

Abstract
CRISPR-Cas9-mediated gene editing has vast applications in basic and clinical research and is a promising tool for several disorders. Our lab previously developed a non-integrating RNA virus, measles virus (MeV), as a single-cycle reprogramming vector by replacing the viral attachment protein with the reprogramming factors for induced pluripotent stem cell generation. Encouraged by the MeV reprogramming vector efficiency, in this study, we develop a single-cycle MeV vector to deliver the gRNA(s) and Cas9 nuclease to human cells for efficient gene editing. We show that the MeV vector achieved on-target gene editing of the reporter (mCherry) and endogenous genes (HBB and FANCD1) in human cells. Additionally, the MeV vector achieved precise knock-in via homology-directed repair using a single-stranded oligonucleotide donor. The MeV vector is a new and flexible platform for gene knock-out and knock-in modifications in human cells, capable of incorporating new technologies as they are developed.
Keywords: measles, viral vector, CRISPR, Cas9, gene editing, NHEJ, HDR, iPSC
Graphical abstract
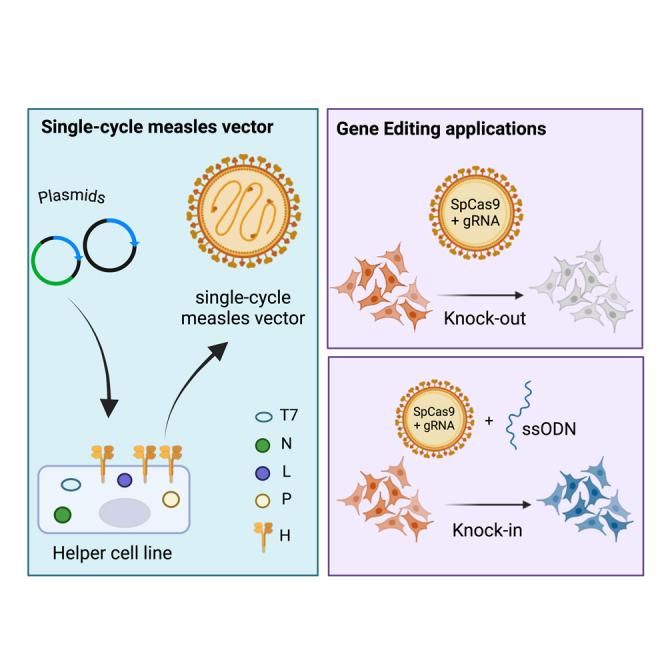
Devaux and colleagues developed a novel single-cycle measles vector allowing gene editing of human cells. They show that Measles can express the CRISPR-Cas9 and gRNA from one genome. Finally, they demonstrate that these vectors can efficiently perform KO and knock-in in human cells without excessive off-target effects.
Introduction
Initially discovered in bacteria and archaeal strains as an adaptive immune response against invading phages and foreign DNA, the CRISPR-Cas9 system was later engineered to target and cleave double-strand DNA (dsDNA).1,2,3,4,5,6 The engineered CRISPR-Cas9 system arrangement comprises of a synthetic guide RNA (gRNA) that leads the Cas9 nuclease to the target complementary strand to cleave the dsDNA.4 The cleavage depends on the presence of a 5′-NGG-3′ protospacer-associated motif (PAM) after the target site.4,7,8,9 The resulting double-strand break is repaired by one of the cell DNA repair pathways, including but not limited to non-homologous end-joining (NHEJ) or homology-directed repair (HDR) pathways.10 Since the NHEJ repair mechanism is error prone, it creates a mixture of insertions and deletions called Indels. Some can disrupt the open reading frame, resulting in a knock-out (KO), while others do not. Hence, the repair pathway determines if the targeted gene is disrupted or the mutation is fixed, restoring functionality.11 The HDR pathway requires an exogenous or endogenous DNA template and occurs less frequently than the NHEJ.11
Adeno-associated virus (AAV) and lentiviral vectors can efficiently infect specific cell types while offering flexibility to incorporate gene editing technologies and, hence, are preferred over non-viral delivery vectors for gene therapy.12,13 However, using DNA or integrating vectors introduces the risk of insertional oncogenesis in the host genome.14,15 Indeed, despite the safer design of these vectors, unwanted integration into the host genome remains a scrutinized risk, affecting the applicability of these vectors.14,16 Non-integrating viral vectors like Sendai virus (SeV) were developed to deliver CRISPR-Cas9 for efficient gene editing.17 However, these vectors are replication-transmission competent, raising concerns about cytotoxicity and ease of vector elimination.17 Taken together, there is a need for a safe, non-integrating vector system ideal for making gene KO and specific knock-in in human cells.
Measles virus (MeV) is a negative-strand, non-integrating virus with no DNA intermediate belonging to the Paramyxoviridae family.18 The virus has a modular gene arrangement with a polar transcriptional gradient and demonstrates broad tropism, making it an ideal vector candidate.19 Indeed, MeV has been extensively validated as a vaccine and oncolytic vector, with its safety as a vaccine platform assessed in more than 1 billion children worldwide.20,21 Our lab has developed a single-cycle MeV vector for reprogramming somatic cells into induced pluripotent stem cells (iPSC).22,23 The validation of MeV vectors in different fields and the ability of MeV vectors to deliver large transgenes safely and effectively have encouraged us to develop MeV as a platform to deliver CRISPR-Cas9 elements for targeted gene editing.
Here, we present the first application of a single-cycle MeV vector platform for targeted gene editing in human cells. We engineered an all-in-one MeV vector platform expressing Cas9 and associated gRNA to genetically modify the reporter gene (mCherry). We also produced two MeV vectors expressing gRNA targeting disease-relevant genes, -HBB (GenBank: NM_000518.5) and FANCD1 (GenBank: NM_000059.4), for which mutations can cause sickle cell disease and Fanconi anemia, respectively.24,25 Using a single-stranded oligonucleotide (ssODN) donor template transfection in conjunction with a MeV vector, we show precise knock-in of a stop codon in the mCherry gene in a reporter cell line. Additionally, we show specific knock-in into the endogenous genes HBB targeting the specific disease-causing site for sickle cell disease. In summary, we have used a single-cycle vaccine strain of MeV to engineer a safe platform ideal for gene editing and to perform KO and knock-in modifications in human cells. Our vector platform offers efficiency and flexibility for investigating new editing technologies and can contribute to the applicability of CRISPR-Cas9 genome editing.
Results
Production of MeV vectors to deliver Cas9 and gRNA targeting mCherry gene in a reporter cell line
To validate the functionality of each editing component in the single-cycle MeV system, we produced two individual MeV vectors, one encoding the SpCas9 (MVCas9) and the other encoding a gRNA targeting the mCherry gene (MVgRNAmch) (Figure 1A; Table S1). In both MVCas9 and MVgRNAmch, the H gene of the MeV is substituted with either the SpCas9 transgene (MVCas9) or GFP (MVgRNAmch) to produce a single-cycle MeV vector. In the case of MVCas9, the genome has two GFP cassettes; one in the additional transcription unit (ATU) after P-(ATU)P and the other in the ATU after the Cas9 transgene-(ATU)H. These two ATUs were introduced for future cloning of gRNA. We produced the MVgRNAmch vector by cloning the gRNAmch from the ATUP. The SpCas9 gene and gRNA sequences used were synthesized by GenScript (Piscataway, NJ). The Cas9 was engineered to contain a nuclear localization signal in the amino-terminal domain of the protein to allow proper translocation into the nucleus. While the transcripts (transgene) inserted in MeV are capped, polyadenylated, and ideal for gene expression, the MeV vector is not naturally equipped to express gRNA and facilitate its precise release. To enable proper gRNA release, it was flanked by two self-cleaving hammerhead ribozymes. The transcript is cleaved at precise locations, releasing a functional gRNA as described previously.17
Figure 1.

Production and characterization of MVCas9 and MVgRNAmch to cause mCherry mutagenesis in a reporter cell line
(A) Schematic of MeV virus and MeV gene editing vectors. (B) Western blot analysis of Cas9 expression in 293T cells after transduction with MVCas9 or LVCas9. Uninfected 293T (negative control), beta-actin (loading control), and MeV N (infection control). (C) Representative confocal images of Cas9 nuclear expression in transduced neonatal human fibroblasts (BJ) and Vero cells (Vero) with MVCas9 vector. Scale bars, 100 μm. (D) Titers of cell-associated and released virus produced upon infection of Vero-H2 cells with MVCas9 (gray) and MVgRNA (black) vectors compared to control vector MVVac2(GFP)H (white) at 24, 48, and 72 h after infection. Error bars represent SD from three individual experiments. A two-way ANOVA was used, followed by Tukey’s multiple comparison test (∗∗∗∗p ≤ 0.0001, all non-significant values are not labeled). (E) Indel (light gray) and KO (gray) efficiency analyzed by ICE-Synthego software created by transfection or/and infection of specified vectors. A two-way ANOVA was used followed by Tukey’s multiple comparison test (∗∗∗∗p ≤ 0.0001, ∗∗∗p ≤ 0.001, all non-significant values are not labeled). n = 3–6.
To confirm the expression of Cas9, western blot analysis of 293T cells transduced with MVCas9 or a lentiviral vector expressing (LVCas9) was performed (Figure 1B). Expression of Cas9 with appropriate molecular weight was observed in both MVCas9 and LVCas9. Confocal analysis confirmed the nuclear localization of Cas9 in both neonatal fibroblasts (BJ) and Vero cells through immunofluorescence staining (Figure 1C). To address the possible effect of Cas9 and gRNA transgenes on the growth and replication of the vector, a growth curve analysis was performed (Figure 1D). All vectors replicated to titers of about 107 tissue culture infectious dose (TCID50)/mL and were comparable to the growth pattern of the control vector, MVvac2(GFP)H at 24 and 48 h. Only a slight decrease was observed at 72 h (Figure 1D, black histogram). These results indicate that the expression of Cas9 and gRNA from the MeV vectors does not affect vector propagation, and high viral stock can be produced. Subsequently, to verify the functionality of both transgenes, Cas9 and gRNAmch expressed from individual vectors, we generated a HEK-293T cell line expressing mCherry using a lentivirus (LV) expressing mCherry (293Tmch). The mCherry expression was confirmed by microscopy and flow cytometry analyses (Figures 2B and S1). Ninety-eight percent of the 293T cell population expressed mCherry with polyclonal expression (Figure S1). This polyclonal cell population was used to validate mCherry gene editing and identify it phenotypically and genotypically. We next verified the functionality of the MVCas9 and MVgRNA. 293Tmch cells were transduced with MVCas9 in combination with a plasmid encoding gRNAmch or with MVgRNAmch (Figure 1E). Five days after transduction, cells were collected, and genomic DNA was extracted. PCR amplification of the mCherry gene was performed, and the amplicon was subjected to Sanger sequencing (Azenta, Burlington, MA, USA). We used ICE Synthego (https://ice.synthego.com) analysis, which utilizes deconvoluted Sanger sequencing trace files to identify indels.26 MVCas9 combined with pgRNAmch or MVgRNAmch achieved indel efficiencies of 3% and 14.5%, respectively (Figure 1E) (MVCas9+pgRNAmch and MVCas9+MVgRNAmch, light gray histogram). No editing (insertions/deletions [indels]) was observed in negative controls transduced with MVCas9 or MVgRNA alone, and 57% of editing was observed in the positive control, transfection with plasmid pgRNAmch-Cas9, encoding for the gRNAmch and spCas9. Additionally, the software can analyze allelic KO efficiencies based on out-of-frame edits due to error-prone cell repair pathways.26 The KO efficiencies with MVCas9 combined with pgRNAmch or MVgRNAmch were 3% and 7.3%, respectively (Figure 1E) (MVCas9+pgRNAmch and MVCas9+MVgRNAmch, dark gray histogram). The pgRNAmch-Cas9 vector, in contrast, achieved 40% KO efficiency (Figure 1E). Together, these data suggest that MeV vectors can deliver Cas9 and gRNA transgenes individually and that MVCas9 and MVgRNAmch are functional in causing on-target mutagenesis in the mCherry gene of the reporter cell line.
Figure 2.

Characterization of all-in-one MeV vectors expressing Cas9 and gRNAmch to cause mCherry KO in a reporter cell line
(A) Schematic of all-in-one MeV gene editing vectors expressing Cas9 and gRNA in a single genome. (B) Representative microscopy images of mCherry and GFP expression in 293Tmch cells after transduction with each MeV gene editing vectors. Scale bar, 100 mm. (C) ICE-Synthego analysis of NHEJ mediated indel and KO percentages on mCherry gene in 293Tmch cells when infected with MV(gRNAmch)PVac(Cas9) (light gray), MV(gRNAmch)PWT(Cas9) (gray), MVPWT(Cas9) (gRNAmch)H (dark gray), and pgRNAmchCas9 (stripe) at an MOI of 0.5 or 1; n = 6. Error bars represent mean ± SD. A two-way ANOVA was used, followed by Tukey’s multiple comparison test (∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001, not significant values are not labeled).
All-in-one MeV vectors for high-efficiency gene editing
To assess whether an all-in-one MeV vector can be used for gene editing, we constructed three independent single-cycle MeV containing both the gRNA and the Cas9 in a single genome. For the first vector, we used the MVvac full-length genome22,23 in which the gRNA (targeting mCherry) (Table S1) was expressed from (ATU)P and Cas9 instead of the MeV-H gene, making the single-cycle vector MV(gRNAmch)Pvac(Cas9). The GFP is placed in the (ATU)H after Cas9 to track the vector infection (Figure 2A, top genome). For the second vector, we used a vaccine backbone in which the P gene was substituted with the P gene of a wild-type strain of measles MV(gRNAmch)Pwt(Cas9), (Figure 2A, middle genome). It is known that the wild-type P (Pwt) gene of MeV induces less interferon (IFN) than the vaccine P (Pvac) gene since the Pwt can better control IFN response.27,28 For example, it was reported that arming the MeV vectors with the Pwt gene (IC-B strain) enhances their oncolytic activity.29 Finally, MeV is equipped with a transcriptional gradient meaning that the transcription efficiency decreases as the polymerase traverses from N to L.30 We took advantage of the transcriptional gradient to assess the effect of location bias in gene editing. For the last construct, we produced a third vector in which the gRNA was expressed from the (ATU)H after Cas9, MVPwt(Cas9) (gRNAmch)H. In this last construct, the GFP is placed in the (ATU)P to track the vector infection (Figure 2A, bottom genome). Nuclear localization and expression of Cas9 with appropriate molecular weight were validated using confocal analysis in both neonatal fibroblasts (BJ) and Vero cells through immunofluorescence staining (Figure S2A) and western blot analysis of 293T cells infected with the three vectors (Figure S2B). A growth curve analysis was performed to address the possible effect of Cas9 and gRNA transgenes on the growth and replication of the vectors (Figure S2C). All vectors replicated to titers of approximately 106–107 TCID50/mL and were comparable with the growth pattern of the control vector, MVVac2(GFP)H (Figure 1D), indicating that the expression of both Cas9 and gRNA from the MeV vector does not affect vector propagation.
We next verified the functionality of the three vectors and compared their efficacy. We transduced 293Tmch cells at a multiplicity of infection (MOI) of 0.5 and 1 for 5 days. To analyze and quantify gene editing, the transduced cells were collected, the genomic DNA extracted, and the mCherry gene amplified for genotypic analysis using ICE-Synthego software. The gene editing was also validated phenotypically using microscopy (Figure 2B). The ICE-Synthego analysis (Figure 2C) revealed that all three vectors performed similarly at low MOI of 0.5 with an indel efficiency of 32%, 28%, and 25% for MV(gRNAmch)Pvac(Cas9) (white), MV(gRNAmch)Pwt(Cas9) (light gray). and MVPwt(Cas9) (gRNAmch)H (dark gray), respectively. When cells were transduced with MOI of 1, both MV(gRNAmch)Pvac(Cas9) (white) and MV(gRNAmch)Pwt(Cas9) (light gray), performed with similar editing efficiencies of 42% and 38% indels, respectively. MVPwt(Cas9) (gRNAmch)H (dark gray) performed slightly better with an indel efficiency of 50% and close to the positive control pgRNAmchCas9, with an indel efficiency of 57%. The analysis of the ICE-Synthego KO score for the three vectors (Figure 2B, right side) showed no significant difference in the allelic KO score between the vectors and the two MOIs. A closer look at the edited sequence revealed that the decrease in KO efficiency compared to indel efficiencies was due to the increased number of in-frame deletions of −6, −12, or a multiple of 3 nucleotides, which do not contribute to the KO score (Figures S2D–S2F). In contrast, the KO score was significantly higher at about 40% for pgRNAmchCas9 due to a 4-fold greater contribution of out-of-frame +1 frameshift insertions in contrast with MVPwt(Cas9) (gRNAmch)H (Figures 2B and S2G compared with Figures S2D–S2F). It is unclear why the MeV vector produces more in-frame indels in 293T compared to a plasmid expressing the same editing components. Taken together, the data demonstrate that, at both MOIs, the IFN modulation, or amount of gRNA, had little to no effect on on-target gene editing and KO efficiencies. Since our data suggest that all vectors performed with similar or equal gene editing efficiencies, we will use MV(gRNAmch)Pvac(Cas9) and MVPwt(Cas9) (gRNAmch)H vectors interchangeably for the rest of the study.
MeV vector platform edits disease-relevant genes HBB and FANCD1
The mutations in the HBB and FANCD1 genes can cause severe disorders like sickle cell anemia, beta-thalassemia, and Fanconi anemia.31,32 We first engineered new MeV vectors MV(gRNAHBB)PVac(Cas9) and MV(gRNAFANCD1)PVac(Cas9) with gRNAs, in the ATU in P position, that target the coding regions of either HBB or FANCD1 genes. The gRNAs (Table S1) were specifically designed to target the Glu to Val mutation responsible for sickle cell anemia, and the 886GT deletion in the Exon 8 within the FANCD1 locus. The targeting efficiency of these vectors was first tested in 293T cells. Cells were transduced with MV(gRNAHBB)PVac(Cas9) or MV(gRNAFANCD1)PVac(Cas9) vectors at MOIs of 0.5 and 1. Five days after transduction, cells were collected, and PCR amplification of HBB and FANCD1 amplicon was performed on extracted genomic DNA, covering the editing site. The analysis, using ICE synthego software, revealed 27% and 11% indel formations at an MOI of 0.5, which increased to 45% and 27% at an MOI of 1 for MV(gRNAHBB)PVac(Cas9) and MV(gRNAFANCD1)PVac(Cas9) vectors, respectively (Figures 3A and 3C, white histograms). As previously seen with mCherry gRNA, the allelic KO score, which contributes to the loss of function of the gene, was nearly one-half of the indel efficiencies at 11% and 4% at MOI of 0.5 and 20% and 13% at MOI of 1, for MV(gRNAHBB)PVac(Cas9) and MV(gRNAFANCD1)PVac(Cas9) vectors, respectively (Figures 3A and 3C, black histograms).
Figure 3.

MeV vector mediated gene editing on endogenous HBB and FANCD1 genes
(A) ICE Synthego analysis of NHEJ-mediated indel (white) and KO (black) percentages on HBB gene of 293T cells when transduced with MV(gRNAHBB)PVac(Cas9) at an MOI of 0.5 or 1.0. Error bars represent mean ± SD of five independent experiments with each containing a biological replicate. A two-way ANOVA was used followed by Sidak post hoc multiple comparisons test (∗∗p ≤ 0.01, not significant values not labeled). (B) ICE Synthego analysis of NHEJ-mediated indel (white) and KO (black) efficiencies on HBB gene of four different primary AHFs (#1, #2, #3, and #4) transduced with MV(gRNAHBB)PVac(Cas9) at an MOI of 0.5 or 1.0. Error bars represent the mean ± SD of three independent experiments with each containing a biological replicate. A two-way ANOVA was used followed by Tukey’s multiple comparisons test (∗∗p ≤ 0.01, not significant values are not labeled). (C) ICE Synthego analysis of NHEJ-mediated indel (white) and KO (black) GE efficiencies on FANCD1 mutagenesis of 293T cells when infected with MV(gRNAFANCD1)PVac(Cas9) at MOI of 0.5 or 1.0. Error bars represent mean ± SD of three independent experiments with each containing a biological replicate. A two-way ANOVA was used followed by Sidak post hoc multiple comparisons test (∗∗p ≤ 0.01, not significant values not labeled). (D) ICE Synthego analysis of NHEJ-mediated indel (white) and KO (black) efficiencies on FANCD1 gene of four different primary AHFs (#1, #2, #3, and #4) when infected with MV(gRNAFANC)PVac(Cas9) at an MOI of 0.5 or 1.0. Error bars represent mean ± SD of three independent experiments with each containing a biological replicate. A two-way ANOVA was used followed by Tukey’s multiple comparisons test (not significant values are not labeled). (E) ICE Synthego analysis of NHEJ-mediated indel (white) and KO (black) efficiencies on HBB gene of iPSC when infected with MV(gRNAHBB)PVac(Cas9) at an MOI of 0.25, 0.5, or 1.0 and collected at days 2, 3 and 5. Error bars represent mean ± SD of three independent experiments with each containing a biological replicate. A two-way ANOVA was used followed by Tukey’s multiple comparisons test (∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001, not significant values are not labeled).
We next confirmed that both vectors can efficiently cause mutagenesis in the HBB and FANCD1 genes of primary adult human fibroblasts (AHF) (Figures 3B and 3D, respectively). We transduced fibroblasts isolated from four different healthy individuals with MV(gRNAHBB)PVac(Cas9) or MV(gRNAFANCD1)PVac(Cas9) vectors at MOIs of 0.5 or 1. While the ICE analysis revealed between 0%–18% and 4%–22% indel formation on the four different primary fibroblasts at MOIs of 0.5 and 1, respectively, for MV(gRNAHBB)PVac(Cas9) (Figure 3B), the level of editing with MV(gRNAFANCD1)PVac(Cas9) was much lower (Figure 3D). Only 0%–2% and 2%–4% indel formation was observed in fibroblasts transduced at MOIs of 0.5 or 1, respectively, indicating a low level of editing at the FANCD1 locus (Figure 3D). Unlike with the 293T cells, for HBB or FANCD1 editing, the KO efficiencies on fibroblasts were the same as that of indel formation. A closer look at the edited sequence revealed that the occurrence of out of frame (100% of −1 or +1 nt for HBB and FANCD1, respectively) versus in frame (22% of -9nts (HBB) or 12% of -3nts plus 3% of -9nts [FANCD1] in 293T) indel population are the main contributors to the difference in indel and KO efficiencies of fibroblasts and 293T (Figures S3A and S3B) (HBB and FANCD1, respectively). This phenomenon could result from more stringent cell repair pathways in primary fibroblasts versus transformed cell lines.33,34 We performed off-target analysis on AHFs transduced with MV(gRNAHBB)PVac(Cas9) or MV(gRNAFANCD1)PVac(Cas9). Using the CRISPOR tool, we identified the top three introns and top three exons for both HBB and FANCD1 gRNA (Table 1). These off-target sites were then amplified via PCR, the DNA purified, sequenced using Sanger sequencing, and analyzed using ICE Synthego software. The results, summarized in Table 1, showed that no off-target was detected after 5 days in MV(gRNAHBB)PVac(Cas9) transduced cells, and only minimal off-target was detected in MV(gRNAFANCD1)PVac(Cas9)-treated AHFs, between 0.167% ± 0.4% and 0.5% ± 1.2% (Table 1). Finally, we performed editing in iPSCs35 to demonstrate the capability of the MeV editing vector in relevant cells. We transduced iPSCs with MV(gRNAHBB)PVac(Cas9) vector at MOIs of 0.25, 0.5, or 1. The ICE analysis revealed an average between 0% and 2%, 0% and 11%, and 1% and 14% indel formation on days 2, 3, and 5, respectively (Figure 3E, white histogram), and a close level of KO compared to the indels formation (Figure 3E, black histogram, 0%–2%, 0%–9%, and 1%–10%, respectively), indicating that the MeV editing vector can efficiently knock out an endogenous gene in iPSC. As for the AHF editing, no off-target was detected after 5 days in the iPSCs (Table 1).
Table 1.
Summary of off-target editing in HBB and FANCD1
| Sample | Off-target genes (average indel %) (n = 4–6) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HBB |
FANCD1 |
|||||||||||
| Introns |
Exons |
Introns |
Exons |
|||||||||
| PCDH19 | DNER | COL6A3 | FSCN3 | C1QTNF6 | KMT2B | XPO4 | LINC00883 | GTDC1 | RP11-353N4.5 | MFAP1 | PAFAH1B2 | |
| AHF #1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| AHF #2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.500 ± 1.225 |
| AHF #3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.333 ± 0.816 | 0.167 ± 0.408 |
| AHF #4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| iPSC | 0 | 0 | 0 | 0 | 0 | 0 | ND | ND | ND | ND | ND | ND |
Values are mean ± SD.
ND, not done.
Altogether, these data demonstrate that MeV-mediated gene editing can efficiently target and knock out disease-relevant genes like HBB and FANCD1 in primary cells and iPSC with minimal or no off-target effect.
MeV gene-editing platform introduces precise knock-in
Since the MeV vector has a solely RNA-based life cycle, it cannot encode a donor template in its genome. Therefore, to achieve homologous recombination for editing, we tested a single-stranded oligodeoxynucleotide (ssODN) donor as a corrective template. To test the ssODNs DNA donor design for MeV-mediated targeting, we utilized the MeV gene editing vector targeting mCherry locus and designed a first set of ssODN donors of variable orientations, sense (80_1S) and antisense (80_1AS).36 These ssODN donors were used at two different concentrations (10 and 20 pM). The ssODN contained an in-frame stop codon (addition of two A’s) instead of a Phe, which should terminate mCherry translation as a result of successful knock-in. We transduced 293Tmch cells with MVPwt(Cas9) (gRNAmch)H vector at an MOI of 1.0, then transfected 8 h later with either 10 or 20 pM of donor nucleotide, as indicated. Five days after infection, ICE analysis revealed knock-in of the donor ssODN at an average of 4%–7% and 1%–2% efficiencies for 10 and 20 pM of 80S_1S and 80_1AS ssODN (Figure 4A, black and gray histograms 80_1S and 80_1AS), indicating that the 80_1S ssODN was the most efficient for knock-in. Since one could argue that the insertion of double A’s in the mCherry sequence to create an in-frame stop codon, could be a result of random insertions from NHEJ and not HDR, we designed a more complex ssODN aimed at knocking in a unique sequence in the original mCherry gene. We added multiple stop codons (ochre and amber) to prevent translation of the mCherry gene and generated ssODN of two different sizes, 60 and 80 nucleotides (60_1S, 60_1AS, 80_2S, and 80_2AS). In the same ssODN, we also introduced PAM-blocking mutations (GGG to AGG). With these modifications, we did not see an improvement in the HDR efficiencies, and the knock-in resulted in a decrease at both concentrations of 10 and 20 pM (Figure 4A, black and gray histograms 80_2S and 80_2 AS). Thus, in the context of mCherry knock-in, the ssODN 80_1S was the most effective.
Figure 4.

MeV editing vectors can promote efficient HDR mediated knock-in mCherry and HBB genes
(A and B) ICE-Synthego GE analysis of mCherry (A) or HBB (B) gene in 293Tmch cells demonstrating the knock-in (KI) (A) in presence of 10 pM or 20 pM of the indicated ssODN. AS, antisense to target region; S, sense. Error bars represent mean ± SD of three independent experiments with each containing a biological replicate. A two-way ANOVA was used followed by Sidak post hoc multiple comparisons test (∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001, not significant values aerw not labeled). (C and D) ICE-Synthego GE analysis of mCherry (A) or HBB (B) gene in 293Tmch cells demonstrating the KO contribution in the HDR experiment for each sample presented in (A) and (B). Error bars represent the mean ± SD of three independent experiments, with each containing a biological replicate. A two-way ANOVA was used followed by Sidak post hoc multiple comparisons test (∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001, not significant values not labeled). (E and F) Optimization of HDR-mediated knock-in HBB gene of 293T using timing and amount of HBB 80_3S ssODN. Cells were transfected with 10 pM (white triangle), 20 pM (white circle), 30 pM (black triangle), 50 (black square), 75 pM (black circle) and 100 pM (white square). Eight hours later, the cells were transduced with MV(gRNAHBB)PVac(Cas9). Cells were collected at 24, 36, 48, 72, 96 h, or 5 days after transfection, n = 4. The percentage of KI was determined after DNA amplification, Sanger sequencing, and ICE-Synthego analysis.
We next designed ssODN for the knock-in into the HBB gene. For the oligo donor, in addition to the PAM blocking silent mutation (CCG to CTG), we also added a silent mutation in the seventh codon of the β-globin gene (GAG to GAA) to test if we could perform site specific correction in the codon that causes sickle cell anemia. We designed ssODN of two different lengths, 60 nts or 80 nts long, to determine if the oligo size could affect knock-in efficiency (60_2S, 60_2AS, 80_3S and 80_3AS). For 80S_3 and 80AS_3 ssODN, at 5 days after transduction of 293T cells, ICE analysis revealed knock-in of an average of 6%–8% and 1%–4% efficiencies, respectively, for 10 and 20 pM (Figure 4B, black and gray histograms, 80_3S and 80_3AS). However, the use of shorter ssODN significantly reduced the knock-in efficiency to an average of approximately 2% (Figure 4B, black and gray histograms, 60Sand 60AS), indicating that, as seen for mCherry, a short ssODN is detrimental to efficient knock-in. However, in contrast to mCherry, for HBB gene, the mutation of the PAM sequence and the 80-nt length resulted in a higher level of knock-in, indicating that the efficiency might be gene and sequence-specific. For all knock-in experiments targeting mCherry or HBB, the level of KO was determined for all ssODN using ICE-Synthego, and there was no significant difference in the amount of KO editing in mCherry or HBB samples (Figures 4C and 4D, mCherry and HBB, respectively). To improve the knock-in efficiency, we also performed a kinetic experiment using the 80_3S ssODN to determine the optimal windows of knock-in efficiency; we additionally tested multiple amounts of ssODN to determine if we could improve knock-in efficiency by increasing the amount of ssODN present in the cell. After transduction, cells were transfected with 10, 20, 30, 50, 75, or 100 pM of ssODN, and samples were collected at 24, 36, 48, 72, 96, and 120 h after transfection. We observed the maximum knock-in efficiency, with an average of 12% at 72 h after transfection with a 20 pM of 80_3S ssODN (Figures 4E and 4F). Overall, these results suggested that the MeV vector can be used concomitantly with an ssODN donor template for HDR-mediated precise knock-in within the target gene with as low as 10 pM of ssODN.
Discussion
This study shows that a non-transmissible MeV vector can deliver CRISPR-Cas9 components for efficient gene editing in human cells. We validate the MeV vector for targeted gene editing by introducing indels in mCherry reporter and endogenous disease-relevant genes, HBB and FANCD1. We also show that the MeV vector in presence of an ssODN donor template can introduce specific knock-ins in the host genome.
In the past few years, there has been a strong effort to develop non-viral vector delivery systems for editing technologies; for example, the use of lipid nanoparticles allows the packaging of editors as transiently expressed messenger RNAs, but their efficiency for in vivo targeting remains a challenge.37,38 More recently, engineered virus-like particles (eVLPs), which consist of the assemblies of viral proteins that can infect cells but lack viral genetic material, have emerged as potentially promising vehicles for delivering gene editing agents as ribonucleoproteins (RNPs). eVLPs are formed spontaneously from the assembly and budding of retroviral proteins and encapsulate cargo molecules from the producer cell (RNP). They are based on murine leukemia virus and retargeted using vesicular stomatitis virus G protein. They do not contain any genetic material, eliminating the risk of prolonged editor expression in the cell, increasing the off-target editing frequencies, and the risk for oncogenic DNA integration.39 They have been used to deliver multiple types of editing cargo from CRISPR-Cas9 to Base Editor and are developed for in vivo editing.40,41,42 While these systems are becoming more efficient, optimizing the editing efficiency of the eVLP in multiple tissues/organs and large-scale production of these eVLPs remains to be established to expand their therapeutic potential. Despite these multiple developments in non-viral vector delivery for CRISPR-Cas9 system, AAV and LV vectors remain favorites for in vivo delivery.43 Specifically, AAV has successfully delivered CRISPR-Cas9 in animal and ex vivo models.44 While initially considered safe and non-immunogenic, recent emerging reports suggest that AAV is not entirely benign. For example, when used to deliver CRISPR-Cas9 to mouse skeletal muscle, AAV resulted in infiltration of CD4+ helper and CD8+ cytotoxic T cells, creating a limitation for the therapy.45 The neutralizing antibodies already prevalent in humans also pose a problem for multiple AAV dosing, which can be addressed by using different serotypes.46,47 Additionally, the packaging size of the AAV vector at 4.7 kb limits gene editing using conventional Cas9 nuclease, which is itself 4.2 kb.48 The other commonly used integrating RNA vector LV has a packaging capacity of 10 kb and demonstrates long-lasting expression of transgenes; however, this attractive quality in a vector is undesired in CRISPR-Cas9 expression.39 When permanently expressed, Cas9 can facilitate unwanted off-target cleavage, affecting their use in clinical applications.49 To circumvent this, integrase-deficient LV was used to deliver Cas9, but the expression level of these vectors is compromised due to a lack of functional integrase.39,50,51 Recently, the wild-type strain of SeV, rSeV-Cas9, was engineered to deliver CRISPR-Cas9.52 However, the rSeV-Cas9 vectors are replication and transmission efficient, posing a problem for vector elimination and cytotoxicity. Additionally, the authors demonstrate rSeV-Cas9 for mutagenesis, but do not demonstrate HDR-mediated knock-in.52
In comparison with the existing vectors, the main advantage of MeV is the safety and flexibility it offers. Here, we show that a single-cycle MeV vector platform can perform CRISPR-Cas9-mediated gene-editing for mutagenesis and HDR-mediated knock-ins. Our study indicates that the non-integrating MeV vector system could be used for future clinical applicability in correcting disease-relevant genes.
Incorporation of gRNA in viral vectors is challenging since it is a small RNA that requires precise ends for effective targeting. Different vectors have implemented several strategies to release gRNA with proper ends. In DNA, viral vectors such as Adeno or AAV, pol III promoters like U3 or U6 drive the expression of the gRNA.53,54,55,56,57 However, U3 and U6 promoters require either an A or G for transcriptional initiation, restricting the gene-editing site to G/AN19NGG.5,58 To overcome these limitations, other strategies like self-cleaving ribozymes, endogenous RNases, or Csy4 have been incorporated into several non-viral or viral systems.59,60,61 For example, the rSeV-Cas9 vector used self-cleaving hammerhead ribozymes to release gRNA and achieved 75%–98% on-target mutagenesis at various endogenous and inducible reporter gene.52 We utilized similar hammerhead ribozymes in the MeV vectors to release a single gRNA and achieved 12%–60% mutagenesis in disease-relevant endogenous (HBB and FANCD1) and mCherry genes in a polyclonal population of reporter cells. In contrast to the wild-type rSeV-Cas9 vector used at high MOIs of 25–50, the MeV vector is based on a vaccine strain used at very low MOIs of 0.5 or 1.0, which could have contributed to the higher efficiency of SeV vectors.52 Additionally, the rSeV-Cas9 vectors have all the viral genes intact, making it a replication and transmission-competent vector, raising the risk of increased cytotoxicity, while the MeV vector is a single-cycle platform with the MeV-H gene replaced by Cas9 transgene.
The MeV platform has a major advantage over other delivery systems due to its transcription gradient. We showed that the expression of the Cas9 from the MeV vector, cloned instead of the H gene, is much greater when compared to an LV expressing Cas9. We had previously observed similar differences in expression between the MeV and LV vectors while developing the MeV reprogramming vectors.22 While there is a possibility that higher expression of Cas9 could increase toxicity in primary cells, we did not record more toxicity with the Cas9 MeV than we had observed with the reprogramming vectors in AHFs.22,35 However, to perform editing in iPSC we required the use of a pan-caspase inhibitor, indicating that either the MeV vector or Cas9 might be causing apoptosis induction in iPSC upon transduction. The potential to modulate the expression of Cas9, by moving the gene downstream in the genome, provides a tool to alleviate any observed toxicity while retaining its editing ability.
We showed that the gene editing from MeV and the plasmid transfection led to different indel formations. As described in the results section, in 293T cells, most of the indels for MeV editing are a multiple of 3 nts, which does not lead to a KO. In contrast, editing with the plasmid led to a majority of indel with an out-of-frame shift Figure S2. In AHF, MeV only led to the formation of −1 nt deletion indels, leading to an out-of-frame shift and KO. Larger indels may exist, but they are only present at lower frequencies in AHF and are not detectable using ICE Synthego analysis. Interestingly, we see an increase in indel species in iPSC. However, the −1 nts remains the major form of indels present in MeV-generated editing. Currently, we do not know why MeV has such a varied pattern of editing and why the range of indel formation seems to be so limited.
Apart from offering safety, another novel feature of our MeV vector platform is flexibility. It can make precise knock-ins, as demonstrated by the knock-in of a stop codon in the mCherry gene or the introduction of a silent mutation in the HBB gene in the Glu amino acid, which is mutated to Val in patients with Sickle cell anemia. In this study, we demonstrate a maximum of 12% knock-in the HBB gene at day 3 with 20 pM of 80_3S ssODN can be achieved with the MeV vector platform. While we did not look for additional optimization, there are multiple ways that can be used to increase efficiency, including inhibition of the NHEJ pathway, regulation of HDR-related factors, cell cycle synchronization, optimal design of the donor DNA template leaving space for additional improvement of the MeV platform in future applications to correct genetic disorders.62,63,64,65
In conclusion, our proof-of-principle study demonstrates MeV as an attractive vector for targeted gene editing. Our single-cycle all-in-one MeV vector is a safe, non-integrating RNA vector used at a very low MOI while achieving comparable efficiencies to other existing vectors in the field. Additionally, the ability of our MeV vector to efficiently perform both KO and knock-in opens the door to incorporating other new CRISPR-Cas technologies and approaches as they are developed, making MeV a highly flexible vector for future clinical applications.
Material and methods
The human cells in this study were obtained from the ATCC (Manassas, VA, USA), while the primary fibroblast cells were obtained from healthy donors and approved through Mayo Clinic Biotrust and approved by Mayo Clinic Institutional Review Board. The Mayo Clinic Institutional Biosafety Committee approved the use of viral vectors used in this study.
Cell culture
Vero (ATCC #CCL81), Vero-H223 and HEK293T (ATCC CRL-3216) were cultured in DMEM with 10% FCS (Life Technologies, Carlsbad, CA, USA) and 1% penicillin/streptomycin (PSP, Corning Mediatech, Manassas, VA, USA) (DMEM 1 media). Adult and neonatal fibroblasts were obtained from healthy donors (Mayo Clinic Biotrust and ATCC [#CRL 2522], respectively). The fibroblasts were maintained in DMEM media with 10% ES-FCS, supplemented with 0.1 mM non-essential amino acid and 1% PSP (DMEM-2 media). Rescue 293-3-46 H2 cells23 were propagated in DMEM 1 media, supplemented with 1.2 mg/mL of G418 (Cardinal Healthcare, Dublin, OH, USA). To generate 293Tmch cells, HEK-293T at low passage cells were transduced with LV expressing mCherry gene and selected using 3 μL puromycin (10 mg/mL) to yield stable polyclonal cells. The 293Tmch cells were maintained in DMEM-1 supplemented with 3 μL puromycin. All cells were cultured in the presence of 5% CO2 and at a temperature of 37°C. iPSCs were maintained in mTeSR1 media (STEMCELL Technologies, Vancouver, Canada).
Full-length MeV cDNA production
MVCas9 was produced by inserting the sequence encoding Cas9 instead of MeV-H gene. The Cas9 fragment was first cloned into EcoRV and SmaI digested the intermediate pCG-H(GFP) vector, making the pCG-ΔH(Cas9) (GFP)H. Then a PacI–SpeI fragment containing the Cas9 and GFP was subcloned back into the MV full-length genome containing a GFP in (ATU)P. The resulting plasmid was called p(+)MVvac2P(GFP)ΔH(Cas9) (GFP)H (MVCas9) (Figure 1A). For MVgRNA construct, the gRNA with hammerhead ribozyme cassette, synthesized at Genescript (Piscataway, NJ) was inserted into (ATU)P of p(+)MVvac2(GFP)PΔH(GFP) by using BsiWI and BssHII restriction enzymes, replacing the existing GFP in (ATU)P. The resulting called p(+)MVvac2P(gRNAmch)ΔH(GFP) (MVgRNA) (Figure 1A). To produce MV(gRNAmch)Pvac(Cas9) we used PacI and SpeI restriction enzymes to transfer Cas9 and GFP from p(+)MVvac2(GFP)PΔH(Cas9) (GFP)H into p(+)MVvac2P(gRNAmch)ΔH(GFP)H. The resulting plasmid was called p(+)MVvac2P(gRNAmch)ΔH(Cas9) (GFP)H (MV(gRNAmch)Pvac(Cas9)) (Figure 2A). The full-length vectors with gRNA specific for HBB and FANCD1 were made in a similar way. MVPwt(Cas9) (gRNAmch)H was made by cloning the mCherry gRNA in a p(+)MVvac2(GFP)PwtΔH(GFP) vector and then introducing the Cas9 and GFP cassettes using PacI-SpeI restriction sites. The plasmid was called p(+)MVvac2Pwt323(gRNAmch)ΔH(Cas9) (GFP)H, (MVPwt(gRNAmch)P(Cas9)) (Figure 2A).
To produce MVPwt(Cas9) (gRNAmch)H, we first cloned the gRNA cassette in to the ATU in H position in the intermediate vector pCG-H(Cas9) (GFP)H to make pCG-H(Cas9) (gRNAmch)H using BsiWI and BssHII. Then a PacI and SpeI fragment containing the Cas9 and gRNA was cloned into the p(+)MVvac2(GFP)PwtΔH(GFP) vector to produce the p(+)MVvac2Pwt323(GFP)ΔH(Cas9) (gRNAmch)H, (MVPwt(Cas9) (gRNAmch)H) (Figure 2A). The Cas9 and gRNA genes were then transferred to full-length MeV plasmid with Pwt containing a GFP in (ATU)P to make MVPwt(Cas9) (gRNAmch)H. The cloning for MeV constructs was performed abiding the rule of six and all constructs were validated by restriction digestions and sequencing.
pgRNAmch-Cas9 and pgRNAmch
For plasmids containing gRNA (pgRNAmch-Cas9 and pgRNAmch), the sgRNA were designed as an oligo and obtained from IDT. The oligonucleotides were annealed in a thermal cycler (37°C for 30 min, 95°C for 5 min, and decrease to 25 °C at a rate of 5°C per minute). Following this, the sgRNA oligos were cloned into the pSpCas9(BB)-2A-Puro (PX459) (Addgene plasmid # 48139; http://n2t.net/addgene:48139; RRID: Addgene_48139) according to the Zhang Lab General Cloning Protocol.66 For pgRNAmch, the pSpCas9(BB)-2A-Puro (PX459) was modified to remove the Cas9 cassette using the KpnI and NotI sites. The transformation of the plasmid was performed using One Shot TOP10 E. coli chemically competent cells (Thermo Fisher Scientific, Waltham, MA, USA) and plasmid DNA was harvested from transformed colonies using a Qiagen plasmid midi kit (Qiagen, Hilden, Germany). HEK-293 cells (2.5 × 105) on a 12-well Matrigel-coated plate were transfected with up to 1 μg plasmid using Lipofectamine 2000 according to the manufacturer’s protocol. The cells are washed with PBS the next day, followed by media change on days 1 and 3 after transduction. The cells were collected on day 5 for the downstream analysis of gene editing.
gRNA design
The gRNA target sequences were chosen based on high predicted specificity using ATUM tool (https://www.atum.bio/eCommerce/cas9/input). The gRNA, the target sequence and the PAM sequence are listed in Table S1.
Vector production and titration
The MeV vectors were rescued as mentioned in Driscoll et al.23 using Rescue 293-3-46 H2 and Vero H2 cells. In brief, Rescue H2 cells were co-transfected with the MeV antigenome plasmid and polymerase plasmid. Three days later, the transfected cells were overlayed on Vero-H2 cells. The GFP expression was used to monitor the rescue and subsequent propagation on Vero-H2 cells. Stocks were produced using standard MeV production techniques to passage 3. All experiments were performed using the third passage of the viral stock, and stocks were titrated on Vero H2 cells. Viral titration was performed using a TCID50 assay and measured using GFP signal 4 days after infection using Spearman-Karber method.67
Growth curves
VeroH2 cells (4 × 105) were infected with MeV vector at an MOI of 0.05 in OptiMEM. Two hours later, the viral inoculum was removed and washed, and 1 mL DMEM 1 media was added to the cells. The spread of GFP was monitored, and subsequently, the infected cells were cultured and collected at 24, 48, and 72 h after infection, along with the media. The samples were later titered using TCID50 titration assay.
Immunofluorescence
BJ or Vero cells were seeded at a density of 2.5x105 on 8-well slides (Lab-Tek, Sigma-Aldrich, St. Louis, MO, USA). The cells were subsequently transduced with MeV vectors at an MOI of 0.5 (BJ) or 0.3 (Vero) for 2 h. The cells were washed and fixed 36–48 h after infection using 2% PFA (Affymetrix, Inc., Cleveland, OH, USA). The cells were subsequently permeabilized (0.1% Triton X- in 2% PFA) and blocked (5% FCS-PBS) O/N at 4°C. The cells were stained with anti-Cas9 antibody (Abcam-ab202580, Cambridge, UK) O/N at 4°C, followed by 594-anti-Rabbit secondary antibody (Life Technologies, A21207) for 1 h at room temperature. Once stained, the cells were washed and mounted using DAPI PROLONG (Life Technologies). The images were taken on LSM 780 and were analyzed on Zeiss black software (Zeiss, Jena, Germany).
Western blot
We transduced 293T cells (3 × 105) with MeV vectors at an MOI of 0.5. The cells were collected 36 h after infection and processed as described by Driscoll et al.23 Protein samples were separated on SDS page gels and transferred on PVDF membranes. Following the transfer, the membranes were blocked and incubated with mouse antibody to SpCas9 (Abcam [7A9-3A3], ab191468, 1/1,000 dilution), and MeV-N (Cl25).23 After subsequent washing with TBS-Tween 0.1%, the membranes were incubated with peroxidase-conjugated secondary anti-Rabbit peroxidase (Calbiochem, 401215, 1/10000), anti-mouse peroxidase (Calbiochem, 401215, ½,500) and a mouse monoclonal antibody anti-β-actin-peroxidase (Sigma-Aldrich A3854, 1/10,000) for 2 h at room temperature. The membranes were washed and subjected to an ECL2 substrate.
Flow cytometry
We transduced 293T and 293Tmch cells or not with MeV vectors (MOI of 0.5). Five days after transduction, cells were collected, washed, and fixed in PBS-2% PFA. Flow cytometry data were acquired on a Becton Dickinson FACS Calibur (Becton Dickinson, Franklin Lakes, NJ, USA) and analyzed by using FlowJo software.
Gene editing
For KO experiments, HEK-293T/293Tmch (2.5 × 105 cells/mL) or BJ (7 × 104 cells/mL) or AHF (5 × 104 cells/mL) cells seeded on Matrigel (Corning, Corning, NY, USA) plate were transduced and subsequently spinoculated with MeV vectors at MOIs of 0.5 or 1 in OptiMEM. The cells were washed with PBS the next day, followed by media change on days 1 and 3 after transduction. The cells were collected on days 2, 3, and 5 for the gene editing downstream analysis. For iPSCs35 KO experiment, iPSCs (2 × 105 cell/mL) were plated onto a 12-well Matrigel-coated plate as single-cell suspension in 1 mL mTeSR1 medium containing 20 μM ROCK inhibitor. After 24 h, the cells were transduced with MeV(gRNAHBB)Pvac(Cas9) at MOIs of 0.25, 0.5, or 1 and spinoculated for 1 h in the presence of 50 μM Z-VAD-FMK. After 16 h, the virus was removed from the cells and washed twice with PBS. The media was changed daily with 1 mL mTeSR1 media containing 50 μM Z-VAD-FMK. The cells were collected on days 2, 3, and 5 for downstream analysis of gene editing.
For HDR-mediated nucleotides knock-in, the 293T or 293Tmch cells were seeded at 2 × 105 cells per well, on a 12-well Matrigel-coated plate. The next day, cells were transduced with MeV gene editing vector at an MOI of 1.0 in OptiMEM and spinoculated for 1 h at 1,100 rpm. The plate was then placed in a 37°C incubator for 8 h. After 8 h, the cells were washed with PBS followed by media change. The cells were next transfected with the indicated concentration (pM) of the specified ssODN oligos Table S1 (Table S3) using Lipofectamine 2000 according to the manufacturer’s protocol. The plate was spinoculated at 1,100 rpm for 30 min and then placed in a 37°C incubator overnight. The cells were washed with PBS the next day, followed by media change on days 1 and 3 after transfection. Cells were collected at the indicated time point.
Gene editing analysis
Following transduction, the cells were collected at the time indicated post-transduction for genomic DNA extraction using DNeasy Blood & Tissue Kits (Qiagen). The target gene was PCR amplified using Promega PCR Master Mix (Thermo Fisher Scientific, Hampton, NH) and pair of primers specified in Table S2, on-target primers (supplemental information). The PCR products were then purified using Qiagen QIAquick PCR Purification Kit (Qiagen). The amplicons were sent for Sanger sequencing through GENEWIZ (South Plainfield, NJ, USA) following the company’s sample submission guidelines using the primers specified in Table S2. We then analyzed the formation of indel or knock-in using the ICE Synthego website (https://ice.synthego.com).26
Off-target analysis
The CRISPOR software tool68 was used to determine off-target genes based on the input the genomic sequence of either HBB or FAND1 genes and gRNA used. The first three introns and exons listed with the highest likelihood of off-target mutations were chosen for each gene of interest. Primers were designed to selectively amplify amplicon of the region targeted by the gRNA to analyze potential off-target effects (Supplemental information, Table S2, off-target primers) The off-target gene was PCR amplified using Promega PCR Master Mix (Thermo Fisher Scientific), purified, sequenced, and analyzed using ICE Synthego as described above.
Statistical analysis
Initial data were processed in Microsoft Excel. The data were graphed using GraphPad Prism 9 software, and further statistical analysis was performed using the same. An unpaired t test was used to analyze single comparisons. One or two-way ANNOVA was used to make multiple comparisons, followed by Sidak and Tukey’s multiple comparison test. The data for all experiments are presented as the average of at least three independent experiments with at least two replicates for each experiment. Data are graphed as mean ± SD. The statistical significance is specified as ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001 and ∗∗∗∗p ≤ 0.0001. Comparisons that are not statistically significant are not presented.
Data and code availability
The article or Supplemental material include all data directly relevant to the study. Further inquiries can be directed to the corresponding author.
Acknowledgments
We thank Dr. Mohni for reading the manuscript. Furthermore, we thank Annaliese Calzadilla and Alex Locsin for their technical assistance. This work was supported by Regenerative Medicine Minnesota (RMM 092319 DS 005 to P.D.), Mayo Graduate School, and the National Institutes of Health (R56HL147852 and R01HL147852-A1 to PD). R.R. was supported by the Mayo Clinic Regenerative Sciences Training Program and the Michael S. and Mary Sue Shannon Foundation. The graphical abstract was created using Biorender.com.
Author contributions
P.D. oversaw the project. P.D. and R.R. designed and conceptualization the project; P.D., R.R., and B.S. designed and produced vector construct; R.R., B.S., S.M., A.R., R.H., and M.I. acquired the data; R.R. wrote the original first draft; P.D., R.R., B.S., A.R., and M.S. reviewed, edited, and rewrote the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2024.101290.
Supplemental information
References
- 1.Ishino Y., Shinagawa H., Makino K., Amemura M., Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987;169:5429–5433. doi: 10.1128/jb.169.12.5429-5433.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mojica F.J., Díez-Villaseñor C., Soria E., Juez G. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol. Microbiol. 2000;36:244–246. doi: 10.1046/j.1365-2958.2000.01838.x. [DOI] [PubMed] [Google Scholar]
- 3.Jansen R., Embden J.D.A.v., Gaastra W., Schouls L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002;43:1565–1575. doi: 10.1046/j.1365-2958.2002.02839.x. [DOI] [PubMed] [Google Scholar]
- 4.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gasiunas G., Barrangou R., Horvath P., Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA. 2012;109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horvath P., Romero D.A., Coûté-Monvoisin A.C., Richards M., Deveau H., Moineau S., Boyaval P., Fremaux C., Barrangou R. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J. Bacteriol. 2008;190:1401–1412. doi: 10.1128/JB.01415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deveau H., Barrangou R., Garneau J.E., Labonté J., Fremaux C., Boyaval P., Romero D.A., Horvath P., Moineau S. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 2008;190:1390–1400. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xue C., Greene E.C. DNA Repair Pathway Choices in CRISPR-Cas9-Mediated Genome Editing. Trends Genet. 2021;37:639–656. doi: 10.1016/j.tig.2021.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nambiar T.S., Baudrier L., Billon P., Ciccia A. CRISPR-based genome editing through the lens of DNA repair. Mol. Cell. 2022;82:348–388. doi: 10.1016/j.molcel.2021.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shahryari A., Jazi M.S., Mohammadi S., Nikoo H.R., Nazari Z., Hosseini E.S., Burtscher I., Mowla S.J., Lickert H. Development and Clinical Translation of Approved Gene Therapy Products for Genetic Disorders. Front. Genet. 2019;10 doi: 10.3389/fgene.2019.00868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zu H., Gao D. Non-viral Vectors in Gene Therapy: Recent Development, Challenges, and Prospects. AAPS J. 2021;23 doi: 10.1208/s12248-021-00608-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen X., Gonçalves M.A.F.V. Engineered Viruses as Genome Editing Devices. Mol. Ther. 2016;24:447–457. doi: 10.1038/mt.2015.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCarty D.M., Young S.M., Samulski R.J. Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annu. Rev. Genet. 2004;38:819–845. doi: 10.1146/annurev.genet.37.110801.143717. [DOI] [PubMed] [Google Scholar]
- 16.McCarty D.M., Young S.M., Samulski R.J. Integration of Adeno-Associated Virus (AAV) and Recombinant AAV Vectors. Annu. Rev. Genet. 2004;38:819–845. doi: 10.1146/annurev.genet.37.110801.143717. [DOI] [PubMed] [Google Scholar]
- 17.Park A., Hong P., Won S.T., Thibault P.A., Vigant F., Oguntuyo K.Y., Taft J.D., Lee B. Sendai virus, an RNA virus with no risk of genomic integration, delivers CRISPR/Cas9 for efficient gene editing. Mol. Ther. Methods Clin. Dev. 2016;3:16057. doi: 10.1038/mtm.2016.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rota P.A., Moss W.J., Takeda M., de Swart R.L., Thompson K.M., Goodson J.L. Measles. Nat. Rev. Dis. Prim. 2016;2 doi: 10.1038/nrdp.2016.49. [DOI] [PubMed] [Google Scholar]
- 19.Hiramoto T., Tahara M., Liao J., Soda Y., Miura Y., Kurita R., Hamana H., Inoue K., Kohara H., Miyamoto S., et al. Non-transmissible MV Vector with Segmented RNA Genome Establishes Different Types of iPSCs from Hematopoietic Cells. Mol. Ther. 2020;28:129–141. doi: 10.1016/j.ymthe.2019.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muhlebach M.D. Measles virus in cancer therapy. Curr. Opin. Virol. 2020;41:85–97. doi: 10.1016/j.coviro.2020.07.016. [DOI] [PubMed] [Google Scholar]
- 21.Moszynski P. Measles campaign's "historic victory" for global public health. Br. Med. J. 2007;334:177. doi: 10.1136/bmj.39101.608762.DB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Q., Vossen A., Ikeda Y., Devaux P. Measles vector as a multigene delivery platform facilitating iPSC reprogramming. Gene Ther. 2019;26:151–164. doi: 10.1038/s41434-019-0058-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Driscoll C.B., Tonne J.M., El Khatib M., Cattaneo R., Ikeda Y., Devaux P. Nuclear reprogramming with a non-integrating human RNA virus. Stem Cell Res. Ther. 2015;6:48. doi: 10.1186/s13287-015-0035-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zittersteijn H.A., Harteveld C.L., Klaver-Flores S., Lankester A.C., Hoeben R.C., Staal F.J.T., Gonçalves M.A.F.V. A Small Key for a Heavy Door: Genetic Therapies for the Treatment of Hemoglobinopathies. Front. Genome. 2020;2 doi: 10.3389/fgeed.2020.617780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moreno O.M., Paredes A.C., Suarez-Obando F., Rojas A. An update on Fanconi anemia: Clinical, cytogenetic and molecular approaches (Review) Biomed. Rep. 2021;15:74. doi: 10.3892/br.2021.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Conant D., Hsiau T., Rossi N., Oki J., Maures T., Waite K., Yang J., Joshi S., Kelso R., Holden K., et al. Inference of CRISPR Edits from Sanger Trace Data. CRISPR J. 2022;5:123–130. doi: 10.1089/crispr.2021.0113. [DOI] [PubMed] [Google Scholar]
- 27.Sparrer K.M.J., Pfaller C.K., Conzelmann K.K. Measles virus C protein interferes with interferon beta transcription in the nucleus. J. Virol. 2012;86:796–805. doi: 10.1128/JVI.05899-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takaki H., Watanabe Y., Shingai M., Oshiumi H., Matsumoto M., Seya T. Strain-to-strain difference of V protein of measles virus affects MDA5-mediated IFN-beta-inducing potential. Mol. Immunol. 2011;48:497–504. doi: 10.1016/j.molimm.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 29.Haralambieva I., Iankov I., Hasegawa K., Harvey M., Russell S.J., Peng K.W. Engineering oncolytic measles virus to circumvent the intracellular innate immune response. Mol. Ther. 2007;15:588–597. doi: 10.1038/sj.mt.6300076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cattaneo R., Rebmann G., Schmid A., Baczko K., ter Meulen V., Billeter M.A. Altered transcription of a defective measles virus genome derived from a diseased human brain. EMBO J. 1987;6:681–688. doi: 10.1002/j.1460-2075.1987.tb04808.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ingram V.M. Gene mutations in human haemoglobin: the chemical difference between normal and sickle cell haemoglobin. Nature. 1957;180:326–328. doi: 10.1038/180326a0. [DOI] [PubMed] [Google Scholar]
- 32.Ceccaldi R., Sarangi P., D'Andrea A.D. The Fanconi anaemia pathway: new players and new functions. Nat. Rev. Mol. Cell Biol. 2016;17:337–349. doi: 10.1038/nrm.2016.48. [DOI] [PubMed] [Google Scholar]
- 33.Eller M.S., Maeda T., Magnoni C., Atwal D., Gilchrest B.A. Enhancement of DNA repair in human skin cells by thymidine dinucleotides: Evidence for a p53-mediated mammalian SOS response. Proc. Natl. Acad. Sci. USA. 1997;94:12627–12632. doi: 10.1073/pnas.94.23.12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Protic M., Roilides E., Levine A.S., Dixon K. Enhancement of DNA-Repair Capacity of Mammalian-Cells by Carcinogen Treatment. Somat. Cell Mol. Genet. 1988;14:351–357. doi: 10.1007/Bf01534643. [DOI] [PubMed] [Google Scholar]
- 35.Rallabandi R., Sharp B., Cruz C., Wang Q., Locsin A., Driscoll C.B., Lee E., Nelson T., Devaux P. miRNA-mediated control of exogenous OCT4 during mesenchymal-epithelial transition increases measles vector reprogramming efficiency. Mol. Ther. Methods Clin. Dev. 2022;24:48–61. doi: 10.1016/j.omtm.2021.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang X., Potter J., Kumar S., Ravinder N., Chesnut J.D. Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J. Biotechnol. 2017;241:136–146. doi: 10.1016/j.jbiotec.2016.11.011. [DOI] [PubMed] [Google Scholar]
- 37.Kazemian P., Yu S.Y., Thomson S.B., Birkenshaw A., Leavitt B.R., Ross C.J.D. Lipid-Nanoparticle-Based Delivery of CRISPR/Cas9 Genome-Editing Components. Mol. Pharm. 2022;19:1669–1686. doi: 10.1021/acs.molpharmaceut.1c00916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loughrey D., Dahlman J.E. Non-liver mRNA Delivery. Acc. Chem. Res. 2022;55:13–23. doi: 10.1021/acs.accounts.1c00601. [DOI] [PubMed] [Google Scholar]
- 39.Dong W., Kantor B. Lentiviral Vectors for Delivery of Gene-Editing Systems Based on CRISPR/Cas: Current State and Perspectives. Viruses. 2021;13 doi: 10.3390/v13071288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mangeot P.E., Risson V., Fusil F., Marnef A., Laurent E., Blin J., Mournetas V., Massouridès E., Sohier T.J.M., Corbin A., et al. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat. Commun. 2019;10:45. doi: 10.1038/s41467-018-07845-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.An M., Raguram A., Du S.W., Banskota S., Davis J.R., Newby G.A., Chen P.Z., Palczewski K., Liu D.R. Engineered virus-like particles for transient delivery of prime editor ribonucleoprotein complexes in vivo. Nat. Biotechnol. 2024 doi: 10.1038/s41587-023-02078-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Banskota S., Raguram A., Suh S., Du S.W., Davis J.R., Choi E.H., Wang X., Nielsen S.C., Newby G.A., Randolph P.B., et al. Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell. 2022;185:250–265.e16. doi: 10.1016/j.cell.2021.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maeder M.L., Gersbach C.A. Genome-editing Technologies for Gene and Cell Therapy. Mol. Ther. 2016;24:430–446. doi: 10.1038/mt.2016.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu C.L., Ruan M.Z.C., Mahajan V.B., Tsang S.H. Viral Delivery Systems for CRISPR. Viruses. 2019;11 doi: 10.3390/v11010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chew W.L., Tabebordbar M., Cheng J.K.W., Mali P., Wu E.Y., Ng A.H.M., Zhu K., Wagers A.J., Church G.M. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat. Methods. 2016;13:868–874. doi: 10.1038/nmeth.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boutin S., Monteilhet V., Veron P., Leborgne C., Benveniste O., Montus M.F., Masurier C. Prevalence of Serum IgG and Neutralizing Factors Against Adeno-Associated Virus (AAV) Types 1, 2, 5, 6, 8, and 9 in the Healthy Population: Implications for Gene Therapy Using AAV Vectors. Hum. Gene Ther. 2010;21:704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 47.Gao G.P., Alvira M.R., Wang L., Calcedo R., Johnston J., Wilson J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Senis E., Fatouros C., Grosse S., Wiedtke E., Niopek D., Mueller A.K., Borner K., Grimm D. CRISPR/Cas9-mediated genome engineering: An adeno-associated viral (AAV) vector toolbox. Biotechnol. J. 2014;9:1402–1412. doi: 10.1002/biot.201400046. [DOI] [PubMed] [Google Scholar]
- 49.Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelson T., Heckl D., Ebert B.L., Root D.E., Doench J.G., Zhang F. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kantor B., Ma H., Webster-Cyriaque J., Monahan P.E., Kafri T. Epigenetic activation of unintegrated HIV-1 genomes by gut-associated short chain fatty acids and its implications for HIV infection. Proc. Natl. Acad. Sci. USA. 2009;106:18786–18791. doi: 10.1073/pnas.0905859106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bayer M., Kantor B., Cockrell A., Ma H., Zeithaml B., Li X., McCown T., Kafri T. A Large U3 Deletion Causes Increased In Vivo Expression From a Nonintegrating Lentiviral Vector. Mol. Ther. 2008;16:1968–1976. doi: 10.1038/mt.2008.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park A., Hong P., Won S.T., Thibault P.A., Vigant F., Oguntuyo K.Y., Taft J.D., Lee B. Sendai virus, an RNA virus with no risk of genomic integration, delivers CRISPR/Cas9 for efficient gene editing. Mol. Ther. Methods Clin. Dev. 2016;3 doi: 10.1038/mtm.2016.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ehrke-Schulz E., Schiwon M., Leitner T., Dávid S., Bergmann T., Liu J., Ehrhardt A. CRISPR/Cas9 delivery with one single adenoviral vector devoid of all viral genes. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-17180-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang W., Zhou H., Bi H., Fromm M., Yang B., Weeks D.P. Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res. 2013;41:e188. doi: 10.1093/nar/gkt780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li J.F., Norville J.E., Aach J., McCormack M., Zhang D., Bush J., Church G.M., Sheen J. Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat. Biotechnol. 2013;31:688–691. doi: 10.1038/nbt.2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nekrasov V., Staskawicz B., Weigel D., Jones J.D.G., Kamoun S. Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013;31:691–693. doi: 10.1038/nbt.2655. [DOI] [PubMed] [Google Scholar]
- 57.Shan Q., Wang Y., Li J., Zhang Y., Chen K., Liang Z., Zhang K., Liu J., Xi J.J., Qiu J.L., Gao C. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat. Biotechnol. 2013;31:686–688. doi: 10.1038/nbt.2650. [DOI] [PubMed] [Google Scholar]
- 58.Ding Q., Regan S.N., Xia Y., Oostrom L.A., Cowan C.A., Musunuru K. Enhanced Efficiency of Human Pluripotent Stem Cell Genome Editing through Replacing TALENs with CRISPRs. Cell Stem Cell. 2013;12:393–394. doi: 10.1016/j.stem.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao Y., Zhao Y. Self- processing of ribozyme- flanked RNAs into guide RNAs in vitro and in vivo for CRISPR- mediated genome editing. J. Integr. Plant Biol. 2014;56:343–349. doi: 10.1111/jipb.12152. [DOI] [PubMed] [Google Scholar]
- 60.Nissim L., Perli S.D., Fridkin A., Perez-Pinera P., Lu T.K. Multiplexed and Programmable Regulation of Gene Networks with an Integrated RNA and CRISPR/Cas Toolkit in Human Cells. Mol. Cell. 2014;54:698–710. doi: 10.1016/j.molcel.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoshioka S., Fujii W., Ogawa T., Sugiura K., Naito K. Development of a mono-promoter-driven CRISPR/Cas9 system in mammalian cells. Sci. Rep. 2015;5 doi: 10.1038/srep18341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maruyama T., Dougan S.K., Truttmann M.C., Bilate A.M., Ingram J.R., Ploegh H.L. Corrigendum: Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2016;34:210. doi: 10.1038/nbt0216-210c. [DOI] [PubMed] [Google Scholar]
- 63.Paulsen B.S., Mandal P.K., Frock R.L., Boyraz B., Yadav R., Upadhyayula S., Gutierrez-Martinez P., Ebina W., Fasth A., Kirchhausen T., et al. Ectopic expression of RAD52 and dn53BP1 improves homology-directed repair during CRISPR-Cas9 genome editing. Nat. Biomed. Eng. 2017;1:878–888. doi: 10.1038/s41551-017-0145-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ferrari S., Jacob A., Beretta S., Unali G., Albano L., Vavassori V., Cittaro D., Lazarevic D., Brombin C., Cugnata F., et al. Efficient gene editing of human long-term hematopoietic stem cells validated by clonal tracking. Nat. Biotechnol. 2020;38:1298–1308. doi: 10.1038/s41587-020-0551-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Renaud J.B., Boix C., Charpentier M., De Cian A., Cochennec J., Duvernois-Berthet E., Perrouault L., Tesson L., Edouard J., Thinard R., et al. Improved Genome Editing Efficiency and Flexibility Using Modified Oligonucleotides with TALEN and CRISPR-Cas9 Nucleases. Cell Rep. 2016;14:2263–2272. doi: 10.1016/j.celrep.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 66.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kärber G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Arch. Exp. Pathol. Pharmakol. 1931;162:480–483. [Google Scholar]
- 68.Concordet J.-P., Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018;46:W242–W245. doi: 10.1093/nar/gky354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The article or Supplemental material include all data directly relevant to the study. Further inquiries can be directed to the corresponding author.