

Abstract
Sensing and characterization of water-soluble peptides is of critical importance in a wide variety of bioapplications. Single molecule nanopore spectrometry (SMNS) is based on the idea that one can use biological protein nanopores to resolve different sized molecules down to limits set by the blockade duration and noise. Previous work has shown that this enables discrimination between polyethylene glycol (PEG) molecules that differ by a single monomer unit. This paper describes efforts to extend SMNS to a variety of biologically relevant, water-soluble peptides. We describe the use of Au25(SG)18 clusters, previously shown to improve PEG detection, to increase the on- and off-rate of peptides to the pore. In addition, we study the role that fluctuations play in the single molecule nanopore spectrometry (SMNS) methodology and show that modifying solution conditions to increase peptide flexibility (via pH or chaotropic salt) leads to a nearly 2-fold reduction in the current blockade fluctuations and a corresponding narrowing of the peaks in the blockade distributions. Finally, a model is presented that connects the current blockade depths to the mass of the peptides, which shows that our enhanced SMNS detection improves the mass resolution of the nanopore sensor more than 2-fold for the largest cationic peptides studied.
Keywords: resistive-pulse nanopore sensing, peptides, angiotensin, neurotensin, alpha hemolysin, Au25(SG)18, thiolate-capped clusters, nanomolecules
Graphical Abstarct
Biosensors offer an enticing direct route for detection and quantification of a host of medically relevant biomarkers (e.g., proteins, heavy metals, DNA, RNA, and peptides).1–10 Detecting, characterizing, and quantifying peptides is of particular interest given the role that proteolysis plays in the onset and development of a wide range of diseases11–13 (e.g., cancer,14–16 Alzheimer’s disease,17,18 and Huntington’s disease19). The vast majority of peptide sensors utilize an immunoassay strategy,20,21 although limitations of this approach have motivated the development of mass spectrometry for peptide detection.22–24 Both approaches utilize costly and time-consuming methodologies, which complicates the development of clinically based peptide sensing. Our goal is to explore alternate means of peptide characterization that could enable rapid and low-cost detection in a clinical setting. This requires the ability to identify label-free peptides in heterogeneous mixtures. One technique that shows promise toward this goal is resistive-pulse nanopore sensing.25–29
While nanopores have previously been used to detect and characterize proteins and peptides,30 further study is required to convert this technology into a robust scheme for identifying specific peptides from heterogeneous mixtures. Nanopore-based detection began in earnest as a method for next-generation DNA sequencing.31 In addition to DNA analysis, there is also a growing body of research into a wide range of other analytes.32–42 For nanopore sensing, the size of the target analyte can dictate the type of nanopore used. For example, full-length proteins have been investigated with solid-state nanopores fabricated in SiN43–45 and pulled glass nanopippettes.46,47 The pores used in these experiments were sufficiently large to enable transport and detection of protein molecules ranging in size from ~30 kDa to ~300 kDa. While these detectors offer an excellent label-free method to quantify protein concentration, it remains difficult to reproducibly fabricate solid state nanopores at the length scales required to distinguish between two similarly sized peptides without internal standards. This can be improved with biological nanopores which possess a high degree of reproducibility and precisely controlled assembly.48,49
One of the first demonstrations of protein analysis with biological nanopores studied polymers interacting with the nanopore and a protein too large to enter the pore’s lumen.50 However, to enable protein sensing directly with nanopores, the molecules must be able to partition into the nanopore. This can be accomplished in several ways. For instance, native peptides and proteins can fold into structures small enough to partition into the pore directly.39,51,52 In some cases the pore can be modified to enhance polypeptide attraction and subsequently lower the energetic barrier to partitioning.53 Proteins have also been chemically denatured,37,54,55 thermally melted,56 and cleaved into smaller fragments38,57–60 to facilitate entry into the pore. To detect specific analytes from a complex sample matrix (i.e., blood serum), peptides can be isolated with affinity tags and extracted for analysis with enzymatic reactions.58 Indirect sensing schemes have been demonstrated in nanopores where specific DNA sequences were detected through interactions with a protein recognition site.61–63 This scheme can be reversed whereby proteins can be detected after binding to a specific DNA molecule.64,65 These examples show the potential of nanopore sensing for proteomics.27 Furthermore, they suggest that nanopore sensors may complement existing analytical modalities because they offer the possibility of quantifying the peptide concentration, observing different peptide conformations,41,66 and measuring a variety of enzymatic reactions.31,67 We propose to advance nanopore-based peptide sensing by adopting single molecule nanopore spectrometry (SMNS).
Previous efforts with SMNS focused on polyethylene glycol (PEG) molecules.68–71 These results showed that biological nanopores are capable of identifying individual PEG molecules based on their size68 and chemistry69,72 with mass resolution to better than a single monomer unit (≈44 g/mol). High-resolution baseline separation has been demonstrated for other analytes,73,74 and SMNS efforts have been extended to include size-dependent detection of short nucleic acids.75 To demonstrate the viability of nanopore sensing for proteomics, one must identify peptide species with indeterminate size and sequence.
While PEG has been used extensively to develop SMNS,68,76–78 peptides present more difficulty given their diverse chemistry (i.e., charge, sequence, secondary structure, etc.). Nevertheless, this diversity has been studied via nanopore sensing in a number of ways including estimating peptide–pore binding interactions of cationic peptides,79 low-resolution structure measurements,80,81 or force-spectroscopy,66 and manipulating peptide-pore kinetics.53,82–84 These results represent important progress in using nanopore sensing to analyze peptides, but demonstrating SMNS-like detection of peptides requires accurate estimates of the current blockade from each peptide interaction with the pore. This can be achieved by manipulating the duration and noise of each blockade.85
There is a precise connection between current blockade depths and corresponding molecular size,68,69 first observed for PEG due to small current fluctuations (ΔiRMS ≈ 2 pA, V = 50–80 mV, bandwidth = 100 kHz) and long residence times (tres ≈ 0.5 ms). Further improvements to PEG-based SMNS were recently demonstrated with the addition of metallic clusters to the cis-side of the alpha-hemolysin (αHL) pore.85,86 The clusters increased the time PEG spent in the pore by more than an order of magnitude while having little effect on the current fluctuations, which led to a reduction in the observed peak widths in the blockade distribution.
Here we report on our efforts to improve the SMNS sensing protocol by using cluster-enhanced SMNS to detect and characterize small water-soluble peptides at the single molecule limit. We present a generalized model that predicts the molecular mass from the magnitude of the peptide-induced current blockade with and without a gold cluster in the pore. Finally, we emphasize how current blockade fluctuations play a critical role in establishing the resolving power of the SMNS technique and that one can further improve peptide analysis by modifying the solution conditions to create more flexible peptides and correspondingly lower noise blockades. This yields narrower peaks in the current blockade distributions. These results demonstrate a viable way forward to perform nanopore analysis of peptide detection and identification at the single molecule limit.
EXPERIMENTAL METHODOLOGY
The experimental procedure and data processing methodology have been described elsewhere.85,86 We briefly summarize the approach here. Unless stated otherwise, all chemicals and peptides were purchased and used as received from Sigma-Aldrich (St. Louis, MO). A 25-μm-thick, polytetrafluoroethylene (PTFE, Teflon) sheet with a 100 μm hole (Eastern Scientific, Rockville, MD, USA) was attached to a PTFE holder with polydimethysiloxane (PDMS, Kwik-Cast, World Precision Instruments, Sarasota, FL, USA). A 1 mg/mL pentane-based prepaint solution (1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhyPC, Avanti Polar Lipids, Alabaster, AL, USA)) was applied to both sides of the 100 μm hole after which the partition was positioned ca. 300 μm from the top of a microscope coverslip. The mounted partition was placed on top of an inverted microscope (AxioObserver D, Zeiss, Germany) for optical access. Approximately 500 and 2500 μL of 3 M KCl, 10 mM Tris electrolyte solution was added above and below the partition, respectively, to support the formation of a lipid bilayer membrane. Unless stated otherwise, the lower compartment (trans-side) was additionally filled with 20 μM of desired analyte.
Lipid bilayer membranes were formed by using a glass bead formed at the end of a glass rod to paint a 10 mg/mL DPhyPC:hexadecane solution over the 100 μm hole. After several seconds or after the application of a sufficient electrical potential (>150 mV) across the hexadecane plug, a stable bilayer membrane formed. Following the formation of the membrane, a 0.1 mg/mL solution of αHL monomers (List Biological Laboratories Inc., Campbell, CA, USA) were ejected in the vicinity of the membrane until several hundred αHL pores were formed across the membrane. The number of αHL pores in the membrane was monitored by applying a small transmembrane voltage (≈1 mV) and comparing the current to the expected conductance of a single channel.87
A Ag/AgCl electrode was inserted into a quartz capillary (1 mm OD, 0.7 mm ID with filament, Sutter Instrument, Novato, CA) patch pipette tip containing 3 M KCl and ca. 40 μM Au25(SG)18 clusters. The cluster synthesis protocol has been described elsewhere.85 The patch pipet was lowered onto the membrane to isolate a single channel. The presence of a single pore can be determined by comparing the time-averaged current with the expected single channel current. Ionic currents were recorded using a patch clamp amplifier (Axopatch 200B, Molecular Devices, Carlsbad, CA) with a built-in 4-pole 10 kHz low-pass Bessel filter. Unless stated otherwise, the ionic current was sampled at 50 kHz using an analog-to-digital converter (Digidata 1440A, Molecular Devices) and processed with commercially available software (pClamp10, Molecular Devices). Data analysis was performed in a manner previously described85,86 with in-house software written in LabView 8.5 (National Instruments, Austin, TX). Briefly, a thresholding algorithm is used to identify blockades with the threshold set at 35% below the averaged open current for a time greater than the minimum cutoff-time. All results reported herein used a value of tcut = 180 μs unless otherwise noted.
RESULTS AND DISCUSSION
Despite the increasingly sophisticated understanding of how proteins and peptides interact with nanopores,79,88–90 little effort has focused on the quantitative connection between blockade depth and peptide size. The goal here is to show that SMNS analysis enables one to estimate the mass of a peptide from the magnitude of the current blockade. To demonstrate proof-of-principle, we selected five biologically relevant water-soluble peptides (leu enkephalin (LE) (Proteochem, Hurricane UT), angiotensin I (A1), angiotensin II (A2), polyglutamine-binding peptide 1 (QBP1) and neurotensin (NT)) whose descriptions, amino acid sequences, molecular weights and calculated charges at pH 7.2 and pH 5.8 are all listed in Section 1 and Table S1 in Supporting Information.
In a typical experiment, ionic current is driven through a nanopore at a fixed applied potential. When the peptide partitions into the trans-side of the pore it causes a short-lived resistive pulse (i.e., a decrease in the observed current). In Figure 1, the pore is tested with NT on the trans-side of the membrane, while anionic Au25(SG)18 clusters (D ≈ 2 nm91,92) are introduced on the cis-side. It was previously shown85 that these clusters are negatively charged with charge z = −5e to −9e, which precludes the entry of multiple clusters into the pore. When there is no cluster in the pore, resistive pulses are deep, short-lived, and infrequent (Figure 1 left and Table S2 in Supporting Information). When a cluster occupies the pore, the peptide-induced blockades remain deep and become longer-lived and more frequent (Figure 1 right and Table S2 in Supporting Information).
Figure 1.
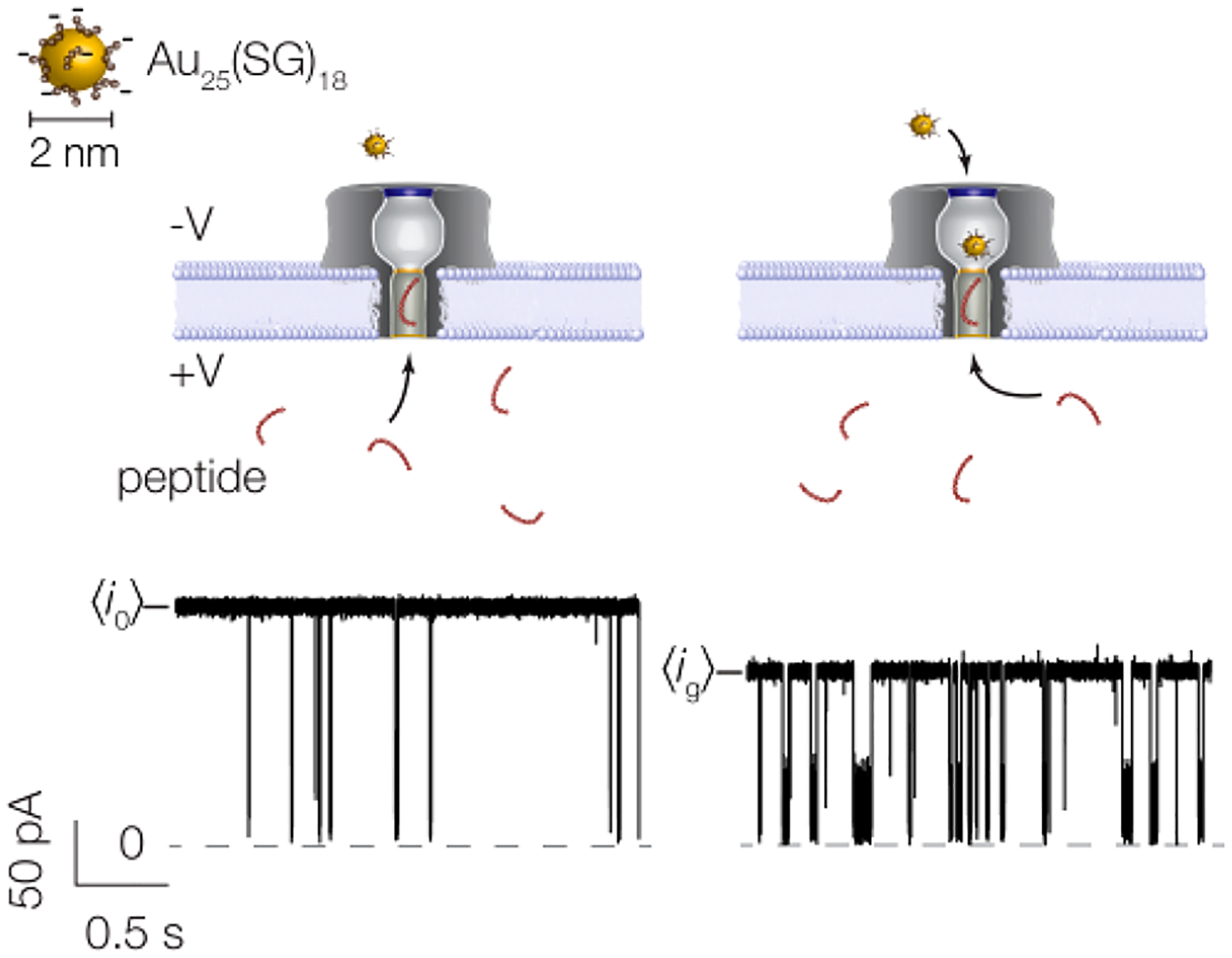
Schematic illustration of the cluster-enhanced nanopore detection with corresponding current traces. (Left) Prior to cluster insertion, the open pore current is stable around pA and short-lived blockades result from peptide (NT) entry and exit from the trans-side of the pore. (Right) Upon insertion of a single cluster from the cis-side of the pore, the current is reduced to pA and both the frequency and residence time of peptide blockades are increased. A large-scale image of a cluster with corresponding scale bar is shown in the upper left corner. The current trace here corresponds to the following conditions: 3 M KCl, pH 7.2, 70 mV applied transmembrane potential, [NT] = 20 μM.
Each of the peptides examined here gives rise to current blockades that exhibit Gaussian fluctuations. In such cases, the most directly accessible blockade characteristic is the magnitude of the current interruption, the average blockade depth, presented as the ratio of the mean blocked current to the mean open channel current with or without a gold cluster in the pore. For each peptide studied, the resistive pulses were characterized and collected into histograms of the blockade depth ( and ) (Figure 2A). Each peptide yielded a blockade depth distribution that is dominated by a single peak. For A1 and NT, there are smaller peaks to the left of the main peaks that may arise from contaminants or secondary conformations of the peptide within the nanopore.45 Figure 2B shows corresponding residence time (toff) and time between event starts (ton) distributions. Fitting single exponential functions to these distributions enables one to extract the mean peptide residence time τres and mean peptide on-rate kon respectively.
Figure 2.
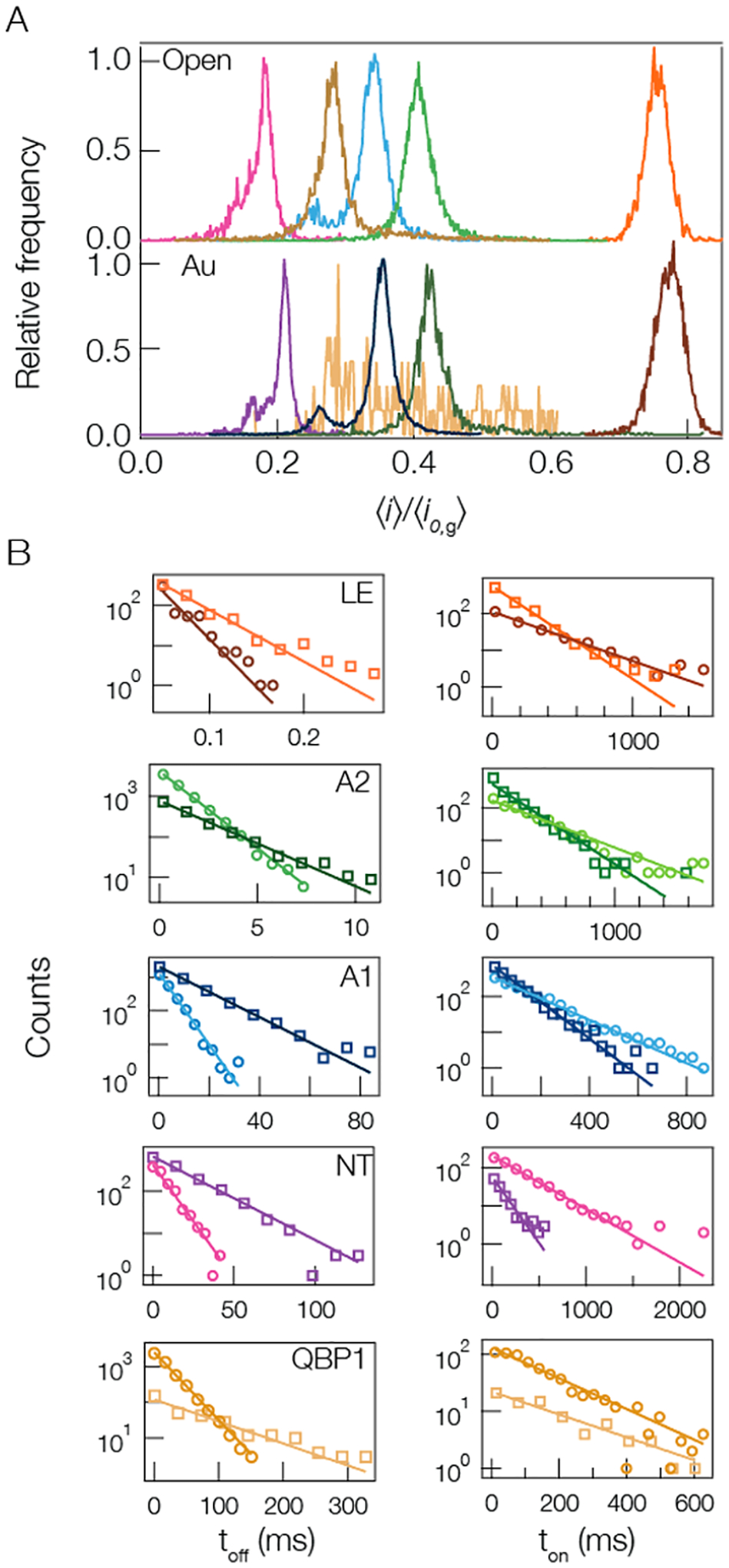
Cluster-enhanced SMNS peptide detection. (A) Representative current blockade distributions for the five different peptides studied ((pink) NT, (gold) QBP1, (blue) A1, (green) A2, and (orange) LE) give rise to current blockade distributions dominated by single peaks with (bottom) and without (top) a gold cluster in the pore. The distributions for the open (gold-occupied) pore configuration are calculated from NT = 2225 (2434), QBP1 = 6649 (177), A1 = 3984 (13130), A2 = 7188 (1845), and LE = 535 (714) blockade events. (B, left column) The corresponding blockade residence time (toff) distributions with (squares) and without (circles) a gold cluster in the pore show that the cluster yields longer lived events. The solid lines correspond to least-squares fits to the data with single exponential functions from which the mean residence time τres is extracted. (B, right column) The time between event starts (ton) distributions, with (squares) and without (circles) a gold cluster in the pore, show that the cluster-based detection yields increased on-rate with a cluster in the pore (except for the QBP1). The solid lines correspond to single exponential functions from which the mean on-rate kon is extracted. All data shown here, unless otherwise noted, was taken under the following conditions: 3 M KCl, pH 7.2, 70 mV applied transmembrane voltage, [peptide] = 20 μM. Each peptide’s open and gold-occupied data was collected on the same pore. See Table S2 in Supporting Information for complete results for peak i/io,g values, τres, and kon.
A cluster in the cis-side of the pore has previously been shown to serve as a localized charge trap that attracts cationic analyte entering from the trans-side.85,86 Cationic peptides will diffuse within the trans-side lumen and interact with the cluster near the constriction separating the lumen and vestibule sections of the pore. This interaction will either slow down the translocation of the peptide through the pore or increase the free energy barrier to escape out the trans-side into which it originally entered. Regardless of the exact mechanism of peptide escape from the pore, the presence of the gold cluster serves to increase the mean residence time and the on-rate of analyte to the pore. Increasing the residence time leads to more accurate estimates of the blockade depth () and this in turn yields narrower peaks in the blockade distributions (fwhm).85,86 Figure 2 and Table S2 in Supporting Information illustrate that this is also true when detecting and analyzing blockades from cationic peptides. In all cases the cluster increases the mean residence time ca. 3-fold. All of these results show that introducing charged metallic clusters in the cis-side of the pore leads to improved sensing of the cationic peptides as evidenced by the increased on-rate of peptides to the pore, residence time of peptides in the pore, and narrowing of the blockade distributions by up to 25% from the open pore system. It is worth noting that while the QBP1 anionic peptide shows an unexpected increase in the mean residence time with a cluster in the pore, the efficacy of sensing the anionic peptide with the gold-occupied pore is reduced because the repulsive interaction between the anionic gold-cluster and QBP1 lowers the peptide on-rate and destabilizes the gold states (see Figure S3 and Table S2 in the Supporting Information). Note that given the shortened time for LE in the pore, all LE data was sampled at 100 kHz and the cutoff duration for each blockade was set to 60 μs. The constant slope seen in Figure 2B for LE’s residence time distributions suggests the 10 kHz filter had little effect on our estimates for τres(LE).
To reduce the reliance on internal standards, and to extend SMNS methodology to analytes beyond regular repeating homopolymers (e.g., PEG, homo-DNA), a connection between current blockade magnitude and the molecular weight of the analyte must be generalized. Figure 3 shows the most probable blockade depth (peak locations from Figure 2A) plotted as a function of corresponding molecular weight for the peptides studied herein and a corresponding distribution of PEG molecules. A clear trend emerges between the current blockade depths and the mass of the sequestered polymer. Already demonstrated for PEG68 and poly dA,75 the trend shown here suggests a connection also exists between the blockade depth and molecular weight for this random collection of peptides. As with previous results, the deepest current blockade peaks correspond to the largest molecules, and the shallowest blockade to the smallest molecules.
Figure 3.
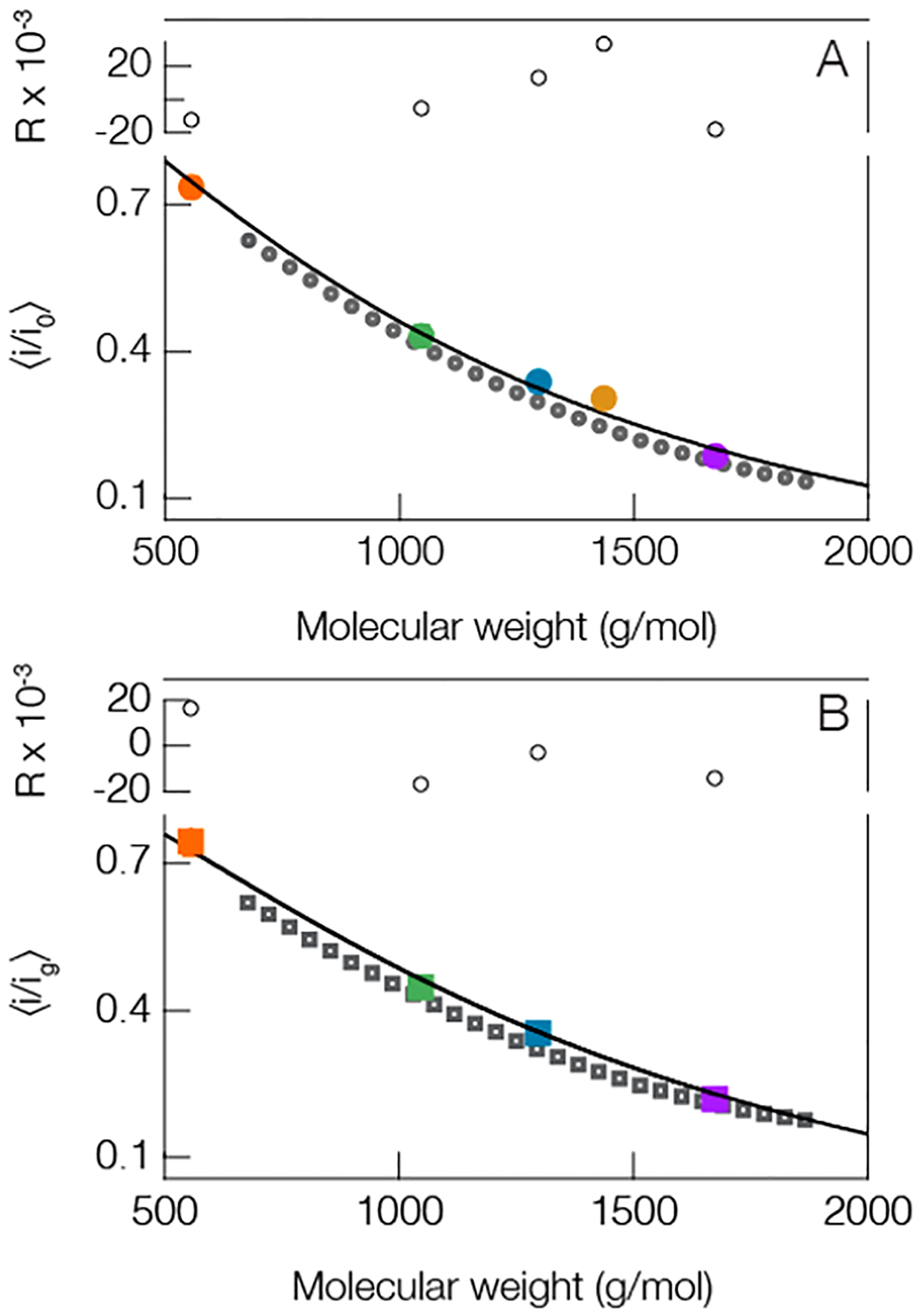
Peptides and PEG show a similar trend between mean current blockade depth and molecular weight. This suggests that one could deduce the size of peptides from the blockade depth. (A, solid circles) open pore and (B, solid squares) gold occupied pore both show a similar trend. The solid line in the open pore case is a least-squares fit to the peptides only (colored circles, same colors used in Figure 2) using eq 1 with fit parameters a = 1.68 ± 0.12 and γ = 0.34 ± 0.06. The solid line in the lower figure corresponds to eq 2 from the main text. This model utilizes one free parameter that quantifies the interaction strength between the peptides and cluster and was found to be d = 0.13 ± 0.04. The QBP1 in the gold cluster case is excluded because of the limited number of gold-state events. This is expected given QBP1, and the gold clusters are negatively charged. Blockade peak positions were calculated from the means of at least three different experiments (see Table S2 in Supporting Information for complete details). The residuals (R) for each fit are shown above each figure. All measurements were performed in 3 M KCl under an applied 70 mV transmembrane potential at pH 7.2 with peptide concentrations of 20 μM. The dark gray open circles in (A) and the dark gray open squares in (B) correspond to PEG blockades which are not included in the fitting protocol. All error bars are smaller than the data points shown, but they are reported in Table S2 in the Supporting Information.
Two of us have previously developed a model that characterized the current blockades (blockade depth and residence time) from PEG using a thermodynamic description for the polymer-pore-electrolyte interaction.69,72,93 In this model, the pore is treated as a piecewise linear combination of open pore and polymer occluded pore. The polymer modifies the current through two mechanisms: volume exclusion and interactions between the ions and the polymer. Here a simplified model is developed that explicitly addresses volume exclusion and treats physical interactions (e.g., electrolyte interactions, electroosmosis, etc.) peripherally in an ad hoc manner. This model connects the peak blockade depth for each polymer to that polymer’s mass
| (1) |
where Mp is the mass of the polymer, Mf is the mass of the smallest polymer that completely fills the nanopore volume, ν is the Flory exponent, and a and γ are free parameters that describe ionic binding to the polymer in the pore. (A derivation of eq 1 with more complete descriptions of the variables can be found in Section 2 of the Supporting Information.)
While the clusters were shown to increase the mean residence time for the cationic peptides (Figure 2B) this enhancement is not useful for routine quantitative identification if the connection between current blockade depth and molecular weight is lost. Figure 3B reports the blockade peak positions as a function of molecular weight for the polydisperse PEG mixture and the four cationic or neutral peptides from Table S1 with a gold cluster in the pore. We find a correlation similar to the open pore case between the mean current blockade depth and molecular weight. To model the data in Figure 3B we transform eq 1 by incorporating a gold cluster into the cis-side of the pore. A complete description of this transformation procedure can be found elsewhere,86 but briefly the gold cluster in the pore leads to a slight modification of the pore resistance that can be taken into account with a piecewiselinear ohmic model that also incorporates an interaction term between the polymer and gold cluster. The overall result transforms the blockade depth without a cluster in the pore into the following expression
| (2) |
where is the experimentally determined ratio of the open pore current to the current through the gold occupied pore and d is a freely adjustable model parameter that characterizes the interaction between cluster and peptide. The data shown in Figure 3A,B was simultaneously fitted with eqs 1 and 2. To minimize the number of free parameters, we fixed the value of Mf = 3000 g/mol77 and assumed that the peptides behave as polymers in a good solvent so the Flory exponent is ν = 0.6.94,95 In addition, we used the experimentally determined value of c = 1.38, which leaves three adjustable parameters a, γ, and d with which to fit the 9 data points plotted across both Figure 3A and B. Least-squares fitting yields values of a = 1.68 ± 0.12, γ = 0.34 ± 0.06, and d = 0.13 ± 0.04 (±1 S.D.). Notably, the d-parameter is in good agreement with previous PEG-based results.86 The quality of the fit, parametrized by the residuals (R) in Figure 3, suggests one can deduce the molecular weight of near-neutral peptides from the magnitude of the corresponding current blockades both with and without a gold cluster in the pore.
The ability to connect the blockade depth to the molecular weight suggests that the SMNS technique should be relevant to a wide range of molecules beyond homopolymers (i.e., PEG).68,69 One reason PEG so clearly illustrates blockade distributions with single monomer resolution is that fluctuations within each blockade are small enough to yield accurate estimates of the mean current. Specifically, the standard deviation of typical current blockades from PEG in 3 M KCl under an applied voltage of 70 mV is on the order of 2 pA. It was previously demonstrated85 that averaging a blockade with this degree of noise over a duration of 1 ms yields a peak in the current blockade distribution with fwhm ~8 × 10–3, which is smaller than the typical peak separation for PEG (). This difference explains why PEG-induced peaks are easily resolved with the SMNS approach. Therefore, in order to make the SMNS approach viable for peptides one must increase the mean blockade time (via gold clusters) and also reduce the fluctuations so the peaks in the current blockade distribution are sufficiently narrow to enable clear distinctions between different mass peptides. Narrowing these blockade distribution peaks enables one to identify different populations of peptides in a mixture and further narrowing of the peaks to baseline resolution would enable one to identify a peptide from a single current blockade. This motivates additional study of methods for reducing the current fluctuations within each peptide induced blockade.
Figure 4 compares the degree of fluctuations between PEG and A1. Typical current blockades for each molecule are shown in Figure 4A and it is clear that A1 produces larger current fluctuations than PEG. This is parametrized in the blockade standard deviation scatter plots shown in Figure 4B. The A1 peptide yields fluctuations ca. 8× greater than the PEG. This discrepancy explains why the peak for A1 is so much wider in Figure 2A than similar peaks for PEG.68 One possibility for why the PEG yields smaller fluctuations than A1 is that PEG molecules are more flexible than peptides so they undergo more rapid fluctuations in the pore that are undetectable at the time scale set by the inverse bandwidth of the measurement (B = 10 kHz). This is supported by the fact that the persistence length of PEG (lp ≈ 0.38 nm96) is much less than typical persistence lengths for homomeric peptides (lp ≈ 4 nm97,98).
Figure 4.
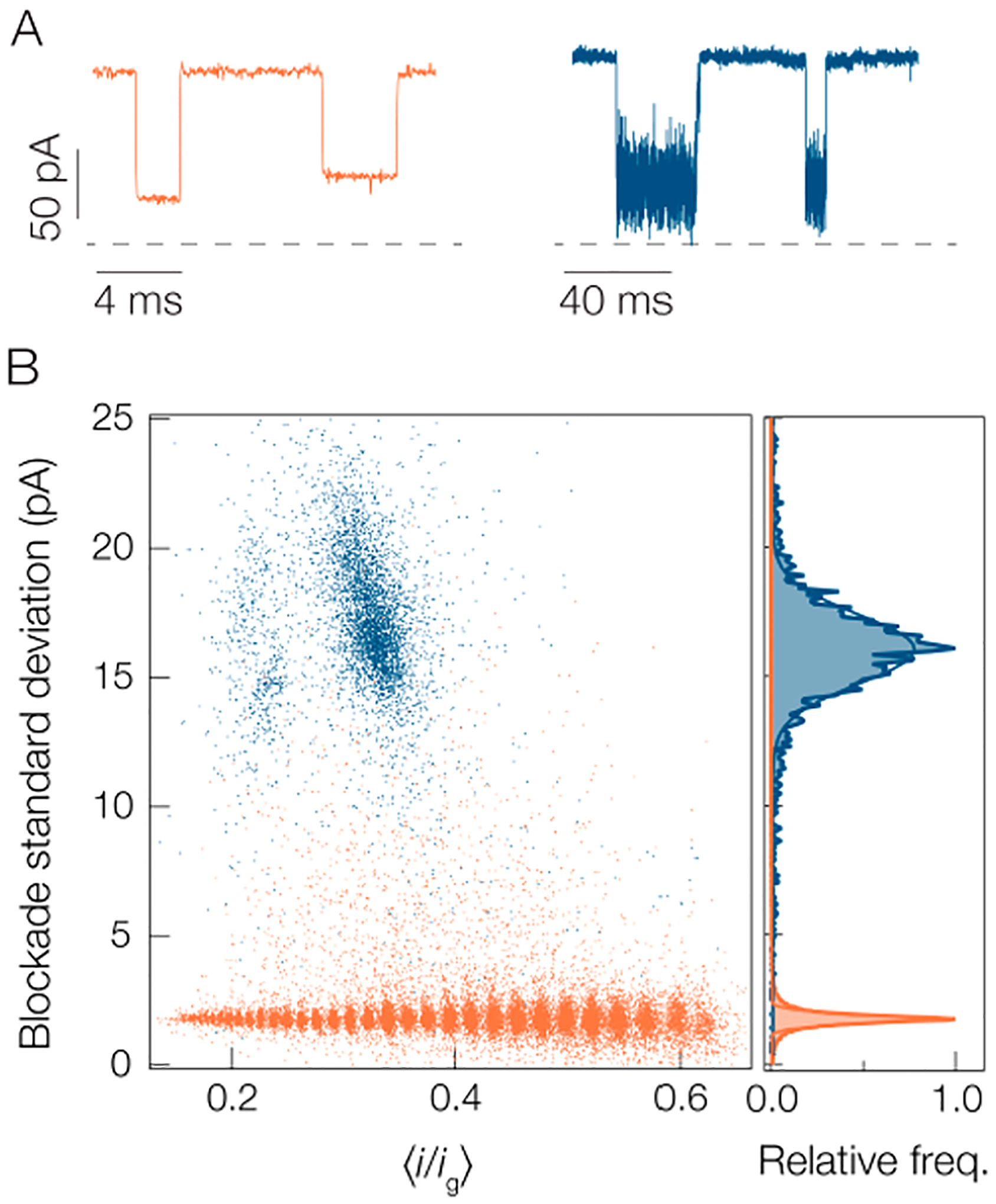
Standard deviation of blockades is greater for peptides than PEG, which complicates the application of the SMNS technique to peptide detection. (A) Sample current traces show that the noise within individual blockades is smaller for the PEG (orange) than the A1 peptide (blue). The apparent difference in noise levels in the open states results from the 10× difference in time scales between the two traces. (B) Scatter distribution of current blockade standard deviations for A1 (blue) and PEG (orange). The right outset shows the distributions for each species from which the peaks of each distribution are found to be (1.75 ± 0.26) pA for PEG and (16.1 ± 1.9) pA for A1. Data was collected in 3 M KCl pH 7.2 solution under a 70 mV applied transmembrane potential with a gold cluster in the pore. The PEG blockades arise from a polydisperse mixture of [PEG 600] = 2 μM, [PEG 1000] = [PEG 1500] = 10 μM, [PEG n = 12] = 0.5 μM, and [PEG n = 28] = 1 μM.
In addition to persistence length, structure also plays a role in establishing polymer dynamics.99 This suggests that modifying the peptide structure either through pH titration or the addition of chaotropic salts should have an effect on the fluctuations within each current blockade. To test this assertion, we first performed a pH titration experiment on A1 and NT to see the effect on current blockade fluctuations.
The expected charge of each peptide can be estimated by assuming a simple acid–base dissociation model. The results for the effective charge calculations of A1 and NT as a function of pH are shown in the Supporting Information (Section 3 and Figure S1 Supporting Information), and they demonstrate that while the NT charge remains nearly constant over the pH range shown, the A1 charge undergoes a dramatic change as expected given its histidine residue (pKa ≈ 6). Circular dichroism and NMR measurements on angiotensin peptides have shown the role that the protonated state of the histidine residue has on modifying the local environment of the nearby tyrosine residue and by extension the structure of angiotensin.100,101 This is consistent with more recent efforts that studied β-hairpin peptide interactions with alpha hemolysin under acidic conditions.41 In that work, protonated histidines were shown to destabilize a β-hairpin into a random coil structure. Therefore, if more flexible peptides reduce the standard deviation of the current blockades, then lowering the solution pH should reduce the fluctuations from A1 induced peptides while having little to no effect on NT induced blockades. This is supported by the data shown in Figure 5 and suggests that modifying the solution conditions and structural state of peptides can reduce the current fluctuations of the current blockades.
Figure 5.
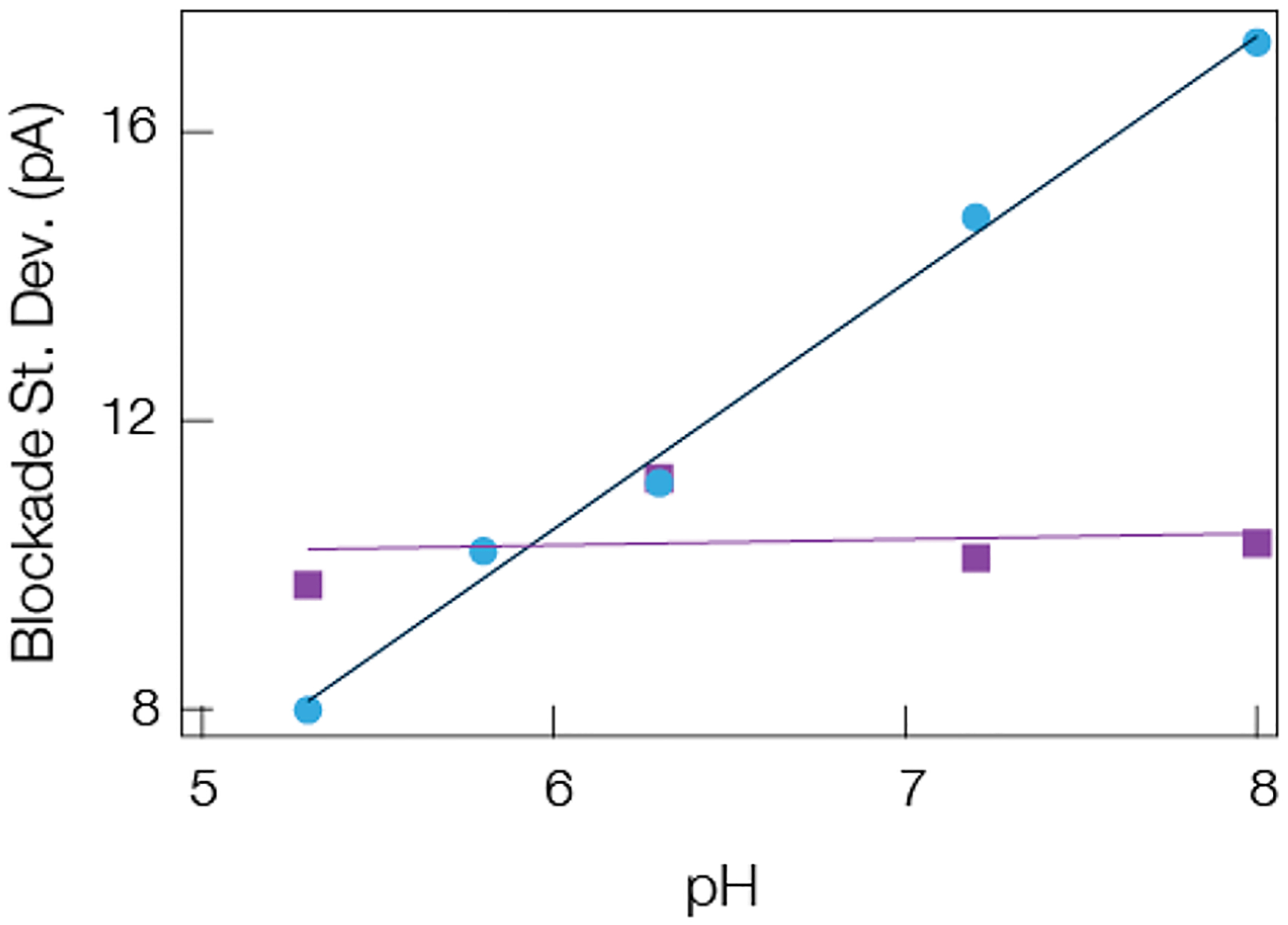
Mean blockade standard deviation depends on the estimated peptide charge, which suggests structural changes can give rise to a reduction in current noise. The mean standard deviation of A1 (blue circles) and NT (purple squares) scales with the electrolyte pH in a manner expected from the corresponding pK values of each peptide. Decreasing the pH leads to a structural change in the A1 peptide which leads to a corresponding reduction in the noise within current blockades. No such behavior is seen or expected for the NT. The solid lines are least-squares linear fits to the data. All data was taken under a 70 mV applied transmembrane potential in 3 M KCl with a gold cluster present in the pore.
In addition to modifying the charge of the peptide with pH, one can also modify the structure of the peptide by disrupting hydrogen bonds that stabilize β-folds with a chaotropic salt such as guanidine hydrochloride (Gdm-HCl).102 If our hypothesis regarding the connection between peptide flexibility and blockade fluctuations is correct then we expect a reduction in blockade standard deviation with the addition of Gdm-HCl due to the fact that the broken hydrogen bonds will yield a more flexible peptide. Figure 6 demonstrates this result for A2, A1, and NT. In all three cases it is clear that the addition of 1 M Gdm-HCl to the trans-side of the pore yields blockades with standard deviations that are nearly halved. This, along with the pH titration experiment, illustrates that modification to the peptide structure via targeted protonation of residues or destabilization of hydrogen bonds can reduce the fluctuations in individual current blockades, which should yield narrower peaks in the current blockade distributions. I–V curves that show 1 M Gdm-HCl on the trans-side of the pore has very little effect on the αHL pore are shown in Section 6 of the Supporting Information.
Figure 6.
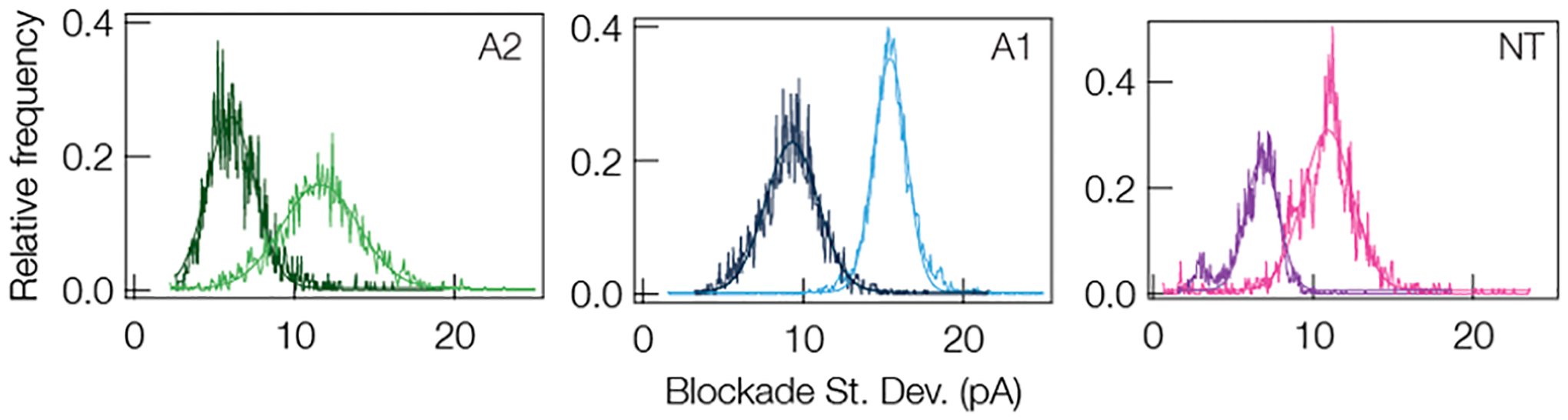
Representative distributions of current blockade standard deviations (SD) for A2, A1, and NT with (dark traces) and without (light traces) 1 M Gdm-HCl on the trans-side of the pore demonstrate that eliminate hydrogen bonding and thus any secondary structure in the peptides leads to an increase in peptide flexibility and reduces the standard deviation. The mean SD without Gdm-HCl for A2, A1, and NT are (11.6 ± 3.4) pA, (15.4 ± 1.5) pA, and (10.9 ± 2.2) pA, respectively. The mean SD with Gdm-HCl for A2, A1, and NT are (6.03 ± 0.03) pA, (9.29 ± 2.33) pA, and (6.81 ± 1.42) pA, respectively. All data was taken under a 70 mV applied transmembrane potential in 3 M KCl, pH 7.2, with a gold cluster present in the pore. The solid lines in all three sets are Gaussian least-squares fits to the data.
Motivated by the results in Figures 5 and 6, we explored the role that reducing the peptide fluctuations have on the quality of the current blockade distributions. Figure 7A,B,C compares the current blockade distributions for the three largest cationic peptides A1, A2, and NT. LE and QBP1 were omitted from this analysis because LE is too small to expect any significant denaturing under Gdm-HCl while QBP1 is anionic and shows no enhancement in the presence of anionic clusters. For all three peptides, the blockade distributions are narrowed from the open pore pH 7.2 case (dashed lines) to the gold-in-pore, pH 5.8, 1 M Gdm-HCl case (solid lines). This is expected given the reduction in blockade fluctuations seen in Figures 5 and 6 and reported in Table S3 of the Supporting Information. It is worth noting that the small secondary peaks in the blockade distributions for A1 and NT remain with the addition of Gdm-HCl, although to a lesser degree, suggesting the presence of contaminant in these samples. In any case, it is clear that decreasing the blockade fluctuations (via lowering pH and adding chaotropic salt) and increasing the residence time (via gold clusters) leads to narrowing of the peak in the current blockade distribution and improved selectivity of the SMNS technique. Figure 7D shows the blockade depth peak positions for the idealized solution conditions is still well described by the model from eqs 1 and 2. Optimizing the solution conditions and introducing gold clusters reduces the overall mass resolution for the three largest cationic peptides ((A1, A2, NT) from Δm = 84.2 ± 9.9 Da to Δm = 37.8 ± 2.8 Da; see Table S3 in Supporting Information) more than 2-fold improvement over the open pore case. See the caption of Table S2 in Supporting Information for a complete description of Δm.
Figure 7.
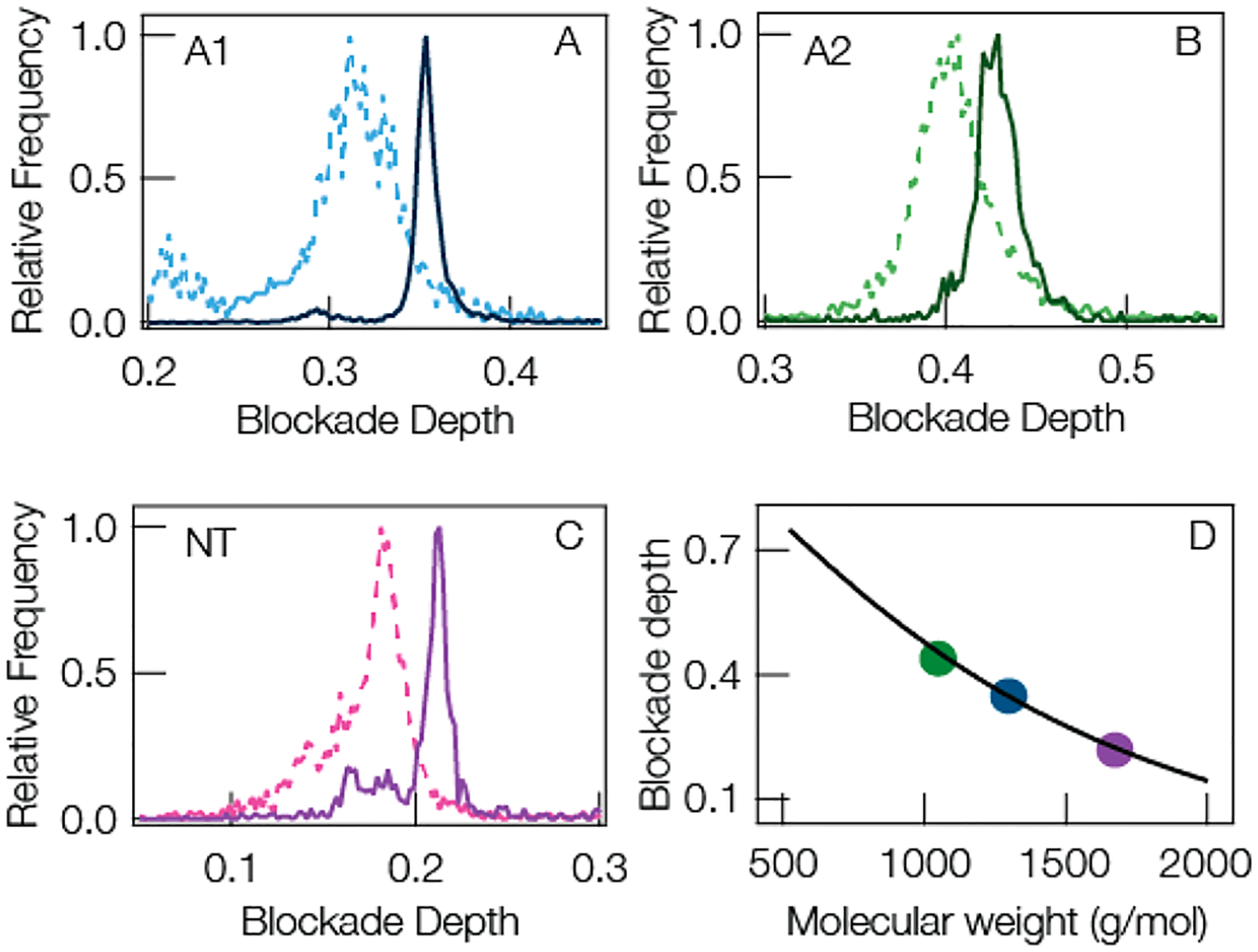
(A–C) Representative current blockade distributions show narrower peaks for (A) angiotensin I, (B) angiotensin II, and (C) neurotensin by increasing the mean residence time with a gold cluster in the pore and reducing fluctuations by modifying solution conditions. The dashed lines show the blockade distributions for the open pore at pH 7.2. The solid lines show pH 5.8 1 M Gdm-HCl added to the trans-side and a gold cluster in pore. (D) The peak positions of the optimized peaks (solid curves in A,B,C) (solid circles) are well described by the model shown in eqs 1 and 2. The solid line is a reproduction of the least-squares fit from Figure 3B. All data was taken under the following conditions: 3 M KCl, 70 mV applied transmembrane potential, [peptide] = 20 μM on the trans-side of the pore.
CONCLUSION
SMNS analysis requires increasing the analyte residence time in the pore while decreasing the current blockade fluctuations. This leads to a narrowing of the peaks in the current blockade distribution. The ultimate goal being baseline-resolved peaks so that one can say for certain that a particular current blockade corresponds to a particular peptide. As has been demonstrated here and elsewhere, the addition of the anionic gold cluster serves to increase the analyte’s time in the pore. For the case of PEG detection, this was shown to increase the quality of the corresponding current blockade distributions. Advancing SMNS detection from PEG to more biologically relevant molecules such as small peptides requires further analysis of the magnitude of the fluctuations within each blockade. The origin of these fluctuations is multifold, but one part that plays an important role is the degree of secondary structure within the peptides. The results shown in Figures 5 and 6 show that manipulating the solution conditions can lead to a significant reduction of the blockade fluctuations, which in turn can reduce the width of the corresponding peaks in the blockade distributions as demonstrated in Figure 7.
In this article we report several important results. For the molecules studied herein, it appears that a connection can be made between the depth of the current-blockade and the corresponding molecular weight, and this connection can be understood with a straightforward linear-ohmic model. Second, the presence of an anionic, thiolate-capped gold cluster increases cationic peptide residence times nearly 3-fold while increasing the peptide on-rate nearly 2-fold. Finally, by modifying the solution conditions and removing the stable secondary structure of a peptide one can achieve significant reduction of the current blockade fluctuations. This in turn yields a more accurate estimate of a given peptide’s current blockade and a better means for characterizing peptides via the SMNS technique.
Supplementary Material
ACKNOWLEDGMENTS
J.E.R. acknowledges funding from the Thomas F. and Kate Miller Jeffress Memorial Trust, VCU start-up funding and a generous equipment donation from NIST in Gaithersburg, MD. N.K. and A.D. were supported by NSF Grant no. 1255519.
ABBREVIATIONS
- A1
angiotensin I
- A2
angiotensin II
- NT
neurotensin
- LE
leuenkephalin polyglutamine-binding peptide 1 (QBP1)
- SMNS
single molecule nanopore spectrometry
- PEG
polyethylene glycol
- αHL
alpha hemolysin
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssensors.7b00362.
Description of the five peptides used; derivation of the Ohmic model; a description of the method used to calculate the effective charge of the peptides; calculated effective charge of the peptides; residence time enhancement; representative current traces; alpha hemolysin I–V curves; supplemental data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Chikkaveeraiah BV; Bhirde AA; Morgan NY; Eden HS; Chen X Electrochemical Immunosensors for Detection of Cancer Protein Biomarkers. ACS Nano 2012, 6, 6546–6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Qureshi A; Gurbuz Y; Niazi JH Biosensors for Cardiac Biomarkers Detection: A review. Sens. Actuators, B 2012, 171–172, 62–76. [Google Scholar]
- (3).Tothill IE Biosensors for Cancer Markers Diagnosis. Semin. Cell Dev. Biol 2009, 20, 55–62. [DOI] [PubMed] [Google Scholar]
- (4).Kierny MR; Cunningham TD; Kay BK Detection of Biomarkers using Recombinant Antibodiies Coupled to Nano-structured Platforms. Nano Rev 2012, 3, 17240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Verma N; Singh M Biosensors for Heavy Metals. BioMetals 2005, 18, 121–129. [DOI] [PubMed] [Google Scholar]
- (6).Tsekenis G; Filippidou MK; Chatzipetrou M; Tsouti V; Zergioti I; Chatzandroulis S Heavy Metal Ion Detection using a Capacitive Micromechanical Biosensor Array for Environmental Monitoring. Sens. Actuators, B 2015, 208, 628–635. [Google Scholar]
- (7).Silvestrini M; Fruk L; Ugo P Functionalized Ensembles of Nanoelectrodes as Affinity Biosensors for DNA Hybridization Detection. Biosens. Bioelectron 2013, 40, 265–270. [DOI] [PubMed] [Google Scholar]
- (8).Sassolas A; Leca-Bouvier BD; Blum LJ DNA Biosensors and Microarrays. Chem. Rev 2008, 108, 109–139. [DOI] [PubMed] [Google Scholar]
- (9).Zhang GJ; Chua JH; Chee RE; Agarwal A; Wong SM Label-Free Direct Detection of MiRNAs with Silicon Nanowire Biosensors. Biosens. Bioelectron 2009, 24, 2504–2508. [DOI] [PubMed] [Google Scholar]
- (10).Afonin KA; Danilov EO; Novikova IV; Leontis NB TokenRNA: A New Type of Sequence-Specific, Label-Free Fluorescent Biosensor for Folded RNA Molecules. ChemBioChem 2008, 9, 1902–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Grune T; Jung T; Merker K; Davies KJA Decreased Proteolysis caused by Protein Aggregates, Inclusion Bodies, Plaques, Lipofuscin, Ceroid, and ‘Aggresomes’ during Oxidative Stress, Aging, and Disease. Int. J. Biochem. Cell Biol 2004, 36, 2519–2530. [DOI] [PubMed] [Google Scholar]
- (12).Bingol B; Sheng M Deconstruction for Reconstruction: The Role of Proteolysis in Neural Plasticity and Disease. Neuron 2011, 69, 22–32. [DOI] [PubMed] [Google Scholar]
- (13).Ehrmann M; Clausen T Proteolysis as a Regulatory Mechanism. Annu. Rev. Genet 2004, 38, 709–724. [DOI] [PubMed] [Google Scholar]
- (14).Frescas D; Pagano M Deregulated Proteolysis by the F-box Proteins SKP2 and Beta-TrCP: Tipping the Scales of Cancer. Nat. Rev. Cancer 2008, 8, 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Garbett EA; Reed MWR; Brown NJ Proteolysis in Human Breast and Colorectal Cancer. Br. J. Cancer 1999, 81, 287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Yamasaki L; Pagano M Cell Cycle, Proteolysis and Cancer. Curr. Opin. Cell Biol 2004, 16, 623–628. [DOI] [PubMed] [Google Scholar]
- (17).Haass C; De Strooper B The Presenilins in Alzheimer’s Disease-Proteolysis Holds the Key. Science 1999, 286, 916–919. [DOI] [PubMed] [Google Scholar]
- (18).De Strooper B Proteases and Proteolysis in Alzheimer Disease: A Multifactorial View on the Disease Process. Physiol. Rev 2010, 90, 465–494. [DOI] [PubMed] [Google Scholar]
- (19).Landles C; Sathasivam K; Weiss A; Woodman B; Moffitt H; Finkbeiner S; Sun B; Gafni J; Ellerby LM; Trottier Y; Richards WG; Osmand A; Paganetti P; Bates GP Proteolysis of Mutant Huntingtin Produces an Exon 1 Fragment that Accumulates as an Aggregated Protein in Neuronal Nuclei in Huntington Disease. J. Biol. Chem 2010, 285, 8808–8823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Gosling JP A Decade of Development in Immunoassay Methodology. Clin. Chem 1990, 36, 1408–1427. [PubMed] [Google Scholar]
- (21).Luppa PB; Sokoll LJ; Chan DW Immunosensors-Principles and Applications to Clinical Chemistry. Clin. Chim. Acta 2001, 314, 1–26. [DOI] [PubMed] [Google Scholar]
- (22).Shi T; Fillmore TL; Sun X; Zhao R; Schepmoes AA; Hossain M; Xie F; Wu S; Kim J–S; Jones N; Moore RJ; Pasa-Tolic L; Kagan J; Rodland KD; Liu T; Tang K; Camp DG II; Smith RD; Qian W–J Antibody-Free, Targeted Mass-Spectrometric Approach for Quantification of Proteins at Low Picogram per Milliliter Levels in Human Plasma/Serum. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 15395–15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hoofnagle AN; Wener MH The Fundamental Flaws of Immunoassays and Potential Solutions Using Tandem Mass Spectrometry. J. Immunol. Methods 2009, 347, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Parker CE; Pearson TW; Anderson NL; Borchers CH Mass-Spectrometry-Based Clinical Proteomics-A Review and Propsective. Analyst 2010, 135, 1830–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Reiner JE; Balijepalli A; Robertson JWF; Campbell J; Suehle J; Kasianowicz JJ Disease Detection and Management via Single Nanopore-Based Sensors. Chem. Rev 2012, 112, 6431–6451. [DOI] [PubMed] [Google Scholar]
- (26).Kasianowicz JJ; Robertson JWF; Chan ER; Reiner JE; Stanford VM Nanoscopic Porous Sensors. Annu. Rev. Anal. Chem 2008, 1, 737–766. [DOI] [PubMed] [Google Scholar]
- (27).Movileanu L Interrogating Single Proteins Through Nanopores: Challenges and Opportunities. Trends Biotechnol 2009, 27, 333–341. [DOI] [PubMed] [Google Scholar]
- (28).Venkatesan BM; Bashir R Nanopore Sensors for Nucleic Acid Analysis. Nat. Nanotechnol 2011, 6, 615–624. [DOI] [PubMed] [Google Scholar]
- (29).Howorka S; Siwy Z Nanopore Analytics: Sensing of Single Molecules. Chem. Soc. Rev 2009, 38, 2360–2384. [DOI] [PubMed] [Google Scholar]
- (30).Oukhaled A; Bacri L; Pastoriza-Gallego M; Betton JM; Pelta J Sensing Proteins through Nanopores: Fundamental to Applications. ACS Chem. Biol 2012, 7, 1935–1949. [DOI] [PubMed] [Google Scholar]
- (31).Kasianowicz JJ; Brandin E; Branton D; Deamer DW Characterization of Individual Polynucleotide Molecules Using a Membrane Channel. Proc. Natl. Acad. Sci. U. S. A 1996, 93, 13770–13773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bezrukov SM; Kullman L; Winterhalter M FEBS Lett 2000, 476, 224–228. [DOI] [PubMed] [Google Scholar]
- (33).Kullman L; Winterhalter M; Bezrukov SM Transport of Maltodextrins through Maltoporin: A Single-Channel Study. Biophys. J 2002, 82, 803–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Berkane E; Orlik F; Charbit A; Danelon C; Fournier D; Benz R; Winterhalter M Nanopores: Maltoporin Channel as a Sensor for Maltodextrin and Lambda-Phage. J. Nanobiotechnol 2005, 3, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Rotem D; Jayasinghe L; Salichou M; Bayley H Protein Detection by Nanopores Equipped with Aptamers. J. Am. Chem. Soc 2012, 134, 2781–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Yusko EC; Johnson JM; Majd S; Prangkio P; Rollings RC; Li J; Yang J; Mayer M Controlling Protein Translocation through Nanopores with Bio-Inspired Fluid Walls. Nat. Nanotechnol 2011, 6, 253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Merstorf C; Cressiot B; Pastoriza-Gallego M; Oukhaled A; Betton JM; Auvray L; Pelta J Wild Type, Mutant Protein Unfolding and Phase Transition Detected by Single-Nanopore Recording. ACS Chem. Biol 2012, 7, 652–658. [DOI] [PubMed] [Google Scholar]
- (38).Zhao Q; de Zoysa RSS; Wang D; Jayawardhana DA; Guan X Real-Time Monitoring of Peptide Cleavage Using a Nanopore Probe. J. Am. Chem. Soc 2009, 131, 6324–6325. [DOI] [PubMed] [Google Scholar]
- (39).Sutherland TC; Long YT; Stefureac RI; Bediako-Amoa I; Kraatz HB; Lee JS Structure of Peptides Investigated by Nanopore Analysis. Nano Lett 2004, 4, 1273–1277. [Google Scholar]
- (40).Mereuta L; Schiopu I; Asandei A; Park Y; Hahm KS; Luchian T Protein Nanopore-Based, Single-Molecule Exploration of Copper Binding to an Antimicrobial-Derived, Histidine-Containing Chimera Peptide. Langmuir 2012, 28, 17079–17091. [DOI] [PubMed] [Google Scholar]
- (41).Mereuta L; Asandei A; Seo CH; Park Y; Luchian T Quantitative Understanding of pH and Salt-Mediated Conformational Folding of Histidine-Containing, Beta Hairpin-Like Peptides, through Single-Molecule Probing with Protein Nanopores. ACS Appl. Mater. Interfaces 2014, 6, 13242–13256. [DOI] [PubMed] [Google Scholar]
- (42).Madampage CA; Andrievskaia O; Lee JS Nanopore Detection of Antibody Prion Interactions. Anal. Biochem 2010, 396, 36–41. [DOI] [PubMed] [Google Scholar]
- (43).Han A; Schürmann G; Mondin G; Bitterli RA; Hegelbach NG; de Rooij NF; Staufer U Sensing Protein Molecules Using Nanofabricated Pores. Appl. Phys. Lett 2006, 88, 093901. [Google Scholar]
- (44).Fologea D; Ledden B; McNabb DS; Li J Electrical Characterization of Protein Molecules by a Solid-State Nanopore. Appl. Phys. Lett 2007, 91, 053901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Cressiot B; Oukhaled A; Patriarche G; Pastoriza-Gallego M; Betton JM; Auvray L; Muthukumar M; Bacri L; Pelta J Protein Transport through a Narrow Solid-State Nanopore at High Voltage: Experiments and Theory. ACS Nano 2012, 6, 6236–6243. [DOI] [PubMed] [Google Scholar]
- (46).Li W; Bell NAW; Hernández-Ainsa S; Thacker VV; Thackray AM; Bujdoso R; Keyser UF Single Protein Molecule Detection by Glass Nanopores. ACS Nano 2013, 7, 4129–4134. [DOI] [PubMed] [Google Scholar]
- (47).Steinbock LJ; Krishnan S; Bulushev RD; Borgeaud S; Blokesch M; Feletti L; Radenovic A Probing the Size of Proteins with Glass Nanopores. Nanoscale 2014, 6, 14380–14387. [DOI] [PubMed] [Google Scholar]
- (48).Song L; Hobaugh MR; Shustak C; Cheley S; Bayley H; Gouaux JE Structure of Staphylococcal Alpha-Hemolysin, a Heptameric Transmembrane Pore. Science 1996, 274, 1859–1866. [DOI] [PubMed] [Google Scholar]
- (49).Faller M; Niederweis M; Schulz GE The Structure of a Mycobacterial Outer-Membrane Channel. Science 2004, 303, 1189–1192. [DOI] [PubMed] [Google Scholar]
- (50).Movileanu L; Howorka S; Braha O; Bayley H Detecting Protein Analytes that Modulate Transmembrane Movement of a Polymer Chain within a Single Protein Pore. Nat. Biotechnol 2000, 18, 1091–1095. [DOI] [PubMed] [Google Scholar]
- (51).Stefureac R; Waldner L; Howard P; Lee JS Nanopore Analysis of a Small 86-Residue Protein. Small 2008, 4, 59–63. [DOI] [PubMed] [Google Scholar]
- (52).Stefureac RI; Lee JS Nanopore Analysis of the Folding of Zinc Fingers. Small 2008, 4, 1646–1650. [DOI] [PubMed] [Google Scholar]
- (53).Wolfe AJ; Mohammad MM; Cheley S; Bayley H; Movileanu L Catalyzing the Translocation of Polypeptides through Attractive Interactions. J. Am. Chem. Soc 2007, 129, 14034–14041. [DOI] [PubMed] [Google Scholar]
- (54).Oukhaled G; Mathé J; Biance A–L; Bacri L; Betton J–M; Lairez D; Pelta J; Auvray L Unfolding of Proteins and Long Transient Conformations Detected by Single Nanopore Recording. Phys. Rev. Lett 2007, 98, 158101. [DOI] [PubMed] [Google Scholar]
- (55).Pastoriza-Gallego M; Rabah L; Gibrat G; Thiebot B; van der Goot FG; Auvray L; Betton J–M; Pelta J Dynamics of Unfolded Protein Transport through an Aerolysin Pore. J. Am. Chem. Soc 2011, 133, 2923–2931. [DOI] [PubMed] [Google Scholar]
- (56).Payet L; Martinho M; Pastoriza-Gallego M; Betton J–M; Auvray L; Pelta J; Mathé J Thermal Unfolding of Proteins Probed at the Single Molecule Level Using Nanopores. Anal. Chem 2012, 84, 4071–4076. [DOI] [PubMed] [Google Scholar]
- (57).Kukwikila M; Howorka S Electrically Sensing Protease Activity with Nanopores. J. Phys.: Condens. Matter 2010, 22, 454103. [DOI] [PubMed] [Google Scholar]
- (58).Kukwikila M; Howorka S Nanopore-Based Electrical and Label-Free Sensing of Enzyme Activity in Blood Serum. Anal. Chem 2015, 87, 9149–9154. [DOI] [PubMed] [Google Scholar]
- (59).Wang Y; Montana V; Grubisic V; Stout RF Jr.; Parpura V; Gu L–Q Nanopore Sensing of Botulinum Toxin Type B by Discriminating an Enzymatically Cleaved Peptide from a Synaptic Protein Synaptobrevin 2 Derivative. ACS Appl. Mater. Interfaces 2015, 7, 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Zhou S; Wang L; Chen X; Guan X Label-Free Nanopore Single-Molecule Measurement of Trypsin Activity. ACS Sens 2016, 1, 607–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Kasianowicz JJ; Henrickson SE; Weetall HH; Robertson B Simultaneous Multianalyte Detection with a Nanometer-Scale Pore. Anal. Chem 2001, 73, 2268–2272. [DOI] [PubMed] [Google Scholar]
- (62).Carlsen AT; Zahid OK; Ruzicka JA; Taylor EW; Hall AR Selective Detection and Quantification of Modified DNA with Solid-State Nanopores. Nano Lett 2014, 14, 5488–5492. [DOI] [PubMed] [Google Scholar]
- (63).Zahid OK; Wang F; Ruzicka JA; Taylor EW; Hall AR Sequence-Specific Recognition of MicroRNAs and Other Short Nucleic Acids with Solid-State Nanopores. Nano Lett 2016, 16, 2033–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Marshall MM; Ruzicka J; Zahid OK; Henrich VC; Taylor EW; Hall AR Nanopore Analysis of Single-Stranded Binding Protein Interactions with DNA. Langmuir 2015, 31, 4582–4588. [DOI] [PubMed] [Google Scholar]
- (65).Bell NAW; Keyser UF Specific Protein Detection Using Designed DNA Carriers and Nanopores. J. Am. Chem. Soc 2015, 137, 2035–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Goodrich CP; Kirmizialtin S; Huyghues-Despointes BM; Zhu A; Scholtz JM; Makarov DE; Movileanu L Single-Molecule Electrophoresis of Beta-Hairpin Peptides by Electrical Recordings and Langevin Dynamics Simulations. J. Phys. Chem. B 2007, 111, 3332–3335. [DOI] [PubMed] [Google Scholar]
- (67).Macrae MX; Blake S; Jiang X; Capone R; Estes DJ; Mayer M; Yang J A Semi-Synthetic Ion Channel Platform for Detection of Phosphatase and Protease Activity. ACS Nano 2009, 3, 3567–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Robertson JWF; Rodrigues CG; Stanford VM; Rubinson KA; Krasilnikov OV; Kasianowicz JJ Single-molecule mass spectrometry in solution using a solitary nanopore. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 8207–8211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Reiner JE; Kasianowicz JJ; Nablo BJ; Robertson JWF Theory for polymer analysis using nanopore-based single-moleucle mass spectrometry. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 12080–12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Baaken G; Ankri N; Schuler A–K; Ruhe J; Behrends JC Nanopore-Based Single-Molecule Mass Spectrometry on a Lipid Membrane Microarray. ACS Nano 2011, 5, 8080–8088. [DOI] [PubMed] [Google Scholar]
- (71).Baaken G; Halimeh I; Bacri L; Pelta J; Oukhaled A; Behrends JC High-Resolution Size-Discrimination of Single Nonionic Synthetic Polymers with a Highly Charged Biological Nanopore. ACS Nano 2015, 9, 6443–6449. [DOI] [PubMed] [Google Scholar]
- (72).Balijepalli A; Robertson JWF; Reiner JE; Kasianowicz JJ; Pastor RW Theory of Polymer-Nanopore Interactions Refined Using Molecular Dynamics Simulations. J. Am. Chem. Soc 2013, 135, 7064–7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Kumar S; Tao C; Chien M; Hellner B; Balijepalli A; Robertson JWF; Zengmin L; Russo JJ; Reiner JE; Kasianowicz JJ; Ju J PEG-Labeled Nucleotides and Nanopore Detection for Single Molecule DNA Sequencing by Synthesis. Sci. Rep 2012, 2, 684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Fuller CW; Kumar S; Porel M; Chien M; Bibillo A; Stranges PB; Dorwart M; Tao C; Li Z; Guo W; Shi S; Korenblum D; Trans A; Aguirre A; Liu E; Harada ET; Pollard J; Bhat A; Cech C; Yang A; Arnold C; Palla M; Hovis J; Chen R; Morozova I; Kalachikov S; Russo JJ; Kasianowicz JJ; Davis R; Roever S; Church GM; Ju J Real-time single-molecule electronic DNA sequencing by synthesis using polymer-tagged nucleotides on a nanopore array. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 5233–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Cao C; Ying Y–L; Hu Z–L; Liao D–F; Tian H; Long Y–T Discrimination of oligonucleotides of different lengths with a wild-type aerolysin nanopore. Nat. Nanotechnol 2016, 11, 713–718. [DOI] [PubMed] [Google Scholar]
- (76).Breton MF; Discala F; Bacri L; Foster D; Pelta J; Oukhaled A Exploration of Neutral Versus Polyelectrolyte Behavior of Poly(ethylene glycol)s in Alkali Ion Solutions using Single-Nanopore Recording. J. Phys. Chem. Lett 2013, 4, 2202–2208. [Google Scholar]
- (77).Krasilnikov OV; Rodrigues CG; Bezrukov SM Single polymer molecules in a protein nanopore in the limit of a strong polymer-pore attraction. Phys. Rev. Lett 2006, 97, 018301. [DOI] [PubMed] [Google Scholar]
- (78).Machado DC; Junior JJS; Melo MCA; Silva AMB; Fontes A; Rodrigues CG Effects of alkali and ammonium ions in the detection of poly(ethylene glycol) by alpha-hemolysin nanopore sensor. RSC Adv 2016, 6, 56647–56655. [Google Scholar]
- (79).Movileanu L; Schmittschmitt JP; Scholtz JM; Bayley H Interactions of Peptides with a Protein Pore. Biophys. J 2005, 89, 1030–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Stefureac R; Long Y–T; Kraatz H–B; Howard P; Lee JS Transport of alpha-Helical Peptides through alpha-Hemolysin and Aerolysin Pores. Biochemistry 2006, 45, 9172–9179. [DOI] [PubMed] [Google Scholar]
- (81).Yusko EC; Bruhn BR; Eggenberger O; Houghtaling J; Rollings RC; Walsh NC; Nandivada S; Pindrus M; Hall AR; Sept D; Li J; Kalonia D; Mayer M Real-time shape approximation and fingerprinting of single proteins using a nanopore. Nat. Nanotechnol 2016, 12, 360. [DOI] [PubMed] [Google Scholar]
- (82).Mohammad M; Prakash S; Matouschek A; Movileanu L Controlling a Single Protein in a Nanopore through Electrostatic Traps. J. Am. Chem. Soc 2008, 130, 4081–4088. [DOI] [PubMed] [Google Scholar]
- (83).Bikwemu R; Wolfe AJ; Xing X; Movileanu L Facilitated translocation of polypeptides through a single nanopore. J. Phys.: Condens. Matter 2010, 22, 454117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Asandei A; Chinappi M; Lee J; Seo CH; Mereuta L; Park Y; Luchian T Placement of oppositely charged aminoacids at a polypeptide termini determines the voltage-controlled braking of polymer transport through nanometer-scale pores. Sci. Rep 2015, 5, 10419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Angevine CE; Chavis AE; Kothalawala N; Dass A; Reiner JE Enhanced Single Molecule Mass Spectrometry via Charged Metallic Clusters. Anal. Chem 2014, 86, 11077–11085. [DOI] [PubMed] [Google Scholar]
- (86).Chavis AE; Brady KT; Kothalawala N; Reiner JE Voltage and blockade state optimization of cluster-enhanced nanopore spectrometry. Analyst 2015, 140, 7718–7725. [DOI] [PubMed] [Google Scholar]
- (87).Meller A; Branton D Single molecule measurements of DNA transport through a nanopore. Electrophoresis 2002, 23, 2583–2591. [DOI] [PubMed] [Google Scholar]
- (88).Vaitheeswaran S; Thirumalai D Interactions between amino acid side chains in cylindrical hydrophobic nanoproes with applications to peptide stability. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 17636–17641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Asandei A; Chinappi M; Kang H–K; Seo CH; Mereuta L; Park Y; Luchian T Acidity-Mediated, Electrostatic Tuning of Asymmetrically Charged Peptides Interactions with Protein Nanopores. ACS Appl. Mater. Interfaces 2015, 7, 16706–16714. [DOI] [PubMed] [Google Scholar]
- (90).Asandei A; Schiopu I; Chinappi M; Seo CH; Park Y; Luchian T Electroosmotic Trap Against the Electrophoretic Force Near a Protein Nanopore Reveals Peptide Dynamics During Capture and Translocation. ACS Appl. Mater. Interfaces 2016, 8, 13166–13179. [DOI] [PubMed] [Google Scholar]
- (91).Ackerson CJ; Jadzinsky PD; Kornberg RD Thiolate Ligands for Synthesis of Water-Soluble Clusters. J. Am. Chem. Soc 2005, 127, 6550–6551. [DOI] [PubMed] [Google Scholar]
- (92).Heaven MW; Dass A; White PS; Holt KM; Murray RW Crystal Structure of the Gold Nanoparticle [N(C8H17)4] [Au25(SCH2CH2Ph)18]. J. Am. Chem. Soc 2008, 130, 3754–3755. [DOI] [PubMed] [Google Scholar]
- (93).Kasianowicz JJ; Reiner JE; Robertson JWF; Henrickson SE; Rodrigues C; Krasilnikov OV Methods Mol. Biol 2012, 870, 3–20. [DOI] [PubMed] [Google Scholar]
- (94).Flory PJ The Configuration of Real Polymer Chains. J. Chem. Phys 1949, 17, 303–310. [Google Scholar]
- (95).Fitzkee NC; Rose GD Reassessing random-coil statistics in unfolded proteins. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 12497–12502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Kienberger F; Pastushenko VP; Kada G; Gruber HJ; Riener C; Schindler H; Hinterdorfer P Static and Dynamical Properties of Single Poly(Ethylene Glycol) Molecules Investigated by Force Spectroscopy. Single Mol 2000, 1, 123–128. [Google Scholar]
- (97).Schuler B; Lipman EA; Steinbach PJ; Kumke M; Eaton WA Polyproline and the “spectroscopic ruler” revisited with single-molecule fluorescence. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 2754–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Lakkaraju SK; Hwang W Critical buckling length versus persistence length: what governs biofilament conformation? Phys. Rev. Lett 2009, 102, 118102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Furó I; Iliopoulos I; Stilbs P Structures and Dynamics of Associative Water-Soluble Polymer Aggregates As Seen by 19F NMR Spectroscopy. J. Phys. Chem. B 2000, 104, 485–494. [Google Scholar]
- (100).Lintner K; Fermandjian S; Fromageot P pH titration effects on the cd spectra of angiotensin II, truncated peptides and other analogues: Aromatic region. FEBS Lett 1975, 56, 366–369. [DOI] [PubMed] [Google Scholar]
- (101).Glickson JD; Cunningham WD; Marshall GR Proton magnetic resonance study of angiotensin II (Asn1 + Val5) in aqueous solution. Biochemistry 1973, 12, 3684–3692. [DOI] [PubMed] [Google Scholar]
- (102).Huerta-Viga A; Woutersen S Protein Denaturation with Guanidinium: A 2D-IR Study. J. Phys. Chem. Lett 2013, 4, 3397–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.