

Abstract

Predicting the activities of new compounds against biophysical or phenotypic assays based on the known activities of one or a few existing compounds is a common goal in early stage drug discovery. This problem can be cast as a “few-shot learning” challenge, and prior studies have developed few-shot learning methods to classify compounds as active versus inactive. However, the ability to go beyond classification and rank compounds by expected affinity is more valuable. We describe Few-Shot Compound Activity Prediction (FS-CAP), a novel neural architecture trained on a large bioactivity data set to predict compound activities against an assay outside the training set, based on only the activities of a few known compounds against the same assay. Our model aggregates encodings generated from the known compounds and their activities to capture assay information and uses a separate encoder for the new compound whose activity is to be predicted. The new method provides encouraging results relative to traditional chemical-similarity-based techniques as well as other state-of-the-art few-shot learning methods in tests on a variety of ligand-based drug discovery settings and data sets. The code for FS-CAP is available at https://github.com/Rose-STL-Lab/FS-CAP.
Introduction
Early stage drug discovery often involves discovering a few “hit” compounds with weak activity against a disease-related target and using these compounds and their known activities to help discover new actives.1−6 A traditional approach at this stage is to use chemical similarity, such as the Tanimoto similarity between structural compound fingerprints7,8 or more complex compound descriptors,9−12 to identify new compounds similar to known actives. These similar compounds tend to have similar activities to the initial hit, but what is desired, ideally, are instead novel compounds with higher activities. While progress has been made on the use of machine learning to predict compound activities using the identity of a designated protein target (e.g., Öztürk et al.,13 Somnath et al.,14 Ragoza et al.,15 Stepniewska-Dziubinska et al.,16 Jones et al.17), many assays are phenotypic and lack a defined target. In addition, even when the target is known, learning from known ligands can act as a starting point in the discovery process.3,4
Here, we frame the search for new, potent compounds, given knowledge of a few active compounds, as a few-shot learning18 challenge. Few-shot learning is a machine learning framework that enables a pretrained model to generalize to a new domain, given a small amount of additional data from the new domain. In the present context, we start with a model trained on a large set of known compound activities and seek to generalize it to a new target given the measured activities of a few compounds against the new target.
While few-shot learning techniques have been developed to use known binary activities of a few compounds (the “context” compounds) to predict the binary activities of new compounds (the “query” compounds),19−26 experimental activity readouts are often continuous,27 so formulating the problem with binary compound activities requires ad hoc activity thresholding and discards potentially useful information. Here, we have developed a few-shot method that instead uses continuous compound activities as input and provides quantitative activity predictions as output. Such quantitative predictions are more useful than mere classification (active/inactive)28−31 but are also considered more difficult to generate.31 Indeed, we are aware of only one prior few-shot learning method that yields continuous, as opposed to binary, predictions in this domain.30 This prior work used an Attentive Neural Process (ANP, Kim et al.32)—a variant of neural processes—and provided encouraging initial results in a limited set of test cases.
Here, we describe and evaluate a novel neural-network-based tool, Few-Shot Compound Activity Prediction (FS-CAP), to predict the activity of a new query compound against a given target based on the known activities of a few existing context compounds against the same target. Our method employs new techniques that improve performance on the compound regression problem, including a new molecular featurization and a new neural encoder design.
In brief, FS-CAP employs a deterministic neural encoder to represent the context compounds and their activities in a given target via a new multiplication-based featurization, while a separate encoder is used to capture the target-independent characteristics of the query compound. The two resulting encodings are concatenated and fed into a predictor network, which predicts the activity of the query compound against the target of interest. The model is pretrained to minimize mean squared error (MSE) across BindingDB,33 a large experimental data set of compound activities. Once trained, the model is tested for its ability to predict compound activities in targets not present in the training data. The model outperforms state-of-the-art few-shot learning baselines in predictive capability and diversity of retrieved compounds across multiple data sets and identifies more diverse and potent actives than a traditional Tanimoto-based similarity approach. We provide the code and model at https://github.com/Rose-STL-Lab/FS-CAP.
Methods
Here we describe the FS-CAP method, the data set, and seven baseline methods.
Problem Statement
We seek to predict the activity of a query compound against a target, such as a binding or phenotypic target, given only a small set of context compounds and their activities against the same target. The neural network constructed to perform this task will have been trained and tested on many artificially constructed cases of the same type (i.e., a query compound and a few context compounds with known activities) for a variety of different targets drawn from publicly available data.
Consider a data set with experimentally
measured activity data for K different training targets.
For each target k ∈ [1, ..., K], we have a set of Nk molecules {m}k with
their respective real-valued assay results {π}k. Thus, the data set for target k comprises
the Nk molecules and
their respective activities Dk = {(m, π)}k. Supposing a hitherto untested query molecule mq has an experimental activity πkq against target k, we aim
to train a model f that learns to predict 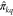 , given the context Ck ⊂ Dk of n ≤ 8 known molecules
and their activities against that target; i.e.,
, given the context Ck ⊂ Dk of n ≤ 8 known molecules
and their activities against that target; i.e., 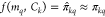 .
.
We train the model against many training examples, each consisting of one query molecule mq ∈ Dk and a context Ck generated by randomly sampling, without replacement, n other molecule–activity pairs in Dk. Thus, f is trained to predict the available experimental results πq across a large number of these training cases. To test the model, we construct analogous test cases, each comprising a query molecule and context molecules with known assay data, with the critical constraint that the targets in the test cases are omitted from the training cases. In this way, we measure the ability of the model to generalize from a large number of training targets to a hitherto unseen target.
Model Architecture
The architecture of FS-CAP is shown in Figure 1. The query molecule is represented by its Morgan fingerprint, which is a binary vector. We chose Morgan fingerprints for their speed of computation, simplicity, and previously reported strong performance on a diverse set of prediction tasks.34,35 We tried a range of fingerprint parameters but found that the exact choice of parameters made little difference (see “Implementation details” in the Supporting Information for more details). We also experimented with the pretrained, deep-learning-based Continuous and Data-Driven Descriptors (CDDD; Winter et al.36) as the molecular representation for FS-CAP but found that the performance matched FS-CAP with Morgan fingerprints. Therefore, we chose to use simpler Morgan fingerprints. Each context molecule is represented by a real-valued vector of the same length as the Morgan fingerprint and given by the product of the Morgan fingerprint and the compound’s experimental activity value π, so that each nonzero element of the fingerprint vector equals π instead of 1. We train one encoder fq for the query molecule and another encoder fc for the context molecules and their associated activities
| 1 |
where (mi, πi) ∈ C, with xq and ri being the encoded representation of the query and i-th context example, respectively. Thus, fq encodes the query molecule into a representation xq that is useful for predicting its activity, while fc captures key information from the context data about what determines a compound’s activity in target k. We use averaging to aggregate the individual context encodings ri into a single real-valued vector xc that represents the context set as a whole:
| 2 |
This maintains permutation invariance, as the order of the context molecules should not affect their encoding.
Figure 1.

Overview of the FS-CAP architecture. The context encoder (left) receives the Morgan fingerprint of each context compound multiplied by its associated activity value. A final context encoding is produced by aggregating the individual encodings of each context compound. The query encoder (right), which has different weights, receives the Morgan fingerprint of the query compound. A predictor network receives the concatenated outputs of each encoder and produces a final scalar activity prediction of the query compound.
The predictor network g combines the query and context encodings to generate an activity prediction for the query molecule
| 3 |
where ⊕ denotes vector concatenation
and 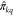 is the model’s prediction of the
true experimental activity in target k, πkq.
is the model’s prediction of the
true experimental activity in target k, πkq.
Training
We use BindingDB, a large experimental data set, for training (Table 1), setting aside some of these targets for testing. FS-CAP is trained in an end-to-end fashion (meaning all components of the model are trained simultaneously), with the loss for each epoch defined in terms of the mean square error (MSE):
| 4 |
Table 1. Summary of Data Setsa.
| Data set | Training targets | Test targets | Unique compounds | Date accessed |
|---|---|---|---|---|
| BindingDB33 | 1,754 | 41 | 609,791 | 12/1/2022 |
| PubChem High-Throughput Screening (PubChemHTS)37 | 0 | 100 | 34,716 | 12/23/2022 |
| Cancer Cell Line Encyclopedia (CCLE)38 | 0 | 275 | 24 | 8/6/2022 |
We report the number of targets in each data set, the number of these targets excluded from training and used for testing, and the number of unique compounds present across both training and test set targets in each data set. We also report the source and access date of the data set, if applicable.
Training and Testing Data Sets
We use three distinct compound activity data sets (Table 1) to evaluate and compare FS-CAP and baseline methods:
BindingDB (Used for Training and Testing)
BindingDB33 is a large experimental binding affinity database comprising thousands of protein targets, each associated with a known amino acid sequence. We initially treated each unique amino acid sequence in BindingDB as a separate training target but found that many of the sequences were very similar, meaning many of the held-out test targets were overly similar to the training targets. This made it relatively easy to predict test-set binders, even when no context compounds were provided. To address this, we clustered all sequences such that any two sequences in separate clusters had at most 0.2% sequence identity and then only kept the cluster midpoints. We did not combine all targets from each cluster into one because the protein targets within each cluster were different enough to make prediction difficult.
PubChemHTS (Used for Testing Only)
In a real-world, early stage drug discovery project, it is common to use information about the activity of a few initial hit compounds to guide the search for new actives in an existing chemical library. We modeled this scenario with a new data set we term PubChemHTS. To assemble this, we collected PubChem BioAssays,37 which is a publicly available data set of assays deposited by experimenters. It contains binary active/inactive classifications of ligands (marked as “Screening” data), for which we could also find at least a few quantitative activity data to use as context compounds. A PubChem Screening assay typically scans a rather generic chemical library for actives by testing each compound at a given concentration such as 10 μM.
Cancer Cell Line Encyclopedia (Used for Testing Only)
The Cancer Cell Line Encyclopedia38 contains cytotoxicity data for 24 drugs against 479 patient-derived cancer cell lines. We used the data set reported in Table S11 of Barretina et al.38 and extracted IC50 measurements for each drug measured against each cell line.
Further data set and preprocessing details are provided in the Supporting Information under “Data set details”. For all data sets, activity data are expressed as pActivity = – log10(C), where C is the characteristic concentration (e.g., IC50), in M, reported against the target.
Baseline Methods
We compare FS-CAP with baseline methods, including standard Tanimoto fingerprint similarity and state-of-the-art approaches in few-shot learning. We applied both optimization-based (MAML, Finn et al.39) and model-based (MetaNet, Munkhdalai and Yu;40 ANP, Kim et al.32) methods to the regression of compound activities. Details on the training and implementation of FS-CAP and baselines are reported in the Supporting Information under “Implementation details”.
Tanimoto similarity. Traditional molecular-structure-based similarity measure based on binary Morgan fingerprints.7,8 When given multiple context compounds, we use the highest similarity score between each of the contexts and the query. We tried a range of fingerprint parameters and chose the best-performing combination.
MolBERT + attentive neural process (ANP). Combines MolBERT, which is a start-of-the-art sequence-based molecular featurizer for property prediction tasks,41 with an attentive neural process model32 for the few-shot prediction of activity values.
• Non-Gaussian Gaussian process (NGGP).(42) Expands on basic Gaussian process techniques for few-shot learning by modeling the posterior distribution with an ODE-based normalizing flow.
MetaNet.(40) Uses two separate learners, the base learner and the meta-learner which utilizes a memory mechanism, to quickly adapt to new tasks in the few-shot setting via fast parametrization.
Meta-MGNN.(25) Uses a graph neural network in combination with a self-supervised module to aid in task adaptation by predicting bond and atom types in the context set.
Model-agnostic meta-learning (MAML).(39) Learns a model that can quickly adapt to a new task by training on a small set of context examples. We use a simple multilayer perceptron that takes a Morgan fingerprint as input for the base model.
MetaDTA.(30) Applies attentive neural processes to the few-shot regression of continuous activity values. We use the MetaDTA(I) variant because its performance is superior to that of the other reported variants.
Results
Few-Shot Learning on the BindingDB Test Set
We trained all few-shot learning methods on the 1,754 training targets in BindingDB and then evaluated them on the 41 held-out test targets. For each method, a separate model was trained for each different number of context compounds (1, 2, 4, 8). For this evaluation, we seek to emulate a hit and lead optimization challenge, where one wishes to use knowledge of a few compounds with modest experimentally determined activities in a binding or phenotypic assay to predict the activities of additional candidate compounds. Compounds with high activity are often not known at the hit stage, so we only sampled context compounds (in both training and testing) with pActivity < 6 (i.e., effective concentrations >1 μM), which is typical of hit compounds.43 Here, every compound in the test-set targets was treated as a query compound, and for each query compound we randomly sampled a different set of 1–8 context compounds from the subset of compounds with pActivity < 6 against the same target.
Table 2 reports the mean correlation of the predicted and ground truth activity values across all test-set targets for each method. We find that FS-CAP consistently outperforms Tanimoto similarity, a de facto standard in medicinal chemistry, as well as the baseline few-shot learning methods across all tested numbers of context compounds. This suggests that FS-CAP may be useful for hit and lead discovery as it is the most successful in predicting the activities of unknown compounds using only weakly active context compounds.
Table 2. Pearson’s Correlation for Few-Shot Learning Methods and Tanimoto Similarity on the BindingDB Test Seta.
| # context compounds | 1 | 2 | 4 | 8 |
|---|---|---|---|---|
| Tanimoto similarity | –0.06 | 0.11 | 0.17 | 0.23 |
| MolBERT + ANP | 0.10 | 0.11 | 0.11 | 0.14 |
| NGGP | 0.05 | 0.11 | 0.15 | 0.20 |
| MetaNet | –0.02 | –0.01 | 0.04 | 0.06 |
| Meta-MGNN | 0.20 | 0.20 | 0.21 | 0.23 |
| MAML | 0.22 | 0.22 | 0.23 | 0.24 |
| MetaDTA | 0.24 | 0.24 | 0.25 | 0.25 |
| FS-CAP | 0.27 | 0.28 | 0.29 | 0.32 |
Results are reported as the mean across all BindingDB targets of the Pearson’s r between predicted and ground-truth pActivity. We ran three replicate training runs with different random number seeds for the three best performing methods (MAML, MetaDTA, FS-CAP) and obtained standard deviations of at most 0.01.
We also evaluated the ability of Tanimoto similarity and the leading few-shot learning methods to correctly identify active compounds that are chemically dissimilar to the one context compound. To do this, we identified the query compounds with the highest predicted activities for each method and computed the average Tanimoto similarities between the context compound and the query compound for the top κ compounds per target with the most favorable predictions from each method. As shown in Table 3, Tanimoto similarity and FS-CAP retrieve compounds with comparable pActivities, while the competing few-shot learning methods retrieve slightly less active compounds. However, the actives retrieved by FS-CAP are much less similar to the context compounds than those retrieved by Tanimoto and the other one-shot learning methods. This suggests that our approach, relative to the baselines, is able to retrieve highly novel compounds that still show strong activity.
Table 3. Activities and Chemical Similarities to Context Compounds of Top κ = 1, 5, or 20 Retrieved Compoundsa.
| pActivity |
Similarity |
|||||
|---|---|---|---|---|---|---|
| 1 | 5 | 20 | 1 | 5 | 20 | |
| Tanimoto | 5.95 ± 0.11 | 5.91 ± 0.08 | 5.79 ± 0.01 | 0.95 ± 0.02 | 0.77 ± 0.01 | 0.55 ± 0.01 |
| Meta-MGNN | 5.73 ± 0.12 | 5.82 ± 0.10 | 5.64 ± 0.05 | 0.33 ± 0.03 | 0.25 ± 0.03 | 0.27 ± 0.01 |
| MAML | 5.89 ± 0.04 | 5.85 ± 0.06 | 5.80 ± 0.03 | 0.55 ± 0.01 | 0.58 ± 0.01 | 0.60 ± 0.00 |
| MetaDTA | 5.88 ± 0.05 | 5.87 ± 0.03 | 5.75 ± 0.02 | 0.38 ± 0.02 | 0.39 ± 0.01 | 0.41 ± 0.00 |
| FS-CAP | 5.93 ± 0.12 | 5.92 ± 0.17 | 5.83 ± 0.07 | 0.21 ± 0.01 | 0.21 ± 0.00 | 0.22 ± 0.02 |
Mean pActivity (−log10 of M) and Tanimoto similarity between the one context compound and the query compound for the top κ scored compounds, averaged across all targets in the testing set. Results are presented for traditional Tanimoto similarity, FS-CAP and the two baseline methods that gave the best results in Table 2.
See the “Activity cliff performance” section in the Supporting Information for additional results measuring the performance of FS-CAP and baselines around activity cliffs in the BindingDB test set. In brief, we found that FS-CAP, compared to baselines, achieved the lowest RMSE among a subset of test compounds deemed to be cliff compounds.
Results on the PubChem High-Throughput Screening Data Set
We evaluated the ability of FS-CAP and baseline
methods to recover active compounds in the PubChemHTS collection,
based on available quantitative activity data for compounds in these
assays (Table 4). We
use the models trained on BindingDB for this challenge (without any
context activity limits as in the previous section) without any further
training on PubChemHTS. However, the few-shot learning models predict
a continuous activity value for each query compound, while the PubChemHTS
data are binary (active vs inactive at a given concentration of compound
in the assay). Therefore, instead of reporting Pearson correlation
coefficients as done for the BindingDB data set, we use the quantitative
predictions to rank-order the compounds in each PubChemHTS assay and
evaluate the quality of the predictions using two other metrics. One
is the area under the receiver-operating characteristic curve (ROC-AUC),
a standard metric in the HTS literature.44 The other is the “enrichment” of true actives in the
compounds top ranked by the few-shot methods, another standard metric
in the HTS literature.45 Given a proportion
of actives ptop among
the top retrieved compounds and a proportion of actives pbase among all compounds, the enrichment
is defined as the ratio 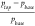 . We looked at enrichment in the top 0.5,
1.0, and 2.0% of compounds. We applied Tanimoto similarity to this
problem by taking the maximum similarity of the query compound to
any of the context compounds as the activity prediction. In other
words, screening compounds most similar to any of the context compounds
were predicted as active, and the least similar compounds were predicted
as inactive.
. We looked at enrichment in the top 0.5,
1.0, and 2.0% of compounds. We applied Tanimoto similarity to this
problem by taking the maximum similarity of the query compound to
any of the context compounds as the activity prediction. In other
words, screening compounds most similar to any of the context compounds
were predicted as active, and the least similar compounds were predicted
as inactive.
Table 4. Average ROC-AUC and Enrichment Statistics across All High-Throughput Screening Assays in PubChemHTSa.
| ROC-AUC | 0.5% | 1% | 2% | |
|---|---|---|---|---|
| Tanimoto similarity | 0.51 | 2.0 | 1.6 | 1.3 |
| MolBERT + ANP | 0.53 | 1.9 | 1.9 | 1.6 |
| NGGP | 0.49 | 1.6 | 1.5 | 1.5 |
| MetaNet | 0.50 | 1.1 | 1.0 | 1.2 |
| Meta-MGNN | 0.53 | 1.5 | 1.5 | 1.4 |
| MAML | 0.54 | 1.3 | 1.2 | 1.3 |
| MetaDTA | 0.52 | 1.9 | 1.5 | 1.4 |
| FS-CAP | 0.56 | 2.3 | 2.1 | 2.0 |
Enrichment is shown as a ratio, where 1 means no enrichment over the base rate. Eight context compounds were used for the few-shot learning methods. We ran three replicate training runs with different random number seeds for the three best performing methods (MAML, MetaDTA, FS-CAP) and obtained standard deviations in ROC-AUC of at most 0.01.
As detailed in Table 4, FS-CAP outperforms baselines in both ROC-AUC and all three enrichment measurements. This suggests that FS-CAP is more capable of predicting compound activities in screening libraries than baseline methods and thus may be used to good effect in screening chemical libraries for compounds with an elevated likelihood of being active against an assay for which one has a few known actives.
In an early stage project, it is often the case that the known actives available as context compounds have only weak activities. Thus, we further analyzed the models by measuring how well they maintain their performance as context compounds with lower and lower activity are allowed. Figure 2 shows the performance, as measured by ROC-AUC, of FS-CAP and the most competitive baselines for different context activities. ROC-AUC was measured across all query compounds for which the context activity was randomly selected to be within the range on the x-axis. As shown, all methods have ROC-AUC > 0.5 when the single context compound is highly active (<1 nM), but performance degrades for all methods as the context activity decreases. FS-CAP is the only method that retains ROC-AUC > 0.5 across all measured context activities, suggesting it may be the most capable at predicting activity regardless of the activity of the context compound. While the other few-shot methods have variable success across different context activities, Tanimoto similarity in particular becomes mostly random as the context activity decreases, confirming that measuring similarity to a weakly active compound is of limited utility. It is not clear why the other three methods show improvement in performance when going from context activities of <102 nM to the less stringent <104 nM, but these results are robust, as evident from the modest standard errors across multiple runs shown in Figure 2.
Figure 2.

FS-CAP excels at recovering true actives given context compounds across a range of activities. ROC-AUC measured on the HTS data set (y-axis) for different context compound activity values (x-axis). The values on the x-axis represent an upper bound; e.g., 101 nM represents activities ≥100 and <101 nM. Each point represents the mean over 10 separate runs and the shaded region the standard error.
Generalization to Cancer Cell Line Encylopedia
We explored how well the models trained on BindingDB generalize to an entirely different challenge, predicting the cytotoxity of query compounds against patient-derived cancer cell lines, given the cytotoxicity data of a few context compounds. This study uses the Cancer Cell Line Encyclopedia (CCLE, Barretina et al.38) data set. Context compounds were randomly sampled from all compounds with activity data against a given cell line and used to predict the activities of query compounds not in the context set against the same cell line. We measured the mean correlation between predicted and experimental IC50 data across compounds instead of across targets as in the previous sections. This means, for each drug compound in the CCLE, we measured the correlation between real and predicted IC50 for each cell line with measured data against that compound and then averaged these correlations across all compounds. This is to avoid the correlation being influenced by the base differences in compound activities and instead to measure the ability of each method to make variable and accurate predictions depending on the cell line.
As shown in Table 5, FS-CAP is better than the baseline methods at predicting cytotoxicity in this setting. Although the number of compounds tested in the CCLE is relatively small, the success of FS-CAP trained only on BindingDB in predicting activity values in this data set suggests that it may learn fundamental relationships between compounds and activities that generalize across data sets. To rule out the possibility that the observed performance is due to simple baseline differences in the sensitivity of each cell line, we measured the performance of a method that simply predicted the mean of the context activities. This approach achieved a correlation of r = 0.13 with 8 context compounds, showing that the observed performance of FS-CAP and the other few-shot learning methods is due to factors beyond the baseline sensitivity of each cell line.
Table 5. Average Correlation per Anti-Cancer Compounda.
| # context compounds | 1 | 2 | 4 | 8 |
|---|---|---|---|---|
| Tanimoto similarity | 0.02 | 0.03 | 0.03 | 0.05 |
| MolBERT + ANP | 0.05 | 0.08 | 0.11 | 0.13 |
| NGGP | 0.08 | 0.09 | 0.11 | 0.12 |
| MetaNet | 0.01 | 0.03 | 0.04 | 0.06 |
| Meta-MGNN | 0.07 | 0.11 | 0.13 | 0.15 |
| MAML | 0.10 | 0.12 | 0.14 | 0.18 |
| MetaDTA | 0.09 | 0.10 | 0.14 | 0.17 |
| FS-CAP | 0.10 | 0.13 | 0.16 | 0.22 |
Mean Pearson’s r between ground truth and predicted drug activity values across all anti-cancer compounds in the CCLE. Experiments were performed using 1, 2, 4, and 8 context compounds. We report the mean from three independent training runs on the BindingDB data set with random seeds for the top three baselines. The standard deviation between runs was at most 0.02.
Ablations of Model Components
We report performance metrics of model ablations to the FS-CAP architecture in Table 6. For each ablation, we trained a separate model and then measured the mean correlation of the predicted and ground truth activity values across all test-set targets in BindingDB. This experiment is similar to that on the BindingDB test set reported at the beginning of the Results. Eight context compounds were used for all of the tests.
Table 6. Model Ablationsa.
| Ablation | Pearson’s r |
|---|---|
| Base model (FS-CAP) | 0.32 ± 0.01 |
| No query encoding | 0.30 ± 0.01 |
| Concatenated context | 0.28 ± 0.00 |
| No context | 0.17 ± 0.03 |
Mean correlation between ground-truth and predicted activities across all test targets in BindingDB using 8 context compounds. We report the mean ± one standard deviation from three independent training runs with random seeds.
We test the significance of using a separate query encoder network (“Base model”) or feeding the query features directly to the predictor network (“No query encoding”). The greater performance of the variation with the query encoder suggests that encoding the query independent of target information is beneficial for prediction. “Concatenated context” means that we feed the context encoder a binary compound fingerprint concatenated with its associated activity value instead of multiplying the two. This is similar to a neural process model. This variation shows inferior performance, suggesting that combining the context compound fingerprint and activity value scalar via multiplication is a useful featurization for the activity prediction task. “No context” denotes that no context was fed to the model at all, and it made activity predictions solely on the basis of the query compound.
Discussion and Conclusions
The proposed few-shot learning model FS-CAP surpasses both a standard chemical similarity metric and prior few-shot learning baselines in multiple tasks of interest in early stage drug discovery. These tasks include prediction of compound activities based on a set of weak-binding context compounds, prediction of screening library compounds as active or inactive, and prediction of antitumor activity in cell-based assays, all performed with models trained on a large activity data set. Together, these results suggest that FS-CAP may be broadly useful for target-free, or ligand-based, drug discovery, which remains a common paradigm in the setting of phenotypic screening and in early stage projects.3,4
FS-CAP shows promise as a tool to leverage the limited compound activity data that are typically available in the earliest stages of drug discovery, focusing attention on candidate compounds that are much more likely than randomly chosen compounds to be active against a target of interest. It thus offers a novel approach to speed drug discovery and reduce its costs. Exploring the use of FS-CAP for other compound properties might open further applications. For example, it may find applications in predicting pharmacokinetic parameters of candidate compounds, such as bioavailability and half-life, metabolic susceptibility, and toxicity.
Limitations of the present implementation of FS-CAP include its use of the simple Morgan fingerprint representation and a context aggregation technique with limited expressiveness. Additionally, the inherent limitations of training on experimental assay data, such as the limited tested dose range31 or systematic biases in which compounds are tested against which targets, may limit the applicability of few-shot methods like FS-CAP trained on these data sets to real-world drug discovery projects. Another limitation of the data set we use for training is that binding data against one target may in fact encompass multiple distinct pockets. While more careful data set curation could alleviate this issue and is an area for future improvement, we do not think it presents a very large concern as experimentalists are usually careful to only include the relevant pocket in their binding assay. Additionally, this issue would only degrade performance and not make the results appear stronger than they actually are, because predicting activities against a target with multiple pockets would be more difficult than a single pocket. Since the performance of our trained model is already fairly strong, the issue of multiple pockets does not seem to be causing a major effect. Future developments could include the exploration of more complex molecular representations (e.g., sequence or graph-based) and the application of more complex context aggregation methods beyond the mean. Finally, research into incorporating target information, when available, with few-shot methods may allow for increased prediction accuracy beyond using target information or context compounds alone.
Data and Software Availability
Our code is available at https://github.com/Rose-STL-Lab/FS-CAP. We provide scripts to preprocess the data set, train FS-CAP, and predict the activities of new compounds. We also provide a pretrained model file and a Docker container for ease of use.
Acknowledgments
This work was supported in part by Army-ECASE award W911NF-23-1-0231, the U.S. Department Of Energy, Office of Science under #DE-SC0022255, IARPA HAYSTAC Program, CDC-RFA-FT-23-0069, NSF Grants #2205093, #2146343, and #2134274. MKG acknowledges funding from National Institute of General Medical Sciences (GM061300). These findings are solely of the authors and do not necessarily represent the views of the NIH.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.4c00485.
Implementation and data set details and additional results (PDF)
The authors declare the following competing financial interest(s): RY has an equity interest and is a scientific advisor of Salient Predictions. MKG has an equity interest in and is a cofounder and scientic advisor of VeraChem LLC.
Supplementary Material
References
- Paul D.; Sanap G.; Shenoy S.; Kalyane D.; Kalia K.; Tekade R. K. Artificial intelligence in drug discovery and development. Drug Discovery Today 2021, 26, 80. 10.1016/j.drudis.2020.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vamathevan J.; Clark D.; Czodrowski P.; Dunham I.; Ferran E.; Lee G.; Li B.; Madabhushi A.; Shah P.; Spitzer M.; et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discovery 2019, 18, 463–477. 10.1038/s41573-019-0024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haasen D.; Schopfer U.; Antczak C.; Guy C.; Fuchs F.; Selzer P. How phenotypic screening influenced drug discovery: lessons from five years of practice. Assay Drug Dev. Technol. 2017, 15, 239–246. 10.1089/adt.2017.796. [DOI] [PubMed] [Google Scholar]
- Swinney D. C.; Lee J. A. Recent advances in phenotypic drug discovery. F1000Research 2020, 9, 944. 10.12688/f1000research.25813.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loew G. H.; Villar H. O.; Alkorta I. Strategies for indirect computer-aided drug design. Pharm. Res. 1993, 10, 475–486. 10.1023/A:1018977414572. [DOI] [PubMed] [Google Scholar]
- Acharya C.; Coop A.; Polli J. E.; MacKerell A. D. Recent advances in ligand-based drug design: relevance and utility of the conformationally sampled pharmacophore approach. Curr. Comput.-Aided Drug Des. 2011, 7, 10–22. 10.2174/157340911793743547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajusz D.; Rácz A.; Héberger K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations?. J. Cheminf. 2015, 7, 1–13. 10.1186/s13321-015-0069-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers D.; Hahn M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- Danishuddin; Khan A. U. Descriptors and their selection methods in QSAR analysis: paradigm for drug design. Drug Discovery Today 2016, 21, 1291–1302. 10.1016/j.drudis.2016.06.013. [DOI] [PubMed] [Google Scholar]
- Li X.; Wang Z.; Liu H.; Yu H. Quantitative structure–activity relationship for prediction of the toxicity of phenols on Photobacterium phosphoreum. Bull. Environ. Contam. Toxicol. 2012, 89, 27–31. 10.1007/s00128-012-0662-3. [DOI] [PubMed] [Google Scholar]
- Kohlbacher S. M.; Langer T.; Seidel T. QPHAR: quantitative pharmacophore activity relationship: method and validation. J. Cheminf. 2021, 13, 1–14. 10.1186/s13321-021-00537-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearnes S.; Pande V. ROCS-derived features for virtual screening. J. Comput.-Aided Mol. Des. 2016, 30, 609–617. 10.1007/s10822-016-9959-3. [DOI] [PubMed] [Google Scholar]
- Öztürk H.; Özgür A.; Ozkirimli E. DeepDTA: deep drug–target binding affinity prediction. Bioinformatics 2018, 34, i821–i829. 10.1093/bioinformatics/bty593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somnath V. R.; Bunne C.; Krause A. Multi-scale representation learning on proteins. Advances in Neural Information Processing Systems 2021, 34, 25244–25255. [Google Scholar]
- Ragoza M.; Hochuli J.; Idrobo E.; Sunseri J.; Koes D. R. Protein–ligand scoring with convolutional neural networks. J. Chem. Inf. Model. 2017, 57, 942–957. 10.1021/acs.jcim.6b00740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepniewska-Dziubinska M. M.; Zielenkiewicz P.; Siedlecki P. Development and evaluation of a deep learning model for protein–ligand binding affinity prediction. Bioinformatics 2018, 34, 3666–3674. 10.1093/bioinformatics/bty374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D.; Kim H.; Zhang X.; Zemla A.; Stevenson G.; Bennett W. D.; Kirshner D.; Wong S. E.; Lightstone F. C.; Allen J. E. Improved protein–ligand binding affinity prediction with structure-based deep fusion inference. J. Chem. Inf. Model. 2021, 61, 1583–1592. 10.1021/acs.jcim.0c01306. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Yao Q.; Kwok J. T.; Ni L. M. Generalizing from a few examples: A survey on few-shot learning. ACM Computing Surveys 2021, 53, 1–34. 10.1145/3386252. [DOI] [Google Scholar]
- Altae-Tran H.; Ramsundar B.; Pappu A. S.; Pande V. Low data drug discovery with one-shot learning. ACS Cent. Sci. 2017, 3, 283–293. 10.1021/acscentsci.6b00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimunek J.; Friedrich L.; Kuhn D.; Rippmann F.; Hochreiter S.; Klambauer G.. A generalized framework for embedding-based few-shot learning methods in drug discovery. ELLIS Machine Learning for Molecules Workshop 2021.
- Wang Y.; Abuduweili A.; Yao Q.; Dou D. Property-aware relation networks for few-shot molecular property prediction. Advances in Neural Information Processing Systems 2021, 34, 17441–17454. [Google Scholar]
- Ding K.; Wang J.; Li J.; Shu K.; Liu C.; Liu H.. Graph prototypical networks for few-shot learning on attributed networks. Proceedings of the 29th ACM International Conference on Information & Knowledge Management 2020, 295–304.
- Vella D.; Ebejer J.-P. Few-Shot Learning for Low-Data Drug Discovery. J. Chem. Inf. Model. 2023, 63, 27. 10.1021/acs.jcim.2c00779. [DOI] [PubMed] [Google Scholar]
- Nguyen C. Q.; Kreatsoulas C.; Branson K. M.. Meta-learning initializations for low-resource drug discovery. ChemRxiv 2020.
- Guo Z.; Zhang C.; Yu W.; Herr J.; Wiest O.; Jiang M.; Chawla N. V. Few-shot graph learning for molecular property prediction. Proceedings of the Web Conference 2021, 2021, 2559–2567. [Google Scholar]
- Lv Q.; Chen G.; Yang Z.; Zhong W.; Chen C. Y.-C. Meta learning with graph attention networks for low-data drug discovery. IEEE Transactions on Neural Networks and Learning Systems 2023, 1. 10.1109/TNNLS.2023.3250324. [DOI] [PubMed] [Google Scholar]
- Chandrasekaran S. N.; Ceulemans H.; Boyd J. D.; Carpenter A. E. Image-based profiling for drug discovery: due for a machine-learning upgrade?. Nat. Rev. Drug Discovery 2021, 20, 145–159. 10.1038/s41573-020-00117-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo M.; Park A.; Kim K.; Son W.-J.; Lee H. S.; Lim G.; Lee J.; Lee D. H.; An J.; Kim J. H.; et al. A deep learning model for cell growth inhibition IC50 prediction and its application for gastric cancer patients. Int. J. Mol. Sci. 2019, 20, 6276. 10.3390/ijms20246276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenhof K.; Eckhart L.; Gerstner N.; Kehl T.; Lenhof H.-P. Simultaneous regression and classification for drug sensitivity prediction using an advanced random forest method. Sci. Rep. 2022, 12, 1–13. 10.1038/s41598-022-17609-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E.; Yoo J.; Lee H.; Hong S.. MetaDTA: Meta-learning-based drug-target binding affinity prediction. ICLR Machine Learning for Drug Discovery Workshop 2022.
- Stanley M.; Bronskill J. F.; Maziarz K.; Misztela H.; Lanini J.; Segler M.; Schneider N.; Brockschmidt M.. Fs-mol: A few-shot learning dataset of molecules. Conference on Neural Information Processing Systems Datasets and Benchmarks Track 2021.
- Kim H.; Mnih A.; Schwarz J.; Garnelo M.; Eslami A.; Rosenbaum D.; Vinyals O.; Teh Y. W.. Attentive neural processes. arXiv.org, e-Print Archive 2019.
- Gilson M. K.; Liu T.; Baitaluk M.; Nicola G.; Hwang L.; Chong J. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. 10.1093/nar/gkv1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riniker S.; Landrum G. A. Open-source platform to benchmark fingerprints for ligand-based virtual screening. J. Cheminf. 2013, 5, 26. 10.1186/1758-2946-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awale M.; Reymond J.-L. Polypharmacology browser PPB2: target prediction combining nearest neighbors with machine learning. J. Chem. Inf. Model. 2019, 59, 10–17. 10.1021/acs.jcim.8b00524. [DOI] [PubMed] [Google Scholar]
- Winter R.; Montanari F.; Noé F.; Clevert D.-A. Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations. Chem. Sci. 2019, 10, 1692–1701. 10.1039/C8SC04175J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Xiao J.; Suzek T. O.; Zhang J.; Wang J.; Zhou Z.; Han L.; Karapetyan K.; Dracheva S.; Shoemaker B. A.; et al. PubChem’s BioAssay database. Nucleic Acids Res. 2012, 40, D400–D412. 10.1093/nar/gkr1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J.; Caponigro G.; Stransky N.; Venkatesan K.; Margolin A. A.; Kim S.; Wilson C. J.; Lehár J.; Kryukov G. V.; Sonkin D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn C.; Abbeel P.; Levine S.. Model-agnostic meta-learning for fast adaptation of deep networks. International Conference on Machine Learning 2017, 1126–1135.
- Munkhdalai T.; Yu H.. Meta networks. International Conference on Machine Learning 2017, 2554–2563. [PMC free article] [PubMed]
- Li J.; Jiang X. Mol-BERT: An Effective Molecular Representation with BERT for Molecular Property Prediction. Wireless Communications and Mobile Computing 2021, 2021, 1. 10.1155/2021/7181815.35573891 [DOI] [Google Scholar]
- Sendera M.; Tabor J.; Nowak A.; Bedychaj A.; Patacchiola M.; Trzcinski T.; Spurek P.; Zieba M. Non-Gaussian Gaussian Processes for Few-Shot Regression. Advances in Neural Information Processing Systems 2021, 34, 10285–10298. [Google Scholar]
- Zhu T.; Cao S.; Su P.-C.; Patel R.; Shah D.; Chokshi H. B.; Szukala R.; Johnson M. E.; Hevener K. E. Hit identification and optimization in virtual screening: Practical recommendations based on a critical literature analysis: Miniperspective. J. Med. Chem. 2013, 56, 6560–6572. 10.1021/jm301916b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triballeau N.; Acher F.; Brabet I.; Pin J.-P.; Bertrand H.-O. Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J. Med. Chem. 2005, 48, 2534–2547. 10.1021/jm049092j. [DOI] [PubMed] [Google Scholar]
- Lopes J. C. D.; Dos Santos F. M.; Martins-José A.; Augustyns K.; De Winter H. The power metric: a new statistically robust enrichment-type metric for virtual screening applications with early recovery capability. J. Cheminf. 2017, 9, 1–11. 10.1186/s13321-016-0189-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Our code is available at https://github.com/Rose-STL-Lab/FS-CAP. We provide scripts to preprocess the data set, train FS-CAP, and predict the activities of new compounds. We also provide a pretrained model file and a Docker container for ease of use.