52, 342–352 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahmood M et al. Mitochondrial DNA mutations drive aerobic glycolysis to enhance checkpoint blockade response in melanoma. Nat. Cancer 10.1038/s43018-023-00721-w (2024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suomalainen A & Battersby BJ Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 19, 77–92 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Rood JE, Maartens A, Hupalowska A, Teichmann SA & Regev A Impact of the human cell atlas on medicine. Nat. Med. 28, 2486–2496 (2022). [DOI] [PubMed] [Google Scholar]
- 25.Rajewsky N et al. LifeTime and improving European healthcare through cell-based interceptive medicine. Nature 587, 377–386 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lodato MA et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350, 94–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luquette LJ et al. Single-cell genome sequencing of human neurons identifies somatic point mutation and indel enrichment in regulatory elements. Nat. Genet. 54, 1564–1571 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ludwig LS et al. Lineage tracing in humans enabled by mitochondrial mutations and single-cell genomics. Cell 176, 1325–1339.e22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu J et al. Single-cell lineage tracing by endogenous mutations enriched in transposase accessible mitochondrial DNA. eLife 8, e45105 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Picelli S et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 9, 171–181 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Hagemann-Jensen M et al. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol. 38, 708–714 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Hagemann-Jensen M, Ziegenhain C & Sandberg R Scalable single-cell RNA sequencing from full transcripts with Smart-seq3xpress. Nat. Biotechnol. 40, 1452–1457 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hahaut V et al. Fast and highly sensitive full-length single-cell RNA sequencing using FLASH-seq. Nat. Biotechnol. 40, 1447–1451 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng GXY et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 1–12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller TE et al. Mitochondrial variant enrichment from high-throughput single-cell RNA sequencing resolves clonal populations. Nat. Biotechnol. 40, 1030–1034 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gier RA et al. Clonal cell states link Barrett’s esophagus and esophageal adenocarcinoma. Preprint at bioRxiv 10.1101/2023.01.26.525564 (2023). [DOI] [Google Scholar]
- 37.Salmen F et al. High-throughput total RNA sequencing in single cells using VASA-seq. Nat. Biotechnol. 40, 1780–1793 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lareau CA et al. Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling. Nat. Biotechnol. 39, 451–461 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stefan Isaac R et al. Single-nucleoid architecture reveals heterogeneous packaging of mitochondrial DNA. Nat. Struct. Mol. Biol. 31, 568–577 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mimitou EP et al. Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nat. Biotechnol. 39, 1246–1258 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fiskin E et al. Single-cell profiling of proteins and chromatin accessibility using PHAGE-ATAC. Nat. Biotechnol. 40, 374–381 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Izzo F et al. Mapping genotypes to chromatin accessibility profiles in single cells. Nature 629, 1149–1157 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weng C et al. Deciphering cell states and genealogies of human hematopoiesis. Nature 10.1038/s41586-024-07066-z (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gerstung M et al. The evolutionary history of 2,658 cancers. Nature 578, 122–128 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sankaran VG, Weissman JS & Zon LI Cellular barcoding to decipher clonal dynamics in disease. Science 378, eabm5874 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spencer Chapman M et al. Lineage tracing of human development through somatic mutations. Nature 595, 85–90 (2021). [DOI] [PubMed] [Google Scholar]
- 47.Mitchell E et al. Clonal dynamics of haematopoiesis across the human lifespan. Nature 606, 343–350 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Picca A, Faitg J, Auwerx J, Ferrucci L & D’Amico D Mitophagy in human health, ageing and disease. Nat. Metab. 5, 2047–2061 (2023). [DOI] [PubMed] [Google Scholar]
- 49.Fellous TG et al. Locating the stem cell niche and tracing hepatocyte lineages in human liver. Hepatology 49, 1655–1663 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Taylor RW et al. Mitochondrial DNA mutations in human colonic crypt stem cells. J. Clin. Invest. 112, 1351–1360 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Teixeira VH et al. Stochastic homeostasis in human airway epithelium is achieved by neutral competition of basal cell progenitors. eLife 2, e00966 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blackwood JK et al. In situ lineage tracking of human prostatic epithelial stem cell fate reveals a common clonal origin for basal and luminal cells. J. Pathol. 225, 181–188 (2011). [DOI] [PubMed] [Google Scholar]
- 53.Cereser B et al. Analysis of clonal expansions through the normal and premalignant human breast epithelium reveals the presence of luminal stem cells. J. Pathol. 244, 61–70 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tempest N et al. Histological 3D reconstruction and in vivo lineage tracing of the human endometrium. J. Pathol. 251, 440–451 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Rodriguez-Fraticelli AE et al. Clonal analysis of lineage fate in native haematopoiesis. Nature 553, 212–216 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li L et al. A mouse model with high clonal barcode diversity for joint lineage, transcriptomic, and epigenomic profiling in single cells. Cell 186, 5183–5199.e22 (2023). [DOI] [PubMed] [Google Scholar]
- 57.Ferrari S et al. Efficient gene editing of human long-term hematopoietic stem cells validated by clonal tracking. Nat. Biotechnol. 38, 1298–1308 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lareau CA, Ludwig LS & Sankaran VG Longitudinal assessment of clonal mosaicism in human hematopoiesis via mitochondrial mutation tracking. Blood Adv. 3, 4161–4165 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu D et al. Intercellular mitochondrial transfer as a means of tissue revitalization. Signal Transduct. Target Ther. 6, 65 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang H et al. Systematic investigation of mitochondrial transfer between cancer cells and T cells at single-cell resolution. Cancer Cell 41, 1788–1802.e10 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beneyto-Calabuig S et al. Clonally resolved single-cell multi-omics identifies routes of cellular differentiation in acute myeloid leukemia. Cell Stem Cell 30, 706–721.e8 (2023). [DOI] [PubMed] [Google Scholar]
- 62.Lee-Six H et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 561, 473–478 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haghverdi L & Ludwig LS Single-cell multi-omics and lineage tracing to dissect cell fate decision-making. Stem Cell Rep. 18, 13–25 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weinreb C, Rodriguez-Fraticelli A, Camargo FD & Klein AM Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 367, eaaw3381 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fearon ER & Vogelstein B A genetic model for colorectal tumorigenesis. Cell 61, 759–767 (1990). [DOI] [PubMed] [Google Scholar]
- 66.Penter L et al. Longitudinal single-cell dynamics of chromatin accessibility and mitochondrial mutations in chronic lymphocytic leukemia mirror disease history. Cancer Discov. 11, 3048–3063 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Poos AM et al. Resolving therapy resistance mechanisms in multiple myeloma by multi-omics subclone analysis. Blood 10.1182/blood.2023019758 (2023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nam AS et al. Single-cell multi-omics of human clonal hematopoiesis reveals that DNMT3A R882 mutations perturb early progenitor states through selective hypomethylation. Nat. Genet. 54, 1514–1526 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adams NM, Grassmann S & Sun JC Clonal expansion of innate and adaptive lymphocytes. Nat. Rev. Immunol. 20, 694–707 (2020). [DOI] [PubMed] [Google Scholar]
- 70.Jenkins MK, Chu HH, McLachlan JB & Moon JJ On the composition of the preimmune repertoire of T cells specific for peptide-major histocompatibility complex ligands. Annu. Rev. Immunol. 28, 275–294 (2010). [DOI] [PubMed] [Google Scholar]
- 71.Schatz DG & Ji Y Recombination centres and the orchestration of V(D)J recombination. Nat. Rev. Immunol. 11, 251–263 (2011). [DOI] [PubMed] [Google Scholar]
- 72.Yost KE et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 25, 1251–1259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Musvosvi M et al. T cell receptor repertoires associated with control and disease progression following Mycobacterium tuberculosis infection. Nat. Med. 29, 258–269 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rückert T, Lareau CA, Mashreghi M-F, Ludwig LS & Romagnani C Clonal expansion and epigenetic inheritance of long-lasting NK cell memory. Nat. Immunol. 23, 1551–1563 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Walker MA et al. Purifying selection against pathogenic mitochondrial DNA in human T cells. N. Engl. J. Med. 383, 1556–1563 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Glynos A et al. High-throughput single-cell analysis reveals progressive mitochondrial DNA mosaicism throughout life. Sci. Adv. 9, eadi4038 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rossignol R et al. Mitochondrial threshold effects. Biochem. J. 370, 751–762 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lareau CA et al. Single-cell multi-omics of mitochondrial DNA disorders reveals dynamics of purifying selection across human immune cells. Nat. Genet. 55, 1198–1209 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Franklin IG et al. T cell differentiation drives the negative selection of pathogenic mitochondrial DNA variants. Life Sci. Alliance 6, e202302271 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang J et al. Antigen receptor stimulation induces purifying selection against pathogenic mitochondrial tRNA mutations. JCI Insight 8, e167656 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cree LM et al. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat. Genet. 40, 249–254 (2008). [DOI] [PubMed] [Google Scholar]
- 82.Cao L et al. The mitochondrial bottleneck occurs without reduction of mtDNA content in female mouse germ cells. Nat. Genet. 39, 386–390 (2007). [DOI] [PubMed] [Google Scholar]
- 83.Burr SP et al. Cell lineage-specific mitochondrial resilience during mammalian organogenesis. Cell 186, 1212–1229.e21 (2023). [DOI] [PubMed] [Google Scholar]
- 84.Trifunovic A et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423 (2004). [DOI] [PubMed] [Google Scholar]
- 85.Kujoth GC et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309, 481–484 (2005). [DOI] [PubMed] [Google Scholar]
- 86.Nissanka N & Moraes CT Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 592, 728–742 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Latorre-Pellicer A et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 535, 561–565 (2016). [DOI] [PubMed] [Google Scholar]
- 88.Scotece M et al. Mitochondrial DNA impact on joint damaged process in a conplastic mouse model after being surgically induced with osteoarthritis. Sci. Rep. 11, 1–12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jacoby E et al. Mitochondrial augmentation of hematopoietic stem cells in children with single large-scale mitochondrial DNA deletion syndromes. Sci. Transl. Med. 10.1126/scitranslmed.abo3724 (2022). [DOI] [PubMed] [Google Scholar]
- 90.Mok BY et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 583, 631–637 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ludwig LS & Lareau CA In Chromatin Accessibility: Methods and Protocols (eds Marinov GK & Greenleaf WJ) 269–282 (Humana, 2023). [Google Scholar]
- 92.Ulirsch JC et al. Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat. Genet. 51, 683–693 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Baysoy A, Bai Z, Satija R & Fan R The technological landscape and applications of single-cell multi-omics. Nat. Rev. Mol. Cell Biol. 24, 695–713 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pai JA & Satpathy AT High-throughput and single-cell T cell receptor sequencing technologies. Nat. Methods 18, 881–892 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lareau CA et al. Mitochondrial single-cell ATAC-seq for high-throughput multi-omic detection of mitochondrial genotypes and chromatin accessibility. Nat. Protoc. 18, 1416–1440 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Buenrostro JD et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]