

Abstract
Impaired telomere length (TL) maintenance in ovarian tissue may play a pivotal role in the onset of epithelial ovarian cancer (OvC). TL in either target or surrogate tissue (blood) is currently being investigated for use as a predictor in anti-OvC therapy or as a biomarker of the disease progression, respectively. There is currently an urgent need for an appropriate approach to chemotherapy response prediction.
We performed a monochrome multiplex qPCR measurement of TL in peripheral blood leukocytes (PBL) and tumor tissues of 209 OvC patients. The methylation status and gene expression of the shelterin complex and telomerase catalytic subunit (hTERT) were determined within tumor tissues by High-Throughput DNA methylation profiling and RNA sequencing (RNA-Seq) analysis, respectively. The patients sensitive to cancer treatment (n = 46) had shorter telomeres in PBL compared to treatment-resistant patients (n = 93; P = 0.037). In the patients with a different therapy response, transcriptomic analysis showed alterations in the peroxisome proliferator-activated receptor (PPAR) signaling pathway (q = 0.001). Moreover, tumor TL shorter than the median corresponded to better overall survival (OS) (P = 0.006). TPP1 gene expression was positively associated with TL in tumor tissue (P = 0.026).
TL measured in PBL could serve as a marker of platinum therapy response in OvC patients. Additionally, TL determined in tumor tissue provides information on OvC patients' OS.
Keywords: Telomere length, Ovarian cancer, Shelterin, Telomerase, Therapy response
Highlights
-
•
Ovarian cancer chemotherapy responders have shorter blood telomeres than those who are treatment resistant.
-
•
Longer telomeres in ovarian tumors may be associated with decreased overall survival.
-
•
CpG methylation patterns in telomere-related genes indicate telomere length.
-
•
TPP1 gene expression positively correlates with telomere length in ovarian carcinomas.
Abbreviations
- ADIPOQ
adiponectin
- AMPK
adenosine monophosphate-activated protein kinase
- ATM kinase
ataxia-telangiectasia mutated kinase
- ATR kinase
ataxia-telangiectasia and Rad3-related kinase
- AUC
area under the curve
- BCL7A
B-cell CLL/lymphoma 7 A
- BH
Benjamini and Hochberg
- bp
base pairs
- BRCA1
breast cancer 1
- BRCA2
breast cancer 2
- c-MYC
c-MYC proto-oncogene
- CASC1
cancer susceptibility candidate gene 1
- CD36
cluster determinant 36
- CFAP45
cilia and flagella associated protein 45
- CLDN11
claudin 11
- CLDN18
claudin 18
- CLPTM1L
cleft lip and palate transmembrane protein 1-like protein
- CpG
cytosine-phosphate-guanine
- DE
differential expression
- DKC1
dyskerin pseudouridine synthase 1
- dsDNA
double-stranded DNA
- EFHC2
EF-hand domain containing 2
- FABP4
fatty acid binding protein 4
- FIGO
International Federation of Gynecology and Obstetrics
- FMR1
fragile X messenger ribonucleoprotein 1
- GO
gene ontology
- GSEA
Gene set enrichment analysis
- GWAS
genome-wide association studies
- HNF4A
hepatocyte nuclear factor 4 alpha
- HNRNPAB
heterogenous nuclear ribonucleoprotein A/B
- hTERC
telomerase RNA component
- hTERT
telomerase reverse transcriptase
- HYDIN
axonemal central pair apparatus protein
- JAM3
junction adhesion molecule 3
- KDM2A
lysine demethylase 2 A
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KLB
klotho beta
- log2 FC
log2 fold change
- LPL
lipoprotein lipase
- NASP
nuclear autoantigenic sperm protein
- NR4A3
nuclear receptor 4A3
- OS
overall survival
- OvC
ovarian cancer
- PBL
peripheral blood leukocytes
- PCA
principal component analysis
- PFI
platinum-free interval
- PIGR
polymeric immunoglobulin receptor
- PLIN2
perilipin 2
- POLE3
DNA polymerase epsilon 3
- POT1
protection of telomeres 1
- PPAR
peroxisome proliferator-activated receptor
- qPCR
quantitative polymerase chain reaction
- R
residuum
- RAP1
repressor/activator protein 1
- RBBP8
retinoblastoma-binding protein 8
- RIBC2
RIB43A domain with coiled-coils 2
- RIN
RNA integrity number
- RNA-Seq
RNA sequencing
- S
single-copy gene
- SMC1A
structural maintenance of chromosomes 1 A
- SNP
single nucleotide polymorphism
- ssDNA
single-stranded DNA
- SSRP1
structure specific recognition protein 1
- T
telomere
- TERF1
telomeric repeat binding factor 1
- TERF2
telomeric repeat binding factor 2
- TIN2
TERF1-interacting nuclear factor 2
- TL
telomere length
- TPP1
telomere protective protein 1
- TSS
transcription start site
- UPF1
DNA helicase and adenosine triphosphatase
- UTR
untranslated region
- YEATS4
YEATS domain containing 4
1. Introduction
Human telomeres are made of highly conserved tandem nucleic acid repeats and associated proteins of the shelterin complex located at the ends of chromosomes [1]. The shelterin subunits, namely telomere protective protein 1 (TPP1), repressor/activator protein 1 (RAP1), telomeric repeat binding factor 1 (TERF1) and 2 (TERF2), TERF1-interacting nuclear factor 2 (TIN2), and protection of telomeres 1 (POT1), together with DNA repair, maintain the protective function of telomeres [2]. The chromosome end-cap structure consists of telomeric single-stranded DNA (ssDNA) invading telomeric double-stranded DNA (dsDNA) sites to hide the 3‘-terminus [3]. The assembly of shelterin at telomeres encompasses the binding of POT1-TPP1 to ssDNA overhang, TERF1 and TERF1-RAP1 to dsDNA, and TIN2 to the whole complex by simultaneous interaction with TERF1, TERF2, and TPP1, which keeps the telomeres intact [4]. Shelterin prevents telomere hyper-resection by suppressing DNA damage response kinases [5]. While TERF2 binds and inhibits ataxia-telangiectasia mutated (ATM) kinase, the POT1-TPP1 heterodimer blocks ataxia-telangiectasia and Rad3-related (ATR) kinase activation [5]. Deregulation of telomere maintenance can give rise to cancer [6]. Cancerous cells, unlike almost all somatic cells under physiological conditions, can fix eroded telomeres via either telomerase reactivation or by a homologous recombination-based pathway known as alternative lengthening of telomeres [7]. The majority of cancers, however, rely on telomerase reactivation [8]. Telomerase is a ribonucleoprotein polymerase composed primarily of the telomerase reverse transcriptase (hTERT) and telomerase RNA component (hTERC), together with other associated proteins [9].
Ovarian cancer (OvC), a heterogeneous group of diseases characterized by distinct clinicopathological and molecular patterns, may be driven by telomere dysfunction [10,11]. According to current knowledge, ovarian tumors appear to be strictly telomerase-dependent [10]. Substantial hTERT expression, a limiting factor for telomerase catalytical activity, exhibits 95 % of ovarian tumors [10]. The harboring of hTERT promoter mutation was reported in 16 % of clear cell OvC [12,13] and 50 % of granulosa cell tumors [14].
Currently, not much is known about shelterin impairment in OvC tissue. POT1 knockdown in OvC-derived SK-OV3 cells reduces c-MYC proto-oncogene (c-MYC) expression, causing temporary inhibition of proliferation, but ultimately results in enhanced cell division, tumorigenicity, and histone deacetylase inhibitor response [15]. c-MYC protein mediates the regulation of telomerase activity, and changes in its transcription upset the balance of the telomere/telomerase system [16]. Among the 22 genes related to telomere structure, length, and maintenance, genome-wide association studies (GWAS) identified single nucleotide polymorphisms (SNPs) rs116895242 in the POT1, rs7717443, rs10866498, rs12655062, rs2736098 in the hTERT, and rs75316749 in the hTERC-CLPTM1L (cleft lip and palate transmembrane protein 1-like protein) regions, to be associated with OvC risk [17].
In this study, we investigated tumor and peripheral blood leukocyte (PBL) telomere length (TL) of patients diagnosed with late-stage OvC, along with hTERT and shelterin genes’ methylation/mRNA expression in the tumor tissue. We were also interested in the possible connection between TL assessed in PBL/tumor tissue and therapy response. In this article, we hypothesize that patients’ response to treatment and survival may be predicted by the TL either in PBL, representing a surrogate tissue of the patients or in the tumor tissue itself. We further hypothesize that the mRNA expression and DNA methylation of shelterin subunits and telomerase may be mirrored by TL.
2. Materials and methods
2.1. Study population
Overall, the study comprised 212 OvC patients, of which 184 were sampled for peripheral blood at the time of diagnosis and 59 for tumor tissue during surgical resection. Thirty-one patients were sampled for both tumor tissue and peripheral blood. Regarding the tumor tissue, data from RNA sequencing (RNA-Seq) were available for all the patients, data from the methylation arrays were available only from 58 patients, and telomeres were successfully measured within 56 patients. As for blood, TL was assessed in all 184 samples. The patients were women with newly diagnosed and histologically confirmed OvC. All patients were diagnosed and treated between 2009 and 2019 at the Motol University Hospital in Prague, and the University Hospital in Pilsen, Czech Republic. Biological samples for genetic analyses were obtained from all patients according to the Helsinki declaration. Table 1 summarizes the characteristics of all 212 patients, Table 2 only of 31 patients with both tumor and blood collected. Sensitivity to the OvC therapy was based on the platinum-free interval (PFI) described by Friedlander et al. [18], separating OvC patients as having platinum-sensitive or platinum-resistant disease. The PFI interval was defined as the time from the last dose of platinum-based chemotherapy to the progression of the disease. Patients with PFI≤6 months were considered platinum-resistant (this group also included OvC patients with intermediate PFI – 6 to 12 months), and patients with PFI≥12 months were platinum-sensitive. Overall survival (OS) of the investigated OvC patients was based on the interval from the surgery to the date of death or last follow-up. The follow-up was conducted over 10 years.
Table 1.
All patients' characteristics. The table shows the characteristics of all patients participating in the study. The number of patients does not always add up to 100 % (n = 212) due to missing data for some attributes.
| A studied cohort of patients | Median age (years) [range] | 60.92 [24–89] |
% |
|---|---|---|---|
| n = 212 | |||
| Tumor type | |||
| low-grade serous | 9 | 4.2 | |
| high-grade serous | 162 | 76.4 | |
| mucinous | 11 | 5.2 | |
| clear cell | 9 | 4.2 | |
| endometrioid | 4 | 1.9 | |
| Brenner | 1 | 0.5 | |
| borderline | 1 | 0.5 | |
| adenocarcinoma | 4 | 1.9 | |
| generalized disease | 1 | 0.5 | |
| Grade | |||
| 1 | 7 | 3.3 | |
| 2 | 19 | 9.0 | |
| 3 | 165 | 77.8 | |
| FIGO stagea | |||
| I | 9 | 4.2 | |
| II | 12 | 5.7 | |
| III | 156 | 73.6 | |
| IV | 15 | 7.1 | |
| Residual tumorb | |||
| yes | 102 | 48.1 | |
| no | 90 | 42.5 | |
| BRCA1/BRCA2 mutationc | |||
| yes | 13 | 6.1 | |
| no | 199 | 93.9 | |
| Therapy regimen | |||
| paclitaxel + platinum derivatives | 162 | 76.4 | |
| platinum monotherapy | 6 | 2.8 | |
| other regimend | 10 | 4.7 | |
| none | 1 | 0.5 | |
| Platinum-based therapy resistance | |||
| yes | 60 | 28.3 | |
| no | 106 | 50.0 | |
| Median platinum-free interval (PFI,months) [range] | |||
| platinum-resistant | 5.31 (0–11.5) | ||
| platinum-sensitive | 29.06 (12–97) | ||
FIGO (International Federation of Gynecology and Obstetrics) staging system is used for ovarian, fallopian tube, and peritoneal cancer classification.
Residual tumor after surgery was defined by a surgeon as “yes” (macroscopic lesions <1 cm and peritoneal metastases >1 cm), or “no” [no macroscopic residuum (R)].
Breast cancer 1 and 2 (BRCA1/2) germline mutations are the most frequent causes of developing hereditary OvC.
Other regimen [paclitaxel + platinum derivate + cyclofosfamid, cisplatin + doxorubicin, oxaliplatin + capecitabine (known as XELOX), oxaliplatin + 5-fluorouracil + folinic acid (known as FOLFOX) administered in a regimen called modified FOLFOX6, or paclitaxel + carboplatin + bevacizumab].
Table 2.
Characteristics of patients with both blood and tumor tissue samples. The table shows the characteristics of all patients from whom we could analyze both tissue and blood. The number of patients does not always add up to 100 % (n = 31) due to missing data for some attributes.
| A studied cohort of patients | Median age (years) [range] | 58.77 [40–77] |
% |
|---|---|---|---|
| n = 31 | |||
| Tumor type | |||
| low-grade serous | 3 | 9.7 | |
| high-grade serous | 21 | 67.7 | |
| mucinous | 3 | 9.7 | |
| clear cell | 3 | 9.7 | |
| endometrioid | 0 | 0.0 | |
| Brenner | 0 | 0.0 | |
| borderline | 0 | 0.0 | |
| adenocarcinoma | 0 | 0.0 | |
| generalized disease | 0 | 0.0 | |
| Grade | |||
| 1 | 2 | 6.5 | |
| 2 | 3 | 9.7 | |
| 3 | 26 | 83.9 | |
| FIGO stagea | |||
| I | 2 | 6.5 | |
| II | 1 | 3.2 | |
| III | 25 | 80.6 | |
| IV | 2 | 6.5 | |
| Residual tumorb | |||
| yes | 18 | 58.1 | |
| no | 13 | 41.9 | |
| BRCA1/BRCA2 mutationc | |||
| yes | 5 | 16.1 | |
| no | 26 | 83.9 | |
| Therapy regimen | |||
| paclitaxel + platinum derivatives | 31 | 100 | |
| platinum monotherapy | 0 | 0 | |
| other regimend | 0 | 0 | |
| none | 0 | 0 | |
| Platinum-based therapy resistance | |||
| yes | 10 | 32.3 | |
| no | 21 | 67.7 | |
| Median platinum-free interval (PFI,months) [range] | |||
| platinum-resistant | 4.35 (0.5–8) | ||
| platinum-sensitive | 38.47 (15–97) | ||
FIGO staging system is used for ovarian, fallopian tube, and peritoneal cancer classification.
Residual tumor after surgery was defined by a surgeon as “yes” (macroscopic lesions <1 cm and peritoneal metastases >1 cm), or “no” (no macroscopic residuum).
BRCA1/2 germline mutations are the most frequent causes of developing hereditary OvC.
Other regimen [paclitaxel + platinum derivate + cyclofosfamid, cisplatin + doxorubicin, oxaliplatin + capecitabine (known as XELOX), oxaliplatin + 5-fluorouracil + folinic acid (known as FOLFOX) administered in a regimen called modified FOLFOX6, or paclitaxel + carboplatin + bevacizumab].
2.2. DNA sample isolation and quantification
Tumor DNA was isolated from fresh frozen tumor tissue after pulverization in liquid nitrogen using AllPrep DNA/RNA/Protein Mini Kit (Qiagen, Hildesheim, Germany). Genomic DNA from 250 μL of peripheral blood was performed by BioSprint DNA Blood Kit (Qiagen, Valencia, CA, USA) using Kingfisher mL instrument (ThermoFisher Scientific, Waltham, MA, USA) according to manufacturer protocol. Isolated DNA was quantified by Quant-iT PicoGreen dsDNA Reagent and Kits (ThermoFisher Scientific, Waltham, MA, USA) and the plate reader Infinite 200 (Tecan Group Ltd., Männedorf, Switzerland).
2.3. Telomere length measurement
TL in PBL and tumor tissues was expressed as relative by the monochrome multiplex quantitative polymerase chain reaction (qPCR) method, as described in detail previously [19,20]. Briefly, Ct values for telomere sequences (T) and reference single-copy gene (S; albumin) were determined simultaneously as a multiplex using ViiA 7 Real-time PCR System (Applied Biosystems, Foster City, CA, USA). The standard curve was used to quantify telomere and albumin genes based on the respective cycle threshold values. TL was expressed as the ratio between the T/S. PBL TL was measured using DNA from the whole blood since erythrocytes do not have a nucleus containing telomeric sequences. The qPCR efficiency for telomere sequences in tissues was 93 % and 91 % for albumin. The reaction efficiency for telomere sequences in blood ranged between 98–110 % and 90–104 % for the albumin gene. All the data were normalized based on the qPCR efficiency, as described previously [21].
2.4. RNA sequencing library preparation and sequencing
The total RNA from 59 epithelial OvC tissues with an RNA integrity number (RIN) > 5.4 (mean RIN 8.5, range 5.4–9.9) was used for RNA-Seq analysis (summarized in detail in Supplementary Table 1). Five hundred ng of the total RNA was used for library preparation with QuantSeq 3′mRNA-Seq Library Prep FWD for Illumina (Lexogen, Vienna, Austria) according to the manufacturer's protocol. The quality of the prepared libraries was checked by Bioanalyzer 2100 using the High Sensitivity DNA kit (Agilent Technologies Inc., Santa Clara, CA, USA), and the quantity was measured by qPCR, KAPA Library Quantification Kit Illumina®Platforms (F.Hoffmann-La Roche AG, Basel, Switzerland). The equimolar pool of prepared libraries was sequenced on the NextSeq 500 platform (Illumina Inc., San Diego, CA, USA) using the NextSeq 500 High Output v2 Kit (75 cycles) in one run, with a 1 × 75 base pairs (bp) setting, aimed at 6–7 million reads per sample.
2.4.1. mRNA expression analysis
Raw RNA-Seq data quality was checked by FastQC [22] v0.12.0 and MultiQC [23] v1.14 tools and the fastp v0.23.3 package, which was also used for quality filtering and adapter trimming [24]. Gene annotation was based on reference transcriptome GENCODE v35 (GRCh38.p13). Protein-coding gene abundance was estimated by kallisto tool v0.50.1 using a pseudoalignment approach [25].
For all RNA-Seq analyses of mRNA transcripts, we used R v4.4.0 (R Core Team). Genes with zero counts or one count across the samples were removed before analysis, resulting in 18,462 protein-coding genes out of 19,972 for further analysis. Differential expression analysis of mRNAs was carried out by the DESeq2 package v1.44.0 with default settings [26]. The false discovery rate was managed by the Benjamini and Hochberg (BH) method for P values and by ashr for log2 fold change (log2 FC) values [27], both in the DESeq2 package. Statistically differentially expressed genes were considered genes with q < 0.05 (P value after BH correction).
Gene set enrichment analysis (GSEA) was performed by clusterProfiler v4.12.0 and DOSE v3.30.1 packages in R v4.4.0 [28, 29]. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases were used to explore the biological function of differentially expressed genes [[30], [31], [32]]. As statistically significant enrichment we counted the results with Bonferonni corrected q < 0.05.
2.5. High-Throughput DNA methylation profiling
The genome-wide DNA methylation profile was analyzed by Infinium MethylationEPIC BeadChip microarrays (Illumina Inc.) in 58 epithelial OvC tissues. Five hundred ng of DNA was used as input for bisulfite conversion according to the manufacturer's manual by EZ DNA Methylation™ Kit (Zymo Research, Irvine, CA, USA). Prepared microarrays were scanned by iSCAN System (Illumina Inc.).
2.5.1. Methylation analysis
Quality control and data normalization were performed by the SWAN approach by the minfi package in the R v4.4.0 (R Core Team) as described previously [[33], [34], [35]]. Raw data were converted to β values for further analyses [[36], [37], [38]]. Probes with annotated SNP were removed before analyses [39]. For analysis of gene regions, the probes were then collapsed into specific gene regions based on the manifest for the microarray – TSS200 [cytosine-phosphate-guanine (CpG) between TSS (transcription start site) and 200bp upstream and TSS itself], TSS1500 (CpG between TSS and 1500–200bp upstream), 5′UTR [CpG in 5′ untranslated region (UTR)], 1stExon (CpG in the first exon), gene body (CpG in other exons or introns), 3′UTR (CpG in 3′UTR region) as described previously [40]. The promoter region defined a combination of CpG in TSS200 and TSS1500. We focused on the whole gene, TSS200, TSS1500, and promoter methylation profiles. The degree of gene methylation was divided according to Bibikova et al. [41]: 0.00–0.29 (unmethylated/hypomethylated), 0.30–0.79 (hemimethylated), and 0.80–1.00 (methylated/hypermethylated). For the correlation with expression, we used β values from the promoter probes (TSS1500 and TSS200 together).
2.6. Statistical analysis
Statistical analyses and graphs were made in the R v4.4.0 (R Core Team) environment. The Dplyr package was used for the data manipulation, survival, and survminer packages for the calculation of survivals, and the ggplot2 package for the plot rendering. For the evaluation of the normal distribution, we used the Shapiro-Wilk test. In the case of normal distribution, parametric tests were used (t-test, ANOVA), and in the case of non-normal distribution, non-parametric tests were used (Mann-Whitney and Kruskal-Wallis tests). For survival, the log-rank test was used. For all tests, a P < 0.05 was considered significant.
3. Results
3.1. Telomere length in peripheral blood leukocytes predicts response to therapy
TL was measured within 184 patients with the availability of PBL samples. However, 22 patients were excluded from the analysis since information about chemotherapy administration was not available. Therefore, the total number of patients with information about blood sampling before chemotherapy (n = 136) and after chemotherapy treatment (n = 26) was 162. TL measured in PBL within the groups of patients sampled before (n = 136) and after chemotherapy (n = 26) did not significantly differ (parametrical t-test, P = 0.15). Importantly, therapy-sensitive individuals (n = 93) were associated with shorter telomeres in PBL compared to PBL in the patients with treatment resistance (n = 46, parametrical t-test, P = 0.037), as seen in Fig. 1. Thirty-nine patients did not have a record of the treatment response, and they were not included in the analysis. Patients with postoperative tumor residues (n = 89) did not differ in their PBL TL from those with tumors completely removed (n = 78, parametrical t-test, P = 0.181). Seventeen patients from 184 had the information about tumor residuum missing. Macroscopic residuum is estimated by the surgeon by the end of the debulking surgery using a combination of visual inspection and palpable examination, and it is characterized into three groups: R0 (no macroscopic residuum), R1 (macroscopic residuum <1 cm), and R2 [macroscopic residuum (metastases) >1 cm].
Fig. 1.
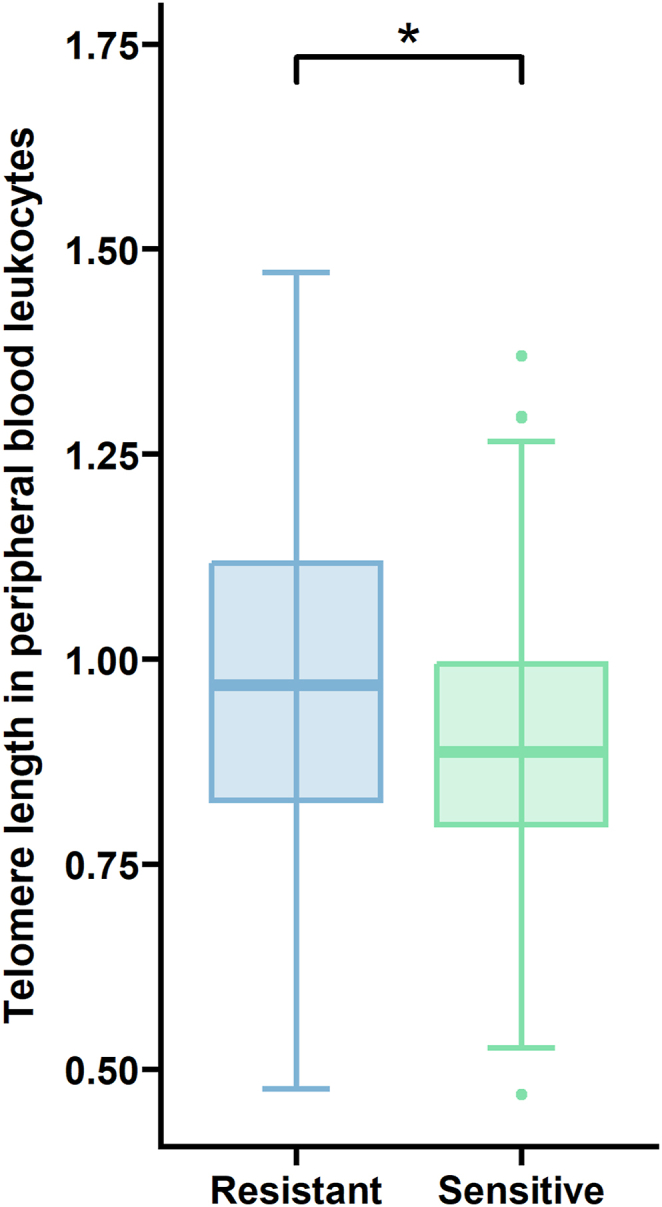
Shorter PBL TL may be coupled with enhanced sensitivity to OvC therapy. Patients responding to treatment (>12 months, n = 93) have shorter telomeres than those therapy-resistant (1–6 months, n = 46, P = 0.037).
3.2. Telomere length in tumor tissue reflects the survival of ovarian cancer patients
TL exceeding the median [0.75 (0.74–1.94)] in tumor tissue was independently associated with OS in our studied group of patients (log-rank test, n = 56, P = 0.006), as illustrated in Fig. 2. However, neither OS (log-rank test, P = 0.39) nor the time to progression (log-rank test, P = 0.18) was associated with PBL TL in our patients (n = 184).
Fig. 2.

OS of patients could be predicted by TL in tumor tissue. A) The group with tumor TL higher than the median had poorer OS (n = 56, P = 0.006). The table illustrates how many patients were at risk in two-year intervals. B) The 56 patients were divided according to their follow-up to those who died (n = 38) and those who were at the time of their last control still alive (n = 18). As a marker, the tumor TL was selected. The area under the curve (AUC) = 0.673 confirmed that the tumor TL might be indicative of OvC patients' OS.
3.3. Association of telomere length in peripheral blood leukocytes and tumor tissue with other clinicopathological characteristics of the patients
No TL differences in either PBL or tumor tissue of the patients diagnosed with different tumor histotypes were observed [high-grade serous carcinoma (n = 145 for PBL, n = 37 for tissue), low-grade serous carcinoma (n = 8 for PBL, n = 4 for tissue), mucinous carcinoma (n = 6 for PBL, n = 8 for tissue), and clear cell carcinoma subtype (n = 7 for PBL, n = 4 for tissue), ANOVA, P = 0.68, and P = 0.94, respectively]. Furthermore, patients diagnosed with high-grade serous carcinoma (n = 145 for PBL, n = 37 for tissue), the most malignant form of the disease, did not have significantly different TL in either PBL or tumor compared to the patients with other subtypes of the tumors with epithelial origin (n = 21 for PBL, n = 16 for tissue, Mann-Whitney U test, P = 0.95, P = 0.11, respectively).
The stage of the disease based on the FIGO system was not associated with TL differences in either PBL or tumor tissue in our studied group of patients [FIGO stage I + II (n = 17 for PBL, n = 7 for tissue), FIGO stage III + IV (n = 148 for PBL, n = 48 for tissue), Mann-Whitney U test, P = 0.44, and P = 0.91, respectively]. The grade of the disease was not associated with TL differences in either PBL or tumor tissue [grade 1 (n = 5 for PBL, n = 4 for tissue), grade 2 (n = 14 for PBL, n = 8 for tissue), grade 2 (n = 145 for PBL, n = 44 for tissue), ANOVA, P = 0.60, P = 1, respectively]. Also, TL in PBL [median (range), 0.91 (0.47–1.29)] did not correlate with TL measured in tumor tissue [median (range), 0.75 (0.74–1.94), n = 31, Pearson correlation coefficient, R = −0,168, P = 0.99].
All TL values measured in PBL or OvC tissue and the results, presented in 3.1–3.3, are displayed in Table 3. The TL data were not age-adjusted due to the lack of relationship between TL in either PBL (n = 184) or tumor tissue (n = 56) and age at the time of diagnosis or clinical surgery (Kendall rank correlation coefficient, R = −0.045, P = 0.37; R = 0.11, P = 0.36 respectively).
Table 3.
PBL and tumor TL in patient subgroups. The table summarizes TL data measured in PBL and tumor tissues of patients diagnosed with OvC, along with various personal and clinicopathological characteristics.
| PBL |
Tumor tissue |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| n = 184 | % | Median TL (range) | P-value | n = 56 | % | Median TL (range) | P-value | ||
| Tumor type | |||||||||
| low-grade serous | 8 | 5 | 0.81 (0.71–1.25) | 0.68 (ANOVA) | 4 | 7.5 | 0.77 (0.69–0.99) | 0.94 (ANOVA) | |
| high-grade serous | 145 | 87 | 0.91 (0.45–1.47) | 37 | 70 | 0.68 (0.13–2.11) | |||
| mucinous | 6 | 4 | 0.91 (0.76–1.47) | 8 | 15 | 0.77 (0.33–1.47) | |||
| clear cell | 7 | 4 | 0.94 (0.80–1.29) | 4 | 7.5 | 0.95 (0.23–1.36) | |||
| Grade | |||||||||
| 1 | 5 | 3 | 0.82 (0.76–1.47) | 0.60 (ANOVA) | 4 | 7 | 0.72 (0.69–0.99) | 1 (ANOVA) | |
| 2 | 14 | 9 | 0.91 (0.63–1.23) | 8 | 14 | 0.87 (0.28–1.47) | |||
| 3 | 145 | 88 | 0.91 (0.46–1.47) | 44 | 79 | 0.75 (0.13–2.11) | |||
| FIGO stage | |||||||||
| I + II | 17 | 10 | 0.85 (0.63–1.47) | 0.44 (Mann-Whitney) | 7 | 13 | 0.69 (0.28–1.54) | 0.91 (Mann-Whitney) | |
| III + IV | 148 | 90 | 0.92 (0.46–1.47) | 48 | 87 | 0.79 (0.13–2.11) | |||
| Residual tumor | |||||||||
| yes | 89 | 53 | 0.90 (0.47–1.40) | 0.18 (t-test) | 31 | 55 | 0.81 (0.17–1.47) | 0.74 (t-test) | |
| no | 78 | 47 | 0.92 (0.46–1.47) | 25 | 45 | 0.69 (0.13–2.11) | |||
| Therapy resistance | |||||||||
| yes | 46 | 33 | 0.97 (0.48–1.47) | 0.037 (t-test) | 23 | 41 | 0.81 (0.17–1.54) | 0.45 (t-test) | |
| no | 93 | 67 | 0.89 (0.47–1.37) | 33 | 59 | 0.72 (0.13–2.11) | |||
3.4. Methylation status of shelterin subunits and hTERT correlates with telomere length in tumors
DNA hypermethylation within the TERF1 body and hypomethylation of the promoter sequence TSS1500 positively correlated with TL (n = 58, Kendall rank correlation coefficient, P = 0.0001, P = 0.0007, respectively). The TPP1 gene body hosting CpG hypomethylation was positively associated with TL (n = 58, Kendall rank correlation coefficient, P = 0.003). RAP1 3′UTR hypermethylation and TL inversely correlated (n = 58, Kendall rank correlation coefficient, P = 0.002). DNA hemimethylated status in hTERT transcriptional start site TSS1500 and TERF2 gene body negatively correlated with TL (n = 58, Kendall rank correlation coefficient, P = 0.0003 and P = 0.0001 for hTERT; and P = 0.0009 for TERF2). Additionally, methylation of hTERT promoter TSS1500 negatively correlated with the gene mRNA expression levels (n = 58, Kendall rank correlation coefficient, R = −0.21, P = 0.025, see Fig. 3). However, the methylation status of none of the shelterin complex genes investigated in the study (TERF1, TERF2, TPP1, RAP1, POT1, and TIN2) was associated with the expression levels of the genes (P values not shown).
Fig. 3.

hTERT is expressed in tumors based on the DNA methylation of the promoter sequence.hTERT mRNA expression is downregulated due to its promoter TSS1500 methylation (n = 58, R = −0.21, P = 0.025).
After the correction for all genes tested, the positive association between TERF1 hemimethylated status and TL remained significant (n = 58, Kendall rank correlation coefficient, P = 0.005). Following the correction for all tested probes, hemimethylated hTERT TSS1500 negatively and hypermethylated TERF1 body positively correlated with TL (n = 58, Kendall rank correlation coefficient, both P = 0.0002).
3.5. mRNA levels of shelterin complex subunit TPP1 correlate with TL in tumor tissue
Analysis of the gene expression data obtained by RNA-seq revealed a positive correlation between the TPP1 mRNA level and the TL measured in the tumor tissue of the patients (n = 52, Kendall rank correlation coefficient, R = 0.21, P = 0.026, see Fig. 4). Further analysis of the RNA-Seq results did not show any correlation between mRNA levels of hTERT, encoding the catalytic subunit of telomerase, or the other shelterin subunits (RAP1, TIN2, TERF1, TERF2, POT1), and TL in the tumor tissue of our studied group of patients (P values not shown).
Fig. 4.

TPP1 mRNA expression is associated with TL in tumor tissue. TL in cancer tissue is positively affected by TPP1 expression (n = 52, R = 0.21, P = 0.026).
3.6. Difference in expression profile regarding platinum-based therapy resistance and telomere length
In the end, the whole transcriptome profile analysis was performed beyond shelterin genes, focusing on the expression differences between OvC patients with good and poor response to platinum-based therapy and between OvC patients with telomeres in tumor tissue shorter and longer than the median TL. Primary analysis of the expression profile of OvC patients by principal component analysis (PCA) showed one outlier sample (depicted in Fig. 5A). For further analysis, the sample was removed (see Fig. 5B).
Fig. 5.

PCA plot of expression distribution across the ovarian carcinoma set. A) PCA plot of all OvC samples (n = 59) that underwent RNA-Seq, showing one outlier sample in the therapy-resistant group (marked by a red circle). B) PCA plot of OvC samples (n = 58) used for further analysis after removing the outlier sample. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Differential expression (DE) analysis between therapy-sensitive and therapy-resistant OvC patients showed overall 59 significantly differentially expressed genes (q < 0.05, see Fig. 6). Expression downregulation was observed for 42 genes and upregulation for 17 genes. All differentially expressed genes are summarized in Supplementary Table 2.
Fig. 6.

Expression differences between OvC patients sensitive and resistant to platinum-based therapy. The volcano plot shows the Top 10 differentially expressed genes in the current comparison of 58 OvC patients.
DE analysis between OvC patients (n = 56) divided into two groups by the median value of TL in tumor tissue showed overall 188 significantly differentially expressed genes (q < 0.05, see Fig. 7). Expression downregulation was observed for 123 genes and upregulation for 65 genes. All differentially expressed genes are in Supplementary Table 3.
Fig. 7.

Expression differences between OvC patients with different lengths of telomeres in tumor tissue. The volcano plot shows the Top 10 differentially expressed genes in the current comparison in the set of 56 OvC patients.
In addition, we performed pathway analysis using GO terms (molecular function, biological process, and cellular component) and KEGG pathways for differentially expressed genes. GSEA of GO terms failed to reveal significant (q < 0.05) differences in OvC patients divided by the therapy response, but the first three Top enriched GO terms (P < 0.001) were transmembrane receptor protein tyrosine kinase signaling pathway (GO:0007169), response to peptide (GO:1901652), and cellular response to peptide (GO:1901653) for the following genes – nuclear receptor 4A3 (NR4A3), adiponectin (ADIPOQ), lipoprotein lipase (LPL), klotho beta (KLB), and polymeric immunoglobulin receptor (PIGR), as shown in Fig. 8A and Supplementary Table 4. The KEGG pathway-focused GSEA showed only one significantly enriched pathway – peroxisome proliferator-activated receptor (PPAR) signaling pathway (hsa03320, q = 0.001) for cluster determinant 36 (CD36), ADIPOQ, perilipin 2 (PLIN2), and fatty acid binding protein 4 (FABP4) genes, as indicated in Supplementary Table 5.
Fig. 8.

Results of GSEA for GO terms. A) The dotplot represents the Top 10 enriched GO terms for differentially expressed genes from the therapy response analysis. B) The dotplot shows the Top 10 enriched GO terms for differentially expressed genes from the tumor TL analysis.
In OvC patients divided by the median TL in tumor tissue, GSEA using GO terms revealed significant (q < 0.05) differences in a total of four significantly enriched terms (q = 0.026, see Fig. 8B and Supplementary Table 6) – three cellular components {axonemal microtubule [GO:0005879, genes: cilia and flagella associated protein 45 (CFAP45), EF-hand domain containing 2 (EFHC2), RIB43A domain with coiled-coils 2 (RIBC2)], axoneme [GO:0005930, genes: CFAP45, axonemal central pair apparatus protein (HYDIN), EFHC2, RIBC2, cancer susceptibility candidate gene 1 (CASC1)] and ciliary plasm (GO:0097014, genes: CFAP45, HYDIN, EFHC2, RIBC2, CASC1)} and one biological process [DNA metabolic process, GO:0006259, genes: retinoblastoma-binding protein 8 (RBBP8), nuclear autoantigenic sperm protein (NASP), B-cell CLL/lymphoma 7 A (BCL7A), YEATS domain containing 4 (YEATS4), structural maintenance of chromosomes 1 A (SMC1A), structure specific recognition protein 1 (SSRP1), heterogenous nuclear ribonucleoprotein A/B (HNRNPAB), lysine demethylase 2 A (KDM2A), dyskerin pseudouridine synthase 1 (DKC1), DNA polymerase epsilon 3 (POLE3), DNA helicase and adenosine triphosphatase (UPF1), fragile X messenger ribonucleoprotein 1 (FMR1)]. GSEA did not identify significant (q < 0.05) KEGG pathway differences between the two groups of OvC patients divided by the TL in tumor tissue. However, the first three Top enriched pathways (listed in Supplementary Table 5) were: adenosine monophosphate-activated protein kinase (AMPK) signaling pathway [hsa04152, genes: CD36, hepatocyte nuclear factor 4 alpha (HNF4A), P = 0.007], leukocyte transendothelial migration [hsa04670, genes: claudin 11 (CLDN11), junction adhesion molecule 3 (JAM3), claudin 18 (CLDN18), P = 0.02], and cell adhesion molecules (hsa04514, genes: CLDN11, JAM3, CLDN18, P = 0.023).
4. Discussion
Earlier studies suggested that OvC, a heterogeneous group of diseases with multifactorial etiopathogenesis, may exhibit telomere dysfunction [10,11]. In addition, ovarian carcinomas appear to be strictly telomerase-dependent. More than 95 % of ovarian tumors are hallmarked with substantial hTERT expression, a limiting factor for telomerase catalytical activity [10]. Harboring of hTERT promoter mutation was reported in 16 % of clear cell OvC [12,13] and 50 % of granulosa cell tumors [14]. Our knowledge of shelterin alteration in OvC is considerably modest. Yet, POT1 knockdown in OvC-derived SK-OV3 cells reduces the expression of c-Myc, triggering temporary inhibition of proliferation but ultimately resulting in enhanced cell division, tumorigenicity, and histone deacetylase inhibitor response [15].
In this, we investigated whether TL in tumor tissue and PBL from patients diagnosed with OvC may relate to their therapy response and prognosis. Here we document, for the first time, that the response of the OvC patients to treatment is ameliorated in the patients with shorter TL in PBL, and patients’ survival is significantly longer in patients with shorter TL in tumor tissue. We also recorded intriguing associations between the mRNA expression and DNA methylation of shelterin subunits, telomerase, and TL.
Consistent with the literature, this study found that prolonged TL in tumors may implicate worse OS of patients diagnosed with OvC. Previously, worse OS in patients with long tumor telomeres (index >1, defined as the ratio of relative mean TL of tumor cells/stromal cells) was confirmed exclusively for clear cell histologic type but not for a whole studied set [42]. On the contrary, no relationship between OvC-specific mortality and TL likely exists in PBL, as previously shown by Kotsopoulos et al. [43].
Our data show that longer PBL TL might be bound up with the onset of therapy resistance. Those patients evaluated as resistant had progressive tumors, low tolerance to neo-/adjuvant chemotherapy, or experienced a rapid relapse. However, conflicting results were described by Falandry et al. [44]. In the study, the OvC patients with longer TL in lymphocytes (>5.77 kb) showed better treatment tolerance (2.7 odds ratio) and a higher probability of therapy completion than those with shorter TL (80 % rate vs. 59 % rate) [44]. In addition, longer lymphocyte TL was associated with less frequent unplanned hospital admissions and lower grade 3–4 non-hematological toxicity (2.14 and 2.04 odds ratio, respectively) [44]. Signs that chemotherapy may impact telomere maintenance machinery, and thus presumably TL, have been proven in vitro. In epithelial OvC cell line HO8910, telomerase activity and hTERT expression alternations have been shown to reflect the effect of cisplatin [45].
It should be emphasized that elevated production of TPP1, which, in complex with POT1 and telomeric DNA, recruits telomerase and stimulates its processivity [46], is connected to telomere elongation. We observed that tumoral TPP1 mRNA expression positively correlates with TL, just as Yang et al. had reported the relationship between TPP1 protein levels in colorectal cancer cell lines [47]. In addition, the study authors stated that TPP1 protein overexpression and longer telomeres are linked to radioresistance. However, we did not find any association between shelterin-subunit mRNA level and therapy response, although the size of the studied group and the OvC heterogeneity might have obscured the association. Furthermore, we observed that hTERT promoter methylation downregulates hTERT mRNA expression and leads to shorter telomeres. Previously, Widschwendter et al. related the hTERT expression in OvC tissue only to the methylation of the whole gene, where the association was not found [48]. Besides hTERT methylation patterns, we associated several methylation statuses of shelterin (TERF1, TERF2, POT1, RAP1) with OvC tissue TL. Our study suggests the importance of telomere biology in the prognosis and treatment prediction of OvC patients.
Analysis of whole transcriptome profiles revealed other important candidates for further detailed studies involving telomerase activity in ovarian tumors. Our data show that differentially expressed genes from analysis focused on OvC patients with different lengths of telomeres in tumor tissue (divided by median value) were enriched in pathways associated with cell adhesion, AMPK signaling pathway, and leukocyte trans-endothelial migration. In our case, the most prominent genes were JAM3, CLDN11, and CLDN18. For the genes, we found no reported connection with ovarian carcinoma therapy resistance or telomerase activity. However, there is evidence of an indirect role of hTERT in cell adhesion, migration (e.g., in sarcoma cancer cell line U2OS) or in cancer stemness and metastasis [49,50]. In breast cancer, in vitro and in vivo models enhanced sensitivity to doxorubicin and alteration in adhesion pathways after hTERT downregulation were observed [51]. These findings show another possible indirect role of telomerase biology in the progression of OvC.
Functional annotation of differentially expressed genes based on therapy response showed alterations in the PPAR signaling pathway (KEGG database) and tyrosine kinase signaling pathway with peptide response (GO terms). The genes enriched in these pathways were PLIN2, ADIPOQ1, FABP4, NR4A3, KLB, LPL, PIGR and CD36. ADIPOQ1 gene is a joint member of all the above mentioned pathways. The PPAR signaling pathway is often altered in cancer tissue, and members of this pathway (PLIN2 and ADIPOQ1) are key parts of adipocyte differentiation. The role of adipocytes in OvC progression is currently being studied, and results are showing their importance in cancer progressiveness [52,53]. In addition, another candidate FABP4, mainly expressed in adipocytes, is connected with OvC progression, resistance to platinum derivatives, and, thus, poor prognosis [54,55]. CD36 molecule, another member of fatty acid metabolism associated with therapy response in our study, is also connected with OvC progression, the metastasis process, and platinum derivatives resistance [[56], [57], [58]]. Moreover, CD36 is considered to be a potential therapeutic target. Novel therapeutics such as photoactivable Pt(IV) metallodrugs targeting CD36-positive OvC cells have already been prepared and tested, and showed their efficiency in eliminating drug-resistant OvC cells [59]. These results support the importance of adipocytes and lipid metabolism in OvC and in cancer in general.
The study offers numerous advantages alongside certain constraints. Our current study's merit lies in comparing TL between blood and tumor tissue DNA in a carefully chosen and similar clinical cohort characterized by advanced stage, grade, well-documented PFI, and shared ethnicity (Slavic Caucasian). This focus is particularly beneficial given the scarcity of studies addressing this aspect. Furthermore, TL examination alongside the gene expression and methylation profile of the shelterin complex within the same patient cohort adds another layer of value to our investigation. However, our study's limitation is its relatively modest sample size, although this is mitigated by the thorough selection and characterization of the patient group.
5. Conclusions
We compared OvC tissue TL with mRNA expression and DNA methylation data of telomerase and shelterin and analyzed TL in PBL. This study found that prolonged TL in tumors may implicate worse OS of patients diagnosed with OvC. Our data showed that longer PBL TL might be bound up with the onset of therapy resistance. DNA methylation analysis of shelterin and telomerase genes identified TERF1, TERF2, TPP1, RAP1, and hTERT CpG sites at which methylation levels are associated with TL in tumors. We also found that TL in tumors positively relates to the gene expression of TPP1 and that hTERT mRNA expression is inversely proportional to its promoter methylation level. The study also showed the possible involvement of telomerase in the migratory potential of cancer cells and the role of adipocytes/lipid metabolism in OvC progression.
Funding
The work was supported by the Czech Science Foundation [19-10543 S (PV), 21-27902 S (LV)]; the Czech Health Research Council grant [NU21-03-00145 (LV, RV)]; and the Cooperatio programs [207035, “Maternal and Childhood Care”, 3rd Faculty of Medicine, Charles University (LR), 207043, “Surgical Disciplines”, Faculty of Medicine in Pilsen, Charles University (KT,LV,RV,PV)]. We also acknowledge the project NPO [LX22NPO5102 (LV)].
Ethics statement and consent to participate
This study was reviewed and approved by the Institutional Review Boards of the National Institute of Public Health in Prague (approval number: IGA NS9803-4, dated January 30, 2008), the University Hospital Kralovske Vinohrady (approval number: EK-VP/25/0/2018, dated June 6, 2018), the University Hospital in Pilsen (approval number: 16–29013 A, dated June 7, 2018), and the University Hospital in Motol (approval number: EK-890/15, dated June 24, 2015). All participants/patients (or their proxies/legal guardians) provided written informed consent to participate in the study.
Data availability statement
RNA-Seq dataset generated and analyzed during the current study is available in the NCBI BioProject repository, https://www.ncbi.nlm.nih.gov/, BioProject ID: PRJNA866991. Methylation and TL datasets generated during the current study are available from the corresponding author on reasonable request.
CRediT authorship contribution statement
Kristyna Tomasova: Writing – original draft, Investigation. Karolina Seborova: Writing – review & editing, Investigation, Formal analysis, Data curation. Michal Kroupa: Writing – original draft, Investigation. Josef Horak: Visualization, Formal analysis. Miriam Kavec: Investigation. Ludmila Vodickova: Project administration, Funding acquisition. Lukas Rob: Resources, Funding acquisition. Martin Hruda: Resources. Marcela Mrhalova: Resources. Alena Bartakova: Resources. Jiri Bouda: Resources. Thomas Fleischer: Investigation, Formal analysis. Vessela N. Kristensen: Supervision. Pavel Vodicka: Writing – review & editing, Supervision, Project administration, Funding acquisition, Conceptualization. Radka Vaclavikova: Writing – review & editing, Supervision, Project administration, Funding acquisition, Conceptualization.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We would like to thank the Genomics Core Facility, Oslo University Hospital (http://oslo.genomics.no/), for performing methylation array analysis.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.heliyon.2024.e33525.
Contributor Information
Kristyna Tomasova, Email: kristyna.tomasova@iem.cas.cz.
Karolina Seborova, Email: karolina.seborova@szu.cz.
Michal Kroupa, Email: michal.kroupa@iem.cas.cz.
Josef Horak, Email: josef.horak@iem.cas.cz.
Miriam Kavec, Email: miriam.kavec@gmail.com.
Ludmila Vodickova, Email: ludmila.vodickova@iem.cas.cz.
Lukas Rob, Email: lukas.rob@lf3.cuni.cz.
Martin Hruda, Email: martin.hruda@lf3.cuni.cz.
Marcela Mrhalova, Email: marcela.mrhalova@lfmotol.cuni.cz.
Alena Bartakova, Email: bartakovaa@fnplzen.cz.
Jiri Bouda, Email: boudaj@fnplzen.cz.
Thomas Fleischer, Email: thomas.fleischer@rr-research.no.
Vessela N. Kristensen, Email: v.n.kristensen@medisin.uio.no.
Pavel Vodicka, Email: pavel.vodicka@iem.cas.cz.
Radka Vaclavikova, Email: radka.vaclavikova@szu.cz.
Appendix ASupplementary data
The following is the Supplementary data to this article:
References
- 1.Srinivas N., Rachakonda S., Kumar R. Telomeres and telomere length: a general overview. Cancers. 2020;12:558. doi: 10.3390/cancers12030558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomasova K., Kroupa M., Forsti A., Vodicka P., Vodickova L. Telomere maintenance in interplay with DNA repair in pathogenesis and treatment of colorectal cancer. Mutagenesis. 2020:35261–35271. doi: 10.1093/mutage/geaa005. [DOI] [PubMed] [Google Scholar]
- 3.de Lange T. Shelterin-mediated telomere protection. Annu. Rev. Genet. 2018;52:223–247. doi: 10.1146/annurev-genet-032918-021921. [DOI] [PubMed] [Google Scholar]
- 4.Hu C., Rai R., Huang C., et al. Structural and functional analyses of the mammalian TIN2-TPP1-TRF2 telomeric complex. Cell Res. 2017;27:1485–1502. doi: 10.1038/cr.2017.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kibe T., Zimmermann M., de Lange T. TPP1 blocks an ATR-mediated resection mechanism at telomeres. Mol. Cell. 2016;61:236–246. doi: 10.1016/j.molcel.2015.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maciejowski J., de Lange T. Telomeres in cancer: tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017;18:175–186. doi: 10.1038/nrm.2016.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cesare A.J., Reddel R.R. Alternative lengthening of telomeres: models, mechanisms and implications. Nat. Rev. Genet. 2010;11:319–330. doi: 10.1038/nrg2763. [DOI] [PubMed] [Google Scholar]
- 8.Hahn W.C., Meyerson M. Telomerase activation, cellular immortalization and cancer. Ann. Med. 2001;33:123–129. doi: 10.3109/07853890109002067. [DOI] [PubMed] [Google Scholar]
- 9.Harley C.B. Telomerase and cancer therapeutics. Nat. Rev. Cancer. 2008;8:167–179. doi: 10.1038/nrc2275. [DOI] [PubMed] [Google Scholar]
- 10.Huda N., Xu Y., Bates A.M., et al. Onset of telomere dysfunction and fusions in human ovarian carcinoma. Cells. 2019;8:414. doi: 10.3390/cells8050414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bojesen S.E., Pooley K.A., Johnatty S.E., et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat. Genet. 2013;45:371–384. doi: 10.1038/ng.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang H.-N., Chiang Y.-C., Cheng W.-F., et al. Molecular alterations in endometrial and ovarian clear cell carcinomas: clinical impacts of telomerase reverse transcriptase promoter mutation. Mod. Pathol. 2015;28:303–311. doi: 10.1038/modpathol.2014.93. [DOI] [PubMed] [Google Scholar]
- 13.Wu R.-C., Ayhan A., Maeda D., et al. Frequent somatic mutations of the telomerase reverse transcriptase promoter in ovarian clear cell carcinoma but not in other major types of gynaecological malignancy. J. Pathol. 2014;232:473–481. doi: 10.1002/path.4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pilsworth J.A., Cochrane D.R., Xia Z., et al. TERT promoter mutation in adult granulosa cell tumor of the ovary. Mod. Pathol. 2018;31:1107–1115. doi: 10.1038/s41379-018-0007-9. [DOI] [PubMed] [Google Scholar]
- 15.Zhou H., Mondal A., Dakic A., et al. Time-dependent effects of POT1 knockdown on proliferation, tumorigenicity, and HDACi response of SK-OV3 ovarian cancer cells. BioMed Res. Int. 2018;2018 doi: 10.1155/2018/7184253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greenberg R.A., O'Hagan R.C., Deng H., et al. Telomerase reverse transcriptase gene is a direct target of c-Myc but is not functionally equivalent in cellular transformation. Oncogene. 1999;18:1219–1226. doi: 10.1038/sj.onc.1202669. [DOI] [PubMed] [Google Scholar]
- 17.Karami S., Han Y., Pande M., et al. Telomere structure and maintenance gene variants and risk of five cancer types. Int. J. Cancer. 2016;139:2655–2670. doi: 10.1002/ijc.30288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friedlander M., Trimble E., Tinker A., et al. Clinical trials in recurrent ovarian cancer. Int. J. Gynecol. Cancer. 2011;21:771–775. doi: 10.1097/IGC.0b013e31821bb8aa. [DOI] [PubMed] [Google Scholar]
- 19.Kroupa M., Rachakonda S.K., Liska V., et al. Relationship of telomere length in colorectal cancer patients with cancer phenotype and patient prognosis. Br. J. Cancer. 2019;121:344–350. doi: 10.1038/s41416-019-0525-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kroupa M., Rachakonda S., Vymetalkova V., et al. Telomere length in peripheral blood lymphocytes related to genetic variation in telomerase, prognosis and clinicopathological features in breast cancer patients. Mutagenesis. 2020;35:491–497. doi: 10.1093/mutage/geaa030. [DOI] [PubMed] [Google Scholar]
- 21.Guenin S., Mauriat M., Pelloux J., et al. Normalization of qRT-PCR data: the necessity of adopting a systematic, experimental conditions-specific, validation of references. J. Exp. Bot. 2009;60:487–493. doi: 10.1093/jxb/ern305. [DOI] [PubMed] [Google Scholar]
- 22.Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010 http://www.bioinformatics.babraham.ac.uk/projects/fastqc Seen 9. 6. 2024. Available online at: [Google Scholar]
- 23.Ewels P., Magnusson M., Lundin S., et al. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinforma Oxf Engl. 2016;32:3047–3048. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen S., Zhou Y., Chen Y., et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:884–890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bray N.L., Pimentel H., Melsted P., et al. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt0816-888d. [DOI] [PubMed] [Google Scholar]
- 26.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stephens M. False discovery rates: a new deal. Biostatistics. 2016;18:275–294. doi: 10.1093/biostatistics/kxw041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu T., Hu E., Xu S., et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2 doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu G., Wang L.-G., Yan G.-R., et al. DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics. 2015;31:608–609. doi: 10.1093/bioinformatics/btu684. [DOI] [PubMed] [Google Scholar]
- 30.Ashburner M., Ball C.A., Blake J.A., et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.The Gene Ontology Consortium. Aleksander S.A., Balhoff J., Carbon S., et al. The gene ontology knowledgebase in 2023. Genetics. 2023;224:31. doi: 10.1093/genetics/iyad031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanehisa M., Furumichi M., Sato Y., et al. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51:587–592. doi: 10.1093/nar/gkac963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.R Core Team European Environment Agency. 2020 https://www.eea.europa.eu/data-and-maps/indicators/oxygen-consuming-substances-in-rivers/r-development-core-team-2006 Seen 9. 6. 2024. Available online at: [Google Scholar]
- 34.Touleimat N., Tost J. Complete pipeline for Infinium® Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics. 2012;4:325–341. doi: 10.2217/epi.12.21. [DOI] [PubMed] [Google Scholar]
- 35.Fleischer T., Frigessi A., Johnson K.C., et al. Genome-wide DNA methylation profiles in progression to in situand invasive carcinoma of the breast with impact on gene transcription and prognosis. Genome Biol. 2014;15:435. doi: 10.1186/PREACCEPT-2333349012841587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fortin J.-P., Triche T.J., Hansen K.D. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics. 2016;33:558–560. doi: 10.1093/bioinformatics/btw691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maksimovic J., Gordon L., Oshlack A. SWAN: subset-quantile within array normalization for Illumina Infinium HumanMethylation450 BeadChips. Genome Biol. 2012;13:44. doi: 10.1186/gb-2012-13-6-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du P., Zhang X., Huang C.-C., et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinf. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pidsley R., Zotenko E., Peters T.J., et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17:208. doi: 10.1186/s13059-016-1066-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seborova K., Hlavac V., Holy P., et al. Complex molecular profile of DNA repair genes in epithelial ovarian carcinoma patients with different sensitivity to platinum-based therapy. Front. Oncol. 2022;12 doi: 10.3389/fonc.2022.1016958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bibikova M., Le J., Barnes B., et al. Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics. 2009;1:177–200. doi: 10.2217/epi.09.14. [DOI] [PubMed] [Google Scholar]
- 42.Kuhn E., Meeker A.K., Visvanathan K., et al. Telomere length in different histologic types of ovarian carcinoma with emphasis on clear cell carcinoma. Mod. Pathol. 2011;24:1139–1145. doi: 10.1038/modpathol.2011.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kotsopoulos J., Prescott J., De Vivo I., et al. Telomere length and mortality following a diagnosis of ovarian cancer. Cancer Epidemiol. Biomark. Prev. 2014;23:2603–2606. doi: 10.1158/1055-9965.EPI-14-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Falandry C., Horard B., Bruyas A., et al. Telomere length is a prognostic biomarker in elderly advanced ovarian cancer patients: a multicenter GINECO study. Aging. 2015;7:1066–1076. doi: 10.18632/aging.100840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun P.-M., Wei L.-H., Luo M.-Y., et al. The telomerase activity and expression of hTERT gene can serve as indicators in the anti-cancer treatment of human ovarian cancer. Eur. J. Obstet. Gynecol. Reprod. Biol. 2007;130:249–257. doi: 10.1016/j.ejogrb.2006.01.028. [DOI] [PubMed] [Google Scholar]
- 46.Zaug A.J., Podell E.R., Nandakumar J., Cech T.R. Functional interaction between telomere protein TPP1 and telomerase. Genes Dev. 2010;24:613–622. doi: 10.1101/gad.1881810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang L., Wang W., Hu L., et al. Telomere-binding protein TPP1 modulates telomere homeostasis and confers radioresistance to human colorectal cancer cells. PLoS One. 2013;8 doi: 10.1371/journal.pone.0081034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Widschwendter A., Muller H., Hubalek M., et al. Methylation status and expression of human telomerase reverse transcriptase in ovarian and cervical cancer. Gynecol. Oncol. 2004;93:407–416. doi: 10.1016/j.ygyno.2004.01.036. [DOI] [PubMed] [Google Scholar]
- 49.Liu H., Liu Q., Ge Y., et al. hTERT promotes cell adhesion and migration independent of telomerase activity. Sci. Rep. 2016;6 doi: 10.1038/srep22886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hannen R., Bartsch J.W. Essential roles of telomerase reverse transcriptase hTERT in cancer stemness and metastasis. FEBS Lett. 2018;592:2023–2031. doi: 10.1002/1873-3468.13084. [DOI] [PubMed] [Google Scholar]
- 51.Romaniuk-Drapała A., Toton E., Konieczna N., et al. hTERT downregulation attenuates resistance to DOX, impairs FAK-mediated adhesion, and leads to autophagy induction in breast cancer cells. Cells. 2021;10:867. doi: 10.3390/cells10040867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chi T., Wang M., Wang X., et al. PPAR-γ modulators as current and potential cancer treatments. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.737776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dai L., Song K., Di W. Adipocytes: active facilitators in epithelial ovarian cancer progression? J. Ovarian Res. 2020;13:115. doi: 10.1186/s13048-020-00718-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mukherjee A., Chiang C.-Y., Daifotis H.A., et al. Adipocyte-induced FABP4 expression in ovarian cancer cells promotes metastasis and mediates carboplatin resistance. Cancer Res. 2020;80:1748–1761. doi: 10.1158/0008-5472.CAN-19-1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gharpure K.M., Pradeep S., Sans M., et al. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat. Commun. 2018;9:2923. doi: 10.1038/s41467-018-04987-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang M., Karsenberg R., Bianchi F., et al. CD36 as a double-edged sword in cancer. Immunol. Lett. 2024;265:7–15. doi: 10.1016/j.imlet.2023.12.002. [DOI] [PubMed] [Google Scholar]
- 57.Ladanyi A., Mukherjee A., Kenny H.A., et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene. 2018;37:2285–2301. doi: 10.1038/s41388-017-0093-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou S., Wang R., Xiao H. Adipocytes induce the resistance of ovarian cancer to carboplatin through ANGPTL4. Oncol. Rep. 2020;44:927–938. doi: 10.3892/or.2020.7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jayawardhana A.M.D.S., Bhandari S., Kaspi-Kaneti A.W., et al. Visible light-activatable platinum(IV) prodrugs harnessing CD36 for ovarian cancer therapy. Dalton Trans. 2023;52:10942–10950. doi: 10.1039/d3dt01292a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq dataset generated and analyzed during the current study is available in the NCBI BioProject repository, https://www.ncbi.nlm.nih.gov/, BioProject ID: PRJNA866991. Methylation and TL datasets generated during the current study are available from the corresponding author on reasonable request.