

RESUME
Les cytopénies auto-immunes se définissent par une destruction immunologique d’un ou plusieurs éléments figurés du sang par des auto-anticorps. Le plus souvent il s’agit d’une anémie hémolytique ou une thrombopénie immunologique ou les deux à la fois définissant un syndrome d’Evans. Chez l’enfant la cytopénie auto-immune peut être secondaire à une infection ou être inaugurale à une pathologie sous-jacente comme une maladie auto-immune systémique ou une immunodéficience primitive en particulier quand elle devient chronique sur plusieurs années.Les déficits immunitaires primitifs ou erreurs innées de l'immunité (IEI) ne sont plus définis uniquement par les infections : l'auto-immunité fait partie du tableau clinique de plusieurs de ces maladies et est dominée par les cytopénies auto-immunes, en particulier, le purpura thrombopénique immunologique (PTI) et les anémies hémolytiques auto-immunes (AHAI).Les problématiques que peut confronter le clinicien sont les situations ou les cytopénies auto-immunes sont chronique, récidivantes et ou réfractaires aux différents moyens thérapeutiques au long cours dont l’action le plus souvent est similaire et consiste généralement en une suppression ou une modulation immunitaire non spécifique, ce qui complique la prise en charge, malgré le panel hétérogène des traitements. Dans ces situations les déficit immunitaires primitifs doivent être diagnostiqués le plus tôt possible pour permettre l'initiation d'un traitement ciblé et éviter plusieurs lignes thérapeutiques inefficaces.
ABSTRACT
Autoimmune cytopenias are defined by autoantibodies' immune destruction of one or more blood elements. Most often it is autoimmune hemolytic anemia or immune thrombocytopenia or both that define Evans syndrome. It may be secondary to infection or to underlying pathology such as systemic autoimmune disease or primary immunodeficiency, especially when it becomes chronic over several years. Primary Immunodeficiencies or inborn errors of immunity (IEI) are no longer defined solely by infections: autoimmunity is part of the clinical features of several of these diseases. It is dominated by autoimmune cytopenias, in particular, immune thrombocytopenia (ITP) and autoimmune hemolytic anaemia (AIHA). The challenges for the clinician are the situations where autoimmune cytopenias are chronic, recurrent and/or refractory to the various long-term therapeutic options. Most of these therapies are similar in action and generally consist of non-mediated immune suppression or modulation. In these situations, primary Immunodeficiencies must be diagnosed as soon as possible to allow the initiation of a targeted treatment and to avoid several ineffective therapeutic lines.
INTRODUCTION
Les cytopénies auto-immunes constituent un groupe d'affections hétérogènes étroitement liées et se définissent par une destruction auto-immune d’une ou plusieurs cellules sanguines. Cette destruction peut être primaire ou secondaire à d'autres maladies (1).
Les cytopénies auto-immunes primaires, anciennement classées comme idiopathiques, consistent en une destruction d'une seule lignée, y compris la thrombocytopénie immunitaire (PTI), l'anémie hémolytique auto-immune (AHAI) et la neutropénie auto-immune (NAI), ainsi qu'une destruction multi-lignée. L’association d’une AHAI et le PTI définissent le syndrome d’Evans (2).
Les cytopénies auto-immunes secondaires résultent d'une autre cause, notamment les maladies auto immunes comme le lupus, les immunodéficiences, les lymphoproliférations, les cancers ou une complication d'une greffe d'organe ou de cellules souches hématopoïétiques(1).
Il existe une continuité entre déficit immunitaire (DIPs) et cytopénie auto-immune (3). En effet les DIPs sont des anomalies génétiques caractérisés par un spectre clinique élargi allant d'une susceptibilité accrue aux infections à une dérégulation immunitaire importante avec auto-immunité et notamment une cytopénie auto-immune, et inversement une cytopénie supposé initialement primaire peut être le premier signe clinique d’un déficit immunitaire qui sera diagnostiqué au cours de l’évolution soit par l’enrichissement de tableau clinique ou par le caractère chronique et rebelle aux thérapeutiques de première lignes. Par conséquent, Le pédiatre ou hématologue pédiatre doit suivre aussi bien une approche immunologique en plus de l’approche hématologique devant une cytopénie surtout quand elle est chronique et de chercher des paramètres faisant suspecter un déficit immunitaire.
LES CYTOPÉNIES AUTO-IMMUNES PRIMAIRES ET LEUR PRISE EN CHARGE
La plupart des enfants atteints de cytopénies auto-immunes sont primaires ou anciennement connues comme idiopathiques et sans cause secondaire et entreront en rémission spontanée avec le temps (5).
Le purpura thrombopénique immunologique
Le PTI est la thrombopénie acquise la plus courante chez les enfants, survenant chez environ 4,2 enfants sur 100 000 par an (6-7). Selon une étude tunisienne (8) sur 140 cas de PTI menée aux services de pédiatrie et d’hématologie du CHU Hédi Chaker de Sfax, L’âge moyen était de 6 ans 7 mois (extrêmes : 3 mois et 15 ans), avec une légère prédominance féminine filles (54 %). Les répartitions saisonnière et mensuelle ont montré un pic de fréquence au printemps (28,4%).
Bien que sa physiopathologie ne soit pas entièrement comprise, le PTI serait dû à des mécanismes complexes qui associent en plus d’une réponse immunitaire humorale et cellulaire inadaptée à des anomalies centrales de production plaquettaire. Les cibles antigéniques principales des auto-Ac sont les glycoprotéines (GP) plaquettaires GPIIb/IIIa (récepteur du fibrinogène), GPIb/ IX (récepteur du facteur von Willebrand) et GPIa/IIa (récepteur du collagène) (9-10)
Figure 1. physiopathologie du PTI; (Référence :Audia et al, Rev Med Int 2011) .
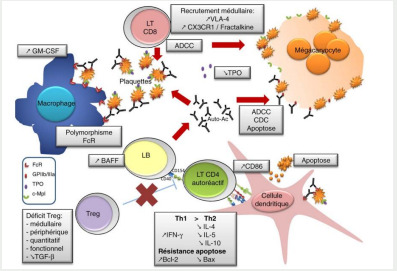
Dans l’histoire naturelle de la maladie trois périodes sont distinguées (11) : - Le PTI depuis moins de 3 mois. - Le PTI persistant entre 3 et 12 mois - Le PTI chronique depuis plus de 12 mois (très faible probabilité de rémission) Selon une étude tunisienne ( 12), les facteurs prédictifs d’évolution vers la chronicité étaient le début insidieux de la maladie, les antécédents hémorragiques personnels et l’échec d’une abstention thérapeutique initiale.
Figure 2. Histoire naturelle du PTI .

80% des enfants atteints entrent en rémission en quelques semaines et même en phase chronique des cas de guérisons peuvent être observés (2).
Le diagnostic de PTI est un diagnostic d’élimination. L’intérêt de l’évaluation initiale est d’importance capitale. Elle a pour objectif d’exclure les diagnostics différentiels et retenir le diagnostic positif afin d’orienter les indications thérapeutiques.
Cliniquement, les patients présentent le plus souvent des saignements cutanéo-muqueux de degrés variables (pétéchies, ecchymoses, épistaxis, bulles hémorragiques….) ces saignements s’accompagnent généralement d’une thrombopénie sévère. La majorité des patients (77 %) n'avaient aucun saignement ou des saignements légers au moment du diagnostic. Les hémorragies sévères sont rares, un seul patient (sur 863 patients évaluables) avait une hémorragie du système nerveux central tandis que 3 % des patients avaient des saignements graves (épistaxis, gsaignements modgérés (11). Le risque hémorragique est apprécié par le score de Buchanan graduant la sévérité globale du saignement de 0 (nulle, aucun signe hémorragique) à 5 Menaçant le pronostic vital.
Tableau 1. score de Buchanan (Evaluation de la gravité hémorragique du PTI) .
| Grade 0 (mineur) | Grade 1 | Grade 2 (moyen) | Grade 3 (modéré) | Grade 4 (sévère) | Grade 5 (pronostic vital mis en jeu) |
|---|---|---|---|---|---|
|
Peau - Rares pétéchies ou ecchymoses |
Pétéchies ou ecchymoses indiscutables |
Nombreuses pétéchies ou ecchymoses |
Pétéchies ou ecchymoses extensives |
- | - |
|
Epistaxis - Sang dans une narine |
Epistaxis ≤15 minutes |
Epistaxis ˃15 minutes |
Epistaxis répétées |
- | - |
|
Buccal - Pétéchies du palais |
Bulles sans saignement actif |
Saignement actif intermittent |
Saignement actif continu |
- | - |
|
Global - Quelques lésions hémorragiques cutanées sans lésions muqueuses |
Lésions hémorragiques cutanées modérées à sévères mais sans lésions muqueuses |
Saignement muqueux ne demandant pas d’intervention médicale |
Saignement muqueux actif nécessitant une intervention médicale |
Saignement documenté du système nerveux central ou hémorragie fatale de n’importe quel site |
- |
Concernant les examens complémentaires, il n’existe pas d’investigation biologique sensible qui permet de poser le diagnostic de PTI. Ce dernier est basé sur un faisceau d’arguments cliniques et biologiques. Parmi les bilans indispensables l’hémogramme, frottis (taille plaquettes, blastes circulants, schizocytes…), TP TCA, bilan hépatique, sérologie virales VIH VHC VHB, le dosage pondéral des immunoglobulines IgG IgA IgM (surtout avant toute perfusion des immunoglobulines intraveineuses). Le myélogramme n’est plus systématique pour les cas typiques de PTI (11). Le reste du bilan n’est pas requis et se fait en fonction du contexte.
La prise en charge du PTI comprend deux grands volets: le premier concerne le mode de vie et l’éducation thérapeutique. C’est l’ensemble des activités ayant pour objectif d’aider le patient et son entourage à s’adapter et mieux comprendre la maladie ainsi que ses thérapeutiques, mais aussi de participer aux soins dans la mesure du possible. Le deuxième volet de traitement médicamenteux qui comprend des différents niveaux depuis l’abstention thérapeutique jusqu’aux médicaments de deuxièmes lignes.
L’abstention thérapeutique avec une surveillance clinique et biologique du patient peut constituer à elle seule une arme thérapeutique en cas de thrombopénie modérée et asymptomatique. Compte tenu de la survenue relativement rare de saignements importants chez les enfants atteints de PTI, la recommandation actuelle est une observation attentive des enfants atteints de PTI nouvellement diagnostiqué et présentant des symptômes de saignement légers ou inexistants.
Chez les enfants nécessitant un traitement pour augmenter le nombre de plaquettes, une dose unique d'IgIV 1g/kg à J1 +/- J3 ou une cure courte de corticoïdes est recommandée en première intention. Les corticoïdes les plus utilisés sont sous forme de prednisone 4mg/kg/j en deux prises pendant 4 jours ou 2mg/kg/j pendant une semaine puis arrêt sur deux semaines, ou dexaméthasone per os 10 mg/m2/j pendant 4 jours (13). La perfusion d'immunoglobuline antiRho(D) à 50 µ/kg peut être considéré comme un traitement alternatif de première intention chez les patients Rh(D) positif les patients non splénectomisés et non anémiques (13).
Les thérapies de deuxième intention comprennent le rituximab, la dexaméthasone à forte dose et la splénectomie. Récemment, les agonistes du récepteur de la thrombopoïétine AR-TPO sont apparus comme de nouvelles thérapies pour le PTI (13). D'autres thérapies immunosuppressives, y compris la mercaptopurine, le danazol, la vincristine, le mycophénolate mofétil et le sirolimus, ont toutes été utilisées hors AMM dans de petites séries de patients.
L’anémie hémolytique auto-immune
L’anémie hémolytique auto-immune (AHAI) se définie par la destruction aiguë de globules rouges par l’intermédiaire d’auto-anticorps fixés sur des antigènes érythrocytaires. Si dans le cas du purpura thrombopénique immunologique (PTI), le mécanisme auto-immun est souvent posé après élimination des autres causes, le diagnostic de l’AHAI repose sur la présence d’une anémie hémolytique avec test de Coombs positif (1 , 2, 14). L’AHAI est classifiée en deux types selon que l’anticorps est actif à 37°C, et on parle d’AHAI à AC chaude ou à 4 °C et on parle d’AHAI à AC froid (15).
Cliniquement, les patients présentent le plus souvent une symptomatologie variable en fonction de type d’hémolyse, et on distingue soit un tableau brutal et aigue en cas de type d’hémolyse intravasculaire avec une symptomatologie bruyante faite d’un syndrome anémique marqué avec pâleur, ictère intense et tachycardie et fièvre, soit un tableau d’apparition plus progressive en cas hémolyse intratissulaire avec un subictère, splénomégalie et un syndrome anémique modéré (14, 16 ).
Les bilans complémentaires pour diagnostiquer l’AHAI sont : un hémogramme avec taux réticulocytes qui objective une régénération, et un frottis qui fournit des informations qualitatives que l’automate ne peut fournir (signes cytologique morphologiques d’hémolyse et des éléments de diagnostic différentiel), un bilan d’hémolyse fait de bilirubine libre (élevé), LDH (élevé) et du taux d’haptoglobine (effondré). La confirmation du mécanisme auto- immun de l’anémie se fait par le test de Coombs direct (IgG, C3d) qui est positif (1 -2, 16).
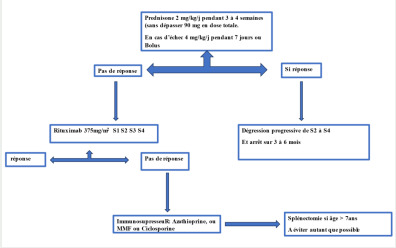
Le traitement de l’AHAI primaire à AC chauds repose sur une corticothérapie en première ligne à base de prednisone. La posologie initiale est de 2 mg/kg/j pendant 3 à 4 semaines, délai souvent nécessaire pour interrompre le processus d’hémolyse et entrainer la correction au moins partielle de l’anémie. Une fois la rémission est obtenue, la dégression de la corticothérapie doit être progressive sur plusieurs semaines jusqu’à la dose minimale efficace et ce pour une durée totale de traitement de 3 à 6 mois minimum.
En cas de cortico-résistance ou de cortico-dépendance d’autres traitements dans le but d’épargner les corticoïdes doivent être envisagés parmi les suivants : le rituximab (hors AMM), les immunosuppresseurs (azathioprine, mycophénolate mofetil…) ou encore la splénectomie. (Voir schéma).
Les IgIV n’ont pas de place dans les AHAI à AC chauds, bien qu'elles puissent constituer une arme thérapeutique utile dans les cas graves sévères, cortico-résistants avec des dépendances transfusionnelles (17).
Arbre décisionnel 1. prise en charge de l’AHAI à AC chauds .
Pour les AHAI à AC froids sont souvent d’origine postinfectieuse (infection à mycoplasme, mononucléose infectieuse) chez l’enfant, et sont d’évolution aigüe et transitoire en quelques semaines. Le traitement est purement symptomatique avec un support transfusionnel si besoin.
Neutropénie auto-immune
La neutropénie se définit par une diminution du nombre absolu de polynucléaires neutrophiles dans le sang circulant. On parle de neutropénie en dessous de 1 500 polynucléaires/mm3 chez l’enfant de plus de 1 an. Il existe plusieurs types de neutropénies, et on distingue les neutropénies acquises, les neutropénies constitutionnelles liées à une pathologie génétique complexe, les neutropénies constitutionnelles primitives (18 ).
Dans les neutropénies acquises, la neutropénie auto-immune primaire constitue la plus fréquente cause de neutropénie chronique de l’enfant. Elle se présente le plus souvent dans la petite enfance après une infection virale, et est souvent spontanément résolutive avec > 95 % des enfants ayant une résolution complète de NAI. La plupart des patients n'ont pas besoin de traitement. Des mesures préventives, y compris une bonne hygiène buccale et des bains de bouche, sont souvent utilisées (4). La détection des AC antipolynucléaires nécessite des examens répétés et n’ont pas de place de fait de l’évolution favorable.
Syndrome d’Evans
Le syndrome d’Evans est défini par une destruction immunologique de deux ou plusieurs lignées, le souvent et l’association AHAI et PTI. C’est un diagnostic d’exclusion et les patients atteints de SE ont tendance à avoir une maladie chronique surtout pour ceux qui se présentent à un jeune âge (1, 2, 4).
LES CYTOPÉNIES AUTO-IMMUNES SECONDAIRES : QUAND PENSER AUX DIP ET COMMENT PRENDRE EN CHARGE ?
Un déficit immunitaire primitif désigne un état dans lequel le système immunitaire est altéré dès la naissance ou tôt dans la vie. Les personnes atteintes de déficits immunitaires primitifs ont une vulnérabilité accrue aux infections et aux maladies auto-immunes. Selon la dernière version de la classification phénotypique de l’union international des sociétés d’immunologie 485 désordres génétiques distincts, ainsi les déficits immunitaires sont classés en 10 groupes en fonction du défaut immunologique impliqué (19).
I. Déficits immunitaires affectant l’immunité cellulaire et humorale II. Déficit immunitaire combiné avec caractéristiques associées ou syndromiques III. Déficiences en anticorps prédominantes. IV. Maladies de la dysrégulation immunitaire. V. Défauts congénitaux du nombre, de la fonction ou des deux des phagocytes. VI. Défauts de l’immunité intrinsèque et innée VII. Troubles auto-inflammatoires. VIII.Déficiences en complément. IX. Troubles de l’insuffisance de la moelle osseuse. X. Phénotypes imitant les déficits immunitaires primitifs. De nombreuses affections et médicaments peuvent entraîner des cytopénies auto-immunes. Parmi ces affections, les maladies rhumatologiques comme le lupus, les immunodéficiences, les lymphoproliférations, les cancers ou une complication d'une greffe d'organe ou de cellules souches hématopoïétiques sont les plus décrites ( 1-2, 4). Le problème devient plus complexe lorsque les cytopénies auto-immunes sont la seule manifestation de la maladie, ce qui rend le diagnostic et la prise en charge aussi complexe. Une anamnèse et un examen physique minutieux s’avère nécessaire pour identifier une cause secondaire chez le patient qui se présente en phase aiguë.
Les déficits immunitaires primitifs sont des troubles génétiques caractérisés par un large éventail de manifestations cliniques, allant d'une susceptibilité accrue aux infections à une dérégulation immunitaire importante ( 20). Les manifestations auto-immunes (MAI) en général et tout particulièrement les cytopénies auto-immunes sont de plus en plus reconnues comme une composante de plusieurs formes de déficits immunitaires primaires ( 21-25). En effet, et il existe des DIPs où l’infection est la composante la plus prédominante et d’autres où l’auto-immunité sans ou avec une composante infectieuse moindre (21). Le risque des cytopénies auto-immunes est 120 fois plus élevé dans les DIPs que la population générale (20). Elles peuvent révéler ou compliquer plusieurs DIPs, parfois plusieurs années après la cytopénie ou l’enfant reçoit divers traitement (corticoïdes, IS, IgIV). Le syndrome lymphoprolifératif auto-immun (ALPS) représente un exemple d’une dysrégulation immunitaire ou l’auto-immunité est au centre des manifestations cliniques le plus souvent sous la forme de cytopénies auto-immunes (anémie hémolytique, thrombopénie et neutropénie) ( 26). Selon Notarangelo et al (3), 80% des ALPS ont des autoAc dont 23% AHAI et 50% PTI. Ce syndrome tumoral bénin, apparait à un âge précoce et associe des adénopathies, une énorme splénomégalie et, éventuellement, une hépatomégalie. Cette lymphoprolifération chronique est dû à l’accumulation dans le sang et les organes lymphoïdes secondaires d’une population de lymphocytes Tαβ matures n’exprimant ni CD4 ni CD8, appelés lymphocytes T « doubles négatifs » (LTαβ DN) (27). L’élévation de la vitamine B12 est un bon indicateur. Sur le plan génétique le syndrome d’ALPS est de transmission autosomique dominante. Les gène mutés concernent la voie Fas-dépendante de l’apoptose et aboutissant à une accumulation des lymphocytes auto-réactifs (26-28). Notarangelo et al a montré également qu’au cours des CVID, des manifestations auto-immunes représentent 22- 48%. Inversement, 65% des syndromes d’Evans de l’enfant ont une anomalie monogénique et 40 % avaient des mutations pathogènes connus pour être impliqués dans les immunodéficiences primaires (TNFRSF6, CTLA4, STAT3, PIK3CD, CBL, ADAR1, LRBA, RAG1 et KRAS) (29). L’atteinte de plusieurs lignées semblent avoir plus de chance de révéler une anomalie monogénique. Plusieurs autres études ont montré l’identification des mutations monogéniques des immunodéficiences primaires au cours du suivi des cytopénies auto-immunes (30-33). En effet, Miano et al (31) dans une étude monocentrique, a montré sur 40 patients suivis pour syndrome d’Evans, 24 cas des anomalies immunologiques, 27 cas de lymphoprolifération. 17 cas avaient des critères d’ALPS, et 18 cas sur 40 qui avaient des mutations génétiques FAS/ CASP10/LRBA/CTLA4/LIG4/STAT3/IKBGK/CARD11/ADA2. Cependant, l'identification d'un DIP sous-jacent dans un groupe hétérogène de patients atteints de divers troubles auto-immuns peut être une tâche ardue. La plupart des pédiatres ou des spécialistes prenant en charge des patients atteints de maladies auto-immunes peuvent ne pas envisager une évaluation immunitaire dans le bilan initial et supposer une faible probabilité. Par conséquent, il n'est pas rare que le diagnostic spécifique des patients très vulnérables atteints d'un déficit immunitaire soit retardé. Il conviendra donc de surveiller l’évolution des patients notamment l’apparition d’autres signes cliniques ou biologiques, y compris après plusieurs années. Parmi les indicateurs alarmants qui font penser au DIP (IEI) au cours du suivi des cytopénies auto-immunes étiquetées initialement comme primaires sont : - Début précoce - Atteinte de plusieurs lignées sanguines - Résistance aux traitements conventionnels - Lymphoprolifération bénigne - Survenue de manifestations évocatrices de déficit immunitaire (infections à répétitions et inhabituelles, diarrhée chronique, retard staturo-pondéral, eczéma…) - Auto-immunité multiple - Bilan immunitaire perturbé L’objectif d’identifier un IEI sous-jacent est de permettre l'initiation des thérapies immuno-modulatrices de deuxième et de troisième ligne ou d'un traitement spécifique ciblé. Les hématologues et les immunologistes devraient par conséquent s'occuper conjointement de ces types de cytopénies auto-immunes et d’élargir le bilan immunitaire voire même moléculaire.
Pour aider le clinicien à mieux déceler ces cytopénies, nous proposons un arbre décisionnel (Figure 1) et le tableau 1 qui regroupe les catégories de DIP et le traitement ciblé pour chacune.
Arbre décisionnel 2. conduite à tenir devant une cytopénie auto-immune.

Tableau 2. catégories de DIP et le traitement ciblé pour catégorie.
|
Catégorie DIP |
Traitement ciblé |
|---|---|
|
ALPS |
Sirolimus (inhibiteur de mTOR) |
|
Déficit en CTLA4 et LRBA |
|
|
STAT1 GOF |
|
|
STAT3 GOF |
|
|
APDS (PIK3CD, PIK3R1) |
|
|
NFATTG variants |
Inhibiteurs de calcineurine (Ciclosporine) |
|
Wiscot Aldrich |
GCSH, Thérapie génique |
|
SCID |
GCSH, Thérapie génique |
CONCLUSION
Les cytopénies auto-immunes peuvent avoir une évolution plus compliquée que celle observée dans la population générale si elles surviennent chez des patients présentant un IEI sous-jacent. Le clinicien doit suivre aussi bien une approche immunologique en plus de l’approche hématologique d’une cytopénie, et suivre son évolution pour repérer un IEI. Les signes alarmant le clinicien sont le multi-linéage et le non réponse aux traitements conventionnels. Le traitement ciblé est le meilleur une fois le diagnostic précis est prouvé.
References
- Taylor O K, Jenny M D. Primary and Secondary Immune Cytopenias: Evaluation and Treatment Approach in Children. Hematol Oncol Clin North Am. 2019 Jun;33:489–506. doi: 10.1016/j.hoc.2019.01.005. [DOI] [PubMed] [Google Scholar]
- Suarez F, Ghez D, Delarue R, Hermine O. Cytopénies auto-immunes périphériques. Réanimation. 2005;14:587–593. [Google Scholar]
- Notarangelo LD. Hematol Am Soc Hematol Educ Progr. 2009. Primary immunodeficiencies (PIDs) presenting with cytopenias; pp. 139–143. [DOI] [PubMed] [Google Scholar]
- Teachey DT, Lambert MP. Diagnosis and management of autoimmune cytopenias in childhood. Pediatr Clin North Am. 2013;60:1489–1511. doi: 10.1016/j.pcl.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Ghaithi I, Wright NA, Breakey VR, Cox K, Warias A, Wong T. Combined Autoimmune Cytopenias Presenting in Childhood. Pediatr Blood Cancer. 2016;63:292–298. doi: 10.1002/pbc.25769. [DOI] [PubMed] [Google Scholar]
- Singh G, Bansal D, Wright NAM. Immune thrombocytopenia in children: consensus and controversies. Indian J Pediatr. 2020;87:150–157. doi: 10.1007/s12098-019-03155-4. [DOI] [PubMed] [Google Scholar]
- Despotovic JM, Grimes AB. Hematology Am Soc Hematol Educ Program. 2018. Pediatric ITP: is it different from adult ITP? pp. 405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfaihi L, Kassar O, Medhaffar O, Kamoun T, Hadiji S, Aloulou H. Primary immune thrombocytopenia in childhood: a regional study in the south of Tunisia. Tunis Med. 2014 Mar;92(3):219–223. [PubMed] [Google Scholar]
- Singh A, Uzun G, Bakchoul T. Primary immune thrombocytopenia: novel insights into pathophysiology and disease management. J Clin Med. 2021;10:789. doi: 10.3390/jcm10040789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audia S, Lorcerie B, Godeau B, Bonnotte B. Physiopathologie du purpura thrombopénique immunologique. La Revue de médecine interne. 2011;32:350–357. doi: 10.1016/j.revmed.2009.05.017. [DOI] [PubMed] [Google Scholar]
- Neunert C, Lim W, Crowther M, et al The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011;117:4190–4207. doi: 10.1182/blood-2010-08-302984. [DOI] [PubMed] [Google Scholar]
- Mazigh MS, Ouederni M, Bouyahia O, Gharsallah L, Boukthir S, Samoud GA. Thrombocytopenic idiopathic purpura: predictive factors for chronic disease. Tunis Med. 2009;87(1):72–75. [PubMed] [Google Scholar]
- Kühne T. Diagnosis and management of immune thrombocytopenia in childhood. Hamostaseologie. 2017;37:36–44. doi: 10.5482/HAMO-16-06-0017. [DOI] [PubMed] [Google Scholar]
- Weli M, Ben Hlima A, Belhadj R, Maalej B, Elleuch A, Mekki N, et al Diagnosis and management of autoimmune hemolytic anemia in children. Transfus Clin Biol. 2020;27:61–64. doi: 10.1016/j.tracli.2020.03.003. [DOI] [PubMed] [Google Scholar]
- Bass GF, Tuscano ET, Tuscano JM. Diagnosis and classification of autoimmune hemolytic anemia. Autoimmun Rev. 2014;13:560–564. doi: 10.1016/j.autrev.2013.11.010. [DOI] [PubMed] [Google Scholar]
- Voulgaridou A, Kalfa TA. Autoimmune Hemolytic Anemia in the Pediatric Setting. J Clin Med. 2021;10:216. doi: 10.3390/jcm10020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalfa TA. Warm antibody autoimmune hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2016;2016:690–697. doi: 10.1182/asheducation-2016.1.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donadieu J, Fenneteau O. Constitutional and acquired neutropenia. EMC-Hématologie. 2005;2:158–186. [Google Scholar]
- Bousfiha A, Moundir A, Tangye SG, Picard C, Jeddane L, Al-Herz W, et al The 2022 update of IUIS phenotypical classification for human inborn errors of immunity. J Clin Immunol. 2022;42:08–20. doi: 10.1007/s10875-022-01352-z. [DOI] [PubMed] [Google Scholar]
- Fischer A, Provot J, Jais JP, Alcais A, Mahlaoui N, et al Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. CEREDIH. 2017 Nov;140:1388–1393. doi: 10.1016/j.jaci.2016.12.978. [DOI] [PubMed] [Google Scholar]
- Walter JE, Ayala IA, Milojevic D. Autoimmunity as a continuum in primary immunodeficiency. Curr Opin Pediatr. 2019 Dec;31:851–862. doi: 10.1097/MOP.0000000000000833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmuganathan Chandrakasan, Chandra Sharat, Davila Saldana Blachy J, Torgerson Troy R, Buchbinder David. Primary immune regulatory disorders for the pediatric hematologist and oncologist: A case-based review. Pediatr Blood Cancer. 2019 May;66:----–----. doi: 10.1002/pbc.27619. [DOI] [PubMed] [Google Scholar]
- Azizi Gholamreza, Ghanavatinejad Alireza, Abolhassani Hassan, Yazdani Reza, Rezaei Nima, Mirshafiey Abbas. Autoimmunity in primary T-cell immunodeficiencies. Expert Rev Clin Immunol. 2016 Sep;12:989–1006. doi: 10.1080/1744666X.2016.1177458. [DOI] [PubMed] [Google Scholar]
- Seide Markus G. Autoimmune and other cytopenias in primary immunodeficiencies: pathomechanisms, novel differential diagnoses, and treatment. Blood. 2014 Oct 9;124:2337–2344. doi: 10.1182/blood-2014-06-583260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter Jolan E, Farmer Jocelyn R, Foldvari Zsofia, Torgerson Troy R, Cooper Megan A. Mechanism-Based Strategies for the Management of Autoimmunity and Immune Dysregulation in Primary Immunodeficiencies. J Allergy Clin Immunol Pract. 2016 Nov-Dec;4:1089–1100. doi: 10.1016/j.jaip.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieux-Laucat Frédéric. Le syndrome lymphoprolifératif avec auto-immunité : Un défaut hérité ou acquis d’apoptose lymphocytaire. MEDECINE/SCIENCES. 2006;22:645–649. doi: 10.1051/medsci/20062267645. [DOI] [PubMed] [Google Scholar]
- Matson Daniel R., Yang David T. Autoimmune Lymphoproliferative Syndrome: An Overview. Arch Pathol Lab Med. 2020 February;144 doi: 10.5858/arpa.2018-0190-RS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira JB, Bleesing JJ, Dianzani U, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood. 2010;116 doi: 10.1182/blood-2010-04-280347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjadj J, Aladjidi N, Fernandes H, Leverger G, Magérus-Chatinet A, Mazerolles F, et al. Pediatric Evans Syndrome Is Associated With a High Frequency of Potentially Damaging Variants in Immune Genes. Blood. 2019;134:9–21. doi: 10.1182/blood-2018-11-887141. [DOI] [PubMed] [Google Scholar]
- Besnard C. Pediatric-onset Evans syndrome: heterogeneous presentation and high frequency of monogenic disorders including LRBA and CTLA4 mutations. Clin Immunol. 2018;188:52–57. doi: 10.1016/j.clim.2017.12.009. [DOI] [PubMed] [Google Scholar]
- Miano M, Guardo D, Grossi A, et al. Underlying inborn errors of immunity in patients with Evans syndrome and multilineage cytopenias. a single centre analysis. Front Immunol. 2022;13 doi: 10.3389/fimmu.2022.869033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Ghaithi Ibrahim, Wright Nicola A M, Breakey Vicky R, Cox Kelly, Warias Ashley, Wong Tiffany, et al. Combined Autoimmune Cytopenias Presenting in Childhood. Pediatr Blood Cancer. 2016 February;:292–298. doi: 10.1002/pbc.25769. [DOI] [PubMed] [Google Scholar]
- Cortesi M, Soresina A, Dotta L, et al. Pathogenesis of autoimmune cytopenias in inborn errors of immunity revealing novel therapeutic targets. Front Immunol. 2022;13:1–10. doi: 10.3389/fimmu.2022.846660. [DOI] [PMC free article] [PubMed] [Google Scholar]