


Abstract
Translational research on the Cre/loxP recombination system focuses on enhancing its specificity by modifying Cre/DNA interactions. Despite extensive efforts, the exact mechanisms governing Cre discrimination between substrates remains elusive. Cre recognizes 13 bp inverted repeats, initiating recombination in the 8 bp spacer region. While literature suggests that efficient recombination proceeds between lox sites with non-loxP spacer sequences when both lox sites have matching spacers, experimental validation for this assumption is lacking. To fill this gap, we investigated target site variations of identical pairs of the loxP 8 bp spacer region, screening 6000 unique loxP-like sequences. Approximately 84% of these sites exhibited efficient recombination, affirming the plasticity of spacer sequences for catalysis. However, certain spacers negatively impacted recombination, emphasizing sequence dependence. Directed evolution of Cre on inefficiently recombined spacers not only yielded recombinases with enhanced activity but also mutants with reprogrammed selective activity. Mutations altering spacer specificity were identified, and molecular modelling and dynamics simulations were used to investigate the possible mechanisms behind the specificity switch. Our findings highlight the potential to fine-tune site-specific recombinases for spacer sequence specificity, offering a novel concept to enhance the applied properties of designer-recombinases for genome engineering applications.
Graphical Abstract
Graphical Abstract.

Introduction
Site-specific recombinases (SSRs) are specialized enzymes that promote site-specific DNA rearrangements between defined target regions (1). Engineering and directed evolution strategies have been successfully used to alter the site-specificity of recombinases, allowing SSRs to be an adaptable tool for precise genome engineering (2). Of particular interest are the tailor-made recombinase systems derived from the Cre/loxP system, where the native DNA specificity is altered to enable the recombination of therapeutically relevant sequences (3–9). In order to accelerate the reprogramming of novel recombinases, it is vital to understand the characteristics that contribute to protein DNA recognition, cleavage, and recombination. Gaining a comprehensive understanding of the factors that govern these mechanisms offers the opportunity to manipulate these features for reprogramming and fine-tuning specificity.
Cre is a member of the tyrosine SSR family and is naturally encoded by bacteriophage P1. Cre recombinase is responsible for excising, exchanging or inverting DNA between a pair of 34 bp loxP target sites. Each loxP site consists of a core 8bp spacer sequence flanked by two 13 bp inverted symmetry regions (half-sites; Figure 1A). Cre/loxP complex formation begins with site-specific binding of a single Cre molecule to each half-site. Once four Cre molecules bound to two loxP sites come together, the tetrameric synaptic complex is formed and poised for catalysis (10). DNA recombination takes place at the spacer region in a stepwise manner, involving cleavage and strand exchange, with the ultimate outcome of the recombination reaction determined by the orientation of the spacer sequence (11–14). Because of the spacer's critical role in site-specific recombination, understanding the spacer characteristics necessary for efficient recombination and contribution to specificity are valuable in studies developing recombinases systems for targeting alternative sequences.
Figure 1.

LoxP spacer library activity screen. (A) LoxP sequence with top (TS) and bottom strand (BS). Bases in the spacer region are labelled and highlighted in teal. The respective attack points of the catalytic tyrosine (Y324) to the DNA strands are indicated by triangles. (B) Schematic of the target site screen. i) Construction of target library around the spacer sequence of the loxP site. Examples of the matching spacers are shown, with alterations to the canonical loxP spacer sequence highlighted in yellow. ii) Library cloning into an E. coli recombinase expression vector. iii) Assembled library vector transformation to E. coli and induced to express Cre. The editing outcomes are indicated by the teal target sites where an active event is recombined and an inactive event is non-recombined. iv) Purification of vectors and subsequent amplification over the target sites with primers containing Illumina indexing sequence. v) Quantification of the recombination rate for each target site calculated from deep sequencing reads (%) displayed in yellow. (C) Target site screen results plotted as activity ratio of Cre recombination (fraction of recombination) for each target site. Activity ratio is calculated for each target site by dividing the activity by the highest activity in the screen. Each dot (teal) represents a unique spacer sequence within the library with wild-type loxP highlighted in yellow. Dotted vertical lines outline the range where the targets are efficiently recombined by Cre, defined by recombination rate ±25% loxP. Plot zoom showing targets with lowest activity. Targets selected for evolution are labeled, loxSE1, loxSE2 and loxSE3. (D) Impact of number of mismatches in the spacer to Cre recombination efficiency. Cre recombination is represented as activity ratio and effects for one (n = 36), two (n = 238), three (n = 831), four (n = 1712), five (n = 1999), six (n = 988), seven (n = 128) and eight (n = 12) mismatches. The wild-type loxP is plotted as a line at 0 mismatches. (E, F) The sequence logos represent spacer preference of Cre. The base height in each logo is calculated by relative frequency in the given subpopulation. The base frequency is normalized to library representation. The logo in E is calculated from a subpopulation of spacer sequences with the highest recombination efficiency or top 10% recombined spacers (n = 595 targets). The logo in F is calculated from a subpopulation of spacer sequences with the lowest recombination efficiency or bottom 10% recombined spacers (n = 595 targets). The loxP bases are colored in teal. (G) Spacer specificity profile of Cre generated from the recombination rates of all target library variants. The heatmap color, and corresponding fold change in each tile, represent the effect of the base change at each position on Cre recombination relative to Cre/loxP recombination. The fold change is calculated from the binomial GLM coefficients. The canonical loxP bases are outlined in black for each base position in the spacer.
Initial investigations in the 1980s regarding the role of the spacer sequence introduced the concept that sequence identity between spacers in each of the target substrates is crucial for recombination (15). These studies were corroborated when efficient recombination was shown to occur between a variety of noncanonical spacers providing the spacer sequences are matching (16). The requirement for identical spacer sequences was further explained as a necessity during strand exchange to facilitate effective ligation of the cleaved strand to its complementary strand (17). Much of our current understanding of lox site preference have been inferred through a series of crystal structures of Cre bound to loxP (2,17–20). Protein–DNA interfaces reveal specific residues crucial for half-site recognition, although, direct base contacts between the protein's amino acid side chains and the substrate spacer region are minimal. Nevertheless, the spacer region plays a pivotal role during recombination catalysis, as it directs the order of strand exchange (2,17–20).
Investigation of the spacer through experimental methods can be challenging due to the repetitive nature of the target sequences. In order to test mutations for matching target sites, each unique sequence must be individually cloned to maintain the repeated targets, which can be time consuming. Alternatively, existing experimental methods using randomized libraries for high-throughput quantification of recombination have been successful in determining half-site recombination requirements and mapping half-site specificity profiles, but are limited to heterologous sites due to the randomization (16,21). Therefore, to study the role of spacer sequences during Cre-mediated recombination, we developed a method to comprehensively quantify recombination efficiencies across a large number of predefined target sequences. We then applied this method with a library of 6000 different loxP-like sites where identity was maintained between the two spacers. Cre demonstrated efficient recombination with 84% of the spacer sequences within the target site library. To assess the feasibility of reprogramming recombinase spacer specificity, we selected three targets with spacer sequences inefficiently recombined by Cre to evolve three Cre-derived recombinase libraries for increased activity. Analysis of recombinase variants from these libraries showed that selectivity of the spacer sequence can indeed be altered. Through comparative screens and molecular dynamics (MD) simulations, we evaluate spacer preferences and structural differences of Cre and an evolved spacer-specific recombinase, denoted RecS3. Our work highlights the ability to leverage spacer specificity to enhance the recombination properties of tailor-made recombinase systems.
Materials and methods
Plasmid construction
The target site library plasmid backbone (pGG) was engineered from the previously described pEVO plasmid modified to replace the pEVO BglII sites with two BbsI restriction sites facing outward (Supplementary Figure S1A). BbsI restriction site (downstream ‘left’ site) designed with the following sequence: 5′-ACACCGGGTCTTC-3′ leaving a GTGG overhang. The BbsI restriction site (upstream ‘right’ site) designed with the following sequence: 5′-GAAGACCTGTTTA-3′ leaving a GTTT overhang. The recombinase to be assayed was cloned into the pGG vector using BsrGI-HF and SbfI (NEB) restriction enzymes. Expression of the recombinase is controlled by an l-arabinose inducible promoter system (araBAD). The target site library was assembled to pGG-recombinase destination vector (Supplementary Figure S1A) following manufacture recommended protocol for setup of type IIs restriction digestion and ligation reaction (22) using T4 DNA Ligase and Type IIS restriction enzyme BbsI (Thermo Scientific).
The evolution plasmids pEVO-loxSE1, pEVO-loxSE2 and pEVO-loxSE3 were generated from the previously described pEVO-loxP plasmid (7,23) (Supplementary Figure S1A) modified to replace the loxP target site with the evolution target sites inserted at the restriction sites PciI and BglII (NEB; Supplementary Figure S1A).
Plasmid-based recombinase activity assay
Successful recombination events result in excision of the DNA flanked by the lox sites. Therefore, to assay the recombination efficiency of either individual recombinase variants or recombinase libraries a simple restriction digest is used to compare plasmid sizes in the sample (3–7) (Supplementary Figure S1B). The recombinase or recombinase library is assembled to either the pEVO or pGG vector containing the target site or library of target sites to be tested. The recombinase or recombinase library is cloned into the vector between the restriction sites BsrGI (NEB) and XbaI (NEB), downstream of the arabinose-inducible araBAD promoter (pBAD) (Supplementary Figure S1A). The assembled plasmids are transformed via electroporation into electrocompetent E. coli X-L1 Blue strain, recovered in SOC medium at 37°C shaking (700 rpm) for 1 h. Cells are plated onto LB-Cm (30 μg/ml chloramphenicol) agar to verify that the ligation was successful and also inoculated in 6ml LB-Cm (15 μg/ml chloramphenicol) containing the desired dose of l-arabinose to induce the expression of the recombinase overnight. The l-arabinose concentrations of 1, 10 and 100 μg/ml were used for the recombinase activity screens. Plasmids were purified from the overnight cultures following the protocol of the Qiagen spin isolation kit (Qiagen Inc.). To visually compare the recombined and non-recombined plasmids in the sample, the purified plasmid DNA is digested using BsrGI-HF and XbaI (NEB; for assaying pEVO) or using BsrGI-HF and PacI (NEB; for assaying pGG) (Supplementary Figure S1B). The digest linearizes the DNA to easily compare the smaller recombined plasmids to the non-recombined plasmids in the sample with gel electrophoresis. Recombination efficiencies were calculated by the ratio of band intensities for each well using GelAnalyzer v23.1 for image processing (GelAnalyzer 23.1, available at www.gelanalyzer.com, by Istvan Lazar Jr., PhD and Istvan Lazar Sr., PhD, CSc). Recombination was calculated by dividing the recombined band intensity by the recombined and the non-recombined band intensity. The calculated recombination was plotted in R 4.0.3 (R Core Team, 2018) with dplyr v1.0.7 (https://dplyr.tidyverse.org/) and visualized with ggplot2 v3.3.5 (https://ggplot2.tidyverse.org/). Bacterial test digests were done in triplicates (n = 3).
Library construction
The library was generated using silicon-based DNA synthesis from Twist Bioscience, providing full control over the sequence design for each oligo in the library. Each oligo was predefined, allowing the unique target site to be encoded twice on the same oligo. To reduce costs, synthesis time, and potential PCR bias during downstream library preparation, we opted to keep the oligo length short. Consequently, each oligo encoded a short distance between the two target sequences.
Previous studies have shown that the efficiency of Cre-mediated intramolecular recombination between two directly repeated loxP sites, depends on the distance between the sites, with a minimum distance of 82 bp required for efficient recombination (24). We considered this when optimizing the screen, comparing recombination of Cre/loxP for different distances between the lox sites: 24 and 700 bp. We also assessed how the recombination rate at these distances changed with increasing concentrations of Cre by using L-arabinose concentrations of 0, 1, 10 and 100 μg/ml in the E. coli plasmid-based excision assay (Supplementary Figure S2A). In agreement with previous findings, our results show that a length of 700 bp between the two lox sites was more efficient than the shorter length of 24 bp. However, the trends of increasing recombination with increasing concentrations of Cre are similar for both lengths. Additionally, sequencing of plasmid DNA from both recombined and non-recombined colonies confirmed that recombination occurred as predicted, with only one loxP site and the full stuffer sequence precisely excised from the DNA of recombined colonies (Supplementary Figure S2B). Excision of DNA between lox sites located a short distance from one another likely occurs in two steps: first, intermolecular integration followed by intramolecular excision.
The target site library for the activity screen comprised 6000 oligonucleotides ordered as oligo pools from Twist Bioscience (Supplementary Table S1). Each oligo sequence is a predefined length of 120nt encoding two copies of the unique 34nt lox-like target sequence (Supplementary Figure S2C). Between the two targets was a 24nt region containing two restriction sites (NdeI and PacI; NEB) used for quality control. Flanking the target sites were 14nt primer binding sites (Supplementary Table S2, primers P1, P2) to amplify the synthesized ssDNA pool for dsDNA. The design of the libraries excluded all restriction enzymes used during the assay and downstream analysis.
Rational design of loxP-like spacer library: In order to understand how changes in the spacer region impact Cre recombination, the library was built on the core 8bp spacer region while the flanking loxP half-sites were kept consistent (Figure 1B, Supplementary Table S1). Because we wanted to fully examine the spacer characteristics contributing to specificity, we designed the libraries to contain target sites with homologous spacers; i.e. each target site variant contains the unique mutation in both spacers of the complex. Another design feature to consider was the size of the library. A library containing all spacer base combinations would consist of 65536 (48) different targets. Instead of having all spacer possibilities, we took a more rational approach to minimize the size of the library, allowing for more reads per target to achieve a higher confidence interval per screen. Because the first and last bases have been reported to influence strand exchange (14,25,26), we mainly designed the spacers with the 6 core nucleotides altered, which we annotate as ANNNNNNC where N refers to the altered position. Additionally, eight different combinations of the four core sequences with the last two and first two spacer nucleotides altered were included in the library (NNGTATNN, NNATACNN, NNCTAANN, NNCTAGNN, NNAATTNN, NNTTAANN, NNGGCCNN, NNCCGGNN). From these combinations, all fully symmetric spacers (e.g. AAAATTTT) were excluded from the library.
Target site library preparation for deep-sequencing
Experiments were conducted in E. coli with Cre under the expression of an inducible promoter for a tighter control of the recombinase/target site exposure (Supplementary Figure S1A and S2D). This gives us the option to adjust the expression of Cre to better distinguish functional diversity due to variation in spacer sequences. The assembled pGG-Recombinase-Library was transformed to E. coli XL1-Blue (Agilent) and the expression of the recombinase was induced with different concentrations of L-arabinose (0, 1, 10 and100 μg/ml l-arabinose; Sigma Aldrich). After growing the cells for 14–16 h in 50 ml LB and in the presence of chloramphenicol (30 mg/ml), plasmid DNA was extracted using the GeneJET Plasmid Miniprep Kit (ThermoFisher). The extracted plasmid DNA was used as a template to amplify over the target sites using primers P3 and P4 (Supplementary Table S2) adding the P5 and P7 indexes for Illumina paired-end sequencing. The PCR was performed using a high-fidelity polymerase (Herculase II Fusion DNA Polymerase, Agilent) carried out with ten PCR cycles. The kit manufacturer protocol was followed using an initial denaturing temperature of 95°C for 3 min and 40 s per cycle, an annealing temperature of 55°C, and an extension temperature of 72°C for 1 minute per cycle. The resulting amplicon length is 220 bp for the non-recombined targets and 162 bp for the recombined targets. The amplicons were purified on a column using the Isolate II PCR and Gel Kit (Bioline) and quantified using the Qubit dsDNA HS Kit (ThermoFisher) according to the manufacturer's instructions. The sequencing of samples was performed using paired-end reads of 150 bases on the Illumina NovaSeq 6000.
Validation and quality control of screen
Before analysis of the results, we confirmed the quality of the screen. Both the Cre and RecS3 activity screen recombination outcomes were quantified with high throughput sequencing of the targets at an average sequencing depth of >1000× per target (Supplementary Figures S3A, S6A). The screen was done in triplicates to determine the consistency of the results. Pearson's correlation coefficient (R) was measured for each replicate and all target sites in the library were plotted to show relationship between the replicate and the pooled replicates recombination rates. For both the Cre and RecS3 activity screens, the value of R ranges between 0.87 and 0.99 indicating a positive linear relationship, and consistent recombination levels among the triplicates (Supplementary Figures S3B, S6B).
Additionally, the activities calculated from the target site screen were validated for a selection of spacers ranging from low to high Cre activity. We compared activities from the screen to activities quantified in triplicates from plasmid-based recombination activity assay. The mean screen activity and mean plasmid-based recombination activity were calculated for the triplicates and plotted to show the relationship between activities. A Pearson's correlation coefficient was computed (R = 0.97; Supplementary Figure S3C) carried out in R (version 4.3.2) showing high correlation between the two assays.
Data analysis
Illumina sequencing data is processed using Cutadapt (27) and R version 4.3.2 (28) to convert to count matrices. Targets with fewer than 100 reads were discarded. Because of symmetry of the recombination reaction, alignment of the spacer either from the top or bottom strand of DNA is arbitrary, therefore, the strand with the highest identity to the wild-type loxP or on-target sequence was considered. To facilitate a comparative analysis between distinct screens, we computed the activity ratio for each dataset by dividing the recombination rate of individual target sites by the highest recombination rate within each dataset.
Sequence logos
Logos were generated for subgroups of target sequences to compare conserved bases associated with the subgroup. Although the generated sequence logos were normalized to the number of sequences, the subgroups that were compared always consisted of the same number of sequences (e.g. subgroup A has n = 10 sequences and subgroup B has n = 10 sequences) in order to avoid effects from sample size.
At each position in the alignment of spacer sequences, the logo plot represents the relative frequency of each base, with the height of each base proportional to its relative frequency (Figures 1E, F and 3B, C). The plots highlight bases that have an observed frequency higher than their expected frequency (frequency in library). Standard logo base height represents observed frequencies p=(pA,pC,pG,pT) compared with a uniform background, q=(0.25,0.25,0.25,0.25). Logos calculated from library subgroups (i.e. top 10% recombined or bottom 10% recombined) were normalized to the library frequency. This normalization is referred to as relative frequency. The relative frequencies were calculated by first calculating the base frequencies for each position in the subgroup (freq. subgroup) and in the library (freq. library).
Figure 3.

Target site library activity screen of RecS3. (A) Comparison of Cre spacer library activity (x-axis) and RecS3 spacer library activity (y-axis). Each dot represents a unique spacer in the library. The activity ratio for each recombinase was calculated by dividing the activity by the highest activity in the respective screen. Activity ratio is plotted on a square root scale for both the x- and y-axis. (B, C) Sequence logos depicting spacer preference of RecS3. The base height in each logo is calculated by relative frequency in the given subpopulation. The base frequency is normalized to library representation. The logo in B is calculated from a subpopulation of spacer sequences with the highest recombination efficiency or top 10% recombined spacers (n = 595 targets). The logo in C is calculated from a subpopulation of spacer sequences with the lowest recombination efficiency or bottom 10% recombined spacers (n = 595 targets). The loxSE3 bases are colored in teal. (D) Spacer specificity profile of RecS3 generated from the recombination rates of all spacer library variants. The heatmap color, and corresponding fold change in each tile, represent the effect of the base change at each position on RecS3 recombination relative to RecS3/loxSE3 recombination. The fold change is calculated from the binomial GLM coefficients. The loxSE3 bases are outlined in black for each base position in the spacer.
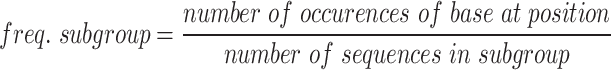 |
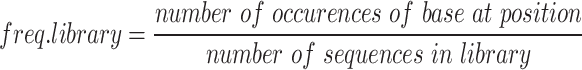 |
The calculated frequencies were then used for relative frequency.
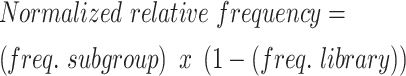 |
The logos were plotted with R package ggseqlogo (29).
Generalized linear model
Effects of individual base changes on recombination levels of different target site sequences were estimated using a logistic regression model, employing the glm function with binomial distribution, both implemented in the stats package for R (28). Bases at each position of spacer sequences, as well as arabinose concentration, were used as independent variables, and log odds of recombination were used as the dependent variable. Visual representation of the model was performed by generating a heatmap of its coefficients for different base-position combinations (Figure 1G and Figure 3D). For ease of interpretation, the coefficients were transformed to fold-change scale relative to on-target (loxP or loxSE3) recombination. Fold change was calculated by exponentiating positive coefficients and taking the negative reciprocal of the exponentiated coefficients for negative coefficients. When referencing the coefficients in the results section, the term ‘odds of recombination’ or ‘chance of recombination’ is used to describe the probability of a recombination event happening divided by the probability of the recombination event not happening.
Substrate-linked directed evolution
New recombinases were evolved using the experimental principles as described previously (3–6) (Figure 2B). To begin the evolution for increased activity on the target sites, a library of Cre mutants was generated by an error-prone PCR. Evolution for each site was done in parallel where the library was subjected to seven rounds of directed evolution and selected for improved activity on the given site. Positive selection pressure for activity on the novel spacer sequences loxSE1, loxSE2 or loxSE3 was achieved through a modified method of substrate-linked directed evolution (Figure 2B). Each cycle of evolution involves the diversification of the libraries through error prone PCR (MyTaq DNA Polymerase, Bioline) and selection of the variants for the desired activity on the presented target site.
Figure 2.

Evolution of Cre for improved activity on unfavorable spacer sequences. (A) Target site sequences used for evolution aligned to loxP. The spacer base sequence of loxP is colored in teal. Base mismatches from loxP are colored in yellow for the loxSE1, loxSE2 and loxSE3 spacer sequences (written in 5′ → 3′ direction). (B) Evolution schematic. i) Recombinase library assembly to evolution vector. ii) Transformation of assembled vectors to E. coli for induction of recombinase expression. iii) Vector purification and selection for active recombinase variants by linearization of non-recombined vectors carrying the inactive recombinase variant. iv) Error-prone amplification with primers p5 and p6 to insert new mutations to the active recombinase library. Only the non-linearized recombined vectors result in an amplification product and are carried on to the next cycle of evolution. (C) The plasmid-based restriction assays show the activities of Cre, LSE1, LSE2 and LSE3 starting libraries (cycle 1) and ending libraries (cycle 11) on target sites loxP, loxSE1, loxSE2 and loxSE3. Two triangles indicate the non-recombined substrate (5kb) and one triangle indicates the recombined substrate (4.2 kb). (D) Violin plots showing the distribution of recombination (%) (y-axis) for each recombinase variant in the final evolved libraries (after 11 rounds of evolution) and Cre activity on target sites loxP, loxSE1, loxSE2 and loxSE3 (x-axis). The width of each curve in the violin plot corresponds to the approximate frequency of recombinase variants with the equivalent recombination (%). Cre replicates are shown in dark blue (n = 25), the library evolved for loxSE1 (LSE1, n = 1782) is shown in yellow, the library evolved for loxSE2 (LSE2, n = 202) is shown in red and the library evolved for loxSE3 (LSE3, n = 2303) is shown in teal. (E) Scatterplots showing the recombination (%) of each library of their evolved target site along the y-axis compared to recombination (%) of loxP along the x-axis. Each dot represents a unique recombinase and the color indicates the library of the recombinase. The upper left gray square in each plot highlights recombinases specific for the evolved site. The recombinases that were further analyzed are labeled: RecS1, RecS2 and RecS3. (F) Plasmid-based restriction assays showing Cre, RecS1, RecS2 and RecS3 activity for loxP and the evolved target sites loxSE1, loxSE2 and loxSE3. M = GeneRuler DNA Ladder Mix 10kb. The bar plots show the mean recombination (%) of the activities quantified from the band intensities of the plasmid-based restriction assay. The restriction assays were done in triplicates (n = 3). The points represent each individual replicate recombination (%).
The diversified libraries were first cloned into the pEVO containing the target site with the restriction sites of BsrGI and XbaI (purchased from NEB). The vector was then transformed into electrocompetent XL1-Blue E. coli and grown overnight in LB/arabinose to induce recombinase expression. To perform selection for recombination of the novel spacer sequence, the purified plasmid was digested with enzymes NdeI and AvrII (NEB) to linearize all non-recombined variants. A PCR was then performed with primers P5 and P6 to amplify only the clones that performed recombination (Figure 2B). Recombination efficiency was monitored through the plasmid-based recombination assay (Supplementary Figure S1B).
Once activity on the given site was achieved, the libraries were enriched for active variants with three rounds of low induction and high-fidelity amplification (Herculase II Fusion DNA Polymerase, Agilent) resulting in increased activity of all libraries compared to Crewt (Figure 2C). The DEQseq method (30) was then used for high-throughput evolution of the activity of individual recombinase variants from each library. The three libraries of active variants were barcoded via amplification, pooled together and then cloned into vectors for the DEQseq protocol. From the pool, around 5000 variants were randomly selected and individual activity was quantified for all four target sites. Although only a portion of variants from each library were assayed, this method provides sequencing and activity of individual variants at multiple target sites, providing us with information, not only on common mutational patterns and epistatic interactions but also a ranking system for activity-determining residue positions.
Further analysis of evolved recombinase variants was done with recombinases selected based on their activity across all four target sites and number of mismatches to Cre. Specific recombinases were defined as those with high on-target activity (the target site where the recombinase was selected for activity during evolution), low activity of the other three targets and high sequence identity to Crewt (amino acid sequence identity greater than 95% to Crewt).
Molecular modelling and dynamics simulations
The three-dimensional (3D) structure of the synaptic Cre/loxP tetrameric complex (PDB ID 3C29, 2.2 Å, (31)) was chosen for modeling based on its high resolution in comparison to other Cre/loxP structures available in the Protein Data Bank. The loxP structure was prepared for simulation purposes (i.e. A1P5’ to A5’) as previously described (32). The structure of loxSE3 was modelled by introducing base-pair mutations on loxPA5’(TS)A1P using the Molecular Operating Environment (MOE, 2023, Chemical Computing Group, Canada) and its DNA builder module operating on double-stranded B-helix with a repack clustering cutoff of 1 Å. PyMOL (Version 2.4, Schrödinger, LLC) was used to introduce the corresponding mutations on Crewt to obtain the mutants RecS3, RecS3S320I and CreI320S as previously described (32). The structure of the complexes of the Cre mutants with loxP and with loxSE3, and the complex of Crewt with loxSE3 were refined by molecular dynamics (MD) simulations in AMBER20 (Case, D. A. et al. (2020) AMBER 2020, University of California, San Francisco). The ff14SB and bsc1 force fields were used for protein and DNA, respectively, as previously described (32). MD trajectories were visualized with VMD (33) and evaluated in terms of B-factors, angles and intermolecular H-bonds by using the CPPTRAJ module implemented in AMBER. A H-bond occupancy of > 10% with a distance acceptor-donor cutoff of 3.5 Å and a 120° angle were taken as criterion for dynamic H-bond formation in the last 100 ns of each MD simulation. The intrinsic flexibility of loxP and loxSE3 as free B-DNA was determined using the TRX scale (34). The DNA structures and their helicoidal parameters were analyzed with the Curves + algorithm and Canal program (35). Origin2023 (OriginPro, Version 2023 (2023) OriginLab, Northampton, MA; http://www.originlab.com) was used for MD-based statistical analysis and the preparation of MD- and DNA-analysis based figures. RMSDCα between MD-based average structures were calculated in PyMOL. All structural figures were created with PyMOL 2.4.
Results
High-throughput analysis of Cre activity on loxP-spacer library
To develop a comprehensive, high-throughput approach for profiling activity of Cre on loxP sites with matching mutant spacers, we designed libraries of oligonucleotides, each 120nt long, encoding two identical mutant lox sites (Figure 1B, Supplementary Figure S2C). Each oligonucleotide in the library was designed with the flanking 13bp half-sites held constant as found in the wild-type loxP sequence while mutations were systematically introduced to the core 8bp spacer region resulting in a target site library of 6000 distinct yet matching spacers (Supplementary Table S1). The library was then cloned to an expression vector encoding for Cre (Supplementary Figure S1A) and subsequently transformed to E. coli for recombinase expression (Figure 1B, Supplementary Figure S2D). To achieve precise quantification of recombination events with high sensitivity, we conducted high-throughput sequencing of the targets after PCR amplification, ensuring an average sequencing depth > 900x coverage per target (Supplementary Figure S3A). Between biological replicates, the quantified recombination of library targets was consistent (median Pearson's R = 0.98, Supplementary Figure S3B), demonstrating the reliability of the approach. These results suggest the data are reproducible, thorough and at a comprehensive scale not previously assessed for recombinase/target activity. Quantification of Cre recombination at each target provided the means to investigate potential sequence preferences.
Systematic characterization of Cre recombinase sequence requirements for functional lox spacers
Consistent with the current literature (15,16,36), we observed that 84% of the matching mutant spacers were efficiently recombined by Cre (Figure 1C). In order to compare the screen results to Crewt/loxP efficiency, we defined efficient recombination as recombination within a range of ±25% wild-type loxP recombination of 87%. With this threshold, 16% of the target sites with mutant spacers were not efficiently recombined by Cre, with some sites showing <5% recombination. These findings underscore that efficient recombination is not guaranteed by spacer identity, in line with previous observations (37,38).
To investigate the results in more detail, we first compared how number of mismatches from the loxP spacer sequence impacted the overall recombination (Figure 1D). The screen results revealed a correlation between an increase in the number of mismatches from the canonical loxP sequence and a subsequent reduction in the potential for Cre recombinase activity. Specifically, target sites where both spacers had 7 or 8 mismatches from loxP exhibited a pronounced decrease in recombination efficiency. Of the targets with 7 or 8 mismatches, 86.7% and 100%, respectively, were inefficiently recombined by Cre (indicated by the shaded region, Figure 1D). Of the spacers with 6, 5, 4 and 3 mismatches, only 30%, 15.8%, 9.4% and 7.6%, respectively, were inefficiently recombined. Nevertheless, the number of mismatches was not the only determinant of recombination efficiency. For instance, the target site with both spacers of sequence 5′-CAGTATTC-3′ (bold font indicating mismatches from loxP), contains only 3 mismatches from the loxP spacer sequence (5′-ATGTATGC-3′) and showed a recombination efficiency below 10%. On the other hand, spacer sequences containing 6–7 mutations from loxP (i.e. 5′-AATGTGTC-3′ and 5′-TGAATTCG-3′), were efficiently recombined by Cre (88% and 78% recombined, respectively). These results suggest that, indeed, Cre reactions accommodate a wide range of spacer sequences for successful recombination. However, there is considerable variation in recombination efficiency across these sequences. This should be taken into consideration when selecting optimal target sites for reprogramming SSRs where, typically, parameters for target site selection focus predominantly on the half-sites, overlooking the specific DNA sequence of the spacer (39).
Cre recombination mismatch sensitivity highlights the sequence dependence of recombination, but it does not detail specific requirements for a functional spacer sequence. Therefore, we further analyzed the relationship between recombination and target sequence composition, as well as the context of base changes. To achieve this, we generated sequence logos for the top 10% recombined spacer sequences (n = 595) and the bottom 10% recombined sequences (n = 595) to visualize conserved patterns in each group (Figure 1E, F). The logo of the top 10% of recombined spacer sequences demonstrates an enrichment of canonical bases A4’, G3 and C4, indicating a preference for the canonical loxP sequence in the flanking regions of the spacers for efficient recombination. Conversely, the logo of the bottom 10% of recombined spacer sequences shows an enrichment of base T3, suggesting that this base change negatively impacts efficient Cre recombination. Collectively, these findings reveal that base identity is an important determinant of efficient Cre-mediated recombination.
To classify the relationship between base substitutions at each position and recombination, we employed a binomial generalized linear model (GLM) using recombination data from the comprehensive loxP spacer mutant library (Figure 1G). We modeled the fold change in recombination rates resulting from single base changes to construct an activity profile for Cre. Analysis of the profile showed a preference for canonical loxP bases at positions A4’, G3 and C4 as illustrated in the logos (Figure 1E, F). However, not all base changes at these positions have the same impact on recombination. For example, a base change from A–G at position 4′ or C-T at position 4 demonstrate a higher chance of recombination compared to other base changes at these sites (10,25,36). These variations can be attributed to the type of mismatch. Generally, base transitions are more likely to be recombined by Cre than base transversions. Notably, a base transversion of G-T at position 3 presents the most significant reduction in successful recombination (–3.8; Figure 1G) compared to any other base change. These results underscore the complex interplay between spacer sequence and efficient recombination. While sequence-based characteristics such as homology and mismatch count to loxP are generally indicative of efficient Cre recombination, the position and type of base change are also critical factors. These results suggest spacer sequence selectivity for efficient recombination in Cre. However, the lack of defined amino acid contacts to bases in the spacer (2) (i.e. indirect readout) makes it necessary to take into account how sequence variations may imply other possible contributing factors that may help to decipher the molecular mechanisms behind spacer selectivity (e.g. influence on the DNA structure; vide infra).
Directed evolution of Cre for altered spacer sequence preference
To test whether we could overcome the restricted activity of Cre, we performed substrate-linked directed evolution (SLiDE) (4) to generated Cre mutants with increased activity on spacer sequences inefficiently recombined by Cre (Figure 1C). Three unique target sites, loxSE1, loxSE2 and loxSE3 (Figure 2A), were selected from the mutant spacer screen based on their low recombination rates compared to wild-type loxP. Three libraries were generated (LSE1, LSE2 and LSE3) by an error prone PCR of Cre followed by testing of the libraries on each of the sites (Figure 2B, C). 11 cycles of SLiDE were performed in parallel for each library on the respective target sequences for increased activity (Figure 2B). For all three libraries we detected increased band intensities representative of the recombined plasmid product, indicating the enrichment of Cre variants with increased activity (Figure 2C). To quantitatively investigate a large number of individual clones in the library, we performed DNA Editing Quantification Sequencing (DEQSeq), a high-throughput Nanopore sequencing method that enables the characterization of thousands of DNA editing enzyme variants on multiple target sites (30). We selected a total of ∼5000 random variants from the three libraries and quantified recombination activity of each of the variants on all four sites (loxP, loxSE1, loxSE2 and loxSE3) at an average sequencing depth of >1000× reads per variant. Experiments using conventional agarose gel quantifications confirmed the validity of the approach (Supplementary Figure S4A, B).
By comparing Cre and the library variants distribution of activities for loxP, loxSE1, loxSE2 and loxSE3, a clear increase in activity of many of the recombinase variants on their respective evolved target site was seen across all libraries while maintaining high activity on the parental loxP wild-type spacer (Figure 2D). Because the selection criteria for evolution was activity and not specificity, many variants emerged with the capacity of efficiently recombining all four spacer sequences. Mutational analysis of recombinase variants with increased activity on all sites showed a frequently occurring mutation at position 86 (K86N) compared to the other variants (2.5-fold more frequent; Supplementary Figure S5A). Across the spectrum of mutations observed at position 86 within the libraries, variants featuring an Asparagine demonstrated higher activity across all four target sites (Supplementary Figure S5B). Furthermore, the K86N mutation has previously been identified in designer recombinases evolved for targeting new substrates (including a new spacer sequence) (3,8). Residue K86 was further described as a key component for novel target recognition and specificity (9). These results suggest that the K86N mutation potentially fosters enhanced recombination efficiency on diverse matching spacer sequences.
Surprisingly, although many recombinase variants evolved an overall increase of activity on all four target sites, <1% of the recombinase variants became unproductive on the parental wild-type loxP spacer (Figure 2E). This result suggested that, although the screen was not designed to produce variants that selectively prefer to recombine a certain spacer sequence, clones had emerged that had lost their ability to efficiently recombine the wild-type loxP spacer, while gaining activity on the mutant spacer. Of the evolved spacer-specific recombinases, we selected three examples (RecS1, RecS2 and RecS3; Figure 2E) to further compare their activity across all targets. Testing the three recombinases in a plasmid-based assay indeed revealed that these enzymes preferentially recombine their selected spacer sequences (Figure 2F), indicating that Cre variants can be generated with spacer sequence selectivity. Of the three variants, RecS3 was the most interesting due to the drastic shift in spacer activity to recombine only the loxSE3 target without prominent activity on any of the other sites (Figure 2F). These results establish the programmability of Cre recombinase to selectively recombine sequences with different spacers.
High-throughput analysis of RecS3 activity on loxP-spacer library
To comprehensively determine the spacer requirements for functional RecS3 recombination (selected for target site loxSE3; Figure 2A) and how those requirements differ from Cre, we applied the same target recombination assay as used with Cre by quantifying RecS3 activity on the 6000 loxP-like spacer variants (Figure 3A, quality control shown in Supplementary Figure S6A, B). Plotting the activities of RecS3 compared to Cre on all target sites in the library (Figure 3A) shows that Cre recombines a wider range of spacer sequences more efficiently than RecS3 (indicated by the density of target sites in the bottom right of the graph; Figure 3A). Nevertheless, RecS3 efficiently recombines numerous spacer sequences, including some that are inefficiently recombined by Cre (upper left quadrant of the graph; Figure 3A). These results provide a direct comparison of spacer sequence selectivity of RecS3 and Cre.
To visualize the selective recombination preferences of RecS3, the conserved characteristics leading to efficient recombination were plotted as logos of the top 10% sequences with the highest activity and the bottom 10% sequences with the lowest activity (Figure 3B, C). Comparing these logos to the Cre logos (Figure 1E, F), a clear difference in spacer sequence preference can be deduced for the two recombinases. In the context of RecS3-mediated recombination, the analysis of base frequencies among the top recombined sequences revealed a pronounced selectivity, with a base frequency exceeding 40%, for loxSE3 at positions A4', G2, T3, and C4. Intriguingly, an additional observation underscores a notable preference for a non-loxSE3 base, T2'. Conversely, examination of base frequencies in the least recombined sequences highlighted a heightened occurrence of bases G2', T1', A1 and T2, all of which are integral components of the wild-type loxP sequence. This notable prevalence within the inefficiently recombined sequences of specific bases in the wild-type loxP sequence may potentially explain the observed reduction in RecS3 activity for loxP. Collectively, these findings emphasize the preference of RecS3 towards the loxSE3 site, particularly underscoring the significance of A4' and G2 as pivotal determinants in fostering efficient recombination. To identify patterns that make certain sequences more favorable for one recombinase and not the other, logos were generated from sequences efficiently recombined by RecS3 and not by Cre (n = 10, top left), sequences recombined efficiently by Cre and not RecS3 (n = 10, bottom right), and sequences efficiently recombined by both recombinases (n = 10; top right, Supplementary Figure S7A). This analysis revealed that spacer position 3 is most prominent, where RecS3 favors base T3 and Cre favors base G3. In contrast, when analyzing sequences that are efficiently recombined by both Cre and RecS3, base changes to A3 and T3’ are most tolerated by both enzymes.
We also evaluated the RecS3 spacer specificity by constructing a profile with recombination rates from all spacer substrates (same application as done with Cre, see GLM model formulation in Methods for calculation, Figure 3D). As observed in the spacer profile of Cre, RecS3 was more permissive to base transitions than base transversions, most notably, the base transversions of C-G2’, decreasing the odds of recombination by 3.5-fold, and the base transversion of A-C4’, decreasing the odds of recombination by 4.4-fold. Additionally, RecS3 displayed a high sensitivity to all base changes at G2. Comparing the specificity profiles of Cre and RecS3 (Supplementary Figure S7B), both recombinases were similarly sensitive to base changes in positions 3′ and 3. In contrast, RecS3 exhibited heightened sensitivity to alterations in positions 4′ and 2, accompanied by a diminished sensitivity to base changes at position 4. A comparative analysis of the two profiles underscored the increased specificity of RecS3, emphasizing the critical roles of bases G2 and T3 in RecS3 recombination, observations not evident in Cre. These findings emphasize the unique and intrinsic specificity shift characterizing RecS3, setting it apart from Cre.
Molecular dissection of specificity switch
RecS3 contains eight mutations compared to Crewt: L5P, V7A, K132E, G246D, T316I, N317T, I320S and N323Y (Supplementary Figure S8). To assess the specific impact of individual mutations on the overall activity of loxP and loxSE3, eight RecS3 mutants were generated, where each contained a single mutation back to the Crewt residue. Among the RecS3 mutants examined, half of the single residue reversion mutants to Crewt (RecS3: P5L, E132K, D246G and T317N) individually demonstrated no significant alteration in the overall activity concerning loxSE3 and loxP (Figure 4A). This is most likely due to the passive nature of these mutations acquired during evolution or that these residue changes are acting in concert with the other mutations. Interestingly, the mutants RecS3A7V and RecS3S320I showed a significant loss of activity on loxSE3 (Figure 4A). Moreover, RecS3S320I significantly increased the activity on loxP, implying that S320I plays a key role in the selectivity for different spacer sequences. In contrast, RecS3A7V showed diminished activity on both loxP and loxSE3, suggesting a role of this mutation for overall recombination function, possibly related to the stability of the recombinase (40). We also analyzed the individual RecS3 mutations in Cre. Interestingly, CreI320S resulted in the strongest decrease in activity on loxP, while an increased activity on loxSE3 was observed (Figure 4B). These results indicate that I320S plays an important role in spacer selectivity.
Figure 4.

Analysis of mutations in RecS3. (A) Restriction assay of RecS3 and indicated RecS3 variants. LoxP and loxSE3 activity of Crewt, RecS3 and eight RecS3 mutants with single residue mutated back to Crewt are shown. Restriction assay displays amount of non-recombined plasmid (indicated by two triangles) and recombined plasmids (indicate by one triangle) in the sample. Bottom graphs plot the correlated mean recombination (%) of the above gel image calculated from gel band intensities. RecS3 recombination in teal and Cre recombination in dark gray. Bar represents mean of n = 3 replicates. (B) Mutational analysis of Cre with RecS3 mutations (analogous to panel A). P values are reported from unpaired two-tailed t-test ns: P > 0.05, *: P < 0.05, **: P < 0.01, ***: P < 0.001, ****: P < 0.0001.
Molecular modelling and dynamics simulations: analysis of the molecular basis for activity and target selectivity
In order to shed light on the molecular mechanisms behind recombination activity and target selectivity in Cre and RecS3 and the most prominent mutation at position 320 with respect to loxP and loxSE3, we built 3D molecular models of their respective complexes: CreI320S/loxP, RecS3/loxP, RecS3S320I/loxP, Crewt/loxSE3, CreI320S/loxSE3, RecS3/loxSE3 and RecS3S320I/loxSE3 (Figure 2A). These models were based on the high resolution synaptic Cre/loxP complex PDB ID 3C29 (see Methods). In this structure, the Cre active (A) monomer exhibits the catalytic residues positioned for top strand (TS) cleavage like in other crystallographic structures available in the Protein Data Bank (41). For the modelling and analysis, only the dimer unit was considered (i.e. two Cre monomers—one active and the other inactive- in complex with one molecule of the corresponding DNA target site (i.e. loxP or loxSE3)). This decision was based on recent work reporting on the Cre/loxP dimeric complex as key functional unit for productive synapsis (10) and own previous MD-based studies using the same constellation (32).
MD simulations of the investigated Cre mutants and the wild-type system and the subsequent comparative H-bond analysis at the catalytic site revealed clear variations in DNA recognition by the catalytic residue Y324 (Figure 5A). In the RecS3/loxP complex, Y324 of the inactive (I) monomer (Y324(I)) exhibits a striking shift with respect to the positioning observed in Crewt (Figure 5A, B) resulting in the loss of the interaction with the DNA phosphate backbone (p) at base G4 of the bottom strand (G4(BS)). Furthermore, no interaction is observed between the catalysis-relevant residue K201(I) and the sugar backbone of base T5(TS). In the context of the active (A) monomer, no interactions of RecS3 with pA5’(TS) and pA4’(TS) are detected. As a consequence, H289(A) is not optimally positioned to facilitate recombination compared to Cre (Supplementary Figure S9A, C). The observed configurational shift in Y324(I) together with the loss of interactions of critical residues for catalysis such as K201(I) and H289(A) could explain the diminished activity of RecS3 towards loxP. Conversely, in the context of the loxSE3 target site, residues Y324, K201 and H289 of RecS3 were found appropriately positioned in both the active and inactive monomers (Figure 5A, Supplementary Figures S10 and S11A). Notably, the recognition of loxSE3 by Crewt was predicted with Y324(I) shifted towards pA3(BS) instead of contacting pG4(BS). Interestingly, Y283(I) was observed establishing contacts with pG4 (BS). Moreover, no interactions with pA5’(BS) were observed, and the interactions of K201(A) with A4’(TS) as well as I320 and N317 with pC2’(TS) were not detected (Supplementary Figure S10A). These differences in the recognition pattern of loxSE3 by Crewt compared to RecS3 could explain why Crewt exhibits inefficiency as recombinase for loxSE3. The observed displacement of Y324(I) suggests its positioning for DNA cleavage, also in the inactive monomer. Recombination occurs through a stepwise cleavage process with synapsis formation (i.e. active tetrameric complex) involving two dimer complexes with cleavage preference for either the BS or TS (i.e. formation of a BS-BS or TS–TS synapsis resulting in a BS or TS first cleavage, respectively). Recent work analyzing pre-synaptic Cre/loxP dimers has suggested that the formation of a BS–TS complex would not be compatible with effective recombination activity (10,42).
Figure 5.

MD-based rationale for activity and selectivity. (A) Summary of MD-based analysis of loxP and loxSE3 recognition by wild-type Cre and mutant variants. The plot shows interactions observed in the last 100 ns of MD simulations between the DNA and Y324 in the active (A) and inactive (I) monomers. DNA numbering according to Figure 2A. (B) Detail of the MD-refined structure of the RecS3/loxP complex. (C) Detail of the MD-refined structure of the RecS3S320I/loxP complex. Active and inactive recombinase monomers are shown in pale and blue cartoons, respectively. Side chains of residues Y324 and 320 and DNA bases are labeled and shown in atom-colored sticks. The surface mesh denotes van der Waals contacts between protein residues at position 320 and DNA bases T3’(TS) and G4(BS). Black dashed indicate H-bonds. (D) Cartoon putty representation of RecS3 in complex with loxP. RecS3 is colored and rendered in accordance to RMSDCα values relative to Cre (as indicated by the gradient side bar). Helices K, L and M are labeled in orange and blue for active (A) and inactive (I) RecS3 monomers, respectively, and helix M(I) is highlighted with a pink square. LoxP is shown in gray ladder representation. (E) TRX score of selected DNA sequences calculated as a sum of individual dinucleotide TRX values according to Heddi et al. (34).
Next, we investigated the molecular basis accounting for the experimentally observed DNA target specificity, in particular the relevance of the residue at position 320 neighboring the catalytic Y324. Our theoretical models predicted a preference for isoleucine at position 320 when the target site is loxP. This preference arises from van der Waals contacts between the side chain of I320(A) and the sugar backbone and base T3’(TS), as observed in Crewt and RecS3S320I (Figure 5C, Supplementary Figure S9A, D). These interactions diminish when isoleucine is substituted by serine. Nevertheless, the recombination activity of RecS3S320I does not reach the levels of Crewt, which may arise from the lack of interactions of I320 with pG2’(TS) and of K201(A) with A4’(TS) (Supplementary Figure S9D). Of particular interest is the behavior of Y324(I) of CreI320S, which interacts with pC3(BS) and pG4(BS) of loxP (53% and 47% of the MD simulation time, respectively; Figure 5A, Supplementary Figures S9B and S11B) and with pA3(BS) and pG4(BS) of loxSE3 (37% and 61% of the MD simulation time, respectively). These interactions in both target sites might further compromise the catalytic activity of these recombinases. Furthermore, residue W315(A), which also supports recombination, does not recognize pT3’(TS) of loxP and pA3’(TS) of loxSE3. For RecS3, residue S320(A) establishes an H-bond with pC2’(TS) in loxSE3, as it is observed in CreI320S. In contrast, such interactions are not observed in the RecS3S320I/loxSE3 complex (Supplementary Figure S10).
The fact that several catalytic residues (i.e. H289, W315, Y324) and some of the mutations introduced in RecS3 with respect to Crewt (i.e. I316T, T317N, S320I, Y323N) are contained in the helices K, L and M of the recombinase, and that the recognition of these regions by the DNA could perhaps account for the predicted displacement of Y324 in the studied systems, led us to investigate potential conformational alterations in these regions. Calculated RMSDCα values from average MD structures showed negligible conformational changes. However, residues Y324(I) and I325(I) of RecS3 exhibited the highest RMSDCα values when complexed to loxP, indicating their displacement in comparison to Cre/loxP (Figure 5D and Supplementary Tables S3 and S4).
The analysis of B-factors per residue for the evaluated systems showed for RecS3/loxP very high fluctuations on helix M of the inactive monomer (helix M(I)), where Y324(I) appeared shifted from its expected position (Supplementary Figure S12A). Intriguingly, RecS3S320I, and to a lesser extent, CreI320S, displayed elevated B-factors for residue 317(I). In contrast, CreI320S, where Y324(A) was not positioned towards pT3’(TS) during the entire MD simulation time, presented high fluctuations spanning along residues at the end of helix K(A) to helix M(A). Conversely, Crewt and RecS3S320I in complex with loxSE3 showed the lowest fluctuations in these regions (Supplementary Figure S12B).
The analysis of the angle formed between helix M(I) and pG4(BS) indicated a pronounced decrease in the RecS3/loxP complex compared to Crewt/loxP, CreI320S/loxP and RecS3S320/loxP (i.e. 55° for Crewtversus 32.1° for RecS3; Supplementary Figure S13A, F). This decrease could also contribute to the substantial displacement of Y324(I) in RecS3/loxP. In the case of the recombinases complexed with loxSE3, the value of this angle was higher than in the complexes with loxP, with RecS3 and CreI320S exhibiting the highest values (Supplementary Figure S13B, F). The comparison of the obtained helix M(I)-pG4(BS)-helix L(I) angles indicated relevant differences (Supplementary Figure S13C–F). RecS3, which exhibits inefficient recombination towards loxP, displayed a 5° increase compared to Crewt in the angle formed between V321(I), pG4(BS) and M310(I). Conversely, CreI320S and RecS3S320I, less efficient than Crewt, presented a 4° decrease with respect to Crewt (Supplementary Figure S13C). These differences were less pronounced in loxSE3 (Supplementary Figure S13D). Nevertheless, RecS3, which has high recombination activity towards loxSE3, displayed the lowest value for the angle formed between Y324(I), pG4(BS) and M310(I) (Supplementary Figure S13E, F). These observations point towards the relative disposition of helices M(I) and L(I) with respect to the DNA accounting for the predicted Y324(I) displacement and recombination efficiency.
Next, we investigated whether the observed variations in angles and recognition profiles could also be related to differences in the intrinsic flexibility of the DNA spacers and flanking regions. Overall, the calculated TRX scores (34) indicated a higher flexibility for the loxSE3 spacer compared to loxP, with the highest flexibility values concentrated in the spacer's core. On the other hand, the sequence asymmetry in the flanking regions of both DNA target sites (i.e. for loxP AATG—CATT and AGCA— TGCT; for loxSE3 AAAC—GTTT and AGAC—GTCT) was reflected in their distinct TRX scores, indicating higher flexibility in loxP than in loxSE3. Nevertheless, in both, the right flanking regions (i.e. where Y324(I) interacts with the DNA) showed greater flexibility than the left ones (i.e. where Y324(A) interacts with the DNA) (Figure 5E).
Although positions 2′, 1′ and 1 of the spacer's core in loxSE3 could include more optimal sequences for RecS3 (see spacer preference analysis for the top 10% recombined; Figure 3B), interestingly, in our flexibility analysis, we appreciate that those sequences exhibit lower flexibility values (i.e. compared to a TXR score value of 127 for the sequence CCGG in loxSE3, TTCG and TTGG have TXR values of 70 and 89, respectively). However, these values still represent much more flexibility than the core of loxP (GTAT = 18). Based on these observations, it could be hypothesized that perhaps RecS3 has certain preference for DNA target sites with particular higher flexibility propensities at the spacer core than loxP.
As a next step in our analysis, and in order to evaluate potential variations in the DNA groove dimensions that could account for DNA-protein recognition and the differences observed in DNA flexibility, we investigated the width and depth of the minor and major grooves. When recognized by Crewt, a comparable profile was obtained for both loxP structures, the crystallographic (PDB ID 3C29) and the MD-simulated (Supplementary Figures S14 and S15). We observe that in loxP and loxSE3 the minor groove width appears wider in the region where Y324(A) interacts with the phosphate group of the base at position 3′ of the TS, while it is tighter in the region where Y324(I) interacts with the DNA. Interestingly, when looking at the width of the minor groove, we observe that the region where Y324(A) interacts appears more sharply defined in loxP than in loxSE3, which exhibits a shallower minor groove (Supplementary Figure S14A, B). This could be related to the higher flexibility observed in the spacer core region for loxSE3 (see Figure 5E). Moreover, we observe a distinct pattern in the recognition region of loxP by K201(I) and Y324(I) of RecS3 in comparison to Crewt, marked by a notable decrease of the minor groove's width and depth, accompanied by an increase of the major groove's depth (see positions C4 and T5 in Supplementary Figures S14A, C, S15C). The observed DNA changes could be considered as additional factors contributing to the predicted lack of interactions of K201(I) with T5(TS) in the minor groove, as well as the displacement of Y324(I) in the RecS3/loxP system. In contrast, the recognition of loxSE3 by the properly aligned Y324(I) in RecS3 was characterized by a wider minor groove compared to Crewt, CreI320S and RecS3S320I (see position C4 in Supplementary Figure S14B), for which their respective Y324(I) were either not positioned correctly or not fully well disposed towards pG4(BS) through the complete MD simulation time (Figure 5A).
Our findings suggest that the distinct specificity of Crewt and RecS3S320I towards loxP, as well as that of RecS3 and CreI320S towards loxSE3, can be attributed to a combination of factors. The specificity for loxP is believed to stem from van der Waals contacts between the side chain of I320 and T3’(TS), while specificity for loxSE3 appears governed by the interaction of S320 with pC2’(TS). The observed positional shifts of the catalytic Y324(I) at the cleavage site observed in CreI320S/loxP, RecS3/loxP, Cre/loxSE3 and CreI320S/loxSE3, the loss of interactions of catalysis-relevant residues such as K201, alongside variations in the DNA spacer sequence influencing DNA flexibility and conformational profiles, may altogether contribute to the molecular mechanisms underlying selectivity and efficiency, as suggested for Cre/loxP and other recombinase systems in previous biophysical studies (43,44).
Discussion
To advance the application of SSRs as genome engineering tools, a comprehensive understanding of the factors dictating target site specificity and proficient recombination is crucial. The spacer region of the target site is of particular interest as it serves as the primary location for the recombination reaction and determines the editing outcome. Former methods for analyzing the spacer region utilized target libraries with randomized spacers, which predominantly test heterologous targets, preventing the analysis of specificity changes that arise from mutant spacers with their native symmetric alignment (21). The high-throughput approach developed in this work allows for profiling recombinase activity of target libraries with matching mutant spacers. Because each target in the library is predetermined and not randomized, it is possible to systematically isolate and compare sequence-based features of the spacer region to the overall effect on recombination, all while maintaining symmetry of the target sites. Although the library used only covers a portion of all possible combinations in the spacer sequence (6000 of the 65536 (48) possibilities), we show that we can build a solid basis to profile the influence of the spacer sequence on recombination activity. Additionally, we anticipate that the data generated can be useful to train deep learning models with the aim of predicting spacer sequence preferences for SSRs (45). Screening the activity of Cre revealed that spacer homology and sequence identity to loxP generally indicate efficient recombination, although certain base changes can overcome this rule, suggesting that Cre recombination is more dependent on spacer sequence than previously thought (14,16,36).
To explore the dependency of the spacer sequence for efficient recombination, we selected spacers that are inefficiently recombined by Cre and applied directed evolution to increase activity on these sites. Surprisingly, through only 11 cycles of evolution achieved solely via positive selection, we not only generated recombinase variants with an overall heightened activity but also uncovered variants that exhibited a switch in specificity. Certain recombinase variants demonstrated a gain of activity on the new spacer sequence, while concurrently losing activity on the parental loxP site, even though no negative selection against loxP was applied. From the variants with altered spacer specificity, we further characterized RecS3. By comprehensively screening the activity of RecS3 using the spacer library, we find that RecS3 has a different set of spacer requirements compared to Cre, which might be related to variations on DNA-protein interactions and differences in the intrinsic flexibility provided by the spacer sequence, making it the first recombinase to be successfully engineered for spacer specificity. This represents a significant finding by providing the opportunity to fine tune activity on a target site uniquely based on the spacer sequence.
Furthermore, comparative mutational analysis of the residue changes in RecS3 with respect to Cre highlights the critical mutation I320S for selective activity between the loxSE3 and loxP sites. MD-based structural analyses suggest that this residue change and other factors are possibly relevant for contributing to specificity and recombination efficiency. First of all, the observed interaction of I320(A) with T3’(TS) of loxP appears responsible for specificity, whereas in the case of the recognition of loxSE3 it is S320(A) interacting with pC2’(TS). Therefore, position 320 seems to be key in the specificity change from Cre/loxP to RecS3/loxSE3. Secondly, the effect of the changes in the spacer sequence on the conformational disposition of catalysis-relevant residues such as K201 and, in particular, Y324 seems to be relevant. We observe clear variations in the flexibility of the DNA depending on the spacer sequence. The spacer of loxSE3 is much more flexible than that of loxP, which could affect its recognition by catalytic residues and, therefore, recombination activity. Our MD simulations predict a positional shifting of Y324(I) towards the DNA in the inefficient recombinase systems studied. This could be attributed to a decrease in the angle formed between the DNA and the helix M(I), where residue Y324 is located. DNA conformational analyses performed on the obtained MD structures also suggest changes in the dimensions of the minor groove as another plausible factor contributing to recombination efficiency. In particular, in the recombination-inefficient system RecS3/loxP, the DNA region that must be recognized by Y324(I) and K201(I) in order to have a functional recombinase, is characterized by a notable decrease of the minor groove's width and depth compared to Cre/loxP. The observed lack of interactions between K201(I) and loxP, as well as the striking predicted shift of Y324(I) could be related to this DNA conformational change. However, a scenario with a wider minor groove is observed in the case of the recombination-efficient systems Cre/loxP and RecS3/loxSE3. All these variations observed in the recognition of the DNA in the studied systems might affect the stepwise cleavage process of recombination.
The obtained results provide a rationale to account for the structure–function relationships and help to interpret the molecular mechanisms behind the experimentally observed changes in specificity and recombination efficiency. Further exploration into key recombinase residue-spacer interactions by molecular modelling and dynamics simulations could be advantageous for unraveling the intricate molecular details that dictate spacer selectivity in Cre-like recombinases and to ultimately be able to design spacer specificity in silico. Moreover, analysis into features such as protein–protein interactions, shape complementarity and water-mediated contacts, may reveal further important determinants for target specificity.
Altogether, our work provides a platform to explore the features associated with the complex nature of Cre-type recombinase specificity. By classifying and understanding the specificity characteristics we can optimize these properties to achieve an additional layer of specificity, increasing the potential for engineering of Cre-type recombinases for therapeutic applications.
Supplementary Material
Acknowledgements
We thank the members of the Buchholz laboratory that have contributed to discussions improving the quality of experiments. Additionally, we thank the High-Performance Computing Center of the TU Dresden (ZIH TUD) for providing computational infrastructure and the DRESDEN-concept Genome Center at the CMCB from TU Dresden for performing the deep sequencing. We thank Roberto Dominguez and Andres Palencia for helpful advice on structural analysis.
Notes
Present address: Maciej Paszkowski-Rogacz, Seamless Therapeutics GmbH, Tatzberg 47/49, 01307 Dresden, Germany.
Present address: Felix Lansing, Seamless Therapeutics GmbH, Tatzberg 47/49, 01307 Dresden, Germany.
Contributor Information
Jenna Hoersten, Medical Faculty and University Hospital Carl Gustav Carus, UCC Section Medical Systems Biology, TU Dresden, 01307 Dresden, Germany.
Gloria Ruiz-Gómez, Structural Bioinformatics, BIOTEC TU Dresden, Tatzberg 47/49, 01307 Dresden, Germany.
Maciej Paszkowski-Rogacz, Medical Faculty and University Hospital Carl Gustav Carus, UCC Section Medical Systems Biology, TU Dresden, 01307 Dresden, Germany.
Giorgio Gilioli, Medical Faculty and University Hospital Carl Gustav Carus, UCC Section Medical Systems Biology, TU Dresden, 01307 Dresden, Germany.
Pedro Manuel Guillem-Gloria, Structural Bioinformatics, BIOTEC TU Dresden, Tatzberg 47/49, 01307 Dresden, Germany.
Felix Lansing, Medical Faculty and University Hospital Carl Gustav Carus, UCC Section Medical Systems Biology, TU Dresden, 01307 Dresden, Germany.
M Teresa Pisabarro, Structural Bioinformatics, BIOTEC TU Dresden, Tatzberg 47/49, 01307 Dresden, Germany.
Frank Buchholz, Medical Faculty and University Hospital Carl Gustav Carus, UCC Section Medical Systems Biology, TU Dresden, 01307 Dresden, Germany.
Data availability
Quality assessment of our molecular models was done with PROCHECK (46).Coordinates are available upon request. DNA-seq data are available from Gene Expression Omnibus (GEO) under accession number GSE254392.
Supplementary data
Supplementary Data are available at NAR Online.
Funding
European Union [ERC 742133, H2020 UPGRADE 825825]; Bundesministerium für Bildung und Forschung [GO-Bio 161B0633 and SaxoCell 03ZU1111FA]; German Research Council [DFG PI 600/4-1]. Funding for open access charge: TU Dresden.
Conflict of interest statement. Technical University (Technische Univeristät) Dresden has filed a patent application based on this work, in which J.H., M.P.R., F.L. and F.B. are listed as inventors. F.L., M.P.R. and F.B. are co-founders and shareholders of Seamless Therapeutics GmbH. The remaining authors declare no competing interests.
References
- 1. Grindley N.D.F., Whiteson K.L., Rice P.A. Mechanisms of site-specific recombination. Annu. Rev. Biochem. 2006; 75:567–605. [DOI] [PubMed] [Google Scholar]
- 2. Meinke G., Bohm A., Hauber J., Pisabarro M.T., Buchholz F. Cre recombinase and other tyrosine recombinases. Chem. Rev. 2016; 116:12785–12820. [DOI] [PubMed] [Google Scholar]
- 3. Lansing F., Paszkowski-Rogacz M., Schmitt L.T., Martin Schneider P., Romanos T.R., Sonntag J., Buchholz F. A heterodimer of evolved designer-recombinases precisely excises a human genomic DNA locus. Nucleic Acids Res. 2019; 48:472–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Buchholz F., Stewart A.F. Alteration of Cre recombinase site specificity by substrate-linked protein evolution. Nat. Biotechnol. 2001; 19:1047–1052. [DOI] [PubMed] [Google Scholar]
- 5. Lansing F., Mukhametzyanova L., Rojo-Romanos T., Iwasawa K., Kimura M., Paszkowski-Rogacz M., Karpinski J., Grass T., Sonntag J., Schneider P.M. et al. Correction of a factor VIII genomic inversion with designer-recombinases. Nat. Commun. 2022; 13:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Karpinski J., Hauber I., Chemnitz J., Schäfer C., Paszkowski-Rogacz M., Chakraborty D., Beschorner N., Hofmann-Sieber H., Lange U.C., Grundhoff A. et al. Directed evolution of a recombinase that excises the provirus of most HIV-1 primary isolates with high specificity. Nat. Biotechnol. 2016; 34:401–409. [DOI] [PubMed] [Google Scholar]
- 7. Sarkar I., Hauber I., Hauber J., Buchholz F. HIV-1 proviral DNA excision using an evolved recombinase. Science. 2007; 316:1912–1915. [DOI] [PubMed] [Google Scholar]
- 8. Rojo-Romanos T., Karpinski J., Millen S., Beschorner N., Simon F., Paszkowski-Rogacz M., Lansing F., Schneider P.M., Sonntag J., Hauber J. et al. Precise excision of HTLV-1 provirus with a designer-recombinase. Mol. Ther. 2023; 31:2266–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abi-Ghanem J., Chusainow J., Karimova M., Spiegel C., Hofmann-Sieber H., Hauber J., Buchholz F., Pisabarro M.T. Engineering of a target site-specific recombinase by a combined evolution- and structure-guided approach. Nucleic Acids Res. 2013; 41:2394–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stachowski K., Norris A.S., Potter D., Wysocki V.H., Foster M.P. Mechanisms of Cre recombinase synaptic complex assembly and activation illuminated by Cryo-EM. Nucleic Acids Res. 2022; 50:1753–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sternberg N., Hamilton D., Hoess R. Bacteriophage P1 site-specific recombination II. Recombination between loxP and the bacterial chromosome. J. Mol. Biol. 1981; 150:487–507. [DOI] [PubMed] [Google Scholar]
- 12. Hoess R.H., Ziese M., Sternberg N. P1 site-specific recombination: nucleotide sequence of the recombining sites. Proc. Natl. Acad. Sci. U.S.A. 1982; 79:3398–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Abremski K., Hoess R., Sternberg N. Studies on the properties of P1 site-specific recombination: evidence for topologically unlinked products following recombination. Cell. 1983; 32:1301–1311. [DOI] [PubMed] [Google Scholar]
- 14. Lee L., Sadowski P.D. Sequence of the loxP site determines the order of strand exchange by the Cre recombinase. J. Mol. Biol. 2003; 326:397–412. [DOI] [PubMed] [Google Scholar]
- 15. Hoess R.H., Wierzbicki A., Abremski K. The role of the loxP spacer region in PI site-specific recombination. Nucleic Acids Res. 1986; 14:2287–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sheren J., Langer S.J., Leinwand L.A. A randomized library approach to identifying functional lox site domains for the Cre recombinase. Nucleic Acids Res. 2007; 35:5464–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Duyne G.D.V. A structural view of Cre- LoxP site-specific recombination. Annu. Rev. Biophys. Biomol. Struct. 2001; 30:87–104. [DOI] [PubMed] [Google Scholar]
- 18. Gopaul D.N., Guo F., Duyne G.D.V. Structure of the Holliday junction intermediate in Cre–loxP site-specific recombination. EMBO J. 1998; 17:4175–4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guo F., Gopaul D.N., Duyne G.D.V. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature. 1997; 389:40–46. [DOI] [PubMed] [Google Scholar]
- 20. Grindley N.D.F. Site-specific recombination: synapsis and strand exchange revealed. Curr. Biol. 1997; 7:R608–R612. [DOI] [PubMed] [Google Scholar]
- 21. Bessen J.L., Afeyan L.K., Dančík V., Koblan L.W., Thompson D.B., Leichner C., Clemons P.A., Liu D.R. High-resolution specificity profiling and off-target prediction for site-specific DNA recombinases. Nat. Commun. 2019; 10:1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Engler C., Kandzia R., Marillonnet S. A one pot, one step, precision cloning method with high throughput capability. PLoS One. 2008; 3:e3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Buchholz F., Hauber J. In vitro evolution and analysis of HIV-1 LTR-specific recombinases. Methods. 2011; 53:102–109. [DOI] [PubMed] [Google Scholar]
- 24. Hoess R., Wierzbicki A., Abremski K. Formation of small circular DNA molecules via an in vitro site-specific recombination system. Gene. 1985; 40:325–329. [DOI] [PubMed] [Google Scholar]
- 25. Abi-Ghanem J., Samsonov S.A., Pisabarro M.T. Insights into the preferential order of strand exchange in the cre/loxP recombinase system: impact of the DNA spacer flanking sequence and flexibility. J. Comput.-Aided Mol. Des. 2015; 29:271–282. [DOI] [PubMed] [Google Scholar]
- 26. Gelato K.A., Martin S.S., Baldwin E.P. Reversed DNA strand cleavage specificity in initiation of cre–LoxP recombination induced by the His289Ala active-site substitution. J. Mol. Biol. 2005; 354:233–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kechin A., Boyarskikh U., Kel A., Filipenko M. cutPrimers: a new tool for accurate cutting of primers from reads of targeted next generation sequencing. J. Comput. Biol. 2017; 24:1138–1143. [DOI] [PubMed] [Google Scholar]
- 28. R Core Team R: A Language and Environment for Statistical Computing. 2023; Vienna, Austria: Foundation for Statistical Computing. [Google Scholar]
- 29. Wagih O. ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics. 2017; 33:3645–3647. [DOI] [PubMed] [Google Scholar]
- 30. Schmitt L.T., Schneider A., Posorski J., Lansing F., Jelicic M., Jain M., Sayed S., Buchholz F., Sürün D. Quantification of evolved DNA-editing enzymes at scale with DEQSeq. Genome Biol. 2023; 24:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ghosh K., Guo F., Duyne G.D.V. Synapsis of loxP Sites by Cre Recombinase. J. Biol. Chem. 2007; 282:24004–24016. [DOI] [PubMed] [Google Scholar]
- 32. Hoersten J., Ruiz-Gómez G., Lansing F., Rojo-Romanos T., Schmitt L.T., Sonntag J., Pisabarro M.T., Buchholz F. Pairing of single mutations yields obligate cre-type site-specific recombinases. Nucleic Acids Res. 2022; 50:1174–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics. 1996; 14:33–38. [DOI] [PubMed] [Google Scholar]
- 34. Heddi B., Oguey C., Lavelle C., Foloppe N., Hartmann B. Intrinsic flexibility of B-DNA: the experimental TRX scale. Nucleic Acids Res. 2010; 38:1034–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lavery R., Moakher M., Maddocks J.H., Petkeviciute D., Zakrzewska K. Conformational analysis of nucleic acids revisited: curves+. Nucleic Acids Res. 2009; 37:5917–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee G., Saito I. Role of nucleotide sequences of loxP spacer region in cre-mediated recombination. Gene. 1998; 216:55–65. [DOI] [PubMed] [Google Scholar]
- 37. Cautereels C., Smets J., Saeger J.D., Cool L., Zhu Y., Zimmermann A., Steensels J., Gorkovskiy A., Jacobs T.B., Verstrepen K.J. Orthogonal LoxPsym sites allow multiplexed site-specific recombination in prokaryotic and eukaryotic hosts. Nat. Commun. 2024; 15:1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Missirlis P.I., Smailus D.E., Holt R.A. A high-throughput screen identifying sequence and promiscuity characteristics of the loxP spacer region in Cre-mediated recombination. BMC Genom. 2006; 7:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Surendranath V., Chusainow J., Hauber J., Buchholz F., Habermann B.H. SeLOX—A locus of recombination site search tool for the detection and directed evolution of site-specific recombination systems. Nucleic Acids Res. 2010; 38:W293–W298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guillén-Pingarrón C., Guillem-Gloria P.M., Soni A., Ruiz-Gómez G., Augsburg M., Buchholz F., Anselmi M., Pisabarro M.T. Conformational dynamics promotes disordered regions from function-dispensable to essential in evolved site-specific DNA recombinases. Comput. Struct. Biotechnol. J. 2022; 20:989–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ennifar E., Meyer J.E.W., Buchholz F., Stewart A.F., Suck D. Crystal structure of a wild-type Cre recombinase– lox P synapse reveals a novel spacer conformation suggesting an alternative mechanism for DNA cleavage activation. Nucleic Acids Res. 2003; 31:5449–5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Martin S.S., Pulido E., Chu V.C., Lechner T.S., Baldwin E.P. The order of strand exchanges in cre-LoxP recombination and its basis suggested by the crystal structure of a cre-LoxP Holliday junction complex. J. Mol. Biol. 2002; 319:107–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guo F., Gopaul D.N., Duyne G.D.V. Asymmetric DNA bending in the cre-loxP site-specific recombination synapse. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:7143–7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luetke K.H., Sadowski P.D. The role of DNA bending in flp-mediated site-specific recombination. J. Mol. Biol. 1995; 251:493–506. [DOI] [PubMed] [Google Scholar]
- 45. Schmitt L.T., Paszkowski-Rogacz M., Jug F., Buchholz F. Prediction of designer-recombinases for DNA editing with generative deep learning. Nat. Commun. 2022; 13:7966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993; 26:283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Quality assessment of our molecular models was done with PROCHECK (46).Coordinates are available upon request. DNA-seq data are available from Gene Expression Omnibus (GEO) under accession number GSE254392.