

Abstract
The number of applications of self-assembled deoxyribonucleic acid (DNA) origami nanoparticles (DNA NPs) has increased drastically, following the development of a variety of single-stranded template DNA (ssDNA) that can serve as the scaffold strand. In addition to viral genomes, such as M13 bacteriophage and lambda DNAs, enzymatically produced ssDNA from various template sources is rapidly gaining traction and being applied as the scaffold for DNA NP preparation. However, separating fully formed DNA NPs that have custom scaffolds from crude assembly mixes is often a multistep process of first separating the ssDNA scaffold from its enzymatic amplification process and then isolating the assembled DNA NPs from excess precursor strands. Only then is the DNA NP sample ready for downstream characterization and application. In this work, we highlight a single-step purification of custom sequence- or M13-derived scaffold-based DNA NPs using photocleavable biotin tethers. The process only requires an inexpensive ultraviolet (UV) lamp, and DNA NPs with up to 90% yield and high purity are obtained. We show the versatility of the process in separating two multihelix bundle structures and a wireframe polyhedral architecture.
Keywords: DNA nanotechnology, DNA origami, asymmetric polymerase chain reaction, purification, magnetic beads, biotin, nanostructures
Graphical Abstract
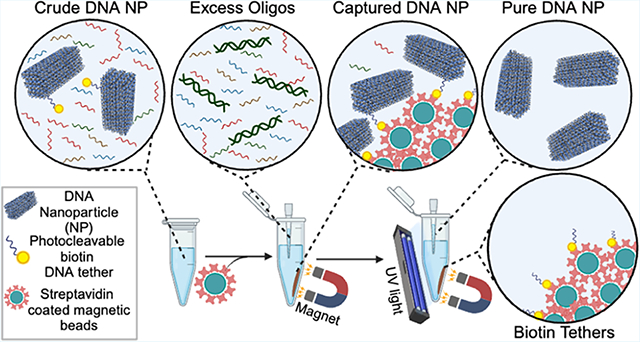
INTRODUCTION
At the turn of 2023, a series of preliminary reports demonstrated that gene-encoded synthetic deoxyribonucleic acid (DNA) nanoparticles (NPs) can enable protein expression via transcription and translation in vitro and in vivo. Simultaneously, custom DNA origami NPs with multiple enzymatically encoded functional modifications are gaining traction for biomedical applications.1–4 In these rapidly growing nanoscale applications of synthetic DNA origami NPs,5 the role of the scaffold strand—which is the long single-stranded template DNA (ssDNA) within these particles—has become multifaceted. The traditional DNA origami NPs—made predominantly with scaffolds sourced from bacteriophage’s canonical genomic ssDNAs—are still widely applied in single molecular studies,6 photonics,7 plasmonic,8 and biomedical applications,9 but a new suite of DNA NPs leverage nonviral sequences as scaffold strands to serve functional roles.1,10–13 These scaffold strands are produced by enzymes (such as asymmetric polymerase chain reaction (aPCR)) by separating ssDNA from double-stranded DNA (dsDNA)14 or phage-based methods, often requiring purification.15 Separation techniques based on molecular weight cutoffs—such as ultrafiltration and precipitation—do not remove higher-molecular-weight contaminants from custom-Synthesized scaffolds such as the strand that is “antisense” or complementary to the scaffold itself and is a common byproduct of custom scaffold synthesis techniques.16 Extraction of specific bands after gel electrophoresis removes aggregates and misfolded structures, but the overall yield is not ideal for scalability.16 The current paradigm in creating purified custom-scaffolded DNA NPs is to first purify the scaffold strand and then process the annealed NP through a secondary purification step, creating a need for versatile purification techniques for DNA NPs that are prepared from heterogeneous scaffold precursors (M13 or custom).
Streptavidin-coated magnetic beads have shown promise in purifying DNA NPs by attaching biotin-modified capture strands on the beads that are complementary to an extended strand on the DNA NP.17,18 After the residual solution is removed and the beads are rinsed, DNA NPs can be recovered by using strand displacement. Chemically labile linkers—such as dithiol, which can be reduced to thiol using dithiothreitol (DTT)—can capture DNA NPs using biotin on the streptavidin-coated beads and release them using DTT.19 However, this approach leaves a residual extended staple strand on the DNA NP. Additionally, magnetic bead-based separation has yet to be applied to purify custom-scaffolded DNA origami particles from a crude scaffold precursor. Light-induced controlled assembly of small DNA nanostructures was first realized with photocleavable protecting groups on nucleobases.20 Alternatively, biotin can be chemically coupled to ssDNA ends via a photocleavable linker (Figure 1), which is susceptible to cleavage by 365 nm ultraviolet (UV) light, thereby recovering ssDNA with a hydroxyl end. Photocleavable biotin linkers have been successful in the past for RNA aptamer selection.21 Photocleavable biotin was also used for the controlled release of the bound protein from DNA origami substrates.22 Yet their use to purify DNA NPs from complex solutions containing higher (such as dsDNA byproducts) and lower (excess staples) molecular weight contaminants has not been demonstrated.
Figure 1.
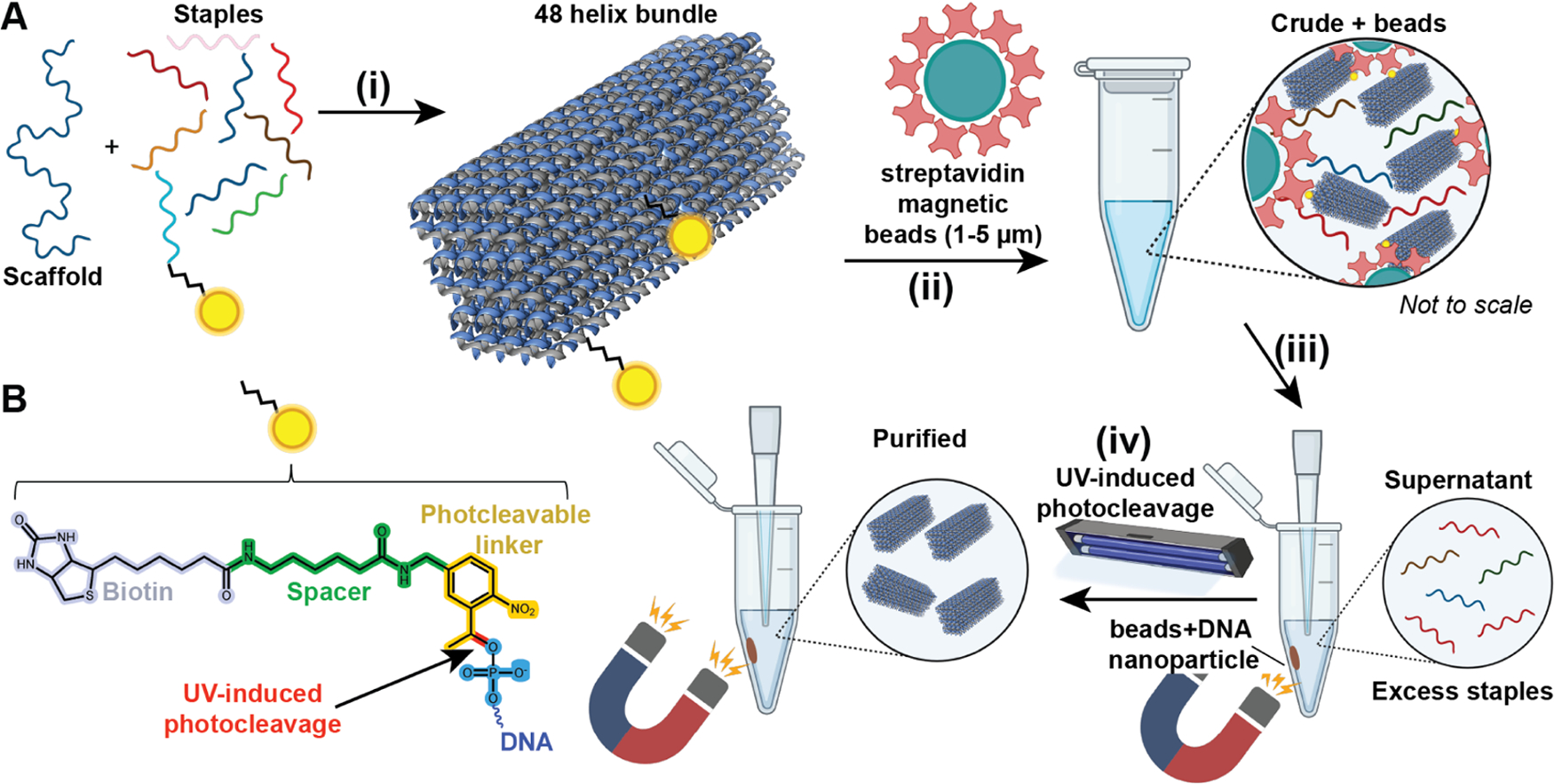
Schematic of the proposed DNA NP purification method. (A-i) A 48 helix bundle (48hb; 53 nm length × 16 nm width × 18 nm depth) DNA NP was self-assembled using the M13mp18 scaffold strand and a set of staple strands. Two staples were modified with biotin attached via a photocleavable linker. One biotin was situated on the longer face and the other on the corner of the 48hb. (A-ii) After thermal annealing, the sample (referred to as “crude”) was incubated with streptavidin-coated magnetic beads for capturing the DNA NPs via biotin–streptavidin binding. (A-iii) Using a magnet, the beads+DNA NPs were isolated or pelleted to a side in the vial, and the remaining supernatant solution was transferred to another vial for analysis and eventually discarded as waste. (A-iv) The pelleted beads with DNA NPs were reconstituted in a target buffer, exposed to UV light for photocleavage, and released from the DNA NPs. The magnet was used this time to pellet the beads and recover the target solution containing released DNA NPs. (B) Chemical structure of photocleavable linker-modified biotin attached to the end of an ssDNA. The site of photocleavage is shown in red.
In this work, streptavidin-coated beads are used to separate DNA origami NPs using photocleavable biotin-labeled (PC-biotin) staple strands and UV-triggered release. PC-biotin comprises biotin attached to a photocleavable 1-(2-nitrophenyl)-ethyl moiety via a spacer arm. The photocleavable linker undergoes photoreaction when illuminated by UV light (300–365 nm), thereby releasing any conjugated molecule in its native form.23 We hypothesize that PC-biotin staple strands will facilitate the capture of folded DNA NPs to the magnetic beads and, after UV-induced photocleavage, the recovery of DNA NPs in their original state without any overhanging staple strands or functional groups. We formulate a methodology that can be applied to separate M13mp18-based and aPCR enzymatically amplified scaffolded DNA origami NPs. In the case of using an aPCR-generated scaffold, we test the extent to which DNA NPs can be purified when a crude aPCR mixture is used directly (containing byproducts and reagents from the PCR reaction), thereby eliminating the need to prepurify the scaffold strand. Results show that in 4 h of the purification process, 57% of DNA NPs can be recovered efficiently. The yield can be increased to 91% by using a longer photocleavable biotin linker, which potentially reduces the effect of the surface charge of the DNA NP. More importantly, the aPCR-amplified scaffolded DNA origami NP does not require an intermediate step to remove the dsDNA byproduct. We anticipate that the purification technique will reduce sample loss compared to gel electrophoresis extraction.
EXPERIMENTAL METHODS
Materials.
All DNA oligonucleotides (staple strands, including PC-biotin modified staples) were purchased from Integrated DNA Technologies (IDT) and suspended in IDTE buffer pH 8.0 up to 100 μM. Staple strands tagged with PC-biotin were purchased lyophilized, and resuspended in Nuclease-Free HyPure Molecular Biology Grade Water (Cytiva, cat no. SD30538.03) to a concentration of 100 μM; concentrations were verified using a NanoQuant plate analyzed by a Tecan Spark Microplate reader. M13mp18 ssDNA (scaffold strand; cat no. P-107) at a concentration of 1.00 μg/μL was purchased from Bayou Biolabs. 1kb DNA ladder at 500 μg/mL (cat no. N3232S) was purchased from New England BioLabs (NEB), and the single-stranded 7K DNA ladder was acquired from PerkinElmer (cat no. CLS157950). Genetic analysis grade agarose (CAS no. 9012-36-6), 10× Tris borate EDTA (CAS no. 10043-35-3), sodium chloride (NaCl; CAS no. 7647-14-5), magnesium chloride hexahydrate (MgCl2; CAS no. 7791-18-6), Tween20 (CAS no. 9005-64-5), and Tris–hydrochloride (Tris–HCl; CAS no. 1185-53-1) were purchased from Fisher Bioreagents. Ethylenediaminetetraacetic acid (EDTA; CAS no. 118432500) was purchased from Thermo Scientific. Dynabeads MyOne Streptavidin C1 beads at 10 mg/mL (cat no. 65002) were purchased from Invitrogen. realUV LED Strip Lights (cat no. PN 702.65) were acquired from Waveform Lighting. A DNA Clean and Concentrator kit was purchased from Zymo Research (cat no. D4033).
DNA NP Assembly.
-
48hb: The 48hb structure used the commercially available M13mp18 scaffold strand. Assembly preparation involved combining the 10 nM M13mp18 ssDNA scaffold, 50 nM per staple strand, 50 nM PC-biotin staples in 1× TBE pH 8.4, 15 mM MgCl2, and Molecular Biology Grade Water. An Analytik Jena Biometra TRIO thermocycler was used for 48hb self-assembly using following the program:
80 °C for 5 min,
26 cycles of −1°C for 10 min from 65 to 40 °C, and 25 °C for 25 min.
The samples were then stored at 4 °C until further processing.
-
6hb and PB: The other two DNA NPs were prepared using aPCR generated scaffold strand. Scaffold synthesis was performed based on Oktay et al.’s recent work.1 There are two ways in which the scaffold strand was applied:
-
One that was gel purified to remove aPCR ingredients and the double stranded DNA (dsDNA) byproduct. Gel purification of the aPCR scaffold was performed using published protocols.1 The concentration of the scaffold was estimated using NanoDrop. Thereafter, 5-fold excess staple strands (including the PC-biotin modified ones) were combined with 10 nM purified scaffold strand in 1× TBE with 12 mM MgCl2, and annealed using the following protocol:
95 °C for 5 min,
80–75 °C at 1 °C per 5 min,
75–30 °C at 1 °C per 15 min, 30–25 at 1 °C per 10 min.
The samples were then stored at 4 °C until further processing.
One that was crude aPCR product that was not gel-purified to separate the dsDNA byproduct. This crude aPCR scaffold sample was cleaned up using Zymo cleanup kit (manufacturer’s protocol). Scaffold concentration determination could not be directly done using NanoDrop as the crude contains the amplified complementary strand apart from the scaffold strand. Scaffold concentration was determined using standard gel band intensity curve shown in Figure S6. Using this, 5-fold excess of staples (including the PC-biotin modified ones) were combined in 1× TBE with 12 mM MgCl2 and annealed using the above-mentioned protocol.
-
Purification with Streptavidin Magnetic Beads.
A wash buffer, recommended by published works,18 was prepared to rinse the streptavidin-coated magnetic beads just before use. Wash buffer comprised 10 mM Tris HCl, 1 mM EDTA, 12 mM MgCl2, 5 mM NaCl, 0.05% Tween20, and Nuclease-Free HyPure Molecular Biology Grade Water. To wash the beads, 1 mL of wash buffer was combined with 0.1 mL of beads in a 1.5 mL Eppendorf vial, mixed by inverting the tube multiple times, and separated to discard the wash by placing in a Cytiva MagRack 6 for 30 s. This wash step was repeated 6 times. We observed that fewer rinses resulted in inconsistent results.
The washed beads were then used for DNA NP (48hb, 6hb, or PB) purification or stored at 4 °C. We observed that washed beads did not require rewashing if used within 4 days. The beads are reconstituted in their original concentration (10 mg/mL). Based on a functional assay by the vendor, the biotin binding capacity (BBC) of the beads is estimated to be 490–750 pmol/mg of beads. We performed our calculations, taking the BBC to be 500 pmol/mg. The purification procedure is as follows and is illustrated in Figure 1.
50- to 200-fold pmol excess bead BBC relative to pmol of PC-biotin staple strands were combined with the annealed crude DNA NP (at 10 nM; 50 nM PC-biotin staples). Generally, the volume of beads added to the crude DNA NP solution was 0.5–2.0 equiv (v/v). 50-fold BBC beads (0.5 equiv v/v) equated to 3.3 mg/mL beads from the stock solution.
The bead+DNA NP mixtures were placed on a Disruptor Genie to shake at 1000 rpm for 1 or 3 h in a cold room.
Magnetic bead precipitation was used to separate the beads +captured DNA NPs from the supernatant containing excess staple strands or other impurities (referred to as S in Figures 2–4) by placing the vial in a MagRack for 30 s. The supernatant was removed and reserved in another vial for analysis (and subsequently discarded).
Beads with captured DNA NPs were rinsed with the target buffer (1× TBE with 15 or 12 mM MgCl2). The target buffer was added to the beads at a 1:1 volume with the original crude DNA NP volume. Beads were concentrated using the magnetic rack, and the target buffer was removed.
To cleave the DNA NP from the beads, the beads were resuspended in target buffer and placed on a Nutating Mixer in a tube rack comprising 365 nm realUV LED Strip Lights for 5–120 min (Figure S3). At this point, the DNA NPs were expected to suspend in the target buffer supernatant. Afterward, the supernatant was removed using magnetic bead precipitation for 30 s (referred to as P in Figures 2–4). The beads were discarded.
Figure 2.
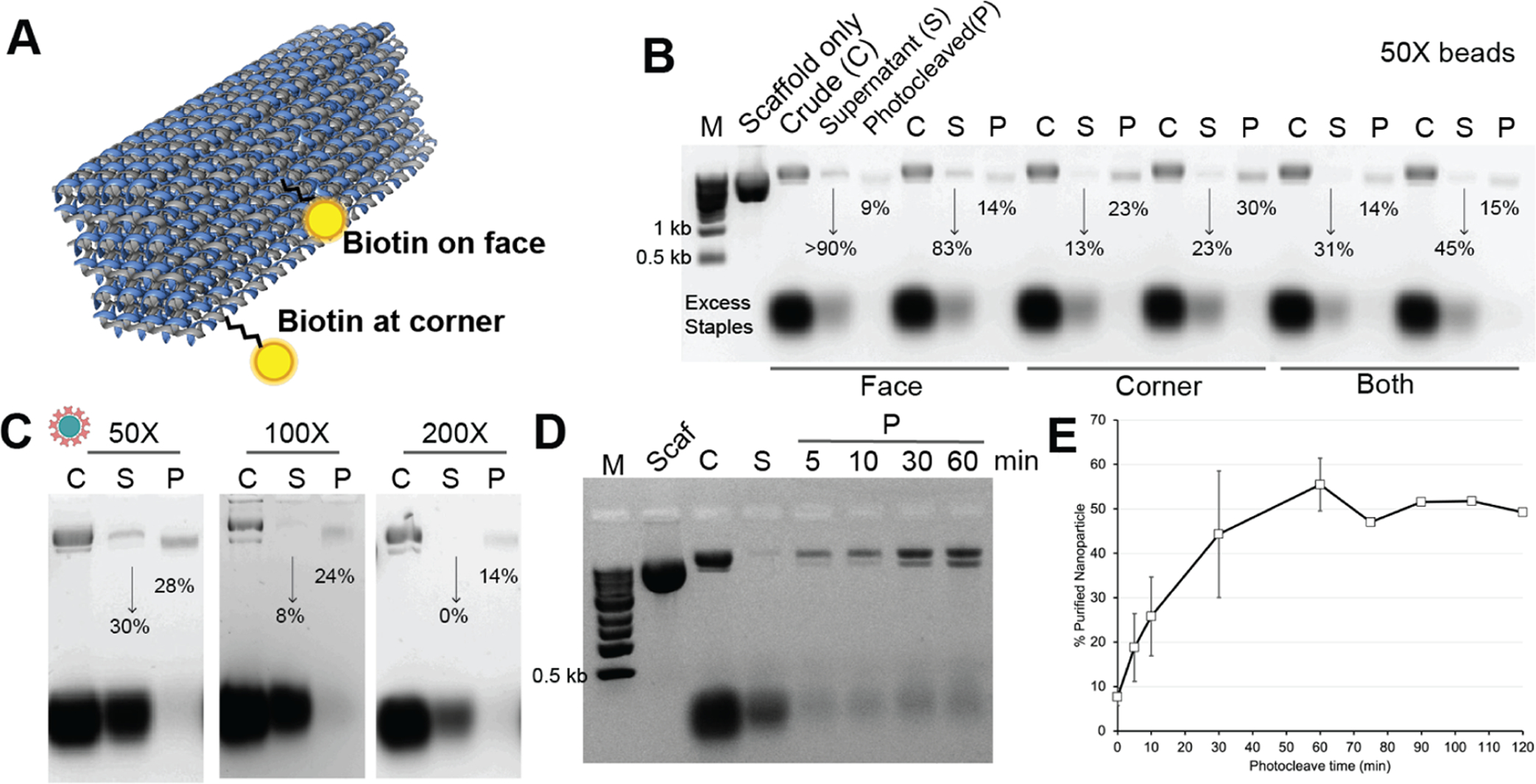
Optimization of parameters for photocleavable linker-enabled DNA NP purification. (A) First, the location of the photocleavable linker-bound biotin relative to the shape of the 48hb was tested wherein biotin in the middle of the long edge was compared to biotin in the corner (attached to the shorter edge). (B) 1% agarose gel electropherogram (AGE; in 1× TBE) showing separation of excess staples from the formed 48hb relative to a molecular marker (M), M13mp18 scaffold only. For different biotin locations present on the 48hb (either middle or corner, or both), the formed 48hb band intensity in the supernatant (S) and photocleaved/purified (P) sample was quantified. The % values below each S and P band represent formed 48hb as a fraction of 48hb in the crude “C” sample. The S lane is 6× dilute owing to the high rinse volume, but our quantification accounts for that. 50-fold excess BBC was used in this experiment. Each biotin location sample is shown in two replicates. (C) 1% AGE showing the efficiency of purification using different equivalents of the streptavidin-coated magnetic beads in proportion to the amount of biotin staples present in the crude mix. Photocleave time was 10 min. The % values below each S and P band quantify formed 48hb as a fraction of crude 48hb. (D) 1% AGE showing the effect of photocleave time (5, 10, 30, and 60 min) on the yield of purified 48hb using 200-fold excess BBC. (E) Purification yield of the method as a function of the photocleave time with 200-fold excess BBC. As seen, the purification yield % has marginal improvement after 60 min.
Figure 4.
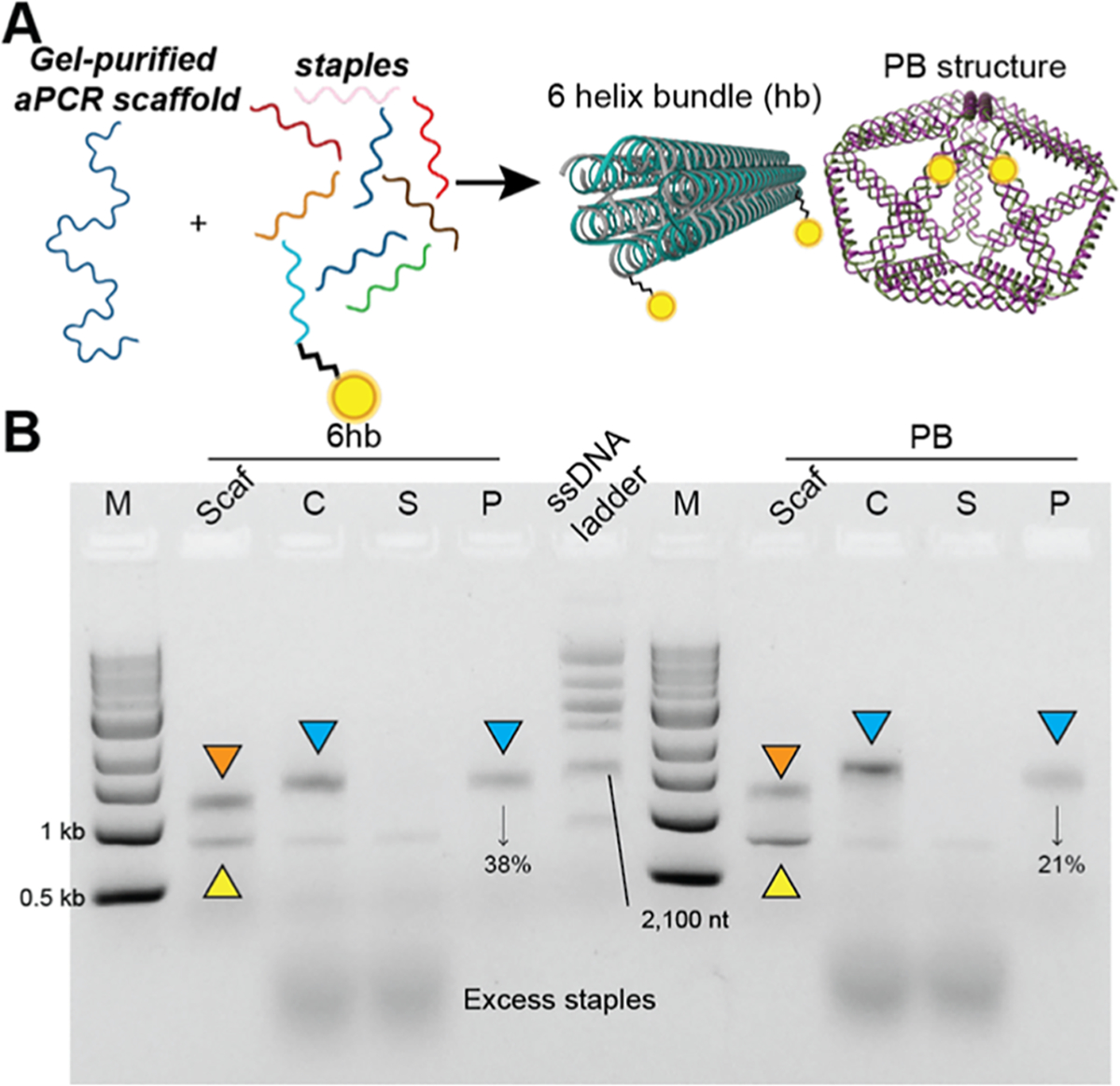
Purification of DNA NPs prepared using the prepurified aPCR-generated scaffold strand. (A) Schematic representation of assembling 6hb (85 nm length × 6 nm diameter)and PB (35 nm diameter) DNA NPs using the gel-purified aPCR-based scaffold strand (1,644 and 1,616 bp, respectively). (B) 1% AGE characterizing the purification of 6hb and PB (modified with 2 PC-biotin staples each) via magnetic bead separation. Lanes are as follows: M (1kb ladder), Scaf (gel-purified aPCR scaffold), C (crude 6hb annealed mix), S (supernatant), P (photocleave released pure 6hb), and ssDNA ladder (indicated band is 2,100 nt), followed by corresponding bands for PB. Bands indicated with orange triangles represent residual dsDNA, yellow triangles represent the aPCR-generated scaffold strand, and blue triangles represent the purified NPs, which were further characterized by AFM/TEM in Figure 6. % values indicate yield (pure/crude × 100).
Transmission Electron Microscopy Imaging.
400-mesh carbon Formvar grids (Electron Microscopy Sciences, cat no. FCF400-Cu-UA) were glow-discharged for 30 s at 15 mA negative polarity using the PELCO easiGlow glow discharge system to render the surface hydrophilic and facilitate efficient sample deposition in the subsequent steps. For optimum results, glow-discharged grids were used within 15 min of treatment.
5 μL of the purified 48hb, 6hb, or PB structures were applied to treated grids and incubated for 5 min. Meanwhile, a fresh 1% uranyl formate solution in water was prepared. To this, 1.25 μL of freshly prepared 1 M NaOH was added, mixed well for 5 min in a thermomixer at 750 rpm, and centrifuged for 30 s using a tabletop microcentrifuge. We observed that preparation of fresh uranyl stain showed dramatic improvement in results when compared to uranyl stains that were stored at −20 °C for extended periods of time.
After the sample was incubated for 5 min, the excess sample was wicked away, and 10 μL of freshly activated uranyl formate was applied to the grid. Incubation was done for 30 s, and excess was wicked away. The grids were then air-dried and imaged using a Tecnai F20 transmission electron microscope operated at 200 kV. Images were analyzed by using Fiji ImageJ analysis software.
Atomic Force Microscopy (AFM) Characterization.
Structural characterization was performed on a JPK Nanowizard 4 fast-scan AFM instrument in a fluid using USC-f0.3-k0.3 tips from NanoWorld. Approximately 30 fmol of each structure in 0.5× TBE and 18 mM MgCl2 (~2 nM structure) was deposited onto a freshly cleaved, 1 cm diameter mica disk and incubated for 5 min. The mica surface was then rinsed twice with the same buffer, and 100 μL of the buffer was split between the AFM tip and the sample for imaging. NiCl2 was added to the mica to improve the adhesion of structures during imaging, and the final imaging buffer was composed of 0.5× TBE, 18 mM MgCl2, and 7 mM NiCl2. Images were acquired with AC mode and fast imaging using the Fast EB scan head, and images were acquired with 6 Hz line scan rates and 1 or 2 nm2 pixel sizes.
Comparison of Different Purification Techniques.
PB was assembled using the aPCR product as the scaffold precursor and then purified using the current method as well as other commonly used techniques, namely, poly(ethylene glycol) (PEG) precipitation, ultrafiltration using two molecular weight cutoff columns (50 and 100 kDa), and gel extraction. In each case, 100 μL of crude PB at 10 nM was put through the purification process. Below are the methods for each technique.
PEG Precipitation.
100 μL of the crude PB origami solution was diluted 2-fold into its respective target buffer (12 mM MgCl2, 1× TBE, pH = 8) to obtain a starting volume of 200 μL. This was then combined in a 1:1 ratio with PEG precipitation buffer (15% PEG 8000 (w/v), 1× TBE, and 505 mM NaCl).24,25 The sample was mixed via pipetting and was then transferred to a centrifuge to spin at 13,000g at room temperature for 30 min. The supernatant was removed using a pipette, and the resulting pellet was resuspended in 100 μL of the target buffer. The solution was equilibrated overnight at room temperature prior to the analysis of the sample.
Ultrafiltration.
100 μL of the crude PB origami solution was purified and concentrated using 50 and 100 kDa Amicon Ultra Centrifugal Filters. The filters were first equilibrated using 500 μL of working buffer (12 mM MgCl2, 1× TBE, pH = 8) and spun at 13,000g for 2 min at 10 °C. Excess liquid was discarded. 100 μL of crude PB origami and 400 μL of working buffer were added to the filter and spun at 9000g for 5 min at 10 °C. The filters were then washed by adding 500 μL of working buffer to the filter and spinning at 9000g for 5 min at 10 °C (washing was repeated a total of three times). Purified PB was eluted by inverting the filter upside down into a clean microcentrifuge tube and centrifuging at 2000g for 3 min at 10 °C.
Gel Extraction.
A 45 μL portion of the crude PB origami solution was run in a 2% agarose gel containing 10 mM MgCl2 and 1× TBE for 2 h (90 V/cm). The desired PB NP band (indicated by a blue triangle in Figure S7) was excised using a razor blade, and excess agarose was trimmed from all sides of the DNA band to maximize yield. The gel was then placed into a Bio-Rad Freeze ‘N Squeeze DNA Gel Extraction Spin Column. The column was then placed into a −20 °C freezer for 5 min. The sample was then spun at 13,000g for 3 min at room temperature using a microcentrifuge. Purified PB origami was collected from the tube, and the filter column was discarded. Subsequently, the PB solution was concentrated using PEG precipitation and was reconstituted in 45 μL of target buffer (12 mM MgCl2, 1× TBE, pH = 8) to maintain the initial volume.
Quantification of Gel Extraction.
Following gel extraction by the Freeze ‘N Squeeze Column, purified PB origami was eluted in a total volume of 220 μL. NanoDrop was used to calculate a concentration of 1.3 ng/μL. Using the molecular weight of the PB nanostructure (998,260.4 g/mol), 0.286 pmol of PB was successfully recovered using this technique. The theoretical amount of recovered origami is 0.450 pmol (45 μL loaded, 10 nM scaffold concentration). In dividing these values, the percent recovery of the PB structure is 63.6%. However, for downstream applications, it is necessary for another method (such as PEG precipitation) to concentrate the origami solution.
RESULTS AND DISCUSSION
To test and optimize our purification method, we first built a 48hb DNA origami NP comprising the native (and commercially available) M13mp18 strand as the scaffold. Figure 1A shows a schematic of the 48hb NP, and Figure S1 represents the caDNAno cross-section design. Two staple strands were modified with PC-biotin (DNA sequences are listed in Table S1 in the Supporting Information (SI) for purification. One strand was located at a corner of the bundle, while the other strand was situated in the middle of the longer faces of the bundle. We hypothesized that the PC-biotin modification on the corner staple would perform better in capturing the DNA NP on the magnetic beads compared to the other staple due to lower electrostatic hindrance from the DNA NP helices. The overall pipeline was to self-assemble the DNA NPs using thermal annealing. Thereafter, the crude annealed mix was combined with prewashed streptavidin-coated magnetic beads (see the Experimental Methods for the washing procedure). The beads were incubated with the crude folding mix in the cold room on a shaker. Next, the beads were collected by magnet concentration, and the supernatant containing excess oligos was removed. Beads were resuspended in the target buffer (typically the same buffer in which the DNA NP was originally assembled) and subjected to UV-induced photocleavage on a rocker. Given the ubiquity of UV gel transilluminators in DNA nanotechnology research laboratories, we first used it as our UV source (302 nm) and photocleaved for 10 min, as recommended in the literature.26 Beads were pelleted again using a magnetic rack, and the supernatant containing the purified 48hb DNA NPs was collected.
To optimize the protocol for separating DNA NPs using this technique, we optimized four independent variables, namely, incubation time with the beads (Figure 1A–ii), stoichiometry of beads to PC-biotin staples in the crude mix, UV treatment time to release the captured DNA NPs via photocleavage (Figure 1A–iv), and the UV source. First, we assembled the 48hb using established thermal annealing procedures by combining scaffold and 5-fold excess staples at final concentrations of 10 and 50 nM, respectively, in 1× TBE and 15 mM MgCl2 buffer. This annealed solution is referred to as crude (or “C”) in the rest of the article. To determine how many beads would be necessary to maximize the capture of the DNA NPs, the vendor-provided biotin binding capacity (BBC) of the beads was used (see the Experimental Methods). A conservative estimate of the bead BBC by the manufacturer is 500 pmol biotin per mg beads. We aimed to have an excess BBC to the total PC-biotin-functionalized staple strands in the assembled crude mix. We therefore tested 50-, 100-, and 200-fold excess bead BBC in relation to moles of PC-biotin staples in the assembly mix to purify the DNA NPs. These quantities generally equated to 0.5–2.0 equiv (v/v) of 10 nM DNA NP with 5-fold excess PC-biotin staple strands.
The incubation time for biotin and PC-biotin-functionalized strands varies considerably based on the quality or age of the streptavidin-coated beads as well as the nature of the analyte; DNA NPs with an extending poly-A staple tail have been incubated overnight on a shaker to be captured by hybridization to biotinylated-capture strands on the beads;17,18 attaching the short biotinylated-capture strands to the beads, however, required only 1 h incubation. The incubation time is shown to be proportionate to the length of the biotinylated DNA; longer biotin-tagged amplified PCR scaffold strands were captured on streptavidin-coated magnetic beads by incubating for 3–4 h.11
Results are shown in Figure 2 for the optimization of each parameter. We started with testing how many PC-biotin staples are necessary on the 48hb for bead capture. We assembled the 48hb with either a face or corner PC-biotin staple or both. After assembly, we combined the samples with 50-fold excess bead BBC compared with the PC-biotin staple amount. The incubation time was kept at 1 h, and photocleavage was done using a UV gel illuminator for 10 min. The crude (C), supernatant (S), and photocleaved/purified (P) in different PC-biotin-configured 48hb NPs were compared via agarose gel electrophoresis, as shown in Figure 2B. We found that 48hb with one PC-biotin on the face resulted in more NPs remaining in the supernatant, indicating poor capture. Using the PC-biotin displayed on the corner of 48hb was sufficient to photocleave and purify the 48hb. Surprisingly, using both PC-biotin staples simultaneously did not improve the purification. The best yield achieved was still 30%.
We next tested altering the equivalency of beads used for NP capture (Figure 2C). When comparing 50-, 100-, and 200-fold excess bead BBC to PC-biotin staples, it was found that 100-fold was sufficient to reduce loss of the NP in the supernatant (14%) and 24% recovery of purified 48hb from photocleavage. It was unclear why adding more beads (200× bead BBC) reduced the purification yield, but it enabled complete capture and zero residual DNA NP in the supernatant. We subsequently tested 200× with different mixing and photocleaving times to further improve yield. For instance, 3 h of constant gentle shaking of the crude 48hb with 200× bead BBC compared to 1 h incubation reduced loss in the supernatant.
As mentioned above, the UV source that we used was a lamp inside a gel transilluminator. However, it was challenging to set up a rocker/shaker in the illuminator box to provide constant shaking to the sample, which was necessary to prevent the beads from settling to the bottom of the vial. We, therefore, assembled another UV source based on Strobel et al.’s work using a 365 nm UV LED light strip (~10 mW/cm2), available for under $50.27 Figure S3 shows the light strip setup that was used. Other notable UV sources applied in the literature can be explored as well.23,26,28,29 UV treatment was then tuned from 10 to 120 min to photocleavage and release. As summarized in Figures 2D–E, the yield plateaued after 60 min of photocleavage at 57 ± 6%. This yield is difficult to compare to the other instances where PC-biotin was used in the context of DNA NPs.22 AFM and TEM images of the purified 48hb confirmed the formation and high purity of the DNA NP (Figure 3). We observed multimer formation in the 48hb images, which we attribute to limited overhangs along the helix ends of the NP.
Figure 3.
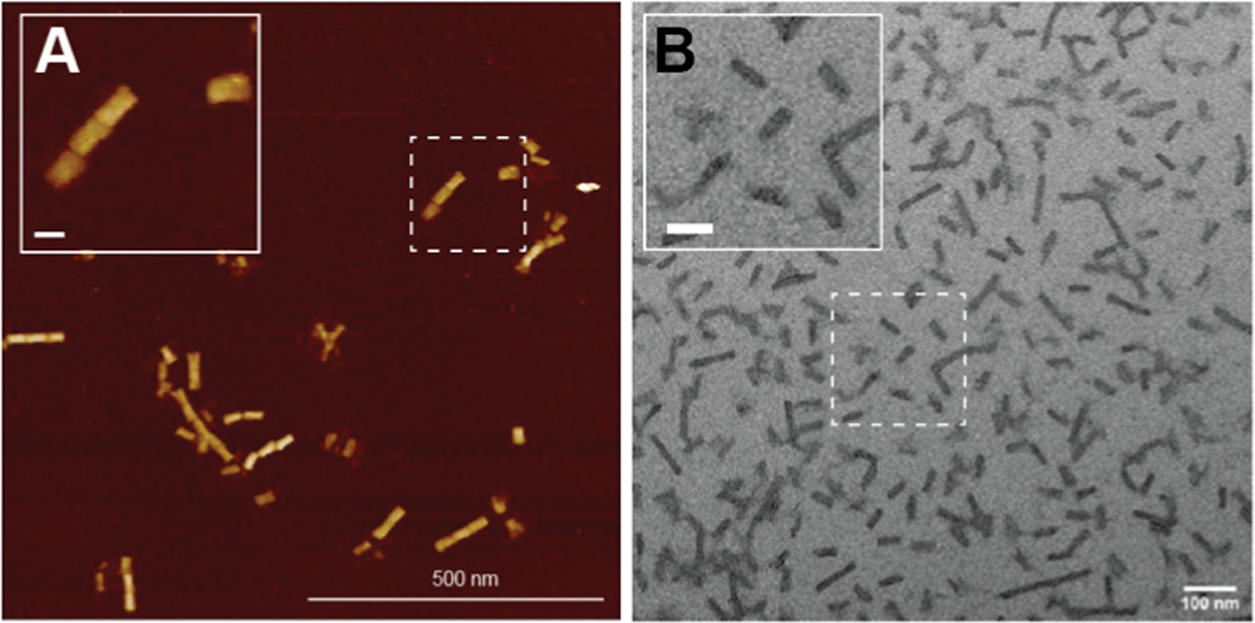
Microscopic characterization of purified 48hb. Representative images of 48hb using (A) AFM and (B) TEM. The Inset scale bar is 50 nm.
Next, we focused on the purification of 6hb and pentagonal bipyramid (PB) NPs that were assembled by using aPCR-generated gel-purified scaffold strands (Figure 4A). These two structures were derived from published works, and the sequences can be accessed from Oktay et al.1 The 6hb uses a 1,644 nucleotide (nt) long scaffold strand, while the PB uses a 1,616 nt long scaffold, both derived by amplifying the requisite length fragment from the M13mp18 template strand. Two staple strands in each structure were modified with a PC-biotin label (Table S1). We followed the recommended protocol to anneal the two structures (see the Experimental Methods) and then subjected them to the purification procedure. Using 50-fold excess bead BBC was found to be sufficient to capture all of the 6hb and PBs, as seen by the absence of NPs in the supernatant “S” lane (Figure 4B). Parameters for the incubation time (3 h) and photocleavage (1 h UV light strip) remained consistent. Impurities such as excess staples and dsDNA byproduct were entirely removed from the photocleaved/purified product (Figure 4B, lane “P”). In the aPCR gel-purified scaffold lane, there was some residual dsDNA byproduct, which was absent from the “S” and “P” lanes, leading us to believe that the scaffold strand within the dsDNA was leveraged and folded into NPs and the antisense strand was left behind. Another point worth highlighting is that this technique successfully purifies helix bundles (6hb and 48hb) and wireframe polyhedral shapes (PB) of DNA NPs, demonstrating added shape-related versatility. For both structures, complete capture by the beads was observed (lanes “S” in Figure 4), but photocleavage and release from the beads yielded PB at 21% and 6hb at 38% efficiency.
Finally, to demonstrate broader applicability, we tested the purification technique to purify the 6hb and PB NPs assembled using a crude aPCR scaffold mixture (Figure 5A). Traditionally, an aPCR-derived scaffold strand is separated from the dsDNA byproduct (and other aPCR-related reagents) inherent to aPCRs before it is used for DNA NP assembly.30 The reason for purifying the scaffold strand from aPCR mixtures is to reduce interference from the dsDNA byproduct in downstream applications. The most established way to isolate and purify scaffold from aPCR is by gel extraction, which is known to provide high-quality separation but low yield.16 If a downstream purification approach bypasses the need for gel-purifying the scaffold, then one could use the crude aPCR mix as the scaffold sample for DNA NP assembly and increase the overall quantity of NP created.
Figure 5.
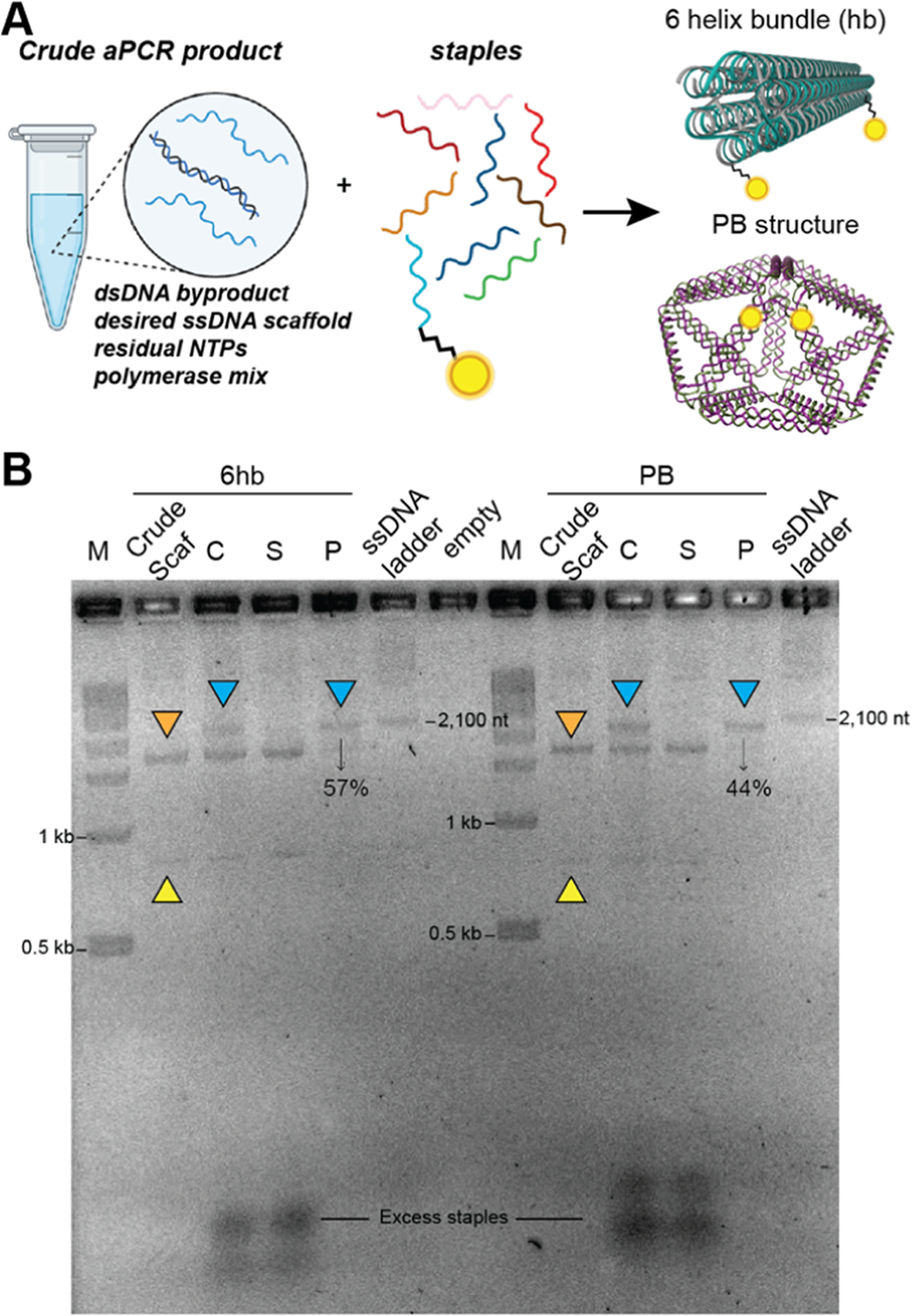
Purification of DNA NPs prepared using the crude aPCR-generated scaffold strand. (A) Schematic representing the assembly of 6hb and PB using the scaffold straight from the aPCR crude mixture that contains the dsDNA byproduct, residual dNTPs, and proteins in addition to the desired scaffold. (B) The NPs were assembled and characterized via 2% AGE postrun stained using 3× GelRed. Lanes are as follows: (M) 1kb dsDNA ladder, 6hb crude scaffold, crude annealed 6hb, supernatant and purified 6hb, ssDNA ladder (2,100 nt band indicated), and empty lane, followed by the same lanes corresponding to PB. Assembled NPs are indicated by blue triangles, the dsDNA byproduct in the crude aPCR mix is indicated by orange triangles, and the scaffold strand is indicated by yellow triangles.
The PC-biotin purification approach designed here is based on the principle of isolating formed DNA NPs from high-molecular-weight dsDNA contaminants or smaller-molecular-weight residual oligos. Therefore, we hypothesized that it would enable isolating formed DNA NPs from a crude mixture containing aPCR byproducts and excess staples without interference from any dsDNA-related hybridization. We therefore used the crude aPCR product (after PCR cleanup for desalting) as the scaffold and combined it with 5× excess staple strands (including the PC-biotin staples) to anneal the PB and 6hb. Thereafter, the samples were subjected to the purification procedure optimized as above. Figure S4 shows that changing the beads amount (50-, 100-, versus 200-fold) did not alter the amount of uncaptured DNA NPs in the supernatant “S” lanes. The gel electropherogram shown in Figure 5B highlights that both 6hb and PB are purified (see lanes labeled as “P”). Microscopy results of the photocleaved product in Figure 6 show well-formed 6hb and PB. The crude scaffold lanes (Figure 5B “crude scaf”) show multiple bands representing the dsDNA byproduct and the scaffold-only band. Lanes “C” show a new band indicating annealed 6hb and PB NPs. In the supernatant “S” lanes, everything but the annealed NP remains, and in the lanes labeled “P,” the purified/photocleaved NP is isolated. We estimated the yield by measuring the band intensity in the gel of purified NP as a fraction of the annealed crude NP (equal volume of sample was loaded in the gel), which was 50 ± 8% and 59 ± 8% for the 6hb and PB, respectively (Figure S5). To put this in context of how much yield can be achieved using the gel-purified aPCR scaffold, we estimated that gel extract results in 16 ± 3% scaffold recovery (Figure S6), which albeit could be assembled into a DNA NP and purified subsequently using higher efficiency techniques. Thus, an overall higher yield of the final NP can be achieved when using the crude aPCR scaffold mix compared to when the gel-purified scaffold is used.
Figure 6.

Microscopic characterization of 6hb and PB post purification. Representative (A, C) AFM and (B, D) TEM images of 6hb and PB, respectively, assembled using the gel-purified aPCR mix and purified using the PC-biotin and magnetic beads. Representative (E, G) AFM and (F, H) TEM images of 6hd and PB, respectively, assembled using the crude aPCR mix and purified using the PC-biotin and magnetic beads. Inset scale bars: 50 nm.
When determining the efficiency of an analytical technique in purifying a DNA NP, four factors are worth considering, namely, the ease of execution, the quality of the purified DNA NP, the quantified yield of the recovered DNA NP, and the extent to which the sample suffers dilution during the purification process.25 Table S3 summarizes our comparative analysis of representative purification techniques and the PC-biotin approach. The PC-biotin purification protocol takes only 4 h to purify a DNA NP sample, making it on par, if not quicker, than other purification techniques while requiring less hands-on time. It eliminates a step necessary for aPCR scaffold purification prior to the DNA NP assembly. The quality of the DNA NP recovered can be considered better than size exclusion (precipitation via PEG or ethanol and ultrafiltration) purifications that retain higher-molecular-weight dsDNA contaminants (Figure S7). The quality of gel-extracted DNA NPs is the highest due to the ability to resolve constructs by their size and selectively excise the band representing formed DNA NPs while discarding higher- and lower-molecular-weight contaminants. However, the sample retains agarose particulates that could compromise downstream application. Like previously described biotin–magnetic bead-based techniques, PC-biotin tends to retain higher-molecular-weight DNA NP aggregates. The DNA NP recovered after photocleavage in the proposed technique is in its native form without any extending DNA overhangs or residual agarose or PEG in the sample.
Compared to other techniques such as PEG/ethanol precipitation (>90%)24,31 and strand displacement-driven magnetic bead separation (>90%),17 the observed yield using the current PC-biotin is admittedly lower (57%), but it can be improved to 90% using a longer linker (as discussed below). PEG-based precipitation was unsuccessful in purifying the wireframe PB NP (Figure S7) but sufficient evidence points to its consistent application to purify a helix bundle and denser DNA NPs with high efficiency. The DNA NP purified using gel extraction can recover up to 20% of the crude NP in pure form (Figure S7 yielded only 11%). Ultrafiltration-based purification yield also varies considerably between 20 and 80%. More critically, gel extraction tends to decrease the concentration of the purified DNA NP due to dilution during the purification process. A subsequent concentration step is often required, which resorts back to PEG or ethanol precipitation or ultrafiltration. By simply reducing the target buffer used during the photocleavage step to reconstitute the DNA NPs in the proposed work, one can change the concentration of the purified sample.
CONCLUSIONS
We demonstrate here a new method for purifying both M13-based scaffolded DNA origami NPs (such as 48hb) and aPCR-generated custom-scaffolded DNA NPs (such as 6hb and PB) through PC-biotin-functionalized staple strands and streptavidin-coated magnetic beads. Gel-purifying the aPCR-generated scaffold prior to NP assembly is no longer necessary, and the crude aPCR product can be directly used as the scaffold to fold NPs and purify in a subsequent single step. The protocol is comparable, if not quicker, in the time required while requiring less hands-on time.
PC-biotin-enabled DNA NP purification helps access more aPCR-synthesized scaffolds than was previously possible. We found that purification of custom scaffolds by gel extraction from aPCR products was low (16 ± 3%), suggesting that there is considerable loss of the scaffold in the putative two-step approach of assembling custom DNA NPs that could otherwise be utilized. Therefore, the proposed technique uniquely allows for augmenting the yield of custom-scaffolded DNA NP by reducing the gel-extracted loss of the scaffold strand.
Collectively, the PC-biotin approach to purifying DNA NPs serves to simplify the process and caters to the growing demand for custom-scaffolded NPs. This method adds to and expands the toolset for purifying these structures, and purification is not an either–or approach where you can only pick and use one technique.25 Techniques can be adapted, adjusted, and combined, and our process allows for more choices and options, which will be useful, since it allows scientists to choose what suits them best. Alterations to the linker length, the number of PC-biotin-functionalized staples, and their location on DNA NPs can increase the overall yield. The current PC-biotin functionalization is connected by a standard linker (supplied by an IDT). A longer linker containing an 18C spacer (referred to as PC-biotin-L), for instance, increased the 48hb yield from 57% to 91 ± 7% and required a smaller proportion of magnetic beads (50-fold, see Figure S8). It is possible that a higher bead concentration becomes inhibitory to effective capturing. The results observed in using a longer linker depended considerably on the location of the staple within the 48hb. This makes sense from a design point of view; the probability of longer tethers being entrapped within the folded DNA NP can be higher than that of shorter tethers. Future work, therefore, is needed to probe the optimum position to display longer PC-biotin linkers, noting that it might also be DNA NP-dependent. Further studies are also warranted on ways to increase the yield by optimizing the position and the number and location of the PC-biotin moieties displayed on the NP, different streptavidin-coated magnetic bead types, and the efficiency of the technique in purifying larger DNA NPs.
Supplementary Material
ACKNOWLEDGMENTS
I.L.M. acknowledges the Office of Naval Research, the U.S. Naval Research Laboratory (NRL), and the NRL’s Nanoscience Institute for programmatic funding. T.L.S. acknowledges the National Institutes of Health/National Institute of General Medical Sciences (1R35GM142706). This work was supported by the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health (R00EB030013). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS
- DNA
deoxyribonucleic acid
- DNA NP
DNA nanoparticle
- PC-biotin
photocleavable-biotin
- PC-biotin-L
photocleavable-biotin with a longer linker
- aPCR
asymmetric polymerase chain reaction
- AFM
atomic force microscopy
- TEM
transmission electron microscopy
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.3c18955.
Quantification estimate of aPCR scaffold recovery; DNA 48hb design and staples; UV photocleavable string of the light setup; 6hb and PB purification; yield estimate for the aPCR-generated scaffold; comparison of the PC-biotin purification method with existing techniques; and 48hb purification using a longer PC-biotin linker (PDF).
Complete contact information is available at: https://pubs.acs.org/10.1021/acsami.3c18955
Contributor Information
Heather R. Everson, Department of Chemistry, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Kayla Neyra, Department of Chemistry, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Dylan V. Scarton, College of Science, Interdisciplinary Program in Neuroscience, George Mason University, Fairfax, Virginia 22030, United States; Institute for Advanced Biomedical Research, George Mason University, Manassas, Virginia 20110, United States
Soumya Chandrasekhar, Department of Physics, Kent State University, Kent, Ohio 44240, United States.
Christopher M. Green, Center for Bio/Molecular Science and Engineering, US Naval Research Laboratory, Washington, District of Columbia 20375, United States
Thorsten-Lars Schmidt, Department of Physics, Kent State University, Kent, Ohio 44240, United States.
Igor L. Medintz, Center for Bio/Molecular Science and Engineering, US Naval Research Laboratory, Washington, District of Columbia 20375, United States
Remi Veneziano, Institute for Advanced Biomedical Research and College of Engineering and Computing, Department of Bioengineering, George Mason University, Manassas, Virginia 20110, United States.
Divita Mathur, Department of Chemistry, Case Western Reserve University, Cleveland, Ohio 44106, United States.
REFERENCES
- (1).Oktay E; Bush J; Vargas M; Scarton DV; O’Shea B; Hartman A; Green CM; Neyra K; Gomes CM; Medintz IL; Mathur D; Veneziano R Customized Scaffolds for Direct Assembly of Functionalized DNA Origami. ACS Appl. Mater. Interfaces 2023, 15 (23), 27759–27773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Oktay E; Alem F; Hernandez K; Girgis M; Green C; Mathur D; Medintz IL; Narayanan A; Veneziano R DNA origami presenting the receptor binding domain of SARS-CoV-2 elicit robust protective immune response. Commun. Biol. 2023, 6 (1), No. 308, DOI: 10.1038/s42003-023-04689-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Narayanan RP; Prasad A; Buchberger A; Zou L; Bernal-Chanchavac J; MacCulloch T; Fahmi NE; Yan H; Zhang F; Webber MJ; Stephanopoulos N High-Affinity Host-Guest Recognition for Efficient Assembly and Enzymatic Responsiveness of DNA Nanostructures. Small 2024, 20, No. e2307585, DOI: 10.1002/smll.202307585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Spratt J; Dias JM; Kolonelou C; Kiriako G; Engstrom E; Petrova E; Karampelias C; Cervenka I; Papanicolaou N; Lentini A; Reinius B; Andersson O; Ambrosetti E; Ruas JL; Teixeira AI Multivalent insulin receptor activation using insulin-DNA origami nanostructures. Nat. Nanotechnol. 2024, 19, 237–245, DOI: 10.1038/s41565-023-01507-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Rothemund PWK Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440 (7082), 297–302. [DOI] [PubMed] [Google Scholar]
- (6).Kramm K; Schroder T; Vera AM; Grabenhorst L; Tinnefeld P; Grohmann D DNA Origami-Based Single-Molecule Force Spectroscopy and Applications. Methods Mol. Biol. 2024, 2694, 479–507. [DOI] [PubMed] [Google Scholar]
- (7).Mathur D; Diaz SA; Hildebrandt N; Pensack RD; Yurke B; Biaggne A; Li L; Melinger JS; Ancona MG; Knowlton WB; Medintz IL Pursuing excitonic energy transfer with programmable DNA-based optical breadboards. Chem. Soc. Rev. 2023, 52 (22), 7848–7948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Liu N; Liedl T DNA-Assembled Advanced Plasmonic Architectures. Chem. Rev. 2018, 118 (6), 3032–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bhatia D; Wunder C; Johannes L Self-assembled, Programmable DNA Nanodevices for Biological and Biomedical Applications. ChemBioChem 2021, 22 (5), 763–778. [DOI] [PubMed] [Google Scholar]
- (10).Kretzmann JA; Liedl A; Monferrer A; Mykhailiuk V; Beerkens S; Dietz H Gene-encoding DNA origami for mammalian cell expression. Nat. Commun. 2023, 14 (1), No. 1017, DOI: 10.1038/s41467-023-36601-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wu X; Yang C; Wang H; Lu X; Shang Y; Liu Q; Fan J; Liu J; Ding B Genetically Encoded DNA Origami for Gene Therapy In Vivo. J. Am. Chem. Soc. 2023, 145 (16), 9343–9353. [DOI] [PubMed] [Google Scholar]
- (12).Wu X; Liu Q; Liu F; Wu T; Shang Y; Liu J; Ding B An RNA/DNA hybrid origami-based nanoplatform for efficient gene therapy. Nanoscale 2021, 13 (30), 12848–12853. [DOI] [PubMed] [Google Scholar]
- (13).Lin-Shiao E; Pfeifer WG; Shy BR; Doost MS; Chen E; Vykunta VS; Hamilton JR; Stahl EC; Lopez DM; Espinoza CRS; Deyanov AE; Lew RJ; Poirer MG; Marson A; Castro CE; Doudna JA CRISPR-Cas9-mediated nuclear transport and genomic integration of nanostructured genes in human primary cells. Nucleic Acids Res. 2022, 50 (3), 1256–1268, DOI: 10.1093/nar/gkac049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Silva-Santos AR; Oliveira-Silva R; Rosa SS; Paulo PMR; Prazeres DMF Affinity-Based Magnetic Particles for the Purification of Single-Stranded DNA Scaffolds for Biomanufacturing DNA-Origami Nanostructures. ACS Appl. Nano Mater. 2021, 4 (12), 14169–14177, DOI: 10.1021/acsanm.1c03623. [DOI] [Google Scholar]
- (15).Bush J; Singh S; Vargas M; Oktay E; Hu CH; Veneziano R Synthesis of DNA Origami Scaffolds: Current and Emerging Strategies. Molecules 2020, 25 (15), No. 3386, DOI: 10.3390/molecules25153386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Mathur D; Medintz IL Analyzing DNA Nanotechnology: A Call to Arms For The Analytical Chemistry Community. Anal. Chem. 2017, 89 (5), 2646–2663. [DOI] [PubMed] [Google Scholar]
- (17).Shaw A; Benson E; Hogberg B Purification of functionalized DNA origami nanostructures. ACS Nano 2015, 9 (5), 4968–4975, DOI: 10.1021/nn507035g. [DOI] [PubMed] [Google Scholar]
- (18).Ye J; Teske J; Kemper U; Seidel R Sequential Pull-Down Purification of DNA Origami Superstructures. Small 2021, 17 (17), No. e2007218, DOI: 10.1002/smll.202007218. [DOI] [PubMed] [Google Scholar]
- (19).Burgahn T; Garrecht R; Rabe KS; Niemeyer CM Solid-Phase Synthesis and Purification of Protein-DNA Origami Nanostructures. Chem. - Eur. J. 2019, 25 (14), 3483–3488, DOI: 10.1002/chem.201805506. [DOI] [PubMed] [Google Scholar]
- (20).Schmidt TL; Koeppel MB; Thevarpadam J; Goncalves DP; Heckel A A light trigger for DNA nanotechnology. Small 2011, 7 (15), 2163–2167, DOI: 10.1002/smll.201100182. [DOI] [PubMed] [Google Scholar]
- (21).Cho S; Lee SH; Chung WJ; Kim YK; Lee YS; Kim BG Microbead-based affinity chromatography chip using RNA aptamer modified with photocleavable linker. Electrophoresis 2004, 25 (21–22), 3730–3739, DOI: 10.1002/elps.200406103. [DOI] [PubMed] [Google Scholar]
- (22).Brglez J; Ahmed I; Niemeyer CM Photocleavable ligands for protein decoration of DNA nanostructures. Org. Biomol. Chem. 2015, 13 (18), 5102–5104. [DOI] [PubMed] [Google Scholar]
- (23).Olejnik J; Sonar S; Krzymanska-Olejnik E; Rothschild KJ Photocleavable biotin derivatives: a versatile approach for the isolation of biomolecules. Proc. Natl. Acad. Sci. U.S.A. 1995, 92 (16), 7590–7594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Stahl E; Martin TG; Praetorius F; Dietz H Facile and scalable preparation of pure and dense DNA origami solutions. Angew. Chem., Int. Ed. 2014, 53 (47), 12735–12740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Neyra K; Everson HR; Mathur D Dominant Analytical Techniques in DNA Nanotechnology for Various Applications. Anal. Chem. 2024, 96 (9), 3687–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bai X; Kim S; Li Z; Turro NJ; Ju J Design and synthesis of a photocleavable biotinylated nucleotide for DNA analysis by mass spectrometry. Nucleic Acids Res. 2004, 32 (2), 535–541, DOI: 10.1093/nar/gkh198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Strobel EJ Preparation of E. coli RNA polymerase transcription elongation complexes by selective photoelution from magnetic beads. J. Biol. Chem. 2021, 297 (1), No. 100812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Shimada YJ; Hasegawa K; Kochav SM; Mohajer P; Jung J; Maurer MS; Reilly MP; Fifer MA Application of Proteomics Profiling for Biomarker Discovery in Hypertrophic Cardiomyopathy. J. Cardiovasc. Transl. Res. 2019, 12 (6), 569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yang H; Zhang Y; Yu Z; Liu S-Y; Xu Y; Dai Z; Zou X A photo-elutable and template-free isothermal amplification strategy for sensitive fluorescence detection of 5-formylcytosine in genomic DNA. Chin. Chem. Lett. 2023, 34 (3), No. 107536. [Google Scholar]
- (30).Veneziano R; Ratanalert S; Zhang K; Zhang F; Yan H; Chiu W; Bathe M Designer nanoscale DNA assemblies programmed from the top down. Science 2016, 352 (6293), No. 1534, DOI: 10.1126/science.aaf4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lei Y; Mei Z; Chen Y; Deng N; Li Y Facile Purification and Concentration of DNA Origami Structures by Ethanol Precipitation. ChemNanoMat 2022, 8 (7), No. e202200161. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.