

Summary
The cystic fibrosis transmembrane conductance regulator (CFTR) is a crucial ion channel whose loss of function leads to cystic fibrosis, while its hyperactivation leads to secretory diarrhea. Small molecules that improve CFTR folding (correctors) or function (potentiators) are clinically available. However, the only potentiator, ivacaftor, has suboptimal pharmacokinetics and inhibitors have yet to be clinically developed. Here we combine molecular docking, electrophysiology, cryo-EM, and medicinal chemistry to identify CFTR modulators. We docked ~155 million molecules into the potentiator site on CFTR, synthesized 53 test ligands, and used structure-based optimization to identify candidate modulators. This approach uncovered mid-nanomolar potentiators as well as inhibitors that bind to the same allosteric site. These molecules represent potential leads for the development of more effective drugs for cystic fibrosis and secretory diarrhea, demonstrating the feasibility of large-scale docking for ion channel drug discovery.
Keywords: Ligand discovery, anion channel, ABC transporter, potentiators, inhibitors
Graphical abstract
In Brief
Large-scale library docking and structure-based optimization are used to identify CFTR modulators, which include a nanomolar potentiator with promising pharmacokinetics and inhibitors that bind to the same allosteric site.
Introduction
CFTR is an anion channel that is widely expressed in epithelial cells of the lung, intestine, pancreas, and reproductive tract, where it regulates salt and fluid homeostasis1. Mutations that disrupt CFTR biosynthesis, folding, trafficking, or ion permeation cause cystic fibrosis (CF), a lethal genetic disease with no cure2. In addition, acquired CFTR dysfunction, through smoking for example, plays an important role in the initiation and progression of the chronic obstructive pulmonary disease (COPD)3,4. On the other hand, excessive activation of CFTR by bacterial pathogens such as Vibrio cholerae and enterotoxigenic Escherichia coli leads to secretory diarrhea, a major cause of mortality in children under the age of five5. Hyperactivity of CFTR is also a key driver in the pathogenesis of the autosomal dominant polycystic kidney disease (ADPKD)6,7. For these reasons, modulators that either up- or downregulate CFTR activity have long been pursued as drug candidates.
Although negative CFTR modulators have not yet been advanced to the clinic, there has been considerable progress in the development of positive modulators, including correctors that increase the abundance of CFTR at the cell surface and potentiators that enhance anion flux8–10. Thus far, one potentiator (ivacaftor or VX-770), two correctors (lumacaftor, tezacaftor), and one dually active potentiator and corrector (elexacaftor) have been made available to CF patients2. Ivacaftor is prescribed for 178 different CFTR mutations – either singly or in combination with correctors11. Although it has certainly improved the health of many CF patients, its physical properties and pharmacokinetics are far from optimal. Ivacaftor is difficult to formulate due to its low water solubility (<0.05 μg/mL; cLogP = 5.6)12. Moreover, its bioavailability is highly variable as 99% is bound to plasma proteins13,14, its usage is limited due to side effects, including liver disease and childhood cataracts15, and off-target modulator activities on ABC transporter paralogues have been reported16–18. In efforts to lower the daily dosing of ivacaftor, new studies are now in progress to evaluate its deuterated version VX-561. Meanwhile, the high expense of the drug (over $200,000 per year) is a strain on public health insurance and puts the drug out of reach of many. Alternative CFTR potentiators, including those inspired by ivacaftor, would therefore be beneficial for CF patients.
Electrophysiological measurements showed that ivacaftor increases the open probability of many mutants, as well as wild-type (WT), CFTR channels19,20. Cryo-EM structures of WT21, ΔF50822, and G551D CFTR23 revealed that ivacaftor binds to all three CFTR variants at the same site, near a hinge region important for gating. The binding-site does not overlap with the positions of disease-causing mutations such as ΔF508 or G551D, indicating that ivacaftor acts as an allosteric modulator. A chemically distinct CFTR potentiator, GLPG1837, binds to the same site on CFTR, indicating that this binding pocket is a hotspot for regulatory ligands. Because the pocket is unique to CFTR, and is not conserved in closely related proteins24, it is an excellent target for the discovery of CFTR modulators with minimal off-target effects.
As a result of recent developments in the use of chemical libraries for molecular docking, it is now feasible to computationally screen large and, more recently, ultra-large chemical libraries to identify potential ligands25–30. For example, nanomolar and sub-nanomolar ligands have been identified for dopamine D4, melatonin MT1, sigma2, and alpha2a adrenergic receptors from structure-based virtual screening25,28–30. Thus far, most of the large-scale library screens have focused on enzymes25,27 and G protein-coupled receptors (GPCRs)26,28–31. Few studies have been performed on membrane transporters or ion channels. Furthermore, the potentiator-binding site in CFTR is shallow and directly exposed to membrane, posing an extra challenge for virtual screening.
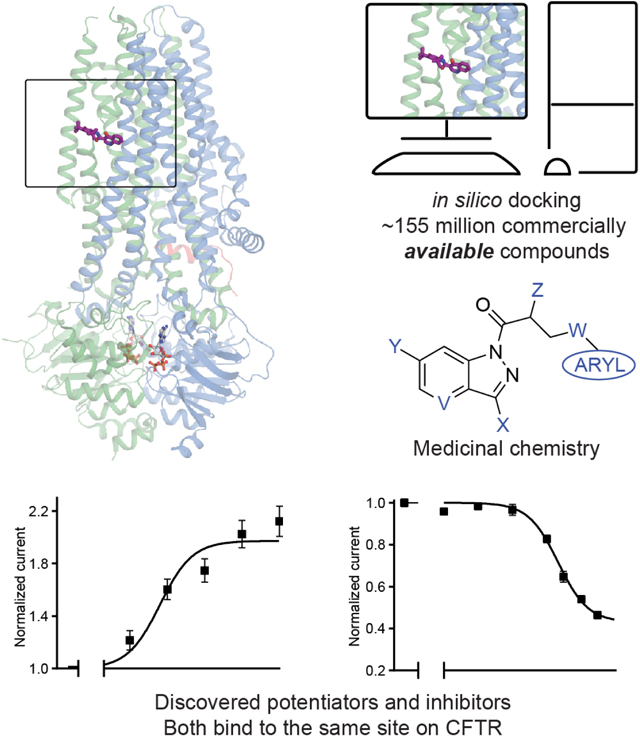
In this study, we sought to identify CFTR ligands by iterative molecular docking, electrophysiology, cryo-EM, and medicinal chemistry efforts (Figure 1A). The structure of CFTR in complex with ivacaftor was used to computationally dock a large virtual library of diverse chemical scaffolds. Through iterative optimization, we identified a potentiator scaffold with mid-nanomolar affinity that is chemically distinct from known CFTR potentiators and that has favorable physical properties and pharmacokinetics. We also discovered modulators that bind to the potentiator site but inhibit CFTR activity, demonstrating that the membrane-exposed allosteric site can be explored to downregulate CFTR activity.
Figure 1: Ultra-large docking screen identifies CFTR potentiators.

(A) The workflow of this study. (B) Compound Z2075279358 (‘358) potentiates ΔF508 CFTR. CFBE41o− cells homozygous for ΔF508 CFTR were cultured with 1 μM lumacaftor to facilitate surface-expression and pre-treated with 20 μM forskolin to activate ΔF508 CFTR by PKA-phosphorylation. The relative potentiation was calculated as the ratio of flux rates with and without 1 μM potentiator. Data points represent the means and standard errors (SEs) of 6 to 8 measurements (each shown as a dot). (C) Potentiation activity of 10 μM GLPG1837 or 5 μM compound against WT CFTR fused to a carboxy-terminal GFP tag. Inside-out membrane patches containing WT CFTR were excised from CHO cells and then fully phosphorylated by protein kinase A (PKA) in the presence of 3 mM ATP. The fold stimulation is defined as the ratio of the current in the presence and absence of added compound. Data represent means and SEs of 3–33 patches with individual measurements shown as dots. Statistical significance relative to absence of compound was tested by two-tailed Student’s t-test with Benjamini-Hochberg correction (ns: not significant; *P < 0.05; **P < 0.01; ****P = 4.2×10−14). (D) The 2D structures of the potentiators GLPG1837, ivacaftor, and the positive hits from the initial screen. (E) Representative macroscopic current trace and dose-response curve of WT CFTR in response to perfusion with ‘358. CFTR-containing membrane patches were fully phosphorylated by PKA. The current in the presence of 3 mM ATP before titration was used to normalize the current potentiated by different concentrations of ‘358. The EC50 is estimated to be 2.2 ± 0.6 μM by fitting the dose-responses with the Hill equation. Data represent means and SEs from 3 patches.
Results
Identification of CFTR potentiators
Seeking new CFTR modulators, we docked a virtual library of ~155 million tangible “drug-like” molecules (molecular weight 300–350 Da; logP ≤ 3.5) from the ZINC database (http://zinc15.docking.org)32,33 to the CFTR/ivacaftor cryo-EM structure (PDB: 6O2P) using DOCK3.7 (Figure 1A)34. Each molecule was sampled in an average of 421 conformations and 3,727 orientations, resulting in over 63 billion complexes being sampled in 76,365 core hours on an in-house cluster. Seeking diverse chemotypes, we clustered the top 100,000 scoring molecules by 2D similarity using ECFP4 fingerprints and a Tanimoto Coefficient (Tc) cutoff of 0.5. The top ranked molecules from each cluster (a total of 1,000) were visually inspected to remove molecules that were conformationally strained or had unsatisfied hydrogen bond acceptors or donors. Compounds engaging the key potentiator-binding residues S308, Y304, F312, and F931 (PDB: 6O2P and 6O1V) were prioritized and 58 of those, each from a different chemotype family, were selected for experimental evaluation.
Of the 58 prioritized compounds 53 were synthesized successfully from the Enamine make-on-demand set, a 91% fulfillment rate; as far as we know, all were new to the planet. They were evaluated for their effects on the CFTR variant found in 90% of CF patients (ΔF508). GLPG1837 was used as a benchmark because ivacaftor has very low solubility35, making it difficult to work with, and studies have shown that GLPG1837, although it was not advanced to the clinic, has a modestly higher efficacy than does ivacaftor for potentiation of both the wild-type CFTR and disease variants36,37. CFTR-mediated ion flux was measured in a bronchial epithelial cell line derived from a patient homozygous for ΔF508 (CFBE41o−) using a well-established halide flux assay38, which was chosen for its experimental throughput and that it has previously been used to identify CFTR modulator leads36. In these experiments surface-expression of ΔF508 CFTR was increased by treatment with the folding corrector lumacaftor. Several of the new compounds potentiated CFTR-mediated ion flux (Figure S1), in particular Z2075279358 (‘358), which increased ion flux to an extent close to that of GLPG1837 (Figure 1B).
To more directly assess whether the effects on ion flux were specific to CFTR, the compounds were also analyzed in inside-out membrane patches containing phosphorylated WT CFTR channels (Figure 1C). At a concentration of 5 μM, 10 compounds increased macroscopic currents by 1.2 to 2.4-fold (Figure 1C). The chemotypes of these 10 docking hits were diverse and distinct compared to known CFTR potentiators such as GLPG1837 and ivacaftor (Figure 1D). In agreement with our cellular assay, the most efficacious compound was ‘358, whose efficacy was comparable to that of GLPG1837 (increased macroscopic current by 2.2-fold)21,37,39 with an EC50 of 2.2 ± 0.6 μM. Furthermore, our large-scale docking screen had a 19% hit rate, further supporting the feasibility of large-scale library screens for identifying allosteric ion channel modulators and resulted in candidate potentiators that are effective on both WT and ΔF508 CFTR (Figure 1C and Figure S1).
Analog screen to identify additional potentiators
To identify more potent ligands for CFTR, we screened the ZINC15 library for analogs of ‘358. These were docked into the allosteric potentiator-binding site and prioritized based on fit. Thirteen high-scoring analogs were selected for synthesis and tested using electrophysiology (Figure 2A). Surprisingly, only one molecule, Z1834339853 (‘853), potentiated WT CFTR currents to the same degree as ‘358 (Figure 2). The other 12 analogs had little or no effect. Concerned by this apparent failure of SAR, we re-examined the structures of the synthesized compounds by detailed NMR spectroscopy. This revealed that both ‘358 and ‘853 are regioisomers of the molecules specified in ZINC15 (Figure 2B). The acyl side chains on the exocyclic nitrogens of the indazole rings in both compounds in the ZINC15 database are instead on the cyclic nitrogens (Figure 2B).
Figure 2: New modulators identified through an analog screen.

(A) Potentiation activity of ‘358 analogs. All compounds were tested at a single concentration of 10 μM in inside-out membrane patches containing fully phosphorylated WT CFTR. The current stimulation levels of GLPG1837 and ‘358 are indicated as dashed lines. (B) Reported structures versus the NMR-determined structures. (C) Docked poses of the reported structure of ‘853 (left) versus the NMR-determined structure of ‘853 (right). (D) (S)-’853 potentiates WT CFTR currents, while (R)-’853 mildly inhibits them in inside-out patches. Both enantiomers were perfused at 100 μM concentration. Data represent means and SEs of 20 ((S)-’853) or 18 ((R)-’853) patches. Statistical significance relative to no effect was tested by two-tailed Student’s t-test (****P = 2.3×10−9 for (S)-’853 and P = 1.6×10−7 for (R)-’853). (E) Competition assay showing that the presence of (R)-’853 (100 μM) diminishes the potentiating effect of (S)-’853 (100 μM). (F) Dose-response curve of (S)-’853 versus the racemic mixture (±)-’853, as described for Figure 1E. The EC50 for (S)-’853 was estimated to be 2.1 ± 0.9 μM. Data represent means and SEs of 2–20 patches. 3 mM ATP was used in all panels.
To analyze how these regioisomeric differences affect binding to CFTR, we compared the docking pose of the synthesized ‘853 with its regioisomer in ZINC15 (Figure 2C). Like the ZINC15 regioisomer, the synthesized ‘853 fits well at the potentiator-binding site, forming several identical interactions: its phenyl ring is located in the hydrophobic pocket defined by F312, F931, G930 and R933; the central carbonyl docks to the main chain of F931 via a hydrogen bond; and the indazole ring is poised to stack with F312. The two poses differ in the orientation of the indazole by ~180° but the docking scores are similar: −34.54 kcal/mol for the synthesized compound and −37.13 kcal/mol for the isomer in the ZINC15 database. This suggests that, despite the side chain difference, the synthesized ‘853 is likely to interact with CFTR at the potentiator-binding site. It is important to note that, although ‘358 is a valid starting point to pursue further optimization, the chemical structure of ‘853 better lent itself to analog synthesis and to exploration of SAR, and so ‘853 was chosen for further optimization.
Because ‘853 contains a chiral center, the pure enantiomers (S)-’853 and (R)-’853 were synthesized and tested (Figure 2D). Interestingly, although the (S)-enantiomer increased the CFTR currents by 1.5-fold, the (R)-enantiomer decreased the currents to 0.89-fold (Figure 2D). Furthermore, when tested together, the presence of (R)-’853 diminished the potentiating effect of (S)-’853 in a competition assay (Figure 2E), and dose-response curves confirmed greater current flow through CFTR in the presence of (S)-’853 compared to the racemic mixture (Figure 2F). Together, these data reveal that the two enantiomers of ‘853 have opposite functional effects: whereas (S)-’853 is a potentiator, (R)-’853 is an inhibitor of CFTR.
Structural investigation of the ‘853 binding site
To confirm the docking predicted binding pose of ‘853 and to guide further optimization for affinity and efficacy, we sought to determine the cryo-EM structure of CFTR in complex with ‘853. Using phosphorylated E1371Q CFTR in the presence of saturating ATP-Mg2+ (10 mM), we obtained a 3.8 Å reconstruction which revealed clear density at the allosteric potentiator-binding site (Figure 3A and Figure S2). The shape and size of the density are consistent with the molecular structure of ‘853 (Figure 3B). Like ivacaftor and GLPG1837, ‘853 binds near the TM8 hinge region at the protein-membrane interface, stabilized by two hydrogen bonds with the side chain of S308 and the main chain carboxyl of F312 (Figure 3C). In addition, the phenol ring on ‘853 is coordinated by pi-pi interactions with F312, and the fluorinated indazole forms pi-pi interactions with F236.
Figure 3: Z1834339853 binds to the same site as ivacaftor and GLPG1837.

(A) Cryo-EM structure of phosphorylated and ATP-bound CFTR (E1371Q) in complex with ‘853. (B) Zoomed-in views of the density of ‘853 (top) and a comparison between the docked pose (salmon) and the cryo-EM pose (magenta). Compared to the experimentally determined pose, the docked ‘853 shifted towards R933. This difference is likely due to the presence of an unknown density between R933 and the potentiator (Liu et al., 2019), which was not modeled for docking. (C) Representative macroscopic current traces and dose-response curves of fully phosphorylated WT, S308A, and Y304A CFTR in response to perfusion of (S)-’853 onto inside-out excised membrane patches. 3 mM ATP was used. Each data point represents the mean and SEs determined from 3 to 12 patches.
Previous mutational scanning experiments identified two polar residues at this location (S308 and Y304) that are most critical for ivacaftor and GLPG1837 recognition21,39. Unlike S308, Y304 does not directly interact with the ligands. Instead, it forms a hydrogen bond with a main chain amide in the TM 8 hinge, thereby stabilizing the structure of the ligand-binding site. Similar to the mutational effects observed for ivacaftor and GLPG183721,39, alanine substitution of either S308 or Y304 also abolished the potentiating effect of (S)-’853 in inside-out patches (Figure 3C), confirming that the modulator discovered by molecular docking interacts with CFTR in the same manner as ivacaftor and GLPG1837.
Structure-based optimization of the CFTR modulators
With the experimental structure of the CFTR/’853 complex in hand, and given the intriguing difference between (S)-’853 and (R)-’853, we carried out further optimization to identify additional potentiators and inhibitors. We used the following medicinal chemistry strategies to synthesize multiple classes of ‘853 analogs (Figure 4A and Figure S3): (1) replacing the fluorine with typical hydrogen bond donor or acceptor functionalities, chlorine or trifluoromethyl (Y); (2) removing or enlarging the methyl substituent (Z) of the linker, or replacing the linker oxygen with a CH2 group (W); (3) removing or substituting the amino group (X); (4) truncating or replacing the terminal phenyl ring with a set of substituted or fused aromatic moieties; (5) introduction of a nitrogen (V) into the heterocyclic ring.
Figure 4: Medicinal chemistry leads to novel CFTR potentiators and inhibitors.

(A) General formula of newly synthesized ‘853 analogs. (B) Effects of ‘853 analogs on currents measured in inside-out excised membrane patches containing fully phosphorylated WT CFTR. Measurements were made with 3 mM ATP. Data represent means and SEs of 2 to 46 patches. (C) The chemical structures (top), dose-response curves (middle), and docked poses (bottom) of three of the most efficacious potentiators. Data represent means and SEs of 5–11 patches. (D) The chemical structures (top), dose-response curves (middle), and docked poses (bottom) of the two most efficacious inhibitors. The dose-responses of the Y304A and S308A variants in response to I1412 perfusion were also shown (left). Data represent means and SEs of 2–8 patches. (E) Pharmacological potentiation and inhibition of iodide flux rates in CFBE41o− cells homozygous for ΔF508 CFTR. 1 μM lumacaftor was used in the culture to facilitate surface-expression. A pre-treatment with 20 μM forskolin was used to activate ΔF508 CFTR. The relative potentiation was calculated as the ratio of flux rates with and without modulator at the indicated concentrations. Data points represent the means and SEs of 2 (CFTRinh-172) or 3 (all other conditions) experiments. Statistical significance relative to a DMSO only treatment was tested by one-way analysis of variance (ANOVA) (*P = 0.049 for I1412, P = 0.041 for I1422, and P = 0.015 for CFTRinh-172; ****P = 9.0×10−7 for GLPG-1837, P = 7.8×10−6 for Ivacaftor, P = 3.0×10−6 for I1421, P = 1.0×10−5 for I1408, P = 6.1×10−7 for (S)-SX263, and P = 3.1 ×10−6 for BBG3). (F) Representative macroscopic current traces of fully phosphorylated WT CFTR in response to perfusion of GLPG-1837, I1421, and Elexacaftor onto inside-out excised membrane patches. 3 mM ATP, 10 μM GLPG-1837, and 1 μM Elexacaftor were used. (G) Relative potentiation of WT CFTR currents by Elexacaftor without or with GLPG1837 or I1421. Data represent means and SEs of 6 to 46 patches. Some data points are replotted from panel B. Statistical significance was tested by one-way ANOVA (*P = 0.026; ****P = 6.3×10−6 for Elexacaftor + GLPG-1837 versus Elexacaftor only, P = 2.6×10−12 for Elexacaftor + GLPG-1837 versus GLPG-1837 only, and P = 9.3×10−5 for Elexacaftor + I1421 versus I1421 only).
Each of the 51 designed compounds was docked into the potentiator-binding site in silico, and a total of 39 compounds were chosen for synthesis based on their docking score. The efficacies of the synthesized analogs were determined by patch clamp experiments, revealing a continuum of CFTR modulation from inhibition to potentiation likely reflecting a range of state-dependent binding energies to the conductive and nonconductive states of CFTR (Figure 4B). The most efficacious analogs, which strongly potentiated CFTR, were the ones in which the para-position in the distal phenyl ring was halogenated and/or the indazole fluorine was replaced by a hydroxyl group. The first modification improved the non-polar complementarity in a hydrophobic subsite of the potentiator-binding pocket (Figure 4C, right panel), while the replacement of the (S)-’853 indazole fluorine with a polar group is predicted to form an additional hydrogen bond with Q237 (Figure 4C, left and middle panels, Figure S3). Three of the most efficacious potentiators, I1421, I1408, and (S)-SX-263, increased currents through WT CFTR with EC50 values of 64 ± 25 nM, 93 ± 42 nM and 136 ± 60 nM, respectively (Figure 4C). As chemotype novelty was important to us, it is useful to note that the modifications that had the biggest impact on potency and efficacy, particularly the conversion of the methoxy to a hydroxymethyl on the indazole ring, but also the addition of a fluorine on the distal end of the molecule, did not make the series more chemically similar to either ivacaftor or to GLPG1837—indeed, neither of those drugs contain a hydroxymethyl, or related group. Meanwhile, these changes had little effect on the overall physical properties of the molecules, which we were trying to optimize relative to those of ivacaftor. For instance, the lipophilicity of I1421 (cLogP 3.0) was actually reduced from that of ‘853 (cLogP 3.4), and naturally remained much lower than that of ivacaftor (clogP 5.1). The improved potentiation of I1421 presumably reflects improved complementarity to the active state of the channel.
Of the 39 analogs tested, several were found to inhibit WT CFTR (Figure 4D and Figure S3), including I1412. This compound is an (R)-enantiomer of the potentiator I1409, providing a second example of (S)- and (R)-enantiomers being positive and negative allosteric modulators, respectively (Figure 4B). Although it is intriguing, and from a selective recognition view perhaps encouraging, that switching from the R to S stereochemistry changes the molecules from positive to negative allosteric modulators, the basis of what distinguishes potentiators from inhibitors from the perspective of CFTR remains unclear to us. Future optimization of this series may illuminate this question. The most efficacious inhibitors in this series inhibited WT CFTR with IC50 values of 21 ± 3 μM (I1412) and 41 ± 17 μM (I1422) (Figure 4D). Similar to the potentiator (S)-’853, single alanine substitutions of either of the binding site residues Y304 or S308 eliminated the activity of I1412 (Figure 4D). This medicinal chemistry approach thus led to the identification of a potentiator (I1421) with a 30-fold greater potency than (S)-’853, as well as negative modulators that bind to the same site on CFTR.
To test the activity of our compounds in a relevant biological system, we measured their effects on CFTR-mediated ion flux in the patient-derived CFBE41o− bronchial epithelial cell line. For these measurements, surface expression of ΔF508 CFTR was rescued by lumacaftor and CFTR activity was stimulated by forskolin. The potentiators I1421, I1408, (S)-SX263, and BBG3 potentiated CFTR-mediated ion flux to similar extents as GLPG-1837 and ivacaftor (Figure 4E). By contrast, I1412 and I1422 inhibited CFTR-mediated ion flux. The inhibitory efficacies of I1412 and I1422 were lesser than the investigational compound CFTRinh-172 (Figure 4E), consistent with their relative efficacies in patch clamp electrophysiology and their distinct mechanisms of action.
The dually active potentiator and corrector, elexacaftor, can potentiate CFTR current additively with ivacaftor40,41. Given the shared binding site of ivacaftor and our potentiator series, we tested whether elexacaftor would co-potentiate with I1421. Elexacaftor was perfused onto inside-out excised patches in which WT CFTR currents had been elicited and potentiated by a saturating dose of GLPG-1837 or I1421 (Figure 4F). As for ivacaftor, the combined efficacies of GLPG-1837 or I1421 with elexacaftor exceeded the efficacies of each potentiator alone (Figure 4G), indicating that our potentiator series can act additively with potentiators acting at the elexacaftor site.
I1421 can rescue multiple CF-causing mutants
Finally, we asked whether the strongest potentiator, I1421, could increase the activity of various disease-causing CFTR mutants. Clones carrying ten of these mutations, distributed at different positions in CFTR, were tested using patch-clamp electrophysiology (Figure 5A). The predominant CF mutation, ΔF508, is defective in both folding and gating. Newly synthesized ΔF508 CFTR is largely retained in the endoplasmic reticulum42 and the few channels that reach the plasma membrane exhibit little activity43,44. I1421 strongly increased currents through ΔF508, indicating that, similar to ivacaftor, it could be used to rescue ΔF508 if used in combination with correctors (Figure 5B, C). Approximately 4% of CF patients carry the G551D mutant, which is expressed on the cell surface but has severe gating defects45. In inside-out membrane patches, I1421 increased the activity of G551D by 25-fold (Figure 5B, C). Potentiation of the other eight mutations by I1421 was also comparable to that of GLPG1837 (Figure 5C). These data indicate that I1421 is a strong potentiator that allosterically activates a wide range of CF-causing mutants.
Figure 5: The activity of I1421 against 10 CF-causing mutations.

(A) The positions of the mutations mapped onto dephosphorylated and ATP-free CFTR (PDB 5UAK). (B) Representative macroscopic current traces in response to I1421 (10 μM) perfusion onto inside-out membrane patches excised from CHO cells. 3 mM ATP was used. (C) Potentiation activity of I1421 versus GLPG1837. The mean and SE values were determined from 2 to 7 patches. (D) Pharmacokinetic analysis of compound I1421. Plasma concentration-time profiles in male C57BL/6N mice following a single subcutaneous (SC), intraperitoneal (IP), per-oral (PO) (dose 10 mg/kg) or intravenous (IV) (3 mg/kg) administration. Data represent means and SDs. (E) Selected pharmacokinetic parameters of I1421. : peak plasma concentration; : the time when the peak plasma concentration was observed; : the areas under the concentration time curve; : terminal half-life; CL: clearance, : steady-state volume of distribution; %F: %bioavailability.
Given these promising results, we investigated the pharmacokinetics of I1421 in C57BL/6 mice following single 10 mg/kg doses administrated by intraperitoneal (IP), oral, or subcutaneous routes (Figure 5D, E). As expected from its cLogP value of 2.5, I1421 was readily formulated in a standard vehicle comprising 10% N-Methyl-Pyrrolidone (NMP), 5% solutol HS-15, and 85% saline. The maximum plasma concentrations (), observed 15 minutes after IP and oral administration, were 7.4 μM and 3.4 μM, respectively, indicating rapid absorption of the compound. Plasma levels following IP administration were higher than the other two routes of administration, as indicated by the areas under the respective plasma concentration curves (AUC) (Figure 5D, E). To determine oral bioavailability, I1421 was intravenously (IV) injected into a group of nine mice at a dose of 3 mg/kg. Comparison of plasma levels from oral versus IV injection yielded a bioavailability of 60%, an encouraging value given that the compound was not subject to special formulation. In contrast, the low solubility of ivacaftor35 has precluded creation of an IV formulation, preventing determination of its oral bioavailability. Low solubility may also affect the non-linear relationship between dose and maximum concentration in vivo, causing ivacaftor’s to plateau with increasing dose46,47, even after extensive formulation optimization. These factors, along with the strong effect of fat-containing food on absorption13, may contribute to the variable effects of ivacaftor.
Importantly, I1421 and other molecules in this series are not clinical candidates, have not been optimized for pharmacokinetics, and retain liabilities such as a short oral T1/2 of 75 minutes. Nevertheless, these results support the notion that a structure-based approach to CFTR drug discovery can identify molecules with improved physical properties, easier formulation, and improved pharmacokinetics.
Discussion
Large library docking has been a disruptive innovation in structure-based ligand discovery, in particular for GPCRs26,28–31 and enzymes25,27. However, such screens, and more broadly structure-based design and discovery, are much less prominent for ion channels. Effective docking campaigns rely heavily on accurate structural insights into the binding pocket and its interaction with ligands, a fact widely acknowledged in the literature25,26,29,31. However, the importance of utilizing known ligands as control molecules in large library docking is often overlooked. While the primary aim of docking is to identify novel molecules, the inclusion of established ligands as controls helps one ensure that the sampling and scoring methods can correctly pose known molecules and can identify them as likely ligands from a large set of decoys 48. While the continual elucidation of ion channel structures and the assembly of comprehensive ligand datasets will undoubtedly advance structure-based discovery against ion channels, a remaining challenge is accurately modeling the contribution of lipids surrounding membrane-exposed sites.
In this study, large library docking against the potentiator-binding site of CFTR revealed potentiators and inhibitors with chemotypes unrelated to known modulators and with better physical properties than the widely used drug ivacaftor. Several aspects of this study are worth highlighting. First, despite the shallow, membrane-exposed binding pocket, the docking hit rate (number of molecules active/number experimentally tested) was substantial at 19%. Second, despite their chemical novelty and dissimilarity to previously known modulators, effective potentiators were still found. For example, I1421 had efficacy comparable to that of GLPG1837 for all ten CF-causing mutants tested. Third, the molecules that we discovered were substantially more polar than the only FDA-approved potentiating drug, ivacaftor, conferring potential benefits to their behavior in vivo. This was borne out in preliminary pharmacokinetic experiments in which I1421 was readily formulated for oral and IV delivery using widely used excipients, resulting in substantial exposure upon oral dosing and a high oral bioavailability of 60%. The good physical properties of this family, and their apparently favorable pharmacokinetics, provide the freedom for further development and optimization. The structure-based approach described here provides multiple points of departure for drug leads for a disease whose treatment remains out of reach for many, and expensive for all. Finally, although our docking campaign used the pore-open structure of CFTR, this study revealed that the allosteric binding site on CFTR can be used to discover not only potentiators but also inhibitors. The allosteric inhibition mechanism differs from commonly used investigational inhibitors like GlyH-101 and CFTRinh-172, both bind inside the pore, potentially offering greater specificity for future optimization 49,50. Future structural investigations of the inhibitor-bound CFTR may inform medicinal chemistry approaches to develop analogs of the present series with improved potency. It is also possible that more potent molecular scaffolds for inhibition may be discovered by using a pore-closed CFTR structure as a target for docking. Such inhibitors may serve as starting points to develop new tools for CFTR research, as well as therapeutic interventions for secretory diarrhea and ADPKD, both of which are life-threatening diseases that lack effective treatment.
Limitations of the Study
We wish to draw attention to certain caveats. In particular, we have not tested our potentiators in an animal model, mainly due to the lack of a model system that fully recapitulates the symptoms of human CF51. Indeed, CF drug discovery, including the development of ivacaftor, has historically relied on in vitro model systems, some of which were used here. In addition, it is important to note that the promising pharmacokinetics of compounds like I1421 are distinct from their pharmacodynamics, so must be viewed cautiously. Moreover, although solubility and oral bioavailability are encouraging, these molecules remain far from optimized and are currently no more than leads. In our docking experiments, the fact that the most potent initial hit represented a different regioisomer to our intended compound tempers the success of the method, even though the two regioisomers occupy similar poses in the binding site and achieve similar docking scores. Finally, although the initial hits sampled a broad range of chemotypes (Figure 1), their potentiating efficacies were modest and have yet to be followed up.
Regardless of these caveats, we have made a number of important observations in this study. By docking a large virtual library against the structure of CFTR, we have uncovered 10 diverse chemical scaffolds that are topologically distinct from known ligands and have favorable physical properties. This wide range of chemotypes holds great promise for new drug discovery efforts against this crucial target. Indeed, the chemical novelty of these compounds supported our discovery of CFTR inhibitors that likely bind to the same allosteric site, and which have the potential to become lead molecules for the treatment of secretory diarrhea. By determining the structure of one of the new ligands in complex with CFTR, we have provided a template for further optimization of this series and for other drug discovery efforts. Encouragingly, the beneficial physical properties of I1421 resulted in favorable pharmacokinetics during animal dosing. In conclusion, our study has revealed that combining structural biology with exploration of a large chemical space can reveal new lead molecules for what remains a crucial drug target for CF, secretory diarrhea, and ADPKD.
Star Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed and will be fulfilled by the lead contact, Jue Chen (juechen@rockefeller.edu).
Materials availability
All materials generated in this study are available from the lead contact upon request.
Data and code availability
The 3D cryo-EM density map of the CFTR-’853 complex generated in this study has been deposited at the Electron Microscopy Data Bank (EMDB), and the coordinates of the CFTR-’853 complex have been deposited at the Protein Data Bank (PDB) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. The identities of the compounds docked in this study are publicly accessible in existing, publicly available databases, which are listed in the key resources table. The docking software, DOCK3.7, is freely available for non-commercial research and is also listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| 1-Ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) | TCI | Cat# D4029 |
| Carbonyldiimidazole | Sigma-Aldrich | Cat# 115533 |
| 3-(1H-Indazol-1-yl)-2-methylpropanoic acid | Enamine | Cat# EN300–206691 |
| 6-Fluoro-1H-indazol-3-amine | Enamine | Cat# EN300–100294 |
| 3-(3-Isopropyl-1,2,4-oxadiazol-5-yl)propanoic acid | Enamine | Cat# EN300–27664 |
| 4-(1-Methyl-1H-pyrazol-4-yl)pyrimidin-2-amine | Enamine | Cat# EN300–187048 |
| 1-(2,6-Difluorophenyl)-1H-pyrazole-3-carboxylic acid | Enamine | Cat# EN300–91144 |
| 3-(Methylamino)benzonitrile | Enamine | Cat# EN300–55410 |
| (R,S)-7-Methyl-5,6,7,8-tetrahydroimidazo[1,2-a]pyridine-2-carboxylic acid | Enamine | Cat# EN300–309016 |
| (R,S)-tert-Butyl (5-amino-5,6,7,8-tetrahydronaphthalen-2-yl)carbamate | Enamine | Cat# EN300–142125 |
| 3-Methoxy-2-naphthoic acid | Enamine | Cat# EN300–00001 |
| 3-(3-Amino-1H-pyrazol-1-yl)propanamide | Enamine | Cat# EN300–80390 |
| Salicylic acid | Enamine | Cat# EN300–16723 |
| 2-(Chloromethyl)-5-(4-chlorophenyl)-1,3,4-oxadiazole | Enamine | Cat# EN300–08138 |
| 5-Amino-4-isopropyl-4H-1,2,4-triazole-3-thiol | Enamine | Cat# EN300–2493721 |
| 6-Chloro-2-(chloromethyl)imidazo[1,2-a]pyridine | Enamine | Cat# EN300–33589 |
| 5-(Benzo[d]thiazol-2-yl)thiophene-2-carboxylic acid | Enamine | Cat# EN300–00707 |
| (4H-1,2,4-Triazol-3-yl)methanamine | Enamine | Cat# EN300–1264530 |
| 2-(Imidazo[1,2-a]pyridin-2-yl)acetonitrile | Enamine | Cat# EN300–25598 |
| 2-Hydroxy-2,3-dihydro-1H-indene-2-carboxylic acid | Enamine | Cat# EN300–82840 |
| 1-(4-Fluorophenyl)-1H-pyrazole-3-carboxylic acid | Enamine | Cat# EN300–36826 |
| N-Methyl-1-(2-methylthiazol-4-yl)methanamine | Enamine | Cat# EN300–27495 |
| 2-Chloro-N-(3-fluoro-4-methylphenyl)propanamide | Enamine | Cat# EN300–23356 |
| N4,N4-Dimethylpyrimidine-4,6-diamine | Enamine | Cat# EN300–187848 |
| N-(3,4-Dimethylbenzyl)cyclopropanamine | Enamine | Cat# EN300–83837 |
| 1-(m-Tolyl)-1H-1,2,3-triazole-4-carboxylic acid | Enamine | Cat# EN300–61349 |
| (2-Chloro-6-fluorophenyl)methanamine | Enamine | Cat# EN300–33074 |
| 2,2,2-trifluoroethyl carbonochloridate | Enamine | Cat# EN300–31105 |
| Phenol | Alfa Aesar | Cat# A15760 |
| Sodium hydride | Sigma Aldrich | Cat# H36490 |
| (R)-3-Bromo-2-methylpropan-1-ol | Manchester Organics | Cat# H39016 |
| (S)-3-Bromo-2-methylpropan-1-ol | Manchester Organics | Cat# R39644 |
| 2-Naphthol | Sigma Aldrich | Cat# 130109 |
| 3-(Trifluormethyl)phenol | SIAL | Cat# 156035 |
| 2,4-Di-tert-butylphenol | SIAL | Cat# 137731 |
| 4-Fluorophenol | Fisher Scientific | Cat# 10723671 |
| 4-Bromophenol | Acros Organics | Cat# 406511000 |
| [1,1’-biphenyl]-4-ol | SIAL | Cat# 371297 |
| Benzofuran-5-ol | BLD Pharmatech | Cat# BD102433 |
| 1-Hydroxy-7-azabenzotriazole (HOAt) | Sigma Aldrich | Cat# 41996 |
| N-Ethyldiisopropylamine (DIPEA) | Acros Organics | Cat# 367841000 |
| Isobutyryl chloride | Sigma Aldrich | Cat# 139122 |
| 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid-hydrochlorid (EDC x HCl) | Sigma Aldrich | Cat# 03450 |
| (R,S)-2-methyl-3-phenoxypropionic acid | Enamine | Cat# EN300–42126 |
| 6-Fluoro-1H-indazole | Sigma Aldrich | Cat# APOH11A9C83A |
| 6-Fluoro-3-methyl-1H-indazole | Sigma Aldrich | Cat# C12H580D8864 |
| 4-Dimethylaminopyridine | Acros Organics | Cat# 148270050 |
| Acetyl chloride | Sigma Aldrich | Cat# 114189 |
| Iodomethan (methyl iodide) | Acros Organics | Cat# 122371000 |
| 3,5-Difluoro-2-pyridinecarbonitrile | TCI | Cat# C2753 |
| Hydrazine hydrate | Acros Organics | Cat# 196711000 |
| Benzotriazol-1-yl-oxytripyrrolidinophosphonium-hexafluorophosphat (PyBOP) | Merck | Cat# 8510090025 |
| 6-Methoxy-1H-indazol-3-amine | abcr | Cat# AB490142 |
| Isopropoxy-1H-indazol-3-amine | A2B Chem | Cat# BE29241 |
| 6-(Trifluoromethyl)-1H-indazol-3-amine | abcr | Cat# AB146621 |
| 6-Chloro-1H-indazol-3-amine | BLD Pharmatech | Cat# BD167849 |
| 1H-Indazole-3,6-diamine | VWR International | Cat# 2C36909 |
| 3-Amino-1H-indazole-6-carboxylic acid | Sigma Aldrich | Cat# AMBH303C4559 |
| 2-Fluoro-4-(hydroxymethyl)benzonitrile | Activate Scientific | Cat# AS38375 |
| 1H-Indazole-6-carboxamide | abcr | Cat# AB527998 |
| 6-Amino-1H-indazole | abcr | Cat# AB136281 |
| 1-Fluoro-3-(prop-2-yn-1-yloxy)benzene (32) | Enamine | Cat# EN300–71767 |
| (Acetonitrile)[2-biphenyl)di-tert-butylphosphine]gold(I) hexafluoroantimonate | Sigma Aldrich | Cat# 697575 |
| Palladium hydroxide 20% on charcoal | Sigma Aldrich | Cat# 21,291–1 |
| N-Bromosuccinimide | Acros Organics | Cat# 107451000 |
| 2.5 M Solution of butyllithium in hexane | Sigma Aldrich | Cat# 230707 |
| 2,2-Dimethylmalononitrile | Tokio Chemical Industry | Cat# D5514 |
| 3-Phenoxypropionic acid | Sigma Aldrich | Cat# P16001 |
| (R,S)-2-(Phenoxymethyl)butanoic acid | Enamine | Cat# EN300–27146916 |
| (R,S)-2-Methoxy-3-phenoxypropanoic acid | CHEM Space | Cat# CSC039927227 |
| (R,S)-2-Methyl-4-phenylbutyric acid | Enamine | Cat# EN300–6496378 |
| Sodium 4-phenylbutyrate | abcr | Cat# AB222448 |
| 2,2-didecylpropane-1,3-bis-β-D-maltopyranoside (LMNG) | Anatrace | NG310 |
| Cholesteryl hemisuccinate (CHS) | Anatrace | CH210 |
| Digitonin | Sigma-Aldrich | D141 |
| sf-900 II SFM medium | Gibco | Cat#10902–088 |
| Cellfectin II reagents | Invitrogen | Cat#10362–100 |
| Freestyle 293 medium | Gibco | Cat#12338–018 |
| PKA | NEB | Cat#P6000L |
| ATP | Sigma-Aldrich | A2383 |
| Critical commercial assays | ||
| CNBR-activated sepharose beads | GE Healthcare | 17–0430–01 |
| Superose 6, 10/300 GL | GE Healthcare | 17–5172–01 |
| Deposited data | ||
| Coordinates of the human CFTR in complex with ligand Z1834339853 | This paper | PDB: 8GLS |
| Cryo-EM map of the human CFTR in complex with ligand Z1834339853 | This paper | EMDB: EMD-40207 |
| Experimental models: Cell lines | ||
| Sf9 | ATCC | CRL-1711 |
| HEK293S GnTI− | ATCC | CRL-3022 |
| CHO-K1 | ATCC | CRL-9618 |
| CFBE41o- cells expressing F508del-CFTR and the fluorescent protein EYFP-H148Q/I152L/F46L | Sondo et al.52 | Gift by Luis J. Galietta |
| Experimental models: Organisms/strains | ||
| C57BL/6 mice | Hylasco Bio | C57BL/6Ncrl Mice |
| Recombinant DNA | ||
| Human CFTR cloned onto a modified pEG Bacmam vector suitable for expression in mammalian cells | This paper | N/A |
| Software and algorithms | ||
| Bruker TopSpin Version 4.2.0 | Bruker | https://www.bruker.com/en/products-and-solutions/mr/nmr-software/topspin.html |
| ChemDraw Version 21.0.0 | Revvity Signals | https://revvitysignals.com/products/research/chemdraw |
| Agilent Chemstation | Agilent | https://www.agilent.com/cs/library/usermanuals/Public/G2070-91126_Understanding.pdf |
| Seriel EM | Mastronarde,74 | http://bio3d.colorado.edu/SerialEM |
| MotionCor2 | Zheng et. al.75 | https://emcore.ucsf.edu/ucsf-motioncor2 |
| RELION-3 | Zivanov et al.78 | http://www3.mrclmb.cam.ac.uk/relion |
| RELION-2 | Kimanius et al.79 | http://www2.mrclmb.cam.ac.uk/relion |
| cryoSPARC | Punjani et al.80 | https://cryosparc.com |
| COOT | Emsley et al.81 | https://www2.mrclmb.cam.ac.uk/personal/pemsley/coot |
| PHENIX | Adams et al.82 | https://www.phenix-online.org |
| MolProbity | Chen et al.83
Davis et al.84 |
http://molprobity.biochem.duke.edu |
| Chimera | Pettersen et al.85 | https://www.cgl.ucsf.edu/chimera |
| Pymol | PyMOL | http://www.pymol.org |
| GraphPad Prism 9 | GraphPad Software | http://www.graphpad.com |
| pCLAMP9 | Molecular Devices | https://mdc.custhelp.com/euf/assets/content/pCLAMP_9.0_Manual.pdf |
| DOCK 3.7 | Coleman et al.34 | https://dock.compbio.ucsf.edu/DOCK3.7/ https://blaster.docking.org/ |
| ZINC15 | Sterling and Irwin,33 | http://zinc15.docking.org/ |
| Reduce | Word et al., 1999 | N/A |
| QNIFFT | Gallagher and Sharp, 1998; Sharp, 1995 | N/A |
| Marvin | https://www.chemaxon.com | version 15.11.23.0, ChemAxon, 2015 |
| Corina | https://www.mn-am.com/products/corina | v.3.6.0026, Molecular Networks GmbH |
| Omega | https://www.eyesopen.com/omega | v.2.5.1.4, OpenEye Scientific Software |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell culture
Sf9 cells were cultured in Sf-900 II SFM medium (GIBCO) supplemented with 5% (v/v) fetal bovine serum (FBS) and 1% (v/v) Antibiotic-Antimycotic. HEK293S GnTl− cells were cultured in Freestyle 293 (GIBCO) supplemented with 2% (v/v) FBS and 1% (v/v) Antibiotic-Antimycotic. Chinese hamster ovary (CHO) cells were cultured in DMEM-F12 (ATCC) supplemented with 10% (v/v) FBS and 1% (v/v) Antibiotic-Antimycotic. CFBE41o- cells expressing F508del-CFTR and the fluorescent protein eYFP-H148Q/I152L/F46L were cultured in MEM-alpha with 10% (v/v) FBS, 1% (v/v) Pen-Strep, 2 mg/mL Puromycin and 0.75 mg/mL G418.
Animals
Animal experiments were performed by Sai Life Sciences (Hyderabad, India) at an AAALAC accredited facility in accordance with the Sai Study Protocol SAIDMPK/PK-22–12–1306 and PK-22–11–1117. International guidelines for animal experiments were followed. Testing was done in healthy male C57BL/6 mice (8–10 weeks old) weighing between 25 ± 5 g (procured from Global, India). Three mice were housed in each cage. Temperature and humidity were maintained at 22 ± 3 ºC and 30–70%, respectively and illumination was controlled to give a sequence of 12 h light and 12 h dark cycle. Temperature and humidity were recorded by an auto-controlled data logger system. All animals were provided laboratory rodent diet (Envigo Research private Ltd, Hyderabad). Reverse osmosis water treated with ultraviolet light was provided ad libitum.
METHOD DETAILS
Ultra-large scale virtual ligand screening
The recently determined CFTR/ivacaftor cryo-EM structure (PDB: 6O2P21) was used to prospectively screen ~155 million “lead-like” molecules (molecular weight 300–350 Da and logP ≤ 3.5), from the ZINC15 database (http://zinc15.docking.org/33), using DOCK3.734. DOCK3.7 fits pregenerated flexible ligands into a small molecule binding site by superimposing atoms of each molecule on local hot spots in the site (“matching spheres”), representing favorable positions for individual ligand atoms. Here, 45 matching spheres were used, drawn from the experimentally determined pose of ivacaftor. The docking model did not explicitly consider the effect of the membrane in calculating the scoring potentials. For the van der Waals calculations, the environmental effects were ignored. For the typically higher-magnitude electrostatic potentials, calculations were performed using Poisson-Boltzmann electrostatics, defining a low and a high-dielectric region without explicit modeling of membrane lipids. Whereas we have found that more detailed modeling of lipids, at least on dielectric boundaries, can affect docking scores, these effects have been modest53, and likely would remain so without detailed modeling of lipid structures. The resulting docked ligand poses were scored by summing the channel–ligand electrostatics and van der Waals interaction energies and corrected for context-dependent ligand desolvation54,55. Channel structures were protonated using Reduce56. Partial charges from the united-atom AMBER force field were used for all channel atoms57. Potential energy grids for the different energy terms of the scoring function were precalculated based on the AMBER potential57 for the van der Waals term and the Poisson–Boltzmann method QNIFFT58,59, for electrostatics. Context-dependent ligand desolvation was calculated using an adaptation of the generalized-Born method54. Ligands were protonated with Marvin (version 15.11.23.0, ChemAxon, 2015; https://www.chemaxon.com), at pH 7.4. Each protomer was rendered into 3D using Corina (v.3.6.0026, Molecular Networks GmbH; https://www.mn-am.com/products/corina) and conformationally sampled using Omega (v.2.5.1.4, OpenEye Scientific Software; https://www.eyesopen.com/omega). Ligand atomic charges and initial desolvation energies were calculated as described33. In the docking screen, each library molecule was sampled in about 3,727 orientations and, on average, 421 conformations. The best scoring configuration for each docked molecule was relaxed by rigid-body minimization. Overall, over 63 billion complexes were sampled and scored; this took 76,365 core hours–spread over 1000 cores, or slightly more than 3 days.
To identify novel and diverse chemotypes, the top 100,000 scoring molecules were clustered based on their 2D structural similarity. This clustering employed Extended-Connectivity Fingerprints with a diameter of 4 bonds (ECFP4), a widely used method to represent molecular topology (2D structure) as readily-compared bit strings60. ECFP4 generates binary fingerprints of small molecules, allowing for categorization based on specified similarity cutoffs. The similarity between molecules, as defined by their fingerprints, was quantified using the Tanimoto Coefficient (Tc = (|A ∩ B|) / (|A B|)), with a chosen cutoff of 0.5 to determine cluster membership. The Tc measures the bit (feature) overlap between two molecule fingerprints, quantifying their similarity, and is widely used. Among the 100,000 molecules 25,714 cluster heads were identified. Subsequently, the top ranked 1,000 cluster heads were visually inspected in their docked poses to remove molecules that are conformationally strained or with unsatisfied hydrogen-bond acceptors or donors. Topologically diverse molecules that adopted favorable geometries and formed specific interactions with the key potentiator-binding residues S308, Y304, F312, and F931 (PDB: 6O2P and 6O1V), were prioritized from among the top 1000 docking-ranked molecules. Ultimately, 58 compounds, each from a different chemotype family, were selected for experimental evaluation.
Synthesis of molecules
Fifty-three molecules prioritized for purchasing were synthesized by Enamine for a total fulfilment rate of 91%. Compounds were sourced from the Enamine REAL database (https://enamine.net/compound-collections/real-compounds). The purities of active molecules synthesized by Enamine were at least 90% and typically above 95%. For compounds synthesized in house purities were at least 95%. The detailed chemical synthesis can be found in the Chemical Synthesis and analytical investigations section.
Hit Optimization
Potential analogs of the hit compound Z2075279358 were identified through a combination of similarity and substructure searches of the ZINC database33. Potential analogs were docked to the CFTR small molecule binding site using DOCK3.754. As was true in the primary screen, the resulting docked poses were manually evaluated for specific interactions and compatibility with the site, and prioritized analogs were acquired and tested experimentally.
Chemical Synthesis and analytical investigations
The library compounds Z2075279358 (abbreviated ‘358) - Z995908944 from Enamine were synthesized taking advantage of the following synthesis protocols61–63.
General materials and methods for organic synthesis of the library compounds Z2075279358 - Z995908944
Method 1:
A solution of amine (100 mg), the carboxylic acid (1.1 mol. eq. to the amine), and DMSO (0.5 mL) were placed into a capped glass vial and the mixture was stirred for 30 min at rt. 1-Ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC, 1.2 mol. eq. to the amine) was added and stirring was continued for 1 h. In case of obtaining a clear solution, the mixture was left overnight at rt. In case of the solution remained opalescent, the vial was placed in an ultrasound bath overnight. The mixture was filtered and the solvent was evaporated to give the crude product, which was further purified by preparative HPLC.
Method 2:
A solution of amine (100 mg), a carboxylic acid (1.1 mol. eq. to the amine), and DMF (0.5 mL) was stirred for 30 min, and subsequently carbonyldiimidazole (CDI, 1.1 mol. eq. to the amine) was added. The mixture was stirred in a sealed vial for 24 h at rt. Chloroform (3 mL) was added to the reaction mixture and a washing step with water (2×1 mL) was performed. The solvent was evaporated under reduced pressure and trifluoroacetic acid (0.6 mL) was added to the residue. The vial was left in a shaker for 12 h at rt, then chloroform (3 mL) was added and evaporation under reduced pressure was performed to give the crude product, which was purified by HPLC.
Method 3:
A solution of amine (100 mg) and DIPEA (1.1 mol. eq. to the amine; additional equivalents were added when the amine used was in a salt form) in DMSO (0.5 mL) was shaken for 20 min at rt and subsequently, the respective alkyl chloride (1 mol. eq. to the amine) was added. The vial was sealed and stirred for 1 h and then heated for 8 h at 100 °C. After cooling down, evaporation under reduced pressure was performed to give the crude product, which was purified by HPLC.
Method 4:
According to a previously published procedure 62: a mixture of of the nitrile derivative (1 mmol), NH2OH·HCl (1.5 mmol), and triethylamine (2 mmol) in ethanol was shaken in a sealed 8 mL vial for 6 h at rt. Then, the reaction mixture was heated for 16 h at 70 °C. The solvent was removed after the heating utilizing a rotary evaporator. To the in situ formed amidoxime, the carboxylic acid derivative (1 mmol), EDC (1.5 mmol), and 0.7 mL of 20 wt % HOAt in DMF were added and the mixture was shaken for 24 h at rt. The cyclodehydration was performed in the presence of triethylamine (1 mmol) by heating the reaction vial for 3 h at 100°C. Then, 3 mL of water was added and the mixture was extracted with CHCl3. The chloroform phase was washed with water thrice and evaporated. The crude product was purified by HPLC.
Method 5:
According to a previously published procedure 63: A mixture of thiourea (1 mmol) and the first alkylating agent (1 mmol) in 0.6 mL of 2-propanol was heated for 2 h at 100 °C in a sealed 8 mL vial. After cooling to room temperature, to the reaction vial was added sequentially KOH (2.5 mmol) as a 4 M solution in methanol and the second alkylating agent (0.95 mmol). The obtained mixture was heated for 2 h at 60 °C in an ultrasonic bath. The resulting sulfides typically precipitate upon cooling the reaction mixture to room temperature. The subsequent filtration and drying of the precipitate resulted in the product. In other cases, chloroform (3 mL) and water (7 mL) were added and the organic phase was washed with water (7 mL), separated, and evaporated to yield a crude sulfide. The crude product was purified by preparative HPLC.
Method 6:
Based on a previously published procedure 61: A mixture of an aromatic amine (1 mmol) and DIPEA (1.5 mmol) in 0.7 mL of acetonitrile was shaken for 30 min at rt in a sealed 8 mL vial. Then trifluoroethylchloroformate (1 mmol) was added dropwise and the resulting mixture was shaken for 30 min at rt. After the addition of the second amine (0.95 mmol) the reaction vial was sealed and heated for 16 h at 60°C. The solvent was evaporated to give a crude product that was dissolved in 0.5 mL of DMSO and purified by HPLC.
Synthesis protocols and analytical data of Z2075279358 - Z995908944
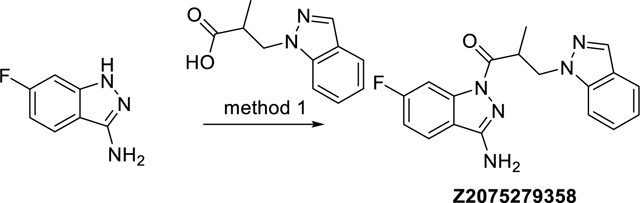
(R,S)-N-(6-Fluoro-1H-indazol-3-yl)-3-(1H-indazol-1-yl)-2-methylpropanamide (Z2075279358)
The synthesis was done according to method 1, starting from 3-(1H-indazol-1-yl)-2-methylpropanoic acid and 6-fluoro-1H-indazol-3-amine to obtain 62% yield. ESI-MS: m/z 338.3 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 338.1412 for C18H17FN5O, found 338.1411. IR (NaCl): 3418, 1676, 1636, 1439, 1425 cm−1. 1H-NMR: (600 MHz, DMSO-d6) δ 1.17 (d, J = 7.0 Hz, 3H), 4.31 (ddq, J = 7.0, 7.0, 7.0 Hz, 1H), 4.53 (dd, J = 14.2, 7.0 Hz, 1H), 4.86 (dd, J = 14.2, 7.0 Hz, 1H), 6.59 (s, 2H), 7.10 – 7.14 (m, 1H), 7.23 (ddd, J = 9.0, 8.8, 2.4 Hz, 1H), 7.36 – 7.40 (m, 1H), 7.71 – 7.74 (m, 2H), 7.89 (dd, J = 9.8, 2.4 Hz, 1H), 7.92 (dd, J = 8.8, 5.2 Hz, 1H), 8.02 – 8.03 (m, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 15.0, 38.4, 50.0, 101.7 (d, J = 29 Hz), 109.8, 112.2 (d, J = 25 Hz), 117.0, 120.4, 120.7, 122.6 (d, J = 11 Hz), 123.4, 126.1, 132.9, 139.6, 139.8 (d, J = 18 Hz), 152.6, 163.2 (d, J = 244 Hz), 172.5. HPLC: system 3: λ = 220 nm, tR = 20.6 min, purity: 99.6%.
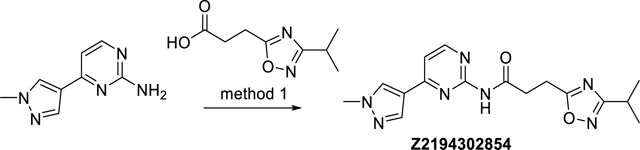
3-(3-Isopropyl-1,2,4-oxadiazol-5-yl)-N-(4-(1-methyl-1H-pyrazol-4-yl)pyrimidin-2-yl) propanamide (Z2194302854)
The synthesis was done according to method 1, starting from 3-(3-isopropyl-1,2,4-oxadiazol-5-yl)propanoic acid and 4-(1-methyl-1H-pyrazol-4-yl)pyrimidin-2-amine, to obtain 47% yield. ESI-MS: m/z 342.2 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 342.1673 for C16H20N7O2, found 342.1672. IR (KBr): 3434, 3150, 3104, 2968, 2934, 1681, 1596, 1581 cm−1. 1H-NMR: (600 MHz, DMSO-d6) δ 1.24 (d, J = 6.9 Hz, 6H), 3.01 (sept, J = 6.9 Hz, 1H), 3.15 – 3.19 (m, 2H), 3.19 – 3.24 (m, 2H), 3.91 (s, 3H), 7.37 (d, J = 5.4 Hz, 1H), 8.11 (s, 1H), 8.41 (s, 1H), 8.53 (d, J = 5.4 Hz, 1H), 10.55 (s, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 20.3, 21.3, 26.0, 32.8, 38.9, 111.1, 120.5, 131.3, 138.2, 157.7, 158.5, 159.4, 170.4, 174.3, 179.1. HPLC: system 3: λ = 220 nm, tR = 13.4 min, purity: 98.8%.
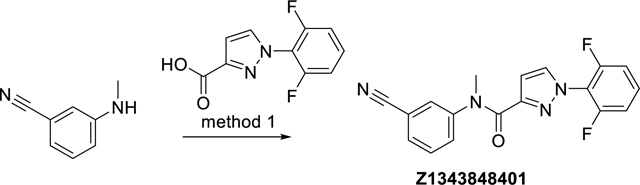
N-(3-Cyanophenyl)-1-(2,6-difluorophenyl)-N-methyl-1H-pyrazole-3-carboxamide (Z1343848401)
The synthesis was done according to method 1, starting from 1-(2,6-difluorophenyl)-1H-pyrazole-3-carboxylic acid and 3-(methylamino)benzonitrile, to obtain 26% yield. ESI-MS: m/z 339.2 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 339.1052 for C18H12F2N4O, found 339.1052. IR (KBr): 3434, 2923, 2227, 1648 cm−1. 1H-NMR: (600 MHz, DMSO-d6) δ 3.43 (s, 3H), 6.67 (s, 1H), 7.32 (dd, J = 8.4, 8.4 Hz, 2H), 7.50 (dd, J = 7.8, 7.8 Hz, 2H), 7.55 – 7.62 (m, 2H), 7.68 (ddd, J = 7.8, 1.3, 1.3 Hz, 1H), 7.74 (s, 1H), 8.08 (s, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 37.6, 108.8, 111.6, 112.6 (dd, J = 19, 4 Hz), 117.3 (t, J = 15 Hz), 118.2, 130.11, 130.14, 130.5, 131.4 (t, J = 10 Hz), 131.9, 134.2, 145.0, 148.2, 156.8 (dd, J = 254, 5 Hz), 162.6. HPLC: system 3: λ = 220 nm, tR = 18.8 min, purity: 99.7%.
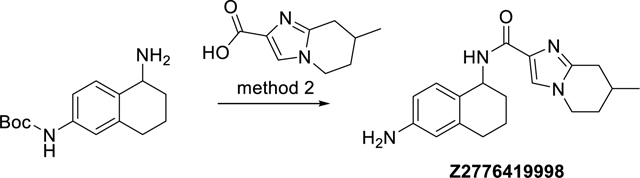
(R,S)-N-((S,R)-6-amino-1,2,3,4-tetrahydronaphthalen-1-yl)-7-methyl-5,6,7,8-tetrahydroimidazo[1,2-a]pyridine-2-carboxamide and (R,S)-N-((R,S)-6-amino-1,2,3,4-tetrahydronaphthalen-1-yl)-7-methyl-5,6,7,8-tetrahydroimidazo[1,2-a]pyridine-2-carboxamide (Z2776419998)
The synthesis was done according to method 2, starting from (R,S)-7-methyl-5,6,7,8-tetrahydroimidazo[1,2-a]pyridine-2-carboxylic acid and (R,S)-tert-butyl (5-amino-5,6,7,8-tetrahydronaphthalen-2-yl)carbamate. The substance was obtained as a mixture of two diastereomers (like and unlike, each racemates), which were not separated to obtain 26% yield. ESI-MS: m/z 325.3 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 325.2023 for C19H25N4O, found 325. 2023. IR (KBr): 3427, 3348, 3170, 1690, 1664, 1567 cm−1. 1H-NMR: (600 MHz, DMSO-d6, diastereomers) δ 1.04 (d, J = 6.6 Hz, 3H), 1.53 – 1.62 (m, 1H), 1.63 – 1.70 (m, 1H), 1.73 – 1.81 (m, 2H), 1.81 – 1.87 (m, 1H), 1.91 – 2.02 (m, 2H), 2.27 (dd, J = 16.4, 10.5 Hz, 1H), 2.51 – 2.57 (m, 1H), 2.59 – 2.66 (m, 1H), 2.80 and 2.81 (2× ddd, each J = 16.4, 4.3, 2.1 Hz, 1H), 3.88 (ddd, J = 12.5, 12.0, 4.7 Hz, 1H), 4.08 (ddd, J = 12.5, 5.6, 3.3 Hz, 1H), 4.90 (brs, 2H), 4.91 – 4.96 (m, 1H), 6.27 (d, J = 2.2 Hz, 1H), 6.350 and 6.354 (2× dd, each J = 8.3, 2.2 Hz, 1H), 6.785 and 6.792 (2× d, each J = 8.3 Hz, 1H), 7.33 and 7.34 (2× d, each J = 9.0 Hz, 1H), 7.52 (s, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6, diastereomers) δ 20.05 and 20.09, 20.8, 27.1, 29.0, 30.0, 30.2, 31.91 and 31.92. 43.8, 45.65 and 45.66, 112.5, 113.2, 120.9, 124.61 and 124.63, 128.68 and 128.72, 135.1. 137.4, 144.3, 147.3, 161.3. HPLC: system 3: λ = 220 nm, tR1 = 10.0 min, tR2 = 10.1 min, purity: 99.1% (both diastereomers).
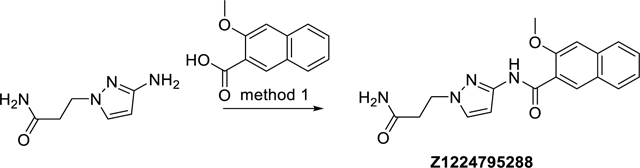
N-(1-(3-Amino-3-oxopropyl)-1H-pyrazol-3-yl)-3-methoxy-2-naphthamide (Z1224795288)
The synthesis was done according to method 1, starting from 3-methoxy-2-naphthoic acid and 3-(3-amino-1H-pyrazol-1-yl)propanamide, to obtain 75% yield. ESI-MS: m/z 339.2 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 339.1452 for C18H19N4O3, found 339. 1452. IR (KBr): 3426, 3348, 3170, 1690, 1664, 1568, 1500 cm−1. 1H-NMR: (600 MHz, DMSO-d6) δ 2.60 (t, J = 7.0 Hz, 2H), 4.01 (s, 3H), 4.23 (t, J = 7.0 Hz, 2H), 6.61 (d, J = 2.1 Hz, 1H), 6.90 (s, 1H), 7.40 (s, 1H), 7.42 (ddd, J = 8.2, 6.9, 1.1 Hz, 1H), 7.51 (s, 1H), 7.56 (ddd, J = 8.0, 6.9, 1.2 Hz, 1H), 7.88 (brd, J = 8.0 Hz, 1H), 7.96 (brd, J = 8.2 Hz, 1H), 8.29 (s, 1H), 10.49 (s, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 35.5, 47.4, 56.0, 96.9, 106.7, 124.3, 125.3, 126.4, 127.5, 127.9, 128.4, 130.5, 130.6, 135.1, 146.6, 154.2, 162.9, 171.5. HPLC: system 3: λ = 220 nm, tR = 16.9 min, purity: 97.7%.
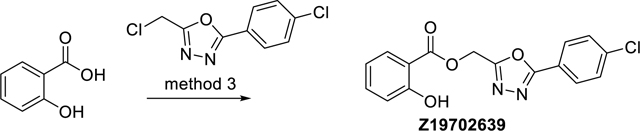
(5-(4-Chlorophenyl)-1,3,4-oxadiazol-2-yl)methyl 2-hydroxybenzoate (Z19702639)
The synthesis was done according to method 3, starting from salicylic acid and 2-(chloromethyl)-5-(4-chlorophenyl)-1,3,4-oxadiazole, to obtain 87% yield. ESI-MS: m/z 331.2 [M+H]+. HR-ESI-MS:m/z [M+H]+ calcd. 331.0480 for C16H12ClN2O4, found 331.0480. IR (KBr): 3448, 3240, 3089, 1690, 1484, 1252 cm−1. 1H-NMR (600 MHz, DMSO-d6) δ 5.69 (s, 2H), 6.96 (ddd, J = 8.1, 7.1, 0.9 Hz, 1H), 7.01 (dd, J = 8.5, 0.9 Hz, 1H), 7.54 (ddd, J = 8.5, 7.1, 1.7 Hz, 1H), 7.67 – 7.71 (m, 2H), 7.84 (dd, J = 8.1, 1.7 Hz, 1H), 8.00 – 8.05 (m, 2H), 10.22 (s, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 56.0, 113.0, 117.6, 119.4, 121.9, 128.5, 129.7, 130.5, 135.9, 137.1, 159.7, 162.2, 164.0, 167.0. HPLC: system 3: λ = 220 nm, tR = 22.7 min, purity: 91.2%.
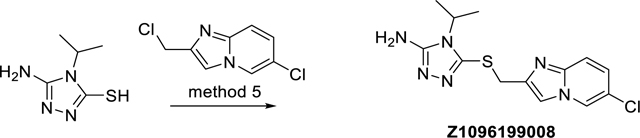
5-(((6-Chloroimidazo[1,2-a]pyridin-2-yl)methyl)thio)-4-isopropyl-4H-1,2,4-triazol-3-amine (Z1096199008)
The synthesis was done according to method 5, starting from 5-amino-4-isopropyl-4H-1,2,4-triazole-3-thiol and 6-chloro-2-(chloromethyl)imidazo[1,2-a]pyridine to obtain 35% yield. ESI-MS: m/z 323.2 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 323.0840 for C13H16ClN6S, found 323.0839. IR (KBr): 3434, 3103, 1654, 1566 cm−1. 1H-NMR: (600 MHz, DMSO-d6) δ 1.25 (d, J = 7.0 Hz, 3H), 4.30 (s, 2H), 4.34 (sept, J = 7.0 Hz, 1H), 5.73 (s, 2H), 7.26 (dd, J = 9.6, 2.1 Hz, 1H), 7.54 (d, J = 9.6 Hz, 1H), 7.78 (s, 1H), 8.78 (dd, J = 2.1, 0.9 Hz, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 20.1, 32.3, 46.2, 111.8, 117.2, 118.8, 124.7, 125.6, 141.9, 142.6, 143.1, 155.4. HPLC: system 3: λ = 220 nm, tR = 10.0 min, purity: 97.0%.
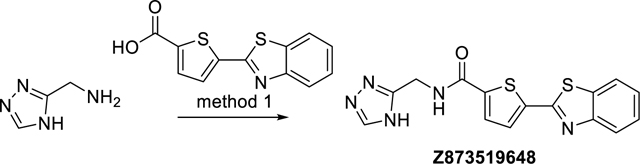
N-((4H-1,2,4-Triazol-3-yl)methyl)-5-(benzo[d]thiazol-2-yl)thiophene-2-carboxamide (Z873519648)
The synthesis was done according to method 1, starting from 5-(benzo[d]thiazol-2-yl)thiophene-2-carboxylic acid and (4H-1,2,4-triazol-3-yl)methanamine to obtain 33% yield. ESI-MS: m/z 342.2 [M+H]+.HR-ESI-MS: m/z [M+H]+ calcd. 342.0478 for C15H12N5OS2, found 342.0477. IR (KBr): 3431, 3379, 3276, 1653, 1624, 1546, 1529 cm−1. 1H-NMR: (600 MHz, DMSO-d6, two isomers were observed) δ 4.57 (brs, 2H), 7.48 (ddd, J = 8.0, 7.3, 1.2 Hz, 1H), 7.56 (ddd, J = 8.0, 7.3, 1.2 Hz, 1H), 7.88 (d, J = 4.2 Hz, 1H), 7.90 (d, J = 4.2 Hz, 1H), 8.05 (ddd, J = 8.0, 1.2, 0.7 Hz, 1H), 8.15 (ddd, J = 8.0, 1.2, 0.7 Hz, 1H), 8.49 (s, 1H), 9.29 (s, 1H), 13.90 (brs, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6, two isomers were observed) δ 35.3 and 36.9, 122.5, 122.8, 125.9, 126.9, 129.1, 129.5, 134.5, 139.7, 142.3 and 143.0, 144.0, 148.8, 151.3, 153.0, 160.3, 160.4 and 160.8. HPLC: system 3: λ = 220 nm, tR = 16.3 min, purity: >99%.
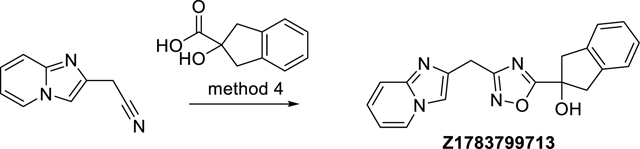
2-(3-(Imidazo[1,2-a]pyridin-2-ylmethyl)-1,2,4-oxadiazol-5-yl)-2,3-dihydro-1H-inden-2-ol (Z1783799713)
The synthesis was done according to method 4, starting from 2-(imidazo[1,2-a]pyridin-2-yl)acetonitrile and 2-hydroxy-2,3-dihydro-1H-indene-2-carboxylic acid to obtain 18% yield. ESI-MS: m/z 333.2 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 333.1348 for C19H17N4O2, found 333.1347. IR (KBr): 3434, 2794, 1637, 1576, 1505 cm−1. 1H-NMR: (600 MHz, DMSO-d6) δ 3.27 (d, J = 16.3 Hz, 2H), 3.53 (d, J = 16.3 Hz, 2H), 4.20 (s, 2H), 6.30 (s, 1H), 6.86 (ddd, J = 6.8, 6.8, 1.2 Hz, 1H), 7.20 (ddd, J = 9.1, 6.8, 1.2 Hz, 1H), 7.16 – 7.20 (m, 2H), 7.23 – 7.27 (m, 2H), 7.47 (dddd, J = 9.1, 1.2, 1.2, 1.2 Hz, 1H), 7.84 (dd, J = 1.2, 0.7 Hz, 1H), 8.49 (ddd, J = 6.8, 1.2, 1.2 Hz, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 25.8, 46.1, 77.0, 110.9, 111.9, 116.3, 124.56, 124.58, 126.7, 139.8, 140.6, 144.1, 168.3, 182.5. HPLC: system 3: λ = 220 nm, tR = 14.0 min, purity: 97.8%.
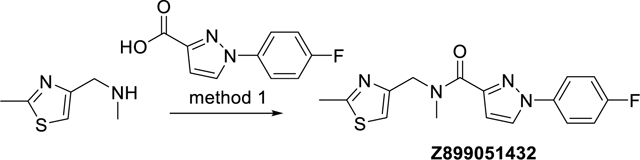
1-(4-Fluorophenyl)-N-methyl-N-((2-methylthiazol-4-yl)methyl)-1H-pyrazole-3-carboxamide (Z899051432)
The synthesis was done according to method 1, starting from 1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid and N-methyl-1-(2-methylthiazol-4-yl)methanamine to obtain 76% yield. ESI-MS: m/z 331.2 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 331.1023 for C16H16FN4OS, found 331.1023. IR (KBr): 3443, 3114, 2926, 1625, 1523, 1509 cm−1. 1H-NMR: (600 MHz, DMSO-d6, two isomers were observed) δ 2.63 and 2.65 (2× s, 3H), 3.01 and 3.07 (2× s, 3H), 4.72 and 5.06 (2× s, 2H), 6.86 and 6.87 (2× d, each J = 2.5 Hz, 1H), 7.25 and 7.29 (2× s, 1H), 7.33 – 7.41 (m, 2H), 7.81 – 7.86 and 7.89 – 7.94 (2× m, 2H), 8.53 and 8.55 (2× d, each J = 2.5 Hz, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6, two isomers were observed) δ 18.8, 33.9 and 36.9, 47.2 and 50.2, 110.1 and 110.2, 115.3 and 115.7, 116.4 (d, J = 23 Hz), 120.8 and 121.0 (2× d, each J = 9 Hz), 128.77 and 128.82, 135.8 and 135. 9 (2× d, each J = 3 Hz), 148.1 and 148.2, 151.6 and 152.5, 160.57 (d, J = 246 Hz) and 160.62 (d, J = 242 Hz), 162.5 and 162.9, 165.6 and 165.7. HPLC: system 3: λ = 220 nm, tR = 17.8 min, purity: >99%.
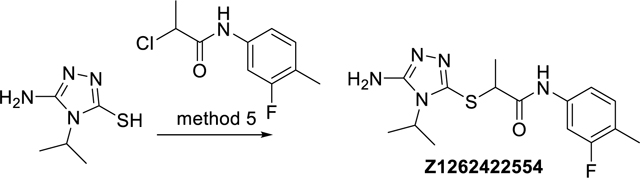
(R,S)-2-((5-Amino-4-isopropyl-4H-1,2,4-triazol-3-yl)thio)-N-(3-fluoro-4-methylphenyl) propanamide (Z1262422554)
The synthesis was done according to method 5, starting from 5-amino-4-isopropyl-4H-1,2,4-triazole-3-thiol and (R,S)-2-chloro-N-(3-fluoro-4-methylphenyl)propanamide, to obtain 37% yield. ESI-MS: m/z 338.2 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 338.1445 for C15H21FN5OS, found 338.1446. IR (KBr): 3316, 3190, 2980, 2933, 1684, 1627, 1551, 1511 cm−1. 1H-NMR (600 MHz, DMSO-d6) δ 1.34 and 1.35 (2× d, each J = 7.1 Hz, 6H), 1.46 (d, J = 7.0 Hz, 3H), 2.17 (d, J = 1.5 Hz, 3H), 4.14 (q, J = 7.0 Hz, 3H), 4.43 (qq, J = 7.1 Hz, 3H), 5.80 (s, 2H), 7.16 (dd, J = 8.3, 1.9 Hz, 1H), 7.20 (dd, J = 8.3, 8.3 Hz, 1H), 7.49 (dd, J = 12.8, 1.9 Hz, 1H), 10.36 (s, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 13.6 (d, J = 2 Hz), 18.1, 20.18, 20.21, 46.4, 47.2, 105.9 (d, J = 27 Hz), 114.8 (d, J = 2 Hz), 118.8 (d, J = 17 Hz), 131.4 (d, J = 7 Hz), 138.1 (d, J = 13 Hz), 140.6, 155.6, 160.2, (d, J = 241 Hz), 169.5. HPLC: system 3: λ = 220 nm, tR = 16.3 min, purity: >99%.
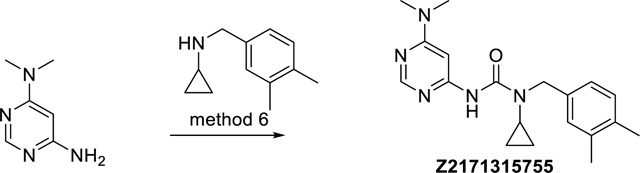
1-Cyclopropyl-3-(6-(dimethylamino)pyrimidin-4-yl)-1-(3,4-dimethylbenzyl)urea (Z2171315755)
The synthesis was done according to method 6, starting from N4,N4-dimethylpyrimidine-4,6-diamine and N-(3,4-dimethylbenzyl)cyclopropanamine to obtain 24% yield. ESI-MS: m/z 340.3 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 340.2132 for C19H26N5O, found 340.2130. IR (KBr): 3422, 2936, 1684, 1608, 1521, 1498 cm−1. 1H-NMR: (600 MHz, DMSO-d6) δ 0.73 – 0.80 (m, 2H), 0.84 – 0.89 (m, 2H), 2.18 (s, 3H), 2.20 (s, 3H), 2.65 – 2.70 (m, 1H), 3.03 (s, 6H), 4.45 (s, 2H), 6.96 (dd, J = 7.8, 1.5 Hz, 1H), 7.02 (brs, 1H), 7.08 (d, J = 7.8 Hz, 1H), 7.16 (d, J = 1.1 Hz, 1H), 8.19 (d, J = 1.1 Hz, 1H), 8.34 (brs, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 8.7, 19.0, 19.4, 28.2, 36.8, 49.2, 86.9, 124.7, 128.4, 129.5, 134.7, 135.8, 136.1, 155.0, 156.9, 157.5, 162.8. HPLC: system 3: λ = 220 nm, tR = 18.5 min, purity: 95.0%.
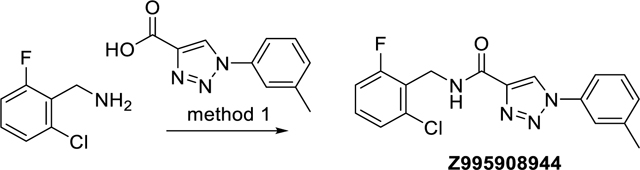
N-(2-Chloro-6-fluorobenzyl)-1-(m-tolyl)-1H-1,2,3-triazole-4-carboxamide (Z995908944)
The synthesis was done according to method 1, starting from 1-(m-tolyl)-1H-1,2,3-triazole-4-carboxylic acid and (2-chloro-6-fluorophenyl)methanamine to obtain 50% yield. ESI-MS: m/z 345.1 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 345.0913 for C17H15ClFN4O, found 345.0912. IR (NaCl): 3415, 3117, 2921, 1685 cm−1. 1H-NMR: (600 MHz, DMSO-d6) δ 2.42 (s, 3H), 4.64 (dd, J = 5.3, 1.0 Hz, 3H), 6.61 (s, 2H), 7.24 (ddd, J = 9.6, 8.3, 1.3 Hz, 1H), 7.32 – 7.35 (m, 1H), 7.34 (dd, J = 8.1, 1.3 Hz, 1H), 7.40 (ddd, J = 8.3, 8.1, 6.1 Hz, 1H), 7.48 (dd, J = 7.8, 7.8 Hz, 1H), 7.71 – 7.74 (m, 1H), 7.78 – 7.80 (m, 1H), 8.87 (t, J = 5.3 Hz, 1H), 9.22 (s, 2H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 20.9, 34.4 (d, J = 4 Hz), 114.5 (d, J = 23 Hz), 117.5, 120.9, 123.7 (d, J = 15 Hz), 124.7, 125.4 (d, J = 3 Hz), 129.7, 130.1 (d, J = 10 Hz), 134.8 (d, J = 8 Hz), 136.2, 139.7, 143.2, 159.1, 61.5 (d, J = 249 Hz). HPLC: system 3: λ = 220 nm, tR = 21.7 min, purity: >99%.
Detailed synthesis procedures of the compounds of type 1–10
The following derivatives of Z1834339853 (abbreviated ‘853) were prepared. 1H- and 13C-NMR spectra and the HPLC-chromatograms demonstrated purities > 95%.
General materials and methods
Reagents and solvents were purchased in their purest grade from abcr, Acros, Alfa Aesar, Chemspace, Enamine, Manchester Organics, Sigma Aldrich, TCI, VWR and were used without further purification. Unless otherwise noted, reactions were performed under nitrogen atmosphere employing dry solvents of commercial quality, used as purchased. All reactions were carried out using a magnetic stirrer with optional aluminum heating block or ice bath for sealed microwave vials and oil or ice bath for round bottom flasks, respectively. Solvents were evaporated by a rotation evaporator equipped with a membrane vacuum pump. Microwave assisted (Discover® microwave oven, CEM Corp.) synthesis was carried out by 20 × irradiating with microwaves (50 W, irradiation time: 10 s). In between each irradiation step, intermittent cooling of the reaction mixture to a temperature of −10°C was achieved by sufficient agitation in an ethanol-ice bath. TLC analyses were performed using Merck 60 F254 aluminum sheets and analyzed by UV light (254 nm). Purification by flash column chromatography was conducted using silica gel 60 (40–63 μm mesh, Merck) and eluents as binary mixtures with the volume ratios indicated. Preparative HPLC was performed on an Agilent 1200 preparative series HPLC system or on an Agilent HPLC 1260 Infinity system combined with an MWD detector and fraction collector, applying a linear gradient and a flow rate as indicated below. As HPLC column, a Zorbax-Eclipse XDB-C8 PrepHT (21.2 mm × 150 mm, 5 μm) was used. Products purified by preparative HPLC using aqueous solvents were lyophilized. For the separation of enantiomers, a Waters preparative HPLC system consisting of the 2545 binary gradient module, 2707 autosampler, 2998 photodiode array detector and the fraction collector III was employed, using a Chiralpak IC column (250 mm × 30 mm, 5 μm), a flow rate of 45 mL/min and an isocratic binary solvent system specified below. Compounds were characterized by NMR-, IR- and high-resolution mass spectra (HRMS). Purity was assessed by RP-HPLC. All assayed compounds were >95% pure. ESI-mass spectra were recorded using LC-MS: Thermo Scientific Dionex Ultimate 3000 UHPLC quarternary pump, autosampler, and RS-diode array detector, column: Zorbax-Eclipse XDB-C8 analytical column, 3.0 mm × 100 mm, 3.5 μm, flow rate 0.4 mL/min using DAD detection (230 nm; 254 nm), coupled to a Bruker Daltonics Amazon mass spectrometer using ESI as the ionization source. High mass accuracy and resolution experiments were performed on a Bruker Daltonics timsTOF Pro spectrometer using electrospray ionization (ESI) as an ionization source. NMR spectra were obtained either on a Bruker Avance III 400 (400 MHz for 1H and 101 MHz for 13C) or a Bruker Avance III 600 (600 MHz for 1H and 151 MHz for 13C) spectrometer, the latter equipped with a Prodigy nitrogen-cooled probe at 297 K, using the deuterated solvents indicated below. For the spectra recorded in organic solvents, the chemical shifts are reported in ppm (δ) relative to TMS. For measurements in D2O, the water peak was used for calibration. IR spectra were performed on a Jasco FT/IR 4100 spectrometer using a KBr pellet or with substance film on a NaCl crystal plate, as specified. Substance purities were assessed by analytical HPLC (Agilent 1100 analytical series, equipped with a quarternary pump and variable wavelength detector; column Zorbax Eclipse XDB-C8 analytical column, 4.6 mm × 150 mm, 5 μm, flow rate 0.5 mL / min, detection wavelengths: 220 nm, 254 nm; system 1: methanol / 0.1% aq. HCOOH, linear gradient: 10% methanol for 3 min, 10% to 100% methanol in 15 min, 100% methanol for 6 min; system 2: acetonitrile / 0.1% aq. HCOOH, linear gradient: 5% acetonitrile for 3 min, 5% to 95% acetonitrile in 15 min, 95% acetonitrile for 6 min; system 3: acetonitrile / 0.1% aq. TFA, linear gradient: 3% to 85% acetonitrile in 26 min, 85% to 95% acetonitrile in 2 min, 95% acetonitrile for 2 min; system 4: acetonitrile / 0.1% aq. HCOOH, linear gradient: 3% to 85% acetonitrile in 26 min, 85% to 95% acetonitrile in 2 min, 95% acetonitrile for 2 min;. chiral analytical HPLC was run on an AGILENT series 1100 system equipped with a VWD and detection at 254 nm. As a chiral column, a DAICEL Chiralpak IC column (4.6 mm × 250 mm, 5 μM) was used at 20 °C and a flow rate 1.0 mL / min with the solvent system as indicated. Specific optical rotation values (°× mL × dm−1 × g−1) were obtained from a Jasco P2000 polarimeter with the solvents indicated. Melting points were determined in open capillaries using a Büchi 510 melting point apparatus and are given uncorrected.
General experimental procedure
General procedure A (GPA): preparation of 1.26 M Jones reagent
To a stirred solution of CrO3 (126 mg) in 500 μL water at 0 °C was slowly added conc. H2SO4 (126 μL). The volume was adjusted to 1 mL using water.
The syntheses of the 1-acyl-3-aminoindazole derivatives of type 1 were performed as shown in scheme 1. Starting from the corresponding phenol derivatives, a recently reported protocol comprising deprotonation with NaH and subsequent SN2-reaction with enantiopure (R)- and (S)-3-bromo-2-methyl propanol,64 respectively, allowed to obtain the enantiopure (R)- and (S)-3-aryloxy-2-methylpropanol derivatives 11a-h and ent11a-d, respectively. Both phenoxy-derivatives 11a/ent11a 64 and the fluoro derivative 11e 65 have been described in the literature. Jones oxidation gave the corresponding carboxylic acid derivatives 12a-h and ent12a-d. It is worthy of note, that the phenoxy derivatives 12a/ent12a,64 the racemic naphthyl derivative of 12b 66 and the biphenyl derivative 12g 67 have been previously published. The coupling reactions using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC × HCl) and 1-hydroxy-7-azabenzotriazole (HOAt) with 6-fluoro-1H-indazole-3-amine (13) proceeded with high regioselectivity to give access to the 1-acyl-3-aminoindazole derivatives 1a-h and ent1a-d in 15–53% yield. Investigation of the alcohol derivatives 11a-h / ent11a-d and the target compounds 1a-h / ent1a-d by chiral HPLC proved, that the synthetic pathway allowed to obtain test compounds with high enantiopurity.
Scheme 1.

Synthesis of the enantiopure derivatives of ‘853, modified at the phenoxy site, reagents and conditions: (a) 1. NaH, DMF, 0 °C, 30 min, 2. (R)-3-bromo-2-methylpropan-1-ol or (S)-3-bromo-2-methylpropan-1-ol, DMF, rt, 18–31 h, (50–72% crude); (b) 1. CrO3, H2SO4, H2O, acetone, 0 °C, 3–5 h, 2. iPrOH, 0 °C -> rt (48–64%, crude); (c) 1. EDC × HCl, HOAt, DMF, rt, 2. 6-fluoro-1H-indazole-3-amine (13), rt, 1–3 h (15–53%).
(R)-2-Methyl-3-phenoxypropan-1-ol (11a):
To a stirred solution of phenol distilled from molecular sieve (187 μL, 2.13 mmol) in DMF (4 mL) in a flame-dried flask, sodium hydride (60% in mineral oil; 97.4 mg, 2.44 mmol) was added. After stirring the suspension for 30 min at rt, (R)-3-bromo-2-methylpropan-1-ol (213 μL, 2.03 mmol) was added. The mixture was stirred for additional 18 h, then 2N NaOH was added and extraction with tert-butyl methyl ether (3×) was performed. The combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated. After purification by flash-chromatography (isohexane / tert-butyl methyl ether, 7:3) compound 11a (202 mg, 1.22 mmol, 60%) was obtained as colorless liquid. ESI-MS:m/z 189.3 [M+Na]+. 1H-NMR: (400 MHz, CDCl3) δ 1.04 (d, J = 7.0 Hz, 3H), 1.90 (dd, J = 5.7, 5.7 Hz, 1H), 2.21 (m, 1H), 3.73 – 3.70 (m, 2H), 3.93 (dd, J = 9.1, 7.0 Hz, 1H), 3.98 (dd, J = 9.1, 5.3 Hz, 1H), 6.89 – 6.93 (m, 2H), 6.93 – 6.98 (m, 1H), 7.32 – 7.26 (m, 2H). 13C-NMR: (DEPTQ, 101 MHz, CDCl3) δ 13.6, 35.7, 66.2, 71.2, 114.5, 120.9, 129.5, 158.8. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 95:5: tR = 6.6 min, 99% ee (254 nm). [α]D24: +2.4 (c = 0.9, chloroform). The analytical data of this compound are in accordance with those reported in the literature.
(S)-2-Methyl-3-phenoxypropan-1-ol (ent-11a):
(S)-2-Methyl-3-phenoxypropan-1-ol (ent-11a) was synthesized analogously as described for (R)-SX224 11a, starting from (S)-3-bromo-2-methylpropan-1-ol. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 95:5: tR = 6.9 min, 99% ee (254 nm). [α]D25: −2.9 (c = 1.6, chloroform). The analytical data of this compound are in accordance with those reported in the literature.
(R)-2-Methyl-3-(naphthalen-2-yloxy)propan-1-ol (11b):
To a stirred solution of 2-naphthol (200 mg, 1.39 mmol) in DMF (4 mL) in a flame-dried flask sodium hydride (60% in mineral oil; 64.0 mg, 1.60 mmol) was added. After stirring the suspension for 30 min at rt, (R)-3-bromo-2-methylpropan-1-ol (138 μL, 1.32 mmol) was added. The mixture was stirred for additional 22 h, then 2N NaOH was added and extraction with tert-butyl methyl ether (3×) was performed. The combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated. After purification by flash-chromatography (isohexane / tert-butyl methyl ether, 7:3) compound 11b (206 mg, 0.95 mmol, 72%) was obtained as pale yellow liquid, which solidified upon standing (white wax). ESI-MS: m/z 238.9 [M+Na]+. HR-ESI-MS: m/z [M+H]+ calcd. 239.1043 for C14H16NaO2, found 239.1044. 1H-NMR: (400 MHz, CDCl3) δ 1.09 (d, J = 7.0 Hz, 3H), 2.22 – 2.34 (m, 1H), 3.74 – 3.78 (m, 2H), 4.06 (dd, J = 9.1, 6.7 Hz, 1H), 4.09 (dd, J = 9.1, 5.5 Hz, 1H), 7.18 – 7.10 (m, 2H), 7.34 (ddd, J = 8.1, 6.9, 1.3 Hz, 1H), 7.44 (ddd, J = 8.2, 6.9, 1.3 Hz, 1H), 7.68 – 7.80 (m, 3H). 13C-NMR: (DEPTQ, 101 MHz, CDCl3) δ 13.7, 35.7, 66.2, 71.2, 106.7, 118.8, 123.7, 126.4, 126.7, 127.6, 129.0, 129.4, 134.5, 156.7. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 98:2: tR = 17.5 min, >99% ee (254 nm). [α]D24: +1.8 (c = 0.7, chloroform).
(S)-2-Methyl-3-(naphthalen-2-yloxy)propan-1-ol (ent-11b):
Methyl-3-(naphthalen-2-yloxy)propan-1-ol (ent-11b) was synthesized analogously as described for 11b, starting from (S)-3-bromo-2-methylpropan-1-ol. The analytical data were in accordance. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 98:2: tR = 18.2 min, >99% ee (254 nm). [α]D23:−3.0 (c = 0.8, chloroform).
(R)-2-Methyl-3-(3-(trifluoromethyl)phenoxy)propan-1-ol (11c):
To a stirred solution of 3-(trifluormethyl)phenol (150 μL, 1.23 mmol) in DMF (4 mL) in a flame-dried flask sodium hydride (60% in mineral oil; 56.2 mg, 1.40 mmol) was added. After stirring the suspension for 30 min at rt, (R)-3-bromo-2-methylpropan-1-ol (123 μL, 1.17 mmol) was added. The mixture was stirred for additional 22 h, then 2N NaOH was added and extraction with tert-butyl methyl ether (3×) was performed. The combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated. Purification by flash-chromatography (isohexane / tert-butyl methyl ether, 7:3) afforded 11c as pale yellow liquid (148 mg, 0.63 mmol) still containing minor amounts of (R)-3-bromo-2-methylpropan-1-ol (confirmed by NMR). The crude product was used for the next step without further purification. ESI-MS: m/z 257.5 [M+Na]+. HR-ESI-MS: m/z [M+H]+ calcd. 257.0760 for C11H13F3NaO2, found 257.0761. 1H-NMR: (400 MHz, CDCl3) δ 1.07 (d, J = 7.0 Hz, 3H), 1.66 – 1.73 (m, 1H), 2.16 – 2.27 (m, 1H), 3.70 – 3.75 (m, 2H), 3.97 – 4.00 (m, 2H), 7.05 – 7.10 (m, 1H), 7.12 – 7.16 (m, 1H), 7.18 – 7.23 (m, 1H), 7.35 – 7.42 (m, 1H). 13C-NMR: (DEPTQ, 101 MHz, CDCl3) δ 13.8, 35.8, 65.7, 71.1, 111.4 (q, J = 4 Hz), 117.6 (q, J = 4 Hz), 118.1, 124.1 (q, J = 272 Hz), 130.1, 132.0 (q, J = 32 Hz), 159.1. Chiral HPLC: isocratic elution with n-hexane/isopropanol, 98:2: tR = 7.0 min, 99% ee (254 nm).
(S)-2-Methyl-3-(3-(trifluoromethyl)phenoxy)propan-1-ol (ent-11c):
(S)-2-Methyl-3-(3-(trifluoromethyl)phenoxy)propan-1-ol (ent-11c) was synthesized analogously as described for 11c, starting from (S)-3-bromo-2-methylpropan-1-ol. The analytical data were in accordance. chiral HPLC: isocratic elution with n-hexane / isopropanol, 98:2: tR = 7.7 min, >99% ee (254 nm).
(R)-3-(2,4-Di-tert-butylphenoxy)-2-methylpropan-1-ol (11d):
To a stirred solution of 2,4-di-tert-butylphenol (215 mg, 1.04 mmol) in DMF (4 mL) in a flame-dried flask sodium hydride (60% in mineral oil; 47.5 mg, 1.19 mmol) was added. After stirring the suspension for 30 min at rt, (R)-3-bromo-2-methylpropan-1-ol (104 μL, 0.99 mmol) was added. The mixture was stirred for additional 22 h, then 2N NaOH was added and extraction with tert-butyl methyl ether (3×) was performed. The combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated. After purification by flash-chromatography (isohexane / tert-butyl methyl ether, 7:3) compound 11d (174 mg, 0.62 mmol, 63%) was obtained as colorless liquid, which solidified upon standing (white wax). ESI-MS: m/z 301.2 [M+Na]+. HR-ESI-MS: m/z [M+H]+ calcd. 301.2138 for C18H30NaO2, found 301.2138. 1H-NMR (400 MHz, CDCl3) δ 1.11 (d, J = 6.9 Hz, 3H), 1.30 (s, 9H), 1.40 (s, 9H), 1.57 (dd, J = 5.6, 5.6 Hz, 1H), 2.13 – 2.32 (m, 1H), 3.69 – 3.81 (m, 2H), 3.94 (dd, J = 9.1, 5.3 Hz, 1H), 3.98 (dd, J = 9.1, 6.1 Hz, 1H), 6.83 (d, J = 8.5 Hz, 1H), 7.17 (dd, J = 8.5, 2.5 Hz, 1H), 7.34 (d, J = 2.5 Hz, 1H). 13C-NMR (DEPTQ, 101 MHz, CDCl3) δ 14.4, 30.1, 31.7, 34.4, 35.2, 36.2, 49.6, 65.8, 70.3, 111.4, 123.5, 124.1, 137.2, 142.7, 155.5. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 98:2: tR = 5.1 min, >99% ee (254 nm). [α]D23: −0.9 (c = 1.1, methanol).
(S)-3-(2,4-Di-tert-butylphenoxy)-2-methylpropan-1-ol (ent-11d):
(S)-3-(2,4-Di-tert-butylphenoxy)-2-methylpropan-1-ol (ent-11d) was synthesized analogously as described for 11d, starting from (S)-3-bromo-2-methylpropan-1-ol. The analytical data were in accordance. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 98:2: tR = 5.4 min, 98% ee (254 nm). [α]D24: +1.6 (c = 1.6, methanol).
(R)-3-(4-Fluorophenoxy)-2-methylpropan-1-ol (11e):
To a stirred solution of 4-fluorophenol (150 mg, 1.34 mmol) in DMF (3 mL) in a flame-dried flask sodium hydride (60% in mineral oil; 61.0 mg, 1.52 mmol) was added. After stirring the suspension for 30 min at rt, (R)-3-bromo-2-methylpropan-1-ol (134 μL, 1.27 mmol) was added. The mixture was stirred for additional 31 h, then 2N NaOH was added and extraction with tert-butyl methyl ether (3×) was performed. The combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated. After purification by flash-chromatography (isohexane / tert-butyl methyl ether, 7:3) compound 11e (125 mg, 0.68 mmol, 53%) was obtained as pale yellow liquid. ESI-MS: m/z 207.5 [M+Na]+. HR-ESI-MS: m/z [M+H]+ calcd. 207.0797 for C10H13FNaO2, found 207.0794. 1H-NMR: (400 MHz, CDCl3) δ 1.04 (d, J = 7.0 Hz, 3H), 1.83 (dd, J = 5.6, 5.6 Hz, 1H), 2.13 – 2.26 (m, 1H), 3.69 – 3.73 (m, 2H), 3.90 (dd, J = 9.0, 6.6 Hz, 1H), 3.93 (dd, J = 9.0, 5.5 Hz, 1H), 6.81 – 6.88 (m, 2H), 6.93 – 7.01 (m, 2H). 13C-NMR: (DEPTQ, 101 MHz, CDCl3) δ 13.7, 35.8, 66.2, 71.9, 115.6 (d, J = 8 Hz), 115.9 (d, J = 23 Hz), 155.1 (d, J = 2 Hz), 157.4 (d, J = 238 Hz). Chiral HPLC: isocratic elution with n-hexane / isopropanol, 98:2: tR = 10.9 min, >99% ee (254 nm). [α]D25: +3.2 (c = 0.9, chloroform).
(R)-3-(4-Bromophenoxy)-2-methylpropan-1-ol (11f):
To a stirred solution of 4-bromophenol (150 mg, 0.87 mmol) in DMF (3 mL) in a flame-dried flask sodium hydride (60% in mineral oil; 39.8 mg, 1.00 mmol) was added. After stirring the suspension for 30 min at rt, (R)-3-bromo-2-methylpropan-1-ol (86.5 μL, 0.83 mmol) was added. The mixture was stirred for additional 31 h, then 2N NaOH was added and extraction with tert-butyl methyl ether (3×) was performed. The combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated. After purification by flash-chromatography (isohexane / tert-butyl methyl ether, 7:3) compound 11f (112 mg, 0.46 mmol, 55%) was obtained as pale yellow liquid. ESI-MS: m/z 274.3 [M+Na]+. HR-ESI-MS: m/z [M+H]+ calcd. 266.9991 for C10H13BrNaO2, found 266.9994. 1H-NMR: (400 MHz, CDCl3) δ 1.04 (d, J = 7.0 Hz, 3H), 1.73 (dd, J = 5.6, 5.6 Hz, 1H), 2.13 – 2.25 (m, 1H), 3.68 – 3.72 (m, 2H), 3.90 – 3.93 (m, 2H), 6.76 – 6.82 (m, 2H), 7.34 – 7.40 (m, 2H). 13C-NMR: (DEPTQ, 101 MHz, CDCl3) δ 13.6, 35.6, 65.8, 71.2, 113.0, 116.3, 132.2, 158.0. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 98:2: tR = 11.8 min, >99% ee (254 nm). [α]D25: +1.7 (c = 0.6, chloroform).
(R)-3-([1,1’-Biphenyl]-4-yloxy)-2-methylpropan-1-ol (11g):
To a stirred solution of [1,1’-biphenyl]-4-ol (150 mg, 0.88 mmol) in DMF (4 mL) in a flame-dried flask sodium hydride (60% in mineral oil; 41.0 mg, 1.01 mmol) was added. After stirring the suspension for 30 min at rt, (R)-3-bromo-2-methylpropan-1-ol (88.0 μL, 0.84 mmol) was added. The mixture was stirred for additional 31 h, then 2N NaOH was added and extraction with tert-butyl methyl ether (3×) was performed. The combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated. After purification by flash-chromatography (isohexane / tert-butyl methyl ether, 7:3) compound 11g (104 mg, 0.42 mmol, 50%) was obtained as white resin. ESI-MS: m/z 243.1 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 265.1199 for C16H18NaO2, found 265.1199. 1H-NMR: (400 MHz, CDCl3) δ 1.06 (d, J = 7.0 Hz, 3H), 1.85 (dd, J = 5.6, 5.6 Hz, 1H), 2.18 – 2.30 (m, 1H), 3.70 – 3.76 (m, 2H), 3.98 (dd, J = 9.1, 6.8 Hz, 1H), 4.02 (dd, J = 9.1, 5.5 Hz, 1H), 6.96 – 7.02 (m, 2H), 7.31 (m, 1H), 7.45 – 7.39 (m, 2H), 7.58 – 7.50 (m, 4H). 13C-NMR: (DEPTQ, 101 MHz, CDCl3) δ 13.6, 35.7, 66.2, 71.3, 114.7, 126.7, 126.7, 128.2, 128.7, 134.0, 140.8, 158.4. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 98:2: tR = 24.3 min, 99% ee (254 nm). [α]D25: +2.1 (c = 0.6, chloroform).
(R)-3-(Benzofuran-5-yloxy)-2-methylpropan-1-ol (11h):
To a stirred solution of benzofuran-5-ol (150 mg, 1.12 mmol) in DMF (3 mL) in a flame-dried flask sodium hydride (60% in mineral oil; 50.9 mg, 1.27 mmol) was added. After stirring the suspension for 30 min at rt, (R)-3-bromo-2-methylpropan-1-ol (112 μL, 1.06 mmol) was added. The mixture was stirred for additional 18 h, then 2N NaOH was added and extraction with tert-butyl methyl ether (3×) was performed. The combined organic layers were washed with water and, brine, dried over Na2SO4 and concentrated. After purification by flash-chromatography (isohexane / tert-butyl methyl ether, 7:3) compound 11h (117 mg, 0.57 mmol, 54%) was obtained as yellow resin. ESI-MS: m/z 207.0 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 229.0835 for C12H14NaO3, found 229.0837. 1H-NMR: (400 MHz, CDCl3) δ 1.05 (d, J = 7.0 Hz, 3H), 2.00 (dd, J = 5.6, 5.6 Hz, 1H), 2.24 (m, 1H), 3.73 (m, 2H), 3.96 (dd, J = 9.1, 7.0 Hz, 1H), 4.01 (dd, J = 9.1, 5.6 Hz, 1H), 6.70 (dd, J = 2.2, 0.9 Hz, 1H), 6.90 (dd, J = 8.9, 2.6 Hz, 1H), 7.08 (d, J = 2.6 Hz, 1H), 7.39 (ddd, J = 8.9, 0.9, 0.5 Hz, 1H), 7.59 (d, J = 2.2 Hz, 1H). 13C-NMR (DEPTQ, 101 MHz, CDCl3) δ 13.6, 35.7, 66.4, 72.4, 104.6, 106.7, 111.8, 113.5, 128.0, 145.8, 150.0, 155.1. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 98:2: tR = 18.5 min, >99% ee (254 nm). [α]D24: +3.8 (c = 1.5, chloroform).
(S)-2-Methyl-3-phenoxypropanoic acid (12a):
11a (171 mg, 1.03 mmol) was dissolved in acetone (9 mL, 0.11 M). Jones reagent (816 μL, 1.26 M, 1.03 mmol, GPA) was added at 0 °C and the mixture was stirred for 3 h at 0 °C. After terminating the reaction by the addition of 2-propanol, the mixture was filtered through a pad of celite, which was further washed with ethyl acetate. The combined organic layers were extracted with 1N NaOH. After acidifying the aqueous layer using 2N HCl and extraction with ethyl acetate (2×), the combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated. Purification by flash-chromatography (CH2Cl2 / MeOH + 0.1% HCOOH, 98:2) afforded compound 12a (105 mg, 0.58 mmol, 57%) as pale yellow liquid, which solidified upon standing (beige resin). ESI-MS: m/z 203.2 [M+Na]+. 1H-NMR: (400 MHz, CDCl3) δ 1.35 (d, J = 7.1 Hz, 3H), 3.00 (ddq, J = 7.1, 6.7, 5.9 Hz, 1H), 4.03 (dd, J = 9.1, 5.9 Hz, 1H), 4.20 (dd, J = 9.1, 6.7 Hz, 1H), 6.89 – 6.93 (m, 2H), 6.94 – 6.98 (m, 1H), 7.26 – 7.31 (m, 2H). 13C-NMR: (DEPTQ, 101 MHz, CDCl3) δ 13.8, 39.7, 69.1, 114.7, 121.1, 129.5, 158.5, 180.2. [α]D22: +2.0 (c = 2.5, chloroform). The analytical data of this compound are in accordance with those reported in the literature.
(R)-2-Methyl-3-phenoxypropanoic acid (ent-12a):
(R)-2-Methyl-3-phenoxypropanoic acid (ent-12a) was synthesized analogously as described for 12a, starting from ent-11a. The analytical data of this compound are in accordance with those reported in the literature.
(S)-2-Methyl-3-(naphthalen-2-yloxy)propanoic acid (12b):
11b (191 mg, 0.88 mmol) was dissolved in acetone (8 mL, 0.11 M). Jones reagent (701 μL, 1.26 M, 0.88 mmol, GPA) was added at 0 °C and the mixture was stirred for 5 h at 0 °C. After terminating the reaction by the addition of 2-propanol, the mixture was filtered through a pad of celite, which was further washed with ethyl acetate. The combined organic layers were extracted with 1N NaOH. After acidifying the aqueous layer using 2N HCl and extraction with ethyl acetate (2×), the combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated. Compound 12b (130 mg, 0.56 mmol, 64%) was obtained as an orange solid, which was used for the next step without further purification. ESI-MS: m/z 252.9 [M+Na]+. HR-ESI-MS: m/z [M+H]+ calcd. 253.0835 for C14H14NaO3, found 253.0837. 1H-NMR: (400 MHz, CDCl3) δ 1.39 (d, J = 7.1 Hz, 3H), 3.06 (ddq, J = 7.1, 6.7, 5.9 Hz, 1H), 4.14 (dd, J = 9.1, 5.9 Hz, 1H), 4.32 (dd, J = 9.1, 6.7 Hz, 1H), 7.12 – 7.18 (m, 2H), 7.34 (ddd, J = 8.2, 6.9, 1.3 Hz, 1H), 7.43 (ddd, J = 8.2, 6.9, 1.3 Hz, 1H), 7.70 – 7.79 (m, 3H). 13C-NMR: (DEPTQ, 151 MHz, CDCl3) δ 13.9, 39.7, 69.2, 107.0, 118.8, 123.8, 126.4, 126.8, 127.6, 129.1, 129.4, 134.5, 156.5, 179.9. [α]D24: −3.5 (c = 0.6, chloroform).
(R)-2-Methyl-3-(naphthalen-2-yloxy)propanoic acid (ent-12b):
(R)-2-Methyl-3-(naphthalen-2-yloxy)propanoic acid (ent-12b) was synthesized analogously as described for 12b, starting from ent-11b. The analytical data were in accordance. [α]D24: +2.5 (c = 0.7, chloroform).
(S)-2-Methyl-3-(3-(trifluoromethyl)phenoxy)propanoic acid (12c):
Crude 11c (141 mg, 0.60 mmol) was dissolved in acetone (6 mL, 0.10 M). Jones reagent (476 μL, 1.26 M, 0.60 mmol, GPA) was added at 0 °C and the mixture was stirred for 4.5 h at 0 °C. After terminating the reaction by the addition of 2-propanol, the mixture was filtered through a pad of celite, which was further washed with ethyl acetate. The combined organic layers were extracted with 1N NaOH. After acidifying the aqueous layer using 2N HCl and extraction with ethyl acetate (2×), the combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated. Compound 12c (65.2 mg, 0.26 mmol) was obtained as yellow liquid, containing a minor amount of a residual impurity (confirmed by NMR). The product was used for the next step without further purification. ESI-MS: m/z 271.0 [M+Na]+. HR-ESI-MS: m/z [M+H]+ calcd. 271.0552 for C11H11F3NaO3, found 271.0553. 1H-NMR: (400 MHz, CDCl3) δ 1.36 (d, J = 7.2 Hz, 3H), 3.01 (ddq, J = 7.2, 6.8, 5.6 Hz, 1H), 4.07 (dd, J = 9.0, 5.6 Hz, 1H), 4.22 (dd, J = 9.0, 6.8 Hz, 1H), 7.05 – 7.10 (m, 1H), 7.12 – 7.15 (m, 1H), 7.20 – 7.24 (m, 1H), 7.36 – 7.43 (m, 1H). 13C-NMR: (DEPTQ, 151 MHz, CDCl3) δ 13.7, 39.6, 69.4, 111.4 (q, J = 4 Hz), 117.81 (q, J = 4 Hz), 118.1, 123.9 (q, J = 272 Hz), 130.0, 131.9 (q, J = 32 Hz), 158.6, 179.4.
(R)-2-Methyl-3-(3-(trifluoromethyl)phenoxy)propanoic acid (ent-12c):
(R)-2-Methyl-3-(3-(trifluoromethyl)phenoxy)propanoic acid (ent-12c) was synthesized analogously as described for 12c, starting from crude ent-11c. The analytical data were in accordance.
(S)-3-(2,4-Di-tert-butylphenoxy)-2-methylpropanoic acid (12d):
11d (82.1 mg, 0.29 mmol) was dissolved in acetone (2.5 mL, 0.12 M). Jones reagent (230 μL, 1.26 M, 0.29 mmol, GPA) was added at 0 °C and the mixture was stirred for 4 h at 0 °C. After terminating the reaction by the addition of 2-propanol, the mixture was filtered through a pad of celite, which was further washed with ethyl acetate. The combined organic layers were extracted with 1N NaOH. After acidifying the aqueous layer using 2N HCl and extraction with ethyl acetate (2×), the combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated. Compound 12d (50.8 mg, 0.17 mmol, 60%) was obtained as a yellow oil, which solidified upon standing (beige wax) and which was used for the next step without further purification. ESI-MS: m/z 315.1 [M+Na]+. HR-ESI-MS: m/z [M+H]+ calcd. 315.1931 for C18H28NaO3, found 315.1932. 1H-NMR: (400 MHz, CDCl3) δ 1.30 (s, 9H), 1.35 (s, 9H), 1.37 (d, J = 7.2 Hz, 3H), 3.04 (ddq, J = 7.2, 6.5, 5.5 Hz, 1H), 4.09 (dd, J = 8.8, 5.5 Hz, 1H), 4.18 (dd, J = 8.8, 6.5 Hz, 1H), 6.79 (d, J = 8.5 Hz, 1H), 7.17 (dd, J = 8.5, 2.5 Hz, 1H), 7.33 (d, J = 2.5 Hz, 1H). 13C-NMR: (DEPTQ, 101 MHz, CDCl3) δ 14.3, 30.0, 31.7, 34.4, 35.1, 40.1, 69.5, 111.1, 123.4, 124.2, 137.3, 143.0, 155.0, 180.1.
(R)-3-(2,4-Di-tert-butylphenoxy)-2-methylpropanoic acid (ent-12d):
(R)-3-(2,4-Di-tert-butylphenoxy)-2-methylpropanoic acid (ent-12d) was synthesized analogously as described for 12d, starting from ent-11d. The analytical data were in accordance.
(S)-3-(4-Fluorophenoxy)-2-methylpropanoic acid (12e):
11e (120 mg, 0.65 mmol) was dissolved in acetone (6 mL, 0.11 M). Jones reagent (516 μL, 1.26 M, 0.65 mmol, GPA) was added at 0 °C and the mixture was stirred for 4.5 h at 0 °C. After terminating the reaction by the addition of 2-propanol, the mixture was filtered through a pad of celite, which was further washed with ethyl acetate. The combined organic layers were extracted with 1N NaOH. After acidifying the aqueous layer using 2N HCl and extraction with ethyl acetate (2×), the combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated. Compound 12e (76.5 mg, 0.39 mmol, 59%) was obtained as a yellow oil. The compound was used for the next step without further purification. ESI-MS: m/z 221.4 [M+Na]+. HR-ESI-MS: m/z [M+H]+ calcd. 221.0584 for C10H11FNaO3, found 221.0586. 1H-NMR: (400 MHz, CDCl3) δ 1.33 (d, J = 7.1 Hz, 3H), 2.97 (ddq, J = 7.1, 6.8, 5.7 Hz, 1H), 3.99 (dd, J = 9.0, 5.7 Hz, 1H), 4.15 (dd, J = 9.0, 6.8 Hz, 1H), 6.81 – 6.88 (m, 2H), 6.93 – 7.00 (m, 2H). 13C-NMR: (DEPTQ, 101 MHz, CDCl3) δ 13.7, 39.7, 69.9, 115.7 (d, J = 2 Hz), 115.9 (d, J = 17 Hz), 154.6 (d, J = 2 Hz), 157.5 (d, J = 239 Hz), 180.2. [α]D24: +3.1 (c = 0.9, chloroform).
(S)-3-(4-Bromophenoxy)-2-methylpropanoic acid (12f):
11f (103 mg, 0.42 mmol) was dissolved in acetone (4 mL, 0.11 M). Freshly prepared Jones reagent (333 μL, 1.26 M, 0.42 mmol, GPA) was added at 0 °C and the mixture was stirred for 5 h at 0 °C. After terminating the reaction by the addition of 2-propanol, the mixture was filtered through a pad of celite, which was further washed with ethyl acetate. The combined organic layers were extracted with 1N NaOH. After acidifying the aqueous layer using 2N HCl and extraction with ethyl acetate (2×), the combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated. Compound 12f (56.4 mg, 0.22 mmol, 52%) was obtained as brown oil, which was used for the next step without further purification. ESI-MS: m/z 281.2 [M+Na]+. HR-ESI-MS: m/z [M+H]+ calcd. 280.9784 for C10H11BrNaO3, found 280.9784. 1H-NMR: (400 MHz, CDCl3) δ 1.33 (d, J = 7.2 Hz, 3H), 2.97 (ddq, J = 7.2, 6.8, 5.8 Hz, 1H), 3.99 (dd, J = 9.0, 5.7 Hz, 1H), 4.15 (dd, J = 9.0, 6.8 Hz, 1H), 6.76 – 6.81 (m, 2H), 7.34 – 7.40 (m, 2H). 13C-NMR: (DEPTQ, 101 MHz, CDCl3) δ 13.7, 39.6, 69.4, 113.3, 116.4, 132.3, 157.6, 180.0. [α]D24: +1.4 (c = 1.0, chloroform).
(S)-3-([1,1’-Biphenyl]-4-yloxy)-2-methylpropanoic acid (12g):
11g (120 mg, 0.50 mmol) was dissolved in acetone (5 mL, 0.10 M). Freshly prepared Jones reagent (393 μL, 1.26 M, 0.50 mmol, GPA) was added at 0 °C and the mixture was stirred for 4.5 h at 0 °C. After terminating the reaction by the addition of 2-propanol, the mixture was filtered through pad of celite, which was further washed with ethyl acetate. The combined organic layers were extracted with 1N NaOH. After acidifying the aqueous layer using 2N HCl and extraction with ethyl acetate (2×), the combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated. Compound 12g (61.4 mg, 0.24 mmol, 48%) was obtained as a white solid, which was used for the next step without further purification. ESI-MS: m/z 279.1 [M+Na]+. HR-ESI-MS: m/z [M+H]+ calcd. 279.0992 for C16H16NaO3, found 279.0994. 1H-NMR: (400 MHz, CDCl3) δ 1.37 (d, J = 7.1 Hz, 3H), 3.03 (ddq, J = 7.1, 6.8, 5.9 Hz, 1H), 4.08 (dd, J = 9.1, 5.9 Hz, 1H), 4.24 (dd, J = 9.1, 6.8 Hz, 1H), 6.96 – 7.02 (m, 2H), 7.28 – 7.34 (m, 1H), 7.38 – 7.45 (m, 2H), 7.49 – 7.58 (m, 4H). 13C-NMR: (DEPTQ, 101 MHz, CDCl3) δ 13.8, 39.6, 69.3, 114.9, 126.7, 126.7, 128.2, 128.7, 134.2, 140.7, 158.0, 179.2. [α]D24: +1.1 (c = 0.5, chloroform).
(S)-3-(Benzofuran-5-yloxy)-2-methylpropanoic acid (12h):
11h (47.0 mg, 0.23 mmol) was dissolved in acetone (2.3 mL, 0.10 M). Freshly prepared Jones reagent (181 μL, 1.26 M, 0.23 mmol, GPA) was added at 0 °C and the mixture was stirred for 4.5 h at 0 °C. After terminating the reaction by the addition of 2-propanol, the mixture was filtered through a pad of celite, which was further washed with ethyl acetate. The combined organic layers were extracted with 1N NaOH. After acidifying the aqueous layer using 2N HCl and extraction with ethyl acetate (2×), the combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated. Compound 12h (26.4 mg, 0.12 mmol) was obtained as an orange oil, still containing a minor amount of residual contaminations (confirmed by NMR). The product was used crude for the next step without further purification. ESI-MS: m/z 243.0 [M+Na]+. 1H-NMR: (400 MHz, CDCl3) δ 1.35 (d, J = 7.1 Hz, 3H), 3.01 (ddq, J = 7.1, 6.8, 5.8 Hz, 1H), 4.06 (dd, J = 9.1, 5.8 Hz, 1H), 4.23 (dd, J = 9.1, 6.8 Hz, 1H), 6.70 (dd, J = 2.2, 0.9 Hz, 1H), 6.91 (dd, J = 8.9, 2.6 Hz, 1H), 7.08 (d, J = 2.6 Hz, 1H), 7.38 (d, J = 8.9 Hz, 1H), 7.59 (d, J = 2.2 Hz, 1H). 13C-NMR: (DEPTQ, 101 MHz, CDCl3) δ 13.8, 39.8, 70.3, 105.0, 106.7, 111.8, 113.7, 127.9, 145.8, 150.2, 154.8, 180.3.
(S)-1-(3-Amino-6-fluoro-1H-indazol-1-yl)-2-methyl-3-phenoxypropan-1-one ((S)-’853, 1a):
To a solution of compound 12a (25.0 mg, 0.14 mmol) in DMF (2 mL) were added HOAt (19.1 mg, 0.14 mmol) and EDC × HCl (27.1 mg, 0.14 mmol) at rt. The resulting solution was stirred for 10 min at rt and then 6-fluoro-1H-indazole-3-amine (13, 17.5 mg, 0.12 mmol) in DMF (0.5 mL) was added. After stirring the reaction mixture at rt for 1 h, the solvent was removed by lyophilization. Purification by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL /min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min (tR = 11.5 min, λ = 254 nm) afforded (S)-’853 (1a) (18.1 mg, 57.9 μmol, 48%) as an off-white solid. ESI-MS: m/z 314.1 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 314.1299 for C17H17FN3O2, found 314.1302. IR (NaCl): 3452, 3350, 3226, 1686, 1628, 1436, 1426 cm−1. M.p.: 45 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 1.29 (d, J = 7.1 Hz, 3H), 4.04 – 4.11 (m, 2H), 4.36 – 4.41 (m, 1H), 6.61 (s, 2H), 6.90 – 6.96 (m, 3H), 7.25 (ddd, J = 9.2, 8.7, 2.2 Hz, 1H), 7.25 – 7.29 (m, 2H), 7.94 (dd, J = 8.7, 5.4 Hz, 1H), 7.96 (dd, J = 9.7, 2.2 Hz, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 13.9, 37.7, 68.8, 101.8 (d, J = 28 Hz), 112.2 (d, J = 24 Hz), 114.5, 117.0, 120.7, 122.7 (d, J = 11 Hz), 129.5, 139.8 (d, J = 13 Hz), 152.6, 158.3, 163.2 (d, J = 244 Hz), 172.4. HPLC: system 2: λ = 254 nm, tR = 18.2 min, purity: 99%. Chiral HPLC: isocratic elution with n-hexane / isopropanol + 0.1% ethylene diamine, 9:1: tR = 7.4 min, 99% ee (254 nm). [α]D23: −4.1 (c = 0.2, methanol).
(R)-1-(3-Amino-6-fluoro-1H-indazol-1-yl)-2-methyl-3-phenoxypropan-1-one ((R)-’853, ent-1a):
(R)-1-(3-Amino-6-fluoro-1H-indazol-1-yl)-2-methyl-3-phenoxypropan-1-one ((S)-’853, ent-1a) was synthesized analogously as described for 1a, starting from ent-12a. The analytical data were in accordance. HR-ESI-MS: m/z [M+H]+ calcd. 314.1299 for C17H17FN3O2, found 314.1300. HPLC: system 2: λ = 254 nm, tR = 18.4 min, purity: >99%. Chiral HPLC: isocratic elution with n-hexane / isopropanol + 0.1% ethylene diamine, 9:1: tR = 6.4 min, 99% ee (254 nm). [α]D24: +4.7 (c = 0.3, methanol).
(S)-1-(3-Amino-6-fluoro-1H-indazol-1-yl)-2-methyl-3-(naphthalen-2-yloxy)propan-1-one ((S)-SX240, 1b):
To a solution of compound 12b (25.2 mg, 0.11 mmol) in DMF (2 mL) were added HOAt (15.3 mg, 0.11 mmol) and EDC × HCl (21.3 mg, 0.11 mmol) at rt. The resulting solution was stirred for 10 min at rt and then 6-fluoro-1H-indazole-3-amine (13, 13.8 mg, 91.2 μmol) in DMF (0.5 mL) were added. After stirring the mixture at rt for 1.5 h, the solvent was removed by lyophilization. Purification by preparative HPLC using a solvent system of CH3OH / 0.1% aq. TFA, a flow rate of 10 mL / min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min, 95% CH3OH for 5 min (tR = 21.0 min, λ = 254 nm) afforded (S)-SX240 (1b, 11.4 mg, 31.3 μmol, 34%) as a white solid. ESI-MS: m/z 364.2 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 364.1456 for C21H19FN3O2, found 364.1460. IR (NaCl): 3458, 2924, 2851, 1629, 1424 cm−1. M.p.: 59 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 1.34 (d, J = 7.0 Hz, 1H), 4.16 (ddq, J = 7.8, 7.0, 5.5 Hz, 1H), 4.21 (dd, J = 9.1, 5.5 Hz, 1H), 4.52 (dd, J = 9.1, 7.8 Hz, 1H), 6.62 (s, 2H), 7.11 (dd, J = 9.0, 2.5 Hz, 1H), 7.26 (ddd, J = 9.0, 8.9, 2.5 Hz, 1H), 7.34 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.39 (d, J = 2.5 Hz, 1H), 7.45 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.78 – 7.83 (m, 3H), 7.94 – 7.99 (m, 2H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 14.0, 37.7, 69.0, 101.8 (d, J = 28 Hz), 106.9, 112.2 (d, J = 25 Hz), 117.0, 118.6, 122.7 (d, J = 11 Hz), 123.6, 126.4, 126.7, 127.5, 128.5, 129.3, 134.2, 139.8 (d, J = 13 Hz), 152.7, 156.2, 163.2 (d, J = 245 Hz), 172.5. 19F-NMR: (377 MHz, DMSO-d6) δ −110.5. HPLC: system 1: λ = 254 nm, tR = 20.5 min, purity: 95%. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 9:1: tR = 9.7 min, >99% ee (254 nm). [α]D23: −15.8 (c = 0.4, methanol).
(R)-1-(3-Amino-6-fluoro-1H-indazol-1-yl)-2-methyl-3-(naphthalen-2-yloxy)propan-1-one ((R)-SX240, ent-1b):
(R)-1-(3-Amino-6-fluoro-1H-indazol-1-yl)-2-methyl-3-(naphthalen-2-yloxy)propan-1-one ((R)-SX240, ent-1b) was synthesized analogously as described for (S)-SX240 (1b), starting from ent-12b. The analytical data were in accordance. HR-ESI-MS: m/z [M+H]+ calcd. 364.1456 for C21H19FN3O2, found 364.1456. HPLC: system 1: λ = 254 nm, tR = 20.9 min, purity: >99%. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 9:1: tR = 8.2 min, 98% ee (254 nm). [α]D23: +21.0 (c = 0.4, methanol).
(S)-1-(3-Amino-6-fluoro-1H-indazol-1-yl)-2-methyl-3-(3-(trifluoromethyl)phenoxy)propan-1-one ((S)-SX245, 1c):
To a solution of crude 12c (14.5 mg, 58.4 μmol) in DMF (1.5 mL) were added HOAt (7.9 mg, 58.4 μmol) and EDC × HCl (11.2 mg, 58.4 μmol) at rt. The resulting solution was stirred for 10 min at rt and then 6-fluoro-1H-indazole-3-amine (13, 10.6 mg, 70.1 μmol) in DMF (0.5 mL) was added. After stirring the reaction mixture at rt for 2 h, the solvent was removed by lyophilization. Purification by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min, 95% CH3OH for 3 min (tR = 18.5 min, λ = 254 nm) yielded (S)-SX245 (1c, 8.2 mg, 21.5 μmol, 37%) as a white solid. ESI-MS: m/z 382.1 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 382.1173 for C18H16F4N3O2, found 382.1172. IR (NaCl): 3401, 3350, 2922, 2850, 1672, 1625, 1423, 1317, 1125 cm−1. M.p.: 121 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 1.30 (d, J = 7.0 Hz, 3H), 4.10 (ddq, J = 8.0, 7.0, 5.4 Hz, 1H), 4.17 (dd, J = 9.3, 5.4 Hz, 1H), 4.47 (dd, J = 9.3, 8.0 Hz, 1H), 6.61 (s, 2H), 7.21 – 7.30 (m, 4H), 7.48 – 7.53 (m, 1H), 7.89 – 7.99 (m, 2H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 13.9, 37.7, 69.4, 101.8 (d, J = 28 Hz), 111.0 (q, J = 4 Hz), 112.3 (d, J = 25 Hz), 117.0, 117.2 (q, J = 4 Hz), 118.8, 122.7 (d, J = 11 Hz), 123.9 (q, J = 273 Hz), 130.3 (q, J = 32 Hz), 130.7, 139.8 (d, J = 14 Hz), 152.7, 158.6, 163.2 (d, J = 245 Hz), 172.2. 19F-NMR: (377 MHz, DMSO-d6) δ −60.6, −110.5. HPLC: system 2: λ = 254 nm, tR = 19.6 min, purity: 98%. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 98:2: tR = 13.4 min, >99% ee (254 nm). [α]D25: −5.1 (c = 0.1, methanol).
(R)-1-(3-Amino-6-fluoro-1H-indazol-1-yl)-2-methyl-3-(3-(trifluoromethyl)phenoxy)propan-1-one ((R)-SX245, ent-1c):
(R)-1-(3-Amino-6-fluoro-1H-indazol-1-yl)-2-methyl-3-(3-(trifluoromethyl)phenoxy)propan-1-one ((R)-SX245, ent-1c) was synthesized analogously as described for (S)-SX245 (1c), starting from crude ent-12c. The analytical data were in accordance. HR-ESI-MS: m/z [M+H]+ calcd. 382.1173 for C18H16F4N3O2, found 382.1176. HPLC: system 2: λ = 254 nm, tR = 19.2 min, purity: >99%. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 98:2: tR = 12.1 min, >99% ee (254 nm). [α]D26: +5.8 (c = 0.2, methanol).
(S)-1-(3-Amino-6-fluoro-1H-indazol-1-yl)-3-(2,4-di-tert-butylphenoxy)-2-methylpropan-1-one ((S)-SX244, 1d):
To a solution of crude 12d (13.6 mg, 46.5 μmol) in DMF (1.5 mL) were added HOAt (6.5 mg, 47.8 μmol), followed by EDC × HCl (9.3 mg, 48.5 μmol) at rt. The resulting solution was stirred for 10 min at rt before adding 6-fluoro-1H-indazole-3-amine (13, 5.9 mg, 38.8 μmol) dissolved in anhydrous DMF (0.5 mL). After stirring the reaction mixture at rt for 2 h, the solvent was removed by lyophilization. Purification by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min, 95% CH3OH for 6 min (tR = 20.5 min, λ = 254 nm) afforded (S)-SX244 (1d, 2.4 mg, 5.64 μmol, 15%) as a white solid. ESI-MS: m/z 426.5 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 426.2551 for C25H33FN3O2, found 426.2558. IR (NaCl): 3463, 3350, 2960, 2864, 1676, 1624, 1423, 1231 cm−1. M.p.: 53 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 1.13 (s, 9H), 1.23 (s, 9H), 1.33 (d, J = 6.7 Hz, 3H), 4.17 (dd, J = 8.0, 5.0 Hz, 1H), 4.23 – 4.17 (m, 1H), 4.27 (dd, J = 8.0, 7.7 Hz, 1H), 6.57 (s, 2H), 6.89 (d, J = 8.5 Hz, 1H), 7.14 (dd, J = 8.5, 2.5 Hz, 1H), 7.16 (d, J = 2.5 Hz, 1H), 7.24 (ddd, J = 9.0, 8.9, 2.3 Hz, 1H), 7.91 – 7.96 (m, 2H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 14.0, 29.5, 31.4, 33.9, 34.3, 37.6, 69.4, 101.7 (d, J = 28 Hz), 111.3, 112.1 (d, J = 25 Hz), 117.1, 122.6 (d, J = 11 Hz), 122.9, 123.4, 136.0, 139.9 (d, J = 13 Hz), 141.8, 152.5, 154.6, 163.1 (d, J = 244 Hz), 172.9. 19F-NMR: (377 MHz, DMSO-d6) δ −110.8. HPLC: system 2: λ = 254 nm, tR = 22.0 min, purity: >99%. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 99:1: tR = 11.4 min, >99% ee (254 nm). [α]D23: −5.7 (c = 0.1, methanol).
(R)-1-(3-Amino-6-fluoro-1H-indazol-1-yl)-3-(2,4-di-tert-butylphenoxy)-2-methylpropan-1-one ((R)-SX244, ent-1d):
(R)-1-(3-Amino-6-fluoro-1H-indazol-1-yl)-3-(2,4-di-tert-butylphenoxy)-2-methylpropan-1-one ((R)-SX244, ent-1d) was synthesized analogously as described for (S)-SX244 (1d), starting from crude ent-12d. The analytical data were in accordance. HR-ESI-MS: m/z [M+H]+ calcd. 426.2551 for C25H33FN3O2, found 426.2552. HPLC: system 2: λ = 254 nm, tR = 22.0 min, purity: >99%. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 99:1: tR = 10.5 min, 98% ee (254 nm). [α]D26: +7.1 (c = 0.1, methanol).
(S)-1-(3-Amino-6-fluoro-1H-indazol-1-yl)-3-(4-fluorophenoxy)-2-methylpropan-1-one ((S)-SX263, 1e):
To a solution of 12e (20.7 mg, 0.10 mmol) in DMF (2.5 mL) were added HOAt (13.6 mg, 0.10 mmol) and EDC × HCl (19.2 mg, 0.10 mmol) at rt. The resulting solution was stirred for 10 min at rt and then 6-fluoro-1H-indazol-3-amine (13, 19.6 mg, 0.13 mmol) in DMF (0.5 mL) was added. After stirring the reaction mixture at rt for 1.5 h, the solvent was removed by lyophilization. Purification by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min, 95% CH3OH for 2 min (tR = 16.0 min, λ = 254 nm) afforded (S)-SX263 (1e, 16.9 mg, 51.0 μmol, 51%) as a white solid. ESI-MS: m/z 332.2 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 332.1205 for C17H16F2N3O2, found 332.1203. IR (NaCl): 3463, 3357, 2925, 2849, 1683, 1625, 1509, 1419 cm−1. M.p.: 148 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 1.28 (d, J = 6.9 Hz, 3H), 4.02 – 4.11 (m, 2H), 4.33 – 4.40 (m, 1H), 6.62 (s, 2H), 6.93 – 6.99 (m, 2H), 7.06 – 7.13 (m, 2H), 7.26 (ddd, J = 9.0, 8.9, 2.3 Hz, 1H), 7.92 – 7.99 (m, 2H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 13.9, 37.7, 69.5, 101.8 (d, J = 28 Hz), 112.2 (d, J = 25 Hz), 115.7 (d, J = 7 Hz), 115.8 (d, J = 8 Hz), 117.0, 122.7 (d, J = 11 Hz), 139.8 (d, J = 13 Hz), 152.7, 155.1 (d, J = 2 Hz), 156.5 (d, J = 236 Hz), 163.2 (d, J = 245 Hz), 172.4. 19F-NMR: (377 MHz, DMSO-d6) δ −110.5, −123.3. HPLC: system 1: λ = 254 nm, tR = 20.1 min, purity: 97%. chiral HPLC: isocratic elution with n-hexane / isopropanol, 95:5: tR = 11.4 min, 99% ee (254 nm). [α]D23: −2.0 (c = 0.9, methanol).
(S)-1-(3-Amino-6-fluoro-1H-indazol-1-yl)-3-(4-bromophenoxy)-2-methylpropan-1-one ((S)-SX264, 1f):
To a solution of 12f (19.2 mg, 74.1 μmol) in DMF (2.5 mL) were added HOAt (10.1 mg, 74.1 μmol and EDC × HCl (14.2 mg, 74.1 μmol) at rt. The resulting solution was stirred for 10 min at rt and then 6-fluoro-1H-indazol-3-amine (13, 13.4 mg, 88.9 μmol) in DMF (0.5 mL) was added. After stirring the reaction mixture at rt for 1.5 h, the solvent was removed by lyophilization. Purification by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min, 95% CH3OH for 4 min (tR = 18.0 min, λ = 254 nm) afforded (S)-SX264 (1f, 8.22 mg, 21.0 μmol, 28%) as a white solid. ESI-MS: m/z 392.3 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 392.0404 for C17H16BrFN3O2, found 392.0402. IR (NaCl): 3466, 3346, 2922, 1680, 1625, 1560, 1423, 1238, 1074 cm−1. M.p.: 112 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 1.28 (d, J = 6.6 Hz, 3H), 4.03 – 4.10 (m, 2H), 4.34 – 4.43 (m, 1H), 6.62 (s, 2H), 6.89 – 6.97 (m, 2H), 7.26 (ddd, J = 9.0, 8.9, 2.3 Hz, 1H), 7.40 – 7.46 (m, 2H), 7.92 – 7.97 (m, 2H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 13.8, 37.6, 69.2, 101.8 (d, J = 28 Hz), 112.1, 112.3 (d, J = 25 Hz), 116.8, 117.0, 122.7 (d, J = 11 Hz), 132.1, 139.8 (d, J = 13 Hz), 152.7, 157.6, 163.2 (d, J = 245 Hz), 172.3. 19F-NMR: (377 MHz, DMSO-d6) δ −110.4. HPLC: system 1: λ = 254 nm, tR = 20.8 min, purity: >99%. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 95:5: tR = 12.0 min, 98% ee (254 nm). [α]D26: −6.1 (c = 0.3, methanol).
(S)-3-([1,1’-Biphenyl]-4-oxy)-1-(3-amino-6-fluoro-1H-indazol-1-yl)-2-methylpropan-1-one ((S)-SX267, 1g):
To a solution of compound 12g (21.5 mg, 83.9 μmol) in DMF (2.5 mL) were added HOAt (11.4 mg, 83.9 μmol) and EDC × HCl (16.1 mg, 83.9 μmol) at rt. The resulting solution was stirred for 10 min at rt and then 6-fluoro-1H-indazol-3-amine (13, 15.2 mg, 101 μmol) in DMF (0.5 mL) was added. After stirring the reaction mixture at rt for 3 h, the solvent was removed by lyophilization. Purification by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min, 95% CH3OH for 4 min (tR = 19.0 min, λ = 254 nm) afforded (S)-SX267 (1g, 17.3 mg, 44.3 μmol, 53%) as a white solid. ESI-MS: m/z 390.3 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 390.1612 for C23H21FN3O2, found 390.1611. IR (NaCl): 3456, 3343, 2925, 1686, 1625, 1440, 1422, 1320, 762 cm−1. M.p.: 101 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 1.31 (d, J = 6.7 Hz, 3H), 4.08 – 4.15 (m, 2H), 4.42 – 4.48 (m, 1H), 6.63 (s, 2H), 7.00 – 7.08 (m, 2H), 7.26 (ddd, J = 9.0, 8.9, 2.4 Hz, 1H), 7.28 – 7.32 (m, 1H), 7.40 – 7.45 (m, 2H), 7.52 – 7.64 (m, 4H), 7.92 – 8.01 (m, 2H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 13.9, 37.7, 69.0, 101.8 (d, J = 28 Hz), 112.2 (d, J = 25 Hz), 115.0, 117.0, 122.7 (d, J = 11 Hz), 126.1, 126.7, 127.7, 128.8, 132.7, 139.8, 139.8 (d, J = 13 Hz), 152.7, 157.9, 163.2 (d, J = 245 Hz), 172.4. 19F-NMR: (377 MHz, DMSO-d6) δ −110.4. HPLC: system 2: λ = 254 nm, tR = 19.6 min, purity: >99%. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 9:1: tR = 11.5 min, 99% ee (254 nm). [α]D25: −11.0 (c = 1.1, methanol).
(S)-1-(3-Amino-6-fluoro-1H-indazol-1-yl)-3-(benzofuran-5-yloxy)-2-methylpropan-1-one ((S)-SX268, 1h):
To a solution of 12h (11.5 mg, 52.2 μmol) in DMF (2.5 mL) were added HOAt (7.1 mg, 52.2 μmol) and EDC × HCl (10.0 mg, 52.2 μmol) at rt. The resulting solution was stirred for 10 min at rt and then 6-fluoro-1H-indazol-3-amine (13, 9.5 mg, 62.7 μmol) in DMF (0.5 mL) was added. After stirring the reaction mixture at rt for 2 h, the solvent was removed by lyophilization. Purification by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min, 95% CH3OH for 4 min (tR = 16.0 min, λ = 254 nm) afforded (S)-SX268 (1h, 3.55 mg, 10.0 μmol, 19%) as a white solid. ESI-MS: m/z 354.2 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 354.1248 for C19H17FN3O3, found 354.1247. IR (NaCl): 3446, 3350, 2922, 1686, 1628, 1436, 1197 cm−1. M.p.: 53 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 1.30 (d, J = 6.6 Hz, 3H), 4.07 – 4.13 (m, 2H), 4.36 – 4.45 (m, 1H), 6.60 (s, 2H), 6.86 (dd, J = 2.2, 0.9 Hz, 1H), 6.87 (dd, J = 7.5, 2.6 Hz, 1H), 7.20 (d, J = 2.6 Hz, 1H), 7.25 (ddd, J = 9.0, 8.9, 2.4 Hz, 1H), 7.45 (dd, J = 7.5, 0.9 Hz, 1H), 7.93 (d, J = 2.2 Hz, 1H), 7.93 – 7.98 (m, 2H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 14.0, 37.8, 69.7, 101.8 (d, J = 28 Hz), 104.8, 106.9, 111.7, 112.2 (d, J = 25 Hz), 113.4, 117.0, 122.7 (d, J = 11 Hz), 127.8, 139.8 (d, J = 14 Hz), 146.7, 149.3, 152.6, 154.6, 163.2 (d, J = 245 Hz), 172.5. HPLC: system 1: λ = 254 nm, tR = 20.1 min, purity: 97%. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 95:5: tR = 18.4 min, 99% ee (254 nm). [α]D25: −9.5 (c = 0.1, methanol).
The fluoroindazole derivative renouncing the phenoxy moiety 2 was obtained in 90% yield by reacting isobutyryl chloride with HOAt, followed by the addition of 6-fluoro-1H-indazole-3-amine (13) (scheme 2).
Scheme 2.

Synthesis of the ‘853 derivative renouncing the phenoxy site, reagents and conditions: (a) 1. isobutyryl chloride, HOAt, DIPEA, DMF, 0 °C, 1 h, 2. 6-fluoro-1H-indazole-3-amine (13), microwave irradiation (90%).
1-(3-Amino-6-methoxy-1H-indazol-1-yl)-2-methylpropan-1-one (I1381, 2):
To a solution of HOAt (54.0 mg, 0.40 mmol) in DMF (1 mL) was added DIPEA (96.5 mg, 130 μL, 0.74 mmol) and then isobutyryl chloride (40.7 mg, 40 μL, 0.38 mmol) at 0 °C. After 1 h of stirring, a solution of 6-fluoro-1H-indazol-3-amine (13, 50.0 mg, 0.33 mmol) in DMF (0.5 mL) at rt was added. Microwave irradiation (described in the general part) in a sealed tube was performed, then the solvent was removed by lyophilization and the residue was purified by preparative HPLC using a solvent system of CH3CN / 0.1% aq. TFA, a flow rate of 10 mL / min and a gradient of 40 to 60% in 15 min (tR: 12.6 min, λ = 220 nm) to afford I1381 (2, 65.6 mg, 90%) as a white solid. ESI-MS: m/z 222.0 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 222.1037 for C11H13FN3O, found 222.1038. IR (KBr): 3460, 3382, 3350, 3211, 1672 cm−1. M.p.: 103 °C. 1H-NMR: (400 MHz, DMSO-d6) δ 1.19 (d, J = 6.9 Hz, 6H), 3.65 (sept, J = 6.9 Hz, 1H), 6.56 (s, 2H), 7.23 (ddd, J = 9.3, 8.8, 2.3 Hz, 1H), 7.92 (dd, J = 8.8, 5.4 Hz, 1H), 7.94 (dd, J = 10.1, 2.3 Hz, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 18.8, 31.8, 101.7 (d, J = 28 Hz), 111.9 (d, J = 24 Hz), 116.8, 122.5 (d, J = 11 Hz), 139.9 (d, J = 10 Hz), 152.4, 163.2 (d, J = 245 Hz), 175.5. HPLC: system 3: λ = 220 nm, tR = 21.2 min, purity: >99%.
The derivatives of ‘853 modified in position 3 of the indazole scaffold were prepared as displayed in scheme S3. The 3-desamino derivative 3a was synthesized as a racemate starting from 6-fluoro-1H-indazole (14) and the commercially available racemic carboxylic acid derivative rac-12a. 6-Fluoro-3-methylindazole (15) gave access to the enantiopure target compound 3b. For the synthesis of the N-methyl derivative 3c, a novel, efficient pathway for the preparation of 3-(N-methylamino)indazole derivatives had to be established. In detail, the diacetyl derivative 16 was prepared from 6-fluoro-1H-indazole-3-amine (13) using acetylchloride in the presence of pyridine and 4-dimethylaminopyridine (DMAP). Deprotonation with sodium hydride (NaH) and subsequent reaction with methyl iodide (MeI) gave rise to the N-methyl derivative 17, which was deprotected with hydrogen chloride (HCl) in methanol (MeOH). The resulting 6-fluoro-N-methyl-1H-indazol-3-amine (18) was acylated with the (S)-carboxylic acid derivative 12a to give the target compound 3c. The aza analog of ‘853 4 was synthesized starting from 6-fluoro-1H-pyrazolo[4,3-b]pyridin-3-amine (20), which was available by a ring closing reaction of the pyridine derivative 19 with hydrazine hydrate according to a previously reported protocol.68 It is noteworthy, that the cyclization reaction also led to the nucleophilic attack of hydrazine in position 5 of the pyridine nucleus. Thus, the respective aryl hydrazine side product was formed, which had to be separated by preparative HPLC. Coupling with the (S)-carboxylic acid derivative 12a resulted in the formation of target compound 4.
Scheme S3.

Synthesis of the ‘853 derivatives with modifications in positions 3 or 4, reagents, and conditions: (a) EDC × HCl, HOAt, DMF, rt (3a: 39%, 3b: 57%, 3c: 29%, 4: 31%); (b) AcCl, pyridine, DMAP, 0 °C to rt, 2 h (72%), (c) NaH, MeI, DMF, rt, 1 h (77%); (d) 1.25 M HCl in MeOH, 115 °C, 2 h (100% crude); (e) N2H4, EtOH, 70 °C, 17 h (15%).
(R,S)-1-(6-Fluoro-1H-indazol-1-yl)-2-methyl-3-phenoxypropan-1-one (I1384, 3a):
To a solution of commercially available (R,S)-2-methyl-3-phenoxypropionic acid (rac-12a, 79.4 mg, 0.44 mmol) and HOAt (60.0 mg, 0.44 mmol) in DMF (1 mL) was added EDC × HCl (84.5 mg, 0.44 mmol). After stirring for 10 min at rt, a solution of commercially available 6-fluoro-1H-indazole (14, 50.0 mg, 0.37 mmol) in DMF (0.5 mL) was added. After stirring the reaction mixture for 4 h, the solvent was removed by lyophilization and the residue was purified by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 20% to 90% CH3OH in 25 min, 90% to 95% CH3OH in 2 min, 95% CH3OH for 2 min (tR: 27.5 min, λ = 220 nm) to afford I1384 (3a, 43.0 mg, 0.14 mmol, 39%) as a yellow-brownish resin. ESI-MS: m/z 299.1 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 299.1190 for C17H16FN2O2, found 299.1191. IR (KBr): 3424, 1715 cm−1. 1H-NMR: (600 MHz, DMSO-d6) δ 1.36 (d, J = 7.1 Hz, 3H), 4.19 (dd, J = 9.6, 5.1 Hz, 1H), 4.31 (ddq, J = 7.4, 5.1, 7.1 Hz, 1H), 4.41 (dd, J = 9.6, 7.4 Hz, 1H), 6.90 – 6.95 (m, 3H), 7.24 – 7.29 (m, 2H), 7.36 (ddd, J = 9.2, 8.8, 2.1 Hz, 1H), 8.00 (dd, J = 8.8, 5.3 Hz, 1H), 8.06 (dd, J = 9.2, 2.1 Hz, 1H), 8.55 (brs, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 14.0, 38.2, 68.9, 101.3 (d, J = 28 Hz), 113.8 (d, J = 24 Hz), 114.5, 120.8, 123.0, 123.8 (d, J = 11 Hz), 129.5, 139.9 (d, J = 13 Hz), 140.8, 158.2, 163.0 (d, J = 245 Hz), 174.3. HPLC: system 3: λ = 220 nm, tR = 27.5 min, purity: 96%.
(S)-1-(6-Fluoro-3-methyl-1H-indazol-1-yl)-2-methyl-3-phenoxypropan-1-one (I1402, 3b):
To a solution of 12a (15.0 mg, 0.08 mmol) and HOAt (11.3 mg, 0.08 mmol) in DMF (0.5 mL) was added EDC × HCl (16.0 mg, 0.08 mmol). After stirring for 10 min at rt, a solution of commercially available 6-fluoro-3-methyl-1H-indazole (15, 15.0 mg, 0.10 mmol) in DMF (0.5 mL) was added. After stirring the reaction mixture for 4 h, the solvent was removed by lyophilization and the residue was purified by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 20% to 90% CH3OH in 22 min, 90% to 95% CH3OH in 2 min, 95% CH3OH for 2 min (tR: 24.0 min, λ = 220 nm) to afford I1402 (3b, 14.9 mg, 47.7 μmol, 57%) as a white solid. ESI-MS: m/z 313.1 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 313.1347 for C18H18FN2O2, found 313.1348. IR (NaCl): 3447, 1707 cm−1. M.p.: 51 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 1.34 (d, J = 7.1 Hz, 3H), 2.57 (s, 3H), 4.15 (dd, J = 9.4, 5.4 Hz, 1H), 4.27 (ddq, J = 8.0, 5.4, 7.1 Hz, 1H), 4.39 (dd, J = 9.4, 8.0 Hz, 1H), 6.91 – 6.94 (m, 3H), 7.24 – 7.29 (m, 2H), 7.35 (ddd, J = 9.2, 8.8, 2.2 Hz, 1H), 7.95 (ddd, J = 8.8, 5.1, 0.3 Hz, 1H), 8.02 (ddd, J = 9.5, 2.2, 0.3 Hz, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 11.8, 13.9, 37.9, 68.8, 101.3 (d, J = 29 Hz), 113.3 (d, J = 25 Hz), 114.4, 120.7, 122.0, 123.0 (d, J = 11 Hz), 129.4, 139.4 (d, J = 17 Hz), 149.1, 158.1, 163.0 (d, J = 256 Hz), 173.8. HPLC: system 3: λ = 220 nm, tR = 28.7 min, purity: 98%. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 99:1: tR = 9.6 min, 99% ee (254 nm). [α]D20: +33.3 (c = 0.5, chloroform).
N-(1-Acetyl-6-fluoro-1H-indazol-3-yl)acetamide (16):
To a solution of 6-fluoro-1H-indazol-3-amine (13, 100.0 mg, 0.66 mmol) and DMAP in pyridine (1.5 mL) was slowly added acetyl chloride (207.7 mg, 189 μL, 2.65 mmol) at 0 °C. After 5 min of stirring at rt, dioxane (3 mL) was added and stirring was continued for 1 h. The solvent was removed in vacuo and flash chromatography was performed (isohexane / ethyl acetate, 9:1 -> 8:2) to afford 16 (11.4 mg, 72%) as a white solid. ESI-MS: m/z 258.1 [M+Na]+. HR-ESI-MS: m/z [M+H]+ calcd. 236.0830 for C11H11FN3O2, found 236.0828. IR (KBr): 3409, 3273, 3232, 1711, 1670 cm−1. M.p.: 193–194 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 2.19 (s, 3H), 2.64 (s, 3H), 7.29 (ddd, J = 9.0, 9.0, 2.3 Hz, 1H), 8.00 (dd, J = 9.7, 2.3 Hz, 1H), 8.08 (dd, J = 9.0, 5.5 Hz, 1H), 10.93 (s, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 22.7, 23.8, 101.0 (d, J = 29 Hz), 112.8 (d, J = 25 Hz), 117.0, 125.6 (brs), 139.7 (d, J = 14 Hz), 145.1, 163.0 (d, J = 246 Hz), 169.1 (brs), 170.2.
N-(1-Acetyl-6-fluoro-1H-indazol-3-yl)-N-methylacetamide (17):
To a solution of N-(1-acetyl-6-fluoro-1H-indazol-3-yl)acetamide (16, 68.1 mg, 0.29 mmol) in DMF (1 mL) was added sodium hydride (8.3 mg, 0.35 mmol) and after 10 min methyl iodide (164.4 mg, 72 μL, 1.16 mmol) at rt. After 1 h of stirring, the solvent was removed in vacuo and flash chromatography was performed (isohexane / ethyl acetate, 9:1 -> 8:2) to afford 17 (55.7 mg, 77%) as a white solid. ESI-MS: m/z 250.1 [M+H]+. HR-ESI-MS: m/z [M+Na]+ calcd. 272.0806 for C12H12FN3NaO2, found 272.0802. IR (KBr): 1728, 1690, 1679 cm−1. M.p.: 98–100 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 2.10 (brs, 3H), 2.68 (s, 3H), 3.37 (brs, 3H), 7.36 (brdd, J = 8.5, 8.5 Hz, 1H), 7.90 (brs, 1H), 8.05 (brd, J = 9.7, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 22.4, 22.6, 35.2 (detected by HSQC), 101.4 (d, J = 28 Hz), 113.8 (brs), 117.9, 124.3 (d, J = 12 Hz), 139.8 (d, J = 18 Hz), 149.0, 163.2 (d, J = 246 Hz), 170.6.
6-Fluoro-N-methyl-1H-indazol-3-amine (18):
A solution of N-(1-acetyl-6-fluoro-1H-indazol-3-yl)-N-methylacetamide (17, 30.0 mg, 0.12 mmol) in 1.25 M HCl in methanol (2 mL) was heated at 115 °C in a sealed tube. After 2 h of stirring, the solvent was removed in vacuo to give crude 18 (21.9 mg, crude) as a brownish oil, which was used for the next reaction without further purification. For analytical purposes, a small sample of the substance was purified by preparative HPLC using a solvent system of CH3OH / 0.1% aq. TFA, a flow rate of 12 mL/min and a gradient of 10 to 50% in 7 min (tR: 8.6 min, λ = 220 nm) to furnish a yellow oil. ESI-MS: m/z 166.1 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 166.0775 for C8H9FN3, found 166.0775. IR (KBr): 3418, 3215, 1651 cm−1. 1H-NMR: (600 MHz, DMSO-d6) δ 2.89 (s, 3H), 6.83–6.89 (m, 1H), 7.06–7.10 (m, 1H), 7.76–7.81 (m, 1H) 11.80 (brs, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 29.8, 95.4 (d, J = 27 Hz), 117.8 (d, J = 27 Hz), 110.0, 122.6 (d, J = 10 Hz), 142.5 (d, J = 13 Hz), 150.2, 162.9 (d, J = 242 Hz).
(S)-1-(6-Fluoro-3-(methylamino)-1H-indazol-1-yl)-2-methyl-3-phenoxypropan-1-one (I1436, 3c):
To a solution of 12a (10.0 mg, 0.055 mmol) in DMF (0.5 mL) was added HOAt (7.6 mg, 0.055 mmol) and EDC × HCl (10.6 mg, 0.055 mmol) at rt. The solution was stirred for 10 min at rt and then crude 6-fluoro-N-methyl-1H-indazol-3-amine (18, 11.0 mg, 0.12 mmol) in DMF (0.5 mL) was added. After stirring the reaction mixture at rt for 14 h, the solvent was removed by lyophilization. Purification by preparative HPLC using a solvent system of CH3OH / 0.1% aq. TFA, a flow rate of 12 mL / min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min (tR = 16.0 min, λ = 220 nm) afforded I1436 (3c, 5.2 mg, 57.9 μmol, 29%) as a white solid. ESI-MS: m/z 328.5 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 328.1456 for C18H19FN3O2, found 328.1455. IR (NaCl): 3376, 1682, cm−1. M.p.: 80 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 1.30 (d, J = 7.0 Hz, 3H), 2.93 (d, J = 4.9 Hz, 3H), 4.09 (dd, J = 9.4, 5.8 Hz, 1H), 4.16 (ddq, J = 7.7, 5.8, 7.0 Hz, 1H), 4.42 (dd, J = 9.4, 7.7 Hz, 1H), 6.91 – 6.95 (m, 1H), 7.18 (q, J = 4.9 Hz, 1H), 7.25 (ddd, J = 8.9, 8.5, 2.3 Hz, 1H), 7.25 – 7.30 (m, 2H), 7.91 (dd, J = 8.5, 5.4 Hz, 1H), 7.97 (dd, J = 9.8, 2.3 Hz, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 13.6, 28.8, 37.6, 68.7, 101.8 (d, J = 28 Hz), 112.3 (d, J = 24 Hz), 114.5, 116.6, 120.6, 122.1 (d, J = 11 Hz), 129.4, 140.0 (d, J = 13 Hz), 153.0, 158.2, 163.1 (d, J = 244 Hz), 172.4. HPLC: system 3: λ = 220 nm, tR = 26.3 min, purity: >99%. [α]D23: −5.7 (c = 0.17, methanol).
6-Fluoro-1H-pyrazolo[4,3-b]pyridin-3-amine (20):
A solution of 3,5-difluoro-2-pyridinecarbonitrile (19 70 mg, 0.50 mmol) and hydrazine hydrate (37.6 mg, 36 μL, 0.75 mmol) in ethanol (1.3 mL) was heated at 70 °C in a sealed tube for 17 h. After cooling to rt, the solvent was removed in vacuo, and the residue was purified by preparative HPLC using a solvent system of CH3CN / 0.1% aq. TFA, a flow rate of 10 mL / min and a gradient of 3% to 18% CH3CN in 12 min (tR: 9.6 min, λ = 220 nm) to afford 20 (11.4 mg, 41.4 μmol, 15%) as a white solid. ESI-MS: m/z 153.0 [M+H]+. IR (KBr): 3444, 3315, 3210 cm−1. 1H-NMR: (600 MHz, DMSO-d6) δ 5.44 (s, 2H), 7.57 (dd, J = 9.6, 2.6 Hz, 1H), 8.26 (d, J = 2.6, 1.2 Hz, 1H), 11.73 (brs, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 102.7 (d, J = 22 Hz), 131.3 (d, J = 29 Hz), 133.5 (d, J = 9 Hz), 148.9, 157.9 (d, J = 249 Hz). The analytical data of this compound are in accordance with those reported in the literature.
(S)-1-(3-Amino-6-fluoro-1H-pyrazolo[4,3-b]pyridin-1-yl)-2-methyl-3-phenoxypropan-1-one (BBG3, 4):
To a solution of 12a (13.5 mg, 0.075 mmol) and HOAt (10.2 mg, 0.075 mmol) in DMF (0.5 mL) was added EDC × HCl (14.4 mg, 0.075 mmol). After 10 min at rt, a solution of 20 (11.4 mg, 0.09 mmol) in DMF (0.5 mL) was added. After stirring for 3 h, the solvent was removed by lyophilization and the residue was purified by preparative HPLC using a solvent system of CH3CN / 0.1% aq. TFA, a flow rate of 10 mL / min and a gradient of, 5% to 76% CH3CN in 23 min, 76% to 95% in 2 min (tR: 23.4 min, λ = 220 nm) to afford BBG3 (4, 7.2 mg, 22.9 μmol, 31%) as a white solid. ESI-MS: m/z 315.1 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 315.1252 for C16H16FN4O2, found 315.1254. IR (NaCl): 3450, 3344, 1694, 1630 cm−1. M.p.: 101 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 1.29 (m, 3H), 4.06 – 4.13 (m, 2H), 4.34 – 4.42 (m, 1H), 6.87 (s, 2H), 6.90 – 6.95 (m, 3H), 7.24 – 7.29 (m, 2H), 8.33 (dd, J = 9.0, 2.5 Hz, 1H), 8.67 (dd, J = 2.5, 0.9 Hz, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 13.7, 37.3, 68.6, 109.4 (d, J = 25 Hz), 114.4, 120.6, 129.4, 132.7 (d, J = 12 Hz), 134.2, 136.1 (d, J = 29 Hz), 152.2, 159.2 (d, J = 254 Hz), 172.9. HPLC: system 3: λ = 220 nm, tR = 23.6 min, purity: >99%. [α]D23: −3.6 (c = 0.3, methanol).
Furthermore, the fluorine in position 6 of the indazole scaffold was replaced by a variety of substituents (scheme 4). The alkoxy substituted compounds 5a,b were synthesized as racemates starting from rac-12a, employing the respective, commercially available 6-alkoxy-indazole derivatives 21 and 22. Benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) in presence of HOAt and N,N-diisopropylethylamine (DIPEA) were used for the coupling reaction under microwave promoted conditions. Starting from the buyable 6-trifluoromethyl-, 6-chloro- or 6-aminoindazole analogs 23, 24 and 25, the EDC/HOAt promoted coupling with the (S)-carboxylic acid derivative 12a furnished the respective target compounds 5c, 5d and 5e, respectively.
Scheme 4.

Synthesis of the ‘853 derivatives with alkoxy substituents in positions 6, reagents and conditions: (a) PyBOP, HOAt, DMF, microwave irradiation (5a: 56%, 5b: 54%); (b) EDC × HCl, HOAt, DMF, rt (5c: 23%, 5d: 54%, 5e: 44%).
(R,S)-1-(3-Amino-6-methoxy-1H-indazol-1-yl)-2-methyl-3-phenoxypropan-1-one (IK195, 5a):
To a solution of commercially available (R,S)-2-methyl-3-phenoxypropionic acid (rac-12, 66.0 mg, 0.37 mmol), PyBOP (194 mg, 0.37 mmol) and HOAt (76 mg, 0.56 mmol) in anhydrous DMF (3 mL) was added DIPEA (64 μL, 0.37 mmol) and then a solution of commercially available 6-methoxy-1H-indazol-3-amine (21, 50.0 mg, 0.26 mmol) in DMF (0.5 mL) at rt. Microwave irradiation (described in the general part) in a sealed tube was performed, then the solvent was removed by lyophilization and the residue was purified by preparative HPLC using a solvent system of CH3CN / 0.1% aq. TFA, a flow rate of 20 mL / min and a gradient of 40% CH3CN for 1 min, 40% to 65% CH3CN in 10 min (tR: 10.2 min, λ = 254 nm) to afford IK195 (5a, 56.3 mg, 0.17 mmol, 56%) as a white solid. ESI-MS: m/z 326.2 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 326.1499 for C18H20N3O3, found 326.1503. IR (KBr): 3446, 3339, 3229, 1676 cm−1. M.p.: 160–163 °C. 1H-NMR: (400 MHz, DMSO-d6) δ 1.28 (d, J = 6.8 Hz, 3H), 3.83 (s, 3H), 4.00 – 4.14 (m, 2H), 4.34 – 4.43 (m, 1H), 6.46 (s, 2H), 6.89 – 6.95 (m, 3H), 6.69 (dd, J = 8.7, 2.2 Hz, 1H), 7.24 – 7.31 (m, 2H), 7.77 (d, J = 8.7 Hz, 1H), 7.79 (d, J = 2.2 Hz, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 14.0, 37.7, 55.5, 68.9, 98.3, 113.2, 114.0, 114.4, 120.6, 121.5, 129.5, 140.9, 152.9, 158.3, 161.2, 172.3. HPLC: system 3: λ = 220 nm, tR = 24.6 min, purity: >99%.
(R,S)-1-(3-Amino-6-isopropoxy-1H-indazol-1-yl)-2-methyl-3-phenoxypropan-1-one (IK192, 5b):
To a solution of commercially available (R,S)-2-methyl-3-phenoxypropionic acid (rac-12, 56.5 mg, 0.31 mmol), PyBOP (163 mg, 0.31 mmol) and HOAt (64 mg, 0.47 mmol) in anhydrous DMF (3 mL) was added DIPEA (54 μL, 0.31 mmol) and then a solution of commercially available 6-isopropoxy-1H-indazol-3-amine (22, 50.0 mg, 0.26 mmol) in DMF (0.5 mL) at ambient temperature. Microwave irradiation (described in the general part) in a sealed tube was performed, then the solvent was removed by lyophilization and the residue was purified by preparative HPLC using a solvent system of CH3CN / 0.1% aq. TFA, a flow rate of 20 mL / min and a gradient of 40% CH3CN for 1 min, 40% to 65% CH3CN in 10 min, 65% to 95% CH3CN in 2 min (tR: 12.9 min, λ = 250 nm) to afford IK192 (5b, 50.0 mg, 0.14 mmol, 54%) as a white solid. ESI-MS: m/z 354.2 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 354.1812 for C18H20N3O3, found 354.1814. IR (KBr): 3438, 3343, 3233, 1679 cm−1. M.p.: 138 °C. 1H-NMR: (400 MHz, DMSO-d6) δ 1.27 (d, J = 6.8 Hz, 3H), 1.30 (d, J = 6.0 Hz, 6H), 4.00 – 4.13 (m, 2H), 4.33 – 4.43 (m, 1H), 4.66 (sept, J = 6.0 Hz, 1H), 6.48 (brs, 2H), 6.90 – 6.96 (m, 3H), 6.92 (dd, J = 8.8, 2.2 Hz, 1H), 7.23 – 7.32 (m, 2H), 7.75 (d, J = 8.8 Hz, 1H), 7.77 (d, J = 2.2 Hz, 1H). 13C-NMR: (DEPTQ, 101 MHz, DMSO-d6) δ 14.1, 21.7, 21.8, 37.8, 68.9, 70.0, 100.2, 113.9, 114.3, 114.5, 120.7, 121.7, 129.5, 140.9, 152.9, 158.3, 159.4, 172.4. HPLC: system 3: λ = 220 nm, tR = 26.7 min, purity: 99%.
(S)-1-(3-Amino-6-(trifluoromethyl)-1H-indazol-1-yl)-2-methyl-3-phenoxypropan-1-one ((S)-KD1, 5c):
To a solution of 12a (10.0 mg, 0.06 mmol) and HOAt (7.7 mg, 0.06 mmol) in anhydrous DMF (1 mL) was added EDC × HCl (11.1 mg, 0.06 mmol). After stirring for 10 min at ambient temperature, a solution of commercially available 6-(trifluoromethyl)-1H-indazol-3-amine (23, 16.7 mg, 0.10 mmol) in DMF (0.5 mL) was added. After stirring the reaction mixture for 2 h, the solvent was removed by lyophilization and the residue was purified by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min, 95% CH3OH for 4 min (tR: 20.6 min, λ = 254 nm) to afford (S)-KD1 (5c, 4.0 mg, 11.0 μmol, 23%) as a white resin. ESI-MS: m/z 364.1 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 364.1267 for C18H17F3N3O2, found 364.1272. IR (KBr): 3441, 3339, 3254, 3229, 1685 cm−1. 1H-NMR: (600 MHz, DMSO-d6) δ 1.28 – 1.34 (m, 3H), 4.05 – 4.17 (m, 2H), 4.35 – 4.44 (m, 1H), 6.82 (s, 2H), 6.90 – 6.97 (m, 3H), 7.24 – 7.31 (m, 2H), 7.75 (dd, J = 8.4, 1.2 Hz, 1H), 8.17 (d, J = 8.4 Hz, 1H), 8.56 (brs, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 13.8, 37.7, 68.7, 111.9 (q, J = 5 Hz), 114.4, 120.3 (q, J = 3 Hz), 120.6, 122.3, 122.7, 124.1 (q, J = 273 Hz), 129.3, 129.7 (q, J = 32 Hz), 138.4, 152.5, 158.2, 172.7. HPLC: system 3: λ = 220 nm, tR = 27.0 min, purity: >99%. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 98:2: tR = 15.3 min, 99% ee (254 nm). [α]D25: +2.2 (c = 0.3, methanol).
(S)-1-(3-Amino-6-chloro-1H-indazol-1-yl)-2-methyl-3-phenoxypropan-1-one (I1414, 5d):
To a solution 12a (15.0 mg, 0.08 mmol) and HOAt (11.3 mg, 0.08 mmol) in DMF (1 mL) was added EDC × HCl (15.9 mg, 0.08 mmol). After stirring for 10 min at rt, a solution of 6-chloro-1H-indazol-3-amine (24, 16.7 mg, 0.10 mmol) in DMF (0.5 mL) was added. After stirring the reaction mixture for 4 h, the solvent was removed by lyophilization and the residue was purified by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min, 95% CH3OH for 2 min, 95% to 50% CH3OH in 2 min (tR: 20.2 min, λ = 220 nm) to afford I1414 (5d, 14.7 mg, 44.7 μmol, 54%) as a grey-white resin. ESI-MS: m/z 330.2 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 330.1004 for C17H17ClN3O2, found 330.1005. IR (KBr): 3429, 3335, 3247, 3223, 1685 cm−1. 1H-NMR: (600 MHz, DMSO-d6) δ 1.27 – 1.39 (m, 3H), 4.03 – 4.11 (m, 2H), 4.35 – 4.41 (m, 1H), 6.65 (s, 2H), 6.90 – 6.95 (m, 3H), 7.24 – 7.30 (m, 2H), 7.43 (dd, J = 8.4, 2.0 Hz, 1H), 7.93 (d, J = 8.4 Hz, 1H), 8.26 (d, J = 2.0 Hz, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 13.9, 37.8, 68.8, 114.5, 114.7, 119.1, 120.7, 122.4, 124.2, 129.5, 139.7, 152.6, 158.3, 172.5. HPLC: system 3: λ = 220 nm, tR = 25.8 min, purity: 96%. chiral HPLC: isocratic elution with n-hexane / isopropanol, 95:5: tR = 13.0 min, 99% ee (254 nm). [α]D20: +45.3 (c = 0.5, chloroform).
(S)-1-(3,6-Diamino-1H-indazol-1-yl)-2-methyl-3-phenoxypropan-1-one ((S)-SX270, 5e):
To a solution of 12a (34.8 mg, 0.19 mmol) in DMF (2.5 mL) were added HOAt (25.9 mg, 0.19 mmol) and EDC × HCl (36.4 mg, 0.19 mmol) at rt. The resulting solution was stirred for 10 min at rt and then 1H-indazole-3,6-diamine (25, 34.3 mg, 0.23 mmol) in DMF (0.5 mL) was added. After stirring the reaction mixture at rt for 2 h, the solvent was removed by lyophilization. Purification by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 12 mL / min and a gradient of 50% to 71% CH3OH in 11 min, 71% to 95% CH3OH in 2 min, 95% CH3OH for 2 min (tR = 8.0 min, λ = 254 nm) afforded (S)-SX270 (5e, 25.7 mg, 82.9 μmol, 44%) as an off-white solid. ESI-MS: m/z 311.1 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 311.1503 for C17H19N4O2, found 311.1505. IR (NaCl): 3452, 3353, 2925, 1676, 1625, 1409, 1238 cm−1. M.p.: 75 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 1.25 (d, J = 6.9 Hz, 3H), 4.00 (dd, J = 8.7, 5.6 Hz, 1H), 4.04 (ddq, J = 7.4, 6.9, 5.6 Hz, 1H), 4.36 (dd, J = 8.7, 7.4 Hz, 1H), 5.66 (s, 2H), 6.15 (s, 2H), 6.55 (dd, J = 8.5, 1.9 Hz, 1H), 6.90 – 6.95 (m, 3H), 7.25 – 7.29 (m, 2H), 7.42 (d, J = 1.9 Hz, 1H), 7.46 (d, J = 8.5 Hz, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 14.1, 37.6, 69.0, 98.1, 110.3, 112.0, 114.5, 120.6, 121.0, 129.5, 141.6, 151.2, 153.1, 158.4, 171.7. HPLC: system 1: λ = 254 nm, tR = 17.3 min, purity: 98%. Chiral HPLC: isocratic elution with n-hexane / isopropanol + 0.1% ethylene diamine, 9:1: tR = 41.4 min, >99% ee (254 nm). [α]D27: +64.3 (c = 0.5, methanol).
The syntheses of the ‘853-congeners bearing an aminocarbonyl or a hydroxymethyl group in position 6 are outlined in scheme 5. The primary amide derivative 27 was obtained from the commercially available carboxylic acid derivative 26 by a one pot coupling protocol employing PyBOP in presence of NH4Cl and DIPEA. The carbinol derivative 29 was synthesized by the cyclization reaction of the commercially available o-fluorobenzonitrile precursor 28 with hydrazine hydrate according to an established method.69 Both scaffolds were coupled with the (S)-carboxylic acid derivative 12a to furnish the both target compounds 5f and 5g. Starting from the (R)-carboxylic acid derivative ent-12a, the corresponding enantiomers ent-5f and ent-5g were prepared as well. Furthermore, the (S)-4-fluorophenoxy analog F-5g was obtained in a similar fashion by the acylation with the (S)-carboxylic acid derivative 12e.
Scheme 5.

Synthesis of novel indazole scaffolds and the corresponding acyl derivatives, reagents and conditions: (a) NH4Cl, PyBOP, DIPEA, DMF, rt, 12 h (83%); (b) EDC × HCl, HOAt, DMF, rt, 3 h (5f: 64%, 5g: 53%, F-5g: 45%); (c) N2H4, BuOH, 120 °C, 4 h (69%).
(S)-3-Amino-1-(2-methyl-3-phenoxypropanoyl)-1H-indazole-6-carboxamide (I1424, 5f):
To a solution of 12a (12.3 mg, 0.07 mmol) and HOAt (9.3 mg, 0.07 mmol) in DMF (0.5 mL) was added EDC × HCl (13.1 mg, 0.09 mmol). After for 10 min at rt, this solution was added to a suspension of 3-amino-1H-indazole-6-carboxamide (27, 12.0 mg, 0.09 mmol) in DMF (0.5 mL). After stirring the reaction mixture for 3 h, the solvent was removed by lyophilization and the residue was purified by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 10% to 45% CH3OH in 22 min (tR: 11.7 min, λ = 220 nm) to afford I1424 (5f, 17.1 mg, 50.6 μmol, 64%) as a white resin. ESI-MS: m/z 339.2 [M+H]+. HR-ESI-MS: m/z [M+Na]+ calcd. 361.1271 for C18H18N4NaO3, found 361.1268. IR (KBr): 3447, 3354, 3310, 3242, 1685 cm−1. 1H-NMR: (600 MHz, DMSO-d6) δ 1.30 (d, J = 7.0 Hz, 3H), 4.08 (dd, J = 9.3, 5.4 Hz, 1H), 4.12 (ddq, J = 7.2, 5.4, 7.0 Hz, 1H), 4.40 (dd, J = 9.3, 7.2 Hz, 1H), 6.63 (brs, 1H), 6.89 – 6.96 (m, 3H), 7.24 – 7.30 (m, 2H), 7.46 (brs, 1H), 7.81 (dd, J = 8.3, 1.5 Hz, 1H), 7.94 (d, J = 8.3 Hz, 1H), 8.14 (brs, 1H), 8.74 (brs, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 14.0, 37.8, 68.9, 114.5, 114.7, 120.4, 120.7, 121.9, 122.9, 129.5, 135.9, 139.0, 152.7, 158.3, 167.8, 172.3. HPLC: system 3: λ = 220 nm, tR = 18.9 min, purity: >99%. [α]D22: +44.5 (c = 0.25, methanol).
(R)-3-Amino-1-(2-methyl-3-phenoxypropanoyl)-1H-indazole-6-carboxamide (I1422, ent-5f):
I1422 (ent-5f) was synthesized analogously as described for I1424 (5f), starting from ent-12a. The analytical data were in accordance. HPLC: system 3: λ = 220 nm, tR = 18.9 min, purity: 98%. [α]D22: −44.5 (c = 0.7, methanol).
(3-Amino-1H-indazol-6-yl)methanol (29):
A solution of commercially available 2-fluoro-4-(hydroxymethyl)benzonitrile (28, 100 mg, 0.66 mmol) and hydrazine hydrate (319 μL, 6.62 mmol) in n-butanol (3 mL) was heated at 120 °C in a sealed tube for 4 h. After cooling to rt, the resulting solution was diluted with a 20% aqueous Na2CO3-solution, extracted with ethyl acetate and dried over MgSO4. After evaporation of the solvent, flash chromatography on silica gel (CH2Cl2 / MeOH 95:5 -> 9:1) was performed to afford 29 (74 mg, 0.45 mmol, 69%) as a white solid. ESI-MS: m/z 163.8 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 164.0818 for C8H10N3O, found 164.0819. IR (KBr): 3438, 3343, 3199 cm−1. M.p.: 214–215 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 4.56 (s, 2H), 5.20 (s, 1H), 5.24 (s, 2H), 6.83 (d, J = 8.3 Hz, 1H), 7.18 (s, 1H), 7.59 (d, J = 8.3 Hz, 1H), 11.28 (brs, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 63.2, 106.5, 113.0, 116.6, 119.7, 140.9, 141.8, 148.9.
(S)-1-(3-Amino-6-(hydroxymethyl)-1H-indazol-1-yl)-2-methyl-3-phenoxypropan-1-one (I1408, 5g):
To a solution of (12a,15.0 mg, 0.08 mmol) and HOAt (13.6 mg, 0.10 mmol) in DMF (0.5 mL) was added EDC × HCl (19.2 mg, 0.10 mmol). After stirring for 10 min at rt, a solution of 29 (16.3 mg, 0.10 mmol) in DMF (0.5 mL) was added. After stirring the reaction mixture for 3 h, the solvent was removed by lyophilization and the residue was purified by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 50% to 80% CH3OH in 15 min (tR: 12.5 min, λ = 220 nm) to afford I1408 (5g, 14.4 mg, 44.3 μmol, 53%) as a white solid. ESI-MS: m/z 326.1 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 326.1499 for C18H19N3O3, found 326.1499. IR (KBr): 3478, 3357, 1636 cm−1. M.p.: 132 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 1.28 (d, J = 7.0 Hz, 3H), 4.05 (dd, J = 8.8, 5.5 Hz, 1H), 4.11 (ddq, J = 8.0, 5.5, 7.0 Hz, 1H), 4.39 (dd, J = 8.8, 8.0 Hz, 1H), 4.64 (d, J = 5.8 Hz, 2H), 5.37 (t, J = 5.8 Hz, 1H), 6.49 (s, 2H), 6.90 – 6.95 (m, 3H), 7.25 – 7.30 (m, 3H), 7.82 (d, J = 8.0 Hz, 1H), 8.27 (brs, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 14.1, 37.8, 63.0, 69.0, 112.8, 114.5, 119.2, 120.3, 122.4, 129.5, 139.7, 145.0, 152.9, 158.4, 172.2. HPLC: system 3: λ = 220 nm, tR = 20.2 min, purity: 99%. Chiral HPLC: isocratic elution with n-hexane / ethanol, 9:1: tR = 16.7 min, 98% ee (254 nm). [α]D22: +27.9 (c = 0.5, methanol).
(R)-1-(3-Amino-6-(hydroxymethyl)-1H-indazol-1-yl)-2-methyl-3-phenoxypropan-1-one (I1411, ent-5g):
I1411 (ent-5g) was synthesized analogously as described for I1408 (5g), starting from ent-12a. The analytical data were in accordance. HPLC:system 3: λ = 220 nm, tR = 20.2 min, purity: 99%. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 99:1: tR = 13.5 min, 98% ee (254 nm). [α]D22: −28.2 (c = 0.4, methanol).
(S)-1-(3-Amino-6-(hydroxymethyl)-1H-indazol-1-yl)-3-(4-fluorphenoxy)-2-methylpropan-1-one (I1421, F-5g):
To a solution of 12e (18.0 mg, 0.09 mmol) and HOAt (12.4 mg, 0.09 mmol) in DMF (0.5 mL) was added EDC × HCl (17.4 mg, 0.09 mmol). After stirring for 10 min at rt, a solution of 29 (14.8 mg, 0.09 mmol) in DMF (0.5 mL) was added. After stirring the reaction mixture for 3 h, the solvent was removed by lyophilization and the residue was purified by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 50% to 80% CH3OH in 15 min (tR: 10.2 min, λ = 220 nm) to afford I1421 (F-5g, 14.2 mg, 41.4 μmol, 45%) as a white solid. ESI-MS: m/z 344.3 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 344.1404 for C18H19FN3O3, found 344.1404. IR (KBr): 3473, 3357, 3314, 1636 cm−1. M.p.: 136 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 1.27 (d, J = 7.3 Hz, 3H), 4.03 (dd, J = 9.1, 5.6 Hz, 1H), 4.09 (ddq, J = 7.9, 5.6, 7.3 Hz, 1H), 4.36 (dd, J = 9.1, 7.9 Hz, 1H), 4.63 (d, J = 5.6 Hz, 2H), 5.36 (t, J = 5.6 Hz, 1H), 6.94 – 6.98 (m, 2H), 7.07 – 7.12 (m, 2H), 7.28 (dd, J = 8.3, 1.1 Hz, 1H), 7.82 (d, J = 8.3 Hz, 1H), 8.26 (brs, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 14.0, 37.7, 62.9, 69.7, 112.7, 115.77 (d, J = 12 Hz), 115.82 (d, J = 19 Hz), 119.1, 120.2, 122.4, 139.6, 145.0, 152.9, 154.7, 156.5 (d, J = 239 Hz), 172.1. HPLC: system 3: λ = 220 nm, tR = 20.4 min, purity: >99%. Chiral HPLC: isocratic elution with n-hexane / ethanol, 9:1: tR = 18.7 min, 99% ee (254 nm). [α]D22: +31.8 (c = 0.5, methanol).
The 6-carboxamide and 6-amino derivatives 7a and 7b renouncing the amino group in position 3 were obtained analogously starting from the respective purchasable precursors 30 and 31 (scheme 6). The enantiomer ent-7a was synthesized employing (R)-carboxylic acid derivative ent-12a.
Scheme 6.

Synthesis of the ‘853 derivatives with modifications in the positions 3 and 6, reagents and conditions: (a) EDC × HCl, HOAt, DMF, rt (7a: 64%, 7b: 14%).
(S)-1-(2-Methyl-3-phenoxypropanoyl)-1H-indazole-6-carboxamide (I1409, 7a):
To a solution of 12a (15.0 mg, 0.08 mmol) and HOAt (13.6 mg, 0.10 mmol) in DMF (0.5 mL) was added EDC × HCl (19.2 mg, 0.10 mmol). After stirring for 10 min at rt, a solution of commercially available 1H-indazole-6-carboxamide (30, 16.1 mg, 0.10 mmol) in DMF (0.5 mL) was added. After stirring the reaction mixture for 3 h, the solvent was removed by lyophilization and the residue was purified by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min (tR: 15.5 min, λ = 220 nm) to afford I1409 (7a, 17.1 mg, 52.9 μmol, 64%) as a white solid. ESI-MS: m/z 324.1 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 324.1343 for C18H17N3O3, found 324.1345. IR (KBr): 3482, 3437, 3397, 3357, 3197, 1718, 1707, 1662 cm−1. M.p.: 141 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 1.38 (d, J = 7.2 Hz, 3H), 4.21 (dd, J = 10.0, 5.3 Hz, 1H), 4.35 (ddq, J = 6.8, 5.3, 7.2 Hz, 1H), 4.43 (dd, J = 10.0, 6.8 Hz, 1H), 6.90–6.94 (m, 3H), 7.24 – 7.28 (m, 2H), 7.53 (brs, 1H), 7.91 (dd, J = 8.3, 1.6 Hz, 1H), 7.98 (dd, J = 8.3, 0.6 Hz, 1H), 8.24 (brs, 1H), 8.60 (d, J = 1.0 Hz, 1H), 8.85 (ddd, J = 1.6, 1.0, 0.6 Hz, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 13.9, 38.2, 68.9, 114.2, 114.4, 120.7, 121.4, 123.9, 129.4, 135.7, 138.2, 140.5, 158.1, 167.6, 174.1. HPLC: system 3: λ = 220 nm, tR = 21.7 min, purity: 98%. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 85:5: tR = 21.6 min, 98% ee (254 nm). [α]D22: +111.4 (c = 0.4, methanol).
(R)-1-(2-Methyl-3-phenoxypropanoyl)-1H-indazole-6-carboxamide (I1412, ent-7a):
I1412 (ent-7a) was synthesized analogously as described for I1409 (7a), starting from ent-12a. The analytical data were in accordance. HPLC: system 3: λ = 220 nm, tR = 21.7 min, purity: 99%. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 85:5: tR = 22.7 min, 98% ee (254 nm). [α]D22: −111.0 (c = 0.65, methanol).
(S)-1-(6-Amino-1H-indazol-1-yl)-2-methyl-3-phenoxypropan-1-one ((S)-SX254, 7b):
To a stirred solution of compound 12a (39.8 mg, 0.22 mmol) in anhydrous DMF (1.5 mL) was added HOAt (29.9 mg, 0.22 mmol), followed by EDC × HCl (42.2 mg, 0.22 mmol) at rt. The resulting solution was stirred for 10 min at rt before adding commercially available 6-amino-1H-indazole (31, 35.9 mg, 0.27 mmol) dissolved in anhydrous DMF (0.5 mL). After stirring the reaction mixture at rt for 40 min., the solvent was removed by lyophilization. Purification by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min, 95% CH3OH for 4 min (tR = 15.0 min, λ = 254 nm) yielded (S)-SX254 (7b, 8.79 mg, 29.8 μmol, 14%) as a colorless resin. ESI-MS: m/z 296.1 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 296.1394 for C17H18N3O2, found 296.1392 [M+H]+. IR (NaCl): 3469, 3368, 2920, 2851, 1698, 1618, 1493, 1424, 1406, 1241 cm−1. 1H-NMR: (400 MHz, DMSO-d6) δ 1.31 (d, J = 7.0 Hz, 1H), 4.12 (dd, J = 9.1, 5.2 Hz, 1H), 4.25 (ddq, J = 7.9, 7.0, 5.2 Hz, 1H), 4.38 (dd, J = 9.1, 7.9 Hz, 1H), 5.86 (s, 2H), 6.68 (dd, J = 8.5, 2.0 Hz, 1H), 6.87 – 6.98 (m, 3H), 7.23 – 7.30 (m, 2H), 7.46 – 7.51 (m, 2H), 8.15 (d, J = 0.7 Hz, 1H). 13C-NMR: (DEPTQ, 101 MHz, DMSO-d6) δ 14.1, 38.1, 69.0, 96.8, 114.0, 114.5, 116.8, 120.8, 121.9, 129.5, 140.8, 140.9, 151.4, 158.3, 174.0. HPLC: system 1: λ = 254 nm, tR = 19.2 min, purity: 97%. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 9:1: tR = 12.3 min, >99% ee (254 nm). [α]D23: +139.1 (c = 0.1, methanol).
The N-formyl congeners 6a, 6b and 8 were prepared starting from the corresponding amine derivatives 5e and 7b taking advantage of activated formic acid (scheme 7).
Scheme 7.

Synthesis of the ‘853 derivatives further modified with a formyl group, reagents and conditions: (a) 1. HCO2H, Ac2O, THF, 60 °, 2 h, 2. 5e, 0 °C, 30 min (42%); (b) 1. HCO2H, Ac2O, THF, 60 °C, 2 h, 2. 5e or 7b, 0 °C to rt, for 6b: 60 min, for 8: 90 min (6b: 37%, 8: 28%).
(S)-N-(3-Amino-1-(2-methyl-3-phenoxypropanoyl)-1H-indazol-6-yl)formamide ((S)-SX276, 6a):
A mixture of formic acid (15.1 μL, 18.5 mg, 401 μmol) and acetic anhydride (25.2 μL, 27.3 mg, 268 μmol) was stirred at 60 °C for 2 h. After cooling to rt, the mixed anhydride was added dropwise to a solution of compound (S)-SX270 (5e, 16.6 mg, 53.5 μmol) in anhydrous THF (1 mL) at 0 °C. After stirring the reaction mixture for 30 min at rt, the mixture was immediately treated with CH3OH / water (1:2) in order to hydrolyze the excess of mixed anhydride and then lyophilization was performed. Purification by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 12 mL / min and a gradient of 50% to 75% CH3OH in 12.5 min, 75% to 95% CH3OH in 0.5 min, 95% CH3OH for 1 min (tR = 9.7 min, λ = 254 nm) afforded (S)-SX276 (6a, 7.64 mg, 22.6 μmol, 42%) as a white solid. ESI-MS: m/z 339.2 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 339.1452 for C18H19N4O3, found 339.1452. IR (NaCl): 3442, 3336, 2925, 1683, 1618, cm−1. M.p.: 77 °C. 1H-NMR: (600 MHz, DMSO-d6, two sets of resonances were observed, rotamers, assignment was confirmed by 2D spectroscopy) δ 1.28 (d, J = 6.8 Hz, 3H), 4.05 (dd, J = 8.8, 5.5 Hz, 1H), 4.07 (ddq, J = 7.4, 6.8, 5.5 Hz 1H), 4.38 (dd, J = 8.8, 7.4 Hz, 1H), 6.47 (s, 1.6 H), 6.49 (s, 0.4 H), 6.90 – 6.96 (m, 3H), 7.24 – 7.29 (m, 2 H and 0.2 H), 7.50 (dd, J = 8.5, 1.8 Hz, 0.8 H), 7.81 (d, J = 8.4 Hz, 0.8 H), 7.82 (d, J = 8.6 Hz, 0.8 H),, 8.04 (d, J = 1.9 Hz, 0.2 H), 8.34 (d, J = 1.8 Hz, 0.8 H), 8.68 (d, J = 1.7 Hz, 0.8 H), 8.89 (d, J = 11.0 Hz, 0.2 H), 10.43 (d, J = 11.0 Hz, 0.2 H), 10.48 (d, J = 1.9 Hz, 0.8 H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6, two sets of resonances were observed, rotamers) δ 14.0 (minor), 14.0 (major), 37.7, 68.9 (minor), 68.9 (major), 103.4 (minor), 105.3 (major), 113.8 (minor), 114.5, 115.7 (major), 116.1 (major), 116.3 (minor), 120.6, 121.1 (major), 121.8 (minor), 129.5, 139.7 (major), 139.9 (major), 140.1 (minor), 140.2 (minor), 152.7 (major), 152.8 (minor), 158.3, 159.8 (major), 162.6 (minor), 172.1 (major), 172.2 (minor). HPLC: system 2: λ = 254 nm, tR = 16.1 min, purity: >99%. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 9:1: tR = 40.7 min, >99% ee (254 nm). [α]D26: +78.1 (c = 0.3, methanol).
(S)-N,N’-(1-(2-Methyl-3-phenoxypropanoyl)-1H-indazole-3,6-diyl)diformamide ((S)-SX272, 6b):
A mixture of formic acid (14.9 μL, 18.1 mg, 394 μmol) and acetic anhydride (24.8 μL, 26.8 mg, 263 μmol) was stirred at 60 °C for 2 h. After cooling to rt, the mixed anhydride was added dropwise to a solution of compound (S)-SX270 (5e, 16.3 mg, 52.5 mmol) in anhydrous THF (2 mL) at 0 °C. After stirring the reaction mixture for 1 h at rt, the solvent was removed under reduced pressure. The residue was treated with a mixture of CH3OH / water (1:2) and lyophilized. After purification by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 12 mL / min and a gradient of 50% to 75% CH3OH in 12.5 min, 75% to 95% CH3OH in 0.5 min, 95% CH3OH for 1 min (tR = 11.0 min, λ = 254 nm), (S)-SX272 (6b, 6.52 mg, 19.3 μmol, 37%) was obtained as a white solid. ESI-MS: m/z 367.2 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 367.1401 for C19H19N4O4, found 367.1404. IR (NaCl): 3354, 2921, 1689, 1632 cm−1. 1H-NMR: (400 MHz, DMSO-d6, two major sets of resonances, rotamers) δ 1.33 (d, J = 6.7 Hz, 3 H), 4.11 – 4.27 (m, 2H), 4.32 – 4.43 (m, 1H), 6.89 – 6.97 (m, 3H), 7.23 – 7.31 (m, 2H), 7.40 (brd, J = 8.9 Hz, 0.2 H), 7.53 (brd, J = 8.8 Hz, 0.8 H), 7.98 (bd, J = 8.5 Hz, 1H), 8.16 – 8.08 (m, 0.2 H), 8.38 (d, J = 1.7 Hz, 0.8 H), 8.55 (s, 0.2 H), 8.87 (brs, 0.8 H), 8.97 (d, J = 10.1 Hz, 0.2 H), 9.10 (brs, 0.8 H), 10.58 (d, J = 10.1 Hz, 0.2 H), 10.68 (brs, 0.8 H), 11.31 (brs, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6, two sets of resonances were observed, rotamers) δ 13.9 (major), 14.0 (minor), 37.7 (minor), 38.0 (major), 68.9, 102.6 (minor), 104.7 (major), 114.5 (minor), 114.5 (major), 114.8 (minor), 116.9 (major), 120.7 (major), 121.2 (minor), 129.4, 139.7, 139.7, 140.6, 145.1, 158.2, 160.1, 162.2, 162.7, 173.3 (major), 173.4 (minor). HPLC: system 1: λ = 254 nm, tR = 18.1 min, purity: 96%. chiral HPLC: isocratic elution with n-hexane / isopropanol + 0.1% ethylene diamine, 8:2: tR = 28.3 min, 97% ee (254 nm). [α]D26: +18.3 (c = 0.1, methanol).
(S)-N-(1-(2-Methyl-3-phenoxypropanoyl)-1H-indazol-6-yl)formamide ((S)-SX258, 8):
A mixture of formic acid (5.9 μL, 7.25 mg, 157 μmol) and acetic anhydride (9.9 μL, 10.7 mg, 105 μmol) was stirred at 60 °C for 2 h. After cooling to rt, the mixed anhydride was added dropwise to a solution of compound (S)-SX254 (7b 6.2 mg, 21.0 μmol) in anhydrous THF (1 mL) at 0 °C. After stirring the reaction mixture for 1.5 h at rt, the solvent was removed under reduced pressure. The residue was treated with CH3OH / water (1:2) and lyophilized. Purification by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min, 95% CH3OH for 1 min (tR = 15.0 min, λ = 254 nm) yielded (S)-SX258 (8, 1.87 mg, 5.78 μmol, 28%) as a white solid. ESI-MS: m/z 324.2 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 324.1343 for C18H18N3O3, found 324.1343. IR (NaCl): 3357, 3275, 2922, 2850, 1700, 1618, 1597 cm−1. 1H-NMR: (600 MHz, DMSO-d6, two sets of signals were observed, rotamers) δ 1.35 (d, J = 7.0 Hz, 3H), 4.17 (dd, J = 9.3, 5.2 Hz, 1H), 4.31 (ddq, J = 7.9, 7.0, 5.2 Hz, 1H), 4.40 (dd, J = 9.3, 7.9 Hz, 1H), 6.89 – 6.95 (m, 3H), 7.23 – 7.30 (m, 2H), 7.38 (dd, J = 8.4, 1.7 Hz, 0.2 H), 7.54 (dd, J = 8.4, 1.7 Hz, 0.8 H), 7.85 (d, J = 8.4 Hz, 0.8 H), 7.86 – 7.87 (m, 0.2 H), 8.12 – 8.14 (m, 0.2 H), 8.37 (d, J = 1.7 Hz, 0.8 H), 8.44 (d, J = 0.9 Hz, 0.8 H), 8.55 – 8.56 (m, 0.2 H), 8.84 – 8.88 (m, 0.8 H), 8.94 (d, J = 10.6 Hz, 0.2 H), 10.51 (d, J = 10.6 Hz, 0.2 H), 10.57 – 10.61 (m, 0.8 H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6, two sets of signals were observed, rotamers) δ 14.0, 38.2, 69.0, 102.5 (minor), 104.5 (major), 114.6 (major), 115.4 (minor), 117.2, 120.8, 122.1 (major), 122.2 (major), 122.4 (minor), 122.8 (minor), 129.5, 139.1 (major), 139.3 (minor), 139.7 (major), 140.2 (minor), 140.6 (major), 140.7 (minor), 158.2, 160.0, 162.7 (major), 165.0 (minor), 174.1 (major), 174.2 (minor). HPLC: system 1: λ = 254 nm, tR = 19.6 min, purity: 97%. chiral HPLC: isocratic elution with n-hexane / isopropanol, 9:1: tR = 28.4 min, 99% ee (254 nm).[α]D23: +74.8 (c = 0.1, methanol).
The preparation of the fused derivative 9 (scheme 8) was achieved starting from the alkine derivative 32, performing a gold(I) promoted hydroarylation reaction to give the chromene derivative 33.70 Since the original cyclization protocol did not work with satisfying yield and purity, we preferred to apply more regioselective conditions for ring closure employing toluene as the solvent and working at 0 °C,71 thus leading to a cleaner formation of only 14% regioisomer instead of 25% reported in the literature. The mixture thus obtained was subjected to hydrogenation to give the chroman derivative 34 (and the respective regioisomer). Reaction with N-bromosuccinimide furnished the 6-brominated derivative 35, when chromatography allowed to reduce the amount of the regioisomer to 6%. Transnitrilation reaction employing n-butyllithium and dimethylmalononitrile allowed to cleanly prepare the cyclization precursor 36,72 which could be subjected to hydrazine hydrate to obtain the fused indazole-amine derivative 37. Acylation with the racemic carboxylic acid derivative rac-12a led to the formation of the target compound 9.
Scheme 8.

Synthesis of the fused acylindazole derivative (a) (acetonitrile)[2-biphenyl)di-tert-butylphosphine]gold(I) hexafluoroantimonate, toluene, 0 °C (crude); (b) H2/Pd(OH)2/C, MeOH, rt, 2 h (crude); (c) N-bromosuccinimide, CH3CN, 0 °C (23% over three steps); (d) 1. BuLi, THF, −79 °C. 2. dimethylmalononitrile, THF, −79 °C (54%); (e) N2H4, BuOH, 120 °C, 22 h (76%); (f) EDC × HCl, HOAt, DMF, microwave irradiation (61%).
7-Fluoro-2H-chromene (33):
(Acetonitrile)[2-biphenyl)di-tert-butylphosphine]gold(I) hexafluoroantimonate (0.50 g, 0.66 mmol) was added slowly to an ice cooled solution of 32 (6.00 g, 39.9 mmol) in toluene (10 mL). After stirring for 90 min the resulting solution was directly subjected to flash chromatography on silica gel (isohexane 100%) to afford crude 33 (5.80 g, 97%) as a volatile, colourless oil, contaminated with residual catalyst and the other ring closing isomer 5-fluoro-2H-chromene (14%, determined by 1H NMR). This crude material was used for the following reaction without further purification. 1H NMR: (600 MHz, CDCl3) δ 4.81 (dd, J = 3.6, 2.0 Hz, 2H), 5.70 (dd, J = 10.0, 3.6 Hz, 2H), 6.38 (dd, J = 10.0, 2.0 Hz, 2H), 6.50 (dd, J = 10.1, 2.7 Hz, 1H), 6.55 (ddd, J = 8.5, 8.4, 2.7 Hz, 1H), 6.89 (dd, J = 8.5, 6.3 Hz, 1H). The NMR data of this compound are in accordance with those reported in the literature.
7-Fluorochromane (34):
To a solution of crude 33 (5.80 g) in methanol / ethyl acetate 1:1 (200 mL) was added palladium hydroxide 20% on charcoal (0.50 g). Hydrogenation by stirring for 2 h under a balloon filled with H2 was performed at rt. The resulting solution was filtered through a pad of celite and the solvent was removed in vacuo, not lower than 200 mbar to afford crude 34 (5.80 g) as a volatile, colourless liquid, still contaminated with residual catalyst and the other ring closing isomer 5-fluorochromane formed by the hydrogenation of the 5-fluoro-2H-chromene byproduct. This crude material was used for the following reaction without further purification. 1H NMR: (600 MHz, CDCl3) δ 1.99 (tt, J = 6.4, 5.2 Hz, 2H), 2.74 (t, J = 6.4 Hz, 2H), 4.17 (t, J = 5.2 Hz, 2H), 6.51 (dd, J = 10.4, 2.5 Hz, 1H), 6.55 (ddd, J = 8.4, 8.3, 2.5 Hz, 1H), 6.96 (ddd, J = 8.3, 6.8, 0.8 Hz, 1H). The NMR data of this compound are in accordance with those reported in the literature.
6-Bromo-7-fluorochromane (35):
To a solution of the crude 34 (5.80 g, 38.1 mmol) in acetonitrile (40 mL) was added slowly N-bromosuccinimide (5.43 g, 30.5 mmol) at 0 °C. After stirring for 2 h, another portion of N-bromosuccinimide (0.68 g, 4,54 mmol) was added. After stirring at 0 °C for 1 h, allowing a full conversion of the educt according to TLC, water was added and the mixture was allowed to warm to ambient temperature. Extraction with ethyl acetate (4x) was performed and the organic layer was washed with water and dried over MgSO4. Gradual evaporation of the solvent was done and the precipitate, which was formed (succinimide), was removed by filtration. Flash chromatography (isohexane 100%) afforded 35 (2.20 g, 9.51 mmol, 23% over the last three steps) as a colourless oil, containing another regioisomer (6% according to NMR) resulting from the bromination of the 5-fluorochromane impurity. This compound was employed for the next reaction without further purification. 1H NMR: (400 MHz, CDCl3) δ 1.98 (tt, J = 6.4, 5.2 Hz, 2H), 2.73 (t, J = 6.4 Hz, 2H), 4.16 (t, J = 5.2 Hz, 2H), 6.58 (d, J = 9.7 Hz, 1H), 7.17 (dd, J = 7.2, 1.0 Hz, 1H). The NMR data of this compound are in accordance with those reported in the literature.
7-Fluorochromane-6-carbonitrile (36):
To a solution of 35 (700 mg, 3.03 mmol) in THF (14 mL) at −79 °C was added slowly a 2.5 M solution of butyllithium in hexane (1.33 mL, 3.33 mmol). After stirring for 20 min, a solution of 2,2-dimethylmalononitrile (428 mg, 4.54 mmol) in THF (2 mL) was added slowly. After stirring at −79 °C for additional 30 min, the reaction was stopped by the addition of a saturated aqueous NH4Cl-solution, warmed to rt, extracted with tert-butyl methyl ether and dried over MgSO4. After evaporation of the solvent, flash chromatography on silica gel (isohexane / ethyl acetate, 99:1 -> 98:2) was performed to afford 36 (290 mg, 1.47 mmol, 54%) as a white solid. ESI-MS: m/z 178.1 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 178.0663 for C10H9FNO, found 178.0663. IR (NaCl): 2229 cm−1. M.p.: 102 °C. 1H-NMR: (400 MHz, CDCl3) δ 2.02 (tt, J = 6.4, 5.2 Hz, 2H), 2.76 (t, J = 6.4 Hz, 2H), 4.25 (t, J = 5.2 Hz, 2H), 6.59 (d, J = 5.2 Hz, 1H), 7.27 (dt, J = 7.2, 1.0 Hz, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 21.5, 24.1, 67.2, 92.5 (d, J = 15 Hz), 104.8 (d, J = 22 Hz), 114.7, 119.4 (d, J = 3 Hz), 134.2 (d, J = 2 Hz), 160.1 (d, J = 12 Hz), 162.6 (d, J = 255 Hz).
1,5,6,7-Tetrahydropyrano[3,2-f]indazol-3-amine (37):
A solution of 36 (200 mg, 1.13 mmol) and hydrazine hydrate (550 μL, 11.3 mmol) in n-butanol (5 mL) was heated at 120 °C in a sealed tube. After 20 h, another portion of hydrazine hydrate (280 μL, 6.64 mmol) was added and heating was continued for additional 2 h. After cooling to ambient temperature, the resulting solution was diluted with a 20% aqueous Na2CO3-solution, extracted with ethyl acetate and dried over MgSO4. After evaporation of the solvent, flash chromatography (CH2Cl2 / CH3OH, 100:0 -> 99:1 -> 98:2) was performed to afford 37 (162 mg, 0.86 mmol, 76%) as a white solid. ESI-MS: m/z 189.8 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 190.0975 for C10H12N3O, found 190.0975. IR (KBr): 3350, 3275, 3172, 1636 cm−1. M.p.: 158–159 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 2.02 (tt, J = 6.3, 5.3 Hz, 2H), 2.82 (t, J = 6.3 Hz, 2H), 4.12 (t, J = 5.3 Hz, 2H), 5.23 (s, 2H), 6.46 (s, 1H), 7.33 (s, 1H), 10.84 (s, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ δ 24.2, 27.3, 69.4, 94.1, 109.3, 114.4, 120.0, 141.5, 148.6, 154.1.
(R,S)-1-(3-Amino-6,7-dihydropyrano[3,2-f]indazol-1(5H)-yl)-2-methyl-3-phenoxypropan-1-one (I1380, 9):
To a solution of commercially available (R,S)-2-methyl-3-phenoxypropionic acid (rac-12a, 57.1 mg, 0.32 mmol), PyBOP (165 mg, 0.32 mmol) and HOAt (60.0 mg, 0.48 mmol) in DMF (1.5 mL) was added DIPEA (55 μL, 0.44 mmol) and then a solution of 32 (50.0 mg, 0.26 mmol) in DMF (0.5 mL) at rt. Microwave irradiation (described in the general part) in a sealed tube was performed, then the solvent was removed by lyophilization and the residue was purified by preparative HPLC using a solvent system of CH3CN / 0.1% aq. TFA, a flow rate of 10 mL / min and a gradient of 40% to 65% CH3CN in 19 min, 65% to 95% CH3CN in 2 min (tR: 19.0 min, λ = 220 nm) to afford I1380 (9, 56.3 mg, 0.16 mmol, 61%) as a white solid. ESI-MS: m/z 352.1 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 352.1656 for C20H22N3O3, found 352.1660. IR (KBr): 3424, 3353, 1647 cm−1. M.p.: 185 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 1.24 – 1.27 (m, 3H), 1.95 (tt, J = 6.3, 5.1 Hz, 1H), 2.86 (t, J = 6.3 Hz, 2H), 4.00 – 4.07 (m, 2H), 4.20 (t, J = 5.1 Hz, 2H), 4.33 – 4.39 (m, 1H), 6.37 (s, 2H), 6.90 – 6.95 (m, 3H), 7.25 – 7.29 (m, 2H), 7.55 (s, 1H), 7.57 (s, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 13.9, 21.5, 24.7, 37.5, 66.4, 68.8, 101.8, 113.8, 114.4, 119.7, 120.5, 121.1, 129.4, 138.8, 152.7, 156.4, 158.2, 171.7. HPLC: system 3: λ = 220 nm, tR = 24.5 min, purity: >99%.
The preparation of the compounds incorporating a modified spacer was performed as shown in scheme 9. Starting from the commercially available carboxylic acids 39–42 or the respective sodium carboxylate 38, coupling with EDC × HCl and HOAt allowed to obtain the target compounds 10a-e. Preparative chiral HPLC gave rise to the separated enantiomers of 10b-d.
Scheme 9.

Synthesis of the ‘853 derivatives with a modified spacer, reagents and conditions: (a) 1. EDC × HCl, HOAt, DMF, rt, 10 min, 2. 6-fluoro-1H-indazole-3-amine (13), rt, 10a and 10b: 3 h, 10c: 2 h, 10d: 1 h, 10e: 1.5 h (10a: 27%, 10b: 45%, 10c: 43%, 10d: 30%, 10e: 42%).
1-(3-Amino-6-fluoro-1H-indazol-1-yl)-3-phenoxypropan-1-one (KD2, 10a):
To a solution of 3-phenoxypropionic acid (38, 52.8 mg, 0.32 mmol) and HOAt (46.1 mg, 0.34 mmol) in DMF (1 mL) was added EDC × HCl (61.6 mg, 0.32 mmol). After stirring for 10 min at rt, a solution of 6-fluoro-1H-indazol-3-amine (13, 40.3 mg, 0.27 mmol) in DMF (0.5 mL) was added. After stirring the reaction mixture for 3 h, the solvent was removed by lyophilization and the residue was recrystallized from methanol. The mother liquor was purified by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min, 95% CH3OH for 4 min (tR: 20.4 min, λ = 254 nm). Combining both fractions afforded KD2 (10a, 21.4 mg, 0.07 mmol, 27%) as a white solid. ESI-MS: m/z 300.1 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 300.1143 for C18H17F3N3O2, found 300.1147. IR (KBr): 3406, 3334, 3209, 1685 cm−1. M.p.: 181 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 3.43 (t, J = 6.0 Hz, 2H), 4.38 (t, J = 6.0 Hz, 2H), 6.59 (s, 2H), 6.92 – 6.97 (m, 3H), 7.25 (ddd, J = 9.1, 8.6, 2.4 Hz, 1H), 7.28 – 7.31 (m, 2H), 7.82 – 7.86 (m, 2H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 34.3, 62.7, 101.5 (d, J = 30 Hz), 112.1 (d, J = 25 Hz), 114.4, 116.9, 120.6, 122.6 (d, J = 11 Hz), 129.4, 139.6 (d, J = 12 Hz), 152.5, 158.2, 163.1 (d, J = 245 Hz), 169.0. HPLC: system 3: λ = 220 nm, tR = 23.6 min, purity: >99%.
1-(3-Amino-6-fluoro-1H-indazol-1-yl)-2-(phenoxymethyl)butan-1-one (KD6, 10b):
To a solution of commercially available (R,S)-2-(phenoxymethyl)butanoic acid (39, 61.9 mg, 0.32 mmol) and HOAt (43.2 mg, 0.32 mmol) in DMF (1 mL) was added EDC × HCl (61.0 mg, 0.32 mmol). After stirring for 10 min at rt, a solution of 6-fluoro-1H-indazol-3-amine (13, 40.6 mg, 0.27 mmol) in DMF (0.5 mL) was added. After stirring the reaction mixture for 3 h, the solvent was removed by lyophilization and the residue was purified by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min, 95% CH3OH for 4 min (tR: 19.7 min, λ = 254 nm) to afford KD6 (10b, (45.5 mg, 0.14 mol, 45%) as a white solid. The enantiomers were separated by performing chiral preparative HPLC (isocratic solvent system: n-hexane / isopropanol, 98:2) to afford the two enantiomers KD6en1 (10b-ent1) (tR: 16.5 min) and KD6en2 (10b-ent2) (tR: 25.1 min). ESI-MS: m/z 328.2 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 328.1456 for C18H19FN3O2, found 328.1456. IR (KBr): 3437, 3340, 3231, 1688, 1642, 1624 cm−1. M.p.: 97 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 0.93 (dd, J = 7.7, 7.3 Hz, 3 H), 1.77 (ddq, J = 14.5, 8.5, 7.3 Hz, 1H), 1.82 (ddq, J = 14.5, 7.7, 5.5 Hz, 1H), 4.05 (dddd, J = 8.5, 8.0, 5.8, 5.5 Hz, 1H), 4.13 (dd, J = 9.3, 5.5 Hz, 1H), 4.37 (dd, J = 9.3, 8.0 Hz, 1H), 6.60 (s, 2H), 6.93 – 6.91 (m, 3H), 7.28 – 7.24 (m, 3H), 7.95 (dd, J = 8.6, 5.3, 1H), 7.98 (dd, J = 9.8, 2.3 Hz). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 11.2, 21.7, 44.0, 67.7, 101.8 (d, J = 28 Hz), 112.2 (d, J = 24 Hz), 114.4, 117.0, 120.6, 122.6 (d, J = 11 Hz), 129.4, 139.6 (d, J = 19 Hz), 152.5, 158.2, 163.1 (d, J = 245 Hz), 171.9. HPLC: system 4: λ = 220 nm, for both enantiomers tR = 25.9 min, KD6en1 (10b-ent1): purity: >99%, KD6en2 (10b-ent2): purity: >99%. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 98:2: KD6en1 (10b-ent1): tR = 17.2 min, >99% ee (254 nm); KD6en2 (10b-ent2): tR = 23.2 min, 98% ee (254 nm).[α]D25: KD6en1 (10b-ent1): +23.6 (c = 0.4, methanol); KD6en2 (10b-ent2): −23.6 (c = 0.2, methanol).
1-(3-Amino-6-fluoro-1H-indazol-1-yl)-2-methoxy-3-phenoxypropan-1-one (KD5, 10c):
To a solution of commercially available (R,S)-2-methoxy-3-phenoxypropanoic acid (40, 62.4 mg, 0.32 mmol) and HOAt (43.4 mg, 0.32 mmol) in DMF (1 mL) was added EDC × HCl (61.4 mg, 0.32 mmol). After stirring for 10 min at rt, a solution of 6-fluoro-1H-indazol-3-amine (13, 40.3 mg, 0.27 mmol) in DMF (0.5 mL) was added. After stirring the reaction mixture for 2 h, the solvent was removed by lyophilization and the residue was purified by preparative HPLC using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 10 mL / min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min (tR: 15.3 min, λ = 254 nm) to afford KD5 (10c, 37.6 mg, 0.11 mmol, 43%) as a white solid. The enantiomers were separated by 2× performing chiral preparative HPLC (isocratic solvent system: n-hexane / isopropanol, 93:7) to afford the two enantiomers KD5en1 (10c-ent1) (tR: 32.6 min) and KD5en2 (10c-ent2) (tR: 35.1 min). ESI-MS: m/z 330.2 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 330.1248 for C17H17FN3O3, found 330.1249. IR (KBr): 3457, 3349, 3227, 1695, 1632 cm−1. M.p.: 134 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 3.41 (s, 3H), 4.26 (dd, J = 10.7, 6.9 Hz), 4.41 (dd, J = 10.7, 2.9 Hz, 1H), 5.21 (dd, J = 6.9, 2.9 Hz, 1H), 6.70 (s, 2H), 6.91 – 6.98 (m, 3H), 7.25 – 7.31 (m, 3H), 7.94 – 7.99 (m, 2H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 57.6, 67.8, 78.1, 101.6 (d, J = 28 Hz), 112.5 (d, J = 25 Hz), 114.4, 116.8, 122.7 (d, J = 12 Hz), 129.4, 139.8 (d, J = 12 Hz), 152.9, 158.0, 163.2 (d, J = 246 Hz), 166.8. HPLC: system 3: λ = 220 nm, for both enantiomers tR = 22.5 min, KD5en1 (10c-ent1): purity: 97%, KD5en2 (10c-ent2): purity: >99%. Chiral HPLC: isocratic elution with n-hexane / isopropanol, 93:7: KD5en1 (10c-ent1): tR = 29.6 min, >99% ee (254 nm); KD5en2 (10c-ent2): tR = 33.7 min, 91% ee (254 nm). [α]D25: KD5en1 (10c-ent1): +33.6 (c = 0.9, methanol); KD5en2 (10c-ent2): n.d.
1-(3-Amino-6-fluoro-1H-indazol-1-yl)-2-methyl-4-phenylbutan-1-one (KD3, 10d):
To a solution of (R,S)-2-methyl-4-phenylbutyric acid (41, 56.7 mg, 0.32 mmol) and HOAt (43.8 mg, 0.32 mmol) in DMF (1 mL) was added EDC x HCl (61.1 mg, 0.32 mmol). After stirring for 10 min at rt, a solution of 6-fluoro-1H-indazol-3-amine (13, 40.2 mg, 0.27 mmol) in DMF (0.5 mL) was added. After stirring the reaction mixture for 1 h, the solvent was removed by lyophilization and the residue was purified by preparative HPLC (Nucleodur C18 HTec, 32 mm × 250 mm, 5 μm) using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 25 mL / min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min, 95% CH3OH for 7 min (tR: 23.0 min, λ = 254 nm) to afford KD3 (10d, 25.1 mg, 0.08 mmol, 30%) as a colorless oil. The enantiomers were separated by 2× performing chiral preparative HPLC (isocratic solvent system: n-hexane / isopropanol, 98:2) to afford the two enantiomers KD3en1 (10d-ent1) (tR: 14.7 min) and KD3en2 (10d-ent2) (tR: 17.2 min). ESI-MS: m/z 312.1 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 312.1507 for C18H19FN3O, found 312.1509. IR (KBr): 3459, 3350, 3227, 1678, 1626 cm−1. 1H-NMR: (600 MHz, DMSO-d6) δ 1.22 (d, J = 6.9 Hz, 3H), 1.76 (dddd, J = 13.3, 9.8, 6.5, 6.4 Hz, 1H), 2.07 (dddd, J = 13.3, 9.8, 7.6, 6.0 Hz, 1H), 2.57 (ddd, J = 13.6, 9.8, 6.0 Hz, 1H), 2.62 (ddd, J = 13.6, 9.8, 6.4 Hz, 1H), 3.64 (ddq, J = 7.6, 6.5, 6.9, 1H), 6.53 (s, 2H), 7.14 – 7.18 (m, 3H), 7.22 – 7.26 (m, 3H), 7.93 (dd, J = 8.7, 5.3, 1H), 7.97 (dd, J = 9.8, 2.3 Hz). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 17.2, 32.7, 34.3, 36.7, 101.7 (d, J = 28 Hz), 111.9 (d, J = 24 Hz), 116.8, 122.5 (d, J = 12 Hz), 125.7, 128.1, 128.2, 139.8 (d, J = 15 Hz), 141.5, 152.3, 163.6 (d, J = 245 Hz), 174.7. HPLC: system 3: λ = 220 nm, for both enantiomers tR = 26.0 min, KD3 10d-ent1: purity: 96%, KD3en2 (10d-ent2): purity: 97%. chiral HPLC: isocratic elution with n-hexane / isopropanol, 98:2: KD3en1 (10d-ent1): tR = 14.3 min, >99% ee (254 nm); KD3en2 (10d-ent2): tR = 16.6 min, 99% ee (254 nm). [α]D25: KD3en1 (10d-ent1): −13.9 (c = 0.9, methanol); KD3en2 (10d-ent2): +14.6 (c = 0.1, methanol).
1-(3-Amino-6-fluoro-1H-indazol-1-yl)-4-phenylbutan-1-one (KD4, 10e):
To a solution of sodium 4-phenylbutyrate (42, 52.5 mg, 0.32 mmol) and HOAt (43.2 mg, 0.32 mmol) in DMF (1 mL) was added EDC × HCl (61.0 mg, 0.32 mmol). After stirring for 10 min at rt, a solution of 6-fluoro-1H-indazol-3-amine (13, 40.7 mg, 0.27 mmol) in DMF (0.5 mL) was added. After stirring the reaction mixture for 1.5 h, the solvent was removed by lyophilization and the residue was purified by preparative HPLC (Nucleodur C18 HTec, 32 mm × 250 mm, 5 μm) using a solvent system of CH3OH / 0.1% aq. HCOOH, a flow rate of 25 mL / min and a gradient of 50% to 80% CH3OH in 15 min, 80% to 95% CH3OH in 2 min, 95% CH3OH for 7 min (tR: 22.8 min, λ = 254 nm) to afford KD4 (10a, 33.9 mg, 0.11 mmol, 42%) as a white solid. ESI-MS: m/z 298.1 [M+H]+. HR-ESI-MS: m/z [M+H]+ calcd. 298.1350 for C18H17F3N3O2, found 298.1353. IR (KBr): 3429, 3341, 3233, 3197, 1674, 1658, 1644 cm−1. M.p.: 82 °C. 1H-NMR: (600 MHz, DMSO-d6) δ 1.99 (tt, J = 7.5, 7.5 Hz, 2H), 2.68 (t, J = 7.5 Hz, 2H), 2.95 (t, J = 7.5 Hz, 2H), 6.52 (s, 2H), 7.16 – 7.24 (m, 4H), 7.27 – 7.31 (m, 2H), 7.91 (dd, J = 8.7, 5.2 Hz, 1H), 7.93 (dd, J = 10.0, 2.5 Hz, 1H). 13C-NMR: (DEPTQ, 151 MHz, DMSO-d6) δ 25.5, 33.5, 34.4, 101.4 (d, J = 27 Hz), 111.8 (d, J = 24 Hz), 116.7, 122.5 (d, J = 11 Hz), 125.7, 128.2, 139.6 (d, J = 13 Hz), 141.5, 152.2, 158.2, 163.1 (d, J = 249 Hz), 171.4. HPLC: system 3: λ = 220 nm, tR = 25.1 min, purity: 97%.
Pharmacokinetic Study
Pharmacokinetic experiments of I1421 were performed by Sai Life Sciences (Hyderabad, India) at an AAALAC accredited facility in accordance with the Sai Study Protocol SAIDMPK/PK-22–12–1306 and PK-22–11–1117. International guidelines for animal experiments were followed. Plasma pharmacokinetics of compound I1421 was measured after a single 10 mg/kg dose, administered intraperitoneal (IP), per-orally (PO) or subcutaneously (SC). Plasma samples were also collected from mice after a single 3 mg/kg intravenous (IV) dose injection to determine oral bioavailability. Both doses were formulated in 10% NMP:5% Solutol HS-15: 85% saline (v/v/v). Testing was done in healthy male C57BL/6 mice (8–10 weeks old) weighing between 25 ± 5 g (procured from Global, India). Three mice were housed in each cage. Temperature and humidity were maintained at 22 ± 3 ºC and 30–70%, respectively and illumination was controlled to give a sequence of 12 h light and 12 h dark cycle. Temperature and humidity were recorded by an auto-controlled data logger system. All animals were provided laboratory rodent diet (Envigo Research private Ltd, Hyderabad). Reverse osmosis water treated with ultraviolet light was provided ad libitum.
For the 10 mg/kg (IP, PO and SC) study, a total of twenty-seven mice were used. Animals in Group 1 (n=9) were administered IP, animals in Group 2 (n=9) were administered PO and animals in Group 3 (n=9) were administered SC with solution formulation of I1421. For the 3 mg/kg (IV) study, animals in Group 1 (n=9) were administered IV with the same solution formulation. Blood samples (approximately 60 μL) were collected from the retro orbital plexus of a set of three mice at 0.083, 0.25, 0.5, 1, 2, 4, 6, 8 and 24 h. Immediately after blood collection, plasma was harvested by centrifugation at 10000 rpm, 10 min at 4 °C and samples were stored at −70±10 ºC until bioanalysis. All samples were processed for analysis by protein precipitation method and analyzed with fit-for-purpose LC-MS/MS method (LLOQ = 1.02 ng/mL for plasma). The pharmacokinetic parameters were estimated using non-compartmental analysis tool of Phoenix® WinNonlin software (Ver 8.3).
Sample Extraction Procedure:
10 μL of study sample plasma or spiked plasma calibration standard was added to individual pre-labeled micro-centrifuge tubes followed by 100 μL of internal standard prepared in Acetonitrile (Cetrizine, 50 ng/mL) except for a blank, where 100 μL of Acetonitrile was added. Samples were vortexed for 5 minutes. Samples were centrifuged for 10 minutes at a speed of 4000 rpm at 4 °C. Following centrifugation, 200 μL of clear supernatant was transferred into 96 well plates and analyzed using LC-MS/MS.
Data Analysis:
Peak plasma concentration and time for the peak plasma concentration were the observed values. The areas under the concentration time curve and were calculated by the linear trapezoidal rule. The terminal elimination rate constant, ke was determined by regression analysis of the linear terminal portion of the log plasma concentration-time curve. The terminal half-life was estimated at 0.693/ke. Mean, SD and %CV was calculated for each analyte. The bioavailability is calculated by , with as 1,211 h*ng/ml and as 610 h*ng/ml with 10 mg/kg and 3 mg/kg dosing respectively.
Protein expression and purification
CFTR E1317Q was expressed and purified as described previously24,73. In summary, bacmids carrying CFTR E1317Q construct were generated in E. Coli DH10Bac cells (Invitrogen). Recombinant baculoviruses were produced and amplified in Sf9 cells. Proteins were expressed in HEK293S GnTl− cells infected with 10% (v/v) baculovirus at a density of 3×106 cells/ml at 37 °C. Cells were induced with 10 mM sodium butyrate 12 hours after infection and cultured at 30 °C for another 48 hours before harvesting.
For protein purification, cell membranes were solubilized in buffer containing 1.25% (w/v) 2,2-didecylpropane-1,3-bis-β-D-maltopyranoside (LMNG) and 0.25% (w/v) cholesteryl hemisuccinate (CHS). Protein was purified via its C-terminal green fluorescence protein (GFP) tag using GFP nanobody-coupled Sepharose beads (GE Healthcare) and eluted by removing the GFP tag with PreScission Protease. The sample was phosphorylated using protein kinase A (NEB) and then further purified on size exclusion chromatography.
EM data acquisition
The phosphorylated E1371Q sample (5.5 mg/mL in 0.06% (w/v) digitonin) was incubated with 10 mM ATP and MgCl2 plus 200 𝜇M Z1834339853 on ice for 15 min. 3 mM fluorinated Fos-choline-8 was added to the sample immediately before freezing on to Quantifoil R1.2/1.3 400 mesh Au grids using a Vitrobot Mark IV (FEI). EM images were collected on a 300 kV Titian Krios (FEI) with a K2 Summit detector (Gatan) in super-resolution mode using SerialEM74. The defocus ranged from 1 to 2.5 μm and the dose rate was 8 e-/pixel/sec. The data were collected in two sessions, with 3,088 and 3,879 movies collected.
EM data processing
The images of the CFTR/Z1834339853 dataset were first corrected for gain reference and binned by 2 to obtain a physical pixel size of 1.03 Å. Beam-induced sample motion was corrected using MotionCor275. CTF estimation was performed using Gctf76. Particles were automatically picked by RELION 377,78.
Two datasets were collected. For the first dataset, a total number of 1,346,123 particles were extracted from 3,879 movies and were subjected to two rounds of 2D classification in RELION 3. 267,967 particles and 190,253 particles were selected and combined. Duplicated molecules were removed to yield 313,972 particles for 3D classification. The best classes from the last 4 iterations of 3D classification were combined; duplicated particles were removed to yield 254,123 particles. For further classification, the best map from 3D classification was low pass filtered to 8, 16, 24, 32, 40, 48 Å and used as reference maps. Local searches were performed at 7.5 and 3.75 degrees for 25 iterations respectively. Thorough searches were done by setting –maxsig 5. Such classification led to 6 classes, with the best class including 40% of the 254,123 particles and having a resolution of 7.92 Å. The best class (102,891 particles) was polished and refined to yield a reconstruction of 4.1 Å. Postprocessing did not improve the resolution further. In the second dataset, a total number of 801,032 particles from 3,088 movies were used for 2D classification, and subsequently 602,374 particles were used for 3D classification in RELION279. 3D classification yielded a best class with 200,815 particles. These particles were further classified with the same six reference models as the previous dataset. The best class contained 106,212 particles and were polished and refined to a 4.9 Å map. The polished particles from the two datasets were combined and imported to cryoSPARC80 for non-uniform refinement to yield a final map of 3.8 Å.
Model building and refinement
The model building and refinement of CFTR were carried out as described24. In summary, the data was randomly split into two halves, one half for model building and refinement (working set) and the other half for validation (free set). The model was built in Coot81 and refined in real space with Phenix82. MolProbity83,84 was used for geometry validation. The final structure contains residues 1–390 of transmembrane domain 1 (TMD1); 391–409, 437–637 of nucleotide binding domain (NBD1); 845–889, 900–1173 of transmembrane domain 2 (TMD2); 1202–1451 of nucleotide binding domain 2 (NBD2); 17 residues of R domain (built as alanines), two ATP and Mg2+ and Z1834339853. Figures were generated with PyMOL and Chimera85.
Inside-out patch clamp recording
All CFTR constructs used for electrophysiology were cloned into the BacMam expression vector. A GFP tag was fused to the C-terminus of CFTR for visualization of transfected cells. Chinese hamster ovary (CHO) cells were plated at 0.4×106 cells per 35-mm dish (Falcon) 12 hours before the transfection. For each dish, cells were transfected with 1 μg BacMam plasmids using Lipofectamine 3000 (Invitrogen) in Opti-MEM media (Gibco). After 12 hours of transfection, media was exchanged to DMEM: F12 (ATCC) supplemented with 2% (v/v) FBS and the cells were incubated at 30 °C for 2 days before recording.
Macroscopic currents were recorded in inside-out membrane patches excised from CHO cells, using buffer compositions, and recording parameters as described37. The culture media were first changed to a bath solution consisting of 145 mM NaCl, 2 mM MgCl2, 5 mM KCl, 1 mM CaCl2, 5 mM glucose, 5 mM HEPES, and 20 mM sucrose, pH 7.4 with NaOH. The pipette solution contained 140 mM N-Methyl-D-glucamine (NMDG), 5 mM CaCl2, 2 mM MgCl2, and 10 mM HEPES (pH 7.4 with HCl). The perfusion solution contained 150 mM NMDG, 2 mM MgCl2, 1 mM CaCl2, 10 mM EGTA, and 8 mM Tris (pH 7.4 with HCl). Borosilicate micropipettes (OD 1.5 mm, ID 0.86 mm, Sutter) were pulled to 2–5 MΩ resistance. After a gigaseal was formed, inside-out patches were excised and exposed to 25 units/ml bovine heart PKA (Sigma-Aldrich, P2645) and 3 mM ATP to phosphorylate CFTR. Currents were recorded at −30 mV and 25 °C using an Axopatch 200B amplifier, a Digidata 1550 digitizer and pCLAMP software (Molecular Devices). The recordings were low-pass filtered at 1 kHz and sampled at 20 kHz. All displayed recordings were further low-pass filtered at 100 Hz. Data were analyzed with Clampfit, GraphPad Prism, and OriginPro. The effect of the compounds were reported as normalized current which was defined as the current level after application of the tested compound divided by the current level before application of the compound. In the reported recordings, membrane disruptions were not observed as the stimulated current was reversed upon removal of ATP.
YFP fluorescent assay
The experiments were performed as described in the literature52. In brief, 50,000 cells (not cultured beyond passage 10) were seeded per well in 96-well plates (GBO 655866) 48 hours before the start of assay and incubated at 37 °C. 24 hours before the start of the assay, the CFTR corrector lumacaftor was added at 1 μM in a final culture volume of 200 μL. The plates were incubated at 30 °C for 24 hours. On the day of experiments, the cells were first washed three times with 200 μL PBS (HyClone SH30264.02), and then incubated with 20 μM forskolin and 1 μM test compound for 25 minutes in a 60 μL total volume. The plate was then transferred to a microplate reader for a 14 second fluorescence reading, split into a 2 second baseline reading, rapid injection of 165 μL iodide containing solution (PBS with 137 mM Cl− replaced by I−), and then a 12-second reading. For the experiments in Figure 4E the method was adapted to 384-well plates (GBO 781091) and fluorescence was imaged with a FDSS/μCell plate imager (Hamamatsu). Data were normalized to initial background-subtracted fluorescence. I− influx rate was determined by fitting the final 11 seconds of data for each well with an exponential function to extrapolate the initial rate of fluorescence quenching (dF/dt). Relative potentiation is defined as the initial rate of fluorescence decay with test compound divided by the initial rate of fluorescence decay without test compound.
Quantification and Statistical Analysis
GraphPad Prism 9 was used to fit the dose response curves of CFTR potentiators and inhibitors and to calculate EC50 values. Statistical significance was calculated by two-tailed Student’s t-test or by one-way analysis of variance (ANOVA) in Prism 9.
Supplementary Material
Table S1. Potentiating activities of the 13 CFTR primary hits, related to Figure 1.
Table S2. Summary of EM data and structure refinement statistics for CFTR in complex with ‘853, related to Figure 3 and Figure S2.
Cells were treated with 20 μM forskolin +/− 1 μM test compound. The relative potentiation was calculated as the ratio of flux rates with and without compound. Data points represent a single measurement for each test compound. For ivacaftor and GLPG1837 data represent means and SEs for 9 or 2 measurements, respectively.
Summary of the image processing procedure.
Figure S3. SAR for optimization of compound ‘853, related to Figure 4.
HIGHLIGHTS.
Large-scale molecular docking of the CFTR potentiator-binding site.
Identified a promising potentiator with a high oral bioavailability of 60%.
Identified allosteric inhibitors that bind to the same site as potentiators.
Acknowledgments
We thank M. Ebrahim and J. Sotiris at Rockefeller’s Evelyn Gruss Lipper Cryo-Electron Microscopy Resource Center for assistance in data collection, Yiming Niu and Chen Zhao for help with cryo-EM data processing, and Iris Torres and Bilge Bebek for technical assistance. We thank Luis J. Galietta for sharing the CFBE41o− cells expressing F508del-CFTR and the fluorescent protein EYFP-H148Q/I152L/F46L.
Funding:
This work is supported by the Howard Hughes Medical Institute (to J.C.), R35GM122481 (to B.K.S.), GM71896 (to J.J.I.) and the Deutsche Forschungsgemeinschaft (to P.G.).
Footnotes
Declaration of interests:
B.K.S. and P.G. are founders of Epiodyne. B.K.S. is a co-founder of BlueDolphin and of Deep Apple Therapeutics, as is J.J.I., and serves on the SRB of Genentech and on the SABs of Vilya Therapeutics and Umbra Therapeutics and consults for Great Point Ventures and for Levator Therapeutics. A patent on the discovery of positive and negative allosteric regulators for CFTR has been filed. The authors declare no other competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cutting GR. (2015). Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet 16, 45–56. 10.1038/nrg3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jaques R, Shakeel A, and Hoyle C. (2020). Novel therapeutic approaches for the management of cystic fibrosis. Multidiscip Respir Med 15, 690. 10.4081/mrm.2020.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dransfield MT, Wilhelm AM, Flanagan B, Courville C, Tidwell SL, Raju SV, Gaggar A, Steele C, Tang LP, Liu B, and Rowe SM. (2013). Acquired cystic fibrosis transmembrane conductance regulator dysfunction in the lower airways in COPD. Chest 144, 498–506. 10.1378/chest.13-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Solomon GM, Fu L, Rowe SM, and Collawn JF. (2017). The therapeutic potential of CFTR modulators for COPD and other airway diseases. Curr Opin Pharmacol 34, 132–139. 10.1016/j.coph.2017.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thiagarajah JR, and Verkman AS. (2012). CFTR inhibitors for treating diarrheal disease. Clin Pharmacol Ther 92, 287–290. 10.1038/clpt.2012.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belibi FA, Reif G, Wallace DP, Yamaguchi T, Olsen L, Li H, Helmkamp GM Jr., and Grantham JJ. (2004). Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int 66, 964–973. 10.1111/j.15231755.2004.00843.x. [DOI] [PubMed] [Google Scholar]
- 7.Davidow CJ, Maser RL, Rome LA, Calvet JP, and Grantham JJ. (1996). The cystic fibrosis transmembrane conductance regulator mediates transepithelial fluid secretion by human autosomal dominant polycystic kidney disease epithelium in vitro. Kidney Int 50, 208–218. 10.1038/ki.1996.304. [DOI] [PubMed] [Google Scholar]
- 8.Hadida S, Van Goor F, Zhou J, Arumugam V, McCartney J, Hazlewood A, Decker C, Negulescu P, and Grootenhuis PD. (2014). Discovery of N-(2,4-di-tert-butyl-5-hydroxyphenyl)-4-oxo-1,4-dihydroquinoline-3-carboxamide (VX-770, ivacaftor), a potent and orally bioavailable CFTR potentiator. J Med Chem 57, 9776–9795. 10.1021/jm5012808. [DOI] [PubMed] [Google Scholar]
- 9.Middleton PG, Mall MA, Drevinek P, Lands LC, McKone EF, Polineni D, Ramsey BW, Taylor-Cousar JL, Tullis E, Vermeulen F, et al. (2019). Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med 381, 1809–1819. 10.1056/NEJMoa1908639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, Griese M, McKone EF, Wainwright CE, Konstan MW, et al. (2011). A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 365, 1663–1672. 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balfour-Lynn IM, and King JA. (2022). CFTR modulator therapies - Effect on life expectancy in people with cystic fibrosis. Paediatr Respir Rev 42, 3–8. 10.1016/j.prrv.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chin S, Hung M, Won A, Wu YS, Ahmadi S, Yang D, Elmallah S, Toutah K, Hamilton CM, Young RN, et al. (2018). Lipophilicity of the Cystic Fibrosis Drug, Ivacaftor (VX-770), and Its Destabilizing Effect on the Major CF-causing Mutation: F508del. Mol Pharmacol 94, 917–925. 10.1124/mol.118.112177. [DOI] [PubMed] [Google Scholar]
- 13.McColley SA. (2016). A safety evaluation of ivacaftor for the treatment of cystic fibrosis. Expert Opin Drug Saf 15, 709–715. 10.1517/14740338.2016.1165666. [DOI] [PubMed] [Google Scholar]
- 14.Wainwright CE. (2014). Ivacaftor for patients with cystic fibrosis. Expert Rev Respir Med 8, 533–538. 10.1586/17476348.2014.951333. [DOI] [PubMed] [Google Scholar]
- 15.Talamo Guevara M, and McColley SA. (2017). The safety of lumacaftor and ivacaftor for the treatment of cystic fibrosis. Expert Opin Drug Saf 16, 1305–1311. 10.1080/14740338.2017.1372419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kinting S, Li Y, Forstner M, Delhommel F, Sattler M, and Griese M. (2019). Potentiation of ABCA3 lipid transport function by ivacaftor and genistein. J Cell Mol Med 23, 5225–5234. 10.1111/jcmm.14397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delaunay JL, Bruneau A, Hoffmann B, Durand-Schneider AM, Barbu V, Jacquemin E, Maurice M, Housset C, Callebaut I, and Ait-Slimane T. (2017). Functional defect of variants in the adenosine triphosphate-binding sites of ABCB4 and their rescue by the cystic fibrosis transmembrane conductance regulator potentiator, ivacaftor (VX-770). Hepatology 65, 560–570. 10.1002/hep.28929. [DOI] [PubMed] [Google Scholar]
- 18.Mareux E, Lapalus M, Amzal R, Almes M, Ait-Slimane T, Delaunay JL, Adnot P, Collado-Hilly M, Davit-Spraul A, Falguieres T, et al. (2020). Functional rescue of an ABCB11 mutant by ivacaftor: A new targeted pharmacotherapy approach in bile salt export pump deficiency. Liver Int 40, 1917–1925. 10.1111/liv.14518. [DOI] [PubMed] [Google Scholar]
- 19.Yu H, Burton B, Huang CJ, Worley J, Cao D, Johnson JP Jr., Urrutia A, Joubran J, Seepersaud S, Sussky K, et al. (2012). Ivacaftor potentiation of multiple CFTR channels with gating mutations. J Cyst Fibros 11, 237–245. 10.1016/j.jcf.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 20.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, et al. (2009). Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A 106, 18825–18830. 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu F, Zhang Z, Levit A, Levring J, Touhara KK, Shoichet BK, and Chen J. (2019). Structural identification of a hotspot on CFTR for potentiation. Science 364, 1184–1188. 10.1126/science.aaw7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiedorczuk K, and Chen J. (2022). Molecular structures reveal synergistic rescue of Delta508 CFTR by Trikafta modulators. Science 378, 284–290. 10.1126/science.ade2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang C, Yang Z, Loughlin BJ, Xu H, Veit G, Vorobiev S, Clarke OB, Jiang F, Li Y, Singh S, et al. (2022). Mechanism of dual pharmacological correction and potentiation of human CFTR. bioRxiv, 2022.2010.2010.510913. 10.1101/2022.10.10.510913. [DOI]
- 24.Zhang Z, and Chen J. (2016). Atomic Structure of the Cystic Fibrosis Transmembrane Conductance Regulator. Cell 167, 1586–1597 e1589. 10.1016/j.cell.2016.11.014. [DOI] [PubMed] [Google Scholar]
- 25.Lyu J, Wang S, Balius TE, Singh I, Levit A, Moroz YS, O’Meara MJ, Che T, Algaa E, Tolmachova K, et al. (2019). Ultra-large library docking for discovering new chemotypes. Nature 566, 224–229. 10.1038/s41586-019-0917-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stein RM, Kang HJ, McCorvy JD, Glatfelter GC, Jones AJ, Che T, Slocum S, Huang XP, Savych O, Moroz YS, et al. (2020). Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature 579, 609–614. 10.1038/s41586-020-2027-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gorgulla C, Boeszoermenyi A, Wang ZF, Fischer PD, Coote PW, Padmanabha Das KM, Malets YS, Radchenko DS, Moroz YS, Scott DA, et al. (2020). An open-source drug discovery platform enables ultra-large virtual screens. Nature 580, 663–668. 10.1038/s41586-020-2117-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alon A, Lyu J, Braz JM, Tummino TA, Craik V, O’Meara MJ, Webb CM, Radchenko DS, Moroz YS, Huang XP, et al. (2021). Structures of the sigma(2) receptor enable docking for bioactive ligand discovery. Nature 600, 759–764. 10.1038/s41586-021-04175-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fink EA, Xu J, Hubner H, Braz JM, Seemann P, Avet C, Craik V, Weikert D, Schmidt MF, Webb CM, et al. (2022). Structure-based discovery of nonopioid analgesics acting through the alpha(2A)-adrenergic receptor. Science 377, eabn7065. 10.1126/science.abn7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sadybekov AA, Sadybekov AV, Liu Y, Iliopoulos-Tsoutsouvas C, Huang XP, Pickett J, Houser B, Patel N, Tran NK, Tong F, et al. (2022). Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature 601, 452–459. 10.1038/s41586-021-04220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaplan AL, Confair DN, Kim K, Barros-Alvarez X, Rodriguiz RM, Yang Y, Kweon OS, Che T, McCorvy JD, Kamber DN, et al. (2022). Bespoke library docking for 5-HT(2A) receptor agonists with antidepressant activity. Nature 610, 582–591. 10.1038/s41586-022-05258-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, and Coleman RG. (2012). ZINC: a free tool to discover chemistry for biology. J Chem Inf Model 52, 1757–1768. 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sterling T, and Irwin JJ. (2015). ZINC 15--Ligand Discovery for Everyone. J Chem Inf Model 55, 2324–2337. 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coleman RG, Carchia M, Sterling T, Irwin JJ, and Shoichet BK. (2013). Ligand pose and orientational sampling in molecular docking. PLoS One 8, e75992. 10.1371/journal.pone.0075992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Csanady L, and Torocsik B. (2019). Cystic fibrosis drug ivacaftor stimulates CFTR channels at picomolar concentrations. Elife 8. 10.7554/eLife.46450. [DOI] [PMC free article] [PubMed]
- 36.Van der Plas SE, Kelgtermans H, De Munck T, Martina SLX, Dropsit S, Quinton E, De Blieck A, Joannesse C, Tomaskovic L, Jans M, et al. (2018). Discovery of N-(3-Carbamoyl-5,5,7,7-tetramethyl-5,7-dihydro-4H-thieno[2,3-c]pyran-2-yl)-lH-pyrazole-5-carboxamide (GLPG1837), a Novel Potentiator Which Can Open Class III Mutant Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Channels to a High Extent. J Med Chem 61, 1425–1435. 10.1021/acs.jmedchem.7b01288. [DOI] [PubMed] [Google Scholar]
- 37.Yeh HI, Sohma Y, Conrath K, and Hwang TC. (2017). A common mechanism for CFTR potentiators. J Gen Physiol 149, 1105–1118. 10.1085/jgp.201711886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galietta LV, Jayaraman S, and Verkman AS. (2001). Cell-based assay for high-throughput quantitative screening of CFTR chloride transport agonists. Am J Physiol Cell 2315 Physiol 281, C1734–1742. 10.1152/ajpcell.2001.281.5.C1734. [DOI] [PubMed] [Google Scholar]
- 39.Yeh HI, Qiu L, Sohma Y, Conrath K, Zou X, and Hwang TC. (2019). Identifying the molecular target sites for CFTR potentiators GLPG1837 and VX-770. J Gen Physiol 151, 912–928. 10.1085/jgp.201912360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shaughnessy CA, Zeitlin PL, and Bratcher PE. (2021). Author Correction: Elexacaftor is a CFTR potentiator and acts synergistically with ivacaftor during acute and chronic treatment. Sci Rep 11, 21295. 10.1038/s41598-021-00539-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Veit G, Vaccarin C, and Lukacs GL. (2021). Elexacaftor co-potentiates the activity of F508del and gating mutants of CFTR. J Cyst Fibros 20, 895–898. 10.1016/j.jcf.2021.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O’Riordan CR, and Smith AE. (1990). Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63, 827–834. [DOI] [PubMed] [Google Scholar]
- 43.Lukacs GL, Chang XB, Bear C, Kartner N, Mohamed A, Riordan JR, and Grinstein S. (1993). The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem 268, 21592–21598. [PubMed] [Google Scholar]
- 44.Dalemans W, Barbry P, Champigny G, Jallat S, Dott K, Dreyer D, Crystal RG, Pavirani A, Lecocq JP, and Lazdunski M. (1991). Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature 354, 526–528. 2335 10.1038/354526a0. [DOI] [PubMed] [Google Scholar]
- 45.https://www.cff.org/sites/default/files/2021-11/Patient-Registry-Annual-Data-Report.pdf (2021). Cystic Fibrosis Foundation Patient Registry Annual Data Report 2021. https://www.cff.org/sites/default/files/2021-11/Patient-Registry-Annual-Data-Report.pdf.
- 46.Fink C, Sun D, Wagner K, Schneider M, Bauer H, Dolgos H, Mader K, and Peters SA. (2020). Evaluating the Role of Solubility in Oral Absorption of Poorly Water-Soluble Drugs Using Physiologically-Based Pharmacokinetic Modeling. Clin Pharmacol Ther 107, 650–661. 10.1002/cpt.1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di L, Fish PV, and Mano T. (2012). Bridging solubility between drug discovery and development. Drug Discov Today 17, 486–495. 10.1016/j.drudis.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 48.Bender BJ, Gahbauer S, Luttens A, Lyu J, Webb CM, Stein RM, Fink EA, Balius TE, Carlsson J, Irwin JJ, and Shoichet BK. (2021). A practical guide to large-scale docking. Nat Protoc 16, 4799–4832. 10.1038/s41596-021-00597-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muanprasat C, Sonawane ND, Salinas D, Taddei A, Galietta LJ, and Verkman AS. (2004). Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J Gen Physiol 124, 125–137. 10.1085/jgp.200409059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Young PG, Levring J, Fiedorczuk K, Blanchard SC, and Chen J. (2024). Structural basis for CFTR inhibition by CFTR(inh)-172. Proc Natl Acad Sci U S A 121, e2316675121. 10.1073/pnas.2316675121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McCarron A, Parsons D, and Donnelley M. (2021). Animal and Cell Culture Models for Cystic Fibrosis: Which Model Is Right for Your Application? Am J Pathol 191, 228–242. 10.1016/j.ajpath.2020.10.017. [DOI] [PubMed] [Google Scholar]
- 52.Sondo E, Tomati V, Caci E, Esposito AI, Pfeffer U, Pedemonte N, and Galietta LJ. (2011). Rescue of the mutant CFTR chloride channel by pharmacological correctors and low temperature analyzed by gene expression profiling. Am J Physiol Cell Physiol 301, C872–885. 10.1152/ajpcell.00507.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gahbauer S, DeLeon C, Braz JM, Craik V, Kang HJ, Wan X, Huang XP, Billesbolle CB, Liu Y, Che T, et al. (2023). Docking for EP4R antagonists active against inflammatory pain. Nat Commun 14, 8067. 10.1038/s41467-023-43506-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mysinger MM, and Shoichet BK. (2010). Rapid context-dependent ligand desolvation in molecular docking. J Chem Inf Model 50, 1561–1573. 10.1021/ci100214a. [DOI] [PubMed] [Google Scholar]
- 55.Wei BQ, Baase WA, Weaver LH, Matthews BW, and Shoichet BK. (2002). A model binding site for testing scoring functions in molecular docking. J Mol Biol 322, 339–355. 10.1016/s0022-2836(02)00777-5. [DOI] [PubMed] [Google Scholar]
- 56.Word JM, Lovell SC, Richardson JS, and Richardson DC. (1999). Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol 285, 1735–1747. 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- 57.Case DA, Berryman JT, Betz RM, Cerutti DS, Cheatham I, T.E., Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, et al. (2015). AMBER 2015. [Google Scholar]
- 58.Gallagher K, and Sharp K. (1998). Electrostatic contributions to heat capacity changes of DNA-ligand binding. Biophys J 75, 769–776. 10.1016/S0006-3495(98)77566-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sharp KA. (1995). Polyelectrolyte electrostatics: Salt dependence, entropic, and enthalpic contributions to free energy in the nonlinear Poisson–Boltzmann model. Biopolymers 36, 227–243. 10.1002/bip.360360210. [DOI] [Google Scholar]
- 60.Rogers D, and Hahn M. (2010). Extended-connectivity fingerprints. J Chem Inf Model 50, 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- 61.Bogolubsky AV, Ryabukhin SV, Pipko SE, Lukin O, Shivanyuk A, Mykytenko D, and Tolmachev A. (2011). A facile synthesis of unsymmetrical ureas. Tetrahedron 67, 3619–3623. 10.1016/j.tet.2011.03.101. [DOI] [Google Scholar]
- 62.Tolmachev A, Bogolubsky AV, Pipko SE, Grishchenko AV, Ushakov DV, Zhemera AV, Viniychuk OO, Konovets AI, Zaporozhets OA, Mykhailiuk PK, and Moroz YS. (2016). Expanding Synthesizable Space of Disubstituted 1,2,4-Oxadiazoles. ACS Comb Sci 18, 616–624. 10.1021/acscombsci.6b00103. [DOI] [PubMed] [Google Scholar]
- 63.Bogolubsky AV, Moroz YS, Mykhailiuk PK, Ostapchuk EN, Rudnichenko AV, Dmytriv YV, Bondar AN, Zaporozhets OA, Pipko SE, Doroschuk RA, et al. (2015). One-Pot Parallel Synthesis of Alkyl Sulfides, Sulfoxides, and Sulfones. ACS Comb Sci 17, 348–354. 10.1021/acscombsci.5b00024. [DOI] [PubMed] [Google Scholar]
- 64.Franchini C, Carocci A, Catalano A, Cavalluzzi MM, Corbo F, Lentini G, Scilimati A, Tortorella P, Camerino DC, and Luca AD. (2003). Optically Active Mexiletine Analogues as Stereoselective Blockers of Voltage-Gated Na+ Channels. Journal of Medicinal Chemistry 46, 5238–5248. 10.1021/jm030865y. [DOI] [PubMed] [Google Scholar]
- 65.Feenstra RW, Visser GM, Kruse CG, Tulp MTM, and Long SK. (1999) Preparation of 5-piperazinotetrahydroquinolines and analogs as 5-HT1 receptor agonists. patent EP900792, patent application Copyright © 2023 American Chemical Society (ACS). All Rights Reserved.
- 66.Colonge J, and Guyot A. (1957). 4-Chromanones. I. 3-Aryloxyalkanoic acids. Bull. Soc. Chim. Fr, 1228.
- 67.Fesik SW, Summers JB Jr., Davidsen SK, Sheppard GS, Steinman DH, Carrera GM Jr., Florjancic A, and Holms JH. (1997) Preparation of biphenyl hydroxamic acids as inhibitors of matrix metalloproteinases. patent WO9718188, patent application Copyright © 2023 American Chemical Society (ACS). All Rights Reserved.
- 68.Hassfeld J, Kinzel T, Koebberling J, Cancho Grande Y, Beyer K, Roehrig S, Koellnberger M, Sperzel M, Burkhardt N, Schlemmer K-H, et al. (2015) Preparation of (aza)pyridopyrazolopyrimidinones and indazolopyrimidinones and their use for treating or preventing diseases. patent US20150126449, patent application Copyright © 2023 American Chemical Society (ACS). All Rights Reserved.
- 69.Sączewski F, Kornicka A, Hudson AL, Laird S, Scheinin M, Laurila JM, Rybczyńska A, Boblewski K, Lehmann A, and Gdaniec M. (2011). 3-[(Imidazolidin-2-yl)imino]indazole ligands with selectivity for the α2-adrenoceptor compared to the imidazoline I1 receptor. Bioorganic & Medicinal Chemistry 19, 321–329. 10.1016/j.bmc.2010.11.020. [DOI] [PubMed] [Google Scholar]
- 70.De La Rosa MA, Johns BA, Velthuisen EJ, Weatherhead J, and Samano V. (2013) Preparation of azaindole compounds and methods for treating HIV. patent US20130018049, patent application Copyright © 2023 American Chemical Society (ACS). All Rights Reserved.
- 71.Lau VM, Pfalzgraff WC, Markland TE, and Kanan MW. (2017). Electrostatic Control of Regioselectivity in Au(I)-Catalyzed Hydroarylation. Journal of the American Chemical Society 139, 4035–4041. 10.1021/jacs.6b11971. [DOI] [PubMed] [Google Scholar]
- 72.Reeves JT, Malapit CA, Buono FG, Sidhu KP, Marsini MA, Sader CA, Fandrick KR, Busacca CA, and Senanayake CH. (2015). Transnitrilation from Dimethylmalononitrile to Aryl Grignard and Lithium Reagents: A Practical Method for Aryl Nitrile Synthesis. Journal of the American Chemical Society 137, 9481–9488. 10.1021/jacs.5b06136. [DOI] [PubMed] [Google Scholar]
- 73.Goehring A, Lee CH, Wang KH, Michel JC, Claxton DP, Baconguis I, Althoff T, Fischer S, Garcia KC, and Gouaux E. (2014). Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat Protoc 9, 2574–2585. 10.1038/nprot.2014.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mastronarde DN. (2005). Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51. 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 75.Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA. (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332. 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang K. (2016). Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1–12. 10.1016/j.jsb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Scheres SH. (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH. (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7. 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kimanius D, Forsberg BO, Scheres SH, and Lindahl E. (2016). Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife 5. 10.7554/eLife.18722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA. (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296. 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 81.Emsley P, Lohkamp B, Scott WG, and Cowtan K. (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC. (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB 3rd, Snoeyink J, Richardson JS, and Richardson DC. (2007). MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35, W375–383. 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE. (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Potentiating activities of the 13 CFTR primary hits, related to Figure 1.
Table S2. Summary of EM data and structure refinement statistics for CFTR in complex with ‘853, related to Figure 3 and Figure S2.
Cells were treated with 20 μM forskolin +/− 1 μM test compound. The relative potentiation was calculated as the ratio of flux rates with and without compound. Data points represent a single measurement for each test compound. For ivacaftor and GLPG1837 data represent means and SEs for 9 or 2 measurements, respectively.
Summary of the image processing procedure.
Figure S3. SAR for optimization of compound ‘853, related to Figure 4.
Data Availability Statement
The 3D cryo-EM density map of the CFTR-’853 complex generated in this study has been deposited at the Electron Microscopy Data Bank (EMDB), and the coordinates of the CFTR-’853 complex have been deposited at the Protein Data Bank (PDB) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. The identities of the compounds docked in this study are publicly accessible in existing, publicly available databases, which are listed in the key resources table. The docking software, DOCK3.7, is freely available for non-commercial research and is also listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| 1-Ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) | TCI | Cat# D4029 |
| Carbonyldiimidazole | Sigma-Aldrich | Cat# 115533 |
| 3-(1H-Indazol-1-yl)-2-methylpropanoic acid | Enamine | Cat# EN300–206691 |
| 6-Fluoro-1H-indazol-3-amine | Enamine | Cat# EN300–100294 |
| 3-(3-Isopropyl-1,2,4-oxadiazol-5-yl)propanoic acid | Enamine | Cat# EN300–27664 |
| 4-(1-Methyl-1H-pyrazol-4-yl)pyrimidin-2-amine | Enamine | Cat# EN300–187048 |
| 1-(2,6-Difluorophenyl)-1H-pyrazole-3-carboxylic acid | Enamine | Cat# EN300–91144 |
| 3-(Methylamino)benzonitrile | Enamine | Cat# EN300–55410 |
| (R,S)-7-Methyl-5,6,7,8-tetrahydroimidazo[1,2-a]pyridine-2-carboxylic acid | Enamine | Cat# EN300–309016 |
| (R,S)-tert-Butyl (5-amino-5,6,7,8-tetrahydronaphthalen-2-yl)carbamate | Enamine | Cat# EN300–142125 |
| 3-Methoxy-2-naphthoic acid | Enamine | Cat# EN300–00001 |
| 3-(3-Amino-1H-pyrazol-1-yl)propanamide | Enamine | Cat# EN300–80390 |
| Salicylic acid | Enamine | Cat# EN300–16723 |
| 2-(Chloromethyl)-5-(4-chlorophenyl)-1,3,4-oxadiazole | Enamine | Cat# EN300–08138 |
| 5-Amino-4-isopropyl-4H-1,2,4-triazole-3-thiol | Enamine | Cat# EN300–2493721 |
| 6-Chloro-2-(chloromethyl)imidazo[1,2-a]pyridine | Enamine | Cat# EN300–33589 |
| 5-(Benzo[d]thiazol-2-yl)thiophene-2-carboxylic acid | Enamine | Cat# EN300–00707 |
| (4H-1,2,4-Triazol-3-yl)methanamine | Enamine | Cat# EN300–1264530 |
| 2-(Imidazo[1,2-a]pyridin-2-yl)acetonitrile | Enamine | Cat# EN300–25598 |
| 2-Hydroxy-2,3-dihydro-1H-indene-2-carboxylic acid | Enamine | Cat# EN300–82840 |
| 1-(4-Fluorophenyl)-1H-pyrazole-3-carboxylic acid | Enamine | Cat# EN300–36826 |
| N-Methyl-1-(2-methylthiazol-4-yl)methanamine | Enamine | Cat# EN300–27495 |
| 2-Chloro-N-(3-fluoro-4-methylphenyl)propanamide | Enamine | Cat# EN300–23356 |
| N4,N4-Dimethylpyrimidine-4,6-diamine | Enamine | Cat# EN300–187848 |
| N-(3,4-Dimethylbenzyl)cyclopropanamine | Enamine | Cat# EN300–83837 |
| 1-(m-Tolyl)-1H-1,2,3-triazole-4-carboxylic acid | Enamine | Cat# EN300–61349 |
| (2-Chloro-6-fluorophenyl)methanamine | Enamine | Cat# EN300–33074 |
| 2,2,2-trifluoroethyl carbonochloridate | Enamine | Cat# EN300–31105 |
| Phenol | Alfa Aesar | Cat# A15760 |
| Sodium hydride | Sigma Aldrich | Cat# H36490 |
| (R)-3-Bromo-2-methylpropan-1-ol | Manchester Organics | Cat# H39016 |
| (S)-3-Bromo-2-methylpropan-1-ol | Manchester Organics | Cat# R39644 |
| 2-Naphthol | Sigma Aldrich | Cat# 130109 |
| 3-(Trifluormethyl)phenol | SIAL | Cat# 156035 |
| 2,4-Di-tert-butylphenol | SIAL | Cat# 137731 |
| 4-Fluorophenol | Fisher Scientific | Cat# 10723671 |
| 4-Bromophenol | Acros Organics | Cat# 406511000 |
| [1,1’-biphenyl]-4-ol | SIAL | Cat# 371297 |
| Benzofuran-5-ol | BLD Pharmatech | Cat# BD102433 |
| 1-Hydroxy-7-azabenzotriazole (HOAt) | Sigma Aldrich | Cat# 41996 |
| N-Ethyldiisopropylamine (DIPEA) | Acros Organics | Cat# 367841000 |
| Isobutyryl chloride | Sigma Aldrich | Cat# 139122 |
| 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid-hydrochlorid (EDC x HCl) | Sigma Aldrich | Cat# 03450 |
| (R,S)-2-methyl-3-phenoxypropionic acid | Enamine | Cat# EN300–42126 |
| 6-Fluoro-1H-indazole | Sigma Aldrich | Cat# APOH11A9C83A |
| 6-Fluoro-3-methyl-1H-indazole | Sigma Aldrich | Cat# C12H580D8864 |
| 4-Dimethylaminopyridine | Acros Organics | Cat# 148270050 |
| Acetyl chloride | Sigma Aldrich | Cat# 114189 |
| Iodomethan (methyl iodide) | Acros Organics | Cat# 122371000 |
| 3,5-Difluoro-2-pyridinecarbonitrile | TCI | Cat# C2753 |
| Hydrazine hydrate | Acros Organics | Cat# 196711000 |
| Benzotriazol-1-yl-oxytripyrrolidinophosphonium-hexafluorophosphat (PyBOP) | Merck | Cat# 8510090025 |
| 6-Methoxy-1H-indazol-3-amine | abcr | Cat# AB490142 |
| Isopropoxy-1H-indazol-3-amine | A2B Chem | Cat# BE29241 |
| 6-(Trifluoromethyl)-1H-indazol-3-amine | abcr | Cat# AB146621 |
| 6-Chloro-1H-indazol-3-amine | BLD Pharmatech | Cat# BD167849 |
| 1H-Indazole-3,6-diamine | VWR International | Cat# 2C36909 |
| 3-Amino-1H-indazole-6-carboxylic acid | Sigma Aldrich | Cat# AMBH303C4559 |
| 2-Fluoro-4-(hydroxymethyl)benzonitrile | Activate Scientific | Cat# AS38375 |
| 1H-Indazole-6-carboxamide | abcr | Cat# AB527998 |
| 6-Amino-1H-indazole | abcr | Cat# AB136281 |
| 1-Fluoro-3-(prop-2-yn-1-yloxy)benzene (32) | Enamine | Cat# EN300–71767 |
| (Acetonitrile)[2-biphenyl)di-tert-butylphosphine]gold(I) hexafluoroantimonate | Sigma Aldrich | Cat# 697575 |
| Palladium hydroxide 20% on charcoal | Sigma Aldrich | Cat# 21,291–1 |
| N-Bromosuccinimide | Acros Organics | Cat# 107451000 |
| 2.5 M Solution of butyllithium in hexane | Sigma Aldrich | Cat# 230707 |
| 2,2-Dimethylmalononitrile | Tokio Chemical Industry | Cat# D5514 |
| 3-Phenoxypropionic acid | Sigma Aldrich | Cat# P16001 |
| (R,S)-2-(Phenoxymethyl)butanoic acid | Enamine | Cat# EN300–27146916 |
| (R,S)-2-Methoxy-3-phenoxypropanoic acid | CHEM Space | Cat# CSC039927227 |
| (R,S)-2-Methyl-4-phenylbutyric acid | Enamine | Cat# EN300–6496378 |
| Sodium 4-phenylbutyrate | abcr | Cat# AB222448 |
| 2,2-didecylpropane-1,3-bis-β-D-maltopyranoside (LMNG) | Anatrace | NG310 |
| Cholesteryl hemisuccinate (CHS) | Anatrace | CH210 |
| Digitonin | Sigma-Aldrich | D141 |
| sf-900 II SFM medium | Gibco | Cat#10902–088 |
| Cellfectin II reagents | Invitrogen | Cat#10362–100 |
| Freestyle 293 medium | Gibco | Cat#12338–018 |
| PKA | NEB | Cat#P6000L |
| ATP | Sigma-Aldrich | A2383 |
| Critical commercial assays | ||
| CNBR-activated sepharose beads | GE Healthcare | 17–0430–01 |
| Superose 6, 10/300 GL | GE Healthcare | 17–5172–01 |
| Deposited data | ||
| Coordinates of the human CFTR in complex with ligand Z1834339853 | This paper | PDB: 8GLS |
| Cryo-EM map of the human CFTR in complex with ligand Z1834339853 | This paper | EMDB: EMD-40207 |
| Experimental models: Cell lines | ||
| Sf9 | ATCC | CRL-1711 |
| HEK293S GnTI− | ATCC | CRL-3022 |
| CHO-K1 | ATCC | CRL-9618 |
| CFBE41o- cells expressing F508del-CFTR and the fluorescent protein EYFP-H148Q/I152L/F46L | Sondo et al.52 | Gift by Luis J. Galietta |
| Experimental models: Organisms/strains | ||
| C57BL/6 mice | Hylasco Bio | C57BL/6Ncrl Mice |
| Recombinant DNA | ||
| Human CFTR cloned onto a modified pEG Bacmam vector suitable for expression in mammalian cells | This paper | N/A |
| Software and algorithms | ||
| Bruker TopSpin Version 4.2.0 | Bruker | https://www.bruker.com/en/products-and-solutions/mr/nmr-software/topspin.html |
| ChemDraw Version 21.0.0 | Revvity Signals | https://revvitysignals.com/products/research/chemdraw |
| Agilent Chemstation | Agilent | https://www.agilent.com/cs/library/usermanuals/Public/G2070-91126_Understanding.pdf |
| Seriel EM | Mastronarde,74 | http://bio3d.colorado.edu/SerialEM |
| MotionCor2 | Zheng et. al.75 | https://emcore.ucsf.edu/ucsf-motioncor2 |
| RELION-3 | Zivanov et al.78 | http://www3.mrclmb.cam.ac.uk/relion |
| RELION-2 | Kimanius et al.79 | http://www2.mrclmb.cam.ac.uk/relion |
| cryoSPARC | Punjani et al.80 | https://cryosparc.com |
| COOT | Emsley et al.81 | https://www2.mrclmb.cam.ac.uk/personal/pemsley/coot |
| PHENIX | Adams et al.82 | https://www.phenix-online.org |
| MolProbity | Chen et al.83
Davis et al.84 |
http://molprobity.biochem.duke.edu |
| Chimera | Pettersen et al.85 | https://www.cgl.ucsf.edu/chimera |
| Pymol | PyMOL | http://www.pymol.org |
| GraphPad Prism 9 | GraphPad Software | http://www.graphpad.com |
| pCLAMP9 | Molecular Devices | https://mdc.custhelp.com/euf/assets/content/pCLAMP_9.0_Manual.pdf |
| DOCK 3.7 | Coleman et al.34 | https://dock.compbio.ucsf.edu/DOCK3.7/ https://blaster.docking.org/ |
| ZINC15 | Sterling and Irwin,33 | http://zinc15.docking.org/ |
| Reduce | Word et al., 1999 | N/A |
| QNIFFT | Gallagher and Sharp, 1998; Sharp, 1995 | N/A |
| Marvin | https://www.chemaxon.com | version 15.11.23.0, ChemAxon, 2015 |
| Corina | https://www.mn-am.com/products/corina | v.3.6.0026, Molecular Networks GmbH |
| Omega | https://www.eyesopen.com/omega | v.2.5.1.4, OpenEye Scientific Software |