

Abstract

Cyclic oligomeric depsipeptides (COD) are a structural class within naturally occurring compounds with a wide range of biological activity. Verticilide is a COD (24-membered ring) that was identified by its inhibition of insect ryanodine receptor (RyR). We have since found that the enantiomer of verticilide (ent-verticilide, 1) is a potent inhibitor of mammalian RyR2, a cardiac calcium channel, and therefore a potential antiarrhythmic agent. Oddly, nat-verticilide does not inhibit RyR2. To further develop ent-verticilide as an antiarrhythmic, we explored potential SAR through systematic modification of the ester’s functionality to both N–H and N–Me amides. The syntheses of these ent-verticilide-inspired analogs are detailed using a monomer-based platform enabled by enantioselective catalysis. Two analogs among 23 exhibited measurable reduction of calcium sparks in a functional assay of RyR2 activity. These findings illustrate the value of natural product-inspired therapeutic development, but the less-studied approach where the non-natural enantiomeric series harbors important SAR.
Introduction
The natural product verticilide A1 was discovered by Omura in 20041 and reported in 20062 to have inhibitory activity against insect ryanodine receptor (RyR). In 2019, we reported that its non-natural enantiomer (1) exhibits activity at mammalian ryanodine receptor 2 (RyR2), while nat-verticilide was inactive, recording an evidently unprecedented case where all detectable activity against a specific target is harbored by the non-natural enantiomer of a natural product.3,4 RyR2 is a target for antiarrhythmic development, and selective inhibitors promise to develop a more complete picture of its role in heart disease.5−7 We have since discovered a second example of activity harbored entirely by the non-natural enantiomer in the antiarrhythmic ent-verticilide B1—the 18-membered oligomer of 1.8 These discoveries provide a basis for the isoform-selective RyR inhibitors recently described, collectively spanning a broad range of molecular size and structure.9−11 Moreover, their non-natural configuration could lead to favorable pharmacokinetics and pharmacodynamics.12
The verticilides are cyclic depsipeptides, and verticilide A1 is a 24-membered cyclic oligomeric depsipeptide (COD) comprised of alternating l-alanine and (R)-α-hydroxy heptanoic acid residues. The surprising activity of ent-verticilide is accented by its Beyond Rule of 5 (bRo5) classification, and its oligomeric nature suggests that the pharmacophore may be a fraction of the molecule’s total size. We have previously reported activity at RyR2 as a function of macrocycle size by examining COD ranging in ring size from 6 to 36, where the nature of the polar backbone is modified only by depsipeptide chain length.13 In this study, the aliphatic side chains thought to drive target engagement are held constant, while modifications to the polar backbone are explored, in order to determine the degree to which the backbone structure and functionality contribute to antiarrhythmic activity. The cyclic oligomeric nature of 1 lends itself to the systematic substitution of ester for N–H and N–Me amide (Figure 1, Series 1–5). This includes a few oligomeric analogs, but mostly analogs that require construction from didepsipeptide (3) or dipeptide (4) building blocks. The didepsi/peptides 3–4 simplify further to α-hydroxy acid 5 and N-protected α-amino acids 6 and 7. While 7 was available commercially, both 5 and 6 required preparations by enantioselective synthesis. Overall, a collection of 23 compounds was prepared based on the ent-verticilide structure. Key to this approach was preservation of the methyl and pentyl side chains across all backbone modifications to isolate the contribution of backbone functionality to activity, either directly by contact (e.g., hydrogen bonding) or indirectly (e.g., conformational effects, permeability14).
Figure 1.
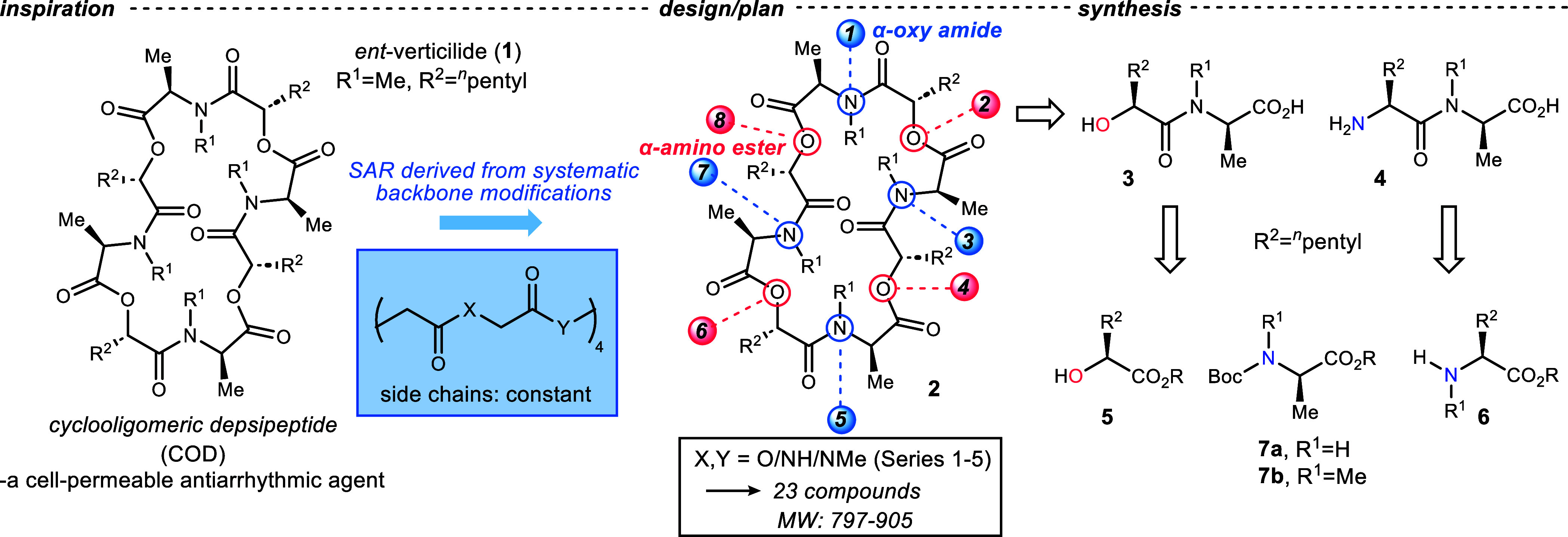
ent-Verticilide cyclooligomeric depsipeptide structure, backbone modification plan, and key retrosynthetic modules for the determination of the effect of backbone modifications on antiarrhythmic activity.
To date, ent-verticilide is not known to be produced by a natural source, but three distinct syntheses by us and two by Omura and Sunazuka (nat-verticilide)2a,2c provide a robust platform for the study of this natural product class, including the non-natural enantiomers described here. The first synthesis of nat-verticilide was reported by Omura in 2006 using a chiral auxiliary to establish the (R)-configuration of the heptanoic acid residue needed for nat-verticilide.2 We subsequently reported the use of an enantioselective Henry reaction to prepare the epimeric residue in order to use a Mitsunobu-based macrocyclooligomerization for nat-verticilide synthesis.15,16 This Henry reaction is equally effective for (R)- or (S)-α-hydroxy heptanoic acid residue preparation, and it has been used in our later preparations of ent-verticilide at larger scales.12,17
Figure 2 summarizes five series of analogs (Series 1–5). Series 1 holds the ester functionality constant while converting each N–Me amide to N–H amide. Series 2 systematically substitutes each ester with N–H amide, while Series 3 similarly substitutes each ester with N–Me amide. Series 4 substitutes all esters in 1 with N–Me amide while systematically alternating the alanine N–Me amides to N–H amides. Series 5 is analogous to Series 4, except all esters are first substituted by N–Me amides, and then the N–Me alanines are systematically replaced by N–H alanines. In each series, there is a pair of adjacent and alternating oligomer isomers that offer an opportunity to examine the effect of symmetry on activity.
Figure 2.
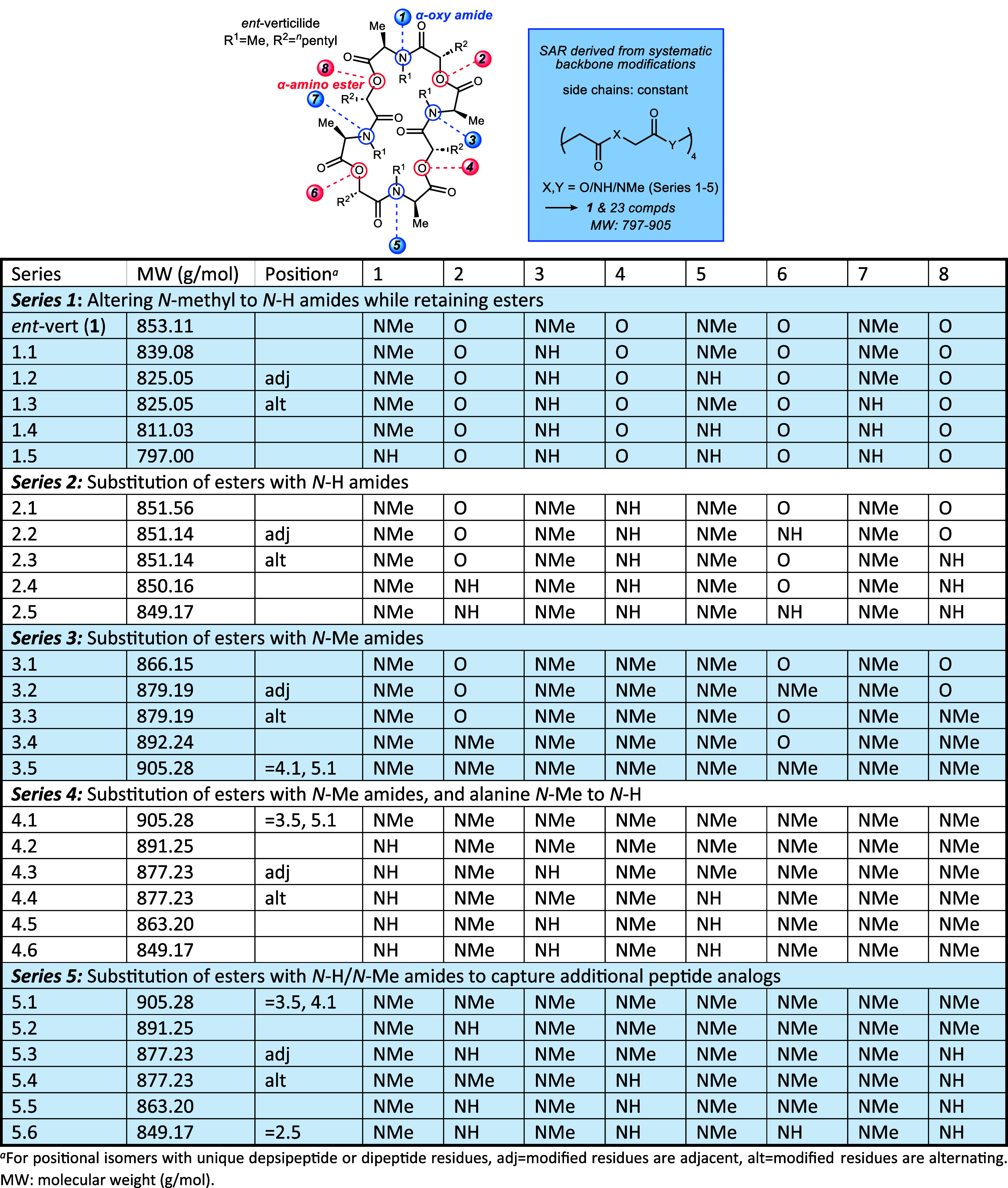
Series 1–5 analogs to probe contributions of the macrocycle backbone to antiarrhythmic activity.
The analogs in Series 1–5 vary in complexity, varying in the number of distinct monomers required for synthesis. Since this analog campaign required (S)-α-hydroxy heptanoic acid ester and amide preparation (e.g., 3), we chose to exploit the versatility of the Henry products, expecting that the nitroalkanes might bifurcate readily to a carboxylic acid18 for conventional ester and amide synthesis or be used directly to amide products (Figure 1).19,20 The development of a robust enantioselective synthesis of (S)-α-hydroxy acid was a critical centerpiece and its use to prepare α-hydroxy benzyl ester 5, N–H amide (3a), and N–Me amide (3b). An enantioselective synthesis of α-amino ester 7 for both N–H and N–Me functionality was equally critical. Successful incorporation of these monomers into a sound synthesis scheme to prepare all members of Series 1–5 to drive a comprehensive study of their antiarrhythmic structure–activity relationships has now been accomplished (Figure 2, Series 1–5).
Results and Discussion
Monomer (M)21 Synthesis
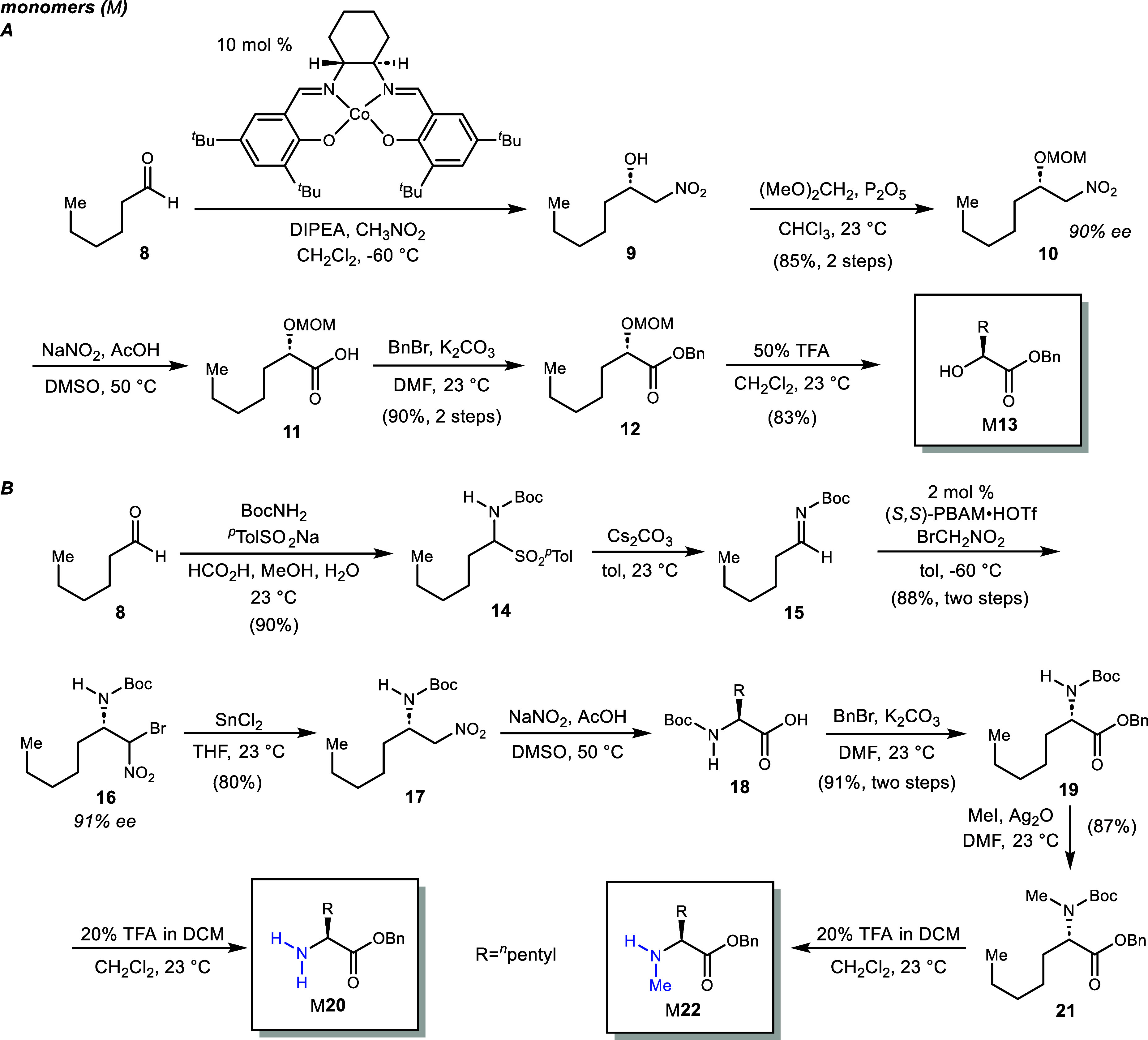
Beginning from hexanal (8), α-hydroxy ester M13 (5, R = Bn) was prepared by enantioselective Henry addition of nitromethane and immediate transacetalization with dimethoxy methane to give terminal nitroalkane 10 in 90% ee and 85% yield over 2 steps.16 The (S)-enantiomer was prepared using the cobalt(II)-salen ligand complex, as described by Yamada.22 Treatment of nitroalkane 10 using the conditions outlined by Mioskowski18 produced the terminal carboxylic acid (11) which was immediately esterified to 12 in 90% yield (2 steps) under mildly basic conditions. Deprotection of the acetal using trifluoroacetic acid yielded α-hydroxy ester M13 in 83% yield. Hexanal was also the starting point for α-amino esters M20 and M22 (7, R = Bn). Conversion of hexanal to α-amido sulfone 14(23) preceded elimination to the N-Boc imine substrate (15)23b for the aza-Henry reaction.24 Catalyzed addition of bromonitromethane25 using the chiral proton catalyst (S,S)-PBAM·HOTf26 furnished β-amino nitroalkane 16 in 91% ee as a 1:1 mixture of diastereomers. These diastereomers converged to a single terminal nitroalkane (17) in 90% ee upon treatment with stannous chloride. This two-step procedure using bromonitromethane provided high overall yields relative to the use of nitromethane in a single-step procedure to prepare 17 from 15.27 Conversion of nitroalkane 17 to acid 18 using the Mioskowski-Nef reaction18 was followed by base-promoted esterification to give 19 and then M20 after acid treatment with TFA to remove the N-Boc protecting group. Similarly, methylation of the N-Boc group gave 21 from 19,28 which was also subjected to acid treatment to give α-amino ester M22 for analogs containing N–Me amide(s). Monomer M13 was routinely prepared at 16 g-scale and M20 and M22 at 5 g-scale (Scheme 1).
Scheme 1. Enantioselective Synthesis of α-Hydroxy Ester and α-Amino Esters for ent-Verticilide Analog Synthesis.
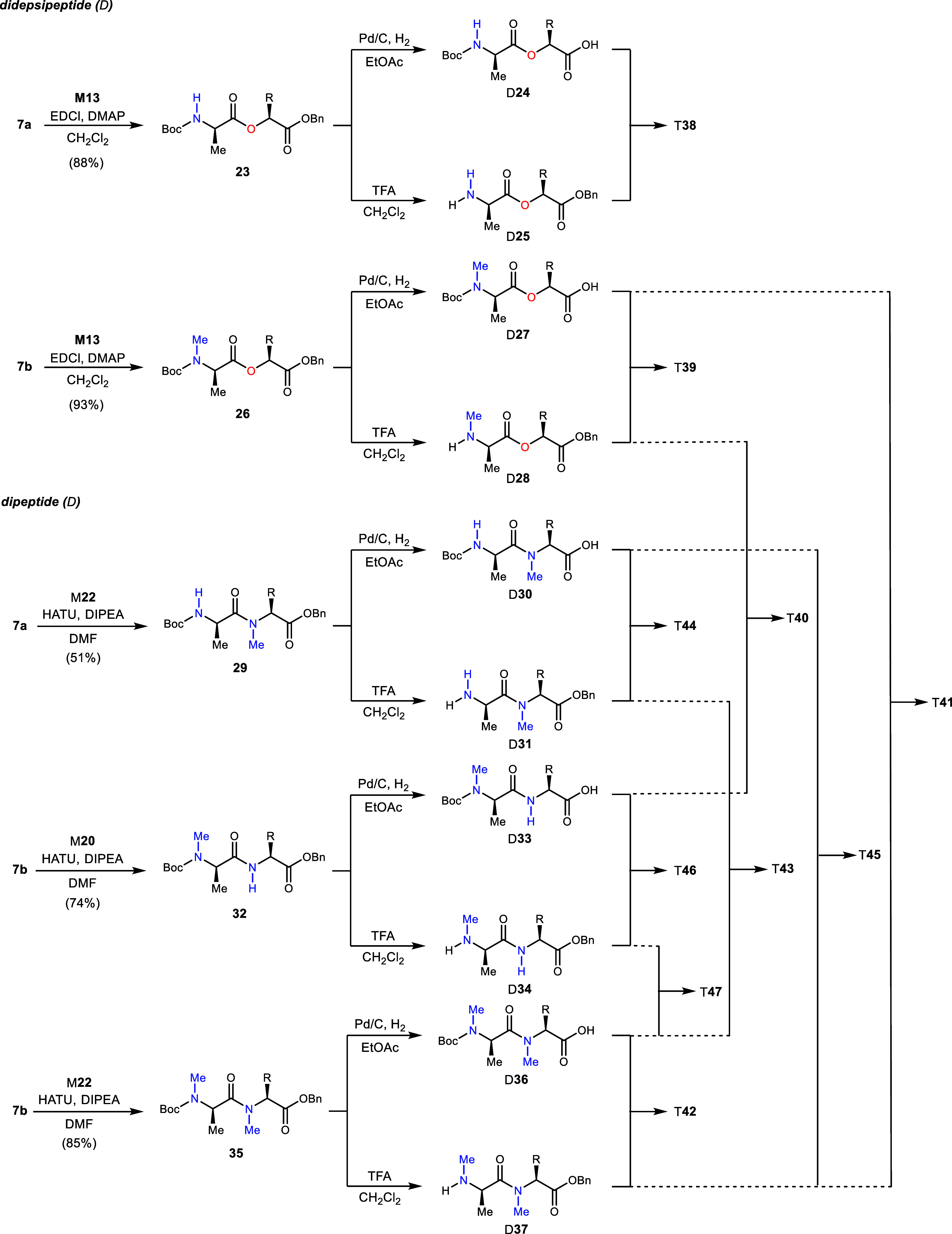
Didepsi/Peptide Synthesis (D)
Depsipeptides 23 and 26 were identified as common components needed for Series 1–3 (Scheme 2). Didepsipeptide 23 was prepared from α-hydroxy ester M13 and N-Boc-protected d-alanine using EDCI29 and DMAP, in 88% yield.13 The preparation of N–Me didepsipeptide 26 followed the same procedure and was prepared in 93% yield. Amounts of didepsipeptides 23 and 26 could be converted to acids D24 and D27, respectively, by hydrogenolysis. In parallel, deprotection to the terminal amines D25 and D28 was accomplished by standard treatment with TFA. Without purification, these units were coupled as needed to build toward the octadepsipeptides. A similar approach was used to prepare dipeptide units (Scheme 2), with dipeptide 29 formed by a coupling of 7a with M22 in 51% yield using HATU and DIPEA in DMF. N–Me dipeptides 32 and 35 were formed from 7b using identical conditions. Also, analogous was the bifurcation of each doubly protected dipeptide into C-deprotected dipeptides D30, D33, and D36, as well as N-deprotected dipeptides D31, D34, and D37. Scheme 2 also diagrams the eventual use of each didepsi/peptide in subsequent conversions to tetradepsi/peptides, as detailed in Scheme 3.
Scheme 2. Conversion of Monomers into Didepsipeptide and Dipeptide Donors for Construction of ent-Verticilide Analogs.
Scheme 3. Construction of Tetradepsipeptide and Tetrapeptide Units for Efficient Construction of Series 1–5 Analogs Using Didepsipeptide and Dipeptide Precursors.
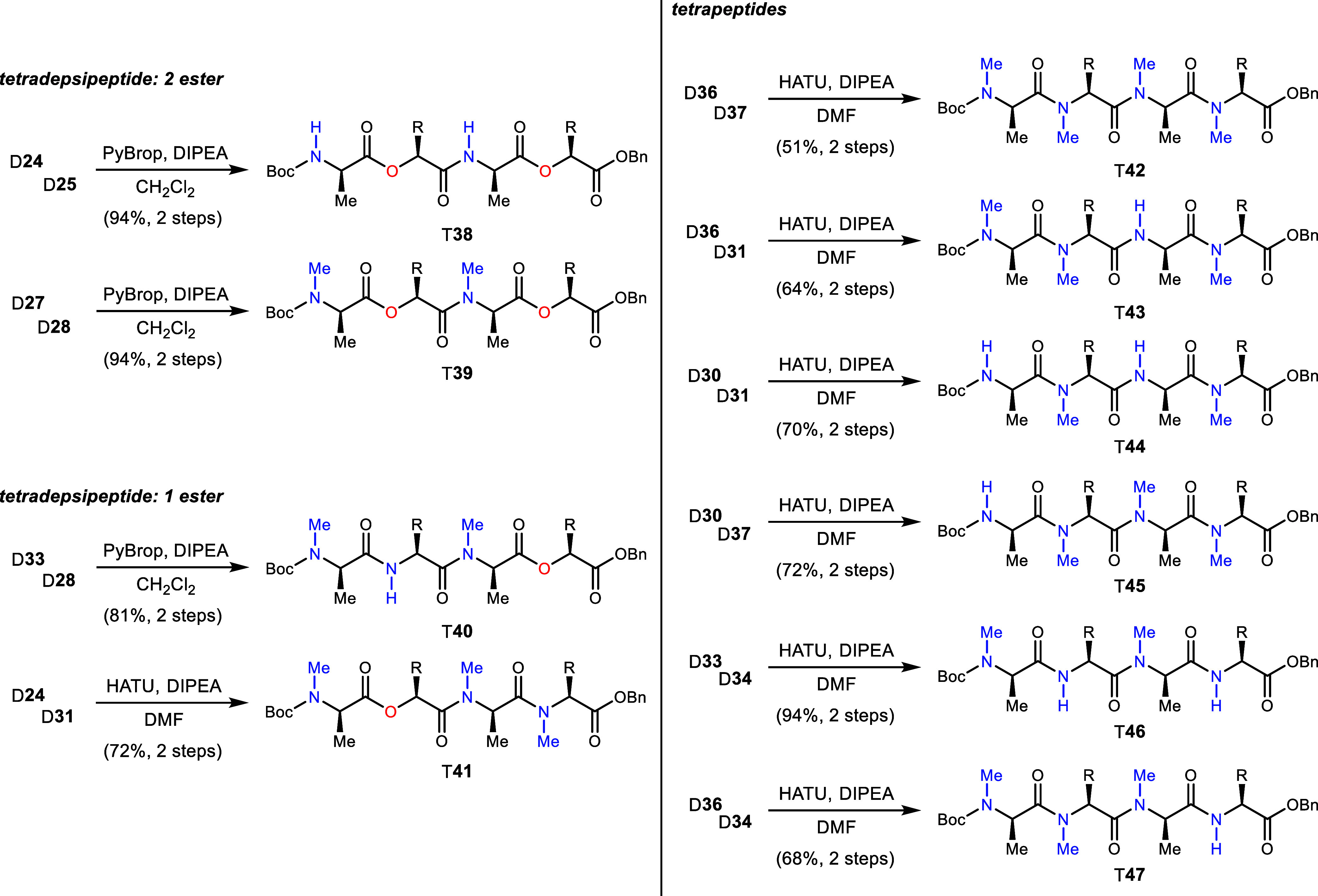
Tetradepsi/Peptide Synthesis (T)
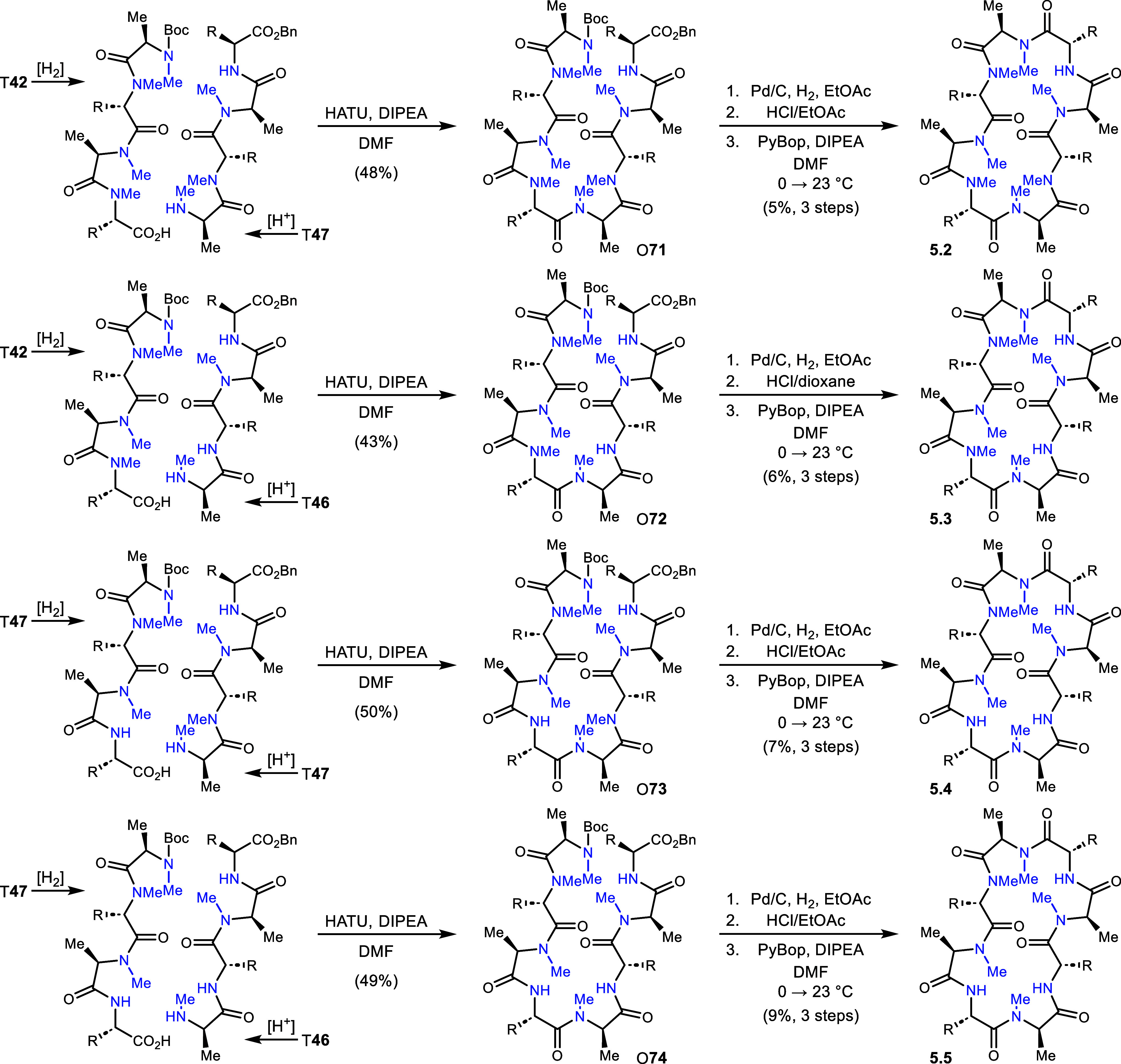
Preparation of a collection of tetradepsipeptides and tetrapeptides was also needed for efficient construction of the final octadepsi/peptides. This was accomplished using the series of dipeptide couplings, as summarized in Scheme 3. Tetradepsipeptides containing two esters were prepared using PyBrop. Didepsipeptides D24 and D25 were joined to form T38, while D27 and D28 coupled to produce T39 in 94% yield, respectively.30 Tetradepsipeptides T40 and T41 containing a single ester each were prepared from D33/D28 and D24/D31, respectively. The latter required a change in coupling reagent from PyBrop to HATU in order to address poor solubility and slow reaction of D31.31,32 Tetrapeptides were synthesized uniformly using HATU in DMF. D36 was coupled to N–Me amine D37 to form T42 in 51% yield overall. N–H amine D31 was joined with D36 and D30 to prepare T43 and T44, respectively, in 64–70% yield. N–Me amine D37 was coupled to D30 to form T45 in 72% yield. Finally, D34 was coupled to both D33 and D36 to prepare T46 and T47, respectively, in good overall yield.
Final Steps of Analog Synthesis (H,O)
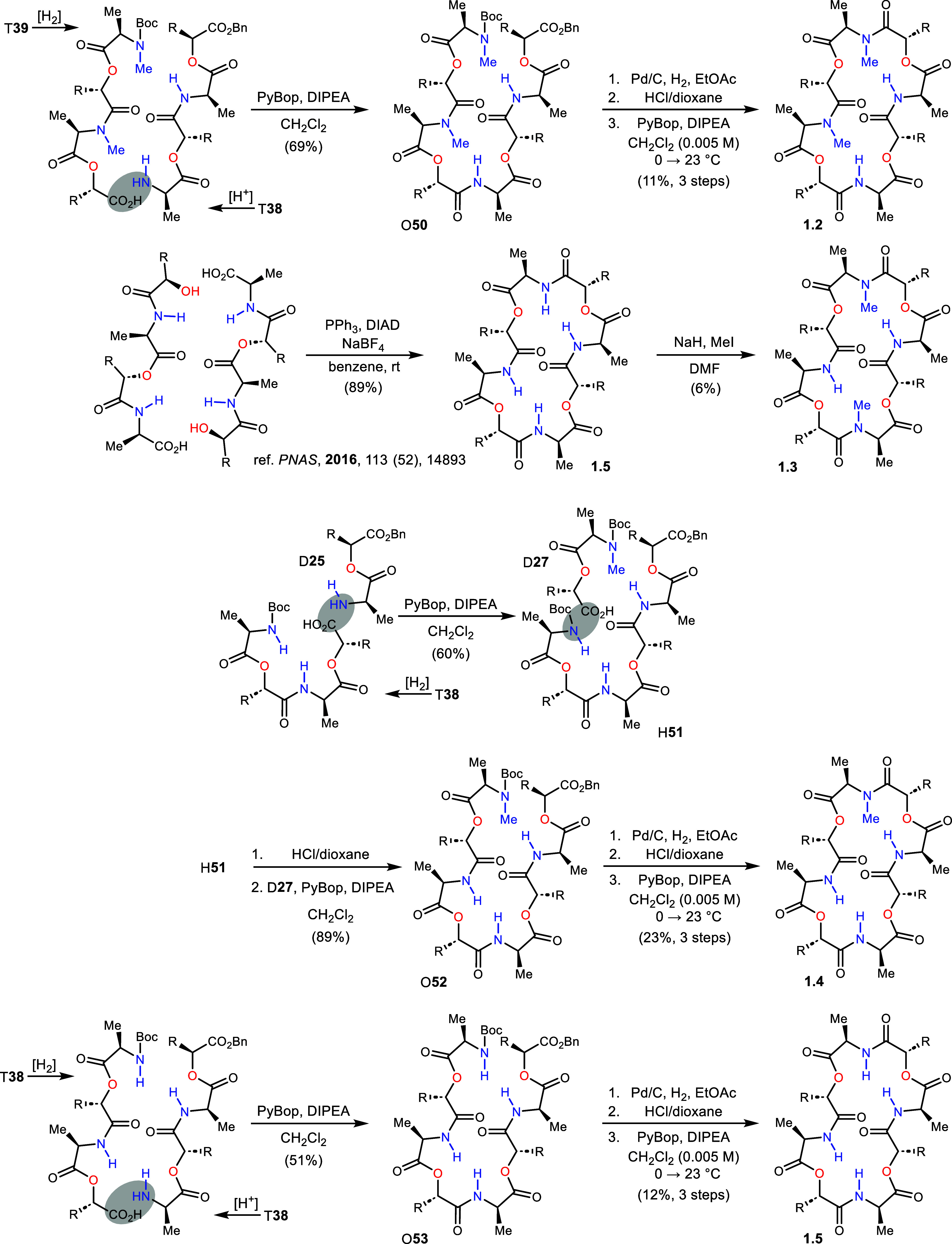
The final phase of each analog synthesis varied considerably to account for relative differences in pseudooligomer simplicity. Analog 1.1 was prepared from T39 (after hydrogenolysis) by extension of the C-terminus with didepsipeptide D28 in 96% yield (Scheme 4). This hexadepsipeptide (H48) was Boc-deprotected and coupled with D24 to give octadepsipeptide O49. The final steps converting O49 to 1.1 involved hydrogenolysis, Boc-deprotection, and PyBop-mediated macrocyclization at high dilution.2 Analog 1.1 was produced in 18% yield (3 steps), after purification by reverse phase HPLC. While 1.1 was prepared using a [4+2]+2 approach to octadepsipeptide O49, analog 1.2 used a 4+4 build from T38 and T39, proceeding in 69% yield (Scheme 5). The final steps were identical to those for analog 1.1, furnishing 1.2 in 11% yield (3 steps). Of the five Series 1 analogs, 1.3 alone had been prepared previously.15 The approach was quite different, using a Mitsunobu-driven cyclodimerization reaction followed by base-promoted methylation (NaH, MeI). Alternating N–Me/N–H amide analog 1.3 was produced in only 6% yield, but the original goal (and major product) of this reaction was permethylation. Analog 1.4 proceeded from T38 and D25 in a [4+2]+2 build similar to 1.1 (Scheme 5). This led to hexadepsipeptide H51 in 60% yield. Subsequent N-deprotection with HCl in dioxane and coupling with carboxylic acid D27 provided O52 in 89% yield overall. Hydrogenolysis and N-deprotection prepared the octamer for macrocyclization, which proceeded in 23% yield (3 steps) to furnish 1.4 in 23% yield. Depsipeptide 1.5, the final analog in this series, was prepared from tetradepsipeptide T38, with half deprotected at the N-terminus and the other at the C-terminus (Scheme 5). These fragments were joined in 51% yield to O53. Deprotection at each end, and macrocyclization, provided 1.5 in 12% yield (3 steps).
Scheme 4. Final Steps to Complete the Synthesis of Analog 1.1.

Scheme 5. Completion of Analogs 1.2–1.5.
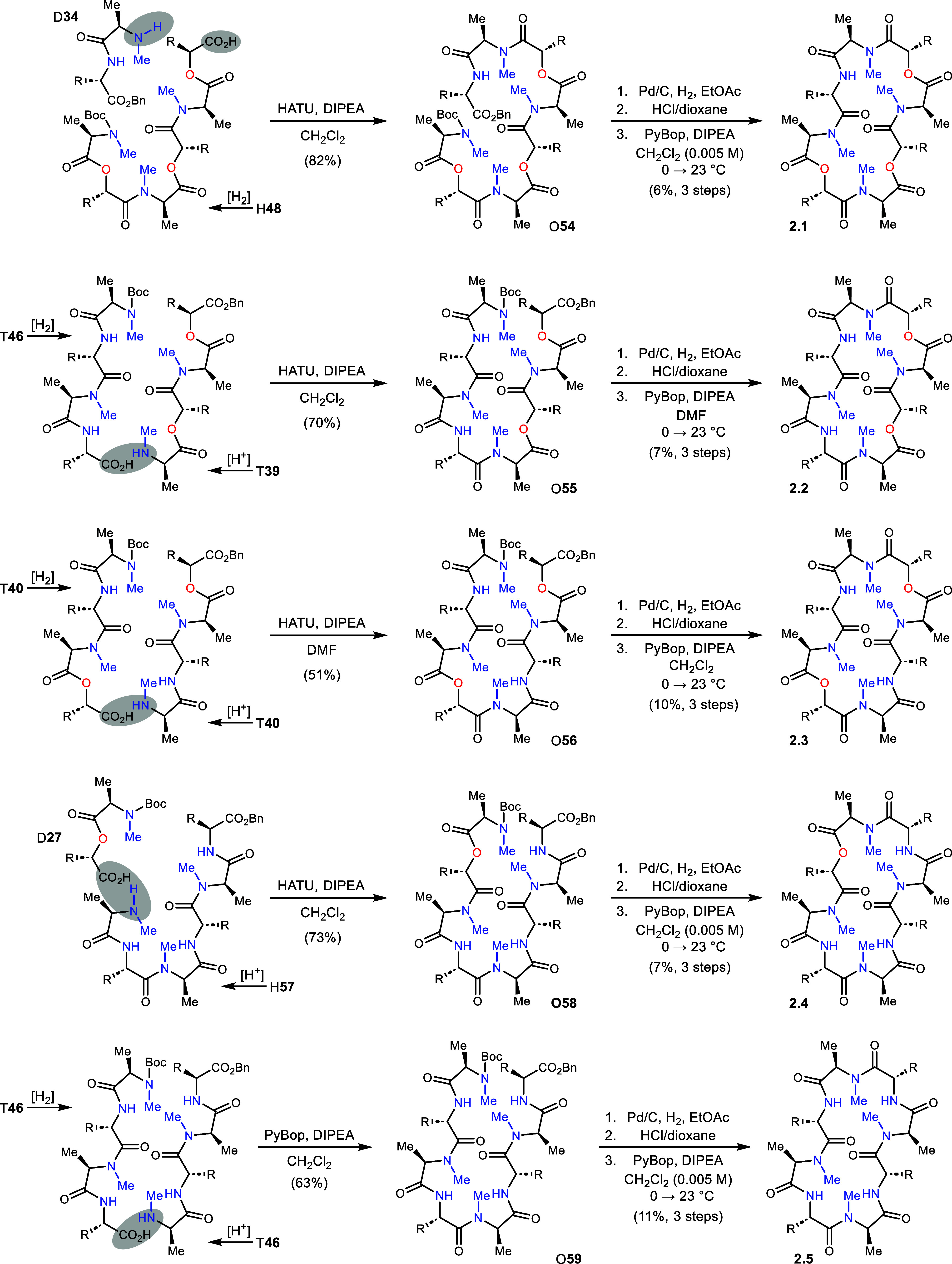
All analogs in Series 2 were prepared from tetradepsi/peptide precursors in a [4+4] build, except 2.1 and 2.4 which required a [2+6] approach (Scheme 6). Analog 2.1 evolved from H48 which, after C-deprotection, was coupled to N–Me amine D34 to furnish O54 in 82% yield. Deprotection of the termini in O54, and macrocyclization, provided 2.1 in 6% yield (3 steps).30 The low yield was a harbinger of future observations for most macrocyclizations, but reasons were not investigated.33 Analog 2.2 was constructed from depsipeptide T39 and peptide T46 to give intermediate O55. This octadepsipeptide was deprotected and macrocyclized to 2.2 in 7% yield (3 steps). Similarly, analog 2.3 began from tetradepsipeptide T40 with half C-deprotected, and the other half N-deprotected. The coupling product O56 was formed in 51% yield but required the solvent DMF for improved solubility. Deprotection at each terminus, and then macrocyclization, provided 2.3 in 10% yield (3 steps). Analog 2.4 was constructed from peptide H57 and depsipeptide D27 which were conjoined after deprotection to give depsipeptide O58 in 70% yield. The standard deprotection and macrocyclization protocols gave 2.4 in 7% yield (3 steps). Analog 2.5 was prepared from peptide T46 after its bifurcation to N- and C-deprotected fragments and coupling to O59. This peptide was deprotected and cyclized to 2.5 in 11% yield (3 steps).
Scheme 6. Construction of Series 2 Analogs (2.1–2.5).
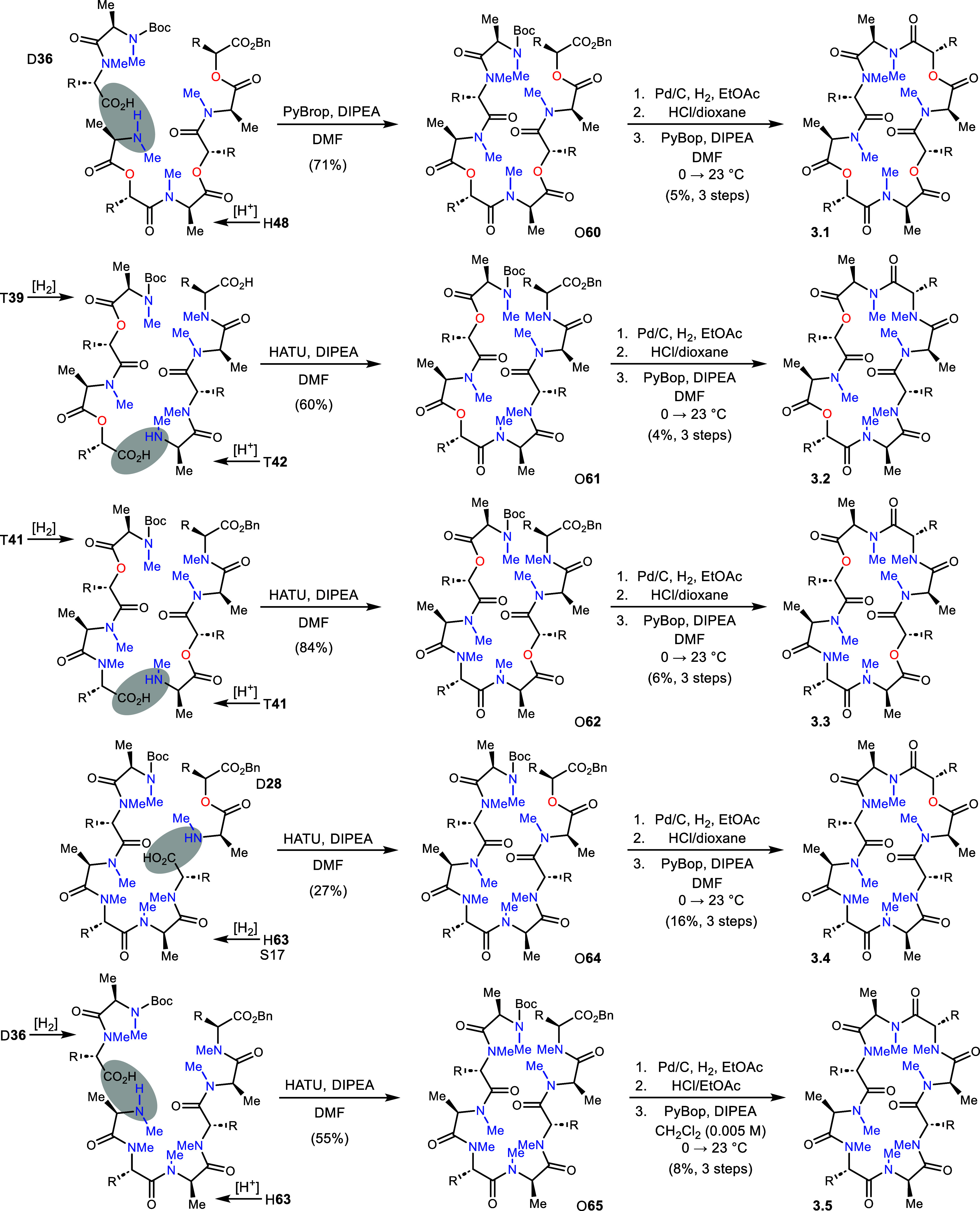
Construction of Series 3 analogs followed a similar approach. A [6+2] strategy for 3.1 required the hexadepsipeptide (H48) first used in the preparation of analog 1.1 (Scheme 7). After N-deprotection of H48, it was coupled with D36 to form O60 in 71% yield. Deprotection of the octadepsipeptide and cyclization of the seco-amide provided analog 3.1 in 5% yield. For analog 3.2, tetrapeptide T42 and depsipeptide T39 were deprotected at N- and C-termini, respectively. The octadepsipeptide O61 resulting from their coupling (60% yield) was converted to analog 3.2 in a 4% yield (3 steps). Octadepsipeptide O62 needed to prepare analog 3.3 was synthesized in 84% yield from two units of T41 using the orthogonal deprotection approach used previously. It was then deprotected and macrocyclized to 3.3 in 6% yield (3 steps). The [6+2] approach was used for analog 3.4, beginning from H63 and its eventual coupling with D28. This maneuver suffered from an unusually low yield (27%) but provided sufficient quantity of octadepsipeptide O64 for finishing to analog 3.4 (16% yield, 3 steps). To finish Series 3, hexapeptide H63 was N-deprotected and then coupled with dipeptide D36 to form octapeptide O65. This intermediate was finished along standard lines to give analog 3.5 in 8% yield (3 steps).
Scheme 7. Construction of Series 3 Analogs.
The first member of Series 4, analog 4.1, is identical to analog 3.5 (and 5.1) whose preparation was described above (Scheme 8). The remaining members were prepared by a [4+4] build. Analog 4.2 was constructed beginning with peptides T42 and T45 through a coupling that provided octapeptide O66 (57% yield). Conversion of O66 to analog 4.2 leveraged the established three step procedure culminating in 8% yield. Analog 4.3 was sourced from peptides T44 and T42, leading to a coupling yield of 53% to give peptide O67. Standard completion steps yielded analog 4.3 in 8% yield (3 steps). Peptide T43 was dimerized after selective N-deprotection and, separately, C-deprotection. This provided peptide O68 in 63% yield and ultimately analog 4.4 in 7% yield (3 steps). The penultimate analog in the series began from T44 (N-deprotected) and T43 (C-deprotected), providing peptide O69 in 46% yield. From O69, analog 4.5 was formed in 5% yield (3 steps) following the standard sequence of finishing steps. The final Series 4 analog (4.6) used the homopeptide bifurcation approach using T44, providing peptide O70 in 42% yield. The finishing sequence provided analog 4.6 in 7% yield (3 steps).
Scheme 8. Construction of Series 4 analogs.
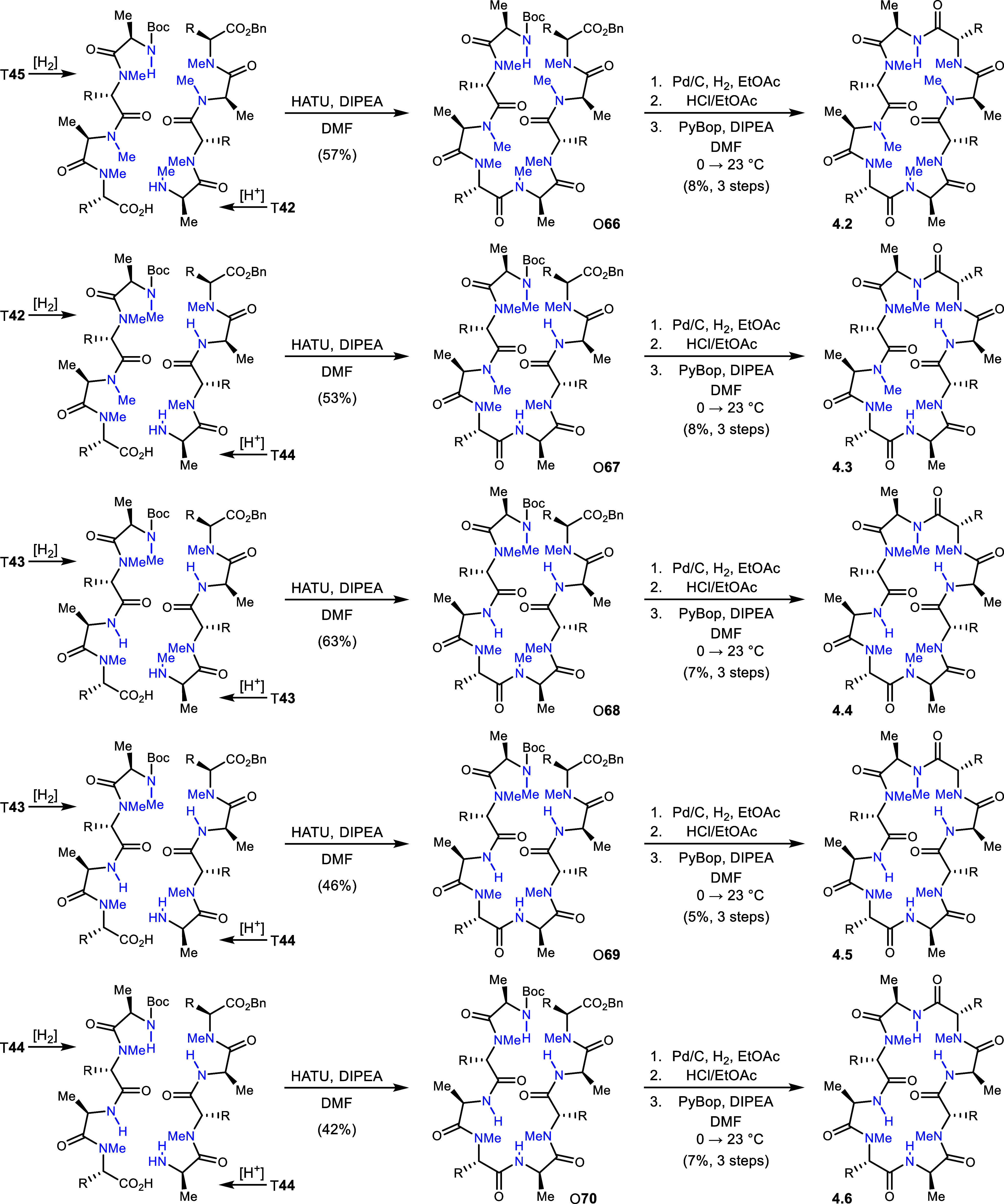
The first analog of Series 5 is identical to 3.5 (and 4.1), as described earlier (Scheme 9). Analog 5.2 was prepared from T47 and T42, leading to peptide O71 in 48% yield. Deprotection of both termini was followed by macrocyclization and 5.2 in 5% yield (3 steps). Analog 5.3 was built using the [4+4] approach as well, from T46 and T42. Peptide O72 was synthesized in 43% and then converted to 5.3 in 6% yield over the standard 3 final steps. Tetrapeptide T47 was the bifurcation point to prepare O73 in 50% yield. To this peptide was applied the standard finishing steps as well, leading to analog 5.4 in 7% yield (3 steps). The final analog (5.5) was prepared from T46 and T47 after N- and C-deprotections, respectively, to give O74 in 49% yield. This peptide was converted to analog 5.5 in 9% yield (3 steps). The final analog of Series 5 is identical to 2.5, as described earlier.
Scheme 9. Construction of Series 5 Analogs.
All analogs were measured to >95% purity using a final purification by reverse phase HPLC. All analogs were noted to be oils at the point of preparation, and none have exhibited a tendency to solidify as of yet. Not unexpectedly, the 1H NMR spectra suggest more than one averaged conformation in each case, although some (e.g 2.2, 3.4) appeared more complex than others. A noteworthy finding is that the nonsymmetrical and highly N-methylated analogs displayed more complex 1H NMR spectra. In most cases, the region between 2.5 and 3.5 ppm highlights the presence of many different conformations. A key example of this is analog 1.2, as there are 2 major conformations, as well as many less prominent conformations seen by 1H NMR. Solubility issues became more apparent in the later series (3–5), but this was managed by using DMF as the reaction solvent. With the amide analogs, it was also shown that using HATU (rather than PyBrop) gave more efficient reactivity and conversion to product.
Evaluation of Series 1–5 in the Calcium Sparks Assay
With a wide range of ent-verticilide analogs in hand, organized by five series that systematically change the backbone functionality without modifying the alternating methyl and pentyl side chains, we pursued their evaluation in the calcium sparks assay for insight into potential antiarrhythmic activity.3 Left ventricular myocytes were isolated from C57BL/6J mice and subjected to brief incubation with saponin, which permeabilizes the sarcolemma and enables equivalent access of compounds to RyR2, irrespective of their permeability.14 Cardiomyocytes were then incubated in an internal solution designed to promote spontaneous RyR2-mediated calcium release events, known as sparks,34 and the spark frequency was recorded following a 15 min incubation with 25 μM of each compound. Figure 3 summarizes these findings; no inhibition of RyR2 (lack of activity) is reflected as a value of 1 (dotted line). The relative activity of Series 1 compounds was reported previously, finding that introduction of one or more N–H amide(s) to the structure leads to loss of measurable activity.35 In that study, we noted that the 18-membered oligomer was more forgiving. Returning to the 24-membered ring oligomers studied here, Series 2 showed a significant trend, with introduction of a single N–H amide as a replacement for an ester (2.1) producing an analog with modest potency. A second ester conversion to an N–H amide was less tolerated (2.2), and further analogs with increased N–H amide:ester enrichment led to loss of activity. The SAR for Series 3 was less apparent, with 3.1 exhibiting partial inhibition, but less so than analog 2.1. Series 4–5 analogs produced nearly flat SAR, with no notable inhibition by any of the analogs. Although no rigorous examination of solubility under these conditions was made, solutions were freshly prepared for each assay and did not produce any observable precipitation.
Figure 3.

RyR2-mediated calcium spark frequency in permeabilized left ventricular myocytes incubated with 25 μM drug for 15 min. All data normalized to vehicle (DMSO) condition. Data displayed as interquartile range with median (boxes) and range (error bars). (A) *P < 0.0001 vs vehicle. N = 29 (2), 102 (5), 105 (5), 108 (5), 119 (4), and 87 cells (mice). Previously reported in Johnston et al. ACS Med. Chem. Lett., 2022, 13(11), 1755–1762. (B) *P < 0.0001 vs vehicle. # P = 0.00041 vs vehicle. N = 38, 46, 54, 50, and 50 cells, respectively, from 2 mice. (C) *P < 0.0001 vs vehicle. # P = 0.034 vs vehicle. N = 79 (4), 58 (3), 79 (4), 80 (5), and 91 (5) cells (mice). (D) P > 0.05 for all groups vs vehicle. N = 66 (4), 42 (2), 37 (2), 38 (2), 43(2), and 37 (2) cells (mice). (E) *P < 0.0001 vs vehicle. N = 66 (4), 85 (5), 93 (5), 100 (5), 89 (5), and 67 (4) cells (mice).
Conclusions
These results suggest that the native conformation of ent-verticilide is likely critical for target engagement. Insofar as increasing N–H amide content is somewhat tolerated with a single N–H amide/ester replacement, but less so with additional replacements, the single N–H amide may provide a transannular hydrogen bond that lightly affects the active conformation and pharmacophore. The ester content of the most active compounds (2.1, 3.1) appears beneficial when compared to those analogs enriched in N–Me or N–H amides. This finding suggests that replacement of an amide by an ester functionality can be beneficial. Our companion study that evaluates the permeability of Series 1–5 analogs using the PAMPA assay provides additional potential insight into the physicochemical impact of changes to the polar backbone.14 Importantly, the effects of these changes on activity do not overlay perfectly with changes to passive permeability, suggesting that the latter is not responsible for activity loss in Series 1–5. Overall, these analogs provide a picture of the structural and functional landscape for further optimization of the ent-verticilide template for antiarrhythmic drug development.
In conclusion, we explored the contribution of backbone functionality to activity using a systematic approach to comparison of ester, N–Me amide, and N–H amide functionality throughout a COD. This required the enantioselective synthesis of essential α-amino acid and α-oxy acid monomers and their use in the synthesis of ent-verticilide and 23 analogs. Evaluation of all compounds in the calcium sparks assay uncovered two analogs (2.1, 3.1) with measurable activity. Interestingly, both exhibit favorable passive permeability as well when measured by PAMPA.14 Further studies aimed at understanding the confluence of permeability and activity of these potential therapeutic will be reported in due course.
Experimental Section
Methods
Materials and Methods
Glassware was flame-dried under vacuum for all nonaqueous reactions. All reagents and solvents were commercial grade and purified prior to use when necessary. Toluene, THF, and dichloromethane (CH2Cl2) were dried by passage through a column of activated alumina, as described by Grubbs.36 Flash column chromatography was performed using Sorbent Technologies 230–400 mesh silica gel with solvent systems indicated. Analytical thin layer column chromatography was performed using Sorbent Technologies 250 μm glass backed UV254 silica gel plates and was visualized by fluorescence upon 250 nm radiation and/or the by use of TLC stain. Solvent removal was effected by rotary evaporation under vacuum (∼25–40 mmHg). All extracts were dried with Na2SO4 unless otherwise noted. Preparative HPLC was performed on an Agilent 1260 system (column: Zorbax Eclipse XDB-C18; 21.2 mm × 150 mm, 5 μm, flow rate 8 mL/min) with 210 nm monitoring wavelength and acetonitrile/water (+0.1% TFA) gradient as indicated. Nuclear magnetic resonance spectra (NMR) were acquired on a Bruker AV-400 (400 MHz) or Bruker AV II-600 (600 MHz) instrument. Mass spectra were recorded by use of electron impact ionization (EI) or electro-spray ionization (ESI) on a high-resolution TQ-Orbitrap 3 XL Penn or Orbitrap 2 Classic FPG in the Vanderbilt Mass Spectrometry Core Laboratory. IR spectra were recorded on a Nicolet Avatar 360 spectrophotometer and are reported in wavenumbers (cm–1) as neat films on a NaCl plate (transmission). Melting points were measured using an OptiMelt automated melting point system (Stanford Research Systems) and are not corrected. Chiral HPLC analysis was conducted on an Agilent 1200 series Infinity instrument using a ChiralPak column. Optical rotations were measured on a Jasco P-2000 polarimeter.
Chemistry
The following procedures describe the synthesis of analog 2.5 and are representative. See the Supporting Information for complete experimental procedures and spectral data.
General Procedure for tert-Butyloxycarbonyl (Boc) Deprotection
A round bottom flask was charged with the depsipeptide (1 equiv) and dissolved in either 4 M HCl/ethyl acetate (1 M in depsipeptide) or 20% TFA in DCM. The reaction was allowed to stir for 1–3 h at ambient temperature. The crude reaction mixture was concentrated, ether was added, and the mixture was then reconcentrated. This procedure was repeated 3 times with diethyl ether.
General Procedure for Benzyl Deprotection
A round bottom flask was charged with the depsipeptide (1 equiv) dissolved in methanol or ethyl acetate (0.1 M) and treated with 10% Pd/C (20 mol %). The reaction flask was evacuated with a light vacuum (∼40 Torr). Hydrogen (balloon) was added, and then, the flask was cycled through a light vacuum three times. The reaction was stirred for 1.5–3 h. After purging the flask with argon, the crude reaction mixture was filtered through Celite and concentrated to afford the carboxylic acid.
1-Tosylhexan-1-amine (14)
A round bottom flask was charged with hexanal (9.0 mL, 73 mmol), tert-butyl carbamate (5.7 g, 49 mmol), and MeOH (73 mL) and stirred until it became a homogeneous solution. NaSO2pTol (17.4 g, 97.6 mmol) was added, along with enough water to dissolve the solids (100 mL). Then, formic acid (3.68 mL, 97.6 mmol) was added, and the mixture was allowed to stir at ambient temperature under argon for 4 days. The reaction mixture was filtered and washed with H2O and hexanes to afford the product as a white solid (15.5 g, 90%). M.p. 107–110 °C; Rf = 0.26 (10% EtOAc/hexanes); IR (film) 3334, 2958, 2930, 2861, 1721, 1597, 1518, 1456, 1392, 1316, 1245, 1167, 1142, 1085 cm–1; 1H NMR (600 MHz, CDCl3) the small doubling of peaks is due to cis and trans amide rotamers. The largely favored isomer is listed here: δ 7.76 (d, J = 8.8 Hz, 2H), 7.30 (d, J = 7.9 Hz, 2H), 5.10 (d, J = 11.0 Hz, 1H), 4.79 (td, J = 10.9, 3.4 Hz, 1H), 2.38 (s, 3H), 2.24–2.16 (m, 1H), 1.74–1.65 (m, 1H), 1.55–1.23 (m, 6H), 1.19 (s, 9H) 0.86 (t, J = 6.5 Hz, 3H); 13C NMR (150 MHz, CDCl3) ppm 154.0, 144.9, 134.0, 129.7, 129.4, 80.6, 31.2, 28.0, 27.7, 26.4, 25.0, 22.4, 21.7, 14.0; HRMS (EI): exact mass calcd for C18H29NnaO4S [M + Na]+ 378.1710, found 378.1719.
tert-Butyl((2S)-1-bromo-1-nitroheptan-2-yl)carbamate (16)
A round bottom flask was charged with sulfone 14 (6.00 g, 16.8 mmol), Cs2CO3 (27.5 g, 84.4 mmol), and toluene (160 mL). The mixture was allowed to stir at ambient temperature under argon for 6 h. The reaction mixture was filtered through a pad of Celite and concentrated. The crude oil was then dissolved in toluene (248 mL) and to it was added (S,S)-PBAM26 as its triflic acid salt (217.5 mg, 331 μmol). The reaction was cooled to −60 °C, and bromonitromethane (1.74 mL, 24.8 mmol) was added. The reaction was allowed to stir for 48 h. The reaction mixture was quenched by running it through a short silica plug while still cold, with 100% EtOAc. The fractions were combined and concentrated. The crude residue was subjected to flash column chromatography (SiO2, 5% ethyl acetate in hexanes) to afford the α-bromo nitroalkane (1:1 dr (1H NMR)) as a white solid (4.5 g, 88% 2-step); the diastereomers were determined to be 90 and 89% ee by chiral HPLC analysis (Chiralcel AD-H, 1% iPrOH/hexanes, 1.0 mL/min, tr(d1, major) = 19.6 min, tr(d1, minor) = 21.1 min, tr(d2, minor) = 25.1 min, tr(d2, major) = 29.4 min). [α]23D −20 (c 0.53, CHCl3); m.p. 62–66 °C; Rf = 0.60 (10% EtOAc/hexanes); IR (film) 3332, 2958, 2931, 2861, 1706, 1567, 1501, 1458, 1392, 1367, 1248, 1165, 1045 cm–1; 1H NMR (600 MHz, CDCl3, 1:1 mixture of diastereomers) δ 6.19 (d, J = 4.4 Hz, 1H), 6.17 (d, J = 3.0 Hz, 1H), 4.83 (d, J = 8.8 Hz, 1H), 4.73 (d, J = 9.1 Hz, 1H), 4.34 (dddd, J = 9.2, 9.2, 3.9, 3.9 Hz, 1H), 4.24 (dddd, J = 9.2, 9.2, 4.6, 4.6 Hz, 1H), 1.90–1.52 (series of m, 4H), 1.46 (s, 9H), 1.44 (s, 9H), 1.40–1.22 (m, 12H), 0.90 (t, J = 7.0 Hz, 3H), 0.88 (t, J = 6.9 Hz, 3H); 13C NMR (150 MHz, CDCl3, 1:1 mixture of diastereomers) ppm 155.2, 155.1, 84.5, 83.2, 80.9, 80.8, 55.0, 54.5, 31.32, 31.28, 30.5, 28.5, 28.4, 28.3, 25.6, 25.5, 22.52, 22.51, 14.0(2C); HRMS (EI): exact mass calcd for C12H23BrN2NaO4 [M + Na]+ 361.0733, found 361.0737.
Benzyl (S)-2-((tert-Butoxycarbonyl)amino)heptanoate (19)
A flame-dried round bottom flask was charged with halo-nitro alkane 16 (383 mg, 1.13 mmol), tin(II) chloride (428 mg, 2.26 mmol), and THF (11.3 mL). The mixture was allowed to stir at ambient temperature under argon for 12 h. The reaction mixture was then poured into H2O, and diethyl ether was added into the separatory funnel. The organic layer was filtered through a pad of Celite, washed with H2O (three times), dried, and concentrated to afford a pale yellow oil. The crude oil was then dissolved in DMSO (8.7 mL) and to it were added NaNO2 (234 mg, 3.39 mmol) and AcOH (885 μL, 17.0 mmol). The reaction was heated to 60 °C and allowed to stir for 12 h. The reaction mixture was allowed to cool to ambient temperature, quenched with 1 M aq HCl, poured into a separatory funnel, and extracted with EtOAc. The organic layers were washed with ice water (four times), dried, and concentrated. The crude reaction mixture was dissolved in DMF (2.1 mL), and to it were added K2CO3 (427 mg, 3.09 mmol) and BnBr (148 μL, 1.24 mmol). The reaction was allowed to stir at ambient temperature under argon for 12 h. The reaction was quenched with 1 M aq HCl and then extracted with EtOAc. The organic layers were then dried and concentrated. The crude residue was subjected to flash column chromatography (SiO2, 10% ethyl acetate in hexanes) to afford the product as a colorless oil (366 mg, 59% (3 steps)); [α]23D −5.39 (c 1.08, CHCl3); Rf = 0.33 (10% EtOAc/hexanes); IR (film) 3365, 2957, 2931, 2861, 1717, 1499, 1456, 1366, 1249, 1161, 1048 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.40–7.28 (m, 5H), 5.21 (d, J = 12.4 Hz, 1H), 5.12 (d, J = 12.4 Hz, 1H), 5.02 (d, J = 7.8 Hz, 1H), 4.35 (ddd, J = 11.7, 6.2, 6.2 Hz, 1H), 1.85–1.70 (m, 1H), 1.68–1.55 (m, 1H), 1.43 (s, 9H), 1.36–1.17 (m, 6H), 0.85 (t, J = 6.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) ppm 172.9, 156.5, 136.6, 128.7, 128.4, 128.3, 79.8, 66.9, 53.7, 32.7, 31.4, 28.4, 24.9, 22.5, 14.0; HRMS (EI): exact mass calcd for C19H29NNaO4 [M + Na]+ 358.1989, found 358.1993.
Benzyl(S)-2-((R)-2-((tert-butoxycarbonyl)(methyl)amino)propanamido)heptanoate (32)
A round bottom flask was charged with amine M20 (990 mg, 4.21 mmol), N-Boc-N-methyl alanine (855 mg, 4.21 mmol), and DMF (4.2 mL). The mixture was cooled to 0 °C, and then, DIPEA (2.20 mL, 12.6 mmol) and HATU (2.40 g, 6.32 mmol) were added. The reaction was stirred at 0 °C for 30 min, then warmed to room temperature, and stirred overnight. The reaction mixture was poured into cold 10% aq citric acid and extracted with EtOAc. The combined organic layers were washed with satd aq NaHCO3, brine, dried, and concentrated. The crude residue was subjected to flash column chromatography (SiO2, 5–30% ethyl acetate in hexanes) to afford the product as a white amorphous solid (1.15 g, 74%). [α]24D + 29 (c 0.43, CHCl3); Rf = 0.42 (30% EtOAc/hexanes); IR (film) 3330, 2957, 2932, 2861, 1739, 1692, 1525, 1456, 1368, 1154, 1091, 1047 cm–1; 1H NMR (600 MHz, CDCl3) δ 7.37–7.32 (series of m, 5H), 6.72 (br m, 1H), 5.15–3.77 (series of m, 4H), 3.03–2.71 (series of m, 3H), 1.82 (br m, 1H), 1.65 (br m, 1H), 1.48–1.43 (series of m, 9H), 1.32–1.23 (series of m, 9H), 0.85 (br t, J = 6.9 Hz, 3H); 13C NMR (150 MHz, CDCl3) ppm 172.8, 172.2, 159.9, 130.7, 129.1, 128.9, 128.7, 81.0, 67.6, 52.8, 46.3, 32.7, 31.9, 30.4, 25.6, 25.5, 24.9, 23.0, 14.5; HRMS (EI): exact mass calcd for C23H37N2O5 [M + H]+ 421.2702, found 421.2684.
Benzyl(6R,9S,12R,15S)-2,2,5,6,11,12-hexamethyl-4,7,10,13-tetraoxo-9,15-dipentyl-3-oxa-5,8,11,14-tetraazahexadecan-16-oate (T46)
A round bottom flask was charged with amine D34 (254 mg, 793 μmol), acid D33 (262 mg, 793 μmol), and DCM (7.0 mL). The mixture was cooled to 0 °C, and then, DIPEA (414 μL, 2.38 mmol) and PyBrop (581 mg, 1.19 mmol) were added. The reaction was stirred at 0 °C for 30 min and then warmed to 23 °C for 2 h. The reaction mixture was poured into cold 10% aq citric acid and extracted with DCM. The combined organic layers were washed with satd aq NaHCO3, brine, dried, and concentrated. The crude residue was subjected to flash column chromatography (SiO2, 5–30% ethyl acetate in hexanes) to afford the product as a light yellow oil (611 mg, 94%). [α]24D + 36 (c 0.86, CHCl3); Rf = 0.26 (30% EtOAc/hexanes); IR (film) 3318, 2956, 2931, 2860, 1742, 1661, 1530, 1456, 1367, 1256, 1154, 1090 cm–1; 1H NMR (600 MHz, CDCl3 δ 7.41–7.28 (series of m, 7H), 7.00 (br s, 1H), 6.70 (br s, 1H), 5.26 (br m, 1H), 5.13 (d, J = 12.1 Hz, 1H), 5.00 (d, J = 12.1 Hz, 1H), 4.71 (br m, 1H), 4.46 (m, 2H), 2.86–2.56 (series of m, 6H), 1.79 (br m, 1H), 1.64 (br m, 1H), 1.54 (br m, 1H), 1.43 (s, 9H), 1.37 (m, 3H), 1.27–1.17 (series of m, 16H), 0.83 (br t, J = 6.1 Hz, 3H), 0.78 (br t, J = 6.3 Hz, 3H); 13C NMR (150 MHz, CDCl3) ppm 172.5, 172.4, 172.0, 170.6, 161.0, 134.9, 128.7, 128.6, 128.5, 79.9, 68.9, 52.5, 52.0, 50.0, 42.2, 32.0, 31.9, 31.6, 31.5, 31.2, 30.8, 30.7, 29.9, 29.7, 29.4, 25.4, 25.0, 22.4, 14.0, 13.4; HRMS (EI): exact mass calcd for C34H60N5O7 [M + NH4]+ 650.4493, found 650.4487.
Benzyl(6R,9S,12R,15S,18R,21S,24R,27S)-2,2,5,6,11,12,17,18,23,24-decamethyl-4,7,10,13,16,19,22,25-octaoxo-9,15,21,27-tetrapentyl-3-oxa-5,8,11,14,17,20,23,26-octaazaoctacosan-28-oate (O59)
A round bottom flask was charged with amine T46-Bn (93.3 mg, 175 μmol), acid T46-Boc (95.0 mg, 175 μmol), and DMF (1.8 mL). The mixture was cooled to 0 °C, and then, DIPEA (91.5 μL, 525 μmol) and HATU (200 mg, 525 μmol) were added. The reaction was stirred at 0 °C for 30 min and then warmed to 23 °C for 18 h. The reaction mixture was poured into cold 10% aq citric acid and extracted with EtOAc. The combined organic layers were washed with satd aq NaHCO3, brine, dried, and concentrated. The crude residue was subjected to flash column chromatography (SiO2, 5–40% ethyl acetate in hexanes) to afford the product as a light yellow oil (117 mg, 63%). [α]24D + 37 (c 0.45, CHCl3); Rf = 0.47 (30% EtOAc/hexanes); IR (film) 3405, 2956, 2929, 2859, 1741, 1652, 1540, 1457, 1391, 1260, 1155, 1092 cm–1; 1H NMR (600 MHz, CDCl3). This compound exists as a mixture of rotamers causing significant peak broadening and overlap. Refer to the image of the 1H NMR spectrum; 13C NMR (150 MHz, CDCl3) This compound is a mixture of rotamers causing significant peak broadening and overlap. Refer to the image of the 13C NMR spectrum; HRMS (EI): exact mass calcd for C56H100N9O11 [M + NH4]+ 1074.7542, found 1074.7544.
(3S,6R,9S,12R,15S,18R,21S,24R)-1,6,7,12,13,18,19,24-Octamethyl-3,9,15,21-tetrapentyl-1,4,7,10,13,16,19,22-octaazacyclotetracosan-2,5,8,11,14,17,20,23-octaone (2.5)
A round bottom flask was charged with depsipeptide O59 (116 mg, 110 μmol), dissolved in EtOAc (1.1 mL), and treated with 10% Pd/C (2.4 mg). The reaction flask was evacuated with light vacuum. Hydrogen (balloon) was added, and the flask was cycled once more. The reaction was allowed to stir for 1 h. After purging the flask with argon, the crude reaction mixture was filtered through Celite. To the crude material was added 4 M HCl/dioxane (1.00 mL), and the reaction mixture was allowed to stir for 30 min. The reaction mixture was concentrated and added to a flame-dried round bottom flask. DMF (24.0 mL) was added, and the reaction was cooled to 0 °C. Once at 0 °C, DIPEA (43.4 μL, 330 μmol) and PyBop (60.0 mg, 115 μmol) were added. The reaction was stirred at 0 °C for 1.5 h and then allowed to warm to ambient temperature to stir for an additional 1 h. The reaction mixture was poured into cold 10% aq citric acid and extracted with EtOAc. The combined organic layers were washed with satd aq NaHCO3, brine, and then dried and concentrated. Preparative HPLC purification (5–95% aqueous acetonitrile, 210 nm, flow rate: 18 mL/min, Rt = 23.1 m) afforded the 24-membered macrocycle (10.2 mg, 11.0%) as a light yellow oil. [α]24D − 11 (c 0.29, CHCl3); Rf = 0.33 (50% EtOAc in hexanes); IR (film) 3292, 2929, 1654, 1637, 1560, 1458 cm–1; 1H NMR (600 MHz, CDCl3). This compound exists in multiple conformations, causing significant peak overlap. Refer to image of the 1H NMR spectrum; 13C NMR (150 MHz, CDCl3) This compound exists in multiple conformations, causing significant peak overlap. Refer to image of the 13C NMR spectrum; HRMS (EI): exact mass calcd for C44H84N9O8 [M+NH4]+ 866.6443, found 866.6440.
Biology
Ethics Statement
All animal work was approved (protocol number M1900081) and carried out in accordance with the guidelines and procedures set forth by the Vanderbilt Division of Animal Care.
Calcium Spark Assay
Calcium spark measurements were made, as previously described.5 Briefly, ventricular cardiomyocytes were isolated from C57Bl/6J mice, adhered to laminin-coated glass bottom culture dishes, permeabilized with saponin, and incubated with an internal solution containing the calcium sensitive dye, Fluo-4. Elementary spark recordings were made using an Olympus inverted microscope equipped with a solid-state diode laser at 488 nm for excitation, 40× silicone objective (1.25 NA), and Hamamatsu CMOS camera for detection. Spark detection analysis was performed using SparkMaster2.37
Statistics and Reproducibility
Statistical analysis was carried out in R using a hierarchical clustering model to cluster data by experiment and account for mouse-to-mouse variability.38 Bonferroni-adjusted P values are reported in the figure legends. A cutoff value of 0.05 was used as the threshold to reject the null hypothesis. The numbers of cells and replicates (mice) are indicated in the figure legend for each compound.
Acknowledgments
Research reported in this publication was supported by the National Heart, Blood, and Lung Institute of the National Institutes of Health (NIH R01 HL151223, HL164064 (F31 support for MPT)), and the PhRMA Foundation Postdoctoral Fellowship (DJB support). We are grateful to Abigail Smith for her contributions to synthesis planning at the initiation of this study.
Glossary
Abbreviations
- Boc
tert-butoxy carbonyl
- bRo5
beyond rule of 5
- COD
cyclic oligomeric depsipeptide
- DIPEA
diisopropyl ethyl amine
- DMAP
dimethylamino pyridine
- DMF
dimethylformamide
- EDCI
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- HATU
hexafluorophosphate azabenzotriazole tetramethyl uronium
- PBAM
pyrollidine bis(amidine)
- PyBrop
bromotripyrrolidinophosphonium hexafluorophosphate
- RyR
ryanodine receptor
- RyR2
ryanodine receptor 2
- SAR
structure–activity relationship
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c00923.
The authors declare no competing financial interest.
Special Issue
Published as part of Journal of Medicinal Chemistryvirtual special issue “Natural Products Driven Medicinal Chemistry”.
Supplementary Material
References
- Omura M.; Shiomi K.; Masuma R.. Novel substance fki-1033 and process for producing the same. WO/2004/044214, 2004.
- a Monma S.; Sunazuka T.; Nagai K.; Arai T.; Shiomi K.; Matsui R.; O̅mura S. Verticilide: Elucidation of Absolute Configuration and Total Synthesis. Org. Lett. 2006, 8, 5601. 10.1021/ol0623365. [DOI] [PubMed] [Google Scholar]; b Shiomi K.; Matsui R.; Kakei A.; Yamaguchi Y.; Masuma R.; Hatano H.; Arai N.; Isozaki M.; Tanaka H.; Kobayashi S.; Turberg A.; Omura S. Verticilide, a new ryanodine-binding inhibitor, produced by Verticillium sp. FKI-1033. J. Antibiot. 2010, 63, 77. 10.1038/ja.2009.126. [DOI] [PubMed] [Google Scholar]; c Watanabe A.; Noguchi Y.; Hirose T.; Monma S.; Satake Y.; Arai T.; Masuda K.; Murashima N.; Shiomi K.; O̅mura S.; Sunazuka T. Efficient synthesis of a ryanodine binding inhibitor verticilide using two practical approaches. Tetrahedron Lett. 2020, 61, 151699 10.1016/j.tetlet.2020.151699. [DOI] [Google Scholar]
- Batiste S. M.; Blackwell D. J.; Kim K.; Kryshtal D. O.; Gomez-Hurtado N.; Rebbeck R. T.; Cornea R. L.; Johnston J. N.; Knollmann B. C. Unnatural verticilide enantiomer inhibits type 2 ryanodine receptor-mediated calcium leak and is antiarrhythmic. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 4810. 10.1073/pnas.1816685116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šeflová J.; Schwarz J. A.; Smith A. N.; Svensson B.; Blackwell D. J.; Phillips T. A.; Nikolaienko R.; Bovo E.; Rebbeck R. T.; Zima A. V.; Thomas D. D.; Van Petegem F.; Knollmann B. C.; Johnston J. N.; Robia S. L.; Cornea R. L. RyR2 Binding of an Antiarrhythmic Cyclic Depsipeptide Mapped Using Confocal Fluorescence Lifetime Detection of FRET. ACS Chem. Biol. 2023, 18, 2290. 10.1021/acschembio.3c00376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kim K.; Blackwell D. J.; Yuen S. L.; Thorpe M. P.; Johnston J. N.; Cornea R. L.; Knollmann B. C. The selective RyR2 inhibitor ent-verticilide suppresses atrial fibrillation susceptibility caused by Pitx2 deficiency. J. Mol. Cell. Cardiol. 2023, 180, 1. 10.1016/j.yjmcc.2023.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schmeckpeper J.; Kim K.; George S. A.; Blackwell D. J.; Brennan J. A.; Efimov I. R.; Knollmann B. C. RyR2 inhibition with dantrolene is antiarrhythmic, prevents further pathological remodeling, and improves cardiac function in chronic ischemic heart disease. J. Mol. Cell. Cardiol. 2023, 181, 67. 10.1016/j.yjmcc.2023.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Schmeckpeper J.; Kim K.; George S. A.; Blackwell D.; Brennan J. A.; Efimov I. R.; Knollmann B. C. RyR2 inhibition with dantrolene is antiarrhythmic, prevents further pathological remodeling, and improves cardiac function in chronic ischemic heart disease. J. Mol. Cell. Cardiol. 2023, 181, 67. 10.1016/j.yjmcc.2023.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opbergen C. J. M. v.; Bagwan N.; Maurya S. R.; Kim J.-C.; Smith A. N.; Blackwell D. J.; Johnston J. N.; Knollmann B. C.; Cerrone M.; Lundby A.; Delmar M. Exercise Causes Arrhythmogenic Remodeling of Intracellular Calcium Dynamics in Plakophilin-2-Deficient Hearts. Circulation 2022, 145, 1480–1496. 10.1161/CIRCULATIONAHA.121.057757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryshtal D. O.; Blackwell D. J.; Egly C. L.; Smith A. N.; Batiste S. M.; Johnston J. N.; Laver D. R.; Knollmann B. C. RYR2 Channel Inhibition Is the Principal Mechanism of Flecainide Action in CPVT. Circ. Res. 2021, 128, 321. 10.1161/CIRCRESAHA.120.316819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gochman A.; Do T. Q.; Kim K.; Schwarz J. A.; Thorpe M. P.; Blackwell D. J.; Ritschel P.; Smith A. N.; Rebbeck R. T.; Akers W. S.; Cornea R. L.; Laver D. R.; Johnston J. N.; Knollmann B. C. ent-Verticilide B1 inhibits type 2 ryanodine receptor channels and is antiarrhythmic in Casq2–/– mice. Mol. Pharmacol. 2024, 105, 194. 10.1124/molpharm.123.000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukawa H.; Murayama T. Development of Ryanodine Receptor (RyR) Inhibitors for Skeletal Muscle and Heart Diseases. Juntendo Medical Journal 2023, 69, 180. 10.14789/jmj.JMJ22-0045-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka M.; Kodama M.; Murayama T.; Ishigami-Yuasa I.-Y.; Mori S.; Ishida R.; Suzuki J.; Kanemaru K.; Sugihara M.; Iino I.; Miura A.; Nishio H.; Morimoto S.; Kagechika H.; Sakurai T.; Kurebayashi N. Screening for Novel Type 2 Ryanodine Receptor Inhibitors by Endoplasmic Reticulum Ca Monitoring. Mol. Pharmacol. 2023, 104, 275–286. 10.1124/molpharm.123.000720. [DOI] [PubMed] [Google Scholar]
- Mitronova G. Y.; Quentin C.; Belov V. N.; Wegener J. W.; Kiszka K. A.; Lehnart S. E. 1,4-Benzothiazepines with Cyclopropanol Groups and Their Structural Analogues Exhibit Both RyR2-Stabilizing and SERCA2a-Stimulating Activities. J. Med. Chem. 2023, 66, 15761. 10.1021/acs.jmedchem.3c01235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell D. J.; Smith A. N.; Do T.; Gochman A.; Schmeckpeper J.; Hopkins C. R.; Akers W. S.; Johnston J. N.; Knollmann B. C. In Vivo Pharmacokinetic and Pharmacodynamic Properties of the Antiarrhythmic Molecule ent-Verticilide. J. Pharmacol. Exp. Ther. 2023, 385, 205. 10.1124/jpet.122.001455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. N.; Blackwell D. J.; Knollmann B. C.; Johnston J. N. Ring Size as an Independent Variable in Cyclooligomeric Depsipeptide Antiarrhythmic Activity. ACS Med. Chem. Lett. 2021, 12, 1942–1947. 10.1021/acsmedchemlett.1c00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a companion study focusing on backbone contributions to passive permeability, see:; Thorpe M. P.; Smith A. N.; Blackwell D. J.; Hopkins C. R.; Knollmann B. C.; Akers W. S.; Johnston J. N.. The Backbone Constitution Drives Passive Permeability Independent of Side Chains in Depsipeptide and Peptide Macrocycle 2024.
- Batiste S. M.; Johnston J. N. Rapid synthesis of cyclic oligomeric depsipeptides with positional, stereochemical, and macrocycle size distribution control. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 14893. 10.1073/pnas.1616462114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leighty M. W.; Shen B.; Johnston J. N. Enantioselective Synthesis of α-Oxy Amides via Umpolung Amide Synthesis. J. Am. Chem. Soc. 2012, 134, 15233. 10.1021/ja306225u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. N.; Johnston J. N. The Formation of Impossible Rings in Macrocyclooligomerizations for Cyclodepsipeptide Synthesis: The 18-from-12 Paradox. J. Org. Chem. 2021, 86, 7904. 10.1021/acs.joc.0c03069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gissot A.; N’Gouela S.; Matt C.; Wagner A.; Mioskowski C. NaNO2-mediated transformation of aliphatic secondary nitroalkanes into ketones or oximes under neutral, aqueous conditions: How the nitro derivative catalyzes its own transformation. J. Org. Chem. 2004, 69, 8997. 10.1021/jo0489824. [DOI] [PubMed] [Google Scholar]; b Ballini R.; Petrini M. The Nitro to Carbonyl Conversion (Nef Reaction): New Perspectives for a Classical Transformation. Adv. Synth. Catal. 2015, 357, 2371. 10.1002/adsc.201500008. [DOI] [Google Scholar]
- a Shen B.; Makley D. M.; Johnston J. N. Umpolung reactivity in amide and peptide synthesis. Nature 2010, 465, 1027. 10.1038/nature09125. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shackleford J. P.; Shen B.; Johnston J. N. Discovery of competing anaerobic and aerobic pathways in umpolung amide synthesis allows for site-selective amide 18O-labeling. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 44. 10.1073/pnas.1113553108. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Makley D. M.; Johnston J. N. Silyl Imine Electrophiles in Enantioselective Catalysis: A Rosetta Stone for Peptide Homologation, Enabling Diverse N-Protected Aryl Glycines from Aldehydes in Three Steps. Org. Lett. 2014, 16, 3146. 10.1021/ol501297a. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Schwieter K. E.; Johnston J. N. Enantioselective synthesis of D-α-amino amides from aliphatic aldehydes. Chem. Sci. 2015, 6, 2590. 10.1039/C5SC00064E. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Crocker M. S.; Foy H.; Tokumaru K.; Dudding T.; Pink M.; Johnston J. N. Direct Observation and Analysis of the Halo-Amino-Nitro Alkane Functional Group. Chem. 2019, 5, 1248. 10.1016/j.chempr.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Crocker M. S.; Deng Z.; Johnston J. N. Preparation of N-Aryl Amides by Epimerization-Free Umpolung Amide Synthesis. J. Am. Chem. Soc. 2022, 144, 16708. 10.1021/jacs.2c05986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecchini P.; Fochi M.; Bartoccini F.; Piersanti G.; Bernardi L. Enantioselective organocatalytic strategies to access noncanonical α-amino acids. Chem. Sci. 2024, 15, 5832–5868. 10.1039/D4SC01081G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conventional compound numbering is used, but a single letter is added for clarity to denote a compound as a Monomer, Didepsi/peptide, Tetradepsi/peptide, Hexadepsi/peptide, or Octadepsipeptide.
- Kogami Y.; Nakajima T.; Ikeno T.; Yamada T. Enantioselective Henry Reaction Catalyzed by Salen-Cobalt Complexes. Synthesis 2004, 12, 1947–1950. 10.1055/s-2004-829157. [DOI] [Google Scholar]
- a Engberts J. B. F. N.; Strating J. The mannich condensation of sulfinic acids, aldehyde, and ethyl carbamate II.: The use of higher aldehydes. Recl. Trav. Chim. Pays-Bas 1965, 84, 942. 10.1002/recl.19650840714. [DOI] [Google Scholar]; b Petrini M. α-Amido Sulfones as Stable Precursors of Reactive N-Acylimino Derivatives. Chem. Rev. 2005, 105, 3949. 10.1021/cr050528s. [DOI] [PubMed] [Google Scholar]
- a Petrini M.; Torregiani E. Recent Advances in Stereoselective Syntheses Using N-Acylimines. Synthesis 2007, 2007, 159. 10.1055/s-2006-958957. [DOI] [Google Scholar]; b Petrini M.; Torregiani E. Aza-Henry reaction of substituted nitro alkanes using a-formamido aryl sulfones as N-acylimino equivalents. Tetrahedron Lett. 2006, 47, 3501. 10.1016/j.tetlet.2006.03.114. [DOI] [Google Scholar]; c Marcantoni E.; Palmieri A.; Petrini M. Recent synthetic applications of α-amido sulfones as precursors of N-acylimino derivatives. Org. Chem. Front. 2019, 6, 2142. 10.1039/C9QO00196D. [DOI] [Google Scholar]
- Thorpe M. P.; Smith A. N.; Crocker M. S.; Johnston J. N. Resolving Bromonitromethane Sourcing by Synthesis: Preparation at the Decagram Scale. J. Org. Chem. 2022, 87, 5451. 10.1021/acs.joc.2c00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Davis T. A.; Dobish M. C.; Schwieter K. E.; Chun A. C.; Johnston J. N. Preparation of H,4PyrrolidineQuin-BAM (PBAM). Org. Synth. 2012, 89, 380–393. 10.15227/orgsyn.089.0380. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nugent B. M.; Yoder R. A.; Johnston J. N. Chiral proton catalysis: A catalytic enantioselective direct aza-Henry reaction. J. Am. Chem. Soc. 2004, 126, 3418. 10.1021/ja031906i. [DOI] [PubMed] [Google Scholar]; c Davis T. A.; Wilt J. C.; Johnston J. N. Bifunctional Asymmetric Catalysis: Amplification of Bronsted Basicity Can Orthogonally Increase the Reactivity of a Chiral Bronsted Acid. J. Am. Chem. Soc. 2010, 132, 2880. 10.1021/ja908814h. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Schwieter K. E.; Johnston J. N. Enantioselective Addition of Bromonitromethane to Aliphatic N-Boc Aldimines Using a Homogeneous Bifunctional Chiral Organocatalyst. ACS Catal. 2015, 5, 6559. 10.1021/acscatal.5b01901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobish M. C.; Villalta F.; Waterman M. R.; Lepesheva G. I.; Johnston J. N. Organocatalytic, Enantioselective Synthesis of VNI: A Robust Therapeutic Development Platform for Chagas, a Neglected Tropical Disease. Org. Lett. 2012, 14, 6322. 10.1021/ol303092v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurelio L.; Brownlee R. T. C.; Hughes A. B. Synthetic Preparation of N-Methyl-α-amino Acids. Chem. Rev. 2004, 104, 5823. 10.1021/cr030024z. [DOI] [PubMed] [Google Scholar]
- Kurzer F.; Douraghi-Zadeh K. Advances in the Chemistry of Carbodiimides. Chem. Rev. 1967, 67, 107. 10.1021/cr60246a001. [DOI] [PubMed] [Google Scholar]
- Throughout these studies, no evidence was observed for epimerization in an amide coupling step.
- Al-Warhi T. I.; Al-Hazimi H. M. A.; El-Faham A. Recent development in peptide coupling reagents. J. Saudi Chem. Soc. 2012, 16, 97. 10.1016/j.jscs.2010.12.006. [DOI] [Google Scholar]
- El-Faham A.; Albericio F. Peptide Coupling Reagents, More than a Letter Soup. Chem. Rev. 2011, 111, 6557. 10.1021/cr100048w. [DOI] [PubMed] [Google Scholar]
- Paquette A. R.; Boddy C. N. Macrocyclization strategies for the total synthesis of cyclic depsipeptides. Org. Biomol. Chem. 2023, 21, 8043–8053. 10.1039/D3OB01229H. [DOI] [PubMed] [Google Scholar]
- Cheng H.; Lederer M. R.; Lederer W. J.; Cannell M. B. Calcium sparks and [Ca2+]i waves in cardiac myocytes. American Journal of Physiology-Cell Physiology 1996, 270, C148. 10.1152/ajpcell.1996.270.1.C148. [DOI] [PubMed] [Google Scholar]
- Smith A. N.; Thorpe M. P.; Blackwell D. J.; Batiste S. M.; Hopkins C. R.; Schley N. D.; Knollmann B. C.; Johnston J. N. Structure–Activity Relationships for the N-Me- Versus N-H-Amide Modification to Macrocyclic ent-Verticilide Antiarrhythmics. ACS Med. Chem. Lett. 2022, 13, 1755. 10.1021/acsmedchemlett.2c00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangborn A. B.; Giardello M. A.; Grubbs R. H.; Rosen R. K.; Timmers F. J. Safe and Convenient Procedure for Solvent Purification. Organometallics 1996, 15, 1518. 10.1021/om9503712. [DOI] [Google Scholar]
- Tomek J.; Nieves-Cintron M.; Navedo M. F.; Ko C. Y.; Bers D. M. SparkMaster 2: A New Software for Automatic Analysis of Calcium Spark Data. Circ. Res. 2023, 133, 450. 10.1161/CIRCRESAHA.123.322847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikkel M. B.; Francis D. P.; Howard J.; Gordon F.; Rowlands C.; Peters N. S.; Lyon A. R.; Harding S. E.; MacLeod K. T. Hierarchical statistical techniques are necessary to draw reliable conclusions from analysis of isolated cardiomyocyte studies. Cardiovasc. Res. 2017, 113, 1743. 10.1093/cvr/cvx151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.