

Abstract
Background
Toll-like receptor 9 (TLR9) agonists induce inflammatory responses that promote the killing of infectious micro-organisms, cancer cells and develop adaptive immune responses. Their ability as immunomodulators to enhance the activity of checkpoint inhibitors (CPI) in treating liver tumors is limited in part by the distinctive biology of intrahepatic myeloid-derived suppressor cells (MDSC) and challenges with tumor-specific therapeutic delivery. We have shown that the regional delivery of type C TLR9 agonist via pressure-enabled drug delivery (PEDD) system improves delivery to the tumor, enhances depletion of MDSCs and overall, stimulates the immune system in combination with or without CPI. Currently, CPIs are delivered intravenously, although there is a growing interest in its subcutaneous (SQ) administration. We compared nelitolimod formerly known as SD-101 administered using PEDD in combination with systemic (Sys) or SQ CPI in murine liver metastases (LM).
Methods
The LM model was developed by injecting MC38-Luc cells via the spleen of 8–12 week old male C57/BL6 mice followed by splenectomy. After a week, fluorescently labeled nelitolimod (10 µg/mouse) was delivered via PEDD and co-administered anti-programmed cell death-1 (α-PD-1) either via Sys or SQ. Tumor burden was monitored by in vivo imaging system. Serum cytokine levels were analyzed by Luminex. Tissues were harvested on Day 3 (D3) or Day 10 (D10) post-PEDD to enrich CD45+ cells and were analyzed via NanoString targeted transcriptomics (D3) or flow cytometry (FC, D10) to interrogate immune cell populations (D10). For NanoString analysis, the innate immune panels were selected, and for FC, MDSCs (CD11b+Gr1+), B cells (B220+), dendritic cells (DC, CD11c+), T (CD3+) cells, and M1-like macrophages (F4/80+CD38+Egr2−) were quantified.
Results
Nelitolimod delivered via PEDD resulted in changes in innate and adaptive immune cells within LM, including depletion of liver MDSC and increased M1-like macrophages in the liver, which are supportive of antitumor immunity. While CPI monotherapy failed to control tumor progression, nelitolimod and CPI combination improved LM control, survival and antitumor immunity beyond the nelitolimod monotherapy effect, irrespective of CPI delivery route.
Conclusion
The SQ route of CPI delivery was equivalent to Sys in combination with nelitolimod, suggesting SQ-CPI may be a rational choice in combination with PEDD of nelitolimod for liver tumor treatment.
Keywords: Toll-like receptor - TLR, Myeloid-derived suppressor cell - MDSC, Tumor microenvironment - TME, Immune Checkpoint Inhibitor
WHAT IS ALREADY KNOWN ON THIS TOPIC
Toll-like receptor 9 class C (TLR9C) agonists delivered regionally using an intravascular approach with pressure-enabled drug delivery (PEDD), in combination with systemic (Sys) checkpoint inhibitors (CPI) can reduce suppressive myeloid cells in the tumor microenvironment, while providing broad immunostimulation.
WHAT THIS STUDY ADDS
Monotherapy with α-PD-1 for liver metastasis (LM) is ineffective in controlling tumor progression in most patients, and subcutaneous administration is emerging as an alternative to intravenous infusion. Here we show in a murine LM model, that the PEDD of a class C TLR9 agonist, nelitolimod, is associated with the depletion of liver myeloid-derived suppressor cell, increases in T-cell infiltration and M1-like macrophage expansion. Control of LM in combination with α-PD-1 was shown to be independent of the CPI delivery route, with equivalent effects for intravenous and subcutaneous (SQ) infusions.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The clinical development of SQ CPI for combinatorial regimens may increase patient satisfaction and compliance, reduce treatment-related costs, lower administrative costs and reduce resource utilization as compared with intravenous administration.
Introduction
The negative regulatory checkpoints such as programmed cell death-1 (PD-1) receptor and cytotoxic T-lymphocyte associate protein (CTLA-4) expressed on T cells engage with cognate ligands (PD-1: programmed death-ligand 1 (PD1: PD-L1/B7-H1, PD-L2/B7-DC; CTLA-4: CD80/B7-1, CD86/B7-2), and represent means by which antigen-specific immune responses can be restrained peripherally.1,3 In multiple solid tumors, PD-L1 is upregulated on tumor cells, which restrains effector T cells (Teff) and results in tumor-mediated immune exhaustion.4 Checkpoint inhibitors (CPIs) block these interactions, permitting an influx of tumor-antigen-specific Teff, resulting in antitumor responses. Currently, there are five CPIs targeting PD-1 (pembrolizumab, nivolumab, cemiplimab, dostarlimab, and retifanlimab), three targeting PD-L1 (atezolizumab, avelumab and durvalumab) and two targeting CTLA-4 (tremelimumab, and ipilimumab) that are Food and Drug Administration-approved via intravenous infusion to treat a variety of solid tumors across several settings including adjuvant, a neoadjuvant and advanced disease both singly and in a variety of combinations.5,8
The liver represents a uniquely immunosuppressive microenvironment for several reasons. In multiple solid tumors wherein CPIs are efficacious, the presence of liver metastases (LM) reduces response rate, progression-free and overall survival.9,11 In patients with melanoma, LM is associated with reduced numbers and function of CD8+ tumor-infiltrating lymphocytes.12 13 In general, myeloid-derived suppressor cells (MDSCs) accumulate in the liver in response to malignant disease progression and orchestrate an immunosuppressive environment limiting T-cell infiltration and activity.14 Circulating MDSCs have been associated with a lack of response to CPI, and the presence of LM is an independent predictor of CPI treatment failure in multiple indications.15 16 Moreover, macrophages promote phagocytosis, the release of inflammatory mediators, and antigen presentation, thus driving inflammatory responses and promoting antitumor immunity in liver metastasis.17
In CPI-refractory melanoma, intratumoral delivery of toll-like receptor 9 (TLR9) agonists is associated with durable responses and a manageable safety profile.18 19 Further, in CPI-naïve melanoma, response to intratumoral TLR9 agonist is associated with macrophage polarization and increase in CD8+ tumor infiltrating leukocytes.18 19 We and others have previously shown that pressure-enabled drug delivery (PEDD) improved delivery of therapeutics and reduced off-target tissue exposure, particularly in the liver.20,22 To this end, PEDD of nelitolimod represents a novel means of addressing the uniquely immunosuppressive tumor microenvironment (TME) within the liver. PEDD of nelitolimod delivers effectively despite high interstitial pressures within the liver.22 Additionally, PEDD of nelitolimod reprograms hepatic MDSCs resulting in increased macrophage-1 like/macrophage-2 like (M1/M2-like) ratio,23 and synergy with systemic α-PD-1 in preclinical models of LM.24 The efficacy of PEDD of nelitolimod in combination with systemic CPI is being evaluated in a variety of indications, including metastatic uveal melanoma (mUM, NCT04935229) and intrahepatic cholangiocarcinoma and hepatocellular carcinoma (NCT05220722). In a phase I dose-escalation trial in mUM, we reported that PEDD of nelitolimod was safe at the optimal biologic dose (nelitolimod 2 mg) in combination with intravenous nivolumab.25 At this dose level, the median progression-free survival was 11.7 months, with 81% disease control which favorably compared with studies of CPI in this patient population.26 27
Given the durable responses and long-term survival seen in a subset of CPI-treated patients, the aggregate time required for clinic-based intravenous infusions result in reduced productivity and is consistently cited as a driver of the exponentially increasing costs associated with these agents.28 This has led to a growing interest in evaluating SQ administration of CPIs to improve patient satisfaction, lower administrative costs and reduce resource utilization compared with intravenous administration, as has been demonstrated with multiple other oncologic agents including rituximab and trastuzimab.29 To this end, multiple programs are evaluating the safety and clinical efficacy of SQ CPIs including PF-06801591,30 envafolimab31 32 and subcutaneous nivolumab.33 Notably, the SQ treatment was preferred by 80%–90% of patients and reduced the median infusion time (up to threefold) which led to a reduction in the treatment cost.34,37 The development of SQ α-PD-1 may facilitate similar patient care and compliance, and to this end, multiple SQ α-PD-1 formulations are currently in various phases of testing.30 In general, for patients with LM, the inherent strong liver tolerance diminishes the ability of α-PD-1 or α -PD-L1 to enhance antitumor immunity and hence combinatorial treatment strategies have become imminent to improve the PD-1/PD-L1 pathway blockade. The present study was undertaken to determine if SQ CPI delivery would negatively impact the control of LM in combination with a class C TLR9 agonist.
We sought to characterize further the effect of the route of administration (systemic (Sys) vs SQ) of α-PD-1 on the efficacy of regional PEDD of nelitolimod in a preclinical model of LM. We observed that the antitumor effects of PEDD of nelitolimod with α-PD-1 were similar in Sys and SQ settings. We further observed that following regional PEDD administration of nelitolimod, multiple pathways, including angiogenesis, cell migration, myeloid cell differentiation, extracellular matrix (ECM) remodeling, helper T cell (Th) programming, antigen presentation, cytokine signaling, and T-cell activation were impacted, regardless of the route of administration of α-PD-1. These data support the further evaluation of SQ α-PD-1 in combination with PEDD of nelitolimod in LM.
Results
Antitumor activity of PEDD of nelitolimod on LM was enhanced by CPIs irrespective of the delivery route
To evaluate whether the route of administration of α-PD-1 antibody influenced combinatorial activity with nelitolimod, LM-bearing mice were treated with 10 µg of nelitolimod delivered regionally via PEDD followed by α-PD-1 treatment (125 µg/mouse) delivered Sys or SQ as shown in the treatment schema (figure 1A). We found that PEDD nelitolimod as monotherapy and in combination with α-PD-1, irrespective of the route of administration, reduced LM progression compared with vehicle (Veh) at 7 days post-treatment as measured by bioluminescence (D7, mean fold change±SEM over D0 bioluminescence—SQ/Sys/nelitolimod: 9.7±4.7/6.5±3.6/25.9±6.2 vs Veh: 85.6±27.8; p<0.0001) (figure 1B). Similar differences in LM burden decreases were noted at day 10 (D10, mean fold change±SEM over D0 bioluminescence—SQ/Sys/nelitolimod: 10.8±6.6/9.9±5.2/45.9±10.2 vs Veh: 98.70±19.92; p<0.0001) (figure 1B). Notably, unlike the PEDD of nelitolimod monotherapy, α-PD-1 monotherapy did not significantly impact LM progression (online supplemental figure 1A). Moreover, at D10 combination of PEDD-nelitolimod and α-PD-1 significantly controlled tumor progression as compared with PEDD-nelitolimod monotherapy (nelitolimod vs SQ; p<0.01 and nelitolimod vs Sys; p<0.0001, respectively). In summary, nelitolimod reduced LM burden compared with Veh and the addition of Sys or SQ α-PD-1 to nelitolimod further augmented tumor control over nelitolimod alone without any apparent negative impacts on the animals (figure 1B–C, online supplemental figure 1B-C).
Figure 1. Antitumor activity of PEDD of nelitolimod on LM was enhanced by checkpoint inhibitors irrespective of delivery route. (A) Schematic representation of the experimental procedures. 8–12-week-old C57/BL6 mice were challenged with 106 MC38-Luc cells/mouse and delivered via spleen followed by splenectomy. After 7 days mice were treated with nelitolimod via PEDD in combination with α-PD-1 administered Sys or SQ on days mentioned in the schema. (B) Bioluminescence values were determined by IVIS on D0, D2, D4, D7 and D10. Phosphate-buffered saline delivered via PV (Portal Vein) using PEDD served as Veh control. Fold change of the tumor burden was calculated based on D0 baseline bioluminescence (total flux/sec). Two-way ANOVA with Tukey’s multiple comparison test was performed to compare the tumor progression among the groups. (C) Mice were sacrificed on D10, and representative images (n=3–4 per group) depicting the bioluminescence at D10 and gross images of the harvested livers. (D) i Representative H&E images show (a–d) the morphology of tumor-bearing liver tissue followed by treatment (n=3); (e–h) depicting portal tract inflammation (white arrowheads) (n=3); (i–l) demonstrating lobular inflammation (black arrowheads) (n=3). ii Graphical representation of tumor burden data, iii table showing the presence or absence of portal tract or lobular inflammations within tumor-bearing liver treated with nelitolimod±α-PD-1, respectively. One-way ANOVA was performed to determine statistical differences among multiple groups. Data presented as the presence and absence of inflammation in the tissue obtained from representative mice per group are mentioned in the figure. Results are representative of at least three independent experiments with the cumulative n reported in the respective figures. Data is presented as mean±SEM; and p value is mentioned in the graph. ANOVA, analysis of variance; IVIS, in vivo imaging system; PEDD, pressure-enabled drug delivery; PD-1, programmed cell death-1; Veh, vehicle.

Histologically, the tumors were composed of epithelioid, and in some areas spindled cells were growing as small nodules and confluent sheets of cells (figure 1Di a and b) with stromal component demonstrated by trichrome stain in the Sys group (online supplemental figure 1D). Similar to the bioluminescence and the gross images, no tumors were identified histologically in the SQ and Sys groups as shown in the representative images (figure 1Di c and d). The histologic changes were consistent with LM bioluminescence data where the monotherapy effect of nelitolimod via PEDD was demonstrated and enhanced in combination with α-PD-1, irrespective of the route of administration. The mean tumor burden was significantly higher in the Veh group as opposed to the nelitolimod, SQ, and Sys group (p<0.01, figure 1Dii).
Normal liver parenchyma on treated and control animals was assessed to determine the broad impact of nelitolimod and CPI on the intrahepatic environment. The portal areas in the Veh and nelitolimod groups did not contain significant inflammatory cells (figure 1Di e and f). In contrast, portal inflammation composed mainly of lymphocytes and histiocytes was present in SQ and Sys groups (figure 1Di g and h). Lobular inflammation was absent in Veh group (figure 1Di i), while scattered lymphocytes and histiocytes were seen in nelitolimod (figure 1Di j), and more prominent lymphoid aggregates were seen in SQ and Sys groups (figure 1Di k and l). Figure 1Diii illustrates the presence of portal tract and lobular inflammation in the LM tissues of each group. There was no evidence of significant hepatocellular or biliary necrosis or organ damage.
Modulation of liver myeloid and lymphoid compartments by nelitolimod via PEDD was preserved in combination with Sys or SQ α-PD-1
To investigate the differential effects of α-PD-1 route of administration on the liver TME programming effects of nelitolimod, livers with established LM were harvested on D10, and CD45+ cells were isolated from bulk hepatic non-parenchymal cells. We have previously shown that monocytic-MDSCs (M-MDSCs) were the dominant subset of MDSCs in the setting of LM.24 The percentage of MDSCs, M-MDSCs, granulocytic-MDSCs (G-MDSCs), DC, B cells, T cells and, M1-like macrophages within the TME were quantified as per the gating strategy shown in online supplemental figure 2A. We found that treatment with PEDD of nelitolimod as monotherapy reduced MDSCs within TME (trending towards significance mean % of MDSCs±SEM—nelitolimod 15.7±2.8 vs Veh 25.9±4.5; p=0.07) compared with Veh, further significantly augmented by Sys or SQ α-PD-1 (figure 2A, mean % of MDSCs±SEM – SQ/Sys – 8.3±1.4/10.9±2.4 vs Veh 25.9±4.5; p<0.01). The ratio of M/G-MDSC was also decreased in all treatment groups compared with Veh (figure 2B, p<0.01), suggesting the reduction of the highly immunosuppressive M-MDSCs within the LM. We also found that PEDD of nelitolimod significantly increased CD11c+ DCs within TME, irrespective of Sys or SQ α-PD-1 (figure 2C, p<0.05) as compared with Veh control. The reduction in M-MDSCs and increase in CD11c+ DCs, was accompanied by upregulation of B cells, CD3+ T cells and M1-like macrophages (figure 2D–F, p<0.05). Immunofluorescence further confirmed that nelitolimod monotherapy and in combination with α-PD-1 (SQ or Sys) significantly decreased in MDSCs within the tumor (figure 2G i a–d and figure 2G ii) and drove robust CD3+CD8+ T-cell infiltration (figure 2G i e–h and figure 2G iii) significantly as compared with Veh. No significant differences were observed between Sys and SQ α-PD-1 groups (figure 2G).
Figure 2. Modulation of liver myeloid and lymphoid compartments by nelitolimod via pressure-enabled drug delivery was preserved in combination with Sys or SQ checkpoint inhibitor. Liver of tumor-bearing mice were harvested 10 days post-treatment. CD45+ cells were isolated from non-parenchymal cells. (A) MDSC cell population (CD11b+Gr1+), (B) monocytic MDSCs (M-MDSC; CD11b+Ly6C+/hiLy6G−/lo), (C) dendritic (CD11c+) cells, (D) B cells (B220+), (E) T cells (CD3+) and (F) M1-like macrophage (F/4/80+CD38+EGR2−) were quantified by flow cytometry. (G) i (a–h) Tumors were isolated from each group, OCT-mounted tissues were sectioned, fixed, and stained for CD3 (green), CD8 (red), CD11b (green), and Gr1 (red). (G) ii–iii Quantification of CD11b+Gr1+ MDSCs and CD3+CD8+T cells and from tumors of mice were performed across five fields/mouse and n=3 mice were used per group. Scale (20 µm). Animal data were presented as mean±SEM from and n was mentioned in the individual graph. One-way analysis of variance was performed to determine statistical differences among multiple groups. MDSC, myeloid-derived suppressor cells; M-MDSC, monocytic MDSC; G-MDSC, granulocytic MDSC; OCT, Optimal temperature cutting compound; DAPI, 4′,6-Diamidino-2-phenylindole; SQ, subcutaneous; Sys, systemic; Veh, vehicle.

Peripheral effects of PEDD of nelitolimod in combination with Sys or SQ α-PD-1
In vitro cultures of normal human peripheral blood mononuclear cells have demonstrated that TLR9 agonists broadly stimulate the production of interferons (IFN), including IFN-α by DCs and IFN-γ by T cells.38 C-X-C motif chemokine ligand 10/Interferon-gamma-induced protein 10 (CXCL10/IP-10) is a type I IFN–induced chemokine that mediates T-cell migration and has been used as the primary pharmacodynamic marker for TLR9 agonist-induced plasmacytoid dendritic cells (pDC) activation in vivo.39 To evaluate the peripheral pharmacodynamic effects of PEDD of nelitolimod, circulatory cytokine profiling using Luminex was performed on serum collected on D3. PEDD of nelitolimod resulted in increased IFN-γ and CXCL10 compared with Veh and there were no significant differences between PEDD of nelitolimod monotherapy and the addition of Sys or SQ α-PD-1 at D3 (figure 3A–B). These results are concordant with prior observations with intratumoral administration of TLR9 agonist and suggest that the changes in IFN-γ and CXCL10 represent pharmacodynamic effects of PEDD of nelitolimod and fortify the effect of α-PD-1 exerted after D3.
Figure 3. Peripheral effects of pressure-enabled drug delivery of nelitolimod in combination with Sys or SQ α-programmed cell death-1. The serum collected on D3 was analyzed for (A) IFN-γ and (B) CXCL10 (IP-10) were measured by Luminex and reported as fold change compared with Veh. Liver function tests were performed to monitor the level of (C) AST, (D) total bilirubin, (E) total protein, and (F) BUN. Normal ranges for C57/Bl6 AST: 46–221 U/L, bilirubin: 0.5–1.1 mg/dL, total protein: 4.6–7.3 g/dL and BUN: 2–71 mg/dL. Animal data were presented as mean±SEM and n was mentioned in the individual graph. One-way analysis of variance and multiple t-test was performed to determine statistical differences among multiple groups. CXCL10, C-X-C motif chemokine ligand 10; IP-10, Interferon-gamma-induced protein 10; AST, aspartate aminotransferase; IFN, interferon; SQ, subcutaneous; Sys, systemic; Veh, vehicle.

We also performed liver and kidney function tests to assess the potential impact of treatment on critical organ function in the three treatment groups and compared the values with Veh group. By measuring liver-related assays such as aspartate aminotransferase (AST), total bilirubin, and total protein, we found treatment with nelitolimod alone or in combination with aPD-1 did not adversely impact hepatic function (figure 3C–E).40 Alkaline phosphatase and alanine transaminase (ALT) levels remained within the normal range (online supplemental figure 2B). The kidney function surrogates similarly and did not demonstrate evidence of organ damage in any of the three treatment groups (figure 3F).
PEDD of nelitolimod in combination with Sys or SQ α-PD-1 promoted transcriptomic changes consistent with enhancement of antitumor immunity in the liver TME
Intratumoral nelitolimod promoted IFN production and cellular composition and overcome resistance to α-PD-1, resulting in durable antitumor immunity.38 Compared with Veh, PEDD of nelitolimod administration resulted in increased expression of genes associated with IFN signaling (Ifn-γ, Isg15, Ifit1bl1, Irf7), T-cell function (granzyme (Gzma)), and cellular immunity (CD274/Pd-l1, Cxcl9, Cxcl10, Cxcl11) (figure 4A). The addition of Sys or SQ α-PD-1 to PEDD of nelitolimod did not dramatically change the transcriptional profile of combination therapy (figure 4A). Mice treated with nelitolimod in combination with α-PD-1, delivered either via Sys or SQ affected most of the genes of the innate and myeloid panels (data not shown). As illustrated by the Venn diagram, approximately 70% of the genes were common among PEDD of nelitolimod, SQ and Sys groups significantly modulated compared with Veh at D3 (figure 4B).
Figure 4. PEDD-nelitolimod in combination with Sys or SQ α-PD-1 promoted transcriptomic changes consistent with enhancement of antitumor immunity in the liver tumor microenvironment. Total RNA was isolated from the CD45+ cells of liver metastases, and nCounter analysis was performed using myeloid and innate panel. (A) Heat map of genes that were significantly upregulated/downregulated followed by α-PD-1 Ctrl, nelitolimod, Sys and SQ treatment compared with Veh control were plotted. (B) The Venn diagram compared genes significantly modulated by PEDD of nelitolimod/SQ/Sys compared with Veh. (C) i–ii Expression of Ifn-γ and granzyme were quantified by qRT PCR. (D) Pathways that were altered by nelitolimod, α-PD-1 monotherapy or combination therapy compared with Veh, evaluated by using nSolver advanced analysis module and plotted, respectively. Data presented as mean±SEM and n are mentioned in the individual graph. One-way analysis of variance was performed to determine statistical differences among multiple groups. qRT, quantitative reverse transcription; IFN, interferon; ECM, extracellular matrix; PEDD, pressure-enabled drug delivery; PD-1, programmed cell death-1; SQ, subcutaneous; Sys, systemic; Veh, vehicle.

Genes such as Gzma, Ifn-γ, Fpr2, Cxcl11, Cxcl9, IP-10 (Cxcl10) were upregulated and Fn1, Pf4, Elane, Arg1, and Encep were significantly downregulated as compared to the Veh group in all treatment groups (figure 4A and online supplemental figure 4A). Quantitative Reverse transcription (RT)-PCR was performed to evaluate the expression Gzma and Ifn-γ in CD45+ cells isolated from LM-bearing tissue. We found that nelitolimod with or without α-PD-1 enhanced the expression of Gzma (figure 4Ci) and Ifn-γ (figure 4Cii). Figure 4D illustrates the impact of treatment on critical cellular pathways. Gene signatures related to cell migration and adhesion, differentiation, and maintenance of myeloid cell, Th2 and metabolism were downregulated following treatment with nelitolimod via PEDD alone or in combination with α-PD-1 compared with Veh or α-PD-1 Ctrl mice (figure 4D, online supplemental figure 4A). However, pathways related to angiogenesis and ECM remodeling were enhanced in the combinatorial treatment of nelitolimod via PEDD with α-PD-1. Increased gene expression in relation to antigen presentation, IFN signaling, T-cell activation and checkpoint, and Th1 activation were upregulated following treatment with nelitolimod via PEDD alone or in combination with α-PD-1. Combination of PEDD nelitolimod with α-PD-1 either Sys or SQ increased cytokine signaling-related gene expression. There were no differences in the gene expression or pathway score profiles in Veh versus α-PD-1 Ctrl or Sys versus SQ groups. Notably, the cytotoxic T-cell score was elevated in both nelitolimod monotherapy and in combination with α-PD-1 compared with Veh (online supplemental figure 4B).
PEDD of nelitolimod in combination with α-PD-1 improved the survival of tumor-bearing mice irrespective of the route of administration
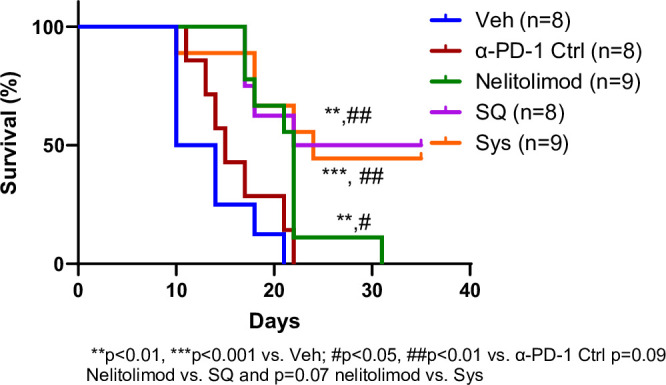
We showed that PEDD of nelitolimod monotherapy promoted antitumor immunity by modulating critical immunological pathways in LM associated with decreased intrahepatic tumor burden. To assess the durability of these effects in mice with LM, we conducted a survival study. The median survival of mice treated with nelitolimod monotherapy was significantly different compared with α-PD-1 Ctrl (p<0.05) and Veh (p<0.01) groups. The median survival days for nelitolimod, α-PD-1 Ctrl and Veh groups were 22, 15 and 12 days, respectively. Moreover, the survival curves for SQ (p<0.01 vs. Veh and α-PD-1 Ctrl) and Sys groups (p<0.001 vs Veh and p<0.01 vs α-PD-1 Ctrl) were significantly different compared with Veh and α-PD-1 Ctrl. The median survival days for SQ and Sys groups were 28.5 and 24 days, respectively, and there were no significant differences between SQ and Sys groups (figure 5). The addition of Sys or SQ α-PD-1 to PEDD of nelitolimod further improved survival significantly, with 44% (4/9) of Sys and 50% (4/8) of SQ mice alive at D35, compared with no surviving animals in the other groups (figure 5). The corresponding tumor burden was similar to figure 1B with individual time-dependent data points up to D35 as shown in online supplemental figure 5, demonstrating antitumor effects of PEDD of nelitolimod monotherapy and in combination with α-PD-1. We concluded that in this aggressive LM model, survival was extended by nelitolimod monotherapy and was further augmented by α-PD-1 combination therapy.
Figure 5. PEDD-nelitolimod in combination with α-PD-1 improved the survival of tumor-bearing mice irrespective of the route of administration. Kaplan-Meier survival curve of liver metastases-bearing mice followed by intravascular PEDD nelitolimod with α-PD-1 administered either Sys or SQ. n denotes the number of mice in each group. Log-rank test was performed to determine the statistical differences among groups. n is mentioned in the individual graph. PEDD, pressure-enabled drug delivery; PD-1, programmed cell death-1; SQ, subcutaneous; Sys, systemic; Veh, vehicle.
Discussion
The present study was conducted to determine if SQ CPI could enhance the monotherapy effect of nelitolimod delivered via PEDD in a murine model of LM as previously reported for systemic α-PD-1.24 41 This study could impact combinatorial immunotherapy approaches to treat intrahepatic tumors. We demonstrated that the augmentation of antitumor efficacy of PEDD nelitolimod in combination with SQ α-PD-1 is similar to Sys α-PD-1. The differential effects across treatment groups on LM progression and outcome were associated with liver TME modulation, with favorable changes including liver MDSC depletion, enhanced T-cell infiltration, and transcriptomic changes across various immunologic pathways. Nelitolimod monotherapy initiated the TME programming effects, and the addition of SQ or Sys α-PD-1 significantly enhanced the control of LM in conjunction with the prolongation of animal survival times.
Nelitolimod is currently undergoing clinical evaluation in three-phase 1/1b trials for liver (NCT04935229, NCT05220722) and pancreatic tumor (NCT05607953) patients. The selection of nelitolimod for these indications was based on the ability of this class C TLR9 agonist to exert a dual mechanism of action in the liver TME: (1) Liver MDSC depletion with favorable myeloid compartment reprogramming and (2) broad TME modulation to support T-cell infiltration and enhanced CPI performance. As a class C TLR9 agonist, nelitolimod may potentially have specific properties, rendering it more suitable for addressing the distinct immunosuppressive pathways present in the setting of LM.42 43 Here, we confirmed that the route of α-PD-1 administration did not impact the ability of combination with PEDD of nelitolimod to control LM in a murine model.
The favorable antitumor effects of PEDD of nelitolimod in combination with α-PD-1 in the murine LM model align well with early clinical results from a phase 1 trial for patients with uveal melanoma LM.44 Although the projected cost reduction, including reduced infusion time and drug manipulation costs are appealing, the SQ formulation might require higher doses. Prolonged disease control, favorable myeloid reprogramming, enhancement of T-cell antitumor functionality, and favorable change in circulating tumor DNA seen in phase 1 trial patients align with the bioluminescence data reported herein (figure 1B). In addition, liver MDSC depletion, induction of T-cell activation in LM, and increase in peripheral cytokine have also been seen in the clinic.25 Importantly, the lack of liver or renal toxicity seen in the murine model resonates with a severe treatment-related adverse event rate of 5% in phase 1 patients receiving nelitolimod+intravenous α-PD-1 therapy, which was recently reported.44
A limitation of this work is that we did not conduct pharmacokinetic (PK) studies to compare the absorption profiles of Sys or SQ α-PD-1 in the presence of nelitolimod. Johnson et al performed a safety, efficacy, and PK study of PF-06801591, where an intravenous dose of 0.5, 1, 3, or 10 mg/kg was administered every 3 weeks, or a 300 mg/patient via SQ was administered every 4 weeks.30 Intravenous PF-06801591 showed linear PKs over the dose range of 0.5–10 mg/kg, but the first SQ dose was absorbed slowly, and an additional monthly dose resulted in a drug level comparable to the intravenous infusion.30 Notably, the antitumor efficacy of Sys and SQ α-PD-1 combined with nelitolimod were equivalent. In this study, we have shown that nelitolimod via PEDD was crucial to alter the TME and demonstrated monotherapy activity, but the addition of α-PD-1, irrespective of the route of administration, further potentiated LM control. SQ administration of CPIs is convenient and economical compared with the recommended intravenous delivery of CPIs.
While combining PEDD of nelitolimod with Sys or SQ α-PD-1 resulted in superior outcomes, nelitolimod monotherapy significantly diminished LM progression. Nelitolimod also appeared to be largely responsible for liver MDSC depletion, particularly the M-MDSC subset, along with the broad TME effects documented earlier.44 Nelitolimod activates the TLR9 pathway via Nuclear factor-κB (NF-κB) activation and deactivates signal transducer and activator of transcription 3 (STAT3) in myeloid cells which induces MDSC apoptosis and a shift toward M1-like macrophage programming.24 It has been shown that MDSCs expand both in human and mouse liver cancers, correlating with poor clinical outcomes.45 In general, MDSCs can be found in circulation and within LM.46 Also, the infiltration of B cells is associated with the survival of patients with colorectal cancers (CRC) and activated B cells can inhibit LM in murine CRC models.47 Nelitolimod via PEDD promoted liver B-cell expansion in this model. As such, the dual mechanism of action of nelitolimod in LM, with its effect on myeloid cells and broad TME programming, may be well suited to enhancing α-PD-1 performance in intrahepatic malignancy.
Tumors secrete diverse circulatory factors that promote the migration and accumulation of MDSCs into the hepatic tissues. Moreover, tumor-induced liver MDSCs are more immunosuppressive than peripheral MDSCs.48 The TME myeloid programming effect of nelitolimod extends beyond MDSC. Nelitolimod promotes the expansion of M1-like macrophages and drives the contraction of the M2 subset.24 There were no additive effects of α-PD-1 with nelitolimod in reducing MDSCs and in upregulating the infiltration of B, CD11c+, and T cells to the LM compared with nelitolimod monotherapy, suggesting that PEDD is primarily responsible for the favorable TME effects demonstrated in this model. Based on clinical trial LM biopsy sample analysis, nelitolimod monotherapy has similar effects in patients.44
Transcriptomic analysis revealed that while α-PD-1 treatment alone had minimal impact on critical TME immune signatures, PEDD nelitolimod in combination with either Sys or SQ α-PD-1 favorably modulated multiple immune signatures in the TME as evidenced by the increases in Gzma, and chemokines critical to monocyte (Cxcl10 and Cxcl11) and DC (Fpr2) traffic.
This study also highlights that PEDD-based delivery of nelitolimod effectively delivers the drug throughout the liver in the setting of LM, which may be associated with favorable effects on intravascular pressure and flow.21 In clinical trials, nelitolimod delivered by PEDD has been associated with a tolerable safety profile with or without CPI, and PK data demonstrate high intrahepatic drug levels with low systemic exposure.20 44 49 Immune-related adverse events (irAEs) of CPIs are widespread and both Sys and SQ delivery demonstrated equivalent serious irAEs,30 50
Our data demonstrates that either SQ or Sys α-PD-1 enhanced the monotherapy effect of PEDD of nelitolimod and were similar regarding safety, immunological responses, and antitumor efficacy. Data from the murine LM PEDD model presented herein aligns with recent phase 1 clinical data.25 PEDD of nelitolimod had favorable immunomodulatory effects on the liver TME, and this addition of CPI, irrespective of administration route, has the potential to support durable disease control.
Materials and methods
Mice, in vivo model, and treatment
We purchased C57BL/6J (stock number: 000664), aged 8–12 weeks from Jackson Laboratories (Bar Harbor, Maine, USA) and housed under pathogen-free conditions in the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-approved Animal Research Facility at Lifespan, Coro East (Rhode Island, USA). All surgical procedures were performed as per Lifespan’s Institutional Animal use and Care Committee’s approved protocol (Protocol#: 5010–22). To develop LM model, we injected 1.0 e6 MC38- Luc cells (Ubigene, China) via the spleen, followed by splenectomy as described previously.24 After 7 days mice were randomized into different treatment groups based on their tumor burden and treated with 10 µg nelitolimod labeled with IRD-800 delivered via portal vein (PV) using PEDD with or without α-PD-1 antibody (BE0146; clone: RMP1-14, Bio X Cell, New Hampshire, USA) administered either via intraperitoneally (Sys: PEDD SD101+IP α-PD-1 antibody) or subcutaneously (SQ: PEDD SD101+SQ α-PD-1 antibody). The α-PD-1 antibody was administered on D0; D2; D4 and D7. Tumor progression was monitored by using IVIS Lumina III Imaging System (PerkinElmer, Connecticut, USA) on D0, D2, D4, D7 and D10 described before.49 Briefly, tumor-bearing mice were injected with D-luciferin (150 mg kg−1, PerkinElmer, Connecticut, USA) via intraperitoneal (IP) route and luminescence-positive signals were detected by IVIS Spectrum CT system after 10 min injection. For some experiments, dissected liver tissues were subjected to imaging analysis. Metastatic lesions were further validated by H&E histological analysis. Masson’s trichrome staining against collagen fibers.
Mice were sacrificed on D3 and D10, blood and liver were collected. Non-parenchymal cells (NPCs) were isolated from the liver, followed by isolating CD45+ cells using immuno-magnetic beads (Miltenyi Biotech, Massachusetts, USA) as described earlier.43 Flow cytometry (CytoFLEX, Beckman Coulter, Indiana, USA) was performed to determine MDSCs, T, B, dendritic (CD11c+) cells, total RNA collected from CD45+ cells at D3 was used for NanoString analysis (nCounter, NanoString, Washington, USA) and circulatory cytokines such as IFN-γ, CXCL10 (IP-10), interleukin (IL)-6, IL12, IL-18, IL-2R and Monocyte chemoattractant protein-1 (MCP1) were analyzed using ProcartaPlex Luminex kit (Thermo Fisher Scientific, Massachusetts, USA) and were measured by Magpix (Luminex Corp, Texas, USA). Only detectable cytokines were reported. For the survival assay upto D35, the α-PD-1 antibody was administered on D0, D2, D4, D7, and D10 and in vivo imaging system was performed on D0, D2, D4, D7, D10, D14 and twice a week afterwards upto D35.
H&E, trichrome mason and immunofluorescence staining
H&E-stained liver tissues were evaluated under the light microscope for tumor burden, tumor necrosis, portal inflammation, and lobular injury by two observers (gastrointestinal and liver pathologist). In addition, fibrosis was evaluated on trichrome-stained slides. Images were taken (Olympus BX41, Japan) and the tumor burden was calculated as a percentage of the tumor per whole tissue section. Cryosectioned tissue (5 µm thick) was made and fixed on slides by iHisto (Massachusetts, USA). Primary antibodies and their corresponding clones used for immunofluorescence (IF) staining: CD11b; E6E1M (Cell Signaling, Massachusetts, USA), GR1; RB6-8C5 (BioLegend, California, USA), CD3; SP7 (Novus Biologicals, USA) and CD8a; 53–6.7 µ (BioLegend, California, USA). For DAPI (4′,6-Diamidino-2-phenylindole) staining and mounting, Molecular Probes Prolong Diamond Antifade Mountant with DAPI from Thermo Fisher Scientific (Massachusetts, USA) was used. Slides were imaged using the PhenoImager from Akoya Biosciences (Massachusetts, USA). Images were quantified by ImageJ software.
Antibodies for flow cytometry
Antibodies used to determine the following antigens along with clones are given below.
Gr1 (Ly6G/Ly6C, RB6-8C5), CD11b (M1/170), Ly6C (AL-21), and Ly6G (1A8) obtained from BD Biosciences (California, USA), F/480 (BM8), CD3 (17A2clones), B220 (B cell) and CD38 (90) obtained from BioLegend (California, USA) and EGR2 (ERONGR2) procured from Thermo Fisher Scientific (Massachusetts, USA). Zombie NIR (BioLegend, California, USA) was used as a cell viability dye. Single-cell suspensions of samples were acquired using a CytoFLEX LX Flow Cytometer and analyzed by CytExpert (Beckman Coulter, Indiana, USA).
NanoString analysis
Total RNA was isolated from immune magnetically separated CD45+ cells from LM. At D3 of the treatment LM was harvested followed by NPC isolation.24 46 Mouse CD45+ microbeads were added to the single cells and CD45+ cells were isolated using LS columns (Miltenyi Biotec, Massachusetts, USA). By using the RNeasy Plus Kit (Qiagen, Maryland, USA), total RNAs from the CD45+ cells were isolated. Mouse nCounter Myeloid Innate Immunity V.2 (NanoString, Washington, USA) panel was used to evaluate the RNA samples. NanoString—nCounter Sprint Profiler was used at the Genomics core, Brown University (Providence, Rhode Island, USA). The nSolver V.4.0 software package was used to perform advanced analysis.
qRT-PCR
Isolated RNAs from CD45+ cells were reverse transcribed by using an iScript cDNA Synthesis Kit (Bio-Rad, California, USA), and SYBR Master Mix (Bio-Rad) was used for quantitative PCR by using the following primers:
| Primers | Forward | Reverse |
| Ifn-γ | TGCCAAGTTTGAGGTCAACAAC | GTGGACCACTCGGATGAGC |
| Gzma | CAAACGTGCTTCCTTTCGGG | CGTGGAGGTGAACCATCCTT |
| Rpl27 | GAGGAGCGATACAAGACAGG | CCCAGTCTCTTCCCACACAAA |
For all samples, ΔCt values were calculated, and Rpl27 (mouse) was used to normalize the gene expression.
Statistics
Statistical significance was analyzed by using one-way analysis of variance (ANOVA), two-way ANOVA and multiple t-tests as mentioned in figure legends. For one-way and two-way ANOVA Tukey’s post hoc multiple comparison test was performed. For multiple t-test, two-stage step-up procedure of Benjamini, Krieger, and Yekutieli, with Q=1% was performed. Prism (V.10) software (GraphPad, San Diego, California, USA) was used to analyze data. For all studies, values of p<0.05 was considered statistically significant.
supplementary material
Footnotes
Provenance and peer review: Not commissioned; externally peer reviewed.
Patient consent for publication: Not applicable.
Ethics approval: Not applicable.
Contributor Information
Chandra C Ghosh, Email: chandra.ghosh@trisaluslifesci.com.
Lauren Cournoyer, Email: lcournoyer@lifespan.org.
Yujia Liu, Email: jennie.liu@trisaluslifesci.com.
Alizee Ballarin, Email: alizee.ballarin@trisaluslifesci.com.
Ilan B Layman, Email: ilayman@lifespan.org.
Jason LaPorte, Email: jason.laporte@trisaluslifesci.com.
Molly Morrissey, Email: molly.morrissey@trisaluslifesci.com.
Kayla Fraser, Email: kayla.fraser@trisaluslifesci.com.
Shriya Perati, Email: shriya.perati@trisaluslifesci.com.
Bryan F Cox, Email: bryan.cox@trisaluslifesci.com.
Evgeny Yakirevich, Email: evgeny_yakirevich@brown.edu.
Diana O Treaba, Email: DTreaba@Lifespan.org.
Timothy D Murtha, Email: timothy.murtha@brownphysicians.org.
Prajna Guha, Email: prajna.guha@trisaluslifesci.com.
Steven C Katz, Email: steven.katz@trisaluslifesci.com.
Diwakar Davar, Email: davard@upmc.edu.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
References
- 1.Brunet JF, Denizot F, Luciani MF, et al. A new member of the immunoglobulin superfamily--CTLA-4. Nature New Biol. 1987;328:267–70. doi: 10.1038/328267a0. [DOI] [PubMed] [Google Scholar]
- 2.Dong H, Zhu G, Tamada K, et al. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and Interleukin-10 secretion. Nat Med. 1999;5:1365–9. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 3.Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 Immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–34. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ai L, Chen J, Yan H, et al. Research status and outlook of PD-1/PD-L1 inhibitors for cancer therapy. Drug Des Devel Ther. 2020;14:3625–49. doi: 10.2147/DDDT.S267433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang C. Retifanlimab: first approval. Drugs (Abingdon Engl) 2023;83:731–7. doi: 10.1007/s40265-023-01884-7. [DOI] [PubMed] [Google Scholar]
- 7.Mirza MR, Chase DM, Slomovitz BM, et al. Dostarlimab for primary advanced or recurrent endometrial cancer. N Engl J Med. 2023;388:2145–58. doi: 10.1056/NEJMoa2216334. [DOI] [PubMed] [Google Scholar]
- 8.Korman AJ, Garrett-Thomson SC, Lonberg N. The foundations of immune checkpoint blockade and the Ipilimumab approval decennial. Nat Rev Drug Discov. 2022;21:509–28. doi: 10.1038/s41573-021-00345-8. [DOI] [PubMed] [Google Scholar]
- 9.Sasaki A, Nakamura Y, Mishima S, et al. Predictive factors for hyperprogressive disease during Nivolumab as anti-Pd1 treatment in patients with advanced gastric cancer. Gastric Cancer. 2019;22:793–802. doi: 10.1007/s10120-018-00922-8. [DOI] [PubMed] [Google Scholar]
- 10.Topalian SL, Hodi FS, Brahmer JR, et al. Five-year survival and correlates among patients with advanced melanoma, renal cell carcinoma, or non-small cell lung cancer treated with Nivolumab. JAMA Oncol. 2019;5:1411–20. doi: 10.1001/jamaoncol.2019.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tumeh PC, Hellmann MD, Hamid O, et al. Liver metastasis and treatment outcome with anti-PD-1 monoclonal antibody in patients with melanoma and NSCLC. Cancer Immunol Res. 2017;5:417–24. doi: 10.1158/2326-6066.CIR-16-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daud AI, Loo K, Pauli ML, et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human Melanoma. J Clin Invest. 2016;126:3447–52. doi: 10.1172/JCI87324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loo K, Tsai KK, Mahuron K, et al. Partially exhausted tumor-infiltrating lymphocytes predict response to combination immunotherapy. JCI Insight. 2017;2:e93433. doi: 10.1172/jci.insight.93433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ostrand-Rosenberg S, Fenselau C. Myeloid-derived Suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol. 2018;200:422–31. doi: 10.4049/jimmunol.1701019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hou A, Hou K, Huang Q, et al. Targeting myeloid-derived suppressor cell, a promising strategy to overcome resistance to immune checkpoint inhibitors. Front Immunol. 2020;11:783. doi: 10.3389/fimmu.2020.00783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer C, Cagnon L, Costa-Nunes CM, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with Ipilimumab. Cancer Immunol Immunother. 2014;63:247–57. doi: 10.1007/s00262-013-1508-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu J, Green MD, Li S, et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat Med. 2021;27:152–64. doi: 10.1038/s41591-020-1131-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ribas A, Medina T, Kirkwood JM, et al. Overcoming PD-1 blockade resistance with Cpg-A toll-like receptor 9 agonist vidutolimod in patients with metastatic melanoma. Cancer Discov. 2021;11:2998–3007. doi: 10.1158/2159-8290.CD-21-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ribas A, Medina T, Kummar S, et al. SD-101 in combination with pembrolizumab in advanced melanoma: results of a phase IB, multicenter study. Cancer Discov. 2018;8:1250–7. doi: 10.1158/2159-8290.CD-18-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katz SC, Moody AE, Guha P, et al. HITM-SURE: hepatic immunotherapy for metastases phase IB anti-CEA CAR-T study utilizing pressure enabled drug delivery. J Immunother Cancer. 2020;8:e001097. doi: 10.1136/jitc-2020-001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaroch DB, Ghosh CC, Guha P, et al. Abstract 1858: enhanced delivery of anti PD-1 antibody to liver tumors in oncopig using pressure enabled drug delivery (PEDD) versus a systemic delivery. Cancer Res. 2023;83:1858. doi: 10.1158/1538-7445.AM2023-1858. [DOI] [Google Scholar]
- 22.Capacio BA, Shankara Narayanan JS, Vicente DA, et al. Pressure-enabled drug delivery (PEDD) of a class C Tlr9 agonist in combination with checkpoint inhibitor therapy in a murine pancreatic cancer model. Surgery. 2023;174:666–73. doi: 10.1016/j.surg.2023.05.011. [DOI] [PubMed] [Google Scholar]
- 23.Mehta AK, Kadel S, Townsend MG, et al. Macrophage biology and mechanisms of immune suppression in breast cancer. Front Immunol. 2021;12:643771. doi: 10.3389/fimmu.2021.643771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh CC, Heatherton KR, Connell KPO, et al. Regional infusion of a class C Tlr9 agonist enhances liver tumor microenvironment reprogramming and MDSC reduction to improve responsiveness to systemic checkpoint inhibition. Cancer Gene Ther. 2022;29:1854–65. doi: 10.1038/s41417-022-00484-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel SP, Carvajal R, Montazeri K, et al. 1534 Clinical activity of sd-101 with immune checkpoint inhibition (ici) in metastatic uveal melanoma liver metastasis (mum-lm) from the perio-01 phase 1 trial. SITC 38th Annual Meeting (SITC 2023) Abstracts Supplement 2; Nov, 2023. [DOI] [Google Scholar]
- 26.Pelster MS, Gruschkus SK, Bassett R, et al. Nivolumab and Ipilimumab in metastatic uveal melanoma: results from a single-arm phase II study. J Clin Oncol. 2021;39:599–607. doi: 10.1200/JCO.20.00605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piulats JM, Espinosa E, de la Cruz Merino L, et al. Nivolumab plus Ipilimumab for treatment-naive metastatic uveal melanoma: an open-label, multicenter, phase II trial by the Spanish multidisciplinary melanoma group (GEM-1402) J Clin Oncol. 2021;39:586–98. doi: 10.1200/JCO.20.00550. [DOI] [PubMed] [Google Scholar]
- 28.Shin YE, Kumar A, Guo JJ. Spending, utilization, and price trends for immune checkpoint inhibitors in US medicaid programs: an empirical analysis from 2011 to 2021. Clin Drug Investig. 2023;43:289–98. doi: 10.1007/s40261-023-01254-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franken MG, Kanters TA, Coenen JL, et al. Potential cost savings owing to the route of administration of oncology drugs: a microcosting study of intravenous and subcutaneous administration of trastuzumab and rituximab in the Netherlands. Anticancer Drugs. 2018;29:791–801. doi: 10.1097/CAD.0000000000000648. [DOI] [PubMed] [Google Scholar]
- 30.Johnson ML, Braiteh F, Grilley-Olson JE, et al. Assessment of subcutaneous vs intravenous administration of anti-PD-1 antibody PF-06801591 in patients with advanced solid tumors: a phase 1 dose-escalation trial. JAMA Oncol. 2019;5:999–1007. doi: 10.1001/jamaoncol.2019.0836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papadopoulos KP, Harb W, Peer CJ, et al. First-in-human phase I study of envafolimab, a novel subcutaneous single-domain anti-PD-L1 antibody, in patients with advanced solid tumors. Oncologist. 2021;26:e1514–25. doi: 10.1002/onco.13817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shimizu T, Nakajima TE, Yamamoto N, et al. Phase I study of envafolimab (Kn035), a novel subcutaneous single-domain anti-PD-L1 monoclonal antibody, in Japanese patients with advanced solid tumors. Invest New Drugs. 2022;40:1021–31. doi: 10.1007/s10637-022-01287-7. [DOI] [PubMed] [Google Scholar]
- 33.Zhao Y, Sanghavi K, Roy A, et al. Model-based dose selection of subcutaneous nivolumab in patients with advanced solid tumors. Clin Pharmacol Ther. 2024;115:488–97. doi: 10.1002/cpt.3148. [DOI] [PubMed] [Google Scholar]
- 34.Davies A, Berge C, Boehnke A, et al. Subcutaneous Rituximab for the treatment of B-cell hematologic malignancies: a review of the scientific rationale and clinical development. Adv Ther. 2017;34:2210–31. doi: 10.1007/s12325-017-0610-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Cock E, Kritikou P, Sandoval M, et al. Time savings with rituximab subcutaneous injection versus rituximab intravenous infusion: a time and motion study in eight countries. PLoS One. 2016;11:e0157957. doi: 10.1371/journal.pone.0157957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Cock E, Pivot X, Hauser N, et al. A time and motion study of subcutaneous versus intravenous trastuzumab in patients with Her2-positive early breast cancer. Cancer Med. 2016;5:389–97. doi: 10.1002/cam4.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rummel M, Kim TM, Aversa F, et al. Preference for subcutaneous or intravenous administration of rituximab among patients with untreated Cd20+ diffuse large B-cell lymphoma or follicular lymphoma: results from a prospective, randomized, open-label, crossover study (Prefmab) Ann Oncol. 2017;28:836–42. doi: 10.1093/annonc/mdw685. [DOI] [PubMed] [Google Scholar]
- 38.Wang S, Campos J, Gallotta M, et al. Intratumoral injection of a Cpg oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional Cd8+ T cells. Proc Natl Acad Sci U S A. 2016;113:E7240–9. doi: 10.1073/pnas.1608555113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blackwell SE, Krieg AM. Cpg-A-induced monocyte IFN-gamma-inducible Protein-10 production is regulated by plasmacytoid dendritic cell-derived IFN-alpha. J Immunol. 2003;170:4061–8. doi: 10.4049/jimmunol.170.8.4061. [DOI] [PubMed] [Google Scholar]
- 40.UCLA . DLAM. Pathology & Laboratory Medicine Services U, editor; 2013. Serum chemistry reference range - Mouse. [Google Scholar]
- 41.Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125:3384–91. doi: 10.1172/JCI80011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghosh C, Katz S, Ghosh C. Impact of SD-101, a toll-like receptor 9 class C (Tlr9C), agonist on myeloid derived Suppressor cells. In: Ghosh CC, LaPorte J, Guha P, editors. SITC 37th Annual Meeting (SITC 2022) Abstracts; Nov, 2022. In. [DOI] [Google Scholar]
- 43.Guha P, Gardell J, Darpolor J, et al. Stat3 inhibition induces bax-dependent apoptosis in liver tumor myeloid-derived Suppressor cells. Oncogene. 2019;38:533–48. doi: 10.1038/s41388-018-0449-z. [DOI] [PubMed] [Google Scholar]
- 44.2023 American society of clinical oncology, annual meeting. Safety and early biologic effects of phase 1 PERIO-01 trial of pressure-enabled drug delivery (PEDD) of TLR9 agonist SD-101 and immune checkpoint inhibition (ICI) in uveal melanoma metastatic to the liver (MUM); 2023. [Google Scholar]
- 45.Ma C, Zhang Q, Greten TF. Mdscs in liver cancer: a critical tumor-promoting player and a potential therapeutic target. Cell Immunol. 2021;361:104295. doi: 10.1016/j.cellimm.2021.104295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thorn M, Point GR, Burga RA, et al. Liver metastases induce reversible hepatic B cell dysfunction mediated by Gr-1+Cd11B+ myeloid cells. J Leukoc Biol. 2014;96:883–94. doi: 10.1189/jlb.3A0114-012RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu Y, Wei Z, Feng M, et al. Tumor-infiltrated activated B cells suppress liver metastasis of colorectal cancers. Cell Rep. 2022;40:111295. doi: 10.1016/j.celrep.2022.111295. [DOI] [PubMed] [Google Scholar]
- 48.Li K, Shi H, Zhang B, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Sig Transduct Target Ther. 2021;6:362. doi: 10.1038/s41392-021-00670-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chai LF, Hardaway JC, Heatherton KR, et al. Regional delivery of anti-PD-1 agent for colorectal liver metastases improves therapeutic index and anti-tumor activity. Vaccines (Basel) 2021;9:807. doi: 10.3390/vaccines9080807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martins F, Sofiya L, Sykiotis GP, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16:563–80. doi: 10.1038/s41571-019-0218-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data relevant to the study are included in the article or uploaded as supplementary information.