

Keywords: cardiovascular disease, cyclic nucleotides, heart failure, phosphodiesterase, vessels
Abstract
Phosphodiesterases (PDEs) are a superfamily of enzymes that hydrolyze cyclic nucleotides, including cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP). Both cyclic nucleotides are critical secondary messengers in the neurohormonal regulation in the cardiovascular system. PDEs precisely control spatiotemporal subcellular distribution of cyclic nucleotides in a cell- and tissue-specific manner, playing critical roles in physiological responses to hormone stimulation in the heart and vessels. Dysregulation of PDEs has been linked to the development of several cardiovascular diseases, such as hypertension, aneurysm, atherosclerosis, arrhythmia, and heart failure. Targeting these enzymes has been proven effective in treating cardiovascular diseases and is an attractive and promising strategy for the development of new drugs. In this review, we discuss the current understanding of the complex regulation of PDE isoforms in cardiovascular function, highlighting the divergent and even opposing roles of PDE isoforms in different pathogenesis.
CLINICAL HIGHLIGHTS.
Phosphodiesterases regulate multiple physiological processes in the cardiovascular system, including cardiac and vessel contraction. Phosphodiesterases are critical components in the signalosomes that precisely control cardiac muscle, smooth muscle, and endothelial cell function in physiological conditions.
Many phosphodiesterases display increased expression and activities in a cell- and tissue-specific manner in pathogenesis. Many have sex-, etiology-, and disease stage-specific regulation, which plays distinct, even opposing, roles in the development of cardiovascular diseases.
Phosphodiesterase 5 inhibitors have been proven effective in erectile dysfunction and pulmonary hypertension. Targeting phosphodiesterase 5 remains an area of intensive interest for additional clinical benefits in diseases such as heart failure with preserved ejection fraction.
Phosphodiesterase 3 inhibitors are effective in acute heart failure and end-stage heart failure, and recent studies aim to improve their utility in managing chronic heart failure and other diseases such as stroke.
Phosphodiesterase 1 inhibitors are effective in stroke, and recent studies aim to explore their utility in managing heart failure with reduced ejection fraction.
Emerging insight into phosphodiesterases 2 and 4 in cardiovascular diseases may provide better strategies to target the enzymes in clinical settings. The development of rational strategies for targeting individual isoforms in a disease-specific manner may allow efficacious therapy with minimal side effects.
1. INTRODUCTION
Upon stimulation, activation of G protein-coupled receptors (GPCRs) and natriuretic peptide receptors (NPRs) increases the activity of adenylate cyclases (ACs) and guanylyl cyclases (GCs), which produce cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP), respectively. cAMP and cGMP function as critical secondary messengers in neurohormonal regulation in the cardiovascular system. Phosphodiesterases (PDEs) serve as a vital mechanism to hydrolyze cyclic nucleotides and turn off neurohormonal stimulation. PDE-mediated hydrolysis contributes to the discrete cyclic nucleotide signaling known as the cAMP and cGMP nanodomains. PDEs also define the amplitude and duration of local cyclic nucleotide signaling to regulate a broad range of effectors, including protein kinases, ion channels, and exchange factors. Through local actions of downstream effectors, PDEs regulate specific physiological responses in a cell- and tissue-specific manner. The ubiquitous PDE activity in the heart and vessels has been demonstrated since the discovery of the first enzymatic deconstruction of cAMP into AMP in the 1950s (1–3). Thus, PDEs critically regulate physiological responses in the cardiovascular system, including cardiac inotropic and chronotropic responses, vessel dilation and constriction, and metabolism. Over the past 30 years, there has been significant progress in understanding this family of enzymes in physiology and cardiovascular diseases (4–10). Abnormal PDE expression and activities are involved in structural and functional remodeling in the heart and vessels. Disrupted cyclic nucleotide signals contribute to heart failure (HF), arrhythmia, hypertension, aneurysm, atherosclerosis, and other cardiovascular diseases. PDE inhibitors have been successfully used in clinical settings and present an intensive area of research. This review summarizes the advances in PDE regulation in the heart and vessels and the implications for targeting these enzymes in the therapy of cardiovascular diseases.
2. OVERVIEW OF THE PDE SUPERFAMILY
The PDE superfamily has 11 subfamilies (PDE1–11) distinguished by their unique structure, enzymatic properties, regulation, and pharmacology (11). Each PDE comprises a catalytic domain, a carboxy terminus, and an amino terminus (FIGURE 1). All PDEs share a similar structure with the catalytic domains at the carboxy-terminal regions. Within the PDE isoforms encoded by the same gene, the distinct amino termini define the subcellular location of individual isoforms, while they share identical catalytic and carboxy-terminal domains (7, 12). PDE isoforms are also subjected to unique enzymatic regulation by posttranslation modification and regulatory proteins. There are 21 PDE genes expressed in mammalian cells, including single PDE2A, PDE5A, PDE9A, PDE10A, and PDE11A genes, two PDE3 (PDE3A and 3B), PDE7 (PDE7A and 7B), and PDE8 (PDE8A and 8B) genes, three PDE1 (PDE1A, 1B, and 1C) and PDE6 (PDE6A, 6B, and 6C) genes, and four PDE4 (PDE4A, 4B, 4C, and 4D) genes (4–10). Over 100 different isoforms are expressed in mammalian cells because of alternative splicing and different translation initiation sites. Based on substrate specificity, these enzymes are classified as cAMP-specific, cGMP-specific, and dual-substrate PDEs. PDE4, 7, and 8 are exclusively involved in cAMP hydrolysis, whereas PDE5, 6, and 9 have cGMP-specific enzymatic activities. The remaining PDEs, including PDE1, 2, 3, and the latter members PDE10 and 11, can hydrolyze both cAMP and cGMP. PDE2, PDE3, and, to a lesser extent, PDE1 have been shown to mediate cGMP-cAMP cross talk upon activation or inhibition of the PDE activities via cGMP binding. cGMP competitively inhibits the cAMP hydrolytic activity of PDE1 and PDE3 (7). In contrast, PDE2 is the only member that is activated upon allosteric cGMP binding, increasing its cAMP hydrolytic activity (13).
FIGURE 1.
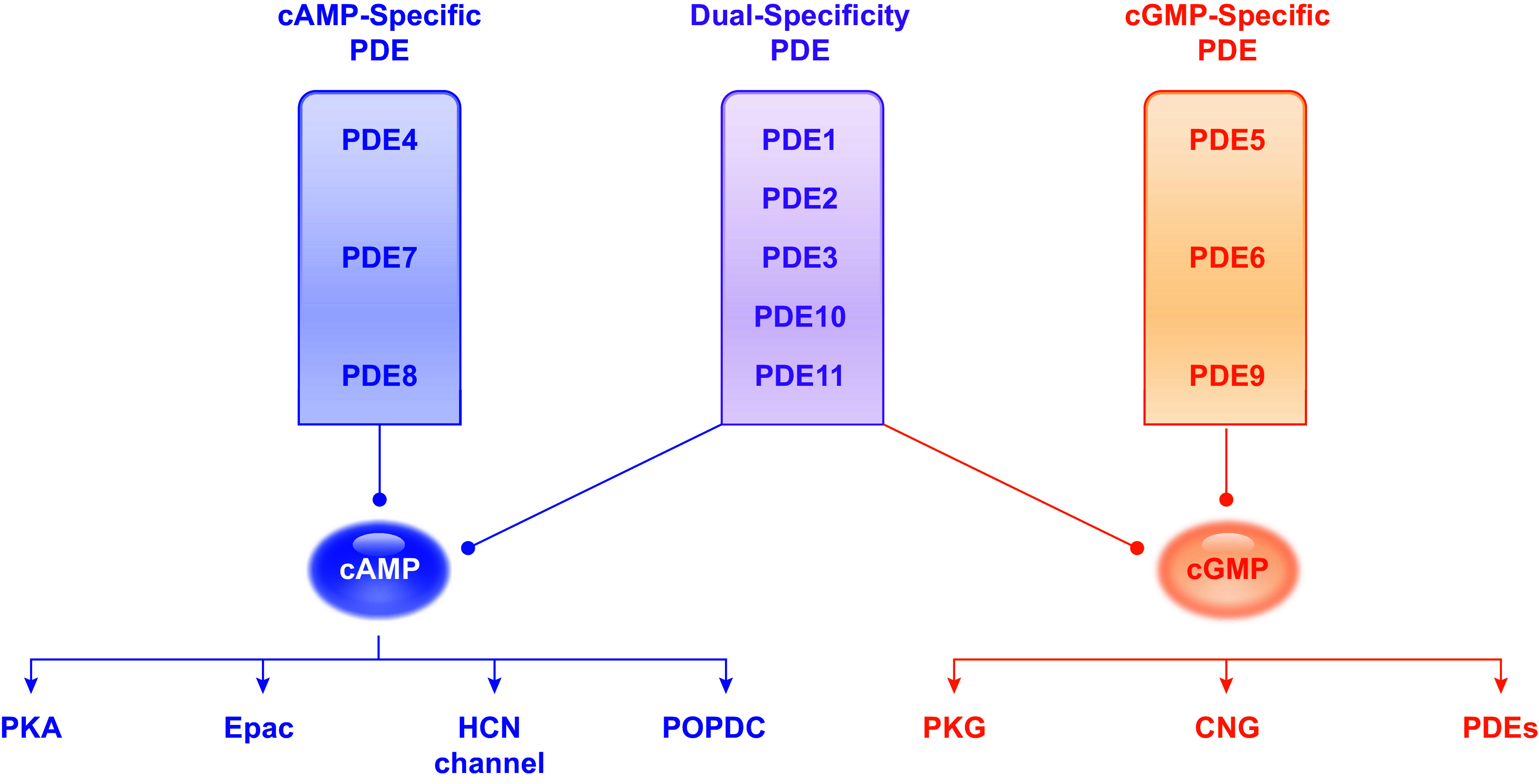
Classification and specificity of phosphodiesterases (PDEs) toward cyclic nucleotides and downstream effectors. PDE4, 7, and 8 hydrolyze cAMP, whereas PDE5, 6, and 9 hydrolyze cGMP. The other PDEs, including PDE1, 2, 3, 10, and 11, hydrolyze both cAMP and cGMP. cAMP targets protein kinase A (PKA), exchange protein directly activated by cAMP (Epac), hyperpolarization-activated cyclic nucleotide-gated channel (HCN), and popeye domain containing protein (POPDC), whereas cGMP targets protein kinase G (PKG), cyclic nucleotide-gated channel (CNG), and PDE.
2.1. The Expression, Biochemical Property, and Regulation of PDEs in the Cardiovascular System
PDEs are expressed in a cell- and tissue-specific manner, with only a few enzymes expressed in any single cell type. Except for PDE6, all PDE families have a broad expression in different tissues, including hearts and vessels (7, 12). Many have different expression and activity in the heart between rodents and humans, such as PDE1, 3, and 4 (14). Of the PDE superfamily, PDE1, 2, 3, 4, 5, 8, and 9 are fundamental components of the cardiac signaling and function regulation under physiological and pathophysiological conditions (8). On the other hand, PDE1, 2, 3, 4, 5, and 7 constitute significant PDE activity in the vasculature (15, 16). PDE1, 2, 3, 4, and 5 are extensively studied in the cardiovascular system (4–9, 16). (FIGURES 2–4 and TABLE 1).
FIGURE 2.
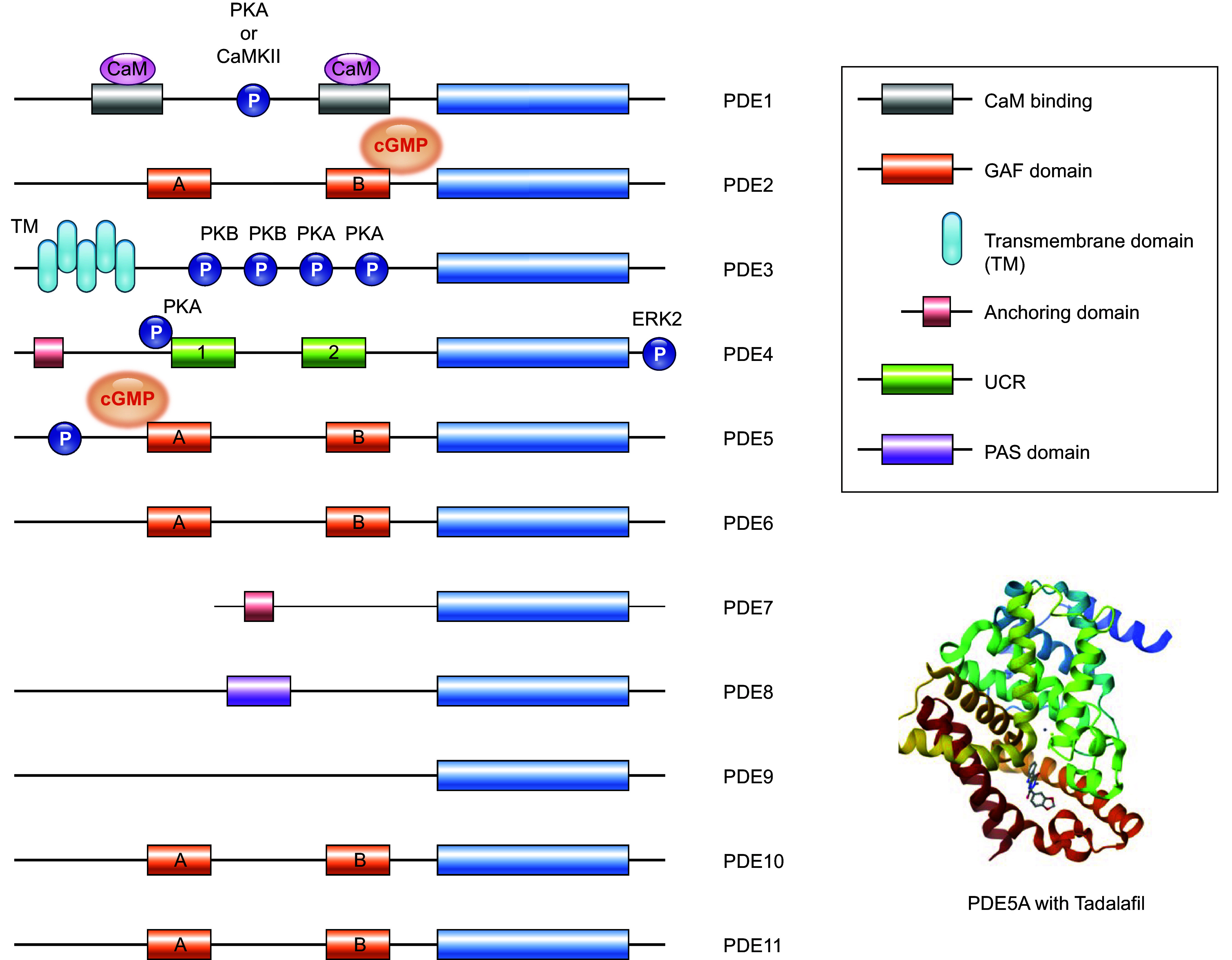
Structure and regulation of phosphodiesterases (PDEs). The catalytic domains conserved in the carboxy terminal of PDEs are depicted as blue cylinders. These enzymes can be precisely regulated by specific targeting domains and modifications, such as phosphorylation, which enables subcellular localization and activity of PDEs. Certain PDEs can dimerize; long isoforms of PDE4, for example, dimerize because of the presence of 2 upstream conserved regions (UCR1 and UCR2). Other PDEs such as PDE2 and PDE5 form a dimer by means of their GAF [cyclic guanylate monophosphate (cGMP)-dependent phosphodiesterase, Anabaena adenylyl cyclase, Escherichia coli FhlA] domains. PDEs with GAF domains, such as PDE2, PDE5, PDE10A and PDE11, are regulated by cyclic nucleotides, with cGMP activating PDE2 and cAMP inhibiting PDE10. The structure of PDE5A with tadalafil is shown at bottom right. CaM, calmodulin; CaMKII, Ca2+/CaM-dependent kinase II; ERK, extracellular signaling-related kinase; PAS, Per-Arnt-Sim; PKA, protein kinase A; PKB, protein kinase B; TM, transmembrane domain.
FIGURE 4.
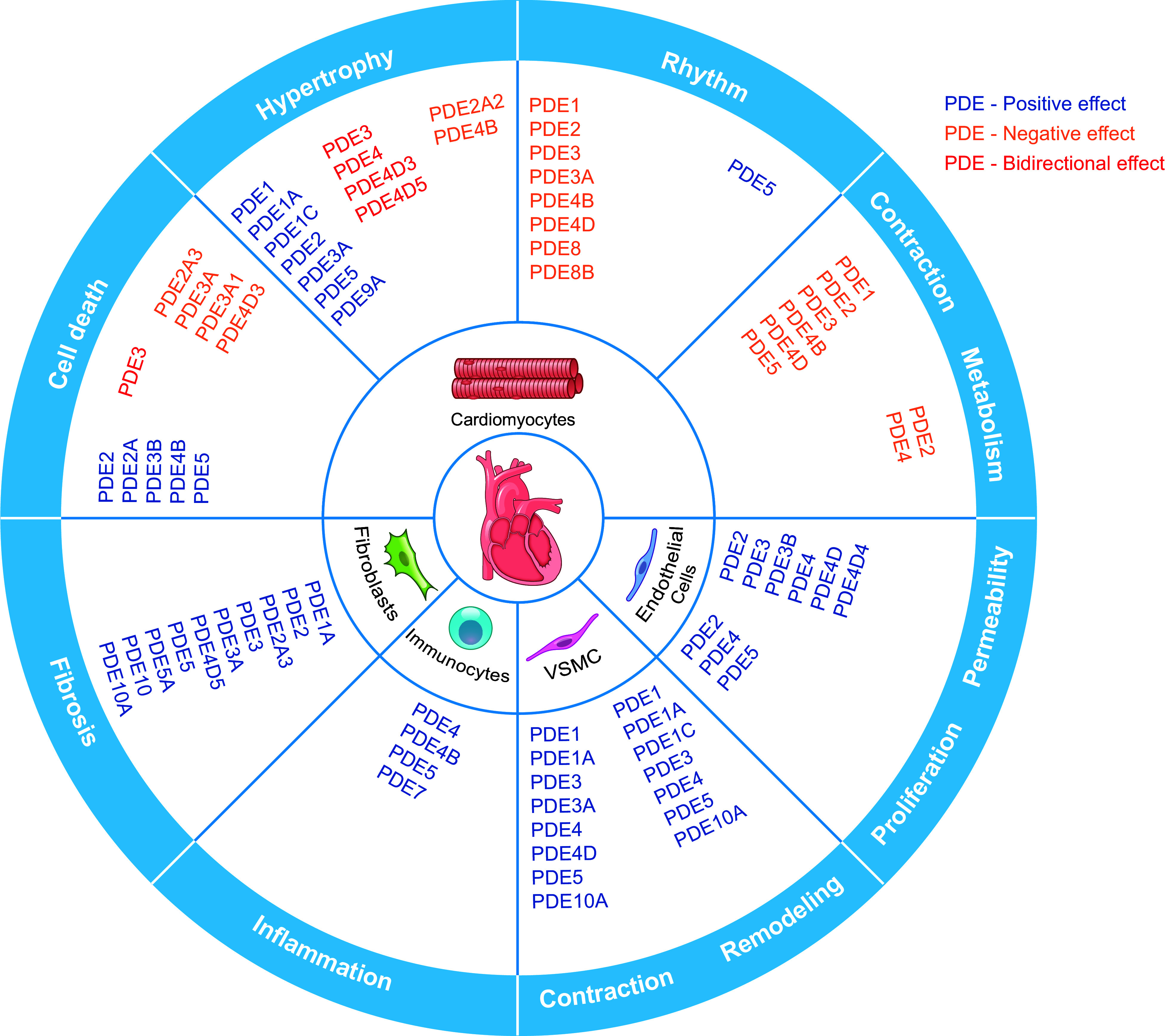
The expression and function of phosphodiesterases (PDEs) in the cardiovascular system. The cartoon highlights the expression of individual isoforms in different cardiovascular cells. The colors indicate the positive, negative, or bidirectional roles of PDE isoforms in specific function in the heart and vessels. VSMC, vascular smooth muscle cell.
Table 1.
The substrates, biochemical properties and subcellular distribution of individual PDE families and isoforms
| PDE Family | Isoform | Variant | Substrate | Regulation | Localization |
|---|---|---|---|---|---|
| PDE1 | CaM-stimulated PDE | ||||
| PDE1A | cGMP>cAMP, cGMP (in vivo) | PKA | |||
| PDE1B | cGMP>cAMP, cGMP (in vivo) | Ca2+/calmodulin | |||
| PDE1C | cAMP/cGMP, cAMP (in vivo) | PKA | Cytoplasm | ||
| PDE2 | cGMP-stimulated PDE | ||||
| PDE2A | |||||
| PDE2A1 | cAMP/cGMP | Cytosol, soluble in the cytoplasm | |||
| PDE2A2 | cAMP/cGMP | Mitochondria | |||
| PDE2A3 | cAMP/cGMP | Golgi body, plasma membrane | |||
| PDE3 | cGMP-inhibited PDE | ||||
| PDE3A | |||||
| PDE3A1 | cAMP>cGMP | PKA | Cytoplasm, particulate fraction microsomal and cytosol | ||
| PDE3A2 | cAMP>cGMP | PKC, PKA | Cytosolic and particulate fractions | ||
| PDE3A3 | cAMP>cGMP | Cytoplasm | |||
| PDE3B | cAMP>cGMP | PKA | T tubules (proximity to mitochondria) | ||
| PDE4 | cAMP-specific PDE | ||||
| PDE4A | cAMP | PKA | Membranes | ||
| PDE4B | cAMP | PKA, ERK | Membranes | ||
| PDE4C | |||||
| PDE4D | cAMP | PKA, CaMKII | Lipid rafts and caveolar membrane, myofilaments, nuclear envelope | ||
| PDE4D3 | RYR2, nuclear envelope | ||||
| PDE4D5 | Nuclear envelope | ||||
| PDE4D8 | |||||
| PDE4D9 | |||||
| PDE5 | cGMP-specific PDE | ||||
| PDE5A1–3 | cGMP | Cytosol, Z lines, caveolae (endothelial cell) | |||
| PDE6 | cGMP-specific PDE | ||||
| PDE6D/H | |||||
| PDE6γ | |||||
| PDE7 | cAMP-specific PDE | ||||
| PDE7A | cAMP | PKA | Cytoplasm | ||
| PDE7A2 | |||||
| PDE7B | |||||
| PDE8 | |||||
| PDE8A | cAMP | PKA | |||
| PDE8B | |||||
| PDE9 | PDE9A | cGMP | |||
| PDE10 | PDE10A2 | cAMP, cGMP | |||
| PDE11 | PDE11A | cAMP, cGMP |
CaM, calmodulin; CaMKII, Ca2+/CaM-dependent kinase II; cAMP, cyclic adenosine monophosphate, cGMP, cyclic guanosine monophosphate; ERK, extracellular signal-regulated kinase; PDE, phosphodiesterase; PKA, protein kinase A, PKC, protein kinase C.
2.1.1. PDE1.
PDE1 constitutes a family of Ca2+/calmodulin (CaM)-dependent enzymes, with all three PDE1 isozymes (PDE1A, PDE1B, and PDE1C) expressed in the heart and vessels (17). PDE1A is expressed in humans (18), bovines (19), canines (20), and rats (21), including both cardiomyocytes (22, 23) and cardiac fibroblasts (24, 25). PDE1C is primarily restricted to cardiomyocytes (19) and is the predominant PDE1 isoform in rabbit, dog, and human hearts (26). The messenger RNA (mRNA) expression levels of PDE1A are sex dependent, with higher levels in males compared with females (27). PDE1 functions as a dual esterase that hydrolyzes cAMP and cGMP, with PDE1A and PDE1B displaying a lower affinity for cAMP, hence favoring cGMP hydrolysis (18, 28). Several studies have demonstrated that PDE1A and PDE1B primarily regulate cGMP in vivo (12, 23, 29). On the other hand, PDE1C hydrolyzes cAMP and cGMP with comparably high affinity and low Michaelis constant (Km) values. Whereas PDE1C has been shown to regulate intracellular cAMP levels in various cell types, a role in cGMP regulation has not been described in vivo (30–32). PDE1 isozymes contain two CaM-binding domains at the amino termini, two phosphorylation sites, and an inhibitory region that keeps the enzymes in an inactive configuration when intracellular Ca2+ is low (33). The binding of CaM to PDE1 increases the activity up to 10-fold in vitro (33). Thus, PDE1 isozymes are important in the cross talk between second messenger Ca2+ and cyclic nucleotide signaling (34). Phosphorylation of PDE1 by either protein kinase A (PKA for PDE1A and PDE1C) (35, 36) or Ca2+/CaM-dependent protein kinase II (CaMKII for PDE1B) (37) reduces the affinity for Ca2+ and CaM, thereby limiting enzymatic activity. Conversely, the binding of CaM to PDE1 elevates hydrolytic activity by preventing PKA- and CaMKII-mediated phosphorylation and promoting a conformational change that raises the maximal catalytic activity (38, 39).
2.1.2. PDE2.
A single PDE2 gene, PDE2A, is expressed with three alternative splicing isoforms (40). PDE2 has been found in human, rat, and mouse hearts (41–43). PDE2A has been detected in both cardiomyocytes and cardiac fibroblasts, with higher expression in the latter (41, 44). The PDE2A isoforms display different subcellular locations, with PDE2A1 being soluble in the cytoplasm and PDE2A2 and PDE2A3 predominantly in a particulate fraction (40). Notably, PDE2A3 is localized in the Golgi (45, 46), whereas PDE2A2 is detected in the mitochondria and regulates local mitochondria-related cAMP pools (47, 48). In addition to cardiomyocytes, PDE2A has been found in neonatal cardiac fibroblasts (44) and endothelial cells (ECs) (49–51). PDE2 shares similarities with PDE1, capable of hydrolyzing both cAMP and cGMP, displaying comparable maximal rates and low Km values for each cyclic nucleotide but with a slight preference for cGMP (52, 53). The amino terminus contains two cGMP-stimulated PDE, Anabaena AC, and Fhla transcription factor (GAF) domains, designated as GAF-A and GAF-B (54). The GAF-B domain selectively binds to cGMP, altering the conformation of PDE2 and raising its esterase activity by a factor of 30 (55, 56). PDE2 is thus uniquely designated as the cGMP-stimulated PDE. By contrast, it is unlikely that cAMP regulates PDE2 activity in vivo in the same manner, given that the affinity of cAMP for the GAF-B site is ∼100-fold lower than that of cGMP (57).
2.1.3. PDE3.
PDE3, the third dual esterase, is a key player in hydrolyzing cAMP and cGMP with high affinities. PDE3 is highly expressed in hearts from small rodents to large mammals, including humans, and constitutes the major cAMP-hydrolyzing activity in the latter (58, 59). PDE3 activity is present in cytosolic and particulate fractions (60). PDE3 enzymes are encoded by two genes, PDE3A and PDE3B. PDE3A is the predominant gene expressed in the heart (61, 62) and is responsible for the tonic effects in the myocardium (63, 64). Three splicing isoforms expressed by PDE3A possess different amino-terminal domains that dictate their subcellular locations. The longest isoform, PDE3A1, is concentrated in particulate fraction and contains two amino-terminal hydrophobic domains (NHRs), NHR1 and NHR2. The short isoform, PDE3A3, lacks both domains and resides in the cytoplasm (62). In comparison, the mRNA expression levels of PDE3B vary depending on sex, with lower levels in males than in females (27). Only one PDE3B isoform is identified in the T tubules near the mitochondria (59, 65). PDE3 enzymes hydrolyze cAMP and cGMP with high affinity, but the Vmax for cGMP is 10 times lower than that for cAMP (53). When cGMP binds to the catalytic site of PDE3, it is hydrolyzed very slowly, inhibiting its enzymatic activity toward cAMP metabolism. Hence, PDE3 is often referred to as a cGMP-inhibited PDE. As a result, PDE3 regulates the intricate cGMP-cAMP cross talk, with the binding of cGMP inhibiting PDE3 activity and acting as a positive regulator of cAMP signaling (66, 67). Biochemically, two isoforms of PDE3A, PDE3A1 and PDE3A2, can be phosphorylated at the common amino-terminal sequence. PDE3A1 is subjected to PKA-dependent phosphorylation of Ser 312, and PKC preferentially phosphorylates PDE3A2 at Ser 428. Both phosphorylation events promote the association of PDE3A isoforms with cellular signaling proteins, such as 14-3-3 (22, 60). PKA also phosphorylates PDE3A1 at Ser 292 of the amino terminus, enhancing its hydrolytic activity and association with multiple proteins, like A-kinase anchoring protein 18 (AKAP18), sarco(endo)plasmic reticulum calcium ATPase 2a (SERCA2a), protein phosphatase 2A (PP2A), PP1, and caveolin 3 (60). Mutation of PDE3A1 at Ser 292 remarkably abolishes the PKA-dependent interaction with SERCA2a, whereas Ser 312 and Ser 428 mutation slightly reduced that association (60). Similarly, a PKA-phosphorylated PDE3B at Ser 318 increases its catalytic activity and interaction with 14-3-3 (68).
2.1.4. PDE4.
PDE4 enzymes are highly selective for cAMP, evidenced by their very low Km values for this cyclic nucleotide (69, 70). In rodent hearts, PDE4 is the dominant family for cAMP-hydrolyzing activities, together with PDE3, accounting for ∼90% of overall cAMP-hydrolyzing activities (14). However, in human hearts, PDE3- and PDE4-mediated cAMP-hydrolyzing activity accounts for ∼40% of total cAMP-hydrolyzing activities, with other non-PDE4 such as PDE1 being much more active (14). Three PDE4 genes are reported in human and rodent hearts, namely PDE4A, PDE4B, and PDE4D (14, 71, 72). PDE4D has higher expression in female than male mouse hearts (73), whereas ovariectomy (OVX) promotes the expression of PDE4B mRNA in rat hearts (74). Both may contribute to the sex-dependent difference in excitation-contraction coupling (ECC) (75). PDE4 provides an important feedback mechanism to control cAMP levels in various cell types, including cardiomyocytes (76–78), smooth muscle cells (SMCs) (79, 80), ECs (27, 81), and fibroblasts (82, 83). Alternative gene splicing produces >20 PDE4 isoforms (PDE4A1–PDE4A8, PDE4B1–PDE4B6, and PDE4D1–PDE4D11), which can exist in short and long forms depending on the presence of upstream conserved regions (UCRs) within the amino terminus (84). Of the nine PDE4D isoforms, at least four PDE4D variants have been detected in human and rodent hearts, including long isoforms PDE4D3, 4D5, 4D8, and 4D9 (14). Whereas the long versions contain UCR1 and UCR2, short PDE4 isoforms possess a single UCR2 domain, and supershort isoforms have none. UCR1 and UCR2 are required for dimerization (85, 86), whereas short and supershort forms without UCR1 remain monomers. PDE4 possesses a PKA phosphorylation site within UCR1, which elevates esterase activity by precluding UCR1-UCR2 interactions (87). CaMKII phosphorylation has been shown to increase the catalytic activity of PDE4D and control cAMP levels in cardiomyocytes, indicating that PDE4D underlies cross talk between Ca2+ and cAMP signaling (88). Additionally, an extracellular signal-regulated kinase (ERK) phosphorylation site at the carboxy terminus serves as a negative regulation of all isoforms (89).
2.1.5. PDE5.
PDE5 is selective for cGMP hydrolysis. In humans, three isoforms of PDE5A, PDE5A1, A2, and A3, are expressed but with no difference in reported signaling and function (22, 90–92). PDE5 is detected in cardiomyocytes (93–95) despite early negative reports on the PDE5 expression in the heart (96–98). PDE5 localizes to the cytoplasm and the Z lines of cardiomyocytes and controls a pool of cGMP produced by soluble GC (sGC), whereas PDE2 and PDE9 regulate cGMP produced by the particulate GC activity of NPRs (91, 99). PDE5 is also present in cardiac fibroblasts and has been shown to participate in fibroblast transformation and proliferation (97, 100). In vascular ECs, PDE5 is in caveolae and negatively modulates nitric oxide synthase 3 (NOS3) signaling (101). PDE5 is also abundantly expressed in vascular SMCs, where it plays a central role in regulating vascular tone (96, 102). PDE5 contains GAF-A and GAF-B domains within its amino terminus. The binding of cGMP to the GAF-A domain enhances the allosteric activation of PDE5 (103, 104), whereas GAF-B contributes to the dimerization of the enzyme (105). Phosphorylation of PDE5 by PKA and protein kinase G (PKG) amplifies the cGMP affinity of the GAF-A domain, thereby stabilizing the active enzyme conformation and maintaining hydrolytic activity (106–108). Thus, cGMP fosters its own degradation through negative feedback.
2.1.6. PDE6–11.
PDE6 is a cGMP-specific enzyme primarily expressed in retinal rod and cone cells and is not present in the heart (109). Recently, PDE6 isoforms have been identified outside the eye, especially PDE6γ in mouse lungs (110), which plays a critical role in regulating p42/p44 mitogen-activated protein kinase (MAPK) signaling. PDE6γ has been observed in rat pulmonary vessels and in human pulmonary SMCs (111). These findings suggest that certain PDE6 subtypes could be therapeutic targets in pulmonary hypertension.
PDE7 comprises two genes, PDE7A and PDE7B, both detectable within the heart (112–114). PDE7 contains an amino-terminal sequence with a PKA phosphorylation site, a catalytic domain with two ion (Zn2+ and Mg2+)-binding sites, and a carboxy-terminal sequence (115). PDE7A2 is present in skeletal and cardiac muscles; however, its cardiac function has not been well defined.
PDE8 comprises two genes, PDE8A and 8B, which are present in human and mouse hearts as well as mouse cardiomyocytes (116–118). PDE8 has at least nine splice variants insensitive to the nonselective PDE inhibitor 3-isobutyl-1-methylxanthine (IBMX) (119). PDE8 contains a receiver (REC) and a Per, Arnt, and Sim (PAS) domain at the amino terminus involved in protein-protein interactions. PDE8A is activated by PKA phosphorylation (120).
PDE9 is encoded by a single gene, PDE9A, which has >20 variants (121, 122). PDE9 has a remarkably high affinity for cGMP, with a Km of 170 nM, much lower than the Km of 230 µM for cAMP. PDE9 thus is emerging as an essential regulator of cGMP signaling (121). The enzyme is about twice as active in the presence of 1–10 mM Mn2+ than in the presence of the same concentration of Mg2+ or Ca2+. PDE9A is insensitive (up to 100 µM) to various PDE inhibitors, including rolipram, vinpocetine, SKF-94120, dipyridamole, and IBMX, but is inhibited by zaprinast (IC50 = 35 µM), a PDE5 inhibitor. PDE9A lacks a region homologous to the allosteric cGMP-binding domains found in other cGMP-binding PDEs. Myocardial PDE9 preferentially hydrolyzes natriuretic peptide (NP)-mediated increases in cGMP but minimally affects the nitric oxide (NO)-soluble GC (sGC)-generated cGMP (123). PDE9 is also expressed in SMCs, where it metabolizes NO-sGC-generated cGMP, in contrast to its role in the myocardium (124).
PDE10, like PDE1, 2, and 3, hydrolyzes both cAMP and cGMP. PDE10 contains two amino-terminal GAF domains and a PKA phosphorylation site (125). cAMP binding to the catalytic domain of PDE10 inhibits its cGMP hydrolysis (126, 127); however, cAMP binding to the GAF domain activates PDE10. Thus, cAMP can have biphasic effects on cGMP hydrolysis depending on its concentration, in which low levels of cAMP binding to the GAF domain increase PDE10 activity for cGMP hydrolysis and high levels of cAMP decrease PDE10 activity for cGMP hydrolysis (128). PDE10A2 is the major PDE10A isoform expressed in the heart (129). Whereas PDE10 expression is low in healthy hearts, it is upregulated in mouse and human failing hearts (129).
PDE 11A is a dual esterase hydrolyzing both cAMP and cGMP and has four splice variants (130, 131). PDE11A mRNA expression has been detected in the heart (130, 132). Whereas the PDE11A protein is not detected in blood vessels or cardiomyocytes, weak expression has been found in neuronal cells within parasympathetic ganglia in the heart (132). At the amino terminus, PDE11A1 contains a single GAF domain, which is similar to those found in other PDEs. This domain constitutes a potential allosteric binding site for cGMP or another small ligand (130). PDE11A1 hydrolyzes both cGMP and cAMP with similar Vmax values but with Km values of 0.52 μM and 1.04 μM, respectively (130). Although PDE11A can be potently inhibited by the PDE5 inhibitor tadalafil, its function in the cardiovascular system remains unknown (133).
2.2. Subcellular-Localized Specific PDE-Containing Signalosomes and Cyclic Nucleotide Nanodomains
Localizing PDE enzymes within signalosomes and nanodomains is a fundamental mechanism for regulating cAMP and cGMP signaling in cells. Hormones and neurotransmitters initiate intracellular signaling by activating membrane receptors and using second messengers such as cAMP and cGMP. cAMP targets two primary downstream effectors, PKA, including type 1 and 2 PKA (134), and exchange protein directly activated by cAMP (Epac) (135), to modulate cellular responses. cGMP primarily activates PKG1 and PKG2 to enhance downstream substrate phosphorylation (136). Additionally, cAMP and cGMP can directly bind to and activate ion channels at the plasma membrane (PM) (137, 138). Sutherland raised a model that synthesizes cAMP from the PM and diffuses it throughout the cell (139). However, such a model fails to explain the hormone-specific cellular effects in response to cyclic nucleotides. For instance, stimulation of β-adrenoceptor (βAR) but not prostaglandin EP4 receptor triggers inotropic effects in cardiomyocytes even though both GPCRs induce similar levels of cAMP signals and PKA activities (140, 141). Despite being ubiquitous messengers diffusible inside cells, experimental (6–8) and computational studies (142–146) support that PDEs compartmentalize the cyclic nucleotide signals into nanodomains in a cell to achieve specific cellular responses. The precise mechanisms by which PDEs accomplish this remain an active area of investigation (143, 146). In this paradigm, the ubiquitous second messengers are responsible for diverse downstream effects and cellular responses at distinct subcellular compartments, which results in precise physiological stimulation in a cell- and tissue-specific manner. Hence, subcellular-localized PDEs control the amplitude, duration, and localization of cAMP and cGMP, including spatiotemporally hydrolyzing these cyclic nucleotides, preventing the diffusion of the cyclic nucleotide, and creating the discrete cyclic nucleotide nanodomains in a cell (10). Disruption of PDE subcellular localization contributes to pathophysiological processes, including hypertension and HF (4–10). Here, we summarize cyclic nucleotide nanodomains in subcellular compartments.
PDEs are distributed within distinct subcellular compartments, granting precise control of cyclic nucleotide and subsequent protein kinase signaling. For example, PDE4 isoforms are enriched in membranes, facilitating receptor signaling at the PM (147–149). Similarly, PDE4D isoforms are localized within lipid rafts and caveolar membranes and control local signaling at the dyad region in the transverse (T)-tubular membrane and the sarcoplasmic reticulum (SR) (150–153). PDE4D is also localized to the myofilaments (152) and the nuclear envelope (46, 154–156). PDE3A1 contains a unique amino-terminal extension of hydrophobic loops embedded in intracellular membranes and associated with SERCA2a and AKAP18. PDE3A2 presents only in the cytoplasm, whereas PDE3A3 exists at both cytoplasm and intracellular membrane locations. PDE2 isoforms are distributed in the cytoplasm (PDE2A1), at the PM (PDE2A3), and in the mitochondria (PDE2A2) (40, 157). PDE5 is localized at the Z band (158). In isolated rat cardiomyocytes, NO promotes the synthesis of a cytoplasmic pool of cGMP that is hydrolyzed specifically by PDE5, whereas NPs generate a separate juxtamembrane cGMP pool that is regulated by PDE2 (99). Functional β3ARs are localized exclusively within the T-tubular membrane and stimulate a cGMP pool that is predominantly regulated by PDE2 and PDE5 (159). Additionally, PDE2 confines the membrane-associated pool of cGMP generated via NP-GC-A signaling within the region of the T tubules in isolated cardiomyocytes (160). Furthermore, PDE1A is expressed in the cytoplasm in contractile SMCs but shifted to the nucleus in pathological proliferative SMCs (29). The general idea is that strategically located PDEs in subcellular compartments dictate the spatiotemporal pattern of cAMP and cGMP in a cell (7), which forms distinct nanodomains by specific intracellular location and proximity with a limited subset of effectors in the local vicinity.
Moreover, PDEs are assembled into isoform-specific macromolecular signalosomes within distinct subcellular nanodomains, granting a precise control of cyclic nucleotide and subsequent protein kinase signaling (FIGURE 3). The PDE supramolecular signalosomes are organized by anchoring proteins, including AKAPs and membrane-associated guanylate kinases (MAGUKs), which can be remodeled in pathogenesis. The AKAP family comprises >50 proteins, known for their ability to bring together PKA and the cAMP-termination PDEs into distinct subcellular locations. Although PKA is a ubiquitous enzyme that phosphorylates a broad range of substrates, the association of PKA with AKAP facilitates PKA phosphorylation of local substrates in the vicinity. AKAPs interact with many other signaling molecules, including receptors, PDEs, kinases, and PPs, and downstream targets such as ion channels and myofilaments. These complexes position all the signaling molecules at the regions of cAMP production. The PDEs in the complexes physically limit cAMP access and restrict phosphorylation of a subset of substrates at specific subcellular locations, such as PM, junctional SR, and myofilament, etc. At the PM, various PDE isoforms have been shown to couple with hormone receptors, for example, βARs (148, 161–166), prostaglandin EP4 receptors (141, 148), adenosine A2R (167), and NPRs (99, 123, 160, 168). Without affecting the global cAMP level, PDE4D deletion increases the localized cAMP response to β-stimulation by more than twofold (169). PDEs also associate with ion channels (170, 171) and the Na+-K+-transporting ATPase (172). PDE3 and PDE4 control Ca2+ release and reuptake in the SR by ryanodine receptor 2 (RyR2) and SERCA2a, respectively (46, 63, 169, 173). Conversely, AKAP79 (147), AKAP150 (174), AKAP5 (175), and AKAP9 (171) are distributed at the PM and proximate with various membrane proteins, including βARs, L-type calcium channel (LTCC), and potassium channel (KCNQ2). AKAP250 (AKAP12) associates with β2AR and PDE4 at the peripheral actin cytoskeleton near the PM (149). AKAP6, also known as mAKAP, has been shown to colocalize with PDE4D3, PKA, PP2A, exchange protein directly activated by cAMP 1 (EPAC1), and ERK at the cardiomyocyte nuclear envelope (155, 156). On the other hand, AKAP6 and AKAP7 have been identified to interact with either RyR2 or SERCA2a at junctional or nonjunctional SR, respectively (176, 177). AKAP350 (also named Yatiao, AKAP450, AKAP9) tethers PDE4D3 and PP2A with signaling molecules, including protein kinase C (PKC), protein kinase N (PKN), and casein kinase-1, at the junctional region, centrosomes, or Golgi (175). AKAP95 (AKAP8) targets PDE4D and PKA in the nuclear matrix (178). In comparison, although PKG is also believed to be anchored at the subcellular complexes via its amino-terminal leucine zipper domain in a cell, only a few anchoring proteins have been identified for PKG (179). These signalosomes affirm the significance of PDE-mediated hydrolysis in the regulation of discrete cyclic nucleotide signaling, known as cAMP and cGMP nanodomains.
FIGURE 3.
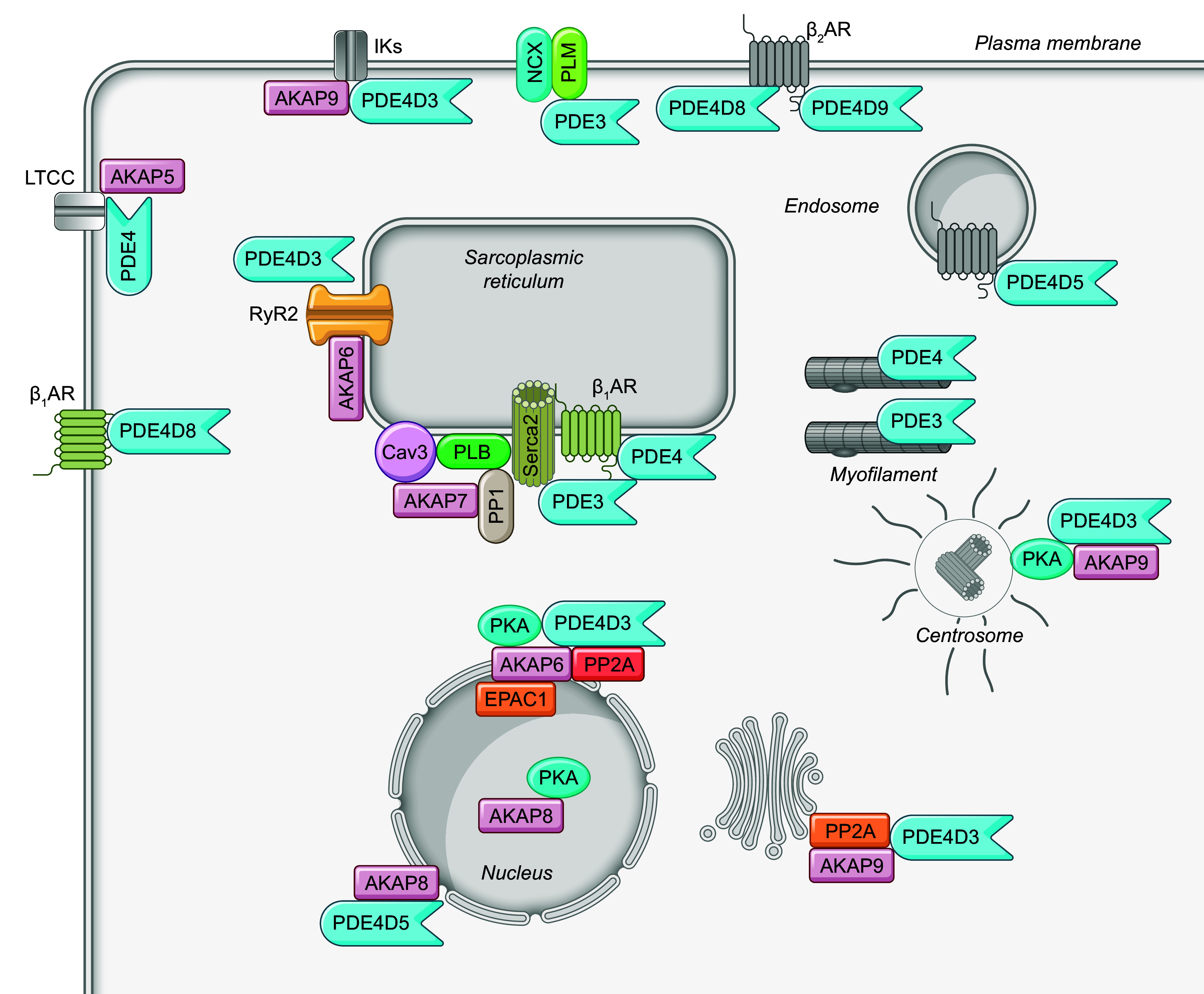
Subcellular localization of phosphodiesterase (PDE) signalosomes in cardiomyocytes. PDEs form signalosomes with various protein partners, such as protein kinase A (PKA), PKA anchoring proteins, and receptors, which are localized in distinct subcellular compartments of cardiomyocytes. AKAP, A-kinase anchoring protein; β1AR, β1 adrenergic receptor; β2AR, β2 adrenergic receptor; Cav3, caveolin 3; Epac, exchange protein activated by cAMP; ERK, extracellular signaling-related kinase; IK, calcium-activated potassium channel; LTCC, L-type calcium channel; NCX, sodium/calcium exchanger; PKG, protein kinase G; PLB, phospholamban; PP1, protein phosphatase 1; PP2A, protein phosphatase 2A; RyR2, ryanodine receptor 2; SERCA, sarco(endo)plasmic reticulum calcium ATPase.
2.3. Detection of Subcellular-Localized Specific PDE-Controlled Cyclic Nucleotide Nanodomains
Our comprehension of compartmentalized cAMP distribution within a cell has been validated with visualization techniques such as Förster resonance energy transfer (FRET)-based biosensors (146). The nanometer-scale resolution of FRET imaging demonstrates the heterogeneous distribution of cAMP within subcellular compartments (180–185). The distinctive membrane architecture and geometry of cardiomyocytes provide a framework for specific cAMP signaling domains. A typical human cardiomyocyte is 100 µm long and 10–25 µm in diameter, with abundant lipid rafts and caveolae constituting various local vicinities for signaling domains. Furthermore, cardiomyocytes present extensive T tubules resulting from deep invagination and extension of the PM. T tubules are at the proximity to the SR networks, the calcium storage, and the myofilament. The maximum distance between the PM, the SR, and the myofilament is ∼300 nM (184). Notably, most FRET-based studies have been performed in cardiomyocytes, and information regarding cyclic nucleotide signaling nanodomains in other cell types, such as SMCs, remains scarce.
FRET-based biosensors have significantly enhanced our understanding of cyclic nucleotide signaling regulation and cAMP-dependent protein kinase activity at the nanoscale (180–183). The spatial confinement of cAMP nanodomains is disrupted by the inhibition of PDEs, indicating the key role of these enzymes in cAMP compartmentalization (10). Recently, the cAMP sensors have been genetically modified to target them to different subcellular compartments and monitor cAMP signaling events at specific sites (141, 152, 184, 186–192). These targeted tools have uncovered novel complexity and interconnectivity that regulate cAMP subcellular domains and reveal the functional relevance of individual cAMP pools. One emerging essential point is that the radius of individual cAMP domains can be as small as a few tens of nanometers (185). Various cAMP nanodomains controlled by highly localized signalosomes have been uncovered in cardiomyocytes. For example, these sensors reveal that the PM, SR, and myofilaments experience distinct amplitudes and kinetics of cAMP signals in response to catecholamine stimulation, even if they are <300 nm apart (184). Similarly, the β-agonist isoproterenol (ISO) triggers a higher and more sensitive PKA response at the PM than at SR and myofilaments, measured by subcellular anchored PKA biosensors (152). These discrete cAMP pools rely on PDE-mediated cAMP degradation. Inhibition of PDE by IBMX normalizes all the cAMP responses at different subcellular sites (184). Intriguingly, the homogeneous cAMP signals induced by the AC agonist forskolin in the presence of IBMX generate a higher troponin I phosphorylation but compromise contractile response relative to β-adrenergic stimulation, suggesting that the compartmentalization of cAMP is essential for the optimal inotropic response in cardiomyocytes (152).
In comparison, the local distribution of cGMP remains less investigated. One of the possible reasons is that cGMP content is 10-fold lower relative to cAMP, which poses a significant challenge for detecting subtle cGMP signals with traditional experimental approaches such as enzymatic quantification. Although the FRET-based biosensors have been developed to detect cGMP, these sensors either are not specific or have lower affinities, making them ineffective for detecting cGMP in cardiomyocytes (193). Thus, the next generation of sensors is needed to detect dynamic cGMP nanodomains in cardiovascular cells. Even with the successful application of cAMP biosensors, there are drawbacks associated with delivering biosensors to targeted cells in the cardiovascular system. The application relies on either transgenic expression of the sensors in vivo or viral infection of isolated primary cells or tissues. The transgenic approach is time-consuming, and the overexpressed biosensors may cause a baseline phenotype by altering cardiovascular function in the transgenic mice. The viral infection approach is limited to in vitro primary culture, which may encounter primary cell dedifferentiation during extended culture periods. Additionally, detection of the cyclic nucleotides in local nanodomains is still beyond the optical resolution of the fluorescence microscope and relies on targeting the sensors with anchor proteins to subcellular domains. Such a strategy could interfere with the local balance of cAMP signaling. Furthermore, during neurohormonal stimulation and under pathological conditions, the anchoring proteins may be relocated and lose fidelity in nanodomain targeting. Therefore, the signals detected with biosensors need to be corroborated with other independent evidence. Novel tools and techniques are needed to detect subcellular local cyclic nucleotide signals with high sensitivity and precision (185). As of today, most studies are based on isolated cells in vitro, and only one recent study shows the cAMP signals detected with a FRET-based sensor in the whole heart with optical mapping (73). Additionally, a recently reported single fluorophore-based cAMP biosensor has been successfully applied to detect cAMP in brain tissues in vivo (194). Technically, it is much simpler to perform single-fluorophore detection compared with the detection of FRET-based biosensors. This sensor could present a better tool for visualizing cAMP signals in the cardiovascular system in vivo. Overall, many technical challenges remain in visualizing cyclic nucleotide signaling in the cardiovascular system in vivo.
In summary, with their distinct localization, dynamics, and functions, compartmentalized cAMP and cGMP pools work in concert to regulate cardiovascular functions, from myocardial ECC to gene expression and pathological remodeling. Because of the pervasive functional role of cAMP and cGMP in the cardiovascular system, small molecules and proteins that modulate cyclic nucleotide signaling have attracted attention as the center of cardiovascular drug development programs. Hence, a detailed understanding of the PDEs in organizing and regulating the function of cAMP and cGMP nanodomains in the heart and vessels becomes increasingly essential (FIGURE 4).
3. PDEs IN THE HEART
The heart functions as a pump, continuously circulating blood through each cardiac contraction and relaxation. The contraction and relaxation cycle is achieved through cardiac ECC, a process predominantly controlled by intracellular Ca2+ (ICa) (195). The depolarized plasma membrane activates voltage-gated LTCCs upon electrical excitation from pacemaker cells, causing Ca2+ influx (ICa,L) in myocytes (195). This Ca2+ entry triggers more Ca2+ release from the SR through RyR2 (196). The Ca2+ influx and release raise ICa, which binds to myofilament proteins such as troponin C to activate the myocyte contractile cycle (195). After the peaking of ICa concentration, ICa declines through SERCA2a- and sodium/calcium exchanger-mediated calcium uptake and efflux, respectively, which leads to Ca2+ myocyte relaxation (197).
β-Adrenergic stimulation plays a critical role in the sympathetic regulation of cardiac output as part of the fight-or-flight response (FIGURE 5) (198). Sympathetic stress releases norepinephrine and epinephrine from the sympathetic nerve and adrenal gland to activate the βARs in cardiomyocytes (199). Activation of βARs increases cAMP and cGMP signals, which play a critical role in regulating cardiac ECC (200). For example, cAMP-activated PKA phosphorylates various downstream targets involved in calcium cycling or myofilament bridge formation (195). PKA can phosphorylate LTCC, RyR2, and phospholamban (PLB) to enhance ICa,L, Ca2+ release, and Ca2+ uptake, yielding increases in ICa (199). The PKA-mediated phosphorylation of myofilament proteins, including troponin I (TnI) and myofilament binding protein C (MyBPC), modulates myocyte contractility without affecting Ca2+ cycling (199). Although the elevation of cAMP is prominent in enhancing ECC, the role of cGMP signal in ECC is less convincing. The literature suggests that cGMP negatively regulates contractility by suppressing the activity of Ca2+-handling proteins, including LTCC, RyR2, and SERCA2a, and myofilament Ca2+ sensitivity (201).
FIGURE 5.
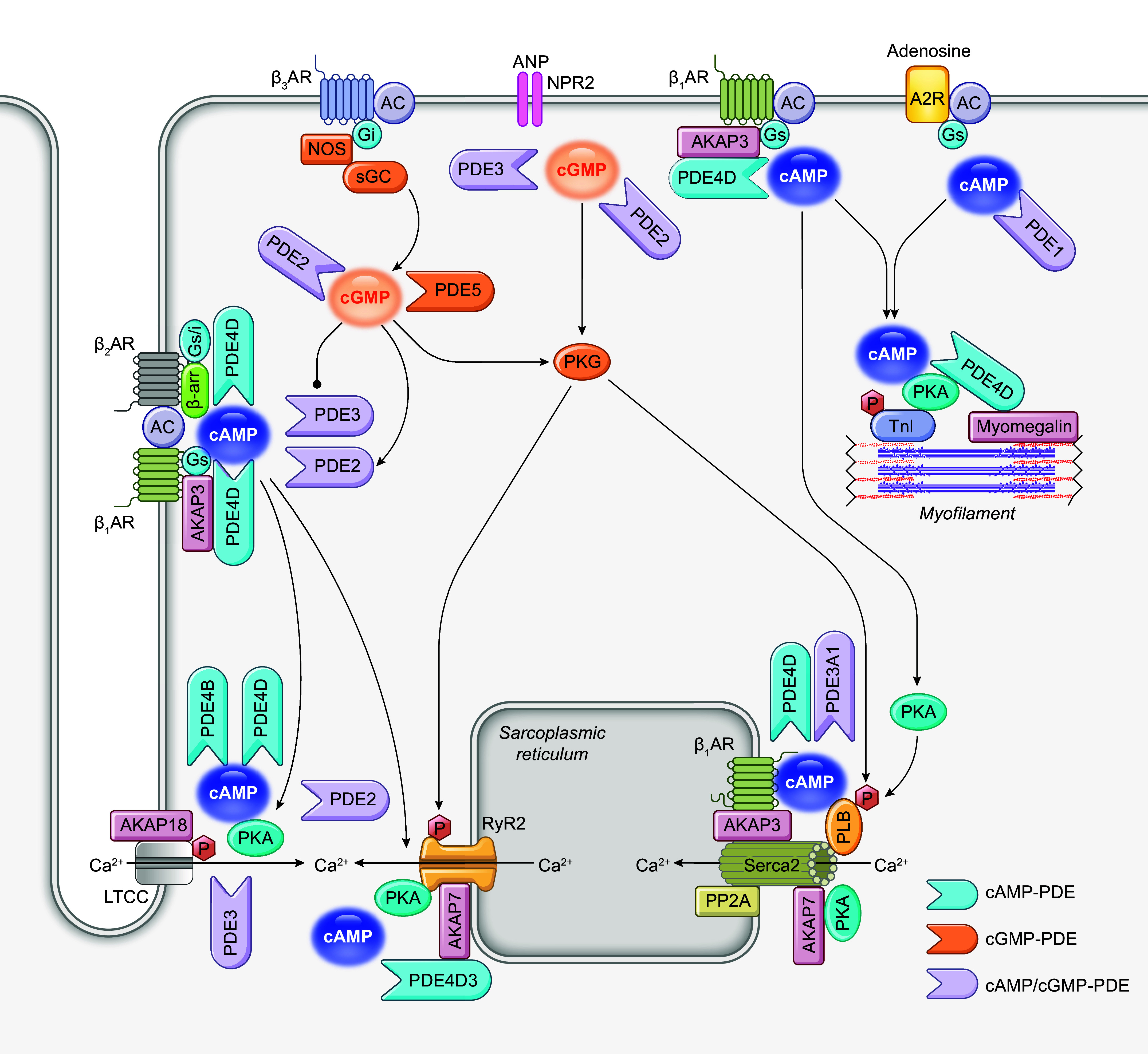
Schematic illustration of phosphodiesterases (PDEs) in cardiac excitation-contraction coupling. A2R, adenosine receptor 2, AC, adenylyl cyclase; ANP, atrial natriuretic peptide; AKAP, A-kinase anchoring protein; β-AR, β adrenergic receptor; cAMP, cyclic adenosine monophosphate, cGMP, cyclic guanosine monophosphate; Gi, inhibitor G protein; Gs, stimulatory G protein; LTCC, L-type calcium channel; NPR2, natriuretic peptide receptor 2; PKA, protein kinase A; PKG, protein kinase G; PLB, phospholamban; PP2A, protein phosphatase 2A; RyR2, ryanodine receptor 2; SERCA, sarco(endo)plasmic reticulum calcium ATPase; sGC, soluble guanylyl cyclase; TnI, troponin I.
Reduced global cAMP is a hallmark of various cardiac diseases, including hypertrophy, myocardial infarction (MI), and HF (134). Local cAMP and cGMP undergo distinct alterations at subcellular compartments in different etiologies (152, 190, 202–204). In this section, we discuss the roles of PDEs in regulating cardiac contractile function and electrical rhythm, as well as in the pathogenesis of cardiac diseases.
3.1. PDE in the Regulation of Cardiac Contractility
3.1.1. PDE in the regulation of Ca2+ influx by LTCC.
Among PDEs expressed in cardiomyocytes, PDE3 and PDE4 are pivotal regulators of myocyte ICa,L by controlling cAMP nanodomains at the sarcolemma because of their association or proximity to βAR or LTCC (76, 164, 165, 170, 205, 206). Whereas PDE3 and PDE4 are the dominant PDE subtypes involved in the regulation of basal ICa, all four PDEs (PDE1–4) determine the response of ICa,L to a stimulus activating cAMP production, with the rank order of potency PDE4 > PDE3 > PDE2 > PDE1 (207). In addition, the deficiency of PDE8A potentiates an adrenergic-induced pool of cAMP to increase ICa,L and ICa (116).
PDE4 is the major PDE expressed in rodent hearts. PDE4A, PDE4B, and PDE4D have been identified in hearts. Specifically, PDE4D3, PDE4D5, PDE4D8, and PDE4D9 have been demonstrated to regulate β-adrenergic signaling and ECC (14, 208). At the sarcolemma membrane, PDE4D isoforms are physically associated with cardiac βARs, either through direct binding or with the assistance of arresting scaffolding (148, 161–166). The association modulates homologous and heterologous receptor desensitization. The interaction with PDE4D5 also prevents the Epac-dependent stimulation of CaMKII. Nonselective inhibition of PDE4 with rolipram potentiates the amplitude and duration of cAMP in cardiomyocytes in response to βAR stimulation (205). Rolipram also induces a drastic increase in PKA activity at the PM and a lesser significant increase at the myofilament and SR and affects myocardial cAMP-PKA-dependent phosphorylation of Ca2+-handling proteins and contractility (152). Consequently, βAR stimulation of ICa,L, Ca2+ transient, and contractility is enhanced by PDE4 inhibition, indicating that PDE4 is a potent negative regulator of LTCC in hearts (76, 209). Meanwhile, PDE4B is also functionally distributed at the sarcolemma membrane (210) and couples with LTCC in myocytes (170). At the baseline, deleting PDE4B but not PDE4D increases ICa,L, Ca2+ transient, and contraction (170).
PDE3 blockade alone has a minor effect on subsarcolemmal cAMP and LTCC, which is also unrelated to inotropy (211). However, when both PDE3 and PDE4 are inhibited, or when all PDEs are blocked with 3-isobutyl-1-methylxanthine, cAMP signal and ICa,L are sustained for an extended period (76). Phosphoinositide 3-kinase γ (PI3Kγ) is found to participate in multiprotein complexes linking PKA to the activation of PDE3A, PDE4A, and PDE4B but not of PDE4D. These PI3Kγ-regulated PDEs lower cAMP and limit PKA-mediated phosphorylation of LTCC and PLB, leading to increased Ca2+ spark occurrence and amplitude on adrenergic stimulation (77). In cardiac hypertrophy, PDE3 inhibition has an enhanced role in promoting ICa,L induced by βARs (209).
Despite its minimal expression in cardiomyocytes and modest contribution to the total cAMP hydrolytic activity (∼3%), PDE2 also modulates cardiac contractility (9, 212). The regulatory function of PDE2 in shaping βAR responses is closely linked to the refined phosphorylation of downstream targets, such as LTCC (213, 214). PDE2 attenuates the cAMP signal, reducing LTCC phosphorylation at PKA-specific sites and ICa,L in ventricular myocytes (214–216). PDE2 inhibition increases the inotropic effects of β2AR stimulation in rat left ventricular myocardium ex vivo, likely via potentiating the β2AR-cAMP signaling (217). Transgenic mice with cardiac-specific overexpression of PDE2 exhibit lower heart rates and display attenuated isoprenaline-induced increases in cAMP signal, ICa,L, Ca2+ transient, sarcomere shortening, and arrhythmia (218). Moreover, PDE2 and PDE3 within βAR nanodomains undergo redistribution in HF animal models, influencing cardiac responses and contractility (172, 219).
Meanwhile, PDE2 can be activated by cGMP binding to the GAF domain. Multiple groups have reported that a cGMP-induced activation of PDE2 reduces ICa,L (220). For example, activation of β3AR transduces the NOS3-NO-cGMP signaling cascade and activates PDE2 (45). In this case, PDE2 inhibition markedly increases norepinephrine-induced cAMP responses, even though PDE2 inhibition alone leads to a minor increase in intracellular cAMP levels (45). Although the expression of β3AR in the myocardium remains contentious (221), the observation suggests that a β3AR-NO-cGMP-PDE2 axis confines the distribution of βAR-generated cAMP (45), consistent with a negative inotrope of NO (43). However, the PDE2-mediated cGMP-cAMP cross talk downstream of β3AR signaling is impaired in an MI-induced failing HF, because of the altered distribution of β3AR and sGC, leading to a decrease in cGMP level and impaired cGMP-cAMP cross talk (159). NPR1 is found in T-tubular membranes, and atrial natriuretic peptide (ANP)-stimulated cGMP is usually constrained at the membrane and does not affect cardiac inotropy and contractility (222). However, PDE2 inhibition augments the NPR-induced cGMP at the PM in adult cardiomyocytes (99). In this scenario, the ANP-stimulated cGMP also diffuses into the cytoplasm to promote the phosphorylation of PLB and regulate Ca2+ cycling (160). PDE2 expression is increased in human and pig models of dilated cardiomyopathy (42) and in a chronic ISO-induced rat HF model (42). The upregulation of PDE2A desensitizes acute βAR responsiveness in failing hearts (42). Additionally, PDE2 is relocalized from β1AR-associated noncaveolar into β2AR-containing caveolar fractions in cardiac hypertrophy after transaortic constriction (TAC) (219).
Thus, PDE4 appears to be the most crucial regulator in enhancing LTCC activity in physiological response, whereas PDE2 and PDE3 play additional roles in coordinating the regulation. Meanwhile, PDE2 and PDE3 may have enhanced roles in regulating cAMP, cGMP, and LTCC activity in cardiac diseases.
3.1.2. PDE in the regulation of SR Ca2+ release and uptake.
PDE4, PDE3, and PDE2 are implicated in regulating SERCA2a and RyR2 activities and SR Ca2+ release and uptake (214, 223, 224). With FRET biosensors, PDE4 and PDE3 are shown to control the baseline PKA activity at the RyR2 and SERCA2a nanodomains and prevent β2AR signaling from reaching these nanodomains in mice, rat, and rabbit myocytes (152). PDE4 is the most determinant when cAMP levels are elevated upon βAR stimulation (214). However, PDE2 and PDE3 also play a prominent role in regulating cardiac contraction and Ca2+ transients at the baseline and after βAR stimulation (214, 223, 224).
PDE4 isoforms associated with SERCA2a affect the Ca2+ pump activity and SR Ca2+ uptake. PDE4D coimmunoprecipitates with SERCA2a in murine and failing human hearts (173) and negatively regulates βAR stimulation of PLB phosphorylation and SR Ca2+ load (206). PI3Kγ is required for PDE4, not PDE3, activity in the subcellular SERCA2a nanodomains in cardiomyocytes, and loss of PI3Kγ selectively abolishes PDE4 activity in the SERCA2a nanodomains (206). Recently, a pool of intracellular β1AR has been identified with SERCA2a (199, 203, 225), suggesting that a local β1AR signaling machinery at the SR regulates cAMP levels at the SERCA2a nanodomains. Mice lacking PDE4D have an enhanced baseline cardiac contractility associated with increased PLB phosphorylation, SR Ca2+ content, and Ca2+ transients (173). The phosphorylation of PLB and SERCA2a function is depressed in HF (203, 226–228). PDE4 is reduced in hypertrophic cardiomyocytes (190), which may be a compensatory adaptation and may help to increase the phosphorylation of PLB and SR Ca2+ uptake. In comparison, the contribution of PDE2 to cAMP hydrolysis is increased in the vicinity of SERCA2a in hypertrophied myocytes, which may affect Ca2+ cycling and contractility (190).
PDE4D3 forms a complex with RyR2 at the SR in human and mouse hearts and regulates the PKA-dependent phosphorylation of RyR2 (169). Selective inhibition of PDE4 enhances the adrenergic stimulation of local cAMP signals at the RyR2 (202). Deficiency of PDE4D leads to PKA hyperphosphorylation of RyR2, promoting RyR2 dissociation from the stabilizing protein calstabin2 (FKBP1.2), aberrant calcium release, arrhythmias, and the development of dilated cardiomyopathy (169). In hypertrophic hearts, the RyR2-associated PDE4 is reduced, leading to an increased RyR2 phosphorylation to β2AR stimulation (169).
Deleting PDE3A increases PLB phosphorylation, SERCA2a activity, SR Ca2+ uptake, and Ca2+ contents and promotes cardiac contractility and relaxation through cAMP-dependent elevations of Ca2+ transients (63). Consequently, PDE3 inhibition enhances contractility in vitro and in vivo (63). Although PDE3A and PDE3B are expressed in the heart (61, 62), knockout experiments suggest that PDE3A but not PDE3B regulates cardiac contractility (63, 64). PDE3A1 associates with SERCA2a in human hearts, forming a signalosome consisting of multiple proteins, including AKAP18, PP2A, PP1, PLB, and caveolin 3 (60, 63). Disrupting the association between PDE3A and SERCA2a with a disruptor peptide increases the Ca2+ pump activity in normal and failing cardiomyocytes (229). After aortic banding, mice injected with recombinant adeno-associated virus 9 (rAAV9) expressing the disruptor peptide have improved contractility but no difference in cardiac remodeling compared to mice with control rAAV9 (229). A recent study shows that the hypertension-related gain-of-function mutations of PDE3A do not affect overall adrenergic stimulation and cAMP levels but reduce PLB phosphorylation, which leads to adaptive change in Ca2+ cycling and protects against hypertension-induced cardiac damage in the hearts (230).
3.1.3. PDE in the regulation of Na+-K+-ATPase activity.
PDEs also affect cardiac contractility by modulating phospholemman-mediated Na+-K+-ATPase activity. Na+-K+-ATPase indirectly affects myocyte ICa through the sodium/calcium exchanger. Phosphorylation of phospholemman, the Na+-K+-ATPase regulator, enhances the pump activity and results in a decrease of intracellular Na+. The decrease of intracellular Na+ reduces ICa and myocyte contractility. PDEs fine-tune the PKA phosphorylation of phospholemman and, therefore, the changes in contractility. PDE3 has been shown to control cAMP in a nanodomain around the phospholemman-Na+-K+-ATPase complex and preferentially regulates this cAMP pool induced by β2AR (172). Moreover, NPR2 stimulation by C-type natriuretic peptide (CNP) enhances cAMP signaling through the cGMP-mediated inhibition of PDE3 and improves contractility in normal and failing hearts (231). In comparison, NPR1 stimulation by ANP or B-type natriuretic peptide (BNP) does not affect βAR signaling (232). In rats after MI-induced chronic HF, a significant increase in PDE2-mediated hydrolysis of cAMP downstream of β2AR is observed within the phospholemman Na+-K+-ATPase nanodomains, whereas the PDE3-induced hydrolysis of cAMP is diminished (172). These data demonstrate that PDEs act together to fine-tune cAMP at the Na+-K+-ATPase nanodomains in the heart.
3.1.4. PDE in the regulation of myofilament cGMP signaling.
The literature points out the prominent roles of PDE1 in regulating cGMP and PKG activity and myofilament relaxation. PDE1C expression is conserved in rabbits, dogs, and humans, which opposes the PDE1A expression in mice and rats (233). A selective PDE1 inhibitor, ITI-214, evokes positive inotropic, lusitropic, chronotropic, and vasodilator effects in normal-conscious dogs (233). These responses are preserved except for heart rate in the same animals when their hearts are induced into failure after 3–4 wk of tachycardia pacing. These cardiac and systemic arterial effects of ITI-214 have been linked to adenosine A2B receptor-induced cAMP signaling (233). A2B receptor stimulation is well known to protect against ischemic injury (234); the protective effects of the A2B receptor may be leveraged by PDE1 inhibition. In cardiomyocytes, PDE1 inhibition augments the resting cell contraction without increasing Ca2+ (235). By contrast, β1AR stimulation or PDE3 inhibition increases cAMP, Ca2+ transients, and cell contraction. The PDE1 inhibitor ITI-214 thus may selectively target myofilaments to induce positive inotropy and lusitropy, differing from that coupled to β1AR-cAMP signaling. Similarly, biased β1AR-Gi signaling induced by carvedilol selectively promotes cGMP-PKG-mediated phosphorylation of myofilament proteins and cardiac contractility without increasing Ca2+ cycling (200). The mechanism underlying PDE1-induced contraction remains to be explored. PDE1 inhibition may have a safety profile different from those observed with PDE3 inhibitors that induce cardiac arrhythmia.
PDE3 regulates global cellular cGMP levels in normal and failing hearts (236). PDE3 has been shown to constrain CNP-stimulated cGMP at the PM, PLB, and myofilament compartments (237). CNP also promotes cGMP-dependent inhibition of PDE3, which increases cAMP through the cGMP-cAMP cross talk. NPR2-mediated cGMP production causes a negative inotropic and a positive lusitropic response in failing hearts (238). Inhibition of PDE3 enhances the CNP-mediated lusitropic response in normal heart muscle and the CNP-mediated negative inotropic and positive lusitropic responses in HF models. CNP also enhances β1AR- and β2AR-mediated inotropic and β1AR-mediated lusitropic responses in nonfailing and failing hearts through the cGMP-cAMP cross talk where cGMP increases cAMP signal by inhibiting PDE3 (231). Furthermore, CNP also enhances the serotonin type 4 (5-HT4)-mediated inotropic response in failing rat heart ventricles via PDE3-mediated cGMP-cAMP cross talk (239). In the long term, this CNP-cGMP-PDE3 inhibitory pathway may be detrimental to failing hearts through mechanisms like those treated with PDE3 inhibitors or β-adrenergic stimulation (238).
Inhibition of PDE5A attenuates the catecholamine-induced contractile function through cGMP-PKG signaling. For instance, a PDE5 inhibitor, sildenafil, attenuates dobutamine and ISO-stimulated cardiac contractility (232, 240, 241). Interestingly, deletion of β3AR abolishes the sildenafil-mediated negative regulation of contractility (242), indicating that activation of the β3AR-NOS pathway is necessary for the sildenafil effect on increasing cGMP signal. The effects of PDE5 inhibitors on cardiac contractility are largely diminished in hypertrophic failing hearts, which is associated with reduced Z-band localization of PDE5A (241). Another report shows that PDE5 is coupled to β2AR in cardiomyocytes isolated from mice after high-fat diet feeding. Inhibition of PDE5 enhances the β2AR-induced cAMP and cGMP signals and PKG-dependent myocyte contractility (243). These findings implicate a shift coupling of PDE5 to different βAR subtypes to regulate highly localized cGMP pools and cardiac contractility in normal and pathological hearts.
PKA-dependent phosphorylation is also critical in modulating myofilament contraction in physiological and disease states (244–247). However, more information is needed to understand the PDE regulation of cAMP signaling and function on the myofilaments. PDE3 and, to a lesser extent, PDE4 and PDE2 regulate the baseline PKA activity at the myofilaments in rabbit myocytes, whereas inhibition of PDE3 and PDE4 enhances the βAR-induced PKA activity (152). In failing rabbit hearts, loss of caveolin 3 leads to reduced impacts of PDE3 and PDE2 on myofilament PKA activity, whereas no change is observed in the impact of PDE4 (152).
3.2. PDE in the Regulation of Heart Rate and Cardiac Arrhythmias
PDEs play a crucial role in regulating the rhythmicity of pacemakers across multiple dimensions (FIGURE 6). Sinoatrial node (SAN) cells initiate and time-keep heartbeat by a precise coupled system of an intracellular Ca2+ clock and a surface membrane voltage clock (248). SAN cells allow individual ion channel currents to ensemble to generate rhythmic action potentials. This ensemble is known as the membrane clock (M clock). The intracellular Ca2+ clock refers to RyR2-mediated Ca2+ release. In spontaneous firing SAN cells, the M clock and Ca2+ clock work in coordination fine-tuned by a multitude of interactions. PDE inhibition in SAN cells markedly augments Ca2+ cycling protein phosphorylation and accelerates the action potential (AP) firing rate by elevating the cAMP signal (249). Consequently, PDEs wield the potential to impact pacemaker activity through diverse mechanisms, encompassing the cAMP-PKA pathway and calcium signaling.
FIGURE 6.
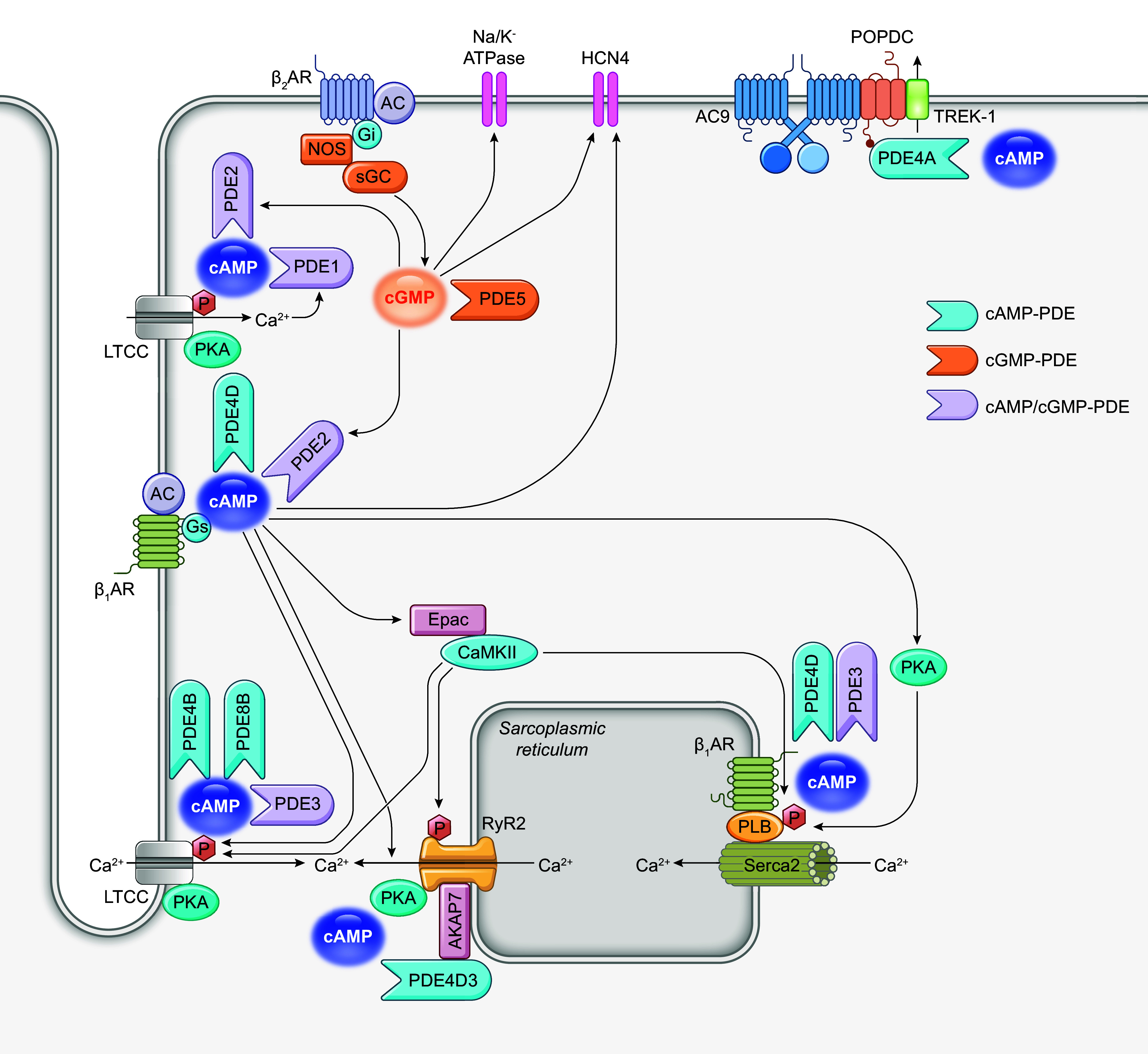
Schematic illustration of phosphodiesterases (PDEs) in the regulation of heart rate and arrhythmia. AC, adenylyl cyclase; AKAP, A-kinase anchoring protein; β1AR, β1 adrenergic receptor; β2AR, β2 adrenergic receptor; β-arr, β-arrestin; CaMKII, Ca2+/calmodulin-dependent kinase II; cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; Epac, exchange protein activated by cAMP; Gi, inhibitor G protein; Gs, stimulatory G protein; HCN, hyperpolarization-activated cyclic nucleotide-gated channel; LTCC, L-type calcium channel; Na/K-ATPase, sodium-potassium-ATPase; NOS, nitric oxide synthase; P, phosphate; PKA, protein kinase A; PLB, phospholamban; POPDC, popeye domain containing protein; RyR2, ryanodine receptor 2; SERCA, sarco(endo)plasmic reticulum calcium ATPase; sGC, soluble guanylyl cyclase.
3.2.1. PDE in the regulation of pacemaker activity.
The modulation of heart rate by PDE entails the precise regulation of cAMP and cGMP through a diverse array of effectors within SAN cells. As a predominant regulatory molecule, cAMP orchestrates alterations in the action potential of SAN cells through downstream effectors: hyperpolarization-activated cyclic nucleotide-gated channel 4 (HCN4) responsible for the “funny” current (If), PKA, EPAC1, and popeye domain-containing (POPDC) proteins (230). Accordingly, manipulating PDEs could enhance or suppress pacemaking activities in SAN cells through these targets. For example, PKA activity underscores the PDE-dependent modulation of pacemaker flexibility. Stimulation of βARs increases cAMP-PKA signaling in the SAN cells to accelerate the spontaneous firing of SAN cells through two parallel mechanisms, which underlies the chronotropic effect under the fight-or-flight response. On one hand, cAMP directly binds to HCN4, increasing the activity of If and enhancing the M clock of SAN pacemaking. On the other hand, the cAMP-PKA signaling promotes PKA phosphorylation and activities of the key downstream proteins of ECC, including LTCC, RyR2, and PLB. These phosphorylation events increase cytosolic Ca2+ entry and enhance SR Ca2+ release through RyR2 and uptake via SERCA2a, promoting the Ca2+ clock to accelerate the action potential firing of SAN cells. The increased Ca2+ signals also activate CaMKII to phosphorylate LTCC, RyR2, and PLB to promote the Ca2+ clock (250).
PDE1 is an essential modulator of heart rate. PDE1 activity accounts for 39% of the total PDE activity in SAN cell lysates, compared to only 4% in left ventricular cardiomyocytes. PDE1 senses Ca2+-CaM signal to regulate the cAMP degradation in lipid raft domains and determine the intensity of Ca2+-AC-cAMP-PKA signaling that drives SAN pacemaker function (251). PDE2 reduces LTCC phosphorylation at PKA-specific sites and ICa,L in atrial cells by attenuating cAMP levels at the PM (213, 252, 253). PDE3 is the most predominantly expressed PDE in human atrial, including SAN, cells. Among PDE3 isoforms, PDE3A is more potent than PDE3B for cAMP degradation in regulating heart rate (64). PDE3 inhibition dramatically increases the basal spontaneous SAN cell beating rate, accompanied by a marked increase in PLB phosphorylation (213, 254). PDE3 inhibitors also enhance the prestimulated ICa,L and Ca2+ uptake and promote heart rate (255). Additionally, POPDC proteins, a group of newly characterized effectors of cAMP, play a crucial role in cardiac pacemaking. POPDC1 preferentially binds to the PDE4A subfamily via a specificity motif in the PDE4A UCR1 region. The PDE4 activity localized to POPDC1 modulates the cycle length of spontaneous Ca2+ transients firing in intact mouse SANs (256).
In SAN cells, cGMP regulates heart rate through various effectors, although the mechanisms in this context have been understudied. NO leads to an increase in If current and heart rate, partially due to NO-stimulated increases in cGMP (257). The stimulatory effects of cGMP on ICa in rabbit atrial cells are likely to be mediated via the PKG-dependent phosphorylation of LTCC or associated proteins. PKG is highly expressed in atrial cells, and PKG-dependent phosphorylation may be necessary for maintaining basal ICa and fully stimulating ICa by β-adrenergic activation (258). Additionally, cGMP can directly bind to HCN4 channels, which is key in determining membrane potential in pacemaker cells (259). Modulating HCN4 and If in SAN cells through phosphorylation or cyclic nucleotides can alter heart rate and cause bradycardia, tachycardia, and SAN dysfunction (259). In pacemaker cells, NO also induces cGMP-dependent activation of PDE2, which attenuates ICa,L and heart rate (201, 260). Accordingly, mice with cardiac-specific PDE2 overexpression exhibit lower heart rates and display blunted isoprenaline-induced increases in cAMP levels and heart rates (218). PDE2 also fine-tunes a specific cAMP pool generated downstream of β2ARs, which PDE5 indirectly controls. Inhibition of PDE5 promotes cGMP-dependent activation of PDE2, which has negative chronotropic effects (261). Epidemiological data indicate a reduction in cardiovascular events and mortality in PDE5 inhibitor users at high cardiovascular risk (262). The antiarrhythmia effects of PDE5 inhibition may involve cGMP, and the underlying mechanism requires further investigation.
3.2.2. PDE in atrial fibrillation.
Atrial fibrillation, which is closely linked to PDE dysfunction, represents the most common arrhythmias. For instance, inhibition of PDE2 increases ICa,L in human atrial myocytes, and PDE2 overexpression attenuates SR Ca2+ release and arrhythmia susceptibility (218). PDE3 is the most abundant PDE in human atria. Chronic treatments with PDE3 inhibitors are an independent risk factor for clinically significant tachyarrhythmias, augmenting the mortality of treated patients (263, 264). Patients with permanent atrial fibrillation have been found to have decreased PDE4 activity (265). PDE4A, PDE4B, and PDE4D isoforms are present in human atrial myocytes, accounting for ∼15% of total PDE activity (265). PDE4B and PDE4D are tethered to LTCC in mouse hearts (170), whereas PDE4D3 is linked to RyR2 (169). PDE4B deletion has been linked with exacerbated βAR stimulation of ICa,L, whereas PDE4D deletion leads to PKA-dependent hyperphosphorylation of RyR2 (169, 173). Consequently, PDE4 inhibition increases the LTCC density and the rate of spontaneous SR Ca2+ release in isolated human atrial myocytes and intact human atrial trabeculae exposed to βAR stimulation (265). PDE5 inhibitors have electrophysiological effects in atrial myocytes, which may contribute to a direct antiarrhythmic action during reperfusion, including reducing ICa,L and intracellular Na+, enhancing Na+-K+-ATPase activity (266), and suppressing β-adrenergic signaling (267). Additionally, PDE8A expression is increased in the human atrium in atrial fibrillation (268). Upregulation of the PDE8B2 isoform in chronic atrial fibrillation reduces ICa,L via direct interaction of PDE8B2 with the LTCC Cav1.2 α1C-subunit. Thus, the upregulated PDE8B2 governs a cAMP-PKA-dependent reduction of ICa,L in human atrial fibrillation, which might be a novel mechanism of the proarrhythmic reduction of ICa,L in chronic atrial fibrillation (269). Collectively, these findings highlight the PDEs’ significant impacts on atrial electrical activity and their potential roles in increasing the risk of arrhythmia. Arrhythmia-related sudden cardiac death accounts for up to 60% of deaths in HF patients (270). Strategies aiming at fine-tuning the activity of individual PDEs may offer effective therapies for cardiac arrhythmia in HF patients.
3.2.3. PDE in ventricular arrhythmias.
PDEs are also involved in ventricular arrhythmias. Genetic ablation of PDE4B or PDE4D enhances the susceptibility to stress-induced ventricular tachycardia (169, 170). PDE2 activation protects against ventricular arrhythmias by preventing Epac- and CaMKII-mediated increases in INa and ICa,L and Ca2+ leakage from the SR (218, 271). In ischemia-reperfusion of isolated rat hearts, pretreatment with sildenafil protects against ventricular fibrillation and reduces infarct size while improving left ventricle recovery, probably through a cGMP signaling pathway (272). Long QT syndrome type 2 (LQT2) arrhythmogenesis is mainly ascribable to the facilitation of Ca2+ waves, reflecting SR instability. An antiarrhythmic effect of PDE5 inhibition has been reported in long QT syndrome by inhibiting the SERCA2a-mediated Ca2+ uptake and reducing SR Ca2+ content (262). Therefore, whereas the elevation of cAMP is linked to increases in ventricular arrhythmicity, the elevation of cGMP has a protective effect.
3.3. PDE in Cardiac Hypertrophy, Apoptosis, and Heart Failure
HF is a severe and complex pathology that remains a major cause of global morbidity and mortality. In response to stressors such as hypertension or neurohumoral activation, the heart undergoes pathological changes, including increased myocyte size and protein synthesis, cell growth, and myocyte apoptosis. These changes lead to cardiac hypertrophy and ventricular remodeling, resulting in decompensation and HF. Current HF treatments primarily aim at preventing ventricular remodeling and improving overall cardiac function.
cAMP and cGMP play a key role in cardiac function in normal and pathological conditions (FIGURE 7). Chronic HF is also characterized by overexcitation of the sympathetic nervous system and the release of catecholamines (262). The subsequent activation of cardiac βARs induces cardiac hypertrophy and apoptosis via the cAMP and PKA-mediated elevation of ICa and CaMKII (263). Chronic β-adrenergic stimulation also activates Epac. Whereas PKA is proapoptotic, Epac protects myocytes via activation of MAPK (273). These distinct pathways may be underlying the observations in which selectively enhancing the type VI AC-induced cAMP-PKA signaling benefits failing hearts (274). Moreover, β-adrenergic stimulation promotes an Epac-phospholipase Cε (PLCε)-dependent phosphatidylinositol 4-phosphate (PI4P) hydrolysis, acting as a critical process for cardiac hypertrophy (275).
FIGURE 7.
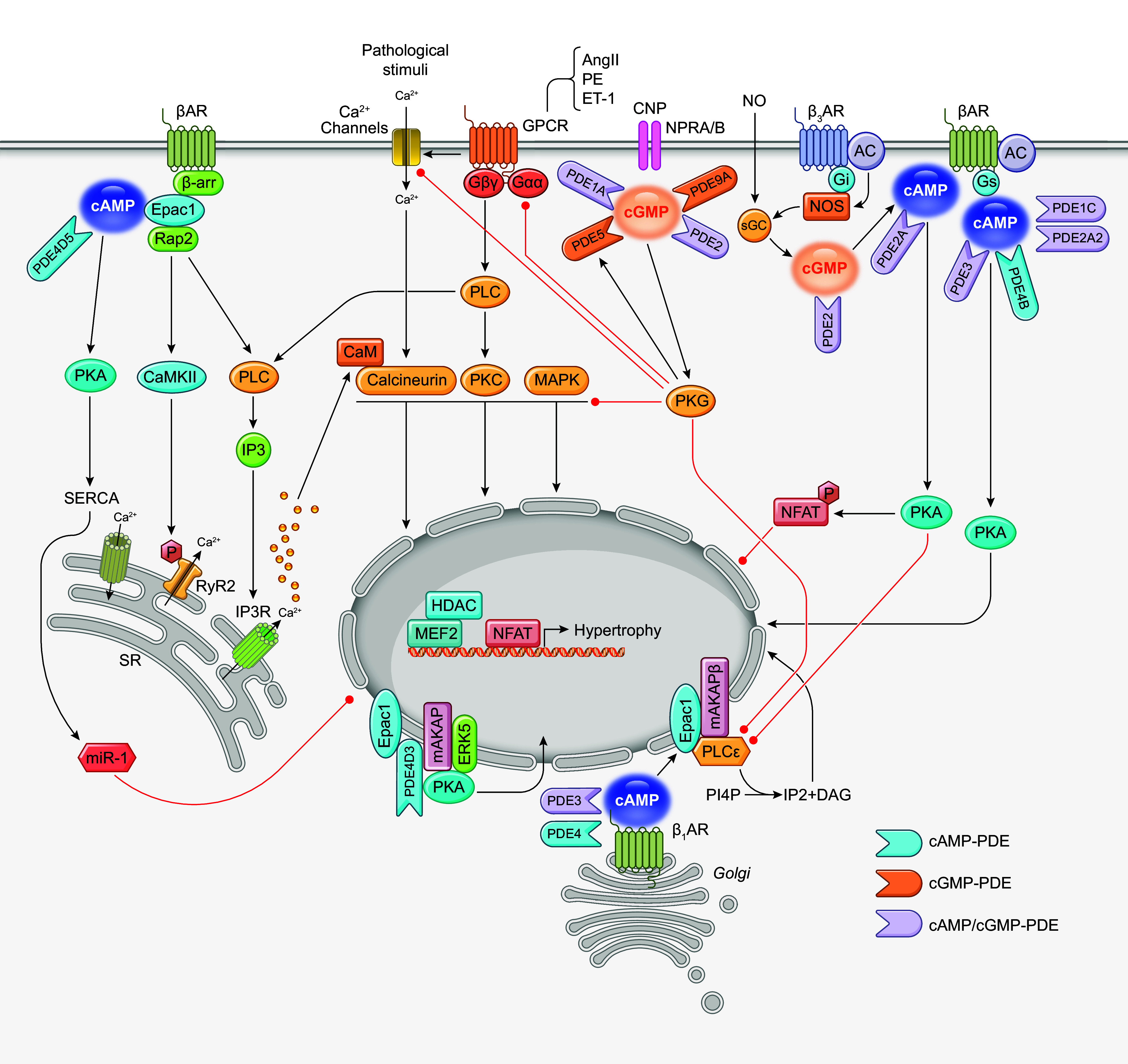
Schematic illustration of phosphodiesterases (PDEs) in the regulation of cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) in cardiac hypertrophy. The cartoon highlights the subcellular regulation of cAMP and cGMP induced by different neurohormonal stimuli in driving cardiac hypertrophy. AC, adenylyl cyclase; AngII, angiotensin II; AKAP, A-kinase anchoring protein; βAR, β adrenergic receptor; β-arr, β-arrestin; CaMKII, Ca2+/calmodulin-dependent kinase II; CNP, C-type natriuretic peptide; DAG, diacylglycerol; Epac, exchange protein activated by cAMP; ET-1, endothelin-1, Gαq, Gαq protein; Gβγ, Gβ and Gγ protein; Gi, inhibitor G protein; GPCR, G protein-coupled receptor; Gs, stimulatory G protein; HDAC, histone deacetylase; IP2, inositol-4,5-bisphosphate; IP3, inositol 1,4,5-trisphosphate; IP3R, inositol 1,4,5-trisphosphate receptor; MAPK, mitogen-activated protein kinase; MEF2, myocyte enhancing factor 2; miR-1, microRNA-1; NFAT, Nuclear factor of activated T cells; NPR2, natriuretic peptide receptor 2; NOS, nitric oxide synthase; PE, phenylephrine; PI4P, phosphatidylinositol-4-phosphate; PKA, protein kinase A, PKC, protein kinase C; PLC, phospholipase C; Rap1, ribosome activated protein 1; RyR2, ryanodine receptor 2; SERCA, sarco(endo)plasmic reticulum calcium ATPase; sGC, soluble guanylyl cyclase.
Despite elevated sympathetic activity, HF is often associated with reduced intracellular content of cAMP and PKA substrate phosphorylation, contributing to impaired cardiac contractility. Clinically, PDE3 inhibitors are developed to attenuate the degradation of cAMP, leading to increased intracellular calcium and improved hemodynamics and exercise capacity in patients with advanced HF (276). However, despite the short-term functional gains, therapy aimed at increasing cAMP levels increases mortality in the long term (277). This raises the question of how we can develop a strategy to trigger only the inotrope response in HF. One can speculate that cAMP signaling controls various and opposing cellular functions via selective activation of different targets residing at subcellular locations. Distinct subcellular cAMP pools and different cAMP levels may have opposing effects on myocyte contractility, growth, and death (155, 278). Another consideration is the interplay between cAMP and cGMP signals. Notably, cGMP can affect cAMP levels in cardiomyocytes by activating or inhibiting cAMP-hydrolyzing PDEs (277). Compared with the cAMP signaling pathway, cGMP mediates the cardiac effects of NO, which has negative inotropic effects. The cGMP-PKG signaling also negatively regulates cardiac hypertrophy (279). Moreover, the modulatory effects of cGMP on cAMP-hydrolyzing PDEs are titrated by subcellular concentrations of cGMP (201). The balance of cAMP and cGMP signals may have a “yin-yang” effect, two opposing forces in harmony, in cardiovascular homeostasis (280). The intricate cross talk between cAMP and cGMP can have divergent downstream actions for a broad range of functional outcomes (FIGURE 7). Accordingly, the functional diversity of individual PDEs could be achieved through the localization, the link to discrete pools of cyclic nucleotides, and the association with downstream signaling molecules. Under pathological environments, the regulation and function of individual PDEs are disrupted, contributing to the progression of cardiac hypertrophy and HF (TABLE 2). In sects. 3.3.1–3.3.3, we discuss how PDEs are involved in the development of hypertrophic HF.
Table 2.
The roles of individual PDE isoforms in cardiac hypertrophy
| PDE Family | Isoform | Variants | Expression | Activity | Pathology | Model in vivo | Model in vitro | Genetic Intervention in vivo | Genetic Intervention in vitro | Inhibitors Used | Positive ↑Negative ↓ | Potential Mechanism | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PDE1 | Ang II infusion mice | vinpocetine | ↓ | ↓PDE1 | (281) | ||||||||
| PDE1A | ↑ | ISO infusion mice | Ang II and ISO-NRCM | PDE1A shRNA | IC86340 | ↓ | ↑cGMP-PKG | (23) | |||||
| PDE1C | ↑ | TAC | PDE1C null mice | ↓ | ↑cAMP-PKA | (26) | |||||||
| PDE2 | ↑ | ↑ | Human HF, TAC | (42) | |||||||||
| AAC | Ang II-NRCM | BAY 60-7550 | ↓ | ↑NO-GC-cGMP signaling | (43) | ||||||||
| TAC | NE-NRVM | BAY 60-7550 | ↓ | ↑PKA-NFAT phosphorylation | (278) | ||||||||
| ET-1-NRVM | BAY60-7550 | ↓ | ↓ cAMP-PKA-dependent inhibition of PLCε; ↓cGMP-PKG-dependent inhibition of PLCε | (275) | |||||||||
| PDE2A | Male rats per se | ARVMs per se | PDE2A overexpression rats | ↑ | ↓PKA-mediated NFAT phosphorylation | (278) | |||||||
| PDE2A2 | NE/PE-ARVM | PDE2A2 adenovirus | ↓ | ↓β-Adrenergic responses | (42) | ||||||||
| PDE3 | Human HF | (282, 283) | |||||||||||
| ↓ | TAC | (284) | |||||||||||
| ↑ | ↑ | Salt-induced HF rat | (285) | ||||||||||
| ↑ | TAC | Milrinone | ↓ | ↓p38 MAPK, ↓calcineurin-NFAT | (286) | ||||||||
| NRCMs per se | Cilostamide | ↑ | ↑ PLCε at the Golgi through Epac →↑PI4P hydrolysis | (275) | |||||||||
| NRCMs per se | Cilostamide | ↑ | ↑ PKA not anchored to AKAPs | (278) | |||||||||
| PDE3A | ↓ | ↓ | Human DCM and IHD, TAC | (58) | |||||||||
| PDE3A | ↓ | TAC | (284) | ||||||||||
| PDE3A | TAC | PDE3A-KO | ↓ | (286) | |||||||||
| PDE3A2 | NRCMs per se | Inactive mutants of PDE3A2 | ↑ | ↑cAMP at sites where prohypertrophic effectors | (278) | ||||||||
| PDE3B | TAC | PDE3B-KO | (286) | ||||||||||
| PDE4 | ↑ | ↑ | Salt-induced HF rat | (285) | |||||||||
| ↑ | Human HF with diabetes, HFD mice | Roflumilast | ↓ | ↑cAMP-CREB-Sirt1- SERCA2a- miR-1 | (83) | ||||||||
| NRCMs per se | Rolipram | ↑ | ↑Epac-PLCε at the Golgi →↑PI4P hydrolysis | (275) | |||||||||
| NRCMs per se | Rolipram | ↑ | ↑PKA not anchored to AKAPs | (278) | |||||||||
| ISO-NRCMs | PDE4 activator UCR1C | ↓ | ↓Nuclear PKA -CREB | (287) | |||||||||
| PDE4A | ↓ | TAC | (284) | ||||||||||
| PDE4B | ↓ | TAC | (284) | ||||||||||
| ↓ | Human HF | ISO infusion, TAC | PDE4B-TG mice, AAV9-PDE4B | ↓ | Blunts β-adrenergic response | (288) | |||||||
| PDE4D | TAC | (284) | |||||||||||
| ↑ | Human HF with diabetes, HFD mice | AAV9-PDE4D shRNA | Roflumilast | ↓ | ↑cAMP-CREB-Sirt1-SERCA2a- miR-1 | (83) | |||||||
| PDE4D3 | LIF-NRCM | Rolipram | ↓ | ↓mAKAP-PDE4D3-Epac1-PKAcomplex anchored ERK5 activity | (155) | ||||||||
| NRVMs per se | Inactive mutants of PDE4D3 | ↑ | (278) | ||||||||||
| PDE4D5 | ↑ | Human HF with diabetes, HFD mice | NRVM | PDE4D5 adenovirus | ↑ | ↓cAMP-CREB-Sirt1-SERCA2a- miR-1 | (83) | ||||||
| ISO-NRCM | competitive peptide 58-Pept | ↑ | ↓β-arrestin–PDE4D5 interaction→ ↑ Epac1 recruit to β2AR | (163) | |||||||||
| PDE5 | PDE5A | ↑ | HF (DCM, ICM) | MI | PDE5-TG | ↑ | ↓cGMP | (92) | |||||
| TAC | PDE5-TG | ↑ | ↑Natriuretic peptide-derived cGMP hydrolysis | (289) | |||||||||
| ↑ | CHF, TAC | TAC | Sildenafil | ↓ | ↓Oxidative stress | (290) | |||||||
| TAC | Sildenafil | ↓ | ↑cGMP-↓oxidized PKG1α | (291) | |||||||||
| TAC | Sildenafil | ↓ | Improve T-tubule remodeling | (292) | |||||||||
| PE-NRCM | PDE5A-shRNA | Sildenafil | ↓ | ↑PKG | (91) | ||||||||
| TAC | PE-NRCM | Sildenafil | ↓ | ↓Calcineurin/NFAT, ↓PI3K/Akt, ↓ERK1/2 | (293) | ||||||||
| SHR | Sildenafil | ↓ | ↓Na+/H+ exchanger activity | (294) | |||||||||
| TAC | PE-NRCM | CRD-733 | ↓ | ↑cGMP | (295) | ||||||||
| PDE9 | PDE9A | ↑ | Human (DCM, HFpEF), aortic stenosis, TAC | TAC | PE, ET-1-NRCM | PDE9A-KO | PDE9A-siRNA | PF-9613 | ↓ | ↑natriuretic peptide-cGMP | (123) | ||
| ET-1-NRCM | PF04449613 | ↓ | ↓cGMP-PKG-dependent inhibition of PLCε | (275) | |||||||||
| ISO rat | PE, ISO-NRCM | C33(S) | ↓ | ↑cGMP | (296) |
Boldface indicates that the phosphodiesterase (PDE) plays a negative role in hypertrophy. AAC, abdominal aortic constriction; AAV9, adeno-associated viral vector serotype 9; AKAP, A-kinase anchoring protein; Ang II, angiotensin II; ARVM, adult rat ventricular myocyte; cAMP, cyclic adenosine monophosphate, cGMP, cyclic guanosine monophosphate; CHF, congestive heart failure; CREB, cAMP response element binding protein; DCM, dilated cardiomyopathy; Epac, exchange protein activated by cAMP; ERK, extracellular signal-regulated kinase; ET-1, endothelin-1; GC, guanylyl cyclase; HF, heart failure; HFD, high-fat diet; HFpEF, heart failure with preserved ejection fraction; ICM, ischemic cardiomyopathy; IHD, ischemic heart disease; ISO, isoproterenol; KO, knockout; LIF, leukemia inhibitory factor; miR-1, microRNA-1; NE, norepinephrine; NFAT, Nuclear factor of activated T cells; NO, nitric oxide; NRCM, neonatal rat cardiomyocyte; NRVM, neonatal rat ventricular cardiomyocyte; PE, phenylephrine; PI4P, phosphatidylinositol 4-phosphate; PKA, protein kinase A, PKC, protein kinase C; PKG, protein kinase G; PLC, phospholipase C; SERCA2a, sarco(endo)plasmic reticulum calcium ATPase; SHR, spontaneously hypertensive rat; shRNA, short hairpin RNA; siRNA, short interfering RNA; Sirt1, Sirtuin 1; TAC, transverse aortic constriction; TG, transgenic mice.
3.3.1. cAMP-selective PDEs.
PDE4 is known to have multiple isoforms with distinct subcellular localization in cardiomyocytes, resulting in the tight control of cAMP-dependent events within nanodomains. This diversification has led to divergent reports on the expression and function of PDE4 in cardiac hypertrophy and HF. PDE4D isoforms are associated with the cardiac βAR signaling and prevent catecholamine-induced cardiac hypertrophy. PDE4 activation by overexpression of the carboxy-terminal portion of the UCR1 domain attenuates hypertrophy in response to chronic βAR stimulation by specifically inhibiting nuclear PKA activity in cardiomyocytes (287). The PDE4D5 recruitment to β2AR by β-arrestin prevents activation of EPAC1 and CaMKII and the prohypertrophic effects of the receptor stimulation. Inhibition of PDE4D also leads to activation of CaMKII (88), promoting maladaptive gene expression in cardiomyocytes (297). Dissociation of the PDE4D5-β-arrestin2 complex allows the recruitment of Epac1 to β2AR and induces a switch from β2AR nonhypertrophic signaling to a β1AR-like prohypertrophic signaling cascade (163). PDE4 inhibitors also promote cardiac hypertrophy by enhancing a local Golgi β1AR-cAMP-dependent activation of the Epac-PLCε pathway (275). Meanwhile, PDE4D3 is part of a nuclear envelope mAKAPβ complex that contains PKA, Epac, and ERK5 and regulates cardiomyocyte hypertrophy through PKA and ERK5 (46, 155, 156). This local pool of PDE4D3 controls the β1AR-induced cAMP signaling in the nucleus for PKA regulation of hypertrophic and apoptotic gene expression (176, 298). Overexpressing catalytically inactive mutant PDE4D3 results in cardiomyocyte hypertrophy (278). These findings suggest that cardiac PDE4D protects against catecholamine-induced cardiac hypertrophy.
However, the control of cAMP-regulated hypertrophy by PDE4 is more intricate than previously appreciated. For example, in the same PDE4D3-mAKAPβ complex, inhibition of PDE4 with rolipram also blocks ERK5 activation-enhanced cardiomyocyte cell size (155). Moreover, the small heat shock protein 20 (HSP20) sequesters PDE4D isoforms (299). PDE4D regulates a specific cAMP pool that controls the PKA phosphorylation of HSP20, through which the phosphorylated HSP20 protects against hypertrophic response triggered by chronic β-agonist administration (299). Cardiac PDE4D expression is increased in a mouse model of type 1 diabetes (300). PDE4 activity is increased in an animal model of salt-induced HF and contributes to the development and exacerbation of HF (285). Recent studies have observed increased cardiac PDE4D5 expression in high-fat diet-fed mice and human diabetes-associated HF (83, 301). PDE4D inhibition by rAAV9 injection or the pharmacological inhibitor roflumilast shows protection against cardiac hypertrophy and dysfunction through promoting cAMP, cAMP response element binding (CREB), and NAD-dependent deacetylase sirtuin 1 signaling for SERCA2a expression (83). The increased SERCA2a expression, in turn, downregulates microRNA 1 (miR-1) and inhibits miR-1-targeted hypertrophy-associated genes (83). These findings suggest that cardiac PDE4D upregulation can be deleterious.
Moreover, studies show that the expression of cardiac PDE4D isoforms is either increased or decreased based on disease models (83, 284, 301, 302). The expression and activity of PDE4A and PDE4B are reportedly decreased in failing and hypertrophic hearts (169, 284, 288). A recent study has uncovered that moderate overexpression of PDE4B blunts β-adrenergic response and maladaptive remodeling in HF induced by chronic isoprenaline infusion or TAC, whereas a higher-level overexpression of PDE4B leads to maladaptive remodeling (288). The results also support the concept that the effects of cAMP on hypertrophy differ depending on the cAMP location and concentration (155). Overall, these studies report the bidirectional roles of PDE4 inhibitors in the myocardium, indicating that the effects of PDE4 isoforms may depend on their expression level and subcellular localization, as well as different etiologies and the progression stages of HF.
3.3.2. cGMP-selective PDEs.
Numerous PDE isozymes involved in the cGMP-PKG signaling pathways are implicated in the regulation of cardiac hypertrophy (7, 9, 303), in which inhibiting cGMP-hydrolyzing PDEs can benefit individuals with HF. Two stimulatory pathways, NOS and NP, promote cGMP signals (304). In chronic pressure-induced hypertrophy, the NO-sGC-dependent cGMP declines, whereas the NP-derived cGMP rises (305). PDE5A localizes to cardiomyocyte Z bands in the normal heart and preferentially regulates cGMP through adrenergic stimulation of NOS3 (148). PDE5 expression is generally low in the heart with low activity in myocytes (88) but is elevated in ventricular hypertrophy in mice, rats, and humans (89, 284, 285, 288, 301–303). Myocardial oxidative stress increases PDE5 expression in the failing heart, whereas reducing oxidative stress by M40401 treatment attenuates PDE5 expression (303). Moreover, PDE5 is translocated from sarcomeres to a dispersed distribution in TAC hearts (305), which shifts the cGMP hydrolytic activity from the NOS3-sGC pathway to the NP signaling pathway. Studies have supported the protective actions of PDE5 inhibitors on cardiac hypertrophy by promoting cGMP-PKG signaling (285, 302, 303, 306). Suppressing myocyte PKG activity exacerbates pressure-overload remodeling and abrogates the antihypertrophy effects of the PDE5 inhibitor sildenafil (305, 307), highlighting PKG as a critical downstream effector in preventing pathological remodeling. PDE5 inhibition also reduces pathological hypertrophy by activating the regulator of G protein signaling and inhibiting the transient receptor potential canonical channel (282, 283, 306). The antihypertrophy actions of PDE5 inhibitors are also associated with the inhibition of multiple hypertrophy-signaling pathways, including the calcineurin-nuclear factor of activated T cells (NFAT), PI3K-Akt, p38, and ERK1/2 MAPK signaling pathways (285, 286), myocardial Na+/H+ exchanger (308), and PKG-1α oxidation (309). Additionally, sildenafil attenuates TAC-induced cardiac hypertrophy and ameliorates T-tubule remodeling, suggesting another potential mechanism underlying the therapeutic benefits of PDE5 inhibitors in cardiac hypertrophy (310).
PDE9A is expressed in hearts from mice and humans and is upregulated in myocardial hypertrophy and HF (120, 311). In contrast to PDE5 involvement in the NO-stimulated cGMP, PDE9A preferentially regulates the NP-induced cGMP (120). Because of different subcellular distributions, PDE5 and PDE9 target different intracellular cGMP pools in myocytes. In the TAC model of HF, PDE9A deficiency ameliorates cardiac hypertrophy and ventricular function (120). Selective inhibitors such as PF-9613, CRD-733, and C33(S) augment the antihypertrophic and antifibrotic NP-cGMP pathways (312–314) and protect against pathological cardiac hypertrophy responses to sustained neurohormone stimulation and pressure overload (120, 315, 316). Unlike PDE5A, PDE9A inhibition can reverse preestablished cardiac hypertrophy independent of NOS activity (120). Thus, PDE9A inhibitors may have greater effectiveness than PDE5A inhibitors for treating cardiac hypertrophy when NO production is low. Moreover, inhibition of neprilysin augments the levels of NPs when used together with PDE9 inhibitors and may have additional beneficial hemodynamic and renal effects in HF compared with either PDE9 or neprilysin inhibition alone (317).
3.3.3. Dual-Substrate PDEs.
Although PDE1A plays a crucial role in cardiac hypertrophy by regulating cGMP degradation, evidence suggests that PDE1C is also involved in regulating cardiomyocyte hypertrophy by regulating cAMP degradation (23, 26). PDE1A upregulation has been observed in the hearts of various pathological hypertrophy animal models and isolated cardiomyocytes treated with neurohumoral stimuli such as angiotensin II (Ang II) and ISO (23). PDE1 inhibitors or PDE1A short hairpin RNA can rescue the reduced cGMP levels in cardiomyocytes and reverse myocyte hypertrophy, suggesting that the antihypertrophic effects of PDE1A inhibition are mediated through cGMP-PKG signaling (306). Conversely, PDE1C is highly expressed in cardiomyocytes and upregulated in mouse and human failing hearts (26). Deletion of PDE1C significantly alleviates cardiac hypertrophy and dysfunction in mice induced by TAC. The antihypertrophic effects of PDE1C deficiency or inhibition largely depend on cAMP-PKA and PI3K-AKT signaling but are not coupled to cGMP signaling (26). Studies have identified a protein complex linking PDE1C to transient receptor potential canonical channel type 3 (Trpc3) and type A2 adenosine receptors (A2Rs), in which PDE1C inhibition enhances the A2R-cAMP-induced antiapoptotic protection (167). Further investigation into the differential mechanisms of PDE1A and PDE1C in cardiac hypertrophy and HF will aid in developing PDE1 isozyme-selective inhibitors to achieve specific pharmacological effects.
PDE2 is less abundant in cardiomyocytes than in fibroblasts and ECs under normal conditions (44). PDE2 expression and cAMP-hydrolyzing activity significantly increase in left ventricular myocardium from patients with terminal HF and in various experimental animal models. The upregulated PDE2 desensitizes cardiomyocytes against acute βAR stimulation (42, 43). PDE2A is also upregulated in inducible pluripotent stem cell-derived cardiomyocytes from patients with familial hypertrophic cardiomyopathy (304). However, in left ventricle (LV) tissues from aortic stenosis patients with myocardial hypertrophy and preserved cardiac function, PDE2 expression remains normal (42). Whereas β-adrenergic stimulation activates the cAMP-Epac-PLCε pathway for cardiac hypertrophy (275), PDE2 limits cAMP-PKA-dependent inhibition of the PLCε hypertrophic pathway. PDE2 also controls a separate PKG-dependent pathway that indirectly inhibits the PLCε hypertrophic pathway (275). However, overexpressing PDE2A yields inconsistent observations on cardiac hypertrophy. One study shows that overexpression of PDE2A is sufficient to induce cardiomyocyte hypertrophy in vitro and in vivo (278). In another report, PDE2A overexpression blunts norepinephrine-induced cellular hypertrophy with a marked decrease in cAMP levels (42). These conflicting results could be due to differential regulation of cAMP and cGMP and the cross talk between cGMP and cAMP that depends on the cGMP concentrations and various stressors. cGMP, at a low concentration, inhibits PDE2A, thereby increasing a local pool of cAMP. A high cGMP concentration activates PDE2 by binding to the regulatory domain of PDE2, allowing a cGMP-mediated decrease in cAMP signaling (305). Nevertheless, PDE2 inhibitors have shown antihypertrophic effects in the heart. In the pressure-load-induced right or left ventricular hypertrophy models, PDE2 inhibition promotes NO-GC-cGMP signaling and PKG-mediated antihypertrophic effects and improves cardiac function (43, 278, 307). Another study shows that the PDE2 inhibitor significantly reduces TAC-induced cardiac hypertrophy by generating a local pool of cAMP that enhances a subset of PKA type II-mediated phosphorylation of NFAT and prevents the NFAT-mediated hypertrophic gene expression (278). Overall, PDE2A plays a pathophysiological role in heart diseases; inhibition of PDE2 in cardiomyocytes might be beneficial to counteract pathological remodeling induced by pressure overload (43, 278, 307). Further studies are needed to determine whether PDE2 inhibition or activation provides therapeutic effects in HF of various etiologies.
Studies have reported inconsistent PDE3 expression and activity in cardiac hypertrophy and HF (7). Early studies show no change in PDE3 activity in human failing hearts (282, 283). However, others report increased activity and expression of PDE3 in the TAC mouse model of HF, streptozotocin-induced diabetic cardiomyopathy, and salt-induced hypertension, hypertrophy, and HF (98, 285, 286, 300). Additional studies have shown that PDE3 activity and PDE3A expression are decreased in mouse and rat hypertrophic and failing hearts induced by pressure overload (58, 284), in pacing-induced chronic HF dogs (58, 284, 302, 308), and in human HF (302). Moreover, Ang II and ISO induce a sustained reduction of PDE3A expression in cultured rat neonatal cardiomyocytes (58). These discrepancies may be due to differences in species and underlying causes of HF. Nevertheless, PDE3 inhibitors have been shown to induce cardiac hypertrophy by increasing a cAMP pool that promotes the Epac-PLCε-PKC-dependent hypertrophic growth (275). The cAMP generated via inhibition of PDE3 has prohypertrophic effects via activation of a subset of PKA that is not anchored to AKAPs (278). Moreover, PDE3A2 isoform operates in a nuclear nanodomain that involves mothers against decapentaplegic homolog 4 (SMAD4) and histone deacetylase 1 (HDAC-1). Inhibition of PDE3 results in increased HDAC-1 phosphorylation, which reduces its deacetylase activity and increases gene transcription and cardiomyocyte hypertrophic growth (309). PDE3 inhibition also worsens chamber stiffness and increases collagen type I deposition induced by chronic βAR stimulation in rats (310). These mechanisms might contribute to the detrimental effects of long-term PDE3 inhibition, suggesting that increasing PDE3A activity could be a compensatory mechanism for cardiac hypertrophy during the development of HF. PDE3A but not PDE3B ablation protects against TAC-induced adverse ventricular remodeling via reducing p38 MAPK and calcineurin-NFAT activation (286). Moreover, PDE2, PDE3, and PDE4 are localized at distinct subcellular sites and uniquely modulate the cAMP response in intact neonatal rat ventricular myocytes (45, 311). βAR activation and other pathological stimuli can generate spatially distinct pools of cAMP with opposing effects on myocyte size (278).
HF has been associated with changes in expression and localization of PDEs. Yet it remains unclear how one PDE compensates for changes in the activity of another PDE during signaling modulation. Future work will focus on better defining the cAMP-cGMP signaling network and alternations. Additional efforts may be sought to simulate the complex network changes during HF with computation approaches, which may lead to new insights into the pathophysiology of HF and the development of new HF therapies.
3.4. PDE in Ischemic Cardiomyopathy
Compared with hypertrophic cardiomyopathy induced by either hormones or pressure overload, MI presents a fundamentally different etiology in the myocardium. The role of PDEs appears to vary, even opposing, in the pathogenesis of ischemic heart diseases compared with hypertrophic HF. Here, we review the function of PDE isozymes in cell death and ischemic cardiomyopathy (TABLE 3).
Table 3.
The roles of individual PDE isoforms in cardiac ischemia injury
| PDE Family | Isoform | Variants | Model in vivo | Model in vitro | Genetic Intervention in vivo | Genetic Intervention in vitro | Inhibitors Used | Positive ↑Negative ↓ | Signaling Pathway | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| PDE2 | Ex vivo perfused mouse heart-reperfusion injury | BAY 60-7550 | ↓ | ↓Arrhythmic events | (271) | |||||
| PDE2A | Ionomycin-NRCM | PDE2A-siRNA | BAY 60-7550 | ↓ | ↑Drp1 phosphorylation ↑mitochondria dynamics | (48) | ||||
| PDE2A3 | LAD ligation mice-MI | PDE2A3-TG | ↓ | ↑Ca2+ homeostasis ↓contractile dysfunction ↓cardiac arrhythmias | (218) | |||||
| PDE3 | ||||||||||
| NRCM per se | Milrinone, cilostamide | ↑ | ↑ICER | (58) | ||||||
| Ischemia-reperfusion dog | Milrinone, olprinone | ↑ | ↑cAMP-PKA-p38 | (318) | ||||||
| Ischemia-reperfusion rat | Olprinone | ↓ | ↑PI3K-Akt ↓ mPTP | (319) | ||||||
| Ex vivo perfused rabbit heart-reperfusion injury | Cilostazol | ↓ | ↑mitoK(Ca) channels | (320) | ||||||
| Ex vivo perfused rabbit heart-reperfusion injury | Milrinone | No effect | (320) | |||||||
| PDE3A | NRCM per se | Adenovirus- antisense-PDE3A | ↑ | ↑ICER | (58) | |||||
| PDE3A1 | Ang II/ISO-NRCM | Adenovirus- PDE3A1 | ↓ | ↑ICER | (58) | |||||
| Ischemia-reperfusion mice | Cardiac-specific overexpression of PDE3A1 | ↓ | ↓ICER | (321) | ||||||
| PDE3B | Ischemia-reperfusion mice | PDE3B−/− mice | ↓ | ↑Ca2+-activated K+ channels, ↓ROS | (59) | |||||
| PDE4 | ||||||||||
| PDE4B | Ischemia-reperfusion mice | PDE4B−/− mice | ↓ | ↑Cardiac microcirculation ↓inflammation | (312) | |||||
| PDE4D | ||||||||||
| PDE4D3 | LAD ligation mice-MI | PDE4D−/− mice | ↑ | ↑RyR2-phosphorylation ↑Ca2+ leakage | (169) | |||||
| PDE5 | Ischemia-reperfusion rabbit | Sildenafil | ↓ | ↑A(1) adenosine receptor activation | (313) | |||||
| Ex vivo perfused mouse heart-reperfusion injury | Sildenafil | ↓ | ↑Mitochondrial K(ATP) channels | (314) |
Boldface indicates that the phosphodiesterase (PDE) plays a negative role in ischemia injury. cAMP, cyclic adenosine monophosphate, Drp1, dynamin-related protein; ICER, Inducible cAMP early repressor; ISO, isoproterenol; LAD, ligation of left anterior coronary artery; MI, myocardial infarction; mPTP, mitochondrial permeability transition pore; NRCM, neonatal rat cardiomyocyte; p38, p38 mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; PKA, protein kinase A; RyR2, ryanodine receptor 2; TG, transgenic mouse.
3.4.1. cAMP-specific PDEs.
PDE4D3 has been reported to localize within the RyR2 macromolecular complex (169). Deletion of PDE4D leads to spontaneous, age-related cardiomyopathy and exacerbates MI by promoting hyperphosphorylated RyR2 and Ca2+ leakage (169). PDE4 binds to mAKAP to reduce nuclear PKA activity and expression of the proapoptotic factor inducible cAMP early repressor (ICER) (176). Moreover, through the negative feedback between PDE4D and CaMKII (88), inhibition or deletion of PDE4D increases CaMKII activity and ICER expression, thereby promoting myocyte apoptosis. These observations are in contrast to the role of PDE4D in cardiac hypertrophy induced by adrenergic stimulation (176, 298) and high-fat diet feeding (83, 301). Meanwhile, the expression of PDE4B but no other PDE4 subtypes increases in mouse hearts with myocardial infarction-reperfusion (MI/R) injury (312). PDE4B is detected primarily in endothelial and myeloid cells of mouse and human hearts. Deleting PDE4B alleviates infarct size and improves cardiac function after MI/R by suppressing inflammation, enhancing cardiac microcirculation, and protecting against MI/R injury (312). These observations contrast with the findings in mice with moderate overexpression of PDE4B in cardiomyocytes, in which PDE4B blunts β-adrenergic response and maladaptive remodeling in HF induced by chronic isoprenaline infusion or TAC (288). Again, these discrepancies indicate an etiology- and cell context-dependent role of PDE4 isoforms in the pathogenesis of heart diseases.
3.4.2. cGMP-specific PDEs.
Cardiac-specific overexpression of PDE5 does not affect myocardial structure or function at the baseline but significantly exacerbates cardiomyocyte hypertrophy and reduces cardiac function after MI (89). When administered before ischemia and after ischemia, PDE5 inhibitors reduce infarct size and apoptosis, and postreperfusion increases contractile function and survival (313–315). Studies demonstrate that sildenafil has cardioprotective effects against MI and ischemia-reperfusion injury through multiple mechanisms, including increasing NO-cGMP signaling and PKG activation and opening mitochondrial K(ATP) channels and Ca2+-activated potassium channels (313, 314). Clinically, the use of PDE5 inhibitors in type 2 diabetes mellitus (T2DM) is associated with a lower risk of overall morbidity and mortality in those with a history of acute MI (316). Further evidence is required to elucidate the mechanism of PDE5 inhibitor-associated cardioprotection in myocardial ischemia.
3.4.3. Dual-substrate PDEs.
In contrast to the pressure-stress hypertrophy model of HF, PDE2 activation in cardiomyocytes might benefit ischemic HF by improving Ca2+ homeostasis, limiting contractile dysfunction, and preventing cardiac arrhythmias (218, 271). Studies suggest that cardiomyocyte-specific overexpression of PDE2A3 has no significant cardiac hypertrophy but improves contraction force and protects against arrhythmia after MI (218, 271). In mice with myocyte-targeted PDE2A3 overexpression, the resting heart rate is reduced, the catecholamine-stimulated arrhythmia is abated, and the cardiac function after MI is improved over littermate control mice (42, 271). In contrast, reports show that inhibition of PDE2A2 affects mitochondria dynamics and protects from apoptotic cell death by enhancing the cAMP-dependent phosphorylation of dynamin-related protein 1 (48). These studies reveal the complex roles of PDE2A isoforms in heart diseases, including the cyclic nucleotide substrate selectivity, the source and location of the substrate, and the etiologies of cardiac diseases (317). Thus, the role of PDE2 depends very much on the ambient conditions in vivo, which may alter the location and balance between cGMP and cAMP and determine whether blocking or activating PDE2 is beneficial.
A decrease of PDE3A with consequent ICER induction is a critical event in Ang II- and ISO-induced cardiomyocyte apoptosis and may contribute to the development of HF (58). Reduction in PDE3A has been shown to upregulate ICER, a negative regulator of B cell lymphoma 2 (Bcl-2)-associated agonist of cell death and a potent proapoptotic factor in cardiomyocytes (322). A decreased PDE3 activity may be related to the loss of cardiomyocytes and subsequent replacement with fibrous tissue in end-stage HF (323). In contrast, mice with cardiac-specific overexpression of PDE3A1 are protected against myocardial ischemia-reperfusion, with a reduced infarct size associated with decreased expression of ICER, increased expression of Bcl-2, and resistance to cardiomyocyte apoptosis (321, 324). However, studies have also reported beneficial roles of PDE3 inhibition in myocardial ischemia-reperfusion, like those reported in cardiomyopathy induced by pressure overload (286). PDE3 inhibition decreases infarct size in canine hearts after myocardial ischemia-reperfusion via a mechanism dependent on the cAMP-PKA-p38 MAPK pathway (318). PDE3 inhibitors, like olprinone and cilostazol, also protect against ischemia-reperfusion injury by inhibiting mitochondrial permeability transition pores and direct activation of mitochondrial potassium channels (319, 320). Moreover, in a model of acute ischemia-reperfusion, PDE3B−/− mice but not PDE3A−/− mice had reduced infarct size (59). The cardioprotective effects of PDE3B deficiency depend on cAMP-PKA signaling, which potentiates the opening of Ca2+-activated K+ channels and decreases reactive oxygen species (ROS) formation by mitochondrial fractions (59). These reported opposite effects of PDE3 in the myocardium may be multifactorial. For instance, the expression level and activity of PDE3 could be isoform dependent: PDE3A and PDE3B may play opposite roles during ischemia-reperfusion, which may be linked to their differential localization and the control of discrete cAMP pools in cardiomyocytes (59). The function of overexpressing PDE3 may be two phased: moderate PDE3 overexpression is cardioprotective, whereas excessive PDE3 is detrimental to the heart (321). Although cAMP is essential for cardiomyocyte survival, chronic cAMP elevation can lead to proapoptotic and antiapoptotic pathways during cardiac remodeling and dysfunction (273). Similarly, PDE3 inhibitors have been used for acute HF management, but chronic usage has been associated with increased mortality in human patients.
3.5. PDE in the Regulation of Mitochondria and Cardiac Metabolism
Mitochondria are the source of cellular energy production. Mitochondrial dysfunction is associated with the development of numerous cardiac diseases, such as ischemia-reperfusion injury, cardiac hypertrophy, and HF, due to the uncontrolled production of reactive oxygen species (ROS). cAMP is rate limiting for matrix Ca2+ entry via the Epac1-dependent mitochondrial Ca2+ uniporter and, as a result, abrogates mitochondrial permeability transition (325). The latter effects protect against cardiomyocyte cell death and apoptosis (325). Studies have highlighted that PDE2 accounts for the largest mitochondrial cAMP-degrading activity compared with PDE3 and PDE4 in adult rat ventricular myocytes (326). PDE2 is also associated with critical signaling pathways modulating cardiomyocytes’ energetic capacities and apoptosis. In a study on rat cardiomyoblasts, 17β-estradiol reduced mitochondrial cAMP levels via cGMP-mediated stimulation of PDE2, decreasing cytochrome oxidase activity and mitochondrial membrane potential (327). Upon PDE2 inhibition with BAY60-7550, elevated mitochondrial cAMP levels are implicated in increased oxygen consumption, mitochondrial membrane potential, and ATP production (325, 328). Moreover, PDE2 inhibition limits sepsis-induced myocardial dysfunction and improves mitochondrial respiration in septic cardiac fibers (329). Furthermore, PDE2A2 is the only isoenzyme reported to be localized within subsarcolemmal mitochondria associated with the mitochondrial inner membrane and regulates mitochondria dynamics in myocytes (47). PDE2A2 inhibition affects mitochondrial elongation and increases the transmembrane potential and, thereby, resistance to proapoptotic stimuli in neonatal rat ventricular myocytes (48). These changes are mediated by cAMP-PKA-dependent phosphorylation of dynamin-related protein 1, which diminishes mitochondria fission, protecting the cardiomyocytes against apoptotic cell death (48). Mice with cardiomyocyte-specific PDE2 overexpression exhibit faster mitochondrial membrane potential loss and mitochondrial swelling (47). Additionally, PDE4 has been implicated in mitochondria regulation: a potent PDE4 inhibitor activates AMP-activated protein kinase and Sirt1 to induce mitochondrial biogenesis in myotubes (330).
3.6. PDE in Cardiac Fibroblasts
Myocardial fibrosis is a common pathological change associated with cardiac diseases and is characterized by an imbalance of extracellular matrix (ECM) deposition, leading to cardiac stiffness and severe HF. Cardiac fibroblasts, the major cell type in the heart, are the primary source of ECM deposition in cardiac fibrosis. In response to various pathological stresses such as MI and pressure overload, cardiac fibroblasts are stimulated to proliferate and differentiate into myofibroblasts, producing ECM deposition and fibrosis (331). Cyclic nucleotide signaling has antifibrotic properties by negatively regulating pathways associated with myofibroblast transformation, proliferation, and expression of ECM molecules in injured or stressed hearts (332, 333). Evidence has suggested that PDE expression and activity are involved in various cellular processes in myocardial fibrosis in MI and hypertrophic cardiomyopathy (25, 44). Among the PDEs, PDE1, 2, 3, 4, 5, and 10 families have been shown to modulate profibrotic function in cardiac fibroblasts (25, 43, 129). Inhibition of these PDEs attenuates cardiac fibrosis in various heart disease models.
3.6.1. cAMP-specific PDEs.
The expression of PDE4D has recently been observed in cardiac fibroblasts (83). PDE4D5 is upregulated by insulin in cardiac fibroblasts in vitro and in mouse hearts after high-fat diet feeding (83). PDE4D5 overexpression enhances transforming growth factor β1 (TGF-β1) signaling, reducing miR-1 expression and increasing collagen deposition in cardiac fibroblasts and mouse hearts after high-fat diet feeding (83). Both PKA and Epac, the primary cAMP effectors, inhibit the TGF-β1-induced synthesis of collagen and DNA by fibroblasts (334). However, PKA and Epac have opposing effects on fibroblast migration and morphology. PDE4 inhibition promotes fibroblast migration at lower concentrations but decreases migration at higher concentrations, which is probably correlated to those elicited by activation of Epac1 and PKA, respectively (334). These studies imply that PDE4 inhibition might have dose differences in its effect on cardiac fibroblast proliferation and migration.
3.6.2. cGMP-specific PDEs.
PDE5 has been identified in mouse cardiac myofibroblasts, and PDE5 activity contributes to approximately half of the total cGMP-hydrolyzing activity of myofibroblasts (97). Inhibition of PDE5 with sildenafil significantly reduces ISO-, Ang II-, and TAC-induced fibrosis in mouse hearts (100, 310, 335). Inhibition of PDE5 attenuates cardiac fibrosis by reducing CREB binding protein 1 recruitment to Smad transcriptional complexes by activating CREB in cardiac fibroblasts (100). Another study reveals that PDE5A inhibition with adenoviral short hairpin RNA partially reduces fibrosis in infarcted hearts through activation of the Akt signaling pathway and reduction of inflammatory cytokines (336). The mechanisms underlying the protective effects of PDE5 inhibition on cardiac fibrosis merit further investigation.
3.6.3. Dual-substrate PDEs.
The Ca2+-CaM-stimulated PDE1A isoform is the main cAMP-hydrolyzing enzyme expressed in cardiac myofibroblasts from mouse, rat, and human fibrotic hearts. PDE1A is highly induced in cardiac fibroblasts activated by profibrotic stimuli such as TGF-β and Ang II and in vivo within fibrotic regions of rodent and human failing hearts (25). Inhibiting PDE1A with a PDE1-selective inhibitor or short hairpin RNA reduces myofibroblast activation in vitro and attenuates interstitial fibrosis in mouse hearts (25). Mechanistic studies reveal that PDE1A controls unique spatial and temporal cAMP and cGMP dynamics, predominantly in perinuclear and nuclear regions of cardiac fibroblasts, which may be implicated in the differential regulation of stress-responsive genes (25). Both cAMP-EPAC and cGMP-PKG are responsible for PDE1A-mediated nuclear fibrotic gene activation (25). Targeting PDE1 with vinpocetine inhibits pathological cardiac fibrosis in mice in response to chronic Ang II infusion (281). Conversely, PDE1C expression is upregulated in mouse and failing human hearts, and it is highly expressed in cardiomyocytes but not in fibroblasts. Deletion of PDE1C significantly attenuates cardiac remodeling and dysfunction induced by TAC, including myocardial hypertrophy and cardiac fibrosis (26). Studies have found that conditioned medium taken from PDE1C-deficient cardiomyocytes can partially attenuate TGF-β-stimulated cardiac fibroblast activation, suggesting that PDE1C in myocytes likely regulates secreted factors that are important for fibroblast activation (26). PDE1 inhibition may serve as an effective strategy to alleviate fibrosis and cardiac dysfunction.
PDE2, a dual-substrate PDE that hydrolyzes both cAMP and cGMP, is abundantly expressed in neonatal rat cardiac fibroblasts (44). Overexpression of PDE2 in cardiac fibroblasts enhances basal cAMP degradation, induces fibroblast conversion to myofibroblast even in the absence of exogenous profibrotic stimuli, and drives the stiffness of fibroblast-derived connective tissues (44). PDE2 suppresses the increase in cAMP in response to ISO in cardiac fibroblasts and promotes myofibroblast formation and fibrosis (44, 337). Meanwhile, cardiac fibroblasts can produce large quantities of NO in response to interleukin-1β (IL-1β) stimulation and the consequent induction of inducible NOS2, which stimulates the NO-sGC-cGMP cascade to activate PDE2 and depress cAMP accumulation (337). A selective pharmacological inhibitor of PDE2, BAY 60-7550, reverses the development of cardiac fibrosis induced by pressure overload (43). Despite persistently depressed cAMP levels, ANP- and sodium nitroprusside-mediated cGMP synthesis completely prevents PDE2 overexpression-induced fibroblast conversion (44). Furthermore, parallel studies in animals deficient in either NO-sensitive sGC-1α (sGC-1α−/−) or natriuretic peptide-responsive GC-A (GC-A−/−) show that the beneficial effects of PDE2 inhibition are maintained in GC-A−/− mice but absent in sGC-1α−/− mice. These data indicate that PDE2 inhibition promotes NO-sGC-cGMP signaling to protect cardiac structure and function (43).
The PDE3 inhibitor milrinone attenuates TAC-induced hypertrophy and interstitial fibrosis, macrophage infiltration, and activation of p38 MAP kinase in mice (286). Whole body genetic deletion of PDE3A, but not PDE3B, provides similar protection against TAC-induced adverse ventricular remodeling (286). Milrinone does not provide additional protection to PDE3A−/− mice, suggesting that PDE3A contributes to cardiac fibrosis induced by pressure overload (286). Moreover, milrinone suppresses Ang II-induced transactivation of fibroblasts to myofibroblasts, consistent with reduced ventricular fibrosis in TAC mice with PDE3 blockade and PDE3A ablation. Cilostazol, a selective PDE3 inhibitor with antiplatelet, antimitogenic, and vasodilating properties, also attenuates Ang II-induced cardiac fibrosis in mice by activating the cAMP-PKA pathway (338).
PDE10A, another dual cAMP and cGMP esterase, is markedly upregulated in failing hearts (129). PDE10A deficiency or inhibition of PDE10A with selective inhibitor TP-10 attenuates TAC or Ang II infusion-induced cardiac fibrosis (129). TP-10 treatment elevates cAMP and cGMP levels and reduces TGF-β-stimulated cardiac fibroblast proliferation, migration, and ECM synthesis (129).
Collectively, pharmacological inhibition of PDEs such as PDE1, PDE2, PDE3A, PDE4D, PDE5, and PDE10A is currently being considered as a potential therapeutic approach for the treatment of heart pathologies associated with cardiac fibrosis.
3.7. PDE in Immune Cells and Inflammation
Growing evidence implies that inflammation contributes to cardiovascular diseases, including atherosclerosis, myocardial infarction, and HF. Patients with decompensated HF show marked systemic inflammation and increased production of oxygen free radicals. Studies have shown that the acute inflammatory response induced by the innate immune system is required for tissue repair during injury and is cardioprotective (339). However, sustained activation of inflammatory signaling contributes to the process of LV remodeling, including cardiomyocyte hypertrophy, activation of collagenolytic matrix metalloproteinases, myocardial fibrosis, and progressive myocyte apoptosis (340). Various types of immune cells are implicated in myocardial inflammation, including neutrophils, macrophages, eosinophils, mast cells, natural killer cells, T cells, and B cells. Immune cells coordinate the responses of cardiomyocytes and noncardiomyocytes during maladaptive remodeling (339). Immune cells modulate not only cardiomyocyte function directly but also injury responses involving scar formation and interstitial fibrosis.
Targeting PDEs with small-molecule inhibitors is a promising therapeutic for chronic inflammatory disorders. Despite extensive knowledge of PDE expression and regulation in immune cells such as macrophages and T cells (341), research into the role of PDEs in cardiac inflammatory responses is limited.
PDE4B is expressed in macrophages (312). PDE4B inhibition or deletion suppresses inflammation, enhances cardiac microcirculation, and protects against MI/R injury. Mechanistically, PDE4B facilitates neutrophil-EC interaction by promoting the release of proinflammatory cytokines and PKA-mediated cell adhesion molecule expression (312). Similarly, inhibition of PDE4 protects against doxorubicin-induced cardiomyopathy in rats by reducing oxidative stress-mediated inflammatory reactions (342, 343). Although the PDE7 inhibitor BRL50481 has only a modest inhibitory effect on proinflammatory cells, it acts additively with other cAMP-elevating drugs, such as PDE4 inhibitors, especially when the PDE7A1 isoform is upregulated (344). It is hypothesized that dual inhibitors of PDE4 and PDE7 may offer a therapeutic choice in HF with inflammation and exhibit a lower propensity to cause adverse side effects by PDE4 inhibitors.
PDE5 inhibitor rescues left ventricular dysfunction and cardiac remodeling in Ang II-induced HF accompanied by reducing inflammatory immune response (345). PDE5A knockout suppresses inflammation by downregulating adhesion molecules in cardiac rupture following MI (346). Treatment with the PDE5 inhibitor tadalafil ameliorates circulating inflammatory cytokines and chemokines in a diabetic animal model while improving fasting glucose levels and reducing infarct size after ischemia-reperfusion injury in the heart (347). Patients with both hyperglycemia and inflammation have poor outcomes. Sildenafil reduces C-X-C motif chemokine ligand 10 levels in human cardiomyocytes and T2DM subjects (348). Moreover, in a clinical trial of diabetic cardiomyopathy, tadalafil reduces low-grade chronic inflammation and improves cardiac function (349). These studies underscore the possibility that PDE5 inhibitors can be developed as a pharmacological tool to control inflammation in diabetic cardiomyopathy.
4. PDEs IN VASCULAR BIOLOGY AND DISEASES
4.1. Vascular Structure and Function
Blood vessels (arteries, veins, and capillaries) are vital in maintaining blood circulation by delivering oxygen and nutrients to and removing wastes from the body. Blood vessels are categorized into different types in size, structure, and function. Malfunctions of different blood vessels are associated with distinct vascular diseases. Arteries, including elastic arteries, muscular arteries, and arterioles, carry oxygenated blood away from the heart to various organs and tissues. The size and wall thickness of the arteries decrease as their distance from the heart increases. The elastic arteries have the thickest walls and withstand the highest pressure of blood pumped out by the heart. Vessel wall degeneration of elastic arteries causes dissections and aneurysms (350). Atherosclerosis, characterized by plaque buildup in the vessel wall, primarily occurs in the middle- to large-sized arteries (351). Hypertension (high blood pressure) is often related to abnormalities in small arteries, particularly arterioles between arteries and capillaries (352). Veins usually have thinner walls and carry deoxygenated blood with low pressure from tissues to the heart. Capillaries, the smallest vessels in the body, mediate the gas and nutrient exchange between the blood and tissues. This review mainly focuses on vascular diseases associated with arteries (FIGURE 8).
FIGURE 8.
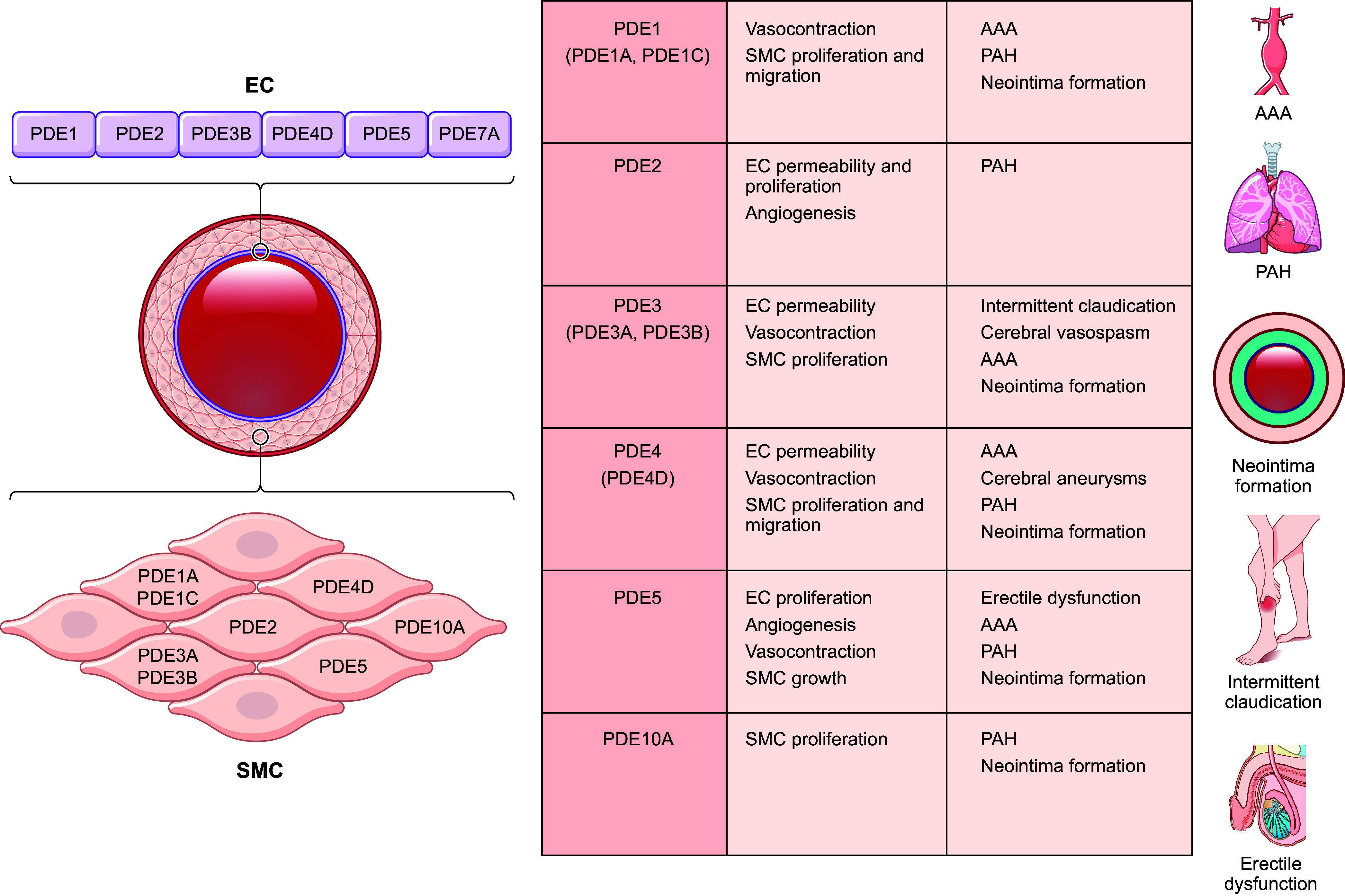
Schematic illustration of the expression and function of phosphodiesterases (PDEs) in the arterial vessel and their involvement in vascular diseases. AAA, abdominal aortic aneurysm; EC, endothelial cell; PAH, pulmonary arterial hypertension; SMC, smooth muscle cell.
Arteries have three layers containing different cell types and performing distinct functions. The innermost layer, also known as intima, is a single-cell layer with ECs that line the surface of blood vessels. The endothelial layer is crucial in sensing biological and mechanical signals in blood and regulating vascular reactivity and permeability. The middle layer, also known as media, mainly contains SMCs and matrixes such as elastin and collagen. SMCs are critical in vasoconstriction or vasodilation. The outer layer, also known as adventitia, is important in maintaining the integrity and elasticity of the arterial wall. Adventitia mainly comprises fibroblasts and connective tissues (such as collagen), and fibroblasts are responsible for producing connective tissue.
4.2. PDE in Vascular Reactivity and Hypertensive Diseases
In response to various biological and environmental factors, blood vessels can change their diameter, which is essential for regulating blood flow to distribute nutrients and oxygen throughout the body and maintaining blood pressure. The cellular and molecular mechanisms underlying vascular reactivity regulation are complex, involving multiple pathways including cyclic nucleotide signaling. Both ECs and SMCs play crucial roles in regulating vascular reactivity. ECs regulate vascular tone by releasing various substances, including vasodilators, such as NO and prostacyclin (PGI2), and vasoconstrictors, such as endothelin (ET) and thromboxane. SMC contraction and relaxation directly control the vessel diameter, which can be regulated by ICa levels. Increased Ca2+ activates myosin light chain kinase (MLCK) that phosphorylates the myosin light chains (MLC) of the myosin molecules, which triggers myosin binding to actin filaments and SMC contraction (353). cAMP and cGMP are key mediators of SMC relaxation. PGI2 can induce cAMP elevation and activate PKA in SMCs, which leads to SMC relaxation by PKA-mediated phosphorylation and activation of MLC phosphatase (MLCP) that dephosphorylates MLC (354, 355). PKA can also increase the activity of the potassium channels to hyperpolarize SMCs, which indirectly reduces Ca2+ entry into SMCs (356, 357). However, a series of recent studies show that glucose can promote ATP-dependent activation of purinergic receptor 11 to induce a discrete pool of cAMP, which can promote PKA activation of LTCC to increase SMC constriction (358–360). Conversely, NO can induce cGMP elevation and activate PKG in SMCs, leading to SMC relaxation via different mechanisms (361), e.g., PKG activates MLCP, like PKA (362); PKG phosphorylates inositol 1,4,5-trisphosphate (IP3) receptors and decreases Ca2+ release from the sarcoplasmic reticulum (363); and PKG-mediated phosphorylation and activation of potassium channels hyperpolarizes SMCs, which indirectly reduces Ca2+ entry into SMCs (357, 364). Thus, through modulating cAMP and cGMP signaling, PDEs play critical roles in regulating SMC contractility. Altering PDE expression and activity may contribute to disorders related to vascular reactivity dysregulation, such as hypertension and erectile dysfunction. This section focuses on the roles of PDE1, 3, 4, and 5 in vascular smooth muscle reactivity (FIGURE 9).
FIGURE 9.

Schematic illustration of phosphodiesterases (PDEs) in smooth muscle cells. The cartoon highlights the regulation of cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) by PDEs in smooth muscle cells to control vessel constriction and proliferation. CaM, calmodulin; GPCR, G protein-coupled receptor; NO, nitric oxide; NP, natriuretic peptide; sGC, soluble guanylyl cyclase.
4.2.1. Dual-substrate PDEs.
In the vasculature, PDE1 is primarily expressed in SMCs but not ECs (365, 366) and regulates vascular reactivity and blood pressure. PDE1A is detected in medial SMCs in many species (18, 33, 367). Human genome-wide association studies (GWAS) show that PDE1A is associated with blood pressure dysregulation (368, 369). In a rat nitrate tolerance model induced by NO donor nitroglycerin infusion, PDE1A expression and activity increase in the aortas (370). Inhibiting PDE1 by vinpocetine partially restores the vasodilatory sensitivity of tolerant vessels to subsequent nitroglycerin exposure. Another PDE1 inhibitor, IC86340, also reduces basal blood pressure in mice (23). Pde1a-null mice had lower aortic blood pressure (371). PDE1B is only reported in SMCs from monkeys and baboons (367, 372), and the expression of PDE1B in contractile medial SMCs is negligible (372, 373). PDE1C expression is almost undetectable in normal medial SMCs (372, 373), which aligns with the fact that PDE1C deficiency in mice does not affect blood pressure (373). These observations suggest that the vasodilatory effect of PDE1 inhibition is primarily through inhibiting PDE1A in SMCs. PDE1A hydrolyzes cGMP with much higher affinity than it hydrolyzes cAMP (18, 28). PDE1 inhibition with vinpocetine increases cGMP levels, accompanied by dilating ex vivo rabbit and rat aortas (370, 374–377). Thus, the effect of PDE1A on vascular relaxation is likely mediated by cGMP-PKG signaling. Vasoconstrictors, such as Ang II, increase PDE1 activity via Ca2+ elevation (34). Ang II decreases cGMP levels (370, 378, 379), and PDE1 inhibition reverses the effect of Ang II on cGMP (370). These observations suggest that PDE1 is a critical mediator in the counterbalance between Ca2+ signaling (vasoconstriction) and cGMP signaling (vasorelaxation).
PDE3A is the major PDE expressed in contractile SMCs and regulates vascular contraction (380). PDE3 inhibitors effectively promote SMC relaxation in various vascular preparations (381), which appears to be independent of endothelium function (382). The drug milrinone, a PDE3 inhibitor, shows powerful vasodilatory effects by reducing total peripheral resistance, enhancing coronary blood flow, and reducing pulmonary vascular resistance in humans (381). Besides the cardiac inotropic effect, the vasodilatory effect significantly contributes to the efficacy of milrinone in the acute treatment of congestive HF. The drug cilostazol, another PDE3 inhibitor, has vasodilatory and antithrombotic properties and is an effective drug for treating chronic peripheral arterial occlusions. These pieces of clinical evidence indicate that PDE3 is important in regulating human vascular reactivity. Both PDE3A and 3B are detected in vascular SMCs (383, 384), with PDE3A predominant in regulating vascular reactivity. For example, PDE3A variants and mutations are linked to autosomal dominant hypertension and brachydactyly syndrome in patients with ischemic stroke caused by spontaneous intracranial artery dissection (385, 386). Rats carrying the human PDE3A mutant with a CRISPR/Cas9 approach recapitulate the vascular phenotypes of human patients (230). Mice with SMC-specific overexpression of mutant PDE3A have increased blood pressure (387). PDE3 inhibition relaxes vascular smooth muscle primarily by attenuating cAMP hydrolysis, thus enhancing the cAMP signaling pathway. PDE3 inhibitors have shown synergistic effects on vascular relaxation with AC activators such as forskolin (388, 389) and ISO (390, 391), which is likely attributable to PDE3A activation by PKA-mediated phosphorylation (392). In addition to posttranslational modifications, PDE3A activity or expression can be regulated by cGMP, which competitively inhibits cAMP hydrolysis by PDE3 (18, 28). Consequently, an increase in cGMP levels leads to a subsequent elevation in cAMP levels. In addition, a recent study reported that the genetic deletion of sGC-1 in vascular SMCs reduces PDE3A expression and activity (393), suggesting a different mechanism by which NO-GC-cGMP signaling negatively regulates PDE3A-modulated cAMP signaling in vascular SMCs.
4.2.2. cAMP-specific PDE4.
Several PDE4 inhibitors are medications used for various medical conditions, including inflammatory diseases like chronic obstructive pulmonary disease, asthma, and psoriasis. These medications moderately affect blood pressure because of their vasodilatory properties (394), indicating the role of PDE4 in regulating vasoreactivity in humans. The effect of PDE4 inhibitors on vasodilation is also seen in rodents (395, 396). The vasorelaxant effect of PDE4 inhibition is largely contributed by PDE4D inhibition in SMCs because SMC-specific PDE4D knockout has an effect similar to PDE4 inhibition on mouse aortic contractility and blood pressure (79). Interestingly, PDE4 inhibitors relax rat aortic rings much better in the presence of a functional endothelium (382). The relaxation in the presence of endothelium is inhibited by the NOS inhibitor NG-monomethyl l-arginine (l-NMMA) and restored by the NOS substrate l-arginine, suggesting that NO is necessary for the relaxing effect of PDE4 inhibitors (382). The effect of NO on potentiating vasorelaxation by PDE4 inhibitors may be mediated by cGMP inhibition of PDE3, stimulation of cAMP-PKA signaling, and PKA phosphorylation and activation of PDE4 (397). The interaction of PDE3 and PDE4 is further supported by the fact that the combination of PDE4 and PDE3 inhibitors produces synergistic effects on SMCs (398). The effects of PDE3 and PDE4 on SMC contractility vary among different vessels. For example, the PDE4 inhibitor alone has relatively poor relaxation effects in large isolated arteries compared with the PDE3 inhibitor (381). However, PDE4 is much better at regulating vascular tone in cerebral vessels. In a canine model of acute cerebral vasospasm, PDE4 inhibitors such as denbufylline and rolipram reverse the basilar artery spasm produced by autologous blood without significantly altering mean arterial blood pressure (399). Consistently, the PDE4D gene variant is a risk factor for ischemic stroke (400). In contrast, siguazodan (a PDE3 inhibitor) produces only weak relaxation of the basilar artery (399). PDE3 inhibitors such as milrinone and amrinone are more potent relaxants of coronary arteries than of cerebral or renal arteries in dogs (401). The differences in PDE3 and PDE4 inhibition on vascular reactivity in different vessels might be due to the differential expression of PDE3 and PDE4 or the distinct endogenous stimulators of AC-cAMP or GC-cGMP signaling in different vascular beds.
4.2.3. cGMP-specific PDE5.
PDE5 is present in almost all types of vascular SMCs. The vasorelaxant effects of PDE5 inhibitors vary depending on the vascular bed, possibly due to differences in local NO release and the relative contribution of PDE5 to the total amount of cGMP-hydrolyzing activity in the vascular beds. PDE5 is highly expressed in the lung and penile corpus cavernosum (402). Thus, PDE5 inhibitors have profound effects on pulmonary artery relaxation and are promising long-term therapy for treating pulmonary hypertension (PH) (403, 404). The PDE5 inhibitor sildenafil has been successfully used to treat erectile dysfunction by reinstating the impaired relaxation of SMCs within the corpus cavernosum of penile vasculature (405). It is known that the erectile response is mediated by corpus cavernosum vascular relaxation, which depends on NO release from ECs and cavernous nonadrenergic and noncholinergic (NANC) nerves in response to sexual stimulation. PDE5 inhibition facilitates a higher and sustained accumulation of cGMP in response to NO, thus enhancing the erectile action (406, 407). PDE5 has low intrinsic catalytic activity when the cGMP level is low, but PDE5 is highly activated by cGMP binding to one of the two amino-terminal GAF domains and PKG-mediated phosphorylation of the regulatory domain of PDE5 when cGMP level is elevated (291). Therefore, PDE5 inhibitors have clinically insignificant effects on systemic blood pressure and minimal effects on erection when used alone. Sexual stimulation (increasing NO-cGMP signaling) is necessary in treating erectile dysfunction with PDE5 inhibitors. Concomitant use of PDE5 inhibitors and nitroglycerin or other organic nitrates may lead to severe hypotension (408).
4.3. PDE in Pathological Vascular Remodeling and Diseases
Pathological vascular remodeling is a process of structural alterations in the vessel wall in response to abnormal mechanical or biological perturbations, which involves cell growth, death, migration, and extracellular matrix modulation (409–411). Pathological vascular remodeling contributes to a wide variety of circulatory system disorders, including coronary artery disease (CAD), peripheral artery disease (PVD), hypertension, aortic aneurysm (AA), and pulmonary arterial hypertension (PAH). An intact and healthy endothelium is pivotal in maintaining vascular integrity through several essential functions, including regulating vascular tone, managing vascular permeability, inhibiting platelet aggregation, preventing leukocyte adhesion and activation, and repressing vascular SMC dedifferentiation. The endothelium resides on the surface of the vessel wall and is most susceptible to a wide range of pathological stimuli and risk factors, including, but not limited to, mechanical stress, smoking, aging, hyperlipidemia, hyperhomocysteinemia, hyperglycemia, obesity, and diabetes (411, 412). Endothelium dysfunction or injury contributes to pathological vascular remodeling and vascular diseases. In a normal vessel wall, SMCs are the prominent cell type of the medial layer. These SMCs express large amounts of myofilament proteins and mainly serve to control contractility and thus are referred to as “contractile” SMCs. However, SMCs have high heterogeneity and plasticity. Upon stimulation by growth factors or inflammatory molecules, medial SMCs can undergo phenotypic modulation (dedifferentiation, e.g., from a quiescent contractile phenotype to an active myofibroblast-like phenotype, frequently referred to as a “synthetic phenotype”) (413–415). Synthetic SMCs are proproliferative and promigrative, contributing to lesion tissue growth. Synthetic SMCs are prone to senescence or apoptosis, leading to ROS production, stress-induced medial degeneration, and lesion rupture. Synthetic SMCs produce proinflammatory mediators, providing an inflammatory microenvironment for leukocyte accumulation and activation. Thus, SMC phenotype switch and aberrant activation play critical roles in pathological vascular remodeling (413–416). This section mainly focuses on the roles of PDE1, 3, 4, and 5 in vascular SMCs that contribute to pathological vascular remodeling (TABLE 4).
Table 4.
The roles of individual PDE isoforms in vascular muscle cell proliferation
| PDE Family | Isoform | Model in vivo | Model in vitro | Genetic Intervention in vivo | Genetic Intervention in vitro | Inhibitors | Inhibitory Effect (↓) | Signaling Pathway | References |
|---|---|---|---|---|---|---|---|---|---|
| PDE1 | |||||||||
| PDE1A | Rat aortic VSMCs | PDE1A shRNA | IC86340 | ↓ | ↑PDE1A nuclear translocation→↓cGMP→↑cyclin D1,↓p27Kip1,↓p53 ↓apoptosis | (29) | |||
| Rat aortic VSMCs | IC86340 | ↓ | ↑nuclear PP2A-GSK3β-β-catenin →↓ β-catenin stability →↓TCF-dependent gene transcription | (417) | |||||
| PDE1C | Left common carotid artery ligation | Mouse SMCs | PDE1C−/− mice | PDE1C−/− SMCs | IC86340 | ↓ | ↑cAMP-PKA→↑LRP1 phosphorylation →↑PDGFRβ degradation | (373) | |
| Human saphenous vein explants ex vivo | Rat aortic VSMCs | IC86340 | ↓ | ↓cAMP→↓ lysosome-mediated collagen I protein degradation | (414) | ||||
| PDE3 | FCS/PDGF-pig aortic SMC | SK&F 94836 | ↓ | ↑cAMP →↓DNA synthesis | (418) | ||||
| FCS/PDGF/IGF-1-rat aortic VSMCs | Cilostazol | ↓ | ↑cAMP →↓DNA synthesis | (419) | |||||
| Common carotid artery balloon injury rat | FCS/growth factors-rat carotid VSMC | Cilostamide | ↓ | ↑cAMP →↓DNA synthesis | (420) | ||||
| PDE4 | FCS/PDGF-pig aortic SMC | Rolipram | ↓ | Combine with PDE3 inhibitor →↑cAMP →↓DNA synthesis | (418) | ||||
| PDE5 | Ang II-rat aortic VSMC | Sildenafil, JNJ-10258859 | ↓ | ↑cGMP | (421) | ||||
| PDGF-bovine coronary artery SMC | Sildenafil | ↓ | ↑cGMP→↓PDE3→↑cAMP →↓DNA synthesis | (422) | |||||
| Human pulmonary artery SMC | Sildenafil | ↓ | ↑cGMP →↓DNA synthesis | (423) | |||||
| PDE10 | PDE10A | Left femoral artery wire injury | PDGF-rat aortic VSMCs | PDE10A-KO | PDE10A shRNAs | MP-10 (Mardepodect, PF-2545920) | ↓ | ↓CNP/NPR2/cGMP/PKG1α | (424) |
cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; CNP, C-natriuretic peptide; FCS, fetal calf serum; GSK3β, glycogen synthase kinase 3β; IGF-I, insulin-like growth factor-I; KO, knockout; LRP1, low-density lipoprotein receptor-related protein-1; NPR2, natriuretic peptide receptor 2; PDE, phosphodiesterase; PDGF, platelet-derived growth factor; PDGFRβ, PDGF receptor β; PKA, protein kinase A; PKGIα, cGMP-dependent protein kinase Iα; shRNA, short hairpin RNA; SMC, smooth muscle cell; TCF, T-cell factor; VSMC, vascular smooth muscle cell.
4.3.1. cAMP-specific PDE4.
PDE4 inhibition reduces neointima formation and inhibits VCAM-1 expression and histone methylation in an Epac-dependent manner (425). The specific PDE4 isozyme contributing to neointima formation remains unknown. In human and mouse abdominal aortic aneurysm (AAA), PDE4B is upregulated primarily in inflammatory cells (426). The pan-PDE4 inhibitor rolipram decreases the incidence, aortic diameter, and rupture of AAA in mice, accompanied by reduced immune cell infiltration (426). Another pan-PDE4 inhibitor, roflumilast, also elicits a protective effect against mouse AAA development (427). PDE4D expression is upregulated in SMCs of AAA tissues (427). SMC-specific PDE4D deficiency decreases AAA in mice (427). PDE4D deficiency or inhibition attenuates SMC apoptosis in a PKA-dependent manner (427). These findings suggest that multiple PDE4 isozymes in different cell types contribute to AAA development. Meanwhile, a PDE4 inhibitor, ibudilast, inhibits cerebral aneurysms by downregulating inflammation-related molecules in the vascular wall (428). PDE4B activation exacerbates PH induced by intermittent hypoxia by enhancing mitochondrial injury and reducing cAMP-PKA-pCREB-peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC-1α) signaling (429). Interestingly, the combination of PDE4 inhibitors with other cAMP-elevating agents has synergistic effects on SMC proliferation (418) and migration (398). For example, an augmented effect on attenuating SMC migration is reported when inhibiting PDE3 and PDE4 together. Inhibiting PDE3 and PDE4 activities also shows significantly improved pulmonary artery relaxation in hypoxia-induced PH (395, 430, 431). Thus, combined PDE3 and PDE4 inhibition should also be tested experimentally in other animal disease models, such as vascular hyperplasia and aortic aneurysms.
4.3.2. cGMP-specific PDE5.
PDE5 is a critical negative regulator of NO-cGMP signaling. Thus, PDE5 is important for various vascular disorders associated with endothelial dysfunctions. There is evidence that PDE5 inhibition potentiates NO-mediated signaling and improves EC function (432). PDE5 inhibitors can restore EC-dependent NO bioavailability and improve the EC function in various vascular disease models, including apolipoprotein knockout (ApoE−/−) mice with atherosclerosis (433–435). Whereas PDE5 expression is high in contractile SMCs and reduced in synthetic SMCs, its expression can be upregulated by various pathological stimuli in synthetic SMCs and regulates the synthetic SMC functions. For example, Ang II upregulates PDE5 expression in growing rat aortic SMCs, and PDE5 inhibition reverses the Ang II-mediated inhibition of cGMP signaling and SMC growth (421). ROS generated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase upregulated PDE5 in human vascular SMCs (436). Sildenafil shows synergistic antiproliferative effects in the presence of NO donors and the GC activator BAY41-2272 in human pulmonary artery cells (423). In bovine coronary artery SMCs, sildenafil acts synergistically with NO donors to reduce platelet-derived growth factor (PDGF)-induced DNA synthesis and cell growth (422). Additionally, decreased NO bioavailability also affects platelet aggregation and vascular wall inflammation (437), which may contribute to vascular diseases and are potentially regulated by PDE5. In animal models, vardenafil attenuated rat carotid artery neointimal hyperplasia induced by endarterectomy that denuded the endothelium (438). Tadalafil inhibited rabbit carotid artery neointimal formation (439). Notably, controversial results are also reported. For example, sildenafil has no protective effects on vascular remodeling in a mouse carotid artery ligation model (440). The discrepancy may be related to the differences in species and injury models with or without EC denudation. Although most studies indicate that increased PDE5 is detrimental to normal vascular function, PDE5A level is lower in the aorta from Marfan, tricuspid, and bicuspid thoracic aneurysms, compared with healthy aorta (441, 442). Many clinical cases have reported aortic dissection after use of PDE5 inhibitor sildenafil or tadalafil (443–445). In a mouse AAA model induced by periaortic elastase treatment, PDE5A expression is significantly reduced in medial SMCs (442). Sildenafil treatment aggravated elastase-induced mouse AAA. These case reports and experimental data strongly suggest a relationship between PDE5 reduction and aortic degenerative diseases. There are many more studies focused on PDE5 in the lung vasculature because of its vasodilating and antiproliferative abilities. In human pulmonary artery SMCs (PASMCs), PDE5 accounts for 80% of cGMP hydrolysis. The inhibition of PDE5 with sildenafil elevates cGMP and attenuates pulmonary artery SMC growth (423, 446). Chronic hypoxia exposure increases PDE5 activity in the first branch and intrapulmonary arteries for cGMP hydrolysis in a rat model of PH (431). PDE5 inhibitors elicit extensive vasorelaxant effects on pulmonary vasculature (404). PDE5 inhibitors are thus a promising long-term therapy for treating PAH (403, 404). In clinical studies of humans with severe primary pulmonary hypertension, oral administration of sildenafil shows a drastic decrease in pulmonary arterial pressure and reduces pulmonary vascular resistance (403, 404). Thus, PDE5 inhibitors, such as sildenafil and tadalafil, are drugs used for treating human pulmonary arterial hypertension (447, 448).
4.3.3. Dual-substrate PDEs.
PDE1A has been found in both contractile and synthetic SMCs (29). PDE1A is localized in the cytoplasm of contractile SMCs, which is primarily responsible for the regulation of SMC contractility (29). However, PDE1A is localized in the nucleus of synthetic SMCs in culture or neointimal lesions. The nuclear PDE1A is critical for synthetic SMC growth and survival (29). The differences between cytoplasmic and nuclear PDE1A and how PDE1A is translocated from the cytoplasm to the nucleus remain unknown. PDE1A promotes SMC proliferation in culture by increasing the protein stability of nuclear β-catenin, a transcription modulator of cell proliferation (417). These findings are consistent with the GWAS showing a significant association of PDE1A single-nucleotide polymorphisms with carotid intima-media thickness (368). However, experimental evidence that directly demonstrates the role of PDE1A in vascular remodeling and diseases in animal models in vivo is still lacking. Unlike PDE1A, PDE1C is barely detected in contractile SMCs. PDE1C expression is drastically elevated in growing SMCs in vitro. PDE1C is highly expressed in SMCs of neointimal and aneurysmal lesions of diseased vessels from mice and humans (372, 373, 449). PDE1C deficiency significantly attenuates SMC growth and migration in vitro (450), hallmarks of vascular hyperplasia remodeling. Indeed, injury-induced neointima formation is significantly reduced in global PDE1C-knockout mice (373). A potential mechanism is that PDE1C interacts with low-density lipoprotein receptor-related protein-1 (LRP1) and platelet-derived growth factor receptor beta (PDGFRβ), thus regulating PDGFRβ endocytosis and lysosome-dependent degradation in a cAMP-PKA-dependent manner. PDE1C inhibition also facilitates lysosome-dependent type I collagen protein degradation and thus decreases collagen contents in SMCs (414). In addition, PDE1C deficiency significantly reduces the formation and development of abdominal AA (AAA) in mice induced by Ang II infusion or periaortic elastase treatment (451). PDE1C inhibition attenuates SMC senescence via cAMP binding and activating Sirtuin 1, which plays an important role in SMC degeneration and AAA development (451). Pan-PDE1 inhibitor IC86340, which targets all PDE1 isoforms, attenuates neointimal hyperplasia and AAA in mice (373). Since PDE1 inhibitors such as ITI-214 have been proven to be safe in clinical trials, PDE1 inhibition may represent a novel therapeutic strategy for combating vascular diseases associated with pathological vascular remodeling. Changes in PDE1 expression have also been reported in PAH. For example, the cGMP-hydrolyzing PDE1 activity increases in the pulmonary artery in chronic hypoxia-induced PH rats (431). PDE1A and PDE1C expression levels are significantly upregulated in a rat pulmonary vascular remodeling model induced by sinoaortic denervation (452) and increased in human pulmonary artery SMCs from patients with idiopathic PH or secondary PH (453). PDE1C contributes to decreased cAMP and increased proliferation of pulmonary artery SMCs in patients with PH (453, 454). Methylxanthine derivatives have shown protection in different preclinical models of PH (454), although methylxanthine derivatives are not selective for PDE1. Moreover, the PDE1 inhibitor vinpocetine, either alone or in combination with other therapies such as prostacyclin analogs and NO, improves hemodynamics in the pulmonary circuit and reduces SMC proliferation in various animal PH models (455–457). More studies with specific PDE1 inhibitors or genetic deficiency mice are needed to further demonstrate the role of PDE1 isozymes in PAH.
PDE3 activity and expression significantly increase in the aortas of atherosclerosis-prone insulin-resistant rats (458). PDE3 inhibitors have been shown to block DNA synthesis in cultured pig aortic SMCs (418) and the growth of cultured SMCs in vitro (418–420). PDE3 inhibitors significantly attenuate neointima formation in a mouse model of photochemically induced vascular injury (420, 459). The PDE3 inhibitor cilostazol is an effective drug for treating symptoms of intermittent claudication caused by peripheral arterial diseases, a manifestation of systemic atherosclerosis (460). In addition, long-term oral administration of a different PDE3 inhibitor, cilostamide, suppresses arterial intimal hyperplasia by 83% in a rat balloon injury model (420). Endothelium-derived NO is critical in attenuating SMC proliferation and migration. The inhibitory effect of NO in vascular SMC mitogenesis is partly mediated by cGMP-dependent inhibition of PDE3 activity, increased cAMP level, and activation of PKA (422). PDE3 inhibition can also potentiate the effects of NO-cGMP on inflammatory molecule expression in vascular SMCs (422, 461). In addition, the PDE3 inhibitor cilostazol attenuates Ang II-induced AAA in mice and elastase-induced AAA in rats (462, 463). These findings support the important role of PDE3 in vascular occlusive diseases such as atherosclerosis and restenosis and in vascular degenerative diseases such as AAA, which suggests that PDE3 inhibition may be useful for the prevention and treatment of these diseases. A vasospasm occurs when a brain blood vessel narrows, blocking blood flow. It can occur in the 2 wk following a subarachnoid hemorrhage or brain aneurysm. PDE3 inhibitors, such as milrinone and cilostazol, effectively prevent chronic cerebral vasospasm in a canine model of chronic cerebral vasospasm, independent of systemic hemodynamic changes (464, 465). Although the increase of PDE3 activity and mRNA expression has been reported in pulmonary artery tissues from PH animals (395, 431), the functional role of PDE3 in PAH has not been well evaluated experimentally.
4.3.4. Other PDEs.
There also is scattered evidence for other PDEs in pathological vascular remodeling and diseases. For example, PDE2 mRNA and protein expression are reduced in pulmonary artery SMCs from PAH patients and rats with hypoxia-induced PH (307). PDE2 inhibition with BAY60-7550 enhances cAMP-induced dilation induced by prostaglandin I2, supporting a potential role of PDE2 in the development of PAH. Additionally, PDE10A expression is induced in cultured SMCs and in neointimal SMC-like cells (466). PDE10A deficiency or inhibition by TP-10 (a selective PDE10 inhibitor) significantly attenuated cultured SMC proliferation (466). Consistent with this, neointima formation in the femoral artery induced by wire injury is suppressed in PDE10A-knockout mice or in mice treated with TP-10 (466). These data support the critical role of PDE10A in pathological remodeling. PDE10A may also participate in PAH. A recent study highlights a central role of PDE10A in progressive pulmonary vascular remodeling, and inhibition of PDE10A induces an accumulation of intracellular cAMP, activates cAMP response element binding protein, and attenuates PASMC proliferation and pulmonary vascular remodeling (467).
4.4. PDE in Endothelial Barrier Function
Endothelial barrier function, such as permeability, plays a crucial role in maintaining the integrity of blood vessels and regulating the movement of substances between the bloodstream and surrounding tissues. When endothelial permeability is dysregulated, it can lead to various pathological conditions, including acute respiratory distress syndrome, sepsis, trauma, cerebral edema, etc. (468, 469). Both cAMP and cGMP signaling are important in the regulation of EC permeability. Various PDEs are expressed in ECs, although their expression may differ depending on the origin of ECs (16, 470, 471). The abnormal expression of PDEs can lead to endothelial permeability dysfunction (16). Targeting three main PDEs, PDE3, PDE4, and PDE5, in ECs can tighten the EC barrier and decrease permeability under pathological conditions (472–475). This section focuses on the roles of cAMP, cGMP, and various PDEs (PDE2, 3, and 4) in regulating EC permeability (FIGURE 10).
FIGURE 10.

Schematic illustration of phosphodiesterases (PDEs) in endothelial cells. The cartoon highlights the roles of cyclic adenosine monophosphate (cAMP), cyclic guanosine monophosphate (cGMP), and various PDEs (PDE2, 3, and 4) in regulating endothelial cell permeability. ANP, atrial natriuretic peptide; Epac, exchange protein activated by cAMP; NO, nitric oxide; PKA, protein kinase A, TGF-β, transforming growth factor β; TNF-α, tumor necrosis factor α; VEGF, vascular endothelial growth factor.
cAMP can regulate endothelial permeability in a PKA-dependent manner. For example, PKA activation promotes cell-cell junctions and decreases the permeability of ECs by phosphorylating vasodilator-stimulated phosphoprotein (476), negatively regulating MLC phosphorylation (477), or modulating Dll4-activated Notch signaling (478). In addition to PKA, Epac also plays an important role in regulating endothelial permeability. For example, cAMP decreases EC permeability by enhancing vascular endothelial cadherin-mediated cell adhesion, through an Epac-Rap1-dependent but PKA-independent mechanism (479). Activating the Epac-Rap1 pathway prevents VEGF-, tumor necrosis factor (TNF)-, or TGF-β-induced increase of permeability (480, 481). The two isoforms of Epac (Epac1 and 2) appear to have different roles in ECs, since genetic depletion of Epac1 but not Epac2 causes an increase in microvascular permeability in mice (482). Stimulating PGI2 and PGE2 with selective agonists improves EC permeability via both PKA- and Epac-dependent pathways (483). The differences between PKA and Epac in EC permeability regulation might be due to the differences in EC sources, such as artery ECs versus microvascular ECs. cGMP is also involved in the modulation of the barrier function of ECs. For example, activating cGMP-PKG signaling by stimulating pGC with ANP, sGC with sodium nitroprusside or a membrane-permeant cGMP analog (8-bromo-cGMP) improves EC permeability in cultured human umbilical vein endothelial cells (HUVECs) (484, 485). In addition, NO-cGMP signaling attenuates hypoxia-induced EC leakiness in vitro (486), and NPR1-cGMP-PKG signaling suppresses histamine-induced EC leakage in vivo (487). However, there are also reports of increased EC permeability by NO and ANP (488–490). Thus, the roles of cGMP signaling in regulating EC barriers could differ, even opposing, which may depend on the origins of ECs and local microenvironmental conditions in different disease statuses. The effects of cGMP on EC permeability can also be mediated through cAMP signaling. For example, cGMP reduces EC permeability through cGMP-dependently inhibiting PDE3 activity and promoting cAMP elevation (485). Interestingly, ANP or NO donors at low and high doses had opposite effects on EC permeability, which appears to be mediated by PDE3 or PDE2 differently. Low doses of ANP or NO potentiated the inhibitory effects of cAMP on thrombin-induced increase of EC permeability through cGMP-mediated inhibition of PDE3 and increase of cAMP (305). However, high doses of ANP or NO oppositely regulate EC permeability through cGMP-mediated activation of PDE2 and decrease of cAMP (305). Hence, the roles of cAMP and cGMP signaling in EC permeability regulation are more complicated by PDE2- or PDE3-mediated cross talk.
4.4.1. cAMP-specific PDE4.
In addition to modulating inflammatory response, PDE4 actively regulates endothelial barrier function. PDE4D tethers Epac1 in vascular endothelial cadherin (VE-cadherin)-based complexes, thus regulating both the activity and subcellular localization of Epac1, thereby affecting EC permeability (491). Knockdown of PDE4D increases EC permeability, which is similar to the effect of Epac1 knockdown (491). PDE4 is centrally involved in the alteration of the permeability of the endothelial barrier caused by inflammation (492), thus suggesting a potential clinical use for PDE4 inhibitors during sepsis. In lung microvascular endothelium, the splice variant PDE4D4 is anchored to spectrin, a cytoskeletal protein located on the inner side of the PM. This PDE4D4 is responsible for cAMP at the membrane domains to activate barrier-enhancing effectors and prevents cAMP from accessing the cytosolic domains, where the phosphorylation of microtubule proteins by PKA induces cell gap formation (493). Importantly, PDE4 plays a critical role in regulating the permeability of the blood-brain barrier (BBB) (494, 495). PDE4 inhibitors such as BBB022 and rolipram reduce cerebrovascular endothelial permeability in the spinal cords of mice with experimental autoimmune encephalomyelitis, preventing the entry of inflammatory cells and factors and reducing tissue edema (496). The PDE4 inhibitor rolipram also maintains the expression of tight junction proteins, such as occludin and claudin-5, avoiding ischemia-induced BBB disruption in mice with stroke (472). However, rolipram can also stabilize ANP-induced disturbance of EC permeability in vivo (473). The discrepancies of PDE4 inhibition on EC permeability may be due to multiple reasons, including the inconsistencies between in vitro and in vivo models, the potential enzyme activity-independent function of PDE4, or the differences in targeting all PDE4 isoforms with pan-PDE4 inhibitors.
4.4.2. Dual-substrate PDE.
PDE2 expression is relatively high in ECs (41, 432, 497). TNF-α further increases PDE2 expression in HUVECs under septic conditions (498). PDE2 inhibition reduces TNF-α- and thrombin-mediated alterations in endothelial F-actins, redistribution of VE-cadherin, and EC permeability (498). PDE2 also participates in regulating pulmonary endothelial permeability (495). Selective inhibition of PDE2 attenuates the endothelial permeability induced by the pneumococcal exotoxin pneumolysin in isolated and perfused mouse lungs and human endothelial cell monolayers (474). Collectively, these studies demonstrate the prominent role of PDE2 in modulating vascular dynamics.
Inhibition of PDE3 activity with cilostazol stabilizes the barrier integrity in primary rat brain capillary endothelial cells and the human brain hCMEC/D3 endothelial cell line (499). Cilostazol protects the BBB from damage induced by oxygen-glucose deprivation (OGD) and reoxygenation by improving the tightness of tight junctions, which PKA mediates. PDE3B is mainly expressed in these cells, presenting a target to protect against BBB pathological permeability. PDE3B tethers Epac1 and p84-regulated PI3Kγ in a signaling complex in human arterial ECs (475). A small peptide displacing Epac1 from PDE3B increases cAMP binding to Epac1 and augments PI3Kγ signaling, which modulates the endothelial barrier (475). Thus, PDE3 inhibitors may prevent the disruption of BBB.
5. DRUGS THAT TARGET PDEs IN CARDIOVASCULAR DISEASES
PDE isoforms control cAMP and cGMP, two critical intracellular second messengers regulating a variety of fundamental cellular processes in the cardiovascular system. Abnormal PDE expression and regulation have been associated with cardiovascular diseases, including cardiac hypertrophy, HF, MI, arrhythmias, hypertension, and atherosclerosis (500). Thus, targeting individual PDEs under pathological conditions is of clinical importance. For example, milrinone was one of the first PDE3 inhibitors used clinically for the therapy of acute HF. Clinical use is limited because of its detrimental side effects, such as arrhythmia in chronic HF. However, the success of sildenafil (Viagra), the first oral PDE5 inhibitor initially developed to treat angina and then approved by the Food and Drug Administration (FDA) for the treatment of male erectile dysfunction, combined with an increasing understanding of PDE biology, has raised new hopes that manipulating PDE activity with greater specificity can yield therapeutic benefits in many diseases (FIGURE 11) (501).
FIGURE 11.
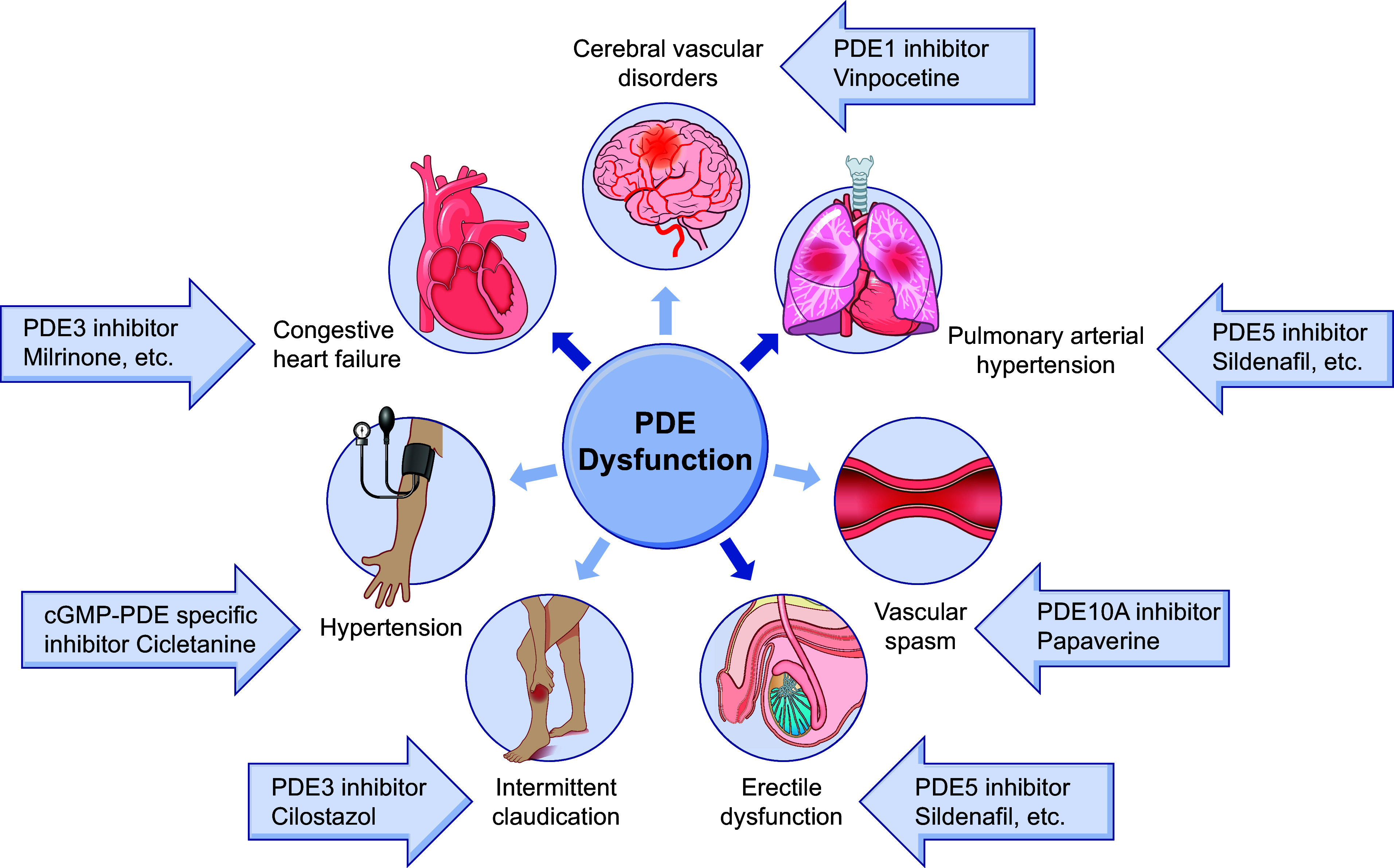
Clinical phosphodiesterase (PDE) inhibitors in cardiovascular diseases.
The contribution of each PDE family varies depending on subcellular compartments. Selectively targeting PDE isoforms within a specific location provides opportunities for greater efficacy and higher specificity in precision therapeutic strategies. Many high-affinity PDE family-selective inhibitors have shown lack of effectiveness and intolerable side effects, resulting in failed clinical trials (7). Despite this, several PDE4 inhibitors have been approved for clinical treatment such as for chronic obstructive pulmonary disorder and psoriasis (500, 502). In the future, determining the isoform-selective distribution, phosphorylation, translocation, and interactome of PDEs under pathological conditions is essential for developing isoform-selective inhibition and therapeutics. The selective PDE inhibitors used clinically or being investigated in clinical trials to treat various cardiovascular diseases are summarized in TABLE 5 and TABLE 6.
Table 5.
Marketed PDE inhibitors in cardiovascular diseases
| Drug Name | Trade Name | PDE Selectivity | Indications |
|---|---|---|---|
| Vinpocetine | Cavinton | PDE1 | Cerebral vascular disorders |
| Cilostazol | Pletal | PDE3A, PDE4A | Intermittent claudication |
| Milrinone | Primacor | PDE3A, PDE4A | Congestive HF |
| Amrinone | Inocor | PDE3A, PDE4 | Congestive HF |
| Enoximone | Perfan | PDE3A | Congestive HF |
| Olprinone | Coretec | PDE3 | HF |
| Pimobendan | Acardi | PDE3 | HF |
| Sildenafil | Viagra | PDE5 | Erectile dysfunctionPulmonary arterial hypertension |
| Tadalafil | Cialis | PDE5 | Erectile dysfunctionPulmonary arterial hypertension |
| Udenafil | Zydena | PDE5 | Pulmonary arterial hypertension |
| Vardenafil | Levitra | PDE5 | Erectile dysfunction |
| Avanafil | Stendra | PDE5 | Erectile dysfunction |
| Lodenafil | Helleva | PDE5 | Erectile dysfunction |
| Mirodenafil | Mvix | PDE5 | Erectile dysfunction |
| Pentoxifylline | Trental | PDE4, PDE5, adenosine 2 receptors | Intermittent claudication |
| Cicletanine | Tenstaten | cGMP-PDE specific | Hypertension |
| Papaverine | Pavabid | PDE10A | Vascular spasm |
FDA, food and drug administration; HF, heart failure; PDE, phosphodiesterase.
Table 6.
Current clinical trials of PDE inhibitors in cardiovascular diseases
| Drug Name | Developer | PDE Selectivity | Indications | Highest CT Phase | |
|---|---|---|---|---|---|
| Ongoing clinical development | ITI-214 | Intra-Cellular Therapies | PDE1 | HF (NCT03387215) | Phase 2 |
| Discontinued or unknown clinical development | Parogrelil | Nissan Chemical Industries | PDE3, PDE5 | Intermittent claudication | Phase 2 |
| Tetomilast | Otsuka Pharmaceutical | PDE4 | HF, reperfusion injury | Phase 2 | |
| SLx-2101 | Surface Logix/Kadmon Pharmaceuticals | PDE5 | Hypertension (NCT00562614) (NCT00562549) | Phase 2 | |
| Zaprinast | Aventis | PDE5, PDE6 | Ischemic heart disease | N/A |
CT, clinical trial; HF, heart failure, N/A, not available; PDE, phosphodiesterase.
5.1. PDE Inhibitors in HFrEF
The clinical syndrome of HF includes fatigue, followed by dyspnea and exercise intolerance. HF can result from disorders of the myocardium, heart valves, or great vessels, with most patients experiencing symptoms due to impaired LV myocardial function filling or ejection of blood (503). HF may be associated with a broad spectrum of LV functional abnormalities, which can be classified as patients with normal LV size and preserved EF (EF ≥ 50; HFpEF) and patients with severe dilation and markedly reduced EF (EF ≤ 40%; HFrEF) (503). Despite sharing similar clinical phenotypes, HFpEF and HFrEF are thought to represent distinct pathophysiological entities and should be studied and treated separately (503).
In patients with HFrEF, abnormalities of systolic and diastolic dysfunction coexist. MI due to coronary artery disease is a major cause of HFrEF, and many other risk factors, such as hypertension and diabetes, may also lead to LV enlargement and HFrEF (503). HFrEF is hallmarked by cAMP and cGMP signaling defects coupled with cardiac dysfunction. PDEs degrade cAMP, restrict the sympathetic stimulation-triggered inotropic effects, and contribute to the development of systolic HF. Inhibitors of PDEs are used as positive inotropes to sensitize the heart to catecholamines (504). PDE3 and PDE4 account for most cAMP hydrolytic activities in hearts. Initial interests focus on the potential of non-isoform-selective inhibitors of PDE3 to enhance cardiac contractility and lusitropy. Although acute cAMP stimulation enhances myocardial contraction and cardiac output (505), chronic elevation of cAMP-PKA leads to cardiac remodeling and dysfunction (273, 506). Moreover, PDE3 inhibition increases the incidence of arrhythmias in HFrEF patients with excess calcium and energy demands and increases sudden death and overall mortality (58, 507–509). As a result, the short-term benefit of PDE3 inhibitors in patients with HFrEF is outweighed by an increase in sudden cardiac death when these drugs are administered chronically (264, 509). The acute benefits and chronic adverse actions of PDE3 inhibitors in HF patients may also result from the phosphorylation of different substrates of PKA in separate intracellular compartments. Meanwhile, a polymorphism in the human PDE3A promoter is reported to regulate PDE3A gene transcription by cAMP-PKA signaling and might explain the variable tolerance to milrinone in patients with HFrEF (510). To offset the unwanted side effects associated with PDE3 inhibitors, a multicenter international trial has been carried out with a combined therapy with enoximone, a PDE3 inhibitor, and beta-blockers. This trial reports no adverse impact on survival or other clinical outcomes and no significant benefit (511). A new extended-release form of milrinone (CRD-102) has been developed for advanced HF, with some early favorable clinical results (512) similar to those reported with enoximone (513). Whether this formulation alters the safety-efficacy profile of PDE3 inhibitors in chronic HFrEF therapy remains to be determined.
Apart from improving contractility in HF patients, PDE3 inhibitors are potent cardiotonic drugs that cause arterial vasodilation and afterload reduction and inhibit platelet aggregation (514). Since milrinone is predominantly excreted renally, the drug is infrequently used in patients with acute renal failure or end-stage renal diseases because of concerns about symptomatic hypotension (515). Milrinone can also be used in end-stage HF for patients resistant to optimal therapy. In addition, short-term oral PDE3 inhibition is used in severe HF patients as a bridge before they receive heart transplantation (513).
Impaired NO-cGMP signaling is known to contribute to LV diastolic abnormalities and remodeling in chronic HFrEF (516). PDE5 expression is increased in the left ventricle of patients with advanced human HF (92, 290), and PDE5 contributes to adverse ventricular remodeling after MI in mice (92). PDE5 inhibition is thus an intriguing pharmacological strategy to enhance NO-cGMP signaling in vivo. In stable HF patients with reduced left ventricle ejection fraction (EF), the PDE5 inhibitor sildenafil significantly improved LV diastolic function and cardiac geometry (516). Other small trials of PDE5 inhibition with sildenafil or udenafil have shown improvements in hemodynamics, exercise capacity, and quality of life without significant adverse events in HFrEF (517–519). However, larger phase III trials are needed to validate these findings.
Sildenafil therapy could effectively improve pulmonary hemodynamics and cardiopulmonary exercise testing measurements in patients with PH due to left heart disease (PH-LHD) with HFrEF, regardless of acute or chronic treatment (520). Additional sildenafil therapy shows symptomatic and functional improvements in PH-LHD with HFrEF, with sustained increases in EF of both the right and left ventricles (516, 519, 520). PDE5 inhibitors appear to have a dual action underlying efficacy in HFrEF: an acute effect predominantly driven by pulmonary vasodilation and a chronic effect resulting from LV remodeling independent of afterload (267). A phase IV clinical trial showed that sildenafil improves diabetic cardiomyopathy (NCT00692237) (521). The early features of diabetic cardiomyopathy are LV concentric hypertrophy associated with altered myocardial contraction dynamics. At this stage, sildenafil improves cardiac kinetics and circulating markers through an antiremodeling effect. This effect is independent of vascular, endothelial, or metabolic factors and is exerted through a direct intramyocardial action (521). Moreover, the sex difference in response to the PDE5 inhibitor tadalafil is being currently trialed in left ventricular hypertrophy associated with diabetic cardiomyopathy (NCT01803828).
Inhibition of PDE5 activity proves protective in ischemia-reperfusion injuries (501). In fact, sildenafil was initially trialed as an antianginal cardiac agent since it increased blood flow through the coronary arteries. The discovery of this “side effect” led investigators to focus on safety rather than benefit in patients with cardiovascular diseases (267). In a population of men with type 2 diabetes, the use of a PDE5 inhibitor is associated with a lower risk of overall mortality and mortality in those with a history of acute MI (316). Similar findings show that treatment with a PDE5 inhibitor might be related to a reduced risk of long-term adverse outcomes after MI (522). However, the SIDAMI trial (NCT01046838) shows that sildenafil does not decrease filling pressure at rest or during exercise in post-MI patients with diastolic dysfunction but there are effects on secondary end points, which require further comparative clinical trial studies (523).
Selective inhibition of PDE1 by ITI-214 confers acute inotropic, lusitropic, and arterial vasodilatory effects in mammals (233). A phase I/II randomized single rising dose study of ITI-214 in patients with systolic HF evaluated safety and tolerability (NCT03387215) (524). ITI-214 is generally well tolerated and acutely reduces arterial vascular resistance while augmenting cardiac inotropy, cardiac output, and heart rate. The effects of ITI-214 are dependent on cAMP modulation in adenosine signaling without calcium increase, potentially having less proarrhythmic risk than PDE3 inhibition in failing hearts (167, 233). PDE9A is upregulated in failing human hearts. PDE9 specifically regulates NPR-cGMP signaling and is independent of NO (123). Augmentation of cGMP and NPR signaling has emerged as a therapeutic strategy in HF (525). An inhibitor of PDE9, PF-04447943, seems well tolerated in humans and is currently being investigated in clinical trials for Alzheimer’s disease (NCT00930059).
5.2. PDE Inhibitors in HFpEF
The American College of Cardiology Foundation-American Heart Association guidelines define HFpEF as clinical signs and symptoms of HF, preserved ejection fraction, and no other apparent cause for symptoms (526). The incidence of HFpEF is increasing, accounting for almost 50% of HF cases and frequently in older women. However, the diagnosis of HFpEF remains a challenge because it is primarily based on exclusion criteria. In contrast to efficacious neurohumoral inhibition for HFrEF, therapies for HFpEF are lacking. Large trials testing neurohumoral inhibition in HFpEF failed to reach a positive primary outcome (527, 528). Recent views attribute this failure to distinct systemic and myocardial signaling in HFpEF and diverse phenotypes within the HFpEF patient population (529).
Patients with HFpEF often include older women with hypertension, T2DM, obesity, and vasculopathy, causing a systemic proinflammatory state and coronary microvascular inflammation (530). It subsequently affects LV diastolic dysfunction through macrophage infiltration, resulting in interstitial fibrosis (531), and alters paracrine communication between ECs and surrounding cardiomyocytes (530). These alterations favor hypertrophy and increase myocyte stiffness by depriving cardiomyocytes of NO and cGMP (532). Thus, organic NO donors could be useful therapeutic tools in HFpEF because they could restore myocardial NO content and concomitantly correct the high arterial load. Unfortunately, in the NEAT-HFpEF trial, the organic nitrate isosorbide mononitrate does not improve but, instead, tends to reduce chronic activity levels measured by accelerometry, with no improvement in quality of life or submaximal exercise capacity in HFpEF patients (533). This result conflicts with the NO hypothesis in HFpEF, but it should be kept in mind that organic nitrates may produce greater than expected hypotensive effects in people with HFpEF or potentially impair cardiac output because of excessive preload reduction (534). Similarly, therapy with PDE3 inhibitors is also being explored in patients with HF with a preserved ejection fraction, which is related to the capacity of PDE3 inhibition to increase cardiac output reserve at lower ventricular filling pressures during exercise (535).
Ventricular hypertrophy with interstitial fibrosis and diastolic chamber stiffening are common pathological changes in HFpEF patients. PKG stimulation has potent antifibrotic and antihypertrophic effects in experimental cardiac disease models. Multiple therapeutic approaches that stimulate PKG are currently in clinical trials or under active investigation. PDE5 primarily impacts NO-sGC-derived cGMP. Unfortunately, the largest randomized RELAX trial with PDE5 inhibition in patients with the greatest comorbidity and broadest inclusion criteria failed to show real advantages on clinical outcomes (536). In another major trial, PDE5 inhibitors had beneficial vascular effects via improved endothelium-dependent vasodilation (537), but these benefits are offset by reductions in contractility (538). Nonetheless, a positive effect of sildenafil has been reported in terms of right ventricular function and remodeling and pulmonary pressure regulation in HFpEF patients with documented PH (539). Thus, future trials should take into consideration the correct patient population based on their clinical indications to fully realize the benefits of PDE5 inhibitors for HFpEF. Meanwhile, given that depressed NO signaling may be the potential reason for the ineffective effects of PDE5 inhibitor to increase myocardial cGMP in HFpEF, inhibition of PDE9 may restore NP efficacy with beneficial effects (123), making PDE9A an attractive alternative for the treatment of HFpEF. LCZ696, a combination of the Ang II receptor inhibitor valsartan and the neprilysin inhibitor sacubitril, augmented active natriuretic peptides, resulting in an increase in cGMP in patients with HFpEF in the PARAMOUNT trial (540) and the PARAGON-HF trial (NCT01920711), offering opportunities for combined treatment with PDE9A inhibitors for HFpEF (123).
5.3. PDE Inhibitors in Pulmonary Hypertension and Vascular Diseases
PDE5 is abundantly expressed in lung tissue and is therefore an ideal target for treating disorders in pulmonary circulation, including PH. PDE5 inhibition promotes cGMP accumulation, enhancing NO-mediated vasodilation, and has antiproliferative effects on pulmonary artery SMCs (446). A series of trials suggest that sildenafil is beneficial in treating PH (541). The improvements in exercise capacity were largely sustained after 3 yr of treatment in the SUPER-2 study (542). Sildenafil was approved by the FDA and the European Medicines Agency (EMEA) in 2005 to treat patients with class II and III PH long term. Tadalafil has also been commercialized for treating PH to improve exercise ability and slow worsening changes in patients’ physical condition (267). In the FUEL trial, treatment with udenafil (87.5 mg twice daily) was not associated with an improvement in oxygen consumption at peak exercise but with improvements in multiple measures of exercise performance at the ventilatory anaerobic threshold (NCT02741115) (543). Meanwhile, the expressions of PDE1, 2, and 3 are increased in pulmonary artery SMCs. The inhibitors for these PDEs are effective in preclinical models of pulmonary hypertension. These PDE families may yield further therapeutic options for treating PH.
Stroke is the third leading cause of death and a major cause of disability worldwide. Endothelial dysfunction is a hallmark of small-vessel and large-artery strokes. Modulating endothelial cAMP and cGMP signaling is a potential therapeutic strategy in stroke. PDE inhibitors may restore cyclic nucleotide signals and cerebral endothelial function. The nonselective PDE1 inhibitor vinpocetine has been approved in European countries for treating dementia and stroke for >30 years. Vinpocetine is also available in the United States as a dietary supplement for improving memory and recovery from stroke through its ability to promote vasodilation by raising cGMP levels and decreasing ICa (544). PDE5A is localized to caveolae and modulates NOS3 activity, which can facilitate the effect of spinal cord stimulation on vasodilation and potentially prevent and treat vasospasm after subarachnoid hemorrhage (545, 546).
Preclinical studies have shown antiplatelet, vasodilator, and antiproliferative actions of the PDE3 inhibitor cilostazol (547, 548). Cilostazol has been shown to protect patients from recurrent cerebral infarction in a multicenter, randomized, placebo-controlled, double-blind clinical trial (549). Additional reports describe the pleiotropic actions of cilostazol, thereby providing a variety of clinical uses, including prevention of recurrent stroke (547, 549), cerebral vasospasm (550, 551), coronary restenosis (552), and peripheral occlusive disease. Moreover, cilostazol received FDA approval in 1999 for intermittent claudication, but off-label uses include secondary prevention of cerebrovascular accidents, percutaneous coronary intervention, and coronary stent stenosis (553). Although it offers functional improvement, cilostazol is associated with severe side effects.
Intimal hyperplasia and luminal stenosis are the key characteristics of several vascular disorders, such as atherosclerosis and postangioplasty restenosis. Several clinical trials have been designed to evaluate the benefits of the PDE3 inhibitor cilostazol. In comparison to standard antiplatelet therapy with aspirin and clopidogrel, cilostazol has been effective in attenuating postangioplasty restenosis, especially in patients at high risk of restenosis 6 mo after stent implantation [CREST trial (554)] and in patients with diabetes mellitus implanted with a drug-eluting stent [DECLARE-DIABETES trial (555)]. Compared with aspirin, cilostazol also reduces the progression of carotid atherosclerosis in patients with T2DM [DAPC trial (556)].
Overall, PDE inhibitors have been valuable tools for improving the functional status of patients with cardiovascular diseases (TABLE 5). Some of the most widely marketed PDE inhibitors fall under the diverse category of cardiovascular drugs, primarily including cardiotonics for congestive HF, vasodilatory agents for hypertension, pulmonary arterial hypertension, intermittent claudication, and cerebrovascular disorders, and antiaggregates for thrombosis-related events (557). Several initially promising therapeutic agents for cardiovascular disease have proved unsuccessful. Recently, the PDE1 inhibitor ITI-214 has completed phase I trials for HF. Additionally, a series of PDE inhibitors are under ongoing investigation in clinical or preclinical studies and predominantly aim at central nervous system (CNS) disorders, solid malignancies, and inflammatory and immune-mediated disorders (TABLE 6) (557).
6. NEW DIRECTIONS AND CHALLENGES IN TARGETING PDEs IN CARDIOVASCULAR DISEASES
The success of PDE5 inhibitors in treating erectile dysfunction has spurred continuous interest in investigating the effects of PDE inhibitors on cardiovascular diseases. Despite encouraging preliminary observations, several PDE inhibitors have failed in clinical trials for potential cardioprotective effects. Several factors may explain these failures.
The relationship between individual PDE isoform expression and diseases is multifaceted and complex, with different stages of cardiac diseases that may require different PDE modulations. Although PDE modulation occurs before or concurrently with the onset of disease in most preclinical studies, patients in clinical studies have established disease status (7). Recent gene profiling shows that the mRNA levels of most PDEs are downregulated in the advanced stage of human HFrEF, whereas they are either little changed or even increased in human HFpEF (558). It is also evident that many individual PDE isoforms have cell- and tissue-specific expression and are selectively localized or recruited to distinct subcellular compartments to regulate local cAMP and cGMP concentrations and specific cell functions. Lack of comprehensive insight into an individual PDE isoform’s distribution, regulation, and function prevents selectively targeting a particular isoform. Current PDE inhibitors generally target a family or subfamily of PDEs without specificity of discriminating multiple isoforms, which can be ineffective for the isoform expressed in specific cellular compartments, and are associated with unwanted side effects like arrhythmias. The overlapping structures of PDE isoforms within a family pose a challenge in designing and targeting one specific isoform with traditional small-molecule inhibitors. Hence, gene manipulation that seeks a selective and efficient approach to abrogate or enhance the activity of single PDE isoforms in a cell type-specific manner may provide a way to combat cardiovascular diseases (229).
We can also develop small molecules or peptides for disrupting specific protein-protein interactions between individual PDE splice variants and their partner, which could modulate distinct PDE pools with potentially fewer adverse effects than the global inhibition of an entire PDE family (501). An alternative approach to combating side effects attributed to systemic PDE inhibitor distribution is to develop targeted delivery systems that can preferentially transport PDE inhibitors to precise tissues or cell types. This approach can specifically disrupt the anchoring of single isoforms or splicing variants in targeted cells and tissues to reduce complications associated with systemic distribution (500). Furthermore, PDE inhibitors can act by allosteric modulation. Structural analysis of PDEs would facilitate the discovery of allosteric PDE modulators and could help correct the detrimental effects of cardiovascular diseases. Future development will aim to improve clinical outcomes with isoform-specific inhibitors and activators and with molecules modulating isoform-specific protein-protein interactions in specific signalosomes (5, 10).
Sex-dependent differences in PDE expression and function are emerging as another critical area to understand the enzymes in pathology and the implications in therapy. The mRNA expression levels of PDE1A and PDE3B are sex dependent, and the levels of PDE1A in males are higher than those in females, whereas the levels of PDE3B in males are lower than those in females (27). A recent study reveals differential regional expression of PDE4D in the apex and base of female hearts and overall higher expression of PDE4D in female hearts relative to male hearts (73). The differential PDE4D expression is linked to cAMP dynamics and action potential under adrenergic stimulation, which may be significant in sex-dependent differences in cardiac arrhythmia (73). Ovariectomy promotes mRNA of PDE4B in rat hearts (74), which may also contribute to the sex-dependent difference in ECC (75). Another report shows that the antihypertrophy effect of PDE5 inhibitor is estrogen dependent in females, though the underlying mechanism is unclear (559). A recent clinical trial indicates that PDE5 inhibitors are more effective in male patients with diabetic cardiomyopathy than in females (349). Parallel comparisons between sexes in aging animals may yield insight into the roles of PDE isoforms in the development of cardiovascular diseases and better strategies to treat patients with PDE inhibitors.
The vascular and lymphatic systems comprise structurally distinct vessels that function to circulate blood and lymph. Recently, emerging attention has been paid to the lymphatic system in health and disease (560), including cardiovascular development and disease (561). A few findings show that PDEs may be involved in lymphatic development. For example, PDE5 is expressed in lymphatic malformation tissues (562), and PDE5 inhibitors are used for severe lymphatic malformations in clinical trials (563). The role of cyclic nucleotides and PDEs in regulating the functions of lymphatic vasculature remains largely unknown, which represents a future direction of research.
The complexity of cardiovascular diseases may require combination therapies, such as multiple PDE inhibitors or simultaneously activating AC and GC and blocking PDEs, to fine-tune cyclic nucleotide levels in specific subcellular compartments, providing more effective therapies with favorable risk-benefit profiles (7, 9). In addition, the discovery of the PDE5 inhibitor sildenafil in erectile dysfunction suggests that repurposing the existing entities is an effective strategy to accelerate the development of PDE inhibitors in cardiovascular diseases. A better understanding of pharmacology and a retrospective analysis of clinical effects during trials or marketed usage for its original indication is a promising approach for finding new indications. It will also facilitate the development of novel PDE inhibitors for treating other diseases while avoiding potential cardiovascular toxicity. The challenge in addressing these issues will determine whether PDE modulators can be successfully translated to the clinical arena for treating patients with cardiovascular diseases (289, 292–296).
GRANTS
This work was supported by National Institute of Health Grants R01GM129376, R01HL162825, and R01HL147263 and Department of Veterans Affairs Grants IK6BX005753 and I01BX005100 to Y.K.X. and by National Natural Science Foundation of China Grants 81773730 and 82273926 to Q.F.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Q.F. and Y.W. prepared figures; Q.F., Y.W., C.Y., and Y.K.X. drafted manuscript; Q.F., Y.W., and Y.K.X. edited and revised manuscript; Q.F., Y.W., C.Y., and Y.K.X. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Victoria Salemme for help in preparation of the manuscript.
REFERENCES
- 1. Rall TW, Sutherland EW. Formation of a cyclic adenine ribonucleotide by tissue particles. J Biol Chem 232: 1065–1076, 1958. doi: 10.1016/S0021-9258(19)77422-5. [DOI] [PubMed] [Google Scholar]
- 2. Sutherland EW, Rall TW. Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J Biol Chem 232: 1077–1091, 1958. doi: 10.1016/S0021-9258(19)77423-7. [DOI] [PubMed] [Google Scholar]
- 3. Butcher RW, Sutherland EW. Adenosine 3′,5′-phosphate in biological materials. I. Purification and properties of cyclic 3′,5′-nucleotide phosphodiesterase and use of this enzyme to characterize adenosine 3′,5′-phosphate in human urine. J Biol Chem 237: 1244–1250, 1962. doi: 10.1016/S0021-9258(18)60316-3. [DOI] [PubMed] [Google Scholar]
- 4. Blair CM, Baillie GS. Reshaping cAMP nanodomains through targeted disruption of compartmentalised phosphodiesterase signalosomes. Biochem Soc Trans 47: 1405–1414, 2019. doi: 10.1042/BST20190252. [DOI] [PubMed] [Google Scholar]
- 5. Chen S, Yan C. An update of cyclic nucleotide phosphodiesterase as a target for cardiac diseases. Expert Opin Drug Discov 16: 183–196, 2021. doi: 10.1080/17460441.2020.1821643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dunkerly-Eyring B, Kass DA. Myocardial phosphodiesterases and their role in cGMP regulation. J Cardiovasc Pharmacol 75: 483–493, 2020. doi: 10.1097/FJC.0000000000000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kamel R, Leroy J, Vandecasteele G, Fischmeister R. Cyclic nucleotide phosphodiesterases as therapeutic targets in cardiac hypertrophy and heart failure. Nat Rev Cardiol 20: 90–108, 2023. doi: 10.1038/s41569-022-00756-z. [DOI] [PubMed] [Google Scholar]
- 8. Kokkonen K, Kass DA. Nanodomain regulation of cardiac cyclic nucleotide signaling by phosphodiesterases. Annu Rev Pharmacol Toxicol 57: 455–479, 2017. doi: 10.1146/annurev-pharmtox-010716-104756. [DOI] [PubMed] [Google Scholar]
- 9. Preedy ME. Cardiac cyclic nucleotide phosphodiesterases: roles and therapeutic potential in heart failure. Cardiovasc Drugs Ther 34: 401–417, 2020. doi: 10.1007/s10557-020-06959-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zaccolo M, Zerio A, Lobo MJ. Subcellular organization of the cAMP signaling pathway. Pharmacol Rev 73: 278–309, 2021. doi: 10.1124/pharmrev.120.000086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Keravis T, Lugnier C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br J Pharmacol 165: 1288–1305, 2012. doi: 10.1111/j.1476-5381.2011.01729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 58: 488–520, 2006. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 13. Pandit J, Forman MD, Fennell KF, Dillman KS, Menniti FS. Mechanism for the allosteric regulation of phosphodiesterase 2A deduced from the X-ray structure of a near full-length construct. Proc Natl Acad Sci USA 106: 18225–18230, 2009. doi: 10.1073/pnas.0907635106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Richter W, Xie M, Scheitrum C, Krall J, Movsesian MA, Conti M. Conserved expression and functions of PDE4 in rodent and human heart. Basic Res Cardiol 106: 249–262, 2011. doi: 10.1007/s00395-010-0138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rybalkin SD, Yan C, Bornfeldt KE, Beavo JA. Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Circ Res 93: 280–291, 2003. doi: 10.1161/01.RES.0000087541.15600.2B. [DOI] [PubMed] [Google Scholar]
- 16. Surapisitchat J, Beavo JA. Regulation of endothelial barrier function by cyclic nucleotides: the role of phosphodiesterases. Handb Exp Pharmacol: 193–210, 2011. doi: 10.1007/978-3-642-17969-3_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Laursen M, Beck L, Kehler J, Christoffersen CT, Bundgaard C, Mogensen S, Mow TJ, Pinilla E, Knudsen JS, Hedegaard ER, Grunnet M, Simonsen U. Novel selective PDE type 1 inhibitors cause vasodilatation and lower blood pressure in rats. Br J Pharmacol 174: 2563–2575, 2017. doi: 10.1111/bph.13868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Loughney K, Martins TJ, Harris EA, Sadhu K, Hicks JB, Sonnenburg WK, Beavo JA, Ferguson K. Isolation and characterization of cDNAs corresponding to two human calcium, calmodulin-regulated, 3',5'-cyclic nucleotide phosphodiesterases. J Biol Chem 271: 796–806, 1996. doi: 10.1074/jbc.271.2.796. [DOI] [PubMed] [Google Scholar]
- 19. Sonnenburg WK, Rybalkin SD, Bornfeldt KE, Kwak KS, Rybalkina IG, Beavo JA. Identification, quantitation, and cellular localization of PDE1 calmodulin-stimulated cyclic nucleotide phosphodiesterases. Methods 14: 3–19, 1998. doi: 10.1006/meth.1997.0561. [DOI] [PubMed] [Google Scholar]
- 20. Clapham JC, Wilderspin AF. Cloning of dog heart PDE1A—a first detailed characterization at the molecular level in this species. Gene 268: 165–171, 2001. doi: 10.1016/s0378-1119(01)00413-9. [DOI] [PubMed] [Google Scholar]
- 21. Yanaka N, Kurosawa Y, Minami K, Kawai E, Omori K. cGMP-phosphodiesterase activity is up-regulated in response to pressure overload of rat ventricles. Biosci Biotechnol Biochem 67: 973–979, 2003. doi: 10.1271/bbb.67.973. [DOI] [PubMed] [Google Scholar]
- 22. Vandeput F, Wolda SL, Krall J, Hambleton R, Uher L, McCaw KN, Radwanski PB, Florio V, Movsesian MA. Cyclic nucleotide phosphodiesterase PDE1C1 in human cardiac myocytes. J Biol Chem 282: 32749–32757, 2007. doi: 10.1074/jbc.M703173200. [DOI] [PubMed] [Google Scholar]
- 23. Miller CL, Oikawa M, Cai Y, Wojtovich AP, Nagel DJ, Xu X, Xu H, Florio V, Rybalkin SD, Beavo JA, Chen YF, Li JD, Blaxall BC, Abe JI, Yan C. Role of Ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circ Res 105: 956–964, 2009. doi: 10.1161/CIRCRESAHA.109.198515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bode DC, Kanter JR, Brunton LL. Cellular distribution of phosphodiesterase isoforms in rat cardiac tissue. Circ Res 68: 1070–1079, 1991. doi: 10.1161/01.res.68.4.1070. [DOI] [PubMed] [Google Scholar]
- 25. Miller CL, Cai Y, Oikawa M, Thomas T, Dostmann WR, Zaccolo M, Fujiwara K, Yan C. Cyclic nucleotide phosphodiesterase 1A: a key regulator of cardiac fibroblast activation and extracellular matrix remodeling in the heart. Basic Res Cardiol 106: 1023–1039, 2011. doi: 10.1007/s00395-011-0228-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Knight WE, Chen S, Zhang Y, Oikawa M, Wu M, Zhou Q, Miller CL, Cai Y, Mickelsen DM, Moravec C, Small EM, Abe J, Yan C. PDE1C deficiency antagonizes pathological cardiac remodeling and dysfunction. Proc Natl Acad Sci USA 113: E7116–E7125, 2016. doi: 10.1073/pnas.1607728113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang J, Bingaman S, Huxley VH. Intrinsic sex-specific differences in microvascular endothelial cell phosphodiesterases. Am J Physiol Heart Circ Physiol 298: H1146–H1154, 2010. doi: 10.1152/ajpheart.00252.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yan C, Zhao AZ, Bentley JK, Loughney K, Ferguson K, Beavo JA. Molecular cloning and characterization of a calmodulin-dependent phosphodiesterase enriched in olfactory sensory neurons. Proc Natl Acad Sci USA 92: 9677–9681, 1995. doi: 10.1073/pnas.92.21.9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nagel DJ, Aizawa T, Jeon KI, Liu W, Mohan A, Wei H, Miano JM, Florio VA, Gao P, Korshunov VA, Berk BC, Yan C. Role of nuclear Ca2+/calmodulin-stimulated phosphodiesterase 1A in vascular smooth muscle cell growth and survival. Circ Res 98: 777–784, 2006. doi: 10.1161/01.RES.0000215576.27615.fd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rybalkin SD, Rybalkina I, Beavo JA, Bornfeldt KE. Cyclic nucleotide phosphodiesterase 1C promotes human arterial smooth muscle cell proliferation. Circ Res 90: 151–157, 2002. doi: 10.1161/hh0202.104108. [DOI] [PubMed] [Google Scholar]
- 31. Han P, Werber J, Surana M, Fleischer N, Michaeli T. The calcium/calmodulin-dependent phosphodiesterase PDE1C down-regulates glucose-induced insulin secretion. J Biol Chem 274: 22337–22344, 1999. doi: 10.1074/jbc.274.32.22337. [DOI] [PubMed] [Google Scholar]
- 32. Dunkern TR, Hatzelmann A. Characterization of inhibitors of phosphodiesterase 1C on a human cellular system. FEBS J 274: 4812–4824, 2007. doi: 10.1111/j.1742-4658.2007.06001.x. [DOI] [PubMed] [Google Scholar]
- 33. Sonnenburg WK, Seger D, Kwak KS, Huang J, Charbonneau H, Beavo JA. Identification of inhibitory and calmodulin-binding domains of the PDE1A1 and PDE1A2 calmodulin-stimulated cyclic nucleotide phosphodiesterases. J Biol Chem 270: 30989–31000, 1995. doi: 10.1074/jbc.270.52.30989. [DOI] [PubMed] [Google Scholar]
- 34. Yan C, Kim D, Aizawa T, Berk BC. Functional interplay between angiotensin II and nitric oxide: cyclic GMP as a key mediator. Arterioscler Thromb Vasc Biol 23: 26–36, 2003. doi: 10.1161/01.atv.0000046231.17365.9d. [DOI] [PubMed] [Google Scholar]
- 35. Sharma RK. Phosphorylation and characterization of bovine heart calmodulin-dependent phosphodiesterase. Biochemistry 30: 5963–5968, 1991. doi: 10.1021/bi00238a021. [DOI] [PubMed] [Google Scholar]
- 36. Ang KL, Antoni FA. Reciprocal regulation of calcium dependent and calcium independent cyclic AMP hydrolysis by protein phosphorylation. J Neurochem 81: 422–433, 2002. doi: 10.1046/j.1471-4159.2002.00903.x. [DOI] [PubMed] [Google Scholar]
- 37. Sharma RK, Wang JH. Calmodulin and Ca2+-dependent phosphorylation and dephosphorylation of 63-kDa subunit-containing bovine brain calmodulin-stimulated cyclic nucleotide phosphodiesterase isozyme. J Biol Chem 261: 1322–1328, 1986. doi: 10.1016/S0021-9258(17)36094-5. [DOI] [PubMed] [Google Scholar]
- 38. Kincaid RL, Stith-Coleman IE, Vaughan M. Proteolytic activation of calmodulin-dependent cyclic nucleotide phosphodiesterase. J Biol Chem 260: 9009–9015, 1985. doi: 10.1016/S0021-9258(17)39450-4. [DOI] [PubMed] [Google Scholar]
- 39. Sonnenburg WK, Seger D, Beavo JA. Molecular cloning of a cDNA encoding the “61-kDa” calmodulin-stimulated cyclic nucleotide phosphodiesterase. Tissue-specific expression of structurally related isoforms. J Biol Chem 268: 645–652, 1993. doi: 10.1016/S0021-9258(18)54200-9. [DOI] [PubMed] [Google Scholar]
- 40. Rosman GJ, Martins TJ, Sonnenburg WK, Beavo JA, Ferguson K, Loughney K. Isolation and characterization of human cDNAs encoding a cGMP-stimulated 3',5'-cyclic nucleotide phosphodiesterase. Gene 191: 89–95, 1997. doi: 10.1016/s0378-1119(97)00046-2. [DOI] [PubMed] [Google Scholar]
- 41. Stephenson DT, Coskran TM, Wilhelms MB, Adamowicz WO, O’Donnell MM, Muravnick KB, Menniti FS, Kleiman RJ, Morton D. Immunohistochemical localization of phosphodiesterase 2A in multiple mammalian species. J Histochem Cytochem 57: 933–949, 2009. doi: 10.1369/jhc.2009.953471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mehel H, Emons J, Vettel C, Wittköpper K, Seppelt D, Dewenter M, Lutz S, Sossalla S, Maier LS, Lechêne P, Leroy J, Lefebvre F, Varin A, Eschenhagen T, Nattel S, Dobrev D, Zimmermann WH, Nikolaev VO, Vandecasteele G, Fischmeister R, El-Armouche A. Phosphodiesterase-2 is up-regulated in human failing hearts and blunts beta-adrenergic responses in cardiomyocytes. J Am Coll Cardiol 62: 1596–1606, 2013. doi: 10.1016/j.jacc.2013.05.057. [DOI] [PubMed] [Google Scholar]
- 43. Baliga RS, Preedy ME, Dukinfield MS, Chu SM, Aubdool AA, Bubb KJ, Moyes AJ, Tones MA, Hobbs AJ. Phosphodiesterase 2 inhibition preferentially promotes NO/guanylyl cyclase/cGMP signaling to reverse the development of heart failure. Proc Natl Acad Sci USA 115: E7428–E7437, 2018. doi: 10.1073/pnas.1800996115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vettel C, Lämmle S, Ewens S, Cervirgen C, Emons J, Ongherth A, Dewenter M, Lindner D, Westermann D, Nikolaev VO, Lutz S, Zimmermann WH, El-Armouche A. PDE2-mediated cAMP hydrolysis accelerates cardiac fibroblast to myofibroblast conversion and is antagonized by exogenous activation of cGMP signaling pathways. Am J Physiol Heart Circ Physiol 306: H1246–H1252, 2014. doi: 10.1152/ajpheart.00852.2013. [DOI] [PubMed] [Google Scholar]
- 45. Mongillo M, Tocchetti CG, Terrin A, Lissandron V, Cheung YF, Dostmann WR, Pozzan T, Kass DA, Paolocci N, Houslay MD, Zaccolo M. Compartmentalized phosphodiesterase-2 activity blunts beta-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ Res 98: 226–234, 2006. doi: 10.1161/01.RES.0000200178.34179.93. [DOI] [PubMed] [Google Scholar]
- 46. Lugnier C, Keravis T, Le Bec A, Pauvert O, Proteau S, Rousseau E. Characterization of cyclic nucleotide phosphodiesterase isoforms associated to isolated cardiac nuclei. Biochim Biophys Acta 1472: 431–446, 1999. doi: 10.1016/s0304-4165(99)00145-2. [DOI] [PubMed] [Google Scholar]
- 47. Liu D, Wang Z, Nicolas V, Lindner M, Mika D, Vandecasteele G, Fischmeister R, Brenner C. PDE2 regulates membrane potential, respiration and permeability transition of rodent subsarcolemmal cardiac mitochondria. Mitochondrion 47: 64–75, 2019. doi: 10.1016/j.mito.2019.05.002. [DOI] [PubMed] [Google Scholar]
- 48. Monterisi S, Lobo MJ, Livie C, Castle JC, Weinberger M, Baillie G, Surdo NC, Musheshe N, Stangherlin A, Gottlieb E, Maizels R, Bortolozzi M, Micaroni M, Zaccolo M. PDE2A2 regulates mitochondria morphology and apoptotic cell death via local modulation of cAMP/PKA signalling. Elife 6: e21374, 2017. doi: 10.7554/eLife.21374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen W, Spitzl A, Mathes D, Nikolaev VO, Werner F, Weirather J, Špiranec K, Röck K, Fischer JW, Kämmerer U, Stegner D, Baba HA, Hofmann U, Frantz S, Kuhn M. Endothelial actions of ANP enhance myocardial inflammatory infiltration in the early phase after acute infarction. Circ Res 119: 237–248, 2016. doi: 10.1161/CIRCRESAHA.115.307196. [DOI] [PubMed] [Google Scholar]
- 50. Favot L, Keravis T, Lugnier C. Modulation of VEGF-induced endothelial cell cycle protein expression through cyclic AMP hydrolysis by PDE2 and PDE4. Thromb Haemost 92: 634–645, 2004. doi: 10.1160/TH03-12-0768. [DOI] [PubMed] [Google Scholar]
- 51. Keravis T, Komas N, Lugnier C. Cyclic nucleotide hydrolysis in bovine aortic endothelial cells in culture: differential regulation in cobblestone and spindle phenotypes. J Vasc Res 37: 235–249, 2000. doi: 10.1159/000025738. [DOI] [PubMed] [Google Scholar]
- 52. Martins TJ, Mumby MC, Beavo JA. Purification and characterization of a cyclic GMP-stimulated cyclic nucleotide phosphodiesterase from bovine tissues. J Biol Chem 257: 1973–1979, 1982. doi: 10.1016/S0021-9258(19)68134-2. [DOI] [PubMed] [Google Scholar]
- 53. Weishaar RE, Burrows SD, Kobylarz DC, Quade MM, Evans DB. Multiple molecular forms of cyclic nucleotide phosphodiesterase in cardiac and smooth muscle and in platelets. Isolation, characterization, and effects of various reference phosphodiesterase inhibitors and cardiotonic agents. Biochem Pharmacol 35: 787–800, 1986. doi: 10.1016/0006-2952(86)90247-9. [DOI] [PubMed] [Google Scholar]
- 54. Aravind L, Ponting CP. The GAF domain: an evolutionary link between diverse phototransducing proteins. Trends Biochem Sci 22: 458–459, 1997. doi: 10.1016/s0968-0004(97)01148-1. [DOI] [PubMed] [Google Scholar]
- 55. Mumby MC, Martins TJ, Chang ML, Beavo JA. Identification of cGMP-stimulated cyclic nucleotide phosphodiesterase in lung tissue with monoclonal antibodies. J Biol Chem 257: 13283–13290, 1982. doi: 10.1016/S0021-9258(18)33443-4. [DOI] [PubMed] [Google Scholar]
- 56. Martinez SE, Wu AY, Glavas NA, Tang XB, Turley S, Hol WG, Beavo JA. The two GAF domains in phosphodiesterase 2A have distinct roles in dimerization and in cGMP binding. Proc Natl Acad Sci USA 99: 13260–13265, 2002. doi: 10.1073/pnas.192374899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wu AY, Tang XB, Martinez SE, Ikeda K, Beavo JA. Molecular determinants for cyclic nucleotide binding to the regulatory domains of phosphodiesterase 2A. J Biol Chem 279: 37928–37938, 2004. doi: 10.1074/jbc.M404287200. [DOI] [PubMed] [Google Scholar]
- 58. Ding B, Abe JI, Wei H, Huang Q, Walsh RA, Molina CA, Zhao A, Sadoshima J, Blaxall BC, Berk BC, Yan C. Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: implication in heart failure. Circulation 111: 2469–2476, 2005. doi: 10.1161/01.CIR.0000165128.39715.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chung YW, Lagranha C, Chen Y, Sun J, Tong G, Hockman SC, Ahmad F, Esfahani SG, Bae DH, Polidovitch N, Wu J, Rhee DK, Lee BS, Gucek M, Daniels MP, Brantner CA, Backx PH, Murphy E, Manganiello VC. Targeted disruption of PDE3B, but not PDE3A, protects murine heart from ischemia/reperfusion injury. Proc Natl Acad Sci USA 112: E2253–E2262, 2015. doi: 10.1073/pnas.1416230112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ahmad F, Shen W, Vandeput F, Szabo-Fresnais N, Krall J, Degerman E, Goetz F, Klussmann E, Movsesian M, Manganiello V. Regulation of sarcoplasmic reticulum Ca2+ ATPase 2 (SERCA2a2) activity by phosphodiesterase 3A (PDE3A) in human myocardium: phosphorylation-dependent interaction of PDE3A1 with SERCA2a2. J Biol Chem 290: 6763–6776, 2015. doi: 10.1074/jbc.M115.638585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hambleton R, Krall J, Tikishvili E, Honeggar M, Ahmad F, Manganiello VC, Movsesian MA. Isoforms of cyclic nucleotide phosphodiesterase PDE3 and their contribution to cAMP hydrolytic activity in subcellular fractions of human myocardium. J Biol Chem 280: 39168–39174, 2005. doi: 10.1074/jbc.M506760200. [DOI] [PubMed] [Google Scholar]
- 62. Wechsler J, Choi YH, Krall J, Ahmad F, Manganiello VC, Movsesian MA. Isoforms of cyclic nucleotide phosphodiesterase PDE3A in cardiac myocytes. J Biol Chem 277: 38072–38078, 2002. doi: 10.1074/jbc.M203647200. [DOI] [PubMed] [Google Scholar]
- 63. Beca S, Ahmad F, Shen W, Liu J, Makary S, Polidovitch N, Sun J, Hockman S, Chung YW, Movsesian M, Murphy E, Manganiello V, Backx PH. Phosphodiesterase type 3A regulates basal myocardial contractility through interacting with sarcoplasmic reticulum calcium ATPase type 2a signaling complexes in mouse heart. Circ Res 112: 289–297, 2013. doi: 10.1161/CIRCRESAHA.111.300003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sun B, Li H, Shakur Y, Hensley J, Hockman S, Kambayashi J, Manganiello VC, Liu Y. Role of phosphodiesterase type 3A and 3B in regulating platelet and cardiac function using subtype-selective knockout mice. Cell Signal 19: 1765–1771, 2007. doi: 10.1016/j.cellsig.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 65. Movsesian M, Ahmad F, Hirsch E. Functions of PDE3 isoforms in cardiac muscle. J Cardiovasc Dev Dis 5: 10, 2018. doi: 10.3390/jcdd5010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Maurice DH, Haslam RJ. Molecular basis of the synergistic inhibition of platelet function by nitrovasodilators and activators of adenylate cyclase: inhibition of cyclic AMP breakdown by cyclic GMP. Mol Pharmacol 37: 671–681, 1990. [PubMed] [Google Scholar]
- 67. Meacci E, Taira M, Moos M, Smith CJ, Movsesian MA, Degerman E, Belfrage P, Manganiello V. Molecular cloning and expression of human myocardial cGMP-inhibited cAMP phosphodiesterase. Proc Natl Acad Sci USA 89: 3721–3725, 1992. doi: 10.1073/pnas.89.9.3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Palmer D, Jimmo SL, Raymond DR, Wilson LS, Carter RL, Maurice DH. Protein kinase A phosphorylation of human phosphodiesterase 3B promotes 14-3-3 protein binding and inhibits phosphatase-catalyzed inactivation. J Biol Chem 282: 9411–9419, 2007. doi: 10.1074/jbc.M606936200. [DOI] [PubMed] [Google Scholar]
- 69. Wang P, Myers JG, Wu P, Cheewatrakoolpong B, Egan RW, Billah MM. Expression, purification, and characterization of human cAMP-specific phosphodiesterase (PDE4) subtypes A, B, C, and D. Biochem Biophys Res Commun 234: 320–324, 1997. doi: 10.1006/bbrc.1997.6636. [DOI] [PubMed] [Google Scholar]
- 70. Huston E, Lumb S, Russell A, Catterall C, Ross AH, Steele MR, Bolger GB, Perry MJ, Owens RJ, Houslay MD. Molecular cloning and transient expression in COS7 cells of a novel human PDE4B cAMP-specific phosphodiesterase, HSPDE4B3. Biochem J 328: 549–558, 1997. doi: 10.1042/bj3280549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lee WS, Tsai WJ, Yeh PH, Wei BL, Chiou WF. Divergent role of calcium on Abeta- and MPTP-induced cell death in SK-N-SH neuroblastoma. Life Sci 78: 1268–1275, 2006. doi: 10.1016/j.lfs.2005.06.036. [DOI] [PubMed] [Google Scholar]
- 72. Kostic MM, Erdogan S, Rena G, Borchert G, Hoch B, Bartel S, Scotland G, Huston E, Houslay MD, Krause EG. Altered expression of PDE1 and PDE4 cyclic nucleotide phosphodiesterase isoforms in 7-oxo-prostacyclin-preconditioned rat heart. J Mol Cell Cardiol 29: 3135–3146, 1997. doi: 10.1006/jmcc.1997.0544. [DOI] [PubMed] [Google Scholar]
- 73. Caldwell JL, Lee IJ, Ngo L, Wang L, Bahriz S, Xu B, Bers DM, Navedo MF, Bossuyt J, Xiang YK, Ripplinger CM. Whole-heart multiparametric optical imaging reveals sex-dependent heterogeneity in cAMP signaling and repolarization kinetics. Sci Adv 9: eadd5799, 2023. doi: 10.1126/sciadv.add5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Parks RJ, Bogachev O, Mackasey M, Ray G, Rose RA, Howlett SE. The impact of ovariectomy on cardiac excitation-contraction coupling is mediated through cAMP/PKA-dependent mechanisms. J Mol Cell Cardiol 111: 51–60, 2017. doi: 10.1016/j.yjmcc.2017.07.118. [DOI] [PubMed] [Google Scholar]
- 75. Farrell SR, Ross JL, Howlett SE. Sex differences in mechanisms of cardiac excitation-contraction coupling in rat ventricular myocytes. Am J Physiol Heart Circ Physiol 299: H36–H45, 2010. doi: 10.1152/ajpheart.00299.2010. [DOI] [PubMed] [Google Scholar]
- 76. Leroy J, Abi-Gerges A, Nikolaev VO, Richter W, Lechêne P, Mazet J-L, Conti M, Fischmeister R, Vandecasteele G. Spatiotemporal dynamics of beta-adrenergic cAMP signals and L-type Ca2+ channel regulation in adult rat ventricular myocytes: role of phosphodiesterases. Circ Res 102: 1091–1100, 2008. doi: 10.1161/CIRCRESAHA.107.167817. [DOI] [PubMed] [Google Scholar]
- 77. Ghigo A, Perino A, Mehel H, Zahradníková A, Morello F, Leroy J, Nikolaev VO, Damilano F, Cimino J, De Luca E, Richter W, Westenbroek R, Catterall WA, Zhang J, Yan C, Conti M, Gomez AM, Vandecasteele G, Hirsch E, Fischmeister R. Phosphoinositide 3-kinase gamma protects against catecholamine-induced ventricular arrhythmia through protein kinase A-mediated regulation of distinct phosphodiesterases. Circulation 126: 2073–2083, 2012. doi: 10.1161/CIRCULATIONAHA.112.114074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Rochais F, Vandecasteele G, Lefebvre F, Lugnier C, Lum H, Mazet JL, Cooper DM, Fischmeister R. Negative feedback exerted by cAMP-dependent protein kinase and cAMP phosphodiesterase on subsarcolemmal cAMP signals in intact cardiac myocytes: an in vivo study using adenovirus-mediated expression of CNG channels. J Biol Chem 279: 52095–52105, 2004. doi: 10.1074/jbc.M405697200. [DOI] [PubMed] [Google Scholar]
- 79. Fan T, Hou Y, Ge W, Fan T, Feng X, Guo W, Song X, Gao R, Wang J. Phosphodiesterase 4D promotes angiotensin II-induced hypertension in mice via smooth muscle cell contraction. Commun Biol 5: 81, 2022. doi: 10.1038/s42003-022-03029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Busch CJ, Liu H, Graveline AR, Bloch KD. Nitric oxide induces phosphodiesterase 4B expression in rat pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 290: L747–L753, 2006. doi: 10.1152/ajplung.00298.2005. [DOI] [PubMed] [Google Scholar]
- 81. Cheng D, Ren J, Gillespie DG, Mi Z, Jackson EK. Regulation of 3',5'-cAMP in preglomerular smooth muscle and endothelial cells from genetically hypertensive rats. Hypertension 56: 1096–1101, 2010. doi: 10.1161/HYPERTENSIONAHA.110.160176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Selige J, Hatzelmann A, Dunkern T. The differential impact of PDE4 subtypes in human lung fibroblasts on cytokine-induced proliferation and myofibroblast conversion. J Cell Physiol 226: 1970–1980, 2011. doi: 10.1002/jcp.22529. [DOI] [PubMed] [Google Scholar]
- 83. Xu R, Fu J, Hu Y, Yang X, Tao X, Chen L, Huang K, Fu Q. Roflumilast-mediated phosphodiesterase 4D inhibition reverses diabetes-associated cardiac dysfunction and remodeling: effects beyond glucose lowering. Diabetes 71: 1660–1678, 2022. doi: 10.2337/db21-0898. [DOI] [PubMed] [Google Scholar]
- 84. Bolger G, Michaeli T, Martins T, St John T, Steiner B, Rodgers L, Riggs M, Wigler M, Ferguson K. A family of human phosphodiesterases homologous to the dunce learning and memory gene product of Drosophila melanogaster are potential targets for antidepressant drugs. Mol Cell Biol 13: 6558–6571, 1993. doi: 10.1128/MCB.13.10.6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Richter W, Conti M. Dimerization of the type 4 cAMP-specific phosphodiesterases is mediated by the upstream conserved regions (UCRs). J Biol Chem 277: 40212–40221, 2002. doi: 10.1074/jbc.M203585200. [DOI] [PubMed] [Google Scholar]
- 86. Cedervall P, Aulabaugh A, Geoghegan KF, McLellan TJ, Pandit J. Engineered stabilization and structural analysis of the autoinhibited conformation of PDE4. Proc Natl Acad Sci USA 112: E1414–E1422, 2015. doi: 10.1073/pnas.1419906112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Beard MB, Olsen AE, Jones RE, Erdogan S, Houslay MD, Bolger GB. UCR1 and UCR2 domains unique to the cAMP-specific phosphodiesterase family form a discrete module via electrostatic interactions. J Biol Chem 275: 10349–10358, 2000. doi: 10.1074/jbc.275.14.10349. [DOI] [PubMed] [Google Scholar]
- 88. Mika D, Richter W, Conti M. A CaMKII/PDE4D negative feedback regulates cAMP signaling. Proc Natl Acad Sci USA 112: 2023–2028, 2015. doi: 10.1073/pnas.1419992112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Baillie GS, MacKenzie SJ, McPhee I, Houslay MD. Sub-family selective actions in the ability of Erk2 MAP kinase to phosphorylate and regulate the activity of PDE4 cyclic AMP-specific phosphodiesterases. Br J Pharmacol 131: 811–819, 2000. doi: 10.1038/sj.bjp.0703636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Vandeput F, Krall J, Ockaili R, Salloum FN, Florio V, Corbin JD, Francis SH, Kukreja RC, Movsesian MA. cGMP-hydrolytic activity and its inhibition by sildenafil in normal and failing human and mouse myocardium. J Pharmacol Exp Ther 330: 884–891, 2009. doi: 10.1124/jpet.109.154468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhang M, Koitabashi N, Nagayama T, Rambaran R, Feng N, Takimoto E, Koenke T, O’Rourke B, Champion HC, Crow MT, Kass DA. Expression, activity, and pro-hypertrophic effects of PDE5A in cardiac myocytes. Cell Signal 20: 2231–2236, 2008. doi: 10.1016/j.cellsig.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pokreisz P, Vandenwijngaert S, Bito V, Van den Bergh A, Lenaerts I, Busch C, Marsboom G, Gheysens O, Vermeersch P, Biesmans L, Liu X, Gillijns H, Pellens M, Van Lommel A, Buys E, Schoonjans L, Vanhaecke J, Verbeken E, Sipido K, Herijgers P, Bloch KD, Janssens SP. Ventricular phosphodiesterase-5 expression is increased in patients with advanced heart failure and contributes to adverse ventricular remodeling after myocardial infarction in mice. Circulation 119: 408–416, 2009. doi: 10.1161/CIRCULATIONAHA.108.822072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kotera J, Fujishige K, Imai Y, Kawai E, Michibata H, Akatsuka H, Yanaka N, Omori K. Genomic origin and transcriptional regulation of two variants of cGMP-binding cGMP-specific phosphodiesterases. Eur J Biochem 262: 866–873, 1999. doi: 10.1046/j.1432-1327.1999.00450.x. [DOI] [PubMed] [Google Scholar]
- 94. Giordano D, De Stefano ME, Citro G, Modica A, Giorgi M. Expression of cGMP-binding cGMP-specific phosphodiesterase (PDE5) in mouse tissues and cell lines using an antibody against the enzyme amino-terminal domain. Biochim Biophys Acta 1539: 16–27, 2001. doi: 10.1016/s0167-4889(01)00086-6. [DOI] [PubMed] [Google Scholar]
- 95. Campolo F, Zevini A, Cardarelli S, Monaco L, Barbagallo F, Pellegrini M, Cornacchione M, Di Grazia A, De Arcangelis V, Gianfrilli D, Giorgi M, Lenzi A, Isidori AM, Naro F. Identification of murine phosphodiesterase 5A isoforms and their functional characterization in HL-1 cardiac cell line. J Cell Physiol 233: 325–337, 2018. doi: 10.1002/jcp.25880. [DOI] [PubMed] [Google Scholar]
- 96. Wallis RM, Corbin JD, Francis SH, Ellis P. Tissue distribution of phosphodiesterase families and the effects of sildenafil on tissue cyclic nucleotides, platelet function, and the contractile responses of trabeculae carneae and aortic rings in vitro. Am J Cardiol 83: 3C–12C, 1999. doi: 10.1016/s0002-9149(99)00042-9. [DOI] [PubMed] [Google Scholar]
- 97. Lukowski R, Rybalkin SD, Loga F, Leiss V, Beavo JA, Hofmann F. Cardiac hypertrophy is not amplified by deletion of cGMP-dependent protein kinase I in cardiomyocytes. Proc Natl Acad Sci USA 107: 5646–51, 2010. doi: 10.1073/pnas.1001360107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Li EA, Xi W, Han YS, Brozovich FV. Phosphodiesterase expression in the normal and failing heart. Arch Biochem Biophys 662: 160–168, 2019. doi: 10.1016/j.abb.2018.12.013. [DOI] [PubMed] [Google Scholar]
- 99. Castro LR, Verde I, Cooper DM, Fischmeister R. Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation 113: 2221–2228, 2006. doi: 10.1161/CIRCULATIONAHA.105.599241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gong W, Yan M, Chen J, Chaugai S, Chen C, Wang D. Chronic inhibition of cyclic guanosine monophosphate-specific phosphodiesterase 5 prevented cardiac fibrosis through inhibition of transforming growth factor beta-induced Smad signaling. Front Med 8: 445–455, 2014. doi: 10.1007/s11684-014-0378-3. [DOI] [PubMed] [Google Scholar]
- 101. Gebska MA, Stevenson BK, Hemnes AR, Bivalacqua TJ, Haile A, Hesketh GG, Murray CI, Zaiman AL, Halushka MK, Krongkaew N, Strong TD, Cooke CA, El-Haddad H, Tuder RM, Berkowitz DE, Champion HC. Phosphodiesterase-5A (PDE5A) is localized to the endothelial caveolae and modulates NOS3 activity. Cardiovasc Res 90: 353–363, 2011. doi: 10.1093/cvr/cvq410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lugnier C, Schoeffter P, Le Bec A, Strouthou E, Stoclet JC. Selective inhibition of cyclic nucleotide phosphodiesterases of human, bovine and rat aorta. Biochem Pharmacol 35: 1743–1751, 1986. doi: 10.1016/0006-2952(86)90333-3. [DOI] [PubMed] [Google Scholar]
- 103. Thomas MK, Francis SH, Corbin JD. Substrate- and kinase-directed regulation of phosphorylation of a cGMP-binding phosphodiesterase by cGMP. J Biol Chem 265: 14971–14978, 1990. doi: 10.1016/S0021-9258(18)77211-6. [DOI] [PubMed] [Google Scholar]
- 104. Rybalkin SD, Rybalkina IG, Shimizu-Albergine M, Tang XB, Beavo JA. PDE5 is converted to an activated state upon cGMP binding to the GAF A domain. EMBO J 22: 469–478, 2003. doi: 10.1093/emboj/cdg051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zoraghi R, Bessay EP, Corbin JD, Francis SH. Structural and functional features in human PDE5A1 regulatory domain that provide for allosteric cGMP binding, dimerization, and regulation. J Biol Chem 280: 12051–12063, 2005. doi: 10.1074/jbc.M413611200. [DOI] [PubMed] [Google Scholar]
- 106. Corbin JD, Turko IV, Beasley A, Francis SH. Phosphorylation of phosphodiesterase-5 by cyclic nucleotide-dependent protein kinase alters its catalytic and allosteric cGMP-binding activities. Eur J Biochem 267: 2760–2767, 2000. doi: 10.1046/j.1432-1327.2000.01297.x. [DOI] [PubMed] [Google Scholar]
- 107. Francis SH, Bessay EP, Kotera J, Grimes KA, Liu L, Thompson WJ, Corbin JD. Phosphorylation of isolated human phosphodiesterase-5 regulatory domain induces an apparent conformational change and increases cGMP binding affinity. J Biol Chem 277: 47581–47587, 2002. doi: 10.1074/jbc.M206088200. [DOI] [PubMed] [Google Scholar]
- 108. Shimizu-Albergine M, Rybalkin SD, Rybalkina IG, Feil R, Wolfsgruber W, Hofmann F, Beavo JA. Individual cerebellar Purkinje cells express different cGMP phosphodiesterases (PDEs): in vivo phosphorylation of cGMP-specific PDE (PDE5) as an indicator of cGMP-dependent protein kinase (PKG) activation. J Neurosci 23: 6452–6459, 2003. doi: 10.1523/JNEUROSCI.23-16-06452.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Taylor RE, Shows KH, Zhao Y, Pittler SJ. A PDE6A promoter fragment directs transcription predominantly in the photoreceptor. Biochem Biophys Res Commun 282: 543–547, 2001. doi: 10.1006/bbrc.2001.4605. [DOI] [PubMed] [Google Scholar]
- 110. Tate RJ, Arshavsky VY, Pyne NJ. The identification of the inhibitory gamma-subunits of the type 6 retinal cyclic guanosine monophosphate phosphodiesterase in non-retinal tissues: differential processing of mRNA transcripts. Genomics 79: 582–586, 2002. doi: 10.1006/geno.2002.6740. [DOI] [PubMed] [Google Scholar]
- 111. Murray F, MacLean MR, Pyne NJ. An assessment of the role of the inhibitory gamma subunit of the retinal cyclic GMP phosphodiesterase and its effect on the p42/p44 mitogen-activated protein kinase pathway in animal and cellular models of pulmonary hypertension. Br J Pharmacol 138: 1313–1319, 2003. doi: 10.1038/sj.bjp.0705190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Han P, Zhu X, Michaeli T. Alternative splicing of the high affinity cAMP-specific phosphodiesterase (PDE7A) mRNA in human skeletal muscle and heart. J Biol Chem 272: 16152–16157, 1997. doi: 10.1074/jbc.272.26.16152. [DOI] [PubMed] [Google Scholar]
- 113. Lakics V, Karran EH, Boess FG. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 59: 367–374, 2010. doi: 10.1016/j.neuropharm.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 114. Hetman JM, Soderling SH, Glavas NA, Beavo JA. Cloning and characterization of PDE7B, a cAMP-specific phosphodiesterase. Proc Natl Acad Sci USA 97: 472–476, 2000. doi: 10.1073/pnas.97.1.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Michaeli T, Bloom TJ, Martins T, Loughney K, Ferguson K, Riggs M, Rodgers L, Beavo JA, Wigler M. Isolation and characterization of a previously undetected human cAMP phosphodiesterase by complementation of cAMP phosphodiesterase-deficient Saccharomyces cerevisiae. J Biol Chem 268: 12925–12932, 1993. doi: 10.1016/S0021-9258(18)31474-1. [DOI] [PubMed] [Google Scholar]
- 116. Patrucco E, Albergine MS, Santana LF, Beavo JA. Phosphodiesterase 8A (PDE8A) regulates excitation-contraction coupling in ventricular myocytes. J Mol Cell Cardiol 49: 330–333, 2010. doi: 10.1016/j.yjmcc.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Soderling SH, Bayuga SJ, Beavo JA. Cloning and characterization of a cAMP-specific cyclic nucleotide phosphodiesterase. Proc Natl Acad Sci USA 95: 8991–8996, 1998. doi: 10.1073/pnas.95.15.8991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Horvath A, Giatzakis C, Tsang K, Greene E, Osorio P, Boikos S, Libè R, Patronas Y, Robinson-White A, Remmers E, Bertherat J, Nesterova M, Stratakis CA. A cAMP-specific phosphodiesterase (PDE8B) that is mutated in adrenal hyperplasia is expressed widely in human and mouse tissues: a novel PDE8B isoform in human adrenal cortex. Eur J Hum Genet 16: 1245–1253, 2008. doi: 10.1038/ejhg.2008.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Wang H, Yan Z, Yang S, Cai J, Robinson H, Ke H. Kinetic and structural studies of phosphodiesterase-8A and implication on the inhibitor selectivity. Biochemistry 47: 12760–12768, 2008. doi: 10.1021/bi801487x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Seliborska Z, Mroczkowski TF, Martin D, Moore L. [Immunotypes of Chlamydia trachomatis and the clinical picture of non-gonorrheal urethritis in men and cervicitis in women]. Przegl Dermatol 77: 272–275, 1990. [PubMed] [Google Scholar]
- 121. Fisher DA, Smith JF, Pillar JS, St Denis SH, Cheng JB. Isolation and characterization of PDE9A, a novel human cGMP-specific phosphodiesterase. J Biol Chem 273: 15559–15564, 1998. doi: 10.1074/jbc.273.25.15559. [DOI] [PubMed] [Google Scholar]
- 122. Rentero C, Monfort A, Puigdomenech P. Identification and distribution of different mRNA variants produced by differential splicing in the human phosphodiesterase 9A gene. Biochem Biophys Res Commun 301: 686–692, 2003. doi: 10.1016/s0006-291x(03)00021-4. [DOI] [PubMed] [Google Scholar]
- 123. Lee DI, Zhu G, Sasaki T, Cho GS, Hamdani N, Holewinski R, Jo SH, Danner T, Zhang M, Rainer PP, Bedja D, Kirk JA, Ranek MJ, Dostmann WR, Kwon C, Margulies KB, Van Eyk JE, Paulus WJ, Takimoto E, Kass DA. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 519: 472–476, 2015. doi: 10.1038/nature14332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Zhang L, Bouadjel K, Manoury B, Vandecasteele G, Fischmeister R, Leblais V. Cyclic nucleotide signalling compartmentation by PDEs in cultured vascular smooth muscle cells. Br J Pharmacol 176: 1780–1792, 2019. doi: 10.1111/bph.14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kotera J, Fujishige K, Yuasa K, Omori K. Characterization and phosphorylation of PDE10A2, a novel alternative splice variant of human phosphodiesterase that hydrolyzes cAMP and cGMP. Biochem Biophys Res Commun 261: 551–557, 1999. doi: 10.1006/bbrc.1999.1013. [DOI] [PubMed] [Google Scholar]
- 126. Fujishige K, Kotera J, Omori K. Striatum- and testis-specific phosphodiesterase PDE10A isolation and characterization of a rat PDE10A. Eur J Biochem 266: 1118–1127, 1999. doi: 10.1046/j.1432-1327.1999.00963.x. [DOI] [PubMed] [Google Scholar]
- 127. Fujishige K, Kotera J, Michibata H, Yuasa K, Takebayashi S, Okumura K, Omori K. Cloning and characterization of a novel human phosphodiesterase that hydrolyzes both cAMP and cGMP (PDE10A). J Biol Chem 274: 18438–18445, 1999. doi: 10.1074/jbc.274.26.18438. [DOI] [PubMed] [Google Scholar]
- 128. Jäger R, Russwurm C, Schwede F, Genieser HG, Koesling D, Russwurm M. Activation of PDE10 and PDE11 phosphodiesterases. J Biol Chem 287: 1210–1219, 2012. doi: 10.1074/jbc.M111.263806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Chen S, Zhang Y, Lighthouse JK, Mickelsen DM, Wu J, Yao P, Small EM, Yan C. A Novel role of cyclic nucleotide phosphodiesterase 10A in pathological cardiac remodeling and dysfunction. Circulation 141: 217–233, 2020. doi: 10.1161/CIRCULATIONAHA.119.042178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Fawcett L, Baxendale R, Stacey P, McGrouther C, Harrow I, Soderling S, Hetman J, Beavo JA, Phillips SC. Molecular cloning and characterization of a distinct human phosphodiesterase gene family: PDE11A. Proc Natl Acad Sci USA 97: 3702–3707, 2000. doi: 10.1073/pnas.97.7.3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Yuasa K, Kotera J, Fujishige K, Michibata H, Sasaki T, Omori K. Isolation and characterization of two novel phosphodiesterase PDE11A variants showing unique structure and tissue-specific expression. J Biol Chem 275: 31469–31479, 2000. doi: 10.1074/jbc.M003041200. [DOI] [PubMed] [Google Scholar]
- 132. Loughney K, Taylor J, Florio VA. 3',5'-cyclic nucleotide phosphodiesterase 11A: localization in human tissues. Int J Impot Res 17: 320–325, 2005. doi: 10.1038/sj.ijir.3901317. [DOI] [PubMed] [Google Scholar]
- 133. Bischoff E. Potency, selectivity, and consequences of nonselectivity of PDE inhibition. Int J Impot Res 16, Suppl 1: S11–S14, 2004. doi: 10.1038/sj.ijir.3901208. [DOI] [PubMed] [Google Scholar]
- 134. Liu Y, Chen J, Fontes SK, Bautista EN, Cheng Z. Physiological and pathological roles of protein kinase A in the heart. Cardiovasc Res 118: 386–398, 2022. doi: 10.1093/cvr/cvab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. McConnachie G, Langeberg LK, Scott JD. AKAP signaling complexes: getting to the heart of the matter. Trends Mol Med 12: 317–323, 2006. doi: 10.1016/j.molmed.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 136. Zhang M, Kass DA. Phosphodiesterases and cardiac cGMP: evolving roles and controversies. Trends Pharmacol Sci 32: 360–365, 2011. doi: 10.1016/j.tips.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Fu Q, Chen X, Xiang YK. Compartmentalization of beta-adrenergic signals in cardiomyocytes. Trends Cardiovasc Med 23: 250–256, 2013. doi: 10.1016/j.tcm.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Takimoto E. Cyclic GMP-dependent signaling in cardiac myocytes. Circ J 76: 1819–1825, 2012. doi: 10.1253/circj.cj-12-0664. [DOI] [PubMed] [Google Scholar]
- 139. Davoren PR, Sutherland EW. The cellular location of adenyl cyclase in the pigeon erythrocyte. J Biol Chem 238: 3016–3023, 1963. doi: 10.1016/S0021-9258(18)51860-3. [DOI] [PubMed] [Google Scholar]
- 140. Di Benedetto G, Zoccarato A, Lissandron V, Terrin A, Li X, Houslay MD, Baillie GS, Zaccolo M. Protein kinase A type I and type II define distinct intracellular signaling compartments. Circ Res 103: 836–844, 2008. doi: 10.1161/CIRCRESAHA.108.174813. [DOI] [PubMed] [Google Scholar]
- 141. Liu S, Li Y, Kim S, Fu Q, Parikh D, Sridhar B, Shi Q, Zhang X, Guan Y, Chen X, Xiang YK. Phosphodiesterases coordinate cAMP propagation induced by two stimulatory G protein-coupled receptors in hearts. Proc Natl Acad Sci USA 109: 6578–6583, 2012. doi: 10.1073/pnas.1117862109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Oliveira RF, Terrin A, Di Benedetto G, Cannon RC, Koh W, Kim M, Zaccolo M, Blackwell KT. The role of type 4 phosphodiesterases in generating microdomains of cAMP: large scale stochastic simulations. PLoS One 5: e11725, 2010. doi: 10.1371/journal.pone.0011725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Greenwald EC, Polanowska-Grabowska RK, Saucerman JJ. Integrating fluorescent biosensor data using computational models. Methods Mol Biol 1071: 227–248, 2014. doi: 10.1007/978-1-62703-622-1_18. [DOI] [PubMed] [Google Scholar]
- 144. Chay A, Zamparo I, Koschinski A, Zaccolo M, Blackwell KT. Control of betaAR- and N-methyl-D-aspartate (NMDA) receptor-dependent cAMP dynamics in hippocampal neurons. PLoS Comput Biol 12: e1004735, 2016. doi: 10.1371/journal.pcbi.1004735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Yang PC, Boras BW, Jeng MT, Docken SS, Lewis TJ, McCulloch AD, Harvey RD, Clancy CE. A computational modeling and simulation approach to investigate mechanisms of subcellular cAMP compartmentation. PLoS Comput Biol 12: e1005005, 2016. doi: 10.1371/journal.pcbi.1005005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Lohse C, Bock A, Maiellaro I, Hannawacker A, Schad LR, Lohse MJ, Bauer WR. Experimental and mathematical analysis of cAMP nanodomains. PLoS One 12: e0174856, 2017. doi: 10.1371/journal.pone.0174856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Halls ML, Cooper DM. Sub-picomolar relaxin signalling by a pre-assembled RXFP1, AKAP79, AC2, beta-arrestin 2, PDE4D3 complex. EMBO J 29: 2772–2787, 2010. doi: 10.1038/emboj.2010.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Rochais F, Abi-Gerges A, Horner K, Lefebvre F, Cooper DM, Conti M, Fischmeister R, Vandecasteele G. A specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circ Res 98: 1081–1088, 2006. doi: 10.1161/01.RES.0000218493.09370.8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Willoughby D, Wong W, Schaack J, Scott JD, Cooper DM. An anchored PKA and PDE4 complex regulates subplasmalemmal cAMP dynamics. EMBO J 25: 2051–2061, 2006. doi: 10.1038/sj.emboj.7601113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Timofeyev V, Myers RE, Kim HJ, Woltz RL, Sirish P, Heiserman JP, Li N, Singapuri A, Tang T, Yarov-Yarovoy V, Yamoah EN, Hammond HK, Chiamvimonvat N. Adenylyl cyclase subtype-specific compartmentalization: differential regulation of L-type Ca2+ current in ventricular myocytes. Circ Res 112: 1567–1576, 2013. doi: 10.1161/CIRCRESAHA.112.300370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Xiang Y, Naro F, Zoudilova M, Jin SL, Conti M, Kobilka B. Phosphodiesterase 4D is required for beta2 adrenoceptor subtype-specific signaling in cardiac myocytes. Proc Natl Acad Sci USA 102: 909–914, 2005. doi: 10.1073/pnas.0405263102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Barbagallo F, Xu B, Reddy GR, West T, Wang Q, Fu Q, Li M, Shi Q, Ginsburg KS, Ferrier W, Isidori AM, Naro F, Patel HH, Bossuyt J, Bers D, Xiang YK. Genetically encoded biosensors reveal PKA hyperphosphorylation on the myofilaments in rabbit heart failure. Circ Res 119: 931–943, 2016. doi: 10.1161/CIRCRESAHA.116.308964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Agarwal SR, Yang PC, Rice M, Singer CA, Nikolaev VO, Lohse MJ, Clancy CE, Harvey RD. Role of membrane microdomains in compartmentation of cAMP signaling. PLoS One 9: e95835, 2014. doi: 10.1371/journal.pone.0095835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Verde I, Pahlke G, Salanova M, Zhang G, Wang S, Coletti D, Onuffer J, Jin SL, Conti M. Myomegalin is a novel protein of the golgi/centrosome that interacts with a cyclic nucleotide phosphodiesterase. J Biol Chem 276: 11189–11198, 2001. doi: 10.1074/jbc.M006546200. [DOI] [PubMed] [Google Scholar]
- 155. Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 437: 574–578, 2005. doi: 10.1038/nature03966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Dodge KL, Khouangsathiene S, Kapiloff MS, Mouton R, Hill EV, Houslay MD, Langeberg LK, Scott JD. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J 20: 1921–1930, 2001. doi: 10.1093/emboj/20.8.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Pyne NJ, Cooper ME, Houslay MD. Identification and characterization of both the cytosolic and particulate forms of cyclic GMP-stimulated cyclic AMP phosphodiesterase from rat liver. Biochem J 234: 325–334, 1986. doi: 10.1042/bj2340325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Takimoto E, Champion HC, Belardi D, Moslehi J, Mongillo M, Mergia E, Montrose DC, Isoda T, Aufiero K, Zaccolo M, Dostmann WR, Smith CJ, Kass DA. cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ Res 96: 100–109, 2005. doi: 10.1161/01.RES.0000152262.22968.72. [DOI] [PubMed] [Google Scholar]
- 159. Schobesberger S, Wright PT, Poulet C, Sanchez Alonso Mardones JL, Mansfield C, Friebe A, Harding SE, Balligand JL, Nikolaev VO, Gorelik J. beta(3)-Adrenoceptor redistribution impairs NO/cGMP/PDE2 signalling in failing cardiomyocytes. eLife 9: e52221, 2020. doi: 10.7554/eLife.52221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Subramanian H, Froese A, Jönsson P, Schmidt H, Gorelik J, Nikolaev VO. Distinct submembrane localisation compartmentalises cardiac NPR1 and NPR2 signalling to cGMP. Nat Commun 9: 2446, 2018. doi: 10.1038/s41467-018-04891-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Shi Q, Li M, Mika D, Fu Q, Kim S, Phan J, Shen A, Vandecasteele G, Xiang YK. Heterologous desensitization of cardiac beta-adrenergic signal via hormone-induced betaAR/arrestin/PDE4 complexes. Cardiovasc Res 113: 656–670, 2017. doi: 10.1093/cvr/cvx036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Mika D, Richter W, Westenbroek RE, Catterall WA, Conti M. PDE4B mediates local feedback regulation of beta1-adrenergic cAMP signaling in a sarcolemmal compartment of cardiac myocytes. J Cell Sci 127: 1033–1042, 2014. doi: 10.1242/jcs.140251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Berthouze-Duquesnes M, Lucas A, Saulière A, Sin YY, Laurent AC, Galés C, Baillie G, Lezoualc'h F. Specific interactions between Epac1, beta-arrestin2 and PDE4D5 regulate beta-adrenergic receptor subtype differential effects on cardiac hypertrophic signaling. Cell Signal 25: 970–980, 2013. doi: 10.1016/j.cellsig.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 164. De Arcangelis V, Liu R, Soto D, Xiang Y. Differential association of phosphodiesterase 4D isoforms with beta2-adrenoceptor in cardiac myocytes. J Biol Chem 284: 33824–33832, 2009. doi: 10.1074/jbc.M109.020388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Richter W, Day P, Agrawal R, Bruss MD, Granier S, Wang YL, Rasmussen SG, Horner K, Wang P, Lei T, Patterson AJ, Kobilka B, Conti M. Signaling from beta1- and beta2-adrenergic receptors is defined by differential interactions with PDE4. EMBO J 27: 384–393, 2008. doi: 10.1038/sj.emboj.7601968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, Miller WE, McLean AJ, Conti M, Houslay MD, Lefkowitz RJ. Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science 298: 834–836, 2002. doi: 10.1126/science.1074683. [DOI] [PubMed] [Google Scholar]
- 167. Zhang Y, Knight W, Chen S, Mohan A, Yan C. Multiprotein complex with TRPC (transient receptor potential-canonical) channel, PDE1C (phosphodiesterase 1C), and A2R (adenosine A2 receptor) plays a critical role in regulating cardiomyocyte cAMP and survival. Circulation 138: 1988–2002, 2018. doi: 10.1161/CIRCULATIONAHA.118.034189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Castro LR, Schittl J, Fischmeister R. Feedback control through cGMP-dependent protein kinase contributes to differential regulation and compartmentation of cGMP in rat cardiac myocytes. Circ Res 107: 1232–1240, 2010. doi: 10.1161/CIRCRESAHA.110.226712. [DOI] [PubMed] [Google Scholar]
- 169. Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M, Marks AR. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 123: 25–35, 2005. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Leroy J, Richter W, Mika D, Castro LR, Abi-Gerges A, Xie M, Scheitrum C, Lefebvre F, Schittl J, Mateo P, Westenbroek R, Catterall WA, Charpentier F, Conti M, Fischmeister R, Vandecasteele G. Phosphodiesterase 4B in the cardiac L-type Ca2+ channel complex regulates Ca2+ current and protects against ventricular arrhythmias in mice. J Clin Invest 121: 2651–2661, 2011. doi: 10.1172/JCI44747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Terrenoire C, Houslay MD, Baillie GS, Kass RS. The cardiac IKs potassium channel macromolecular complex includes the phosphodiesterase PDE4D3. J Biol Chem 284: 9140–9146, 2009. doi: 10.1074/jbc.M805366200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Bastug-Özel Z, Wright PT, Kraft AE, Pavlovic D, Howie J, Froese A, Fuller W, Gorelik J, Shattock MJ, Nikolaev VO. Heart failure leads to altered beta2-adrenoceptor/cyclic adenosine monophosphate dynamics in the sarcolemmal phospholemman/Na,K ATPase microdomain. Cardiovasc Res 115: 546–555, 2019. doi: 10.1093/cvr/cvy221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Beca S, Helli PB, Simpson JA, Zhao D, Farman GP, Jones P, Tian X, Wilson LS, Ahmad F, Chen SR, Movsesian MA, Manganiello V, Maurice DH, Conti M, Backx PH. Phosphodiesterase 4D regulates baseline sarcoplasmic reticulum Ca2+ release and cardiac contractility, independently of L-type Ca2+ current. Circ Res 109: 1024–1030, 2011. doi: 10.1161/CIRCRESAHA.111.250464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Taskén KA, Collas P, Kemmner WA, Witczak O, Conti M, Taskén K. Phosphodiesterase 4D and protein kinase a type II constitute a signaling unit in the centrosomal area. J Biol Chem 276: 21999–22002, 2001. doi: 10.1074/jbc.C000911200. [DOI] [PubMed] [Google Scholar]
- 175. Terrin A, Monterisi S, Stangherlin A, Zoccarato A, Koschinski A, Surdo NC, Mongillo M, Sawa A, Jordanides NE, Mountford JC, Zaccolo M. PKA and PDE4D3 anchoring to AKAP9 provides distinct regulation of cAMP signals at the centrosome. J Cell Biol 198: 607–621, 2012. doi: 10.1083/jcb.201201059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Bedioune I, Lefebvre F, Lechêne P, Varin A, Domergue V, Kapiloff MS, Fischmeister R, Vandecasteele G. PDE4 and mAKAPβ are nodal organizers of β2-ARs nuclear PKA signalling in cardiac myocytes. Cardiovasc Res 114: 1499–1511, 2018. doi: 10.1093/cvr/cvy110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Carlson CR, Aronsen JM, Bergan-Dahl A, Moutty MC, Lunde M, Lunde PK, Jarstadmarken H, Wanichawan P, Pereira L, Kolstad TRS, Dalhus B, Subramanian H, Hille S, Christensen G, Müller OJ, Nikolaev V, Bers DM, Sjaastad I, Shen X, Louch WE, Klussmann E, Sejersted OM. AKAP18delta anchors and regulates CaMKII activity at phospholamban-SERCA2a2 and RYR. Circ Res 130: 27–44, 2022. doi: 10.1161/CIRCRESAHA.120.317976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Clister T, Greenwald EC, Baillie GS, Zhang J. akap95 organizes a nuclear microdomain to control local cAMP for regulating nuclear PKA. Cell Chem Biol 26: 885–891 e4, 2019. doi: 10.1016/j.chembiol.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Prüschenk S, Majer M, Schlossmann J. Novel Functional Features of cGMP Substrate Proteins IRAG1 and IRAG2. Int J Mol Sci 24: 9837, 2023. doi: 10.3390/ijms24129837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Adams SR, Harootunian AT, Buechler YJ, Taylor SS, Tsien RY. Fluorescence ratio imaging of cyclic AMP in single cells. Nature 349: 694–697, 1991. doi: 10.1038/349694a0. [DOI] [PubMed] [Google Scholar]
- 181. Zaccolo M, De Giorgi F, Cho CY, Feng L, Knapp T, Negulescu PA, Taylor SS, Tsien RY, Pozzan T. A genetically encoded, fluorescent indicator for cyclic AMP in living cells. Nat Cell Biol 2: 25–29, 2000. doi: 10.1038/71345. [DOI] [PubMed] [Google Scholar]
- 182. Nikolaev VO, Bünemann M, Hein L, Hannawacker A, Lohse MJ. Novel single chain cAMP sensors for receptor-induced signal propagation. J Biol Chem 279: 37215–37218, 2004. doi: 10.1074/jbc.C400302200. [DOI] [PubMed] [Google Scholar]
- 183. Ponsioen B, Zhao J, Riedl J, Zwartkruis F, van der Krogt G, Zaccolo M, Moolenaar WH, Bos JL, Jalink K. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep 5: 1176–1180, 2004. doi: 10.1038/sj.embor.7400290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Surdo NC, Berrera M, Koschinski A, Brescia M, Machado MR, Carr C, Wright P, Gorelik J, Morotti S, Grandi E, Bers DM, Pantano S, Zaccolo M. FRET biosensor uncovers cAMP nano-domains at beta-adrenergic targets that dictate precise tuning of cardiac contractility. Nat Commun 8: 15031, 2017. doi: 10.1038/ncomms15031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Anton SE, Kayser C, Maiellaro I, Nemec K, Möller J, Koschinski A, Zaccolo M, Annibale P, Falcke M, Lohse MJ, Bock A. Receptor-associated independent cAMP nanodomains mediate spatiotemporal specificity of GPCR signaling. Cell 185: 1130–1142.e11, 2022. doi: 10.1016/j.cell.2022.02.011. [DOI] [PubMed] [Google Scholar]
- 186. DiPilato LM, Cheng X, Zhang J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc Natl Acad Sci USA 101: 16513–16518, 2004. doi: 10.1073/pnas.0405973101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Allen MD, Zhang J. Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem Biophys Res Commun 348: 716–721, 2006. doi: 10.1016/j.bbrc.2006.07.136. [DOI] [PubMed] [Google Scholar]
- 188. Liu S, Zhang J, Xiang YK. FRET-based direct detection of dynamic protein kinase A activity on the sarcoplasmic reticulum in cardiomyocytes. Biochem Biophys Res Commun 404: 581–586, 2011. doi: 10.1016/j.bbrc.2010.11.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Lefkimmiatis K, Leronni D, Hofer AM. The inner and outer compartments of mitochondria are sites of distinct cAMP/PKA signaling dynamics. J Cell Biol 202: 453–462, 2013. doi: 10.1083/jcb.201303159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Sprenger JU, Perera RK, Steinbrecher JH, Lehnart SE, Maier LS, Hasenfuss G, Nikolaev VO. In vivo model with targeted cAMP biosensor reveals changes in receptor-microdomain communication in cardiac disease. Nat Commun 6: 6965, 2015. doi: 10.1038/ncomms7965. [DOI] [PubMed] [Google Scholar]
- 191. Agarwal SR, Miyashiro K, Latt H, Ostrom RS, Harvey RD. Compartmentalized cAMP responses to prostaglandin EP2 receptor activation in human airway smooth muscle cells. Br J Pharmacol 174: 2784–2796, 2017. doi: 10.1111/bph.13904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Pendin D, Greotti E, Lefkimmiatis K, Pozzan T. Exploring cells with targeted biosensors. J Gen Physiol 149: 1–36, 2017. doi: 10.1085/jgp.201611654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Calamera G, Li D, Ulsund AH, Kim JJ, Neely OC, Moltzau LR, Bjørnerem M, Paterson D, Kim C, Levy FO, Andressen KW. FRET-based cyclic GMP biosensors measure low cGMP concentrations in cardiomyocytes and neurons. Commun Biol 2: 394, 2019. doi: 10.1038/s42003-019-0641-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Wang L, Wu C, Peng W, Zhou Z, Zeng J, Li X, Yang Y, Yu S, Zou Y, Huang M, Liu C, Chen Y, Li Y, Ti P, Liu W, Gao Y, Zheng W, Zhong H, Gao S, Lu Z, Ren PG, Ng HL, He J, Chen S, Xu M, Li Y, Chu J. A high-performance genetically encoded fluorescent indicator for in vivo cAMP imaging. Nat Commun 13: 5363, 2022. doi: 10.1038/s41467-022-32994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Bers DM. Cardiac excitation-contraction coupling. Nature 415: 198–205, 2002. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 196. Bers DM. Macromolecular complexes regulating cardiac ryanodine receptor function. J Mol Cell Cardiol 37: 417–429, 2004. doi: 10.1016/j.yjmcc.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 197. Bers DM, Ginsburg KS. Na:Ca stoichiometry and cytosolic Ca-dependent activation of NCX in intact cardiomyocytes. Ann NY Acad Sci 1099: 326–338, 2007. doi: 10.1196/annals.1387.060. [DOI] [PubMed] [Google Scholar]
- 198. Papa A, Zakharov SI, Katchman AN, Kushner JS, Chen BX, Yang L, Liu G, Jimenez AS, Eisert RJ, Bradshaw GA, Dun W, Ali SR, Rodriques A, Zhou K, Topkara V, Yang M, Morrow JP, Tsai EJ, Karlin A, Wan E, Kalocsay M, Pitt GS, Colecraft HM, Ben-Johny M, Marx SO. Rad regulation of CaV1.2 channels controls cardiac fight-or-flight response. Nat Cardiovasc Res 1: 1022–1038, 2022. doi: 10.1038/s44161-022-00157-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Wang Y, Shi Q, Li M, Zhao M, Reddy Gopireddy R, Teoh JP, Xu B, Zhu C, Ireton KE, Srinivasan S, Chen S, Gasser PJ, Bossuyt J, Hell JW, Bers DM, Xiang YK. Intracellular beta1-adrenergic receptors and organic cation transporter 3 mediate phospholamban phosphorylation to enhance cardiac contractility. Circ Res 128: 246–261, 2021. doi: 10.1161/CIRCRESAHA.120.317452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Wang Q, Wang Y, West TM, Liu Y, Reddy GR, Barbagallo F, Xu B, Shi Q, Deng B, Wei W, Xiang YK. Carvedilol induces biased beta1 adrenergic receptor-nitric oxide synthase 3-cyclic guanylyl monophosphate signalling to promote cardiac contractility. Cardiovasc Res 117: 2237–2251, 2021. doi: 10.1093/cvr/cvaa266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Zaccolo M, Movsesian MA. cAMP and cGMP signaling cross-talk: role of phosphodiesterases and implications for cardiac pathophysiology. Circ Res 100: 1569–1578, 2007. doi: 10.1161/CIRCRESAHA.106.144501. [DOI] [PubMed] [Google Scholar]
- 202. Berisha F, Götz KR, Wegener JW, Brandenburg S, Subramanian H, Molina CE, Rüffer A, Petersen J, Bernhardt A, Girdauskas E, Jungen C, Pape U, Kraft AE, Warnke S, Lindner D, Westermann D, Blankenberg S, Meyer C, Hasenfuß G, Lehnart SE, Nikolaev VO. cAMP imaging at ryanodine receptors reveals beta2-adrenoceptor driven arrhythmias. Circ Res 129: 81–94, 2021. doi: 10.1161/CIRCRESAHA.120.318234. [DOI] [PubMed] [Google Scholar]
- 203. Wang Y, Zhao M, Shi Q, Xu B, Zhu C, Li M, Mir V, Bers DM, Xiang YK. Monoamine oxidases desensitize intracellular beta1AR signaling in heart failure. Circ Res 129: 965–967, 2021. doi: 10.1161/CIRCRESAHA.121.319546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Xu B, Wang Y, Bahriz SMFM, Zhao M, Zhu C, Xiang YK. Probing spatiotemporal PKA activity at the ryanodine receptor and SERCA2a2a nanodomains in cardiomyocytes. Cell Commun Signal 20: 143, 2022. doi: 10.1186/s12964-022-00947-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Fu Q, Kim S, Soto D, De Arcangelis V, DiPilato L, Liu S, Xu B, Shi Q, Zhang J, Xiang YK. A long lasting beta1 adrenergic receptor stimulation of cAMP/protein kinase A (PKA) signal in cardiac myocytes. J Biol Chem 289: 14771–14781, 2014. doi: 10.1074/jbc.M113.542589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Kerfant BG, Zhao D, Lorenzen-Schmidt I, Wilson LS, Cai S, Chen SR, Maurice DH, Backx PH. PI3Kgamma is required for PDE4, not PDE3, activity in subcellular microdomains containing the sarcoplasmic reticular calcium ATPase in cardiomyocytes. Circ Res 101: 400–408, 2007. doi: 10.1161/CIRCRESAHA.107.156422. [DOI] [PubMed] [Google Scholar]
- 207. Verde I, Vandecasteele G, Lezoualc’h F, Fischmeister R. Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes. Br J Pharmacol 127: 65–74, 1999. doi: 10.1038/sj.bjp.0702506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Richter W, Jin SL, Conti M. Splice variants of the cyclic nucleotide phosphodiesterase PDE4D are differentially expressed and regulated in rat tissue. Biochem J 388: 803–811, 2005. doi: 10.1042/BJ20050030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Abi-Gerges A, Castro L, Leroy J, Domergue V, Fischmeister R, Vandecasteele G. Selective changes in cytosolic beta-adrenergic cAMP signals and L-type calcium channel regulation by phosphodiesterases during cardiac hypertrophy. J Mol Cell Cardiol 150: 109–121, 2021. doi: 10.1016/j.yjmcc.2020.10.011. [DOI] [PubMed] [Google Scholar]
- 210. Terrin A, Di Benedetto G, Pertegato V, Cheung YF, Baillie G, Lynch MJ, Elvassore N, Prinz A, Herberg FW, Houslay MD, Zaccolo M. PGE1 stimulation of HEK293 cells generates multiple contiguous domains with different [cAMP]: role of compartmentalized phosphodiesterases. J Cell Biol 175: 441–451, 2006. doi: 10.1083/jcb.200605050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Christ T, Galindo-Tovar A, Thoms M, Ravens U, Kaumann AJ. Inotropy and L-type Ca2+ current, activated by beta1- and beta2-adrenoceptors, are differently controlled by phosphodiesterases 3 and 4 in rat heart. Br J Pharmacol 156: 62–83, 2009. doi: 10.1111/j.1476-5381.2008.00015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Weber S, Zeller M, Guan K, Wunder F, Wagner M, El-Armouche A. PDE2 at the crossway between cAMP and cGMP signalling in the heart. Cell Signal 38: 76–84, 2017. doi: 10.1016/j.cellsig.2017.06.020. [DOI] [PubMed] [Google Scholar]
- 213. Vandecasteele G, Verde I, Rücker-Martin C, Donzeau-Gouge P, Fischmeister R. Cyclic GMP regulation of the L-type Ca2+ channel current in human atrial myocytes. J Physiol 533: 329–340, 2001. doi: 10.1111/j.1469-7793.2001.0329a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Mika D, Bobin P, Pomérance M, Lechêne P, Westenbroek RE, Catterall WA, Vandecasteele G, Leroy J, Fischmeister R. Differential regulation of cardiac excitation-contraction coupling by cAMP phosphodiesterase subtypes. Cardiovasc Res 100: 336–346, 2013. doi: 10.1093/cvr/cvt193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Hartzell HC, Fischmeister R. Opposite effects of cyclic GMP and cyclic AMP on Ca2+ current in single heart cells. Nature 323: 273–275, 1986. doi: 10.1038/323273a0. [DOI] [PubMed] [Google Scholar]
- 216. Dittrich M, Jurevicius J, Georget M, Rochais F, Fleischmann B, Hescheler J, Fischmeister R. Local response of L-type Ca2+ current to nitric oxide in frog ventricular myocytes. J Physiol 534: 109–121, 2001. doi: 10.1111/j.1469-7793.2001.00109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Soler F, Fernández-Belda F, Pérez-Schindler J, Handschin C, Fuente T, Hernandez-Cascales J. PDE2 activity differs in right and left rat ventricular myocardium and differentially regulates beta2 adrenoceptor-mediated effects. Exp Biol Med (Maywood) 240: 1205–1213, 2015. doi: 10.1177/1535370214560969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Vettel C, Lindner M, Dewenter M, Lorenz K, Schanbacher C, Riedel M, Lämmle S, Meinecke S, Mason FE, Sossalla S, Geerts A, Hoffmann M, Wunder F, Brunner FJ, Wieland T, Mehel H, Karam S, Lechêne P, Leroy J, Vandecasteele G, Wagner M, Fischmeister R, El-Armouche A. Phosphodiesterase 2 protects against catecholamine-induced arrhythmia and preserves contractile function after myocardial infarction. Circ Res 120: 120–132, 2017. doi: 10.1161/CIRCRESAHA.116.310069. [DOI] [PubMed] [Google Scholar]
- 219. Perera RK, Sprenger JU, Steinbrecher JH, Hübscher D, Lehnart SE, Abesser M, Schuh K, El-Armouche A, Nikolaev VO. Microdomain switch of cGMP-regulated phosphodiesterases leads to ANP-induced augmentation of beta-adrenoceptor-stimulated contractility in early cardiac hypertrophy. Circ Res 116: 1304–1311, 2015. doi: 10.1161/CIRCRESAHA.116.306082. [DOI] [PubMed] [Google Scholar]
- 220. Gauthier C, Leblais V, Kobzik L, Trochu JN, Khandoudi N, Bril A, Balligand JL, Le Marec H. The negative inotropic effect of beta3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J Clin Invest 102: 1377–1384, 1998. doi: 10.1172/JCI2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Heubach JF, Rau T, Eschenhagen T, Ravens U, Kaumann AJ. Physiological antagonism between ventricular beta 1-adrenoceptors and alpha 1-adrenoceptors but no evidence for beta 2- and beta 3-adrenoceptor function in murine heart. Br J Pharmacol 136: 217–229, 2002. doi: 10.1038/sj.bjp.0704700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Stangherlin A, Gesellchen F, Zoccarato A, Terrin A, Fields LA, Berrera M, Surdo NC, Craig MA, Smith G, Hamilton G, Zaccolo M. cGMP signals modulate cAMP levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes. Circ Res 108: 929–939, 2011. doi: 10.1161/CIRCRESAHA.110.230698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Yano M, Kohno M, Ohkusa T, Mochizuki M, Yamada J, Kohno M, Hisaoka T, Ono K, Tanigawa T, Kobayashi S, Matsuzaki M. Effect of milrinone on left ventricular relaxation and Ca2+ uptake function of cardiac sarcoplasmic reticulum. Am J Physiol Heart Circ Physiol 279: H1898–H1905, 2000. doi: 10.1152/ajpheart.2000.279.4.H1898. [DOI] [PubMed] [Google Scholar]
- 224. Malecot CO, Bers DM, Katzung BG. Biphasic contractions induced by milrinone at low temperature in ferret ventricular muscle: role of the sarcoplasmic reticulum and transmembrane calcium influx. Circ Res 59: 151–162, 1986. doi: 10.1161/01.res.59.2.151. [DOI] [PubMed] [Google Scholar]
- 225. Wang Y, Zhao M, Xu B, Bahriz SM, Zhu C, Jovanovic A, Ni H, Jacobi A, Kaludercic N, Di Lisa F, Hell JW, Shih JC, Paolocci N, Xiang YK. Monoamine oxidase A and organic cation transporter 3 coordinate intracellular beta1AR signaling to calibrate cardiac contractile function. Basic Res Cardiol 117: 37, 2022. doi: 10.1007/s00395-022-00944-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Sande JB, Sjaastad I, Hoen IB, Bøkenes J, Tønnessen T, Holt E, Lunde PK, Christensen G. Reduced level of serine16 phosphorylated phospholamban in the failing rat myocardium: a major contributor to reduced SERCA2a2 activity. Cardiovasc Res 53: 382–391, 2002. doi: 10.1016/s0008-6363(01)00489-8. [DOI] [PubMed] [Google Scholar]
- 227. Minamisawa S, Hoshijima M, Chu G, Ward CA, Frank K, Gu Y, Martone ME, Wang Y, Ross J, Kranias EG, Giles WR, Chien KR. Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell 99: 313–322, 1999. doi: 10.1016/s0092-8674(00)81662-1. [DOI] [PubMed] [Google Scholar]
- 228. Schwinger RH, Böhm M, Schmidt U, Karczewski P, Bavendiek U, Flesch M, Krause EG, Erdmann E. Unchanged protein levels of SERCA2a II and phospholamban but reduced Ca2+ uptake and Ca2+-ATPase activity of cardiac sarcoplasmic reticulum from dilated cardiomyopathy patients compared with patients with nonfailing hearts. Circulation 92: 3220–3228, 1995. doi: 10.1161/01.cir.92.11.3220. [DOI] [PubMed] [Google Scholar]
- 229. Aronsen JM, Skogestad J, Albert I, Melleby AO, Carlson CR. Disruption of phosphodiesterase 3A binding to SERCA2a2 increases SERCA2a2 activity and reduces mortality in mice with chronic heart failure. Circulation 148: 857–858, 2023. doi: 10.1161/CIRCULATIONAHA.123.065884. [DOI] [PubMed] [Google Scholar]
- 230. Ercu M, Mücke MB, Pallien T, Markó L, Sholokh A, Schächterle C, et al. Mutant phosphodiesterase 3A protects from hypertension-induced cardiac damage. Circulation 146: 1758–1778, 2022. doi: 10.1161/CIRCULATIONAHA.122.060210. [DOI] [PubMed] [Google Scholar]
- 231. Meier S, Andressen KW, Aronsen JM, Sjaastad I, Hougen K, Skomedal T, Osnes JB, Qvigstad E, Levy FO, Moltzau LR. PDE3 inhibition by C-type natriuretic peptide-induced cGMP enhances cAMP-mediated signaling in both non-failing and failing hearts. Eur J Pharmacol 812: 174–183, 2017. doi: 10.1016/j.ejphar.2017.07.014. [DOI] [PubMed] [Google Scholar]
- 232. Takimoto E, Belardi D, Tocchetti CG, Vahebi S, Cormaci G, Ketner EA, Moens AL, Champion HC, Kass DA. Compartmentalization of cardiac beta-adrenergic inotropy modulation by phosphodiesterase type 5. Circulation 115: 2159–2167, 2007. doi: 10.1161/CIRCULATIONAHA.106.643536. [DOI] [PubMed] [Google Scholar]
- 233. Hashimoto T, Kim GE, Tunin RS, Adesiyun T, Hsu S, Nakagawa R, Zhu G, O’Brien JJ, Hendrick JP, Davis RE, Yao W, Beard D, Hoxie HR, Wennogle LP, Lee DI, Kass DA. Acute enhancement of cardiac function by phosphodiesterase type 1 inhibition. Circulation 138: 1974–1987, 2018. doi: 10.1161/CIRCULATIONAHA.117.030490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Xi J, McIntosh R, Shen X, Lee S, Chanoit G, Criswell H, Zvara DA, Xu Z. Adenosine A2A and A2B receptors work in concert to induce a strong protection against reperfusion injury in rat hearts. J Mol Cell Cardiol 47: 684–690, 2009. doi: 10.1016/j.yjmcc.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Muller GK, Song J, Jani V, Wu Y, Liu T, Jeffreys WPD, O’Rourke B, Anderson ME, Kass DA. PDE1 inhibition modulates Cav1.2 channel to stimulate cardiomyocyte contraction. Circ Res 129: 872–886, 2021. doi: 10.1161/CIRCRESAHA.121.319828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Moltzau LR, Meier S, Aronsen JM, Afzal F, Sjaastad I, Skomedal T, Osnes JB, Levy FO, Qvigstad E. Differential regulation of C-type natriuretic peptide-induced cGMP and functional responses by PDE2 and PDE3 in failing myocardium. Naunyn Schmiedebergs Arch Pharmacol 387: 407–417, 2014. doi: 10.1007/s00210-013-0953-1. [DOI] [PubMed] [Google Scholar]
- 237. Manfra O, Calamera G, Froese A, Arunthavarajah D, Surdo NC, Meier S, Melleby AO, Aasrum M, Aronsen JM, Nikolaev VO, Zaccolo M, Moltzau LR, Levy FO, Andressen KW. CNP regulates cardiac contractility and increases cGMP near both SERCA2a and TnI: difference from BNP visualized by targeted cGMP biosensors. Cardiovasc Res 118: 1506–1519, 2022. doi: 10.1093/cvr/cvab167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Qvigstad E, Moltzau LR, Aronsen JM, Nguyen CH, Hougen K, Sjaastad I, Levy FO, Skomedal T, Osnes JB. Natriuretic peptides increase beta1-adrenoceptor signalling in failing hearts through phosphodiesterase 3 inhibition. Cardiovasc Res 85: 763–772, 2010. doi: 10.1093/cvr/cvp364. [DOI] [PubMed] [Google Scholar]
- 239. Afzal F, Qvigstad E, Aronsen JM, Moltzau LR, Sjaastad I, Skomedal T, Osnes JB, Levy FO. Agents increasing cyclic GMP amplify 5-HT4-elicited positive inotropic response in failing rat cardiac ventricle. Naunyn Schmiedebergs Arch Pharmacol 384: 543–553, 2011. doi: 10.1007/s00210-011-0670-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Borlaug BA, Melenovsky V, Marhin T, Fitzgerald P, Kass DA. Sildenafil inhibits beta-adrenergic-stimulated cardiac contractility in humans. Circulation 112: 2642–2649, 2005. doi: 10.1161/CIRCULATIONAHA.105.540500. [DOI] [PubMed] [Google Scholar]
- 241. Senzaki H, Smith CJ, Juang GJ, Isoda T, Mayer SP, Ohler A, Paolocci N, Tomaselli GF, Hare JM, Kass DA. Cardiac phosphodiesterase 5 (cGMP-specific) modulates beta-adrenergic signaling in vivo and is down-regulated in heart failure. FASEB J 15: 1718–1726, 2001. doi: 10.1096/fj.00-0538com. [DOI] [PubMed] [Google Scholar]
- 242. Lee DI, Vahebi S, Tocchetti CG, Barouch LA, Solaro RJ, Takimoto E, Kass DA. PDE5A suppression of acute beta-adrenergic activation requires modulation of myocyte beta-3 signaling coupled to PKG-mediated troponin I phosphorylation. Basic Res Cardiol 105: 337–347, 2010. doi: 10.1007/s00395-010-0084-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. West TM, Wang Q, Deng B, Zhang Y, Barbagallo F, Reddy GR, Chen D, Phan KS, Xu B, Isidori A, Xiang YK. Phosphodiesterase 5 associates with beta2 adrenergic receptor to modulate cardiac function in type 2 diabetic hearts. J Am Heart Assoc 8: e012273, 2019. doi: 10.1161/JAHA.119.012273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Kötter S, Gout L, Von Frieling-Salewsky M, Müller AE, Helling S, Marcus K, Dos Remedios C, Linke WA, Krüger M. Differential changes in titin domain phosphorylation increase myofilament stiffness in failing human hearts. Cardiovasc Res 99: 648–656, 2013. doi: 10.1093/cvr/cvt144. [DOI] [PubMed] [Google Scholar]
- 245. Kotlo K, Johnson KR, Grillon JM, Geenen DL, deTombe P, Danziger RS. Phosphoprotein abundance changes in hypertensive cardiac remodeling. J Proteomics 77: 1–13, 2012. doi: 10.1016/j.jprot.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Hamdani N, Bishu KG, von Frieling-Salewsky M, Redfield MM, Linke WA. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc Res 97: 464–471, 2013. doi: 10.1093/cvr/cvs353. [DOI] [PubMed] [Google Scholar]
- 247. Gresham KS, Stelzer JE. The contributions of cardiac myosin binding protein C and troponin I phosphorylation to beta-adrenergic enhancement of in vivo cardiac function. J Physiol 594: 669–686, 2016. doi: 10.1113/JP270959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Lakatta EG, Maltsev VA, Vinogradova TM. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ Res 106: 659–673, 2010. doi: 10.1161/CIRCRESAHA.109.206078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Liu J, Sirenko S, Juhaszova M, Ziman B, Shetty V, Rain S, Shukla S, Spurgeon HA, Vinogradova TM, Maltsev VA, Lakatta EG. A full range of mouse sinoatrial node AP firing rates requires protein kinase A-dependent calcium signaling. J Mol Cell Cardiol 51: 730–739, 2011. doi: 10.1016/j.yjmcc.2011.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Curran J, Hinton MJ, Ríos E, Bers DM, Shannon TR. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res 100: 391–398, 2007. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- 251. Lukyanenko YO, Younes A, Lyashkov AE, Tarasov KV, Riordon DR, Lee J, Sirenko SG, Kobrinsky E, Ziman B, Tarasova YS, Juhaszova M, Sollott SJ, Graham DR, Lakatta EG. Ca2+/calmodulin-activated phosphodiesterase 1A is highly expressed in rabbit cardiac sinoatrial nodal cells and regulates pacemaker function. J Mol Cell Cardiol 98: 73–82, 2016. doi: 10.1016/j.yjmcc.2016.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Rivet-Bastide M, Vandecasteele G, Hatem S, Verde I, Bénardeau A, Mercadier JJ, Fischmeister R. cGMP-stimulated cyclic nucleotide phosphodiesterase regulates the basal calcium current in human atrial myocytes. J Clin Invest 99: 2710–2718, 1997. doi: 10.1172/JCI119460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Rozmaritsa N, Christ T, Van Wagoner DR, Haase H, Stasch J-P, Matschke K, Ravens U. Attenuated response of L-type calcium current to nitric oxide in atrial fibrillation. Cardiovasc Res 101: 533–542, 2014. doi: 10.1093/cvr/cvt334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Vinogradova TM, Sirenko S, Lyashkov AE, Younes A, Li Y, Zhu W, Yang D, Ruknudin AM, Spurgeon H, Lakatta EG. Constitutive phosphodiesterase activity restricts spontaneous beating rate of cardiac pacemaker cells by suppressing local Ca2+ releases. Circ Res 102: 761–769, 2008. doi: 10.1161/CIRCRESAHA.107.161679. [DOI] [PubMed] [Google Scholar]
- 255. Osadchii OE. Myocardial phosphodiesterases and regulation of cardiac contractility in health and cardiac disease. Cardiovasc Drugs Ther 21: 171–194, 2007. doi: 10.1007/s10557-007-6014-6. [DOI] [PubMed] [Google Scholar]
- 256. Tibbo AJ, Mika D, Dobi S, Ling J, McFall A, Tejeda GS, Blair C, MacLeod R, MacQuaide N, Gök C, Fuller W, Smith BO, Smith GL, Vandecasteele G, Brand T, Baillie GS. Phosphodiesterase type 4 anchoring regulates cAMP signaling to Popeye domain-containing proteins. J Mol Cell Cardiol 165: 86–102, 2022. doi: 10.1016/j.yjmcc.2022.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Herring N, Rigg L, Terrar DA, Paterson DJ. NO-cGMP pathway increases the hyperpolarisation-activated current, If, and heart rate during adrenergic stimulation. Cardiovasc Res 52: 446–453, 2001. doi: 10.1016/s0008-6363(01)00425-4. [DOI] [PubMed] [Google Scholar]
- 258. Wang Y, Wagner MB, Joyner RW, Kumar R. cGMP-dependent protein kinase mediates stimulation of L-type calcium current by cGMP in rabbit atrial cells. Cardiovasc Res 48: 310–322, 2000. doi: 10.1016/s0008-6363(00)00178-4. [DOI] [PubMed] [Google Scholar]
- 259. Peters CH, Myers ME, Juchno J, Haimbaugh C, Bichraoui H, Du Y, Bankston JR, Walker LA, Proenza C. Isoform-specific regulation of HCN4 channels by a family of endoplasmic reticulum proteins. Proc Natl Acad Sci USA 117: 18079–18090, 2020. doi: 10.1073/pnas.2006238117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Han X, Shimoni Y, Giles WR. A cellular mechanism for nitric oxide-mediated cholinergic control of mammalian heart rate. J Gen Physiol 106: 45–65, 1995. doi: 10.1085/jgp.106.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Isidori AM, Cornacchione M, Barbagallo F, Di Grazia A, Barrios F, Fassina L, Monaco L, Giannetta E, Gianfrilli D, Garofalo S, Zhang X, Chen X, Xiang YK, Lenzi A, Pellegrini M, Naro F. Inhibition of type 5 phosphodiesterase counteracts beta2-adrenergic signalling in beating cardiomyocytes. Cardiovasc Res 106: 408–420, 2015. doi: 10.1093/cvr/cvv123. [DOI] [PubMed] [Google Scholar]
- 262. Hutchings DC, Pearman CM, Madders GW, Woods LS, Eisner DA, Dibb KM, Trafford AW. PDE5 inhibition suppresses ventricular arrhythmias by reducing SR Ca2+ content. Circ Res 129: 650–665, 2021. doi: 10.1161/CIRCRESAHA.121.318473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Smith AH, Owen J, Borgman KY, Fish FA, Kannankeril PJ. Relation of milrinone after surgery for congenital heart disease to significant postoperative tachyarrhythmias. Am J Cardiol 108: 1620–1624, 2011. doi: 10.1016/j.amjcard.2011.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Packer M, Carver JR, Rodeheffer RJ, Ivanhoe RJ, DiBianco R, Zeldis SM, Hendrix GH, Bommer WJ, Elkayam U, Kukin ML. Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N Engl J Med 325: 1468–1475, 1991. doi: 10.1056/NEJM199111213252103. [DOI] [PubMed] [Google Scholar]
- 265. Molina CE, Leroy J, Richter W, Xie M, Scheitrum C, Lee IO, Maack C, Rucker-Martin C, Donzeau-Gouge P, Verde I, Llach A, Hove-Madsen L, Conti M, Vandecasteele G, Fischmeister R. Cyclic adenosine monophosphate phosphodiesterase type 4 protects against atrial arrhythmias. J Am Coll Cardiol 59: 2182–2190, 2012. doi: 10.1016/j.jacc.2012.01.060. [DOI] [PubMed] [Google Scholar]
- 266. Madhani M, Hall AR, Cuello F, Charles RL, Burgoyne JR, Fuller W, Hobbs AJ, Shattock MJ, Eaton P. Phospholemman Ser69 phosphorylation contributes to sildenafil-induced cardioprotection against reperfusion injury. Am J Physiol Heart Circ Physiol 299: H827–H836, 2010. doi: 10.1152/ajpheart.00129.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Hutchings DC, Anderson SG, Caldwell JL, Trafford AW. Phosphodiesterase-5 inhibitors and the heart: compound cardioprotection? Heart 104: 1244–1250, 2018. doi: 10.1136/heartjnl-2017-312865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Garnier A, Bork NI, Jacquet E, Zipfel S, Muñoz-Guijosa C, Baczkó I, Reichenspurner H, Donzeau-Gouge P, Maier LS, Dobrev D, Girdauskas E, Nikolaev VO, Fischmeister R, Molina CE. Mapping genetic changes in the cAMP-signaling cascade in human atria. J Mol Cell Cardiol 155: 10–20, 2021. doi: 10.1016/j.yjmcc.2021.02.006. [DOI] [PubMed] [Google Scholar]
- 269. Grammatika Pavlidou N, Dobrev S, Beneke K, Reinhardt F, Pecha S, Jacquet E, Abu-Taha IH, Schmidt C, Voigt N, Kamler M, Schnabel RB, Baczkó I, Garnier A, Reichenspurner H, Nikolaev VO, Dobrev D, Molina CE. Phosphodiesterase 8 governs cAMP/PKA-dependent reduction of L-type calcium current in human atrial fibrillation: a novel arrhythmogenic mechanism. Eur Heart J 44: 2483–2494, 2023. doi: 10.1093/eurheartj/ehad086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Srinivasan NT, Schilling RJ. Sudden cardiac death and arrhythmias. Arrhythm Electrophysiol Rev 7: 111–117, 2018. doi: 10.15420/aer.2018:15:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Wagner M, Sadek MS, Dybkova N, Mason FE, Klehr J, Firneburg R, Cachorro E, Richter K, Klapproth E, Kuenzel SR, Lorenz K, Heijman J, Dobrev D, El-Armouche A, Sossalla S, Kämmerer S. Cellular mechanisms of the anti-arrhythmic effect of cardiac PDE2 overexpression. Int J Mol Sci 22: 4816, 2021. doi: 10.3390/ijms22094816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Das S, Maulik N, Das DK, Kadowitz PJ, Bivalacqua TJ. Cardioprotection with sildenafil, a selective inhibitor of cyclic 3′,5′-monophosphate-specific phosphodiesterase 5. Drugs Exp Clin Res 28: 213–219, 2002. [PubMed] [Google Scholar]
- 273. Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu Q, Makarewich C, Ai X, Li Y, Tang A, Wang J, Gao H, Wang F, Ge XJ, Kunapuli SP, Zhou L, Zeng C, Xiang KY, Chen X. Cardiotoxic and cardioprotective features of chronic β-adrenergic signaling. Circ Res 112: 498–509, 2013. doi: 10.1161/CIRCRESAHA.112.273896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Roth DM, Gao MH, Lai NC, Drumm J, Dalton N, Zhou JY, Zhu J, Entrikin D, Hammond HK. Cardiac-directed adenylyl cyclase expression improves heart function in murine cardiomyopathy. Circulation 99: 3099–3102, 1999. doi: 10.1161/01.cir.99.24.3099. [DOI] [PubMed] [Google Scholar]
- 275. Nash CA, Brown LM, Malik S, Cheng X, Smrcka AV. Compartmentalized cyclic nucleotides have opposing effects on regulation of hypertrophic phospholipase Cepsilon signaling in cardiac myocytes. J Mol Cell Cardiol 121: 51–59, 2018. doi: 10.1016/j.yjmcc.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Ahmad T, Miller PE, McCullough M, Desai NR, Riello R, Psotka M, Böhm M, Allen LA, Teerlink JR, Rosano GM, Lindenfeld J. Why has positive inotropy failed in chronic heart failure? Lessons from prior inotrope trials. Eur J Heart Fail 21: 1064–1078, 2019. doi: 10.1002/ejhf.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Stangherlin A, Zaccolo M. cGMP-cAMP interplay in cardiac myocytes: a local affair with far-reaching consequences for heart function. Biochem Soc Trans 40: 11–14, 2012. doi: 10.1042/BST20110655. [DOI] [PubMed] [Google Scholar]
- 278. Zoccarato A, Surdo NC, Aronsen JM, Fields LA, Mancuso L, Dodoni G, Stangherlin A, Livie C, Jiang H, Sin YY, Gesellchen F, Terrin A, Baillie GS, Nicklin SA, Graham D, Szabo-Fresnais N, Krall J, Vandeput F, Movsesian M, Furlan L, Corsetti V, Hamilton G, Lefkimmiatis K, Sjaastad I, Zaccolo M. Cardiac hypertrophy is inhibited by a local pool of cAMP regulated by phosphodiesterase 2. Circ Res 117: 707–719, 2015. doi: 10.1161/CIRCRESAHA.114.305892. [DOI] [PubMed] [Google Scholar]
- 279. Ritchie RH, Irvine JC, Rosenkranz AC, Patel R, Wendt IR, Horowitz JD, Kemp-Harper BK. Exploiting cGMP-based therapies for the prevention of left ventricular hypertrophy: NO* and beyond. Pharmacol Ther 124: 279–300, 2009. doi: 10.1016/j.pharmthera.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 280. Goldberg ND, Haddox MK, Nicol SE, Glass DB, Sanford CH, Kuehl FA Jr, Estensen R. Biologic regulation through opposing influences of cyclic GMP and cyclic AMP: the Yin Yang hypothesis. Adv Cyclic Nucleotide Res 5: 307–330, 1975. [PubMed] [Google Scholar]
- 281. Wu MP, Zhang YS, Xu X, Zhou Q, Li JD, Yan C. Vinpocetine attenuates pathological cardiac remodeling by inhibiting cardiac hypertrophy and fibrosis. Cardiovasc Drugs Ther 31: 157–166, 2017. doi: 10.1007/s10557-017-6719-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Movsesian MA, Smith CJ, Krall J, Bristow MR, Manganiello VC. Sarcoplasmic reticulum-associated cyclic adenosine 5'-monophosphate phosphodiesterase activity in normal and failing human hearts. J Clin Invest 88: 15–19, 1991. doi: 10.1172/JCI115272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. von der Leyen H, Mende U, Meyer W, Neumann J, Nose M, Schmitz W, Scholz H, Starbatty J, Stein B, Wenzlaff H. Mechanism underlying the reduced positive inotropic effects of the phosphodiesterase III inhibitors pimobendan, adibendan and saterinone in failing as compared to nonfailing human cardiac muscle preparations. Naunyn Schmiedebergs Arch Pharmacol 344: 90–100, 1991. doi: 10.1007/BF00167387. [DOI] [PubMed] [Google Scholar]
- 284. Abi-Gerges A, Richter W, Lefebvre F, Mateo P, Varin A, Heymes C, Samuel J-L, Lugnier C, Conti M, Fischmeister R, Vandecasteele G. Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on beta-adrenergic cAMP signals. Circ Res 105: 784–792, 2009. doi: 10.1161/CIRCRESAHA.109.197947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Takahashi K, Osanai T, Nakano T, Wakui M, Okumura K. Enhanced activities and gene expression of phosphodiesterase types 3 and 4 in pressure-induced congestive heart failure. Heart Vessels 16: 249–256, 2002. doi: 10.1007/s003800200032. [DOI] [PubMed] [Google Scholar]
- 286. Polidovitch N, Yang S, Sun H, Lakin R, Ahmad F, Gao X, Turnbull PC, Chiarello C, Perry CG, Manganiello V, Yang P, Backx PH. Phosphodiesterase type 3A (PDE3A), but not type 3B (PDE3B), contributes to the adverse cardiac remodeling induced by pressure overload. J Mol Cell Cardiol 132 : 60–70, 2019. doi: 10.1016/j.yjmcc.2019.04.028. [DOI] [PubMed] [Google Scholar]
- 287. Wang L, Burmeister BT, Johnson KR, Baillie GS, Karginov AV, Skidgel RA, O’Bryan JP, Carnegie GK. UCR1C is a novel activator of phosphodiesterase 4 (PDE4) long isoforms and attenuates cardiomyocyte hypertrophy. Cell Signal 27: 908–922, 2015. doi: 10.1016/j.cellsig.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Karam S, Margaria JP, Bourcier A, Mika D, Varin A, Bedioune I, Lindner M, Bouadjel K, Dessillons M, Gaudin F, Lefebvre F, Mateo P, Lechène P, Gomez S, Domergue V, Robert P, Coquard C, Algalarrondo V, Samuel JL, Michel JB, Charpentier F, Ghigo A, Hirsch E, Fischmeister R, Leroy J, Vandecasteele G. Cardiac overexpression of PDE4B blunts β-adrenergic response and maladaptive remodeling in heart failure. Circulation 142: 161–174, 2020. doi: 10.1161/CIRCULATIONAHA.119.042573. [DOI] [PubMed] [Google Scholar]
- 289. Zhang M, Takimoto E, Lee DI, Santos CX, Nakamura T, Hsu S, Jiang A, Nagayama T, Bedja D, Yuan Y, Eaton P, Shah AM, Kass DA. Pathological cardiac hypertrophy alters intracellular targeting of phosphodiesterase type 5 from nitric oxide synthase-3 to natriuretic peptide signaling. Circulation 126: 942–951, 2012. doi: 10.1161/CIRCULATIONAHA.112.090977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Lu Z, Xu X, Hu X, Lee S, Traverse JH, Zhu G, Fassett J, Tao Y, Zhang P, dos Remedios C, Pritzker M, Hall JL, Garry DJ, Chen Y. Oxidative stress regulates left ventricular PDE5 expression in the failing heart. Circulation 121: 1474–1483, 2010. doi: 10.1161/CIRCULATIONAHA.109.906818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Nakamura T, Zhu G, Ranek MJ, Kokkonen-Simon K, Zhang M, Kim GE, Tsujita K, Kass DA. Prevention of PKG-1alpha oxidation suppresses antihypertrophic/antifibrotic effects from PDE5 inhibition but not sGC stimulation. Circ Heart Fail 11: e004740, 2018. doi: 10.1161/CIRCHEARTFAILURE.117.004740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Huang CK, Chen BY, Guo A, Chen R, Zhu YQ, Kutschke W, Hong J, Song LS. Sildenafil ameliorates left ventricular T-tubule remodeling in a pressure overload-induced murine heart failure model. Acta Pharmacol Sin 37: 473–482, 2016. doi: 10.1038/aps.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y, Kass DA. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med 11: 214–222, 2005. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]
- 294. Escudero DS, Brea MS, Caldiz CI, Amarillo ME, Aranda JO, Portiansky EL, Pérez NG, Díaz RG. PDE5 inhibition improves cardiac morphology and function in SHR by reducing NHE1 activity: repurposing sildenafil for the treatment of hypertensive cardiac hypertrophy. Eur J Pharmacol 891: 173724, 2021. doi: 10.1016/j.ejphar.2020.173724. [DOI] [PubMed] [Google Scholar]
- 295. Richards DA, Aronovitz MJ, Liu P, Martin GL, Tam K, Pande S, Karas RH, Bloomfield DM, Mendelsohn ME, Blanton RM. CRD-733, a novel PDE9 (phosphodiesterase 9) inhibitor, reverses pressure overload-induced heart failure. Circ Heart Fail 14: e007300, 2021. doi: 10.1161/CIRCHEARTFAILURE.120.007300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Wang PX, Li ZM, Cai SD, Li JY, He P, Huang Y, Feng GS, Luo HB, Chen SR, Liu PQ. C33(S), a novel PDE9A inhibitor, protects against rat cardiac hypertrophy through upregulating cGMP signaling. Acta Pharmacol Sin 38: 1257–1268, 2017. doi: 10.1038/aps.2017.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson JA, Hill JA, Katus HA, Bassel-Duby R, Maier LS, Olson EN. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci USA 106: 2342–2347, 2009. doi: 10.1073/pnas.0813013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Haj Slimane Z, Bedioune I, Lechêne P, Varin A, Lefebvre F, Mateo P, Domergue-Dupont V, Dewenter M, Richter W, Conti M, El-Armouche A, Zhang J, Fischmeister R, Vandecasteele G. Control of cytoplasmic and nuclear protein kinase A by phosphodiesterases and phosphatases in cardiac myocytes. Cardiovasc Res 102: 97–106, 2014. doi: 10.1093/cvr/cvu029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Sin YY, Edwards HV, Li X, Day JP, Christian F, Dunlop AJ, Adams DR, Zaccolo M, Houslay MD, Baillie GS. Disruption of the cyclic AMP phosphodiesterase-4 (PDE4)-HSP20 complex attenuates the β-agonist induced hypertrophic response in cardiac myocytes. J Mol Cell Cardiol 50: 872–883, 2011. doi: 10.1016/j.yjmcc.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 300. Hanna R, Nour-Eldine W, Saliba Y, Dagher-Hamalian C, Hachem P, Abou-Khalil P, Mika D, Varin A, El Hayek MS, Pereira L, Farès N, Vandecasteele G, Abi-Gerges A. Cardiac phosphodiesterases are differentially increased in diabetic cardiomyopathy. Life Sci 283: 119857, 2021. doi: 10.1016/j.lfs.2021.119857. [DOI] [PubMed] [Google Scholar]
- 301. Wang Q, Liu Y, Fu Q, Xu B, Zhang Y, Kim S, Tan R, Barbagallo F, West T, Anderson E, Wei W, Abel ED, Xiang YK. Inhibiting insulin-mediated beta2-adrenergic receptor activation prevents diabetes-associated cardiac dysfunction. Circulation 135: 73–88, 2017. doi: 10.1161/CIRCULATIONAHA.116.022281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Smith CJ, Huang R, Sun D, Ricketts S, Hoegler C, Ding JZ, Moggio RA, Hintze TH. Development of decompensated dilated cardiomyopathy is associated with decreased gene expression and activity of the milrinone-sensitive cAMP phosphodiesterase PDE3A. Circulation 96: 3116–3123, 1997. doi: 10.1161/01.cir.96.9.3116. [DOI] [PubMed] [Google Scholar]
- 303. Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol 7: 589–600, 2006. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 304. Wu H, Lee J, Vincent LG, Wang Q, Gu M, Lan F, Churko JM, Sallam KI, Matsa E, Sharma A, Gold JD, Engler AJ, Xiang YK, Bers DM, Wu JC. Epigenetic regulation of phosphodiesterases 2A and 3A underlies compromised beta-adrenergic signaling in an iPSC model of dilated cardiomyopathy. Cell Stem Cell 17: 89–100, 2015. doi: 10.1016/j.stem.2015.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Surapisitchat J, Jeon KI, Yan C, Beavo JA. Differential regulation of endothelial cell permeability by cGMP via phosphodiesterases 2 and 3. Circ Res 101: 811–818, 2007. doi: 10.1161/CIRCRESAHA.107.154229. [DOI] [PubMed] [Google Scholar]
- 306. Chen S, Knight WE, Yan C. Roles of PDE1 in pathological cardiac remodeling and dysfunction. J Cardiovasc Dev Dis 5: 22, 2018. doi: 10.3390/jcdd5020022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Bubb KJ, Trinder SL, Baliga RS, Patel J, Clapp LH, MacAllister RJ, Hobbs AJ. Inhibition of phosphodiesterase 2 augments cGMP and cAMP signaling to ameliorate pulmonary hypertension. Circulation 130 : 496–507, 2014. doi: 10.1161/CIRCULATIONAHA.114.009751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Smith CJ, He J, Ricketts SG, Ding JZ, Moggio RA, Hintze TH. Downregulation of right ventricular phosphodiesterase PDE-3A mRNA and protein before the development of canine heart failure. Cell Biochem Biophys 29: 67–88, 1998. doi: 10.1007/BF02737829. [DOI] [PubMed] [Google Scholar]
- 309. Subramaniam G, Schleicher K, Kovanich D, Zerio A, Folkmanaite M, Chao YC, Surdo NC, Koschinski A, Hu J, Scholten A, Heck AJ, Ercu M, Sholokh A, Park KC, Klussmann E, Meraviglia V, Bellin M, Zanivan S, Hester S, Mohammed S, Zaccolo M. Integrated proteomics unveils nuclear PDE3A2 as a regulator of cardiac myocyte hypertrophy. Circ Res 132: 828–848, 2023. doi: 10.1161/CIRCRESAHA.122.321448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Nakata TM, Suzuki K, Uemura A, Shimada K, Tanaka R. Contrasting effects of inhibition of phosphodiesterase 3 and 5 on cardiac function and interstitial fibrosis in rats with isoproterenol-induced cardiac dysfunction. J Cardiovasc Pharmacol 73: 195–205, 2019. doi: 10.1097/FJC.0000000000000652. [DOI] [PubMed] [Google Scholar]
- 311. Mongillo M, McSorley T, Evellin S, Sood A, Lissandron V, Terrin A, Huston E, Hannawacker A, Lohse MJ, Pozzan T, Houslay MD, Zaccolo M. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res 95: 67–75, 2004. doi: 10.1161/01.RES.0000134629.84732.11. [DOI] [PubMed] [Google Scholar]
- 312. Wan Q, Xu C, Zhu L, Zhang Y, Peng Z, Chen H, Rao H, Zhang E, Wang H, Chu F, Ning X, Yang X, Yuan J, Wu Y, Huang Y, Hu S, Liu DP, Wang M. Targeting PDE4B (phosphodiesterase-4 subtype B) for cardioprotection in acute myocardial infarction via neutrophils and microcirculation. Circ Res 131: 442–455, 2022. doi: 10.1161/CIRCRESAHA.122.321365. [DOI] [PubMed] [Google Scholar]
- 313. Salloum FN, Das A, Thomas CS, Yin C, Kukreja RC. Adenosine A1 receptor mediates delayed cardioprotective effect of sildenafil in mouse. J Mol Cell Cardiol 43: 545–551, 2007. doi: 10.1016/j.yjmcc.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Ockaili R, Salloum F, Hawkins J, Kukreja RC. Sildenafil (Viagra) induces powerful cardioprotective effect via opening of mitochondrial K(ATP) channels in rabbits. Am J Physiol Heart Circ Physiol 283: H1263–H1269, 2002. doi: 10.1152/ajpheart.00324.2002. [DOI] [PubMed] [Google Scholar]
- 315. Chau VQ, Salloum FN, Hoke NN, Abbate A, Kukreja RC. Mitigation of the progression of heart failure with sildenafil involves inhibition of RhoA/Rho-kinase pathway. Am J Physiol Heart Circ Physiol 300: H2272–H2279, 2011. doi: 10.1152/ajpheart.00654.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Anderson SG, Hutchings DC, Woodward M, Rahimi K, Rutter MK, Kirby M, Hackett G, Trafford AW, Heald AH. Phosphodiesterase type-5 inhibitor use in type 2 diabetes is associated with a reduction in all-cause mortality. Heart 102: 1750–1756, 2016. doi: 10.1136/heartjnl-2015-309223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Wagner M, Mehel H, Fischmeister R, El-Armouche A. Phosphodiesterase 2: anti-adrenergic friend or hypertrophic foe in heart disease? Naunyn Schmiedebergs Arch Pharmacol 389: 1139–1141, 2016. doi: 10.1007/s00210-016-1289-4. [DOI] [PubMed] [Google Scholar]
- 318. Sanada S, Kitakaze M, Papst PJ, Asanuma H, Node K, Takashima S, Asakura M, Ogita H, Liao Y, Sakata Y, Ogai A, Fukushima T, Yamada J, Shinozaki Y, Kuzuya T, Mori H, Terada N, Hori M. Cardioprotective effect afforded by transient exposure to phosphodiesterase III inhibitors: the role of protein kinase A and p38 mitogen-activated protein kinase. Circulation 104: 705–710, 2001. doi: 10.1161/hc3201.092216. [DOI] [PubMed] [Google Scholar]
- 319. Tosaka S, Makita T, Tosaka R, Maekawa T, Cho S, Hara T, Ureshino H, Sumikawa K. Cardioprotection induced by olprinone, a phosphodiesterase III inhibitor, involves phosphatidylinositol-3-OH kinase-Akt and a mitochondrial permeability transition pore during early reperfusion. J Anesth 21: 176–180, 2007. doi: 10.1007/s00540-006-0485-7. [DOI] [PubMed] [Google Scholar]
- 320. Fukasawa M, Nishida H, Sato T, Miyazaki M, Nakaya H. 6-[4-(1-Cyclohexyl-1H-tetrazol-5-yl)butoxy]-3,4-dihydro-2-(1H)quinolinone (cilostazol), a phosphodiesterase type 3 inhibitor, reduces infarct size via activation of mitochondrial Ca2+-activated K+ channels in rabbit hearts. J Pharmacol Exp Ther 326: 100–104, 2008. doi: 10.1124/jpet.108.136218. [DOI] [PubMed] [Google Scholar]
- 321. Oikawa M, Wu M, Lim S, Knight WE, Miller CL, Cai Y, Lu Y, Blaxall BC, Takeishi Y, Abe JI, Yan C. Cyclic nucleotide phosphodiesterase 3A1 protects the heart against ischemia-reperfusion injury. J Mol Cell Cardiol 64: 11–19, 2013. doi: 10.1016/j.yjmcc.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Yan C, Miller CL, Abe J. Regulation of phosphodiesterase 3 and inducible cAMP early repressor in the heart. Circ Res 100: 489–501, 2007. doi: 10.1161/01.RES.0000258451.44949.d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Altemose GT, Gritsus V, Jeevanandam V, Goldman B, Margulies KB. Altered myocardial phenotype after mechanical support in human beings with advanced cardiomyopathy. J Heart Lung Transplant 16: 765–773, 1997. [PubMed] [Google Scholar]
- 324. Yan C, Ding B, Shishido T, Woo CH, Itoh S, Jeon KI, Liu W, Xu H, McClain C, Molina CA, Blaxall BC, Abe JI. Activation of extracellular signal-regulated kinase 5 reduces cardiac apoptosis and dysfunction via inhibition of a phosphodiesterase 3A/inducible cAMP early repressor feedback loop. Circ Res 100: 510–519, 2007. doi: 10.1161/01.RES.0000259045.49371.9c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Wang Z, Liu D, Varin A, Nicolas V, Courilleau D, Mateo P, Caubere C, Rouet P, Gomez AM, Vandecasteele G, Fischmeister R, Brenner C. A cardiac mitochondrial cAMP signaling pathway regulates calcium accumulation, permeability transition and cell death. Cell Death Dis 7: e2198, 2016. doi: 10.1038/cddis.2016.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Sadek MS, Cachorro E, El-Armouche A, Kämmerer S. Therapeutic implications for PDE2 and cGMP/cAMP mediated crosstalk in cardiovascular diseases. Int J Mol Sci 21: 7462, 2020. doi: 10.3390/ijms21207462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Pozdniakova S, Guitart-Mampel M, Garrabou G, Di Benedetto G, Ladilov Y, Regitz-Zagrosek V. 17β-Estradiol reduces mitochondrial cAMP content and cytochrome oxidase activity in a phosphodiesterase 2-dependent manner. Br J Pharmacol 175: 3876–3890, 2018. doi: 10.1111/bph.14455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Acin-Perez R, Russwurm M, Günnewig K, Gertz M, Zoidl G, Ramos L, Buck J, Levin LR, Rassow J, Manfredi G, Steegborn C. A phosphodiesterase 2A isoform localized to mitochondria regulates respiration. J Biol Chem 286: 30423–30432, 2011. doi: 10.1074/jbc.M111.266379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Neviere R, Delguste F, Durand A, Inamo J, Boulanger E, Preau S. Abnormal mitochondrial cAMP/PKA signaling is involved in sepsis-induced mitochondrial and myocardial dysfunction. Int J Mol Sci 17: 2075, 2016. doi: 10.3390/ijms17122075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Park SJ, Ahmad F, Bahde RJ, Philp A, Kim J, Huang T, Kim MK, Trenkle WC, Chung JH. Potent PDE4 inhibitor activates AMPK and Sirt1 to induce mitochondrial biogenesis. PLoS One 16: e0253269, 2021. doi: 10.1371/journal.pone.0253269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Frangogiannis NG. Cardiac fibrosis. Cardiovasc Res 117: 1450–1488, 2021. doi: 10.1093/cvr/cvaa324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Li P, Wang D, Lucas J, Oparil S, Xing D, Cao X, Novak L, Renfrow MB, Chen YF. Atrial natriuretic peptide inhibits transforming growth factor beta-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ Res 102: 185–192, 2008. doi: 10.1161/CIRCRESAHA.107.157677. [DOI] [PubMed] [Google Scholar]
- 333. Redondo J, Bishop JE, Wilkins MR. Effect of atrial natriuretic peptide and cyclic GMP phosphodiesterase inhibition on collagen synthesis by adult cardiac fibroblasts. Br J Pharmacol 124: 1455–1462, 1998. doi: 10.1038/sj.bjp.0701994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Yokoyama U, Patel HH, Lai NC, Aroonsakool N, Roth DM, Insel PA. The cyclic AMP effector Epac integrates pro- and anti-fibrotic signals. Proc Natl Acad Sci USA 105: 6386–6391, 2008. doi: 10.1073/pnas.0801490105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Patrucco E, Domes K, Sbroggió M, Blaich A, Schlossmann J, Desch M, Rybalkin SD, Beavo JA, Lukowski R, Hofmann F. Roles of cGMP-dependent protein kinase I (cGKI) and PDE5 in the regulation of Ang II-induced cardiac hypertrophy and fibrosis. Proc Natl Acad Sci USA 111: 12925–12929, 2014. doi: 10.1073/pnas.1414364111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Li L, Zhao D, Jin Z, Zhang J, Paul C, Wang Y. Phosphodiesterase 5a inhibition with adenoviral short hairpin RNA benefits infarcted heart partially through activation of Akt signaling pathway and reduction of inflammatory cytokines. PLoS One 10: e0145766, 2015. doi: 10.1371/journal.pone.0145766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Gustafsson AB, Brunton LL. Attenuation of cAMP accumulation in adult rat cardiac fibroblasts by IL-1beta and NO: role of cGMP-stimulated PDE2. Am J Physiol Cell Physiol 283: C463–C471, 2002. doi: 10.1152/ajpcell.00299.2001. [DOI] [PubMed] [Google Scholar]
- 338. Hada Y, Uchida HA, Umebayashi R, Yoshida M, Wada J. Cilostazol attenuates AngII-induced cardiac fibrosis in apoE deficient mice. Int J Mol Sci 23: 9065, 2022. doi: 10.3390/ijms23169065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Strassheim D, Dempsey EC, Gerasimovskaya E, Stenmark K, Karoor V. Role of inflammatory cell subtypes in heart failure. J Immunol Res 2019: 2164017, 2019. doi: 10.1155/2019/2164017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res 116: 1254–1268, 2015. doi: 10.1161/CIRCRESAHA.116.302317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Witwicka H, Kobiałka M, Siednienko J, Mitkiewicz M, Gorczyca WA. Expression and activity of cGMP-dependent phosphodiesterases is up-regulated by lipopolysaccharide (LPS) in rat peritoneal macrophages. Biochim Biophys Acta 1773: 209–218, 2007. doi: 10.1016/j.bbamcr.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 342. Imam F, Al-Harbi NO, Al-Harbi MM, Ansari MA, Al-Asmari AF, Ansari MN, Al-Anazi WA, Bahashwan S, Almutairi MM, Alshammari M, Khan MR, Alsaad AM, Alotaibi MR. Apremilast prevent doxorubicin-induced apoptosis and inflammation in heart through inhibition of oxidative stress mediated activation of NF-κB signaling pathways. Pharmacol Rep 70: 993–1000, 2018. doi: 10.1016/j.pharep.2018.03.009. [DOI] [PubMed] [Google Scholar]
- 343. Mohamed HE, Asker ME, Ali SI, el-Fattah TM. Protection against doxorubicin cardiomyopathy in rats: role of phosphodiesterase inhibitors type 4. J Pharm Pharmacol 56: 757–768, 2004. doi: 10.1211/0022357023565. [DOI] [PubMed] [Google Scholar]
- 344. Smith SJ, Cieslinski LB, Newton R, Donnelly LE, Fenwick PS, Nicholson AG, Barnes PJ, Barnette MS, Giembycz MA. Discovery of BRL 50481 [3-(N,N-dimethylsulfonamido)-4-methyl-nitrobenzene], a selective inhibitor of phosphodiesterase 7: in vitro studies in human monocytes, lung macrophages, and CD8+ T-lymphocytes. Mol Pharmacol 66: 1679–1689, 2004. doi: 10.1124/mol.104.002246. [DOI] [PubMed] [Google Scholar]
- 345. Westermann D, Becher PM, Lindner D, Savvatis K, Xia Y, Fröhlich M, Hoffmann S, Schultheiss HP, Tschöpe C. Selective PDE5A inhibition with sildenafil rescues left ventricular dysfunction, inflammatory immune response and cardiac remodeling in angiotensin II-induced heart failure in vivo. Basic Res Cardiol 107: 308, 2012. doi: 10.1007/s00395-012-0308-y. [DOI] [PubMed] [Google Scholar]
- 346. Li S, Ma Y, Yan Y, Yan M, Wang X, Gong W, Nie S. Phosphodiesterase-5a knock-out suppresses inflammation by down-regulating adhesion molecules in cardiac rupture following myocardial infarction. J Cardiovasc Transl Res 14: 816–823, 2021. doi: 10.1007/s12265-021-10102-2. [DOI] [PubMed] [Google Scholar]
- 347. Varma A, Das A, Hoke NN, Durrant DE, Salloum FN, Kukreja RC. Anti-inflammatory and cardioprotective effects of tadalafil in diabetic mice. PLoS One 7: e45243, 2012. doi: 10.1371/journal.pone.0045243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Di Luigi L, Corinaldesi C, Colletti M, Scolletta S, Antinozzi C, Vannelli GB, Giannetta E, Gianfrilli D, Isidori AM, Migliaccio S, Poerio N, Fraziano M, Lenzi A, Crescioli C. Phosphodiesterase type 5 inhibitor sildenafil decreases the proinflammatory chemokine CXCL10 in human cardiomyocytes and in subjects with diabetic cardiomyopathy. Inflammation 39: 1238–1252, 2016. doi: 10.1007/s10753-016-0359-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Pofi R, Giannetta E, Feola T, Galea N, Barbagallo F, Campolo F, Badagliacca R, Barbano B, Ciolina F, Defeudis G, Filardi T, Sesti F, Minnetti M, Vizza CD, Pasqualetti P, Caboni P, Carbone I, Francone M, Catalano C, Pozzilli P, Lenzi A, Venneri MA, Gianfrilli D, Isidori AM. Sex-specific effects of daily tadalafil on diabetic heart kinetics in RECOGITO, a randomized, double-blind, placebo-controlled trial. Sci Transl Med 14: eabl8503, 2022. doi: 10.1126/scitranslmed.abl8503. [DOI] [PubMed] [Google Scholar]
- 350. Krings T, Mandell DM, Kiehl TR, Geibprasert S, Tymianski M, Alvarez H, terBrugge KG, Hans FJ. Intracranial aneurysms: from vessel wall pathology to therapeutic approach. Nat Rev Neurol 7: 547–559, 2011. doi: 10.1038/nrneurol.2011.136. [DOI] [PubMed] [Google Scholar]
- 351. Libby P. The changing landscape of atherosclerosis. Nature 592: 524–533, 2021. doi: 10.1038/s41586-021-03392-8. [DOI] [PubMed] [Google Scholar]
- 352. Guzik TJ, Touyz RM. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension 70: 660–667, 2017. doi: 10.1161/HYPERTENSIONAHA.117.07802. [DOI] [PubMed] [Google Scholar]
- 353. Sun J, Qiao YN, Tao T, Zhao W, Wei LS, Li YQ, Wang W, Wang Y, Zhou YW, Zheng YY, Chen X, Pan HC, Zhang XN, Zhu MS. Distinct roles of smooth muscle and non-muscle myosin light chain-mediated smooth muscle contraction. Front Physiol 11: 593966, 2020. doi: 10.3389/fphys.2020.593966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Chang AN, Gao N, Liu Z, Huang J, Nairn AC, Kamm KE, Stull JT. The dominant protein phosphatase PP1c isoform in smooth muscle cells, PP1cbeta, is essential for smooth muscle contraction. J Biol Chem 293: 16677–16686, 2018. doi: 10.1074/jbc.RA118.003083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Álvarez-Santos MD, Álvarez-González M, Estrada-Soto S, Bazán-Perkins B. Regulation of myosin light-chain phosphatase activity to generate airway smooth muscle hypercontractility. Front Physiol 11: 701, 2020. doi: 10.3389/fphys.2020.00701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Fong Z, Lall A, Mullins ND, Santana LF, Hollywood MA, Thornbury KD, Sergeant GP. Blockade of Kv7 channels reverses the inhibitory effects of exchange protein directly activated by cAMP activation on purinergic contractions of the murine detrusor. Basic Clin Pharmacol Toxicol 133: 29–42, 2023. doi: 10.1111/bcpt.13881. [DOI] [PubMed] [Google Scholar]
- 357. Jung HS, Seo MS, An JR, Heo R, Kang M, Han ET, Park H, Song G, Son YK, Jung WK, Choi IW, Na SH, Park WS. The anti-diabetic drug alogliptin induces vasorelaxation via activation of Kv channels and SERCA2a pumps. Eur J Pharmacol 898: 173991, 2021. doi: 10.1016/j.ejphar.2021.173991. [DOI] [PubMed] [Google Scholar]
- 358. Prada MP, Syed AU, Buonarati OR, Reddy GR, Nystoriak MA, Ghosh D, Simó S, Sato D, Sasse KC, Ward SM, Santana LF, Xiang YK, Hell JW, Nieves-Cintrón M, Navedo MF. A G(s)-coupled purinergic receptor boosts Ca2+ influx and vascular contractility during diabetic hyperglycemia. Elife 8: e42214, 2019. doi: 10.7554/eLife.42214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Prada MP, Syed AU, Reddy GR, Martín-Aragón Baudel M, Flores-Tamez VA, Sasse KC, Ward SM, Sirish P, Chiamvimonvat N, Bartels P, Dickson EJ, Hell JW, Scott JD, Santana LF, Xiang YK, Navedo MF, Nieves-Cintrón M. AKAP5 complex facilitates purinergic modulation of vascular L-type Ca2+ channel CaV1.2. Nat Commun 11: 5303, 2020. doi: 10.1038/s41467-020-18947-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Syed AU, Reddy GR, Ghosh D, Prada MP, Nystoriak MA, Morotti S, Grandi E, Sirish P, Chiamvimonvat N, Hell JW, Santana LF, Xiang YK, Nieves-Cintrón M, Navedo MF. Adenylyl cyclase 5-generated cAMP controls cerebral vascular reactivity during diabetic hyperglycemia. J Clin Invest 129: 3140–3152, 2019. doi: 10.1172/JCI124705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev 62: 525–563, 2010. doi: 10.1124/pr.110.002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362. Bonnevier J, Arner A. Actions downstream of cyclic GMP/protein kinase G can reverse protein kinase C-mediated phosphorylation of CPI-17 and Ca2+ sensitization in smooth muscle. J Biol Chem 279: 28998–29003, 2004. doi: 10.1074/jbc.M404259200. [DOI] [PubMed] [Google Scholar]
- 363. Murthy KS, Zhou H. Selective phosphorylation of the IP3R-I in vivo by cGMP-dependent protein kinase in smooth muscle. Am J Physiol Gastrointest Liver Physiol 284: G221–G230, 2003. doi: 10.1152/ajpgi.00401.2002. [DOI] [PubMed] [Google Scholar]
- 364. Tanaka Y, Aida M, Tanaka H, Shigenobu K, Toro L. Involvement of maxi-KCa channel activation in atrial natriuretic peptide-induced vasorelaxation. Naunyn Schmiedebergs Arch Pharmacol 357: 705–708, 1998. doi: 10.1007/pl00005228. [DOI] [PubMed] [Google Scholar]
- 365. Lugnier C, Schini VB. Characterization of cyclic nucleotide phosphodiesterases from cultured bovine aortic endothelial cells. Biochem Pharmacol 39: 75–84, 1990. doi: 10.1016/0006-2952(90)90650-a. [DOI] [PubMed] [Google Scholar]
- 366. Kishi Y, Ashikaga T, Numano F. Phosphodiesterases in vascular endothelial cells. Adv Second Messenger Phosphoprotein Res 25: 201–213, 1992. [PubMed] [Google Scholar]
- 367. Rybalkin SD, Bornfeldt KE. Cyclic nucleotide phosphodiesterases and human arterial smooth muscle cell proliferation. Thromb Haemost 82: 424–434, 1999. doi: 10.1055/s-0037-1615862. [DOI] [PubMed] [Google Scholar]
- 368. Tragante V, Barnes MR, Ganesh SK, Lanktree MB, Guo W, Franceschini N, et al. Gene-centric meta-analysis in 87,736 individuals of European ancestry identifies multiple blood-pressure-related loci. Am J Hum Genet 94: 349–360, 2014. doi: 10.1016/j.ajhg.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Bautista Niño PK, Durik M, Danser AH, de Vries R, Musterd-Bhaggoe UM, Meima ME, Kavousi M, Ghanbari M, Hoeijmakers JH, O’Donnell CJ, Franceschini N, Janssen GM, De Mey JG, Liu Y, Shanahan CM, Franco OH, Dehghan A, Roks AJ. Phosphodiesterase 1 regulation is a key mechanism in vascular aging. Clin Sci (Lond) 129: 1061–1075, 2015. doi: 10.1042/CS20140753. [DOI] [PubMed] [Google Scholar]
- 370. Kim D, Rybalkin SD, Pi X, Wang Y, Zhang C, Munzel T, Beavo JA, Berk BC, Yan C. Upregulation of phosphodiesterase 1A1 expression is associated with the development of nitrate tolerance. Circulation 104: 2338–2343, 2001. doi: 10.1161/hc4401.098432. [DOI] [PubMed] [Google Scholar]
- 371. Wang X, Yamada S, LaRiviere WB, Ye H, Bakeberg JL, Irazabal MV, Chebib FT, van Deursen J, Harris PC, Sussman CR, Behfar A, Ward CJ, Torres VE. Generation and phenotypic characterization of Pde1a mutant mice. PLoS One 12: e0181087, 2017. doi: 10.1371/journal.pone.0181087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Rybalkin SD, Bornfeldt KE, Sonnenburg WK, Rybalkina IG, Kwak KS, Hanson K, Krebs EG, Beavo JA. Calmodulin-stimulated cyclic nucleotide phosphodiesterase (PDE1C) is induced in human arterial smooth muscle cells of the synthetic, proliferative phenotype. J Clin Invest 100: 2611–2621, 1997. doi: 10.1172/JCI119805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Cai Y, Nagel DJ, Zhou Q, Cygnar KD, Zhao H, Li F, Pi X, Knight PA, Yan C. Role of cAMP-phosphodiesterase 1C signaling in regulating growth factor receptor stability, vascular smooth muscle cell growth, migration, and neointimal hyperplasia. Circ Res 116: 1120–1132, 2015. doi: 10.1161/CIRCRESAHA.116.304408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Hagiwara M, Endo T, Hidaka H. Effects of vinpocetine on cyclic nucleotide metabolism in vascular smooth muscle. Biochem Pharmacol 33: 453–457, 1984. doi: 10.1016/0006-2952(84)90240-5. [DOI] [PubMed] [Google Scholar]
- 375. Chiu PJ, Tetzloff G, Ahn HS, Sybertz EJ. Comparative effects of vinpocetine and 8-Br-cyclic GMP on the contraction and 45Ca-fluxes in the rabbit aorta. Am J Hypertens 1: 262–268, 1988. doi: 10.1093/ajh/1.3.262. [DOI] [PubMed] [Google Scholar]
- 376. Ahn HS, Crim W, Romano M, Sybertz E, Pitts B. Effects of selective inhibitors on cyclic nucleotide phosphodiesterases of rabbit aorta. Biochem Pharmacol 38: 3331–3339, 1989. doi: 10.1016/0006-2952(89)90631-X. [DOI] [PubMed] [Google Scholar]
- 377. Souness JE, Brazdil R, Diocee BK, Jordan R. Role of selective cyclic GMP phosphodiesterase inhibition in the myorelaxant actions of M&B 22,948, MY-5445, vinpocetine and 1-methyl-3-isobutyl-8-(methylamino)xanthine. Br J Pharmacol 98: 725–734, 1989. doi: 10.1111/j.1476-5381.1989.tb14599.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Molina CR, Andresen JW, Rapoport RM, Waldman S, Murad F. Effect of in vivo nitroglycerin therapy on endothelium-dependent and independent vascular relaxation and cyclic GMP accumulation in rat aorta. J Cardiovasc Pharmacol 10: 371–378, 1987. doi: 10.1097/00005344-198710000-00001. [DOI] [PubMed] [Google Scholar]
- 379. Jaiswal RK. Endothelin inhibits the atrial natriuretic factor stimulated cGMP production by activating the protein kinase C in rat aortic smooth muscle cells. Biochem Biophys Res Commun 182: 395–402, 1992. doi: 10.1016/s0006-291x(05)80158-5. [DOI] [PubMed] [Google Scholar]
- 380. Ercu M, Markó L, Schächterle C, Tsvetkov D, Cui Y, Maghsodi S, et al. Phosphodiesterase 3A and arterial hypertension. Circulation 142: 133–149, 2020. doi: 10.1161/CIRCULATIONAHA.119.043061. [DOI] [PubMed] [Google Scholar]
- 381. Polson JB, Strada SJ. Cyclic nucleotide phosphodiesterases and vascular smooth muscle. Annu Rev Pharmacol Toxicol 36: 403–427, 1996. doi: 10.1146/annurev.pa.36.040196.002155. [DOI] [PubMed] [Google Scholar]
- 382. Komas N, Lugnier C, Stoclet JC. Endothelium-dependent and independent relaxation of the rat aorta by cyclic nucleotide phosphodiesterase inhibitors. Br J Pharmacol 104: 495–503, 1991. doi: 10.1111/j.1476-5381.1991.tb12457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Tilley DG, Maurice DH. Vascular smooth muscle cell phosphodiesterase (PDE) 3 and PDE4 activities and levels are regulated by cyclic AMP in vivo. Mol Pharmacol 62: 497–506, 2002. doi: 10.1124/mol.62.3.497. [DOI] [PubMed] [Google Scholar]
- 384. Liu H, Maurice DH. Expression of cyclic GMP-inhibited phosphodiesterases 3A and 3B (PDE3A and PDE3B) in rat tissues: differential subcellular localization and regulated expression by cyclic AMP. Br J Pharmacol 125: 1501–1510, 1998. doi: 10.1038/sj.bjp.0702227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Maass PG, Aydin A, Luft FC, Schächterle C, Weise A, Stricker S, et al. PDE3A mutations cause autosomal dominant hypertension with brachydactyly. Nat Genet 47: 647–653, 2015. doi: 10.1038/ng.3302. [DOI] [PubMed] [Google Scholar]
- 386. Lee CG, Kang K, Yoon RG, Seo JY, Park JM. PDE3A variant associated with hypertension and brachydactyly syndrome in a patient with ischemic stroke caused by spontaneous intracranial artery dissection: a review of the clinical and molecular genetic features. Eur J Med Genet 63: 103781, 2020. doi: 10.1016/j.ejmg.2019.103781. [DOI] [PubMed] [Google Scholar]
- 387. Ercu M, Walter S, Klussmann E. Mutations in phosphodiesterase 3A (PDE3A) cause hypertension without cardiac damage. Hypertension 80: 1171–1179, 2023. doi: 10.1161/HYPERTENSIONAHA.122.19433. [DOI] [PubMed] [Google Scholar]
- 388. Domenichini G, Diab I, Campbell NG, Dhinoja M, Hunter RJ, Sporton S, Earley MJ, Schilling RJ. A highly effective technique for transseptal endocardial left ventricular lead placement for delivery of cardiac resynchronization therapy. Heart Rhythm 12: 943–949, 2015. doi: 10.1016/j.hrthm.2015.01.038. [DOI] [PubMed] [Google Scholar]
- 389. Lindgren S, Andersson KE, Belfrage P, Degerman E, Manganiello VC. Relaxant effects of the selective phosphodiesterase inhibitors milrinone and OPC 3911 on isolated human mesenteric vessels. Pharmacol Toxicol 64: 440–445, 1989. doi: 10.1111/j.1600-0773.1989.tb00683.x. [DOI] [PubMed] [Google Scholar]
- 390. Lindgren SH, Andersson TL, Vinge E, Andersson KE. Effects of isozyme-selective phosphodiesterase inhibitors on rat aorta and human platelets: smooth muscle tone, platelet aggregation and cAMP levels. Acta Physiol Scand 140: 209–219, 1990. doi: 10.1111/j.1748-1716.1990.tb08993.x. [DOI] [PubMed] [Google Scholar]
- 391. Maurice DH, Crankshaw D, Haslam RJ. Synergistic actions of nitrovasodilators and isoprenaline on rat aortic smooth muscle. Eur J Pharmacol 192: 235–242, 1991. doi: 10.1016/0014-2999(91)90048-u. [DOI] [PubMed] [Google Scholar]
- 392. Murthy KS, Zhou H, Makhlouf GM. PKA-dependent activation of PDE3A and PDE4 and inhibition of adenylyl cyclase V/VI in smooth muscle. Am J Physiol Cell Physiol 282: C508–C517, 2002. doi: 10.1152/ajpcell.00373.2001. [DOI] [PubMed] [Google Scholar]
- 393. Dünnes S, Voussen B, Aue A, Groneberg K, Nikolaev V, Groneberg D, Friebe A. Phosphodiesterase 3A expression and activity in the murine vasculature is influenced by NO-sensitive guanylyl cyclase. Pflugers Arch 470: 693–702, 2018. doi: 10.1007/s00424-017-2106-8. [DOI] [PubMed] [Google Scholar]
- 394. Sandner P, Kornfeld M, Ruan X, Arendshorst WJ, Kurtz A. Nitric oxide/cAMP interactions in the control of rat renal vascular resistance. Circ Res 84: 186–192, 1999. doi: 10.1161/01.res.84.2.186. [DOI] [PubMed] [Google Scholar]
- 395. Wagner RS, Smith CJ, Taylor AM, Rhoades RA. Phosphodiesterase inhibition improves agonist-induced relaxation of hypertensive pulmonary arteries. J Pharmacol Exp Ther 282: 1650–1657, 1997. [PubMed] [Google Scholar]
- 396. Spina D. Theophylline and PDE4 inhibitors in asthma. Curr Opin Pulm Med 9: 57–64, 2003. doi: 10.1097/00063198-200301000-00010. [DOI] [PubMed] [Google Scholar]
- 397. Lugnier C, Keravis T, Eckly-Michel A. Cross talk between NO and cyclic nucleotide phosphodiesterases in the modulation of signal transduction in blood vessel. J Physiol Pharmacol 50: 639–652, 1999. [PubMed] [Google Scholar]
- 398. Palmer D, Tsoi K, Maurice DH. Synergistic inhibition of vascular smooth muscle cell migration by phosphodiesterase 3 and phosphodiesterase 4 inhibitors. Circ Res 82: 852–861, 1998. doi: 10.1161/01.res.82.8.852. [DOI] [PubMed] [Google Scholar]
- 399. Willette RN, Shiloh AO, Sauermelch CF, Sulpizio A, Michell MP, Cieslinski LB, Torphy TJ, Ohlstein EH. Identification, characterization, and functional role of phosphodiesterase type IV in cerebral vessels: effects of selective phosphodiesterase inhibitors. J Cereb Blood Flow Metab 17: 210–219, 1997. doi: 10.1097/00004647-199702000-00011. [DOI] [PubMed] [Google Scholar]
- 400. Gretarsdottir S, Thorleifsson G, Reynisdottir ST, Manolescu A, Jonsdottir S, Jonsdottir T, et al. The gene encoding phosphodiesterase 4D confers risk of ischemic stroke. Nat Genet 35: 131–138, 2003. doi: 10.1038/ng1245. [DOI] [PubMed] [Google Scholar]
- 401. Harris AL, Lemp BM, Bentley RG, Perrone MH, Hamel LT, Silver PJ. Phosphodiesterase isozyme inhibition and the potentiation by zaprinast of endothelium-derived relaxing factor and guanylate cyclase stimulating agents in vascular smooth muscle. J Pharmacol Exp Ther 249: 394–400, 1989. [PubMed] [Google Scholar]
- 402. Corbin JD, Beasley A, Blount MA, Francis SH. High lung PDE5: a strong basis for treating pulmonary hypertension with PDE5 inhibitors. Biochem Biophys Res Commun 334: 930–938, 2005. doi: 10.1016/j.bbrc.2005.06.183. [DOI] [PubMed] [Google Scholar]
- 403. Prasad S, Wilkinson J, Gatzoulis MA. Sildenafil in primary pulmonary hypertension. N Engl J Med 343: 1342, 2000. doi: 10.1056/NEJM200011023431814. [DOI] [PubMed] [Google Scholar]
- 404. Steiner MK, Preston IR, Klinger JR, Hill NS. Pulmonary hypertension: inhaled nitric oxide, sildenafil and natriuretic peptides. Curr Opin Pharmacol 5: 245–250, 2005. doi: 10.1016/j.coph.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 405. Andersson KE. PDE5 inhibitors—pharmacology and clinical applications 20 years after sildenafil discovery. Br J Pharmacol 175: 2554–2565, 2018. doi: 10.1111/bph.14205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406. Boolell M, Gepi-Attee S, Gingell JC, Allen MJ. Sildenafil, a novel effective oral therapy for male erectile dysfunction. Br J Urol 78: 257–261, 1996. doi: 10.1046/j.1464-410x.1996.10220.x. [DOI] [PubMed] [Google Scholar]
- 407. Ballard SA, Gingell CJ, Tang K, Turner LA, Price ME, Naylor AM. Effects of sildenafil on the relaxation of human corpus cavernosum tissue in vitro and on the activities of cyclic nucleotide phosphodiesterase isozymes. J Urol 159: 2164–2171, 1998. doi: 10.1016/S0022-5347(01)63299-3. [DOI] [PubMed] [Google Scholar]
- 408. Kloner RA. Cardiovascular risk and sildenafil. Am J Cardiol 86: 57f–61f, 2000. doi: 10.1016/s0002-9149(00)00895-x. [DOI] [PubMed] [Google Scholar]
- 409. Hutton M, Frazer M, Lin A, Patel S, Misra A. New targets in atherosclerosis: vascular smooth muscle cell plasticity and macrophage polarity. Clin Ther 45: 1047–1054, 2023. doi: 10.1016/j.clinthera.2023.08.015. [DOI] [PubMed] [Google Scholar]
- 410. Borek I, Birnhuber A, Voelkel NF, Marsh LM, Kwapiszewska G. The vascular perspective on acute and chronic lung disease. J Clin Invest 133: e170502, 2023. doi: 10.1172/JCI170502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Pohl L, Schiessl IM. Endothelial cell plasticity in kidney fibrosis and disease. Acta Physiol (Oxf) 239: e14038, 2023. doi: 10.1111/apha.14038. [DOI] [PubMed] [Google Scholar]
- 412. Di Pietrantonio N, Cappellacci I, Mandatori D, Baldassarre MP, Pandolfi A, Pipino C. Role of epigenetics and metabolomics in predicting endothelial dysfunction in type 2 diabetes. Adv Biol (Weinh) 7: e2300172, 2023. doi: 10.1002/adbi.202300172. [DOI] [PubMed] [Google Scholar]
- 413. Bkaily G, Abou Abdallah N, Simon Y, Jazzar A, Jacques D. Vascular smooth muscle remodeling in health and disease. Can J Physiol Pharmacol 99: 171–178, 2021. doi: 10.1139/cjpp-2020-0399. [DOI] [PubMed] [Google Scholar]
- 414. Cai Y, Miller CL, Nagel DJ, Jeon KI, Lim S, Gao P, Knight PA, Yan C. Cyclic nucleotide phosphodiesterase 1 regulates lysosome-dependent type I collagen protein degradation in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 31: 616–623, 2011. doi: 10.1161/ATVBAHA.110.212621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415. Wang G, Luo Y, Gao X, Liang Y, Yang F, Wu J, Fang D, Luo M. MicroRNA regulation of phenotypic transformations in vascular smooth muscle: relevance to vascular remodeling. Cell Mol Life Sci 80: 144, 2023. doi: 10.1007/s00018-023-04793-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Chakraborty A, Li Y, Zhang C, Li Y, Rebello KR, Li S, Xu S, Vasquez HG, Zhang L, Luo W, Wang G, Chen K, Coselli JS, LeMaire SA, Shen YH. Epigenetic induction of smooth muscle cell phenotypic alterations in aortic aneurysms and dissections. Circulation 148: 959–977, 2023. doi: 10.1161/CIRCULATIONAHA.123.063332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417. Jeon KI, Jono H, Miller CL, Cai Y, Lim S, Liu X, Gao P, Abe JI, Li JD, Yan C. Ca2+/calmodulin-stimulated PDE1 regulates the beta-catenin/TCF signaling through PP2A B56 gamma subunit in proliferating vascular smooth muscle cells. FEBS J 277: 5026–5039, 2010. doi: 10.1111/j.1742-4658.2010.07908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418. Souness JE, Hassall GA, Parrott DP. Inhibition of pig aortic smooth muscle cell DNA synthesis by selective type III and type IV cyclic AMP phosphodiesterase inhibitors. Biochem Pharmacol 44: 857–866, 1992. doi: 10.1016/0006-2952(92)90116-z. [DOI] [PubMed] [Google Scholar]
- 419. Takahashi S, Oida K, Fujiwara R, Maeda H, Hayashi S, Takai H, Tamai T, Nakai T, Miyabo S. Effect of cilostazol, a cyclic AMP phosphodiesterase inhibitor, on the proliferation of rat aortic smooth muscle cells in culture. J Cardiovasc Pharmacol 20: 900–906, 1992. doi: 10.1097/00005344-199212000-00009. [DOI] [PubMed] [Google Scholar]
- 420. Inoue Y, Toga K, Sudo T, Tachibana K, Tochizawa S, Kimura Y, Yoshida Y, Hidaka H. Suppression of arterial intimal hyperplasia by cilostamide, a cyclic nucleotide phosphodiesterase 3 inhibitor, in a rat balloon double-injury model. Br J Pharmacol 130: 231–241, 2000. doi: 10.1038/sj.bjp.0703287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421. Kim D, Aizawa T, Wei H, Pi X, Rybalkin SD, Berk BC, Yan C. Angiotensin II increases phosphodiesterase 5A expression in vascular smooth muscle cells: a mechanism by which angiotensin II antagonizes cGMP signaling. J Mol Cell Cardiol 38: 175–184, 2005. doi: 10.1016/j.yjmcc.2004.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422. Osinski MT, Rauch BH, Schrör K. Antimitogenic actions of organic nitrates are potentiated by sildenafil and mediated via activation of protein kinase A. Mol Pharmacol 59: 1044–1050, 2001. doi: 10.1124/mol.59.5.1044. [DOI] [PubMed] [Google Scholar]
- 423. Wharton J, Strange JW, Møller GM, Growcott EJ, Ren X, Franklyn AP, Phillips SC, Wilkins MR. Antiproliferative effects of phosphodiesterase type 5 inhibition in human pulmonary artery cells. Am J Respir Crit Care Med 172: 105–113, 2005. doi: 10.1164/rccm.200411-1587OC. [DOI] [PubMed] [Google Scholar]
- 424. Luo L, Cai Y, Zhang Y, Hsu CG, Korshunov VA, Long X, Knight PA, Berk BC, Yan C. Correction to: Role of PDE10A in vascular smooth muscle cell hyperplasia and pathological vascular remodelling. Cardiovasc Res 119: 879, 2023. doi: 10.1093/cvr/cvac011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425. Lehrke M, Kahles F, Makowska A, Tilstam PV, Diebold S, Marx J, Stöhr R, Hess K, Endorf EB, Bruemmer D, Marx N, Findeisen HM. PDE4 inhibition reduces neointima formation and inhibits VCAM-1 expression and histone methylation in an Epac-dependent manner. J Mol Cell Cardiol 81: 23–33, 2015. doi: 10.1016/j.yjmcc.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 426. Varona S, Puertas L, Galán M, Orriols M, Cañes L, Aguiló S, Camacho M, Sirvent M, Andrés V, Martínez-González J, Rodríguez C. Rolipram prevents the formation of abdominal aortic aneurysm (AAA) in mice: PDE4B as a target in AAA. Antioxidants (Basel) 10: 460, 2021. doi: 10.3390/antiox10030460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Gao R, Guo W, Fan T, Pang J, Hou Y, Feng X, Li B, Ge W, Fan T, Zhang T, Lu J, Jing H, Jin M, Yan C, Wang J. Phosphodiesterase 4D contributes to angiotensin II-induced abdominal aortic aneurysm through smooth muscle cell apoptosis. Exp Mol Med 54: 1201–1213, 2022. doi: 10.1038/s12276-022-00815-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428. Yagi K, Tada Y, Kitazato KT, Tamura T, Satomi J, Nagahiro S. Ibudilast inhibits cerebral aneurysms by down-regulating inflammation-related molecules in the vascular wall of rats. Neurosurgery 66: 551–559, 2010. doi: 10.1227/01.NEU.0000365771.89576.77. [DOI] [PubMed] [Google Scholar]
- 429. Pan Z, Wu X, Zhang X, Hu K. Phosphodiesterase 4B activation exacerbates pulmonary hypertension induced by intermittent hypoxia by regulating mitochondrial injury and cAMP/PKA/p-CREB/PGC-1α signaling. Biomed Pharmacother 158: 114095, 2023. doi: 10.1016/j.biopha.2022.114095. [DOI] [PubMed] [Google Scholar]
- 430. Xu R, Gopireddy RR, Wu Y, Wu L, Tao X, Shao J, Wang W, Li L, Jovanovic A, Xu B, Kenyon NJ, Lu Q, Xiang YK, Fu Q. Hyperinsulinemia promotes heterologous desensitization of beta2 adrenergic receptor in airway smooth muscle in obesity. FASEB J 34: 3996–4008, 2020. doi: 10.1096/fj.201800688RR. [DOI] [PubMed] [Google Scholar]
- 431. Maclean MR, Johnston ED, Mcculloch KM, Pooley L, Houslay MD, Sweeney G. Phosphodiesterase isoforms in the pulmonary arterial circulation of the rat: changes in pulmonary hypertension. J Pharmacol Exp Ther 283: 619–624, 1997. [PubMed] [Google Scholar]
- 432. Kass DA, Takimoto E, Nagayama T, Champion HC. Phosphodiesterase regulation of nitric oxide signaling. Cardiovasc Res 75: 303–314, 2007. doi: 10.1016/j.cardiores.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 433. Dussault S, Maingrette F, Ménard C, Michaud SE, Haddad P, Groleau J, Turgeon J, Perez G, Rivard A. Sildenafil increases endothelial progenitor cell function and improves ischemia-induced neovascularization in hypercholesterolemic apolipoprotein E-deficient mice. Hypertension 54: 1043–1049, 2009. doi: 10.1161/HYPERTENSIONAHA.109.139451. [DOI] [PubMed] [Google Scholar]
- 434. Balarini CM, Leal MA, Gomes IBS, Pereira TM, Gava AL, Meyrelles SS, Vasquez EC. Sildenafil restores endothelial function in the apolipoprotein E knockout mouse. J Transl Med 11: 3, 2013. doi: 10.1186/1479-5876-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435. Teloken PE, Mulhall JP. Impact of phosphodiesterase type 5 inhibitors on endothelial function. Rev Urol 10: 26–30, 2008. [PMC free article] [PubMed] [Google Scholar]
- 436. Muzaffar S, Shukla N, Bond M, Sala-Newby GB, Newby AC, Angelini GD, Jeremy JY. Superoxide from NADPH oxidase upregulates type 5 phosphodiesterase in human vascular smooth muscle cells: inhibition with iloprost and NONOate. Br J Pharmacol 155: 847–856, 2008. doi: 10.1038/bjp.2008.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437. Kaur R, Kaur M, Singh J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: molecular insights and therapeutic strategies. Cardiovasc Diabetol 17: 121, 2018. doi: 10.1186/s12933-018-0763-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438. Hirschberg K, Radovits T, Loganathan S, Entz L, Beller CJ, Gross ML, Sandner P, Karck M, Szabó G. Selective phosphodiesterase-5 inhibition reduces neointimal hyperplasia in rat carotid arteries after surgical endarterectomy. J Thorac Cardiovasc Surg 137: 1508–1514, 2009. doi: 10.1016/j.jtcvs.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 439. Guzeloglu M, Aykut K, Albayrak G, Atmaca S, Oktar S, Bagriyanik A, Hazan E. Effect of tadalafil on neointimal hyperplasia in a rabbit carotid artery anastomosis model. Ann Thorac Cardiovasc Surg 19: 468–474, 2013. doi: 10.5761/atcs.oa.12.02017. [DOI] [PubMed] [Google Scholar]
- 440. Lukowski R, Weinmeister P, Bernhard D, Feil S, Gotthardt M, Herz J, Massberg S, Zernecke A, Weber C, Hofmann F, Feil R. Role of smooth muscle cGMP/cGKI signaling in murine vascular restenosis. Arterioscler Thromb Vasc Biol 28: 1244–1250, 2008. doi: 10.1161/ATVBAHA.108.166405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441. Cesarini V, Pisano C, Rossi G, Balistreri CR, Botti F, Antonelli G, Ruvolo G, Jannini EA, Dolci S. Regulation of PDE5 expression in human aorta and thoracic aortic aneurysms. Sci Rep 9: 12206, 2019. doi: 10.1038/s41598-019-48432-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442. Zhang C, Mohan A, Shi H, Yan C. Sildenafil (Viagra) aggravates the development of experimental abdominal aortic aneurysm. J Am Heart Assoc 11: e023053, 2022. doi: 10.1161/JAHA.121.023053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443. Nachtnebel A, Stöllberger C, Ehrlich M, Finsterer J. Aortic dissection after sildenafil-induced erection. South Med J 99: 1151–1152, 2006. doi: 10.1097/01.smj.0000240732.65859.aa. [DOI] [PubMed] [Google Scholar]
- 444. Lameijer CM, Tielliu IF, van Driel MF, Zeebregts CJ. Type B aortic dissection after the use of tadalafil. Ann Thorac Surg 93: 651–653, 2012. doi: 10.1016/j.athoracsur.2011.07.016. [DOI] [PubMed] [Google Scholar]
- 445. Tiryakioglu SK, Tiryakioglu O, Turan T, Kumbay E. Aortic dissection due to sildenafil abuse. Interact Cardiovasc Thorac Surg 9: 141–143, 2009. doi: 10.1510/icvts.2009.205849. [DOI] [PubMed] [Google Scholar]
- 446. Tantini B, Manes A, Fiumana E, Pignatti C, Guarnieri C, Zannoli R, Branzi A, Galie N. Antiproliferative effect of sildenafil on human pulmonary artery smooth muscle cells. Basic Res Cardiol 100: 131–138, 2005. doi: 10.1007/s00395-004-0504-5. [DOI] [PubMed] [Google Scholar]
- 447. Hoeper MM, Al-Hiti H, Benza RL, Chang S-A, Corris PA, Gibbs JS, et al. Switching to riociguat versus maintenance therapy with phosphodiesterase-5 inhibitors in patients with pulmonary arterial hypertension (REPLACE): a multicentre, open-label, randomised controlled trial. Lancet Respir Med 9: 573–584, 2021. doi: 10.1016/S2213-2600(20)30532-4. [DOI] [PubMed] [Google Scholar]
- 448. Ruopp NF, Cockrill BA. Diagnosis and treatment of pulmonary arterial hypertension: a review. JAMA 327: 1379–1391, 2022. doi: 10.1001/jama.2022.4402. [DOI] [PubMed] [Google Scholar]
- 449. Clouthier DE, Comerford SA, Hammer RE. Hepatic fibrosis, glomerulosclerosis, and a lipodystrophy-like syndrome in PEPCK-TGF-beta1 transgenic mice. J Clin Invest 100: 2697–2713, 1997. doi: 10.1172/JCI119815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450. Brzezinska P, Maurice DH. An EPAC1/PDE1C-signaling axis regulates formation of leading-edge protrusion in polarized human arterial vascular smooth muscle cells. Cells 8: 1473, 2019. doi: 10.3390/cells8121473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451. Zhang C, Zhao H, Cai Y, Xiong J, Mohan A, Lou D, Shi H, Zhang Y, Long X, Wang J, Yan C. Cyclic nucleotide phosphodiesterase 1C contributes to abdominal aortic aneurysm. Proc Natl Acad Sci U S A 118: e2107898118, 2021. doi: 10.1073/pnas.2107898118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452. Tao X, Zhang YJ, Shen FM, Guan YF, Su DF. Fosinopril prevents the pulmonary arterial remodeling in sinoaortic-denervated rats by regulating phosphodiesterase. J Cardiovasc Pharmacol 51: 24–31, 2008. doi: 10.1097/FJC.0b013e318159e097. [DOI] [PubMed] [Google Scholar]
- 453. Murray F, Patel HH, Suda RY, Zhang S, Thistlethwaite PA, Yuan JX, Insel PA. Expression and activity of cAMP phosphodiesterase isoforms in pulmonary artery smooth muscle cells from patients with pulmonary hypertension: role for PDE1. Am J Physiol Lung Cell Mol Physiol 292: L294–L303, 2007. doi: 10.1152/ajplung.00190.2006. [DOI] [PubMed] [Google Scholar]
- 454. Schermuly RT, Pullamsetti SS, Kwapiszewska G, Dumitrascu R, Tian X, Weissmann N, Ghofrani HA, Kaulen C, Dunkern T, Schudt C, Voswinckel R, Zhou J, Samidurai A, Klepetko W, Paddenberg R, Kummer W, Seeger W, Grimminger F. Phosphodiesterase 1 upregulation in pulmonary arterial hypertension: target for reverse-remodeling therapy. Circulation 115: 2331–2339, 2007. doi: 10.1161/CIRCULATIONAHA.106.676809. [DOI] [PubMed] [Google Scholar]
- 455. Schermuly RT, Inholte C, Ghofrani HA, Gall H, Weissmann N, Weidenbach A, Seeger W, Grimminger F. Lung vasodilatory response to inhaled iloprost in experimental pulmonary hypertension: amplification by different type phosphodiesterase inhibitors. Respir Res 6: 76, 2005. doi: 10.1186/1465-9921-6-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456. Phillips PG, Long L, Wilkins MR, Morrell NW. cAMP phosphodiesterase inhibitors potentiate effects of prostacyclin analogs in hypoxic pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol 288: L103–L115, 2005. doi: 10.1152/ajplung.00095.2004. [DOI] [PubMed] [Google Scholar]
- 457. Evgenov OV, Busch CJ, Evgenov NV, Liu R, Petersen B, Falkowski GE, Petho B, Vas A, Bloch KD, Zapol WM, Ichinose F. Inhibition of phosphodiesterase 1 augments the pulmonary vasodilator response to inhaled nitric oxide in awake lambs with acute pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 290: L723–L729, 2006. doi: 10.1152/ajplung.00485.2004. [DOI] [PubMed] [Google Scholar]
- 458. Nagaoka T, Shirakawa T, Balon TW, Russell JC, Fujita-Yamaguchi Y. Cyclic nucleotide phosphodiesterase 3 expression in vivo: evidence for tissue-specific expression of phosphodiesterase 3A or 3B mRNA and activity in the aorta and adipose tissue of atherosclerosis-prone insulin-resistant rats. Diabetes 47: 1135–1144, 1998. doi: 10.2337/diabetes.47.7.1135. [DOI] [PubMed] [Google Scholar]
- 459. Kondo K, Umemura K, Miyaji M, Nakashima M. Milrinone, a phosphodiesterase inhibitor, suppresses intimal thickening after photochemically induced endothelial injury in the mouse femoral artery. Atherosclerosis 142: 133–138, 1999. doi: 10.1016/S0021-9150(98)00203-2. [DOI] [PubMed] [Google Scholar]
- 460. Smith JA. Measuring treatment effects of cilostazol on clinical trial endpoints in patients with intermittent claudication. Clin Cardiol 25: 91–94, 2002. doi: 10.1002/clc.4960250303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461. Aizawa T, Wei H, Miano JM, Abe J, Berk BC, Yan C. Role of phosphodiesterase 3 in NO/cGMP-mediated antiinflammatory effects in vascular smooth muscle cells. Circ Res 93: 406–413, 200 3. doi: 10.1161/01.RES.0000091074.33584.F0. [DOI] [PubMed] [Google Scholar]
- 462. Umebayashi R, Uchida HA, Kakio Y, Subramanian V, Daugherty A, Wada J. Cilostazol attenuates angiotensin II-induced abdominal aortic aneurysms but not atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 38: 903–912, 2018. doi: 10.1161/ATVBAHA.117.309707. [DOI] [PubMed] [Google Scholar]
- 463. Zhang Q, Huang JH, Xia RP, Duan XH, Jiang YB, Jiang Q, Sun WJ. Suppression of experimental abdominal aortic aneurysm in a rat model by the phosphodiesterase 3 inhibitor cilostazol. J Surg Res 167: e385–e393, 2011. doi: 10.1016/j.jss.2011.01.017. [DOI] [PubMed] [Google Scholar]
- 464. Khajavi K, Ayzman I, Shearer D, Jones SC, Levy JH, Prayson RA, Skibinski CI, Hahn JF, Chyatte D. Prevention of chronic cerebral vasospasm in dogs with milrinone. Neurosurgery 40: 354–363, 1997. doi: 10.1097/00006123-199702000-00025. [DOI] [PubMed] [Google Scholar]
- 465. Fraticelli AT, Cholley BP, Losser MR, Saint Maurice JP, Payen D. Milrinone for the treatment of cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Stroke 39: 893–898, 2008. doi: 10.1161/STROKEAHA.107.492447. [DOI] [PubMed] [Google Scholar]
- 466. Luo L, Cai Y, Zhang Y, Hsu CG, Korshunov VA, Long X, Knight PA, Berk BC, Yan C. Role of PDE10A in vascular smooth muscle cell hyperplasia and pathological vascular remodelling. Cardiovasc Res 118: 2703–2717, 2022. doi: 10.1093/cvr/cvab304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467. Tian X, Vroom C, Ghofrani HA, Weissmann N, Bieniek E, Grimminger F, Seeger W, Schermuly RT, Pullamsetti SS. Phosphodiesterase 10A upregulation contributes to pulmonary vascular remodeling. PLoS One 6: e18136, 2011. doi: 10.1371/journal.pone.0018136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468. Thickett DR, Armstrong L, Christie SJ, Millar AB. Vascular endothelial growth factor may contribute to increased vascular permeability in acute respiratory distress syndrome. Am J Respir Crit Care Med 164: 1601–1605, 2001. doi: 10.1164/ajrccm.164.9.2011071. [DOI] [PubMed] [Google Scholar]
- 469. Wautier JL, Wautier MP. Vascular permeability in diseases. Int J Mol Sci 23: 3645, 2022. doi: 10.3390/ijms23073645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470. Miró X, Casacuberta JM, Gutiérrez-López MD, de Landázuri MO, Puigdomènech P. Phosphodiesterases 4D and 7A splice variants in the response of HUVEC cells to TNF-alpha(1). Biochem Biophys Res Commun 274: 415–421, 2000. doi: 10.1006/bbrc.2000.3146. [DOI] [PubMed] [Google Scholar]
- 471. Lugnier C, Meyer A, Talha S, Geny B. Cyclic nucleotide phosphodiesterases: new targets in the metabolic syndrome? Pharmacol Ther 208: 107475, 2020. doi: 10.1016/j.pharmthera.2020.107475. [DOI] [PubMed] [Google Scholar]
- 472. Kraft P, Schwarz T, Göb E, Heydenreich N, Brede M, Meuth SG, Kleinschnitz C. The phosphodiesterase-4 inhibitor rolipram protects from ischemic stroke in mice by reducing blood-brain-barrier damage, inflammation and thrombosis. Exp Neurol 247: 80–90, 2013. doi: 10.1016/j.expneurol.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 473. Lin YC, Samardzic H, Adamson RH, Renkin EM, Clark JF, Reed RK, Curry FR. Phosphodiesterase 4 inhibition attenuates atrial natriuretic peptide-induced vascular hyperpermeability and loss of plasma volume. J Physiol 589: 341–353, 2011. doi: 10.1113/jphysiol.2010.199588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474. Witzenrath M, Gutbier B, Schmeck B, Tenor H, Seybold J, Kuelzer R, Grentzmann G, Hatzelmann A, van Laak V, Tschernig T, Mitchell TJ, Schudt C, Rosseau S, Suttorp N, Schütte H. Phosphodiesterase 2 inhibition diminished acute lung injury in murine pneumococcal pneumonia. Crit Care Med 37: 584–590, 2009. doi: 10.1097/CCM.0b013e3181959814. [DOI] [PubMed] [Google Scholar]
- 475. Wilson LS, Baillie GS, Pritchard LM, Umana B, Terrin A, Zaccolo M, Houslay MD, Maurice DH. A phosphodiesterase 3B-based signaling complex integrates exchange protein activated by cAMP 1 and phosphatidylinositol 3-kinase signals in human arterial endothelial cells. J Biol Chem 286: 16285–16296, 2011. doi: 10.1074/jbc.M110.217026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476. Comerford KM, Lawrence DW, Synnestvedt K, Levi BP, Colgan SP. Role of vasodilator-stimulated phosphoprotein in PKA-induced changes in endothelial junctional permeability. FASEB J 16: 583–585, 2002. doi: 10.1096/fj.01-0739fje. [DOI] [PubMed] [Google Scholar]
- 477. Tang ST, Tang HQ, Su H, Wang Y, Zhou Q, Zhang Q, Wang Y, Zhu HQ. Glucagon-like peptide-1 attenuates endothelial barrier injury in diabetes via cAMP/PKA mediated down-regulation of MLC phosphorylation. Biomed Pharmacother 113: 108667, 2019. doi: 10.1016/j.biopha.2019.108667. [DOI] [PubMed] [Google Scholar]
- 478. Boardman R, Pang V, Malhi N, Lynch AP, Leach L, Benest AV, Bates DO, Machado MJ. Activation of Notch signaling by soluble Dll4 decreases vascular permeability via a cAMP/PKA-dependent pathway. Am J Physiol Heart Circ Physiol 316: H1065–H1075, 2019. doi: 10.1152/ajpheart.00610.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479. Fukuhara S, Sakurai A, Sano H, Yamagishi A, Somekawa S, Takakura N, Saito Y, Kangawa K, Mochizuki N. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol Cell Biol 25: 136–146, 2005. doi: 10.1128/MCB.25.1.136-146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480. Ramos CJ, Lin C, Liu X, Antonetti DA. The EPAC-Rap1 pathway prevents and reverses cytokine-induced retinal vascular permeability. J Biol Chem 293: 717–730, 2018. doi: 10.1074/jbc.M117.815381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481. Sehrawat S, Cullere X, Patel S, Italiano J Jr, Mayadas TN. Role of Epac1, an exchange factor for Rap GTPases, in endothelial microtubule dynamics and barrier function. Mol Biol Cell 19: 1261–1270, 2008. doi: 10.1091/mbc.e06-10-0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482. Kopperud RK, Rygh CB, Karlsen TV, Krakstad C, Kleppe R, Hoivik EA, Bakke M, Tenstad O, Selheim F, Lidén Å, Madsen L, Pavlin T, Taxt T, Kristiansen K, Curry FR, Reed RK, Døskeland SO. Increased microvascular permeability in mice lacking Epac1 (Rapgef3). Acta Physiol (Oxf) 219: 441–452, 2017. doi: 10.1111/apha.12697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483. Birukova AA, Zagranichnaya T, Fu P, Alekseeva E, Chen W, Jacobson JR, Birukov KG. Prostaglandins PGE2 and PGI2 promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Exp Cell Res 313: 2504–2520, 2007. doi: 10.1016/j.yexcr.2007.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484. Westendorp RG, Draijer R, Meinders AE, van Hinsbergh VW. Cyclic-GMP-mediated decrease in permeability of human umbilical and pulmonary artery endothelial cell monolayers. J Vasc Res 31: 42–51, 1994. doi: 10.1159/000159030. [DOI] [PubMed] [Google Scholar]
- 485. Draijer R, Atsma DE, van der Laarse A, van Hinsbergh VW. cGMP and nitric oxide modulate thrombin-induced endothelial permeability. Regulation via different pathways in human aortic and umbilical vein endothelial cells. Circ Res 76: 199–208, 1995. doi: 10.1161/01.RES.76.2.199. [DOI] [PubMed] [Google Scholar]
- 486. Kolluru GK, Tamilarasan KP, Rajkumar AS, Geetha Priya S, Rajaram M, Saleem NK, Majumder S, Jaffar Ali BM, Illavazagan G, Chatterjee S. Nitric oxide/cGMP protects endothelial cells from hypoxia-mediated leakiness. Eur J Cell Biol 87: 147–161, 2008. doi: 10.1016/j.ejcb.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 487. Chen W, Oberwinkler H, Werner F, Gaßner B, Nakagawa H, Feil R, Hofmann F, Schlossmann J, Dietrich A, Gudermann T, Nishida M, Del Galdo S, Wieland T, Kuhn M. Atrial natriuretic peptide-mediated inhibition of microcirculatory endothelial Ca2+ and permeability response to histamine involves cGMP-dependent protein kinase I and TRPC6 channels. Arterioscler Thromb Vasc Biol 33: 2121–2129, 2013. doi: 10.1161/ATVBAHA.113.001974. [DOI] [PubMed] [Google Scholar]
- 488. Di Lorenzo A, Lin MI, Murata T, Landskroner-Eiger S, Schleicher M, Kothiya M, Iwakiri Y, Yu J, Huang PL, Sessa WC. eNOS-derived nitric oxide regulates endothelial barrier function through VE-cadherin and Rho GTPases. J Cell Sci 126: 5541–5552, 2013. doi: 10.1242/jcs.115972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489. Yang B, Cai B, Deng P, Wu X, Guan Y, Zhang B, Cai W, Schaper J, Schaper W. Nitric oxide increases arterial endothelial permeability through mediating VE-cadherin expression during arteriogenesis. PLoS One 10: e0127931, 2015. doi: 10.1371/journal.pone.0127931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490. Tsujimae K, Suzuki S, Murakami S, Kurachi Y. Frequency-dependent effects of various IKr blockers on cardiac action potential duration in a human atrial model. Am J Physiol Heart Circ Physiol 293: H660–H669, 2007. doi: 10.1152/ajpheart.01083.2006. [DOI] [PubMed] [Google Scholar]
- 491. Rampersad SN, Ovens JD, Huston E, Umana MB, Wilson LS, Netherton SJ, Lynch MJ, Baillie GS, Houslay MD, Maurice DH. Cyclic AMP phosphodiesterase 4D (PDE4D) tethers EPAC1 in a vascular endothelial cadherin (VE-Cad)-based signaling complex and controls cAMP-mediated vascular permeability. J Biol Chem 285: 33614–33622, 2010. doi: 10.1074/jbc.M110.140004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492. Flemming S, Schlegel N, Wunder C, Meir M, Baar W, Wollborn J, Roewer N, Germer CT, Schick MA. Phosphodiesterase 4 inhibition dose dependently stabilizes microvascular barrier functions and microcirculation in a rodent model of polymicrobial sepsis. Shock 41: 537–545, 2014. doi: 10.1097/SHK.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 493. Creighton J, Zhu B, Alexeyev M, Stevens T. Spectrin-anchored phosphodiesterase 4D4 restricts cAMP from disrupting microtubules and inducing endothelial cell gap formation. J Cell Sci 121: 110–119, 2008. doi: 10.1242/jcs.011692. [DOI] [PubMed] [Google Scholar]
- 494. Fertig BA, Baillie GS. PDE4-mediated cAMP signalling. J Cardiovasc Dev Dis 5: 8, 2018. doi: 10.3390/jcdd5010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495. Mokra D, Mokry J. Phosphodiesterase inhibitors in acute lung injury: what are the perspectives? Int J Mol Sci 22: 1929, 2021. doi: 10.3390/ijms22041929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496. Folcik VA, Smith T, O’Bryant S, Kawczak JA, Zhu B, Sakurai H, Kajiwara A, Staddon JM, Glabinski A, Chernosky AL, Tani M, Johnson JM, Tuohy VK, Rubin LL, Ransohoff RM. Treatment with BBB022A or rolipram stabilizes the blood-brain barrier in experimental autoimmune encephalomyelitis: an additional mechanism for the therapeutic effect of type IV phosphodiesterase inhibitors. J Neuroimmunol 97: 119–128, 1999. doi: 10.1016/s0165-5728(99)00063-6. [DOI] [PubMed] [Google Scholar]
- 497. Favot L, Keravis T, Holl V, Le Bec A, Lugnier C. VEGF-induced HUVEC migration and proliferation are decreased by PDE2 and PDE4 inhibitors. Thromb Haemost 90: 334–343, 2003. doi: 10.1160/TH03-02-0084. [DOI] [PubMed] [Google Scholar]
- 498. Seybold J, Thomas D, Witzenrath M, Boral S, Hocke AC, Bürger A, Hatzelmann A, Tenor H, Schudt C, Krüll M, Schütte H, Hippenstiel S, Suttorp N. Tumor necrosis factor-alpha-dependent expression of phosphodiesterase 2: role in endothelial hyperpermeability. Blood 105: 3569–3576, 2005. doi: 10.1182/blood-2004-07-2729. [DOI] [PubMed] [Google Scholar]
- 499. Horai S, Nakagawa S, Tanaka K, Morofuji Y, Couraud PO, Deli MA, Ozawa M, Niwa M. Cilostazol strengthens barrier integrity in brain endothelial cells. Cell Mol Neurobiol 33: 291–307, 2013. doi: 10.1007/s10571-012-9896-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500. Baillie GS, Tejeda GS, Kelly MP. Therapeutic targeting of 3',5'-cyclic nucleotide phosphodiesterases: inhibition and beyond. Nat Rev Drug Discov 18: 770–796, 2019. doi: 10.1038/s41573-019-0033-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501. Bobin P, Belacel-Ouari M, Bedioune I, Zhang L, Leroy J, Leblais V, Fischmeister R, Vandecasteele G. Cyclic nucleotide phosphodiesterases in heart and vessels: a therapeutic perspective. Arch Cardiovasc Dis 109: 431–443, 2016. doi: 10.1016/j.acvd.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 502. Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC. Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discov 13: 290–314, 2014. doi: 10.1038/nrd4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, et al. ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 128: e240–327, 2013. doi: 10.1161/CIR.0b013e31829e8776. [DOI] [PubMed] [Google Scholar]
- 504. Eschenhagen T. PDE4 in the human heart–major player or little helper? Br J Pharmacol 169: 524–527, 2013. doi: 10.1111/bph.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505. Jaski BE, Fifer MA, Wright RF, Braunwald E, Colucci WS. Positive inotropic and vasodilator actions of milrinone in patients with severe congestive heart failure. Dose-response relationships and comparison to nitroprusside. J Clin Invest 75: 643–649, 1985. doi: 10.1172/JCI111742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506. Zhang Y, Wang WE, Zhang X, Li Y, Chen B, Liu C, Ai X, Zhang X, Tian Y, Zhang C, Tang M, Szeto C, Hua X, Xie M, Zeng C, Wu Y, Zhou L, Zhu W, Yu D, Houser SR, Chen X. Cardiomyocyte PKA ablation enhances basal contractility while eliminates cardiac β-adrenergic response without adverse effects on the heart. Circ Res 124: 1760–1777, 2019. doi: 10.1161/CIRCRESAHA.118.313417. [DOI] [PubMed] [Google Scholar]
- 507. Amsallem E, Kasparian C, Haddour G, Boissel JP, Nony P. Phosphodiesterase III inhibitors for heart failure. Cochrane Database Syst Rev 2005: CD002230, 2005. doi: 10.1002/14651858.CD002230.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508. Holmes JR, Kubo SH, Cody RJ, Kligfield P. Milrinone in congestive heart failure: observations on ambulatory ventricular arrhythmias. Am Heart J 110: 800–806, 1985. doi: 10.1016/0002-8703(85)90460-0. [DOI] [PubMed] [Google Scholar]
- 509. DiBianco R, Shabetai R, Kostuk W, Moran J, Schlant RC, Wright R. A comparison of oral milrinone, digoxin, and their combination in the treatment of patients with chronic heart failure. N Engl J Med 320: 677–683, 1989. doi: 10.1056/NEJM198903163201101. [DOI] [PubMed] [Google Scholar]
- 510. Sucharov CC, Nakano SJ, Slavov D, Schwisow JA, Rodriguez E, Nunley K, Medway A, Stafford N, Nelson P, McKinsey TA, Movsesian M, Minobe W, Carroll IA, Taylor MR, Bristow MR. A PDE3A promoter polymorphism regulates cAMP-induced transcriptional activity in failing human myocardium. J Am Coll Cardiol 73: 1173–1184, 2019. doi: 10.1016/j.jacc.2018.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511. Metra M, Eichhorn E, Abraham WT, Linseman J, Bohm M, Corbalan R, DeMets D, De Marco T, Elkayam U, Gerber M, Komajda M, Liu P, Mareev V, Perrone SV, Poole-Wilson P, Roecker E, Stewart J, Swedberg K, Tendera M, Wiens B, Bristow MR; ESSENTIAL Investigators. Effects of low-dose oral enoximone administration on mortality, morbidity, and exercise capacity in patients with advanced heart failure: the randomized, double-blind, placebo-controlled, parallel group ESSENTIAL trials. Eur Heart J 30: 3015–3026, 2009. doi: 10.1093/eurheartj/ehp338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512. Nanayakkara S, Mak V, Crannitch K, Byrne M, Kaye DM. Extended release oral milrinone, CRD-102, for advanced heart failure. Am J Cardiol 122: 1017–1020, 2018. doi: 10.1016/j.amjcard.2018.06.009. [DOI] [PubMed] [Google Scholar]
- 513. Shakar SF, Abraham WT, Gilbert EM, Robertson AD, Lowes BD, Zisman LS, Ferguson DA, Bristow MR. Combined oral positive inotropic and beta-blocker therapy for treatment of refractory class IV heart failure. J Am Coll Cardiol 31: 1336–1340, 1998. doi: 10.1016/S0735-1097(98)00077-1. [DOI] [PubMed] [Google Scholar]
- 514. Harrison SA, Reifsnyder DH, Gallis B, Cadd GG, Beavo JA. Isolation and characterization of bovine cardiac muscle cGMP-inhibited phosphodiesterase: a receptor for new cardiotonic drugs. Mol Pharmacol 29: 506–514, 1986. [PubMed] [Google Scholar]
- 515. Chong LY, Satya K, Kim B, Berkowitz R. Milrinone dosing and a culture of caution in clinical practice. Cardiol Rev 26: 35–42, 2018. doi: 10.1097/CRD.0000000000000165. [DOI] [PubMed] [Google Scholar]
- 516. Guazzi M, Vicenzi M, Arena R, Guazzi MD. PDE5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: results of a 1-year, prospective, randomized, placebo-controlled study. Circ Heart Fail 4: 8–17, 2011. doi: 10.1161/CIRCHEARTFAILURE.110.944694. [DOI] [PubMed] [Google Scholar]
- 517. Lewis GD, Lachmann J, Camuso J, Lepore JJ, Shin J, Martinovic ME, Systrom DM, Bloch KD, Semigran MJ. Sildenafil improves exercise hemodynamics and oxygen uptake in patients with systolic heart failure. Circulation 115: 59–66, 2007. doi: 10.1161/CIRCULATIONAHA.106.626226. [DOI] [PubMed] [Google Scholar]
- 518. Lewis GD, Shah R, Shahzad K, Camuso JM, Pappagianopoulos PP, Hung J, Tawakol A, Gerszten RE, Systrom DM, Bloch KD, Semigran MJ. Sildenafil improves exercise capacity and quality of life in patients with systolic heart failure and secondary pulmonary hypertension. Circulation 116: 1555–1562, 2007. doi: 10.1161/CIRCULATIONAHA.107.716373. [DOI] [PubMed] [Google Scholar]
- 519. Kim KH, Kim HK, Hwang IC, Cho HJ, Je N, Kwon OM, Choi SJ, Lee SP, Kim YJ, Sohn DW. PDE 5 inhibition with udenafil improves left ventricular systolic/diastolic functions and exercise capacity in patients with chronic heart failure with reduced ejection fraction: a 12-week, randomized, double-blind, placebo-controlled trial. Am Heart J 169: 813–822.e3, 2015. doi: 10.1016/j.ahj.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 520. Jiang R, Wang L, Zhu CT, Yuan P, Pudasaini B, Zhao QH, Gong SG, He J, Liu JM, Hu QH. Comparative effectiveness of sildenafil for pulmonary hypertension due to left heart disease with HFrEF. Hypertens Res 38: 829–839, 2015. doi: 10.1038/hr.2015.73. [DOI] [PubMed] [Google Scholar]
- 521. Giannetta E, Isidori AM, Galea N, Carbone I, Mandosi E, Vizza CD, Naro F, Morano S, Fedele F, Lenzi A. Chronic inhibition of cGMP phosphodiesterase 5A improves diabetic cardiomyopathy: a randomized, controlled clinical trial using magnetic resonance imaging with myocardial tagging. Circulation 125: 2323–2333, 2012. doi: 10.1161/CIRCULATIONAHA.111.063412. [DOI] [PubMed] [Google Scholar]
- 522. Andersson DP, Trolle Lagerros Y, Grotta A, Bellocco R, Lehtihet M, Holzmann MJ. Association between treatment for erectile dysfunction and death or cardiovascular outcomes after myocardial infarction. Heart 103: 1264–1270, 2017. doi: 10.1136/heartjnl-2016-310746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523. Andersen MJ, Ersbøll M, Axelsson A, Gustafsson F, Hassager C, Køber L, Borlaug BA, Boesgaard S, Skovgaard LT, Møller JE. Sildenafil and diastolic dysfunction after acute myocardial infarction in patients with preserved ejection fraction: the Sildenafil and Diastolic Dysfunction After Acute Myocardial Infarction (SIDAMI) trial. Circulation 127: 1200–1208, 2013. doi: 10.1161/CIRCULATIONAHA.112.000056. [DOI] [PubMed] [Google Scholar]
- 524. Gilotra NA, DeVore AD, Povsic TJ, Hays AG, Hahn VS, Agunbiade TA, DeLong A, Satlin A, Chen R, Davis R, Kass DA. Acute hemodynamic effects and tolerability of phosphodiesterase-1 inhibition with ITI-214 in human systolic heart failure. Circ Heart Fail 14: e008236, 2021. doi: 10.1161/CIRCHEARTFAILURE.120.008236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525. Gheorghiade M, Greene SJ, Butler J, Filippatos G, Lam CS, Maggioni AP, Ponikowski P, Shah SJ, Solomon SD, Kraigher-Krainer E, Samano ET, Müller K, Roessig L, Pieske B; SOCRATES-REDUCED Investigators and Coordinators. Effect of vericiguat, a soluble guanylate cyclase stimulator, on natriuretic peptide levels in patients with worsening chronic heart failure and reduced ejection fraction: the SOCRATES-REDUCED Randomized Trial. JAMA 314: 2251–2262, 2015. doi: 10.1001/jama.2015.15734. [DOI] [PubMed] [Google Scholar]
- 526. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL. ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 128: 1810–1852, 2013. doi: 10.1161/CIR.0b013e31829e8807. [DOI] [PubMed] [Google Scholar]
- 527. Paulus WJ, van Ballegoij JJ. Treatment of heart failure with normal ejection fraction: an inconvenient truth! J Am Coll Cardiol 55: 526–537, 2010. doi: 10.1016/j.jacc.2009.06.067. [DOI] [PubMed] [Google Scholar]
- 528. Holland DJ, Kumbhani DJ, Ahmed SH, Marwick TH. Effects of treatment on exercise tolerance, cardiac function, and mortality in heart failure with preserved ejection fraction. A meta-analysis. J Am Coll Cardiol 57: 1676–1686, 2011. doi: 10.1016/j.jacc.2010.10.057. [DOI] [PubMed] [Google Scholar]
- 529. Shah SJ, Kitzman DW, Borlaug BA, van Heerebeek L, Zile MR, Kass DA, Paulus WJ. Phenotype-specific treatment of heart failure with preserved ejection fraction: a multiorgan roadmap. Circulation 134: 73–90, 2016. doi: 10.1161/CIRCULATIONAHA.116.021884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530. Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62: 263–271, 2013. doi: 10.1016/j.jacc.2013.02.092. [DOI] [PubMed] [Google Scholar]
- 531. Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, von Schlippenbach J, Skurk C, Steendijk P, Riad A, Poller W, Schultheiss HP, Tschöpe C. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail 4: 44–52, 2011. doi: 10.1161/CIRCHEARTFAILURE.109.931451. [DOI] [PubMed] [Google Scholar]
- 532. van Heerebeek L, Hamdani N, Falcão-Pires I, Leite-Moreira AF, Begieneman MP, Bronzwaer JG, van der Velden J, Stienen GJ, Laarman GJ, Somsen A, Verheugt FW, Niessen HW, Paulus WJ. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 126: 830–839, 2012. doi: 10.1161/CIRCULATIONAHA.111.076075. [DOI] [PubMed] [Google Scholar]
- 533. Redfield MM, Anstrom KJ, Levine JA, Koepp GA, Borlaug BA, Chen HH, LeWinter MM, Joseph SM, Shah SJ, Semigran MJ, Felker GM, Cole RT, Reeves GR, Tedford RJ, Tang WH, McNulty SE, Velazquez EJ, Shah MR, Braunwald E; NHLBI Heart Failure Clinical Research Network. Isosorbide mononitrate in heart failure with preserved ejection fraction. N Engl J Med 373: 2314–2324, 2015. doi: 10.1056/NEJMoa1510774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534. Schwartzenberg S, Redfield MM, From AM, Sorajja P, Nishimura RA, Borlaug BA. Effects of vasodilation in heart failure with preserved or reduced ejection fraction implications of distinct pathophysiologies on response to therapy. J Am Coll Cardiol 59: 442–451, 2012. doi: 10.1016/j.jacc.2011.09.062. [DOI] [PubMed] [Google Scholar]
- 535. Kaye DM, Nanayakkara S, Vizi D, Byrne M, Mariani JA. Effects of milrinone on rest and exercise hemodynamics in heart failure with preserved ejection fraction. J Am Coll Cardiol 67: 2554–2556, 2016. doi: 10.1016/j.jacc.2016.03.539. [DOI] [PubMed] [Google Scholar]
- 536. Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 309: 1268–1277, 2013. doi: 10.1001/jama.2013.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537. Borlaug BA, Lewis GD, McNulty SE, Semigran MJ, LeWinter M, Chen H, Lin G, Deswal A, Margulies KB, Redfield MM. Effects of sildenafil on ventricular and vascular function in heart failure with preserved ejection fraction. Circ Heart Fail 8: 533–541, 2015. doi: 10.1161/CIRCHEARTFAILURE.114.001915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538. D’Amario D, Migliaro S, Borovac JA, Restivo A, Vergallo R, Galli M, Leone AM, Montone RA, Niccoli G, Aspromonte N, Crea F. Microvascular dysfunction in heart failure with preserved ejection fraction. Front Physiol 10: 1347, 2019. doi: 10.3389/fphys.2019.01347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539. Hussain I, Mohammed SF, Forfia PR, Lewis GD, Borlaug BA, Gallup DS, Redfield MM. Impaired right ventricular-pulmonary arterial coupling and effect of sildenafil in heart failure with preserved ejection fraction: an ancillary analysis from the Phosphodiesterase-5 Inhibition to Improve Clinical Status And Exercise Capacity in Diastolic Heart Failure (RELAX) Trial. Circ Heart Fail 9: e002729, 2016. doi: 10.1161/CIRCHEARTFAILURE.115.002729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540. Solomon SD, McMurray JJ, Anand IS, Ge J, Lam CS, Maggioni AP, et al. Angiotensin-neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med 381: 1609–1620, 2019. doi: 10.1056/NEJMoa1908655. [DOI] [PubMed] [Google Scholar]
- 541. Galiè N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, Fleming T, Parpia T, Burgess G, Branzi A, Grimminger F, Kurzyna M, Simonneau G; Sildenafil Use in Pulmonary Arterial Hypertension (SUPER) Study Group. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 353: 2148–2157, 2005. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 542. Rubin LJ, Badesch DB, Fleming TR, Galiè N, Simonneau G, Ghofrani HA, Oakes M, Layton G, Serdarevic-Pehar M, McLaughlin VV, Barst RJ; SUPER-2 Study Group. Long-term treatment with sildenafil citrate in pulmonary arterial hypertension: the SUPER-2 study. Chest 140: 1274–1283, 2011. doi: 10.1378/chest.10-0969. [DOI] [PubMed] [Google Scholar]
- 543. Goldberg DJ, Zak V, Goldstein BH, Schumacher KR, Rhodes J, Penny DJ, et al. Results of the FUEL Trial. Circulation 141: 641–651, 2020. doi: 10.1161/CIRCULATIONAHA.119.044352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544. Samidurai A, Xi L, Das A, Iness AN, Vigneshwar NG, Li PL, Singla DK, Muniyan S, Batra SK, Kukreja RC. Role of phosphodiesterase 1 in the pathophysiology of diseases and potential therapeutic opportunities. Pharmacol Ther 226: 107858, 2021. doi: 10.1016/j.pharmthera.2021.107858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545. Yin D, Slavin KV. A hypothesis on possible neurochemical mechanisms of action of cervical spinal cord stimulation in prevention and treatment of cerebral arterial vasospasm after aneurysmal subarachnoid hemorrhage. Med Hypotheses 85: 355–358, 2015. doi: 10.1016/j.mehy.2015.06.014. [DOI] [PubMed] [Google Scholar]
- 546. Washington CW, Derdeyn CP, Dhar R, Arias EJ, Chicoine MR, Cross DT, Dacey RG Jr, Han BH, Moran CJ, Rich KM, Vellimana AK, Zipfel GJ. A Phase I proof-of-concept and safety trial of sildenafil to treat cerebral vasospasm following subarachnoid hemorrhage. J Neurosurg 124: 318–327, 2016. doi: 10.3171/2015.2.JNS142752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547. Lee JH, Park SY, Shin HK, Kim CD, Lee WS, Hong KW. Protective effects of cilostazol against transient focal cerebral ischemia and chronic cerebral hypoperfusion injury. CNS Neurosci Ther 14: 143–152, 2008. doi: 10.1111/j.1527-3458.2008.00042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548. Kambayashi J, Liu Y, Sun B, Shakur Y, Yoshitake M, Czerwiec F. Cilostazol as a unique antithrombotic agent. Curr Pharm Des 9: 2289–2302, 2003. doi: 10.2174/1381612033453910. [DOI] [PubMed] [Google Scholar]
- 549. Gotoh F, Tohgi H, Hirai S, Terashi A, Fukuuchi Y, Otomo E, Shinohara Y, Itoh E, Matsuda T, Sawada T, Yamaguchi T, Nishimaru K, Ohashi Y. Cilostazol stroke prevention study: a placebo-controlled double-blind trial for secondary prevention of cerebral infarction. J Stroke Cerebrovasc Dis 9: 147–157, 2000. doi: 10.1053/jscd.2000.7216. [DOI] [PubMed] [Google Scholar]
- 550. Liu J, He J, Chen X, Feng Y, Wang C, Awil MA, Wang Y, Tian Y, Hou D. Cilostazol for aneurysmal subarachnoid hemorrhage: an updated systematic review and meta-analysis. Cerebrovasc Dis 51: 138–148, 2022. doi: 10.1159/000518731. [DOI] [PubMed] [Google Scholar]
- 551. Suzuki H, Nakatsuka Y, Yasuda R, Shiba M, Miura Y, Terashima M, Suzuki Y, Hakozaki K, Goto F, Toma N. Dose-dependent inhibitory effects of cilostazol on delayed cerebral infarction after aneurysmal subarachnoid hemorrhage. Transl Stroke Res 10: 381–388, 2019. doi: 10.1007/s12975-018-0650-y. [DOI] [PubMed] [Google Scholar]
- 552. Douglas JS Jr, Holmes DR Jr, Kereiakes DJ, Grines CL, Block E, Ghazzal ZM, Morris DC, Liberman H, Parker K, Jurkovitz C, Murrah N, Foster J, Hyde P, Mancini GB, Weintraub WS Jr.. Coronary stent restenosis in patients treated with cilostazol. Circulation 112: 2826–2832, 2005. doi: 10.1161/CIRCULATIONAHA.104.530097. [DOI] [PubMed] [Google Scholar]
- 553. Dalainas I. Cilostazol in the management of vascular disease. Int Angiol 26: 1–7, 2007. [PubMed] [Google Scholar]
- 554. Zhang Z, Foster JK, Kolm P, Jurkovitz CT, Parker KM, Murrah NV, Anderson GT, Douglas JS Jr, Weintraub WS. Reduced 6-month resource use and costs associated with cilostazol in patients after successful coronary stent implantation: results from the Cilostazol for RESTenosis (CREST) trial. Am Heart J 152: 770–776, 2006. doi: 10.1016/j.ahj.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 555. Lee SW, Chun KJ, Park SW, Kim HS, Kim YH, Yun SC, Kim WJ, Lee JY, Park DW, Lee CW, Hong MK, Rhee KS, Chae JK, Ko JK, Park JH, Lee JH, Choi SW, Jeong JO, Seong IW, Jon S, Cho YH, Lee NH, Kim JH, Park SJ. Comparison of triple antiplatelet therapy and dual antiplatelet therapy in patients at high risk of restenosis after drug-eluting stent implantation (from the DECLARE-DIABETES and -LONG Trials). Am J Cardiol 105: 168–173, 2010. doi: 10.1016/j.amjcard.2009.08.667. [DOI] [PubMed] [Google Scholar]
- 556. Katakami N, Kim YS, Kawamori R, Yamasaki Y. The phosphodiesterase inhibitor cilostazol induces regression of carotid atherosclerosis in subjects with type 2 diabetes mellitus: principal results of the Diabetic Atherosclerosis Prevention by Cilostazol (DAPC) study: a randomized trial. Circulation 121: 2584–2591, 2010. doi: 10.1161/CIRCULATIONAHA.109.892414. [DOI] [PubMed] [Google Scholar]
- 557. Bondarev AD, Attwood MM, Jonsson J, Chubarev VN, Tarasov VV, Liu W, Schiöth HB. Recent developments of phosphodiesterase inhibitors: clinical trials, emerging indications and novel molecules. Front Pharmacol 13: 1057083, 2022. doi: 10.3389/fphar.2022.1057083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558. Hahn VS, Knutsdottir H, Luo X, Bedi K, Margulies KB, Haldar SM, Stolina M, Yin J, Khakoo AY, Vaishnav J, Bader JS, Kass DA, Sharma K. Myocardial gene expression signatures in human heart failure with preserved ejection fraction. Circulation 143: 120–134, 2021. doi: 10.1161/CIRCULATIONAHA.120.050498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559. Sasaki H, Nagayama T, Blanton RM, Seo K, Zhang M, Zhu G, Lee DI, Bedja D, Hsu S, Tsukamoto O, Takashima S, Kitakaze M, Mendelsohn ME, Karas RH, Kass DA, Takimoto E. PDE5 inhibitor efficacy is estrogen dependent in female heart disease. J Clin Invest 124: 2464–2471, 2014. doi: 10.1172/JCI70731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560. Mehrara BJ, Radtke AJ, Randolph GJ, Wachter BT, Greenwel P, Rovira II, Galis ZS, Muratoglu SC. The emerging importance of lymphatics in health and disease: an NIH workshop report. J Clin Invest 133: e171582, 2023. doi: 10.1172/JCI171582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561. Liu X, Oliver G. The lymphatic vasculature in cardiac development and ischemic heart disease. Circ Res 132: 1246–1253, 2023. doi: 10.1161/CIRCRESAHA.122.321672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562. Green JS, Prok L, Bruckner AL. Expression of phosphodiesterase-5 in lymphatic malformation tissue. JAMA Dermatol 150: 455–456, 2014. doi: 10.1001/jamadermatol.2013.7002. [DOI] [PubMed] [Google Scholar]
- 563. Swetman GL, Berk DR, Vasanawala SS, Feinstein JA, Lane AT, Bruckner AL. Sildenafil for severe lymphatic malformations. N Engl J Med 366: 384–386, 2012. doi: 10.1056/NEJMc1112482. [DOI] [PubMed] [Google Scholar]