

Abstract
Background
Androgen deprivation therapy (ADT) is a front-line treatment for prostate cancer. In some men, their tumors can become refractory leading to the development of castration-resistant prostate cancer (CRPC). This causes tumors to regrow and metastasize, despite ongoing treatment, and impacts negatively on patient survival. ADT is known to stimulate the accumulation of immunosuppressive cells like protumoral tumor-associated macrophages (TAMs), myeloid-derived suppressor cells and regulatory T cells in prostate tumors, as well as hypofunctional T cells. Protumoral TAMs have been shown to accumulate around tumor blood vessels during chemotherapy and radiotherapy in other forms of cancer, where they drive tumor relapse. Our aim was to see whether such perivascular (PV) TAMs also accumulate in ADT-treated prostate tumors prior to CRPC, and, if so, whether selectively inducing them to express a potent immunostimulant, interferon beta (IFNβ), would stimulate antitumor immunity and delay CRPC.
Methods
We used multiplex immunofluorescence to assess the effects of ADT on the distribution and activation status of TAMs, CD8+T cells, CD4+T cells and NK cells in mouse and/or human prostate tumors. We then used antibody-coated, lipid nanoparticles (LNPs) to selectively target a STING agonist, 2′3′-cGAMP (cGAMP), to PV TAMs in mouse prostate tumors during ADT.
Results
TAMs accumulated at high density around blood vessels in response to ADT and expressed markers of a protumoral phenotype including folate receptor-beta (FR-β), MRC1 (CD206), CD169 and VISTA. Additionally, higher numbers of inactive (PD-1-) CD8+T cells and reduced numbers of active (CD69+) NK cells were present in these PV tumor areas. LNPs coated with an antibody to FR-β selectively delivered cGAMP to PV TAMs in ADT-treated tumors, where they activated STING and upregulated the expression of IFNβ. This resulted in a marked increase in the density of active CD8+T cells (along with CD4+T cells and NK cells) in PV tumor areas, and significantly delayed the onset of CRPC. Antibody depletion of CD8+T cells during LNP administration demonstrated the essential role of these cells in delay in CRPC induced by LNPs.
Conclusion
Together, our data indicate that targeting a STING agonist to PV TAMs could be used to extend the treatment window for ADT in prostate cancer.
Keywords: prostate cancer, immunotherapy, macrophage
WHAT IS ALREADY KNOWN ON THIS TOPIC.
WHAT THIS STUDY ADDS
This study is the first to demonstrate that FR-β+ macrophages with an immunosuppressive phenotype accumulate around blood vessels in mouse and human prostate tumors during ADT, prior to the onset of CRPC. Lipid nanoparticles coated with an antibody to FR-β+ were then used to deliver a STING agonist selectively to these perivascular (PV) cells during ADT. This triggered STING signaling and the release of the potent immunostimulant, interferon beta, by PV macrophages, which then activated tumor-infiltrating CD8+T cells and delayed the onset of CRPC.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The delivery of an immunostimulant specifically to PV regions of tumors represents a new, more targeted form of immunotherapy that ensures the activation of T cells as soon as they cross the vasculature into tumors. This new approach could be used to extend the treatment window for ADT in men with prostate cancer. In doing so, it would delay/circumvent the need for additional treatments like radiotherapy and/or or chemotherapy.
Introduction
Prostate cancer is the second most common cancer in men. Androgens are known to stimulate the progression of this disease via androgen receptors (ARs) overexpressed on cancer cells. This prompted the development of drugs that either reduce/ablate androgen synthesis in the testes or reduce AR signaling. Collectively, these anti-androgens are known as androgen deprivation therapy (ADT).1
Men with prostate cancer are offered ADT at various stages of their disease. Although this initially reduces tumor burden, some patients develop resistance to ADT, a condition called castration resistant prostate cancer (CRPC). Despite the development of various treatment options for CRPC, including androgen/AR signaling inhibitors such as abiraterone and enzalutamide, and PARP inhibitors, the median survival rate for men with metastatic CRPC remains poor at under 3 years.2,4 This highlights the unmet clinical need for novel therapies that can prevent/delay the emergence of CRPC.
Mechanistically, the emergence of CRPC has been strongly linked to aberrant AR signaling, defective DNA damage/repair pathways, and the intratumoral synthesis of androgens.3 However, AR-independent mechanisms can also facilitate CRPC, including loss of function mutations in the tumor suppressor genes, p53 and PTEN, and deregulation of the FGF, TGFβ, RAS/MAPK, hedgehog and Wnt/β-catenin signaling pathways. Although therapeutic agents that target one or more of the above have shown promise in the clinic, outcomes remain poor.4
ADT has been shown to increase the frequency of various immune effector cells in both mouse and human prostate tumors.5,8 For example, several reports have shown that tumor infiltration by CD8+ Τ cells increases with ADT, although this fails to impact favorably on relapse or survival as these cells have reduced cytotoxic function.5,8 Additionally, ADT has been shown to impair T cell priming by antigen-presenting cells and T cell expression of cytotoxicity-related genes (granzymes and perforins).59,11 It is also reported to increase the intratumoral abundance of various immunosuppressor cell types including tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and regulatory T cells (Tregs).512,14
Agonists of stimulator of interferon genes (STING)-signaling pathway in cells are emerging as an exciting form of immunotherapy for cancer.15 16 These include factors that activate STING like cyclic guanosine monophosphate (GMP)–AMP synthase (cGAS) or cyclic dinucleotide 2′3′-cGAMP (cGAMP).17 Activation of STING in cells causes them to upregulate type I interferons (IFNs), including IFNs αandβ.18 In tumors, the latter have been shown to stimulate antitumor immunity via multiple mechanisms including the cross-priming and activation of CD8+T cells by antigen presenting cells, as well as the activation of both CD4+T cells and NK cells.19 20 However, the clinical efficacy of STING agonists administered systemically is compromised by their rapid excretion, low bioavailability, lack of specificity, and adverse, off-target, side effects.21 Furthermore, intratumoral administration is limited by the accessibility of tumors. To overcome these limitations, a number of delivery systems have been used to provide protection for STING agonists in the circulation and enable them to access tumors. These include their encapsulation in lipid nanoparticles (LNPs), exosomes and bacterial vectors.22
In the present study, we show that ADT causes a marked change in the immune landscape immediately adjacent to blood vessels in both mouse and human prostate tumors just prior to the onset of CRPC. For example, a marked increase in the density of protumoral TAMs and naïve (PD-1−) CD8+T cells was seen in these perivascular (PV) areas, indicating the ability of ADT to promote an immunosuppressive, PV niche.
We reasoned that the systemic targeting of a STING agonist LNPs to such protumoral, PV TAMs would be effective due to their close proximity to tumor blood vessels. So, we synthesized LNPs containing cGAMP and coated them with an antibody raised against a receptor we show is expressed by protumoral PV TAMs in ADT-treated tumors—folate receptor-beta (FR-β). Following systemic administration of these LNPs to ADT-treated mice bearing orthotopic prostate tumors, cGAMP was selectively delivered to PV TAMs, triggering STING signaling and their upregulation of IFNβ. This resulted in the activation of tumor-infiltrating CD8+T cells (as well as other effectors like CD4+T cells and NK cells), and delayed the onset of CRPC.
Methods
Orthotopic mouse models of primary prostate cancer
Myc-CaP implants
Myc-CaP cells23 stably expressing luciferase were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin and were tested regularly for mycoplasma. 50,000 Myc-CaP cells (1:1 PBS:Matrigel) were injected into the dorsal prostate lobe of male FVB mice (aged 6–8 weeks). Mice with detectable tumors 7 days later were randomized into experimental groups and injected subcutaneously (once on day 7 alone or with a second dose on day 14) with either vehicle control (PBS) or degarelix (25 mg/kg) (Ferring Pharmaceuticals, Parsippany, New Jersey, USA) (n=5–8/treatment arm). Tumor growth was then monitored every 2–3 days using an IVIS Spectrum imaging system (ie, 30 min after injection with 90 mg/kg d-luciferin).
All Myc-CaP experiments were carried out in compliance with UK Home Office Regulations as specified in the Animals (Scientific Procedures) Act of 1986 and were approved by the Animal Welfare and Ethical Review Body of the University of Sheffield.
Transgenic Pten-deficient mice
PBiCre+/−;Ptenfl/fl mice were generated as described previously.24 At 200 days of age, mice were randomly assigned to sham-castrated or castrated groups. Prostate tumors were harvested 2 weeks after surgery, snap frozen in liquid nitrogen, and optimum cutting temperature (OCT)-embedded before being cryosectioned.
Transgenic mouse experiments adhered to the guidelines outlined in the National Health and Medical Research Council Australian Code of Practice for the Care and Use of Animals for Scientific Purposes and were approved by the Animal Experimentation Ethics Committee at the Peter MacCallum Cancer Centre.
Quantitative immunofluorescence staining of mouse prostate tumors
Cryosections of mouse prostate tumors were fixed with ice-cold acetone for 10 min, blocked with 5% goat serum and 10% mouse FcR blocking solution before being incubated for 40 min with primary antibodies; F4/80 (BIO-RAD, 1:100), FR-β (BioLegend, 1:100), CD169 (BioLegend, 1:100), VISTA (BioLegend, 1:100), CD206 (MRC1) (BioLegend, 1:100), CD31 (BioLegend, 1:100), CD8 (BioLegend, 1:100), PD-1 (BioLegend, 1:100), phopho-STING (ThermoFisher 1:100), NK-1.1 (Biolegend 1:100), CD69 (Biolegend 1:100), CD4 (Biolegend 1:100), and IFNbeta (Invitrogen, 1:100). Primary rabbit antibodies were detected using goat anti-rabbit (IgG) Alexa Fluor 555 (Fisher Scientific 1:400). All sections were counter-stained with 50 ng/mL DAPI solution before washing and mounting.
A Nikon A1 confocal microscope was used to capture five randomly selected images/tumor (×20 magnification). The acquired images were subsequently analyzed using QuPath (V.0.4.3) or ImageJ. For analysis of cell density relative to blood vessels, cells <15 µm from CD31+blood vessels were defined as PV. These were counted in each regions of interest (ROI) and divided by its vessel area to estimate the PV density/ROI. Cells>15 µm from CD31+blood vessels were defined as non-PV, the density of which was calculated by dividing cell numbers in these areas of a given ROI, by the total non-PV area of that ROI.
Human primary prostate tumors
Sections were cut from localized, human prostate tumors removed from men in the following two groups (supplemental Table):
Matched pretreatment biopsies and post-treatment, radical prostatectomies (RPs) from 20 patients who received neoadjuvant ADT for 6 months at the Dana-Farber Cancer Institute, Boston, USA. Additionally, pathological staging of prostatectomy samples (ie, after ADT) enabled us to divide these into “responder” or “non-responder” (NR) groups. Responders were defined as patients with residual prostate cancer of ≤5 mm, T stage=0–2, after ADT. Whereas NRs had tumors that were T stage ≥3 a after ADT (and so were starting to regrow and show signs of CRPC). There was no regional lymph node involvement in either group. These sections were used to assess the coexpression of CD68 and FR-β in PV versus non-PV tumor areas in matched tumor samples before and after ADT. An antibody to CD31 was also used to label blood vessels.
Six “untreated” tumors (ie, not given ADT; four prostatectomies and two transurethral resections of the prostate or “TURPs”), and five TURPs from patients who received ADT (median treatment time, 7 months). Three patients in the latter group had draining lymph node involvement (ie, early metastasis). Both groups of samples were supplied by The Institute of Cancer Research, London. These sections were used to assess T cell subsets relative to CD31+ blood vessels.
Quantitative immunofluorescence staining of human prostate tumors
Antigen retrieval was performed using a pressure cooker in conjunction with Dako 10X retrieval solution (S1699). Subsequently, the sections were blocked to reduce non-specific binding using 10% goat serum and 1% TBS (at room temperature for 30 min.).
For tumor group 1, sections were incubated in primary antibodies diluted in 1% bovine serum albumen (BSA) overnight at 4°C, then washed x3 in TBS, followed by secondary antibodies at room temperature for 1 hour and DAPI for 3 min, before washing and mounting. The primary antibodies used were a mouse monoclonal (IgG3) anti-human CD68 (Dako, Clone PG-M1 1:200), a mouse monoclonal (IgG1) anti-CD31 (Antibodies.com, Clone JC/70A 1:100) and a rabbit polyclonal anti-FR-β (Abcam 1:300). The negative controls for these primary antibodies were species, isotype and concentration-matched antibodies. The secondary antibodies were a goat anti-mouse IgG3-Alexa Fluor 488 (Fisher Scientific 1:200), a goat anti-mouse IgG1-Alexa Fluor 647 (ThermoFisher 1:200) and a goat anti-rabbit IgG-Alexa Fluor 555 (Fisher Scientific 1:100).
For tumor group 2, the “MultiOmyx” procedure was used to detect CD8, PD-1 and CD31 as described by us previously25 in untreated and ADT-treated tumors. The antibodies used were mouse anti-human CD8 (Dako), a rabbit anti-human PD-1 (Abcam), and a mouse anti-human CD31/PECAM-1 (Cell Signaling). All three were conjugated to fluorophores and diluted with 3% (wt/vol) BSA (to working concentrations optimized previously.25 They were applied to sections for 1 hour at RT, then washed in PBS and high-resolution images collected from 20 ROIs across viable tumor areas using a ×20 objective on an INCell analyzer 2200 microscope (GE Healthcare Life Sciences). An AI-based, advanced analytics platform, proprietary to NeoGenomics Labs, called “NeoLYTX”, was used to quantify and analyze subsets of PD-1−CD8+ T cells and PD-1+CD8+ T cells, and CD31+blood vessels in PV (<15 µm from CD31+blood vessels) and non-PV (>15 µm from CD31+blood vessels) areas of tumors.
Generation of LNPs to selectively target PV TAMs in vivo
LNPs containing the STING (STimulator of IFN Genes) agonist, cGAMP, or an inert version of this molecule and coated with one of two antibodies, a rat anti-mouse FR-β or a rat IgG2a (isotype matched), control antibody (both from BioLegend) were synthesized by LipExoGen Biotech (as described in online supplemental figure 6A).
For this, 20 mg/mL D-Lin-MC3-DMA (MC3, MedChemExpress) in ethanol, was mixed 1:1 (v/v) with 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC, Avanti Polar Lipids), cholesterol (Chol, Avanti Polar Lipids), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)- 2000] (DSPE-PEG2000, Avanti Polar Lipids) and OG488 DHPE (AAT Bioquest) in ethanol. The molar ratios of the components for the base formulation were as follows: MC3/Chol/DSPC/DSPE-PEG2000/DHPE-OG488 (50/38.5/9.8/1.5/0.2, mol/mol). cGAMP (cGAMP, InvivoGen, tlrl-nacga23) or its inactive control (InvivoGen, tlrl-nagpap) were dissolved in UltraPure water (Invitrogen) and diluted in acetate buffer, pH 4. LNPs were synthesized using a 3:1 aqueous:organic ratio and subsequently washed in UltraPure water using Amicon centrifugal columns (100 kDa). The encapsulation efficiency of cGAMP (or its inactive control) was determined by HPLC.
Prior to the functionalization of the LNPs with antibodies, 1 mol% of DSPE-PEG2000 was postinserted into LNPs. Fc-specific labeling (ie, to ensure antibody attachment to LNPs in the correct orientation for binding to FR-β on cells) was achieved by performing a click reaction between the DSPE-PEG2000-DBCO (BroadPharm) and terminal GlcNAz residues on the antibody carbohydrate domain. To obtain the latter, the SiteClick Antibody Azido Modification Kit (Invitrogen) was used to replace terminal galactose residues on the N-linked sugars in the Fc region with the azide-containing sugar GalNAz, which is reactive towards DBCO through strain-promoted alkyne-azide cycloaddition. The lipidated antibodies in DPBS(1X) were postinserted into the LNPs according to the methods described by Swart et al.26 Finally, an additional 2 mol% DSPE-PEG2000 was postinserted into the LNPs via thin film hydration to minimize non-specific cellular uptake. The inclusion in LNPs of a phospholipid labeled on the head group with the bright, green-fluorescent fluorinated fluorescein dye (also called Oregon Green 488), “DHPE-OG488”, will enable LNP uptake/distribution in tumors to be tracked by in vivo.
To test the effects of LNPs in vivo, mice received either a subcutaneous (s.c) injection of PBS or degarelix in PBS (“ADT”, Ferring Pharmaceuticals, Parsippany, New Jersey, USA); alone 7 days after inoculation (5–8 mice/group), then divided into the following groups (with LNPs administered by s.c. injection every 2 days starting 2 days after PBS or ADT): (1) control—that is, PBS, alone (no LNPs); (2) PBS plus FR-β-antibody coated LNPs containing inactive (control) cGAMP (called “LNP(C)” in some figures); (3) PBS plus FRβ-antibody-coated plus LNPs containing active cGAMP (called “LNP(E)” in some figures); (4) degarelix alone (no LNPs); (5) degarelix plus FRβ-antibody coated LNPs containing inactive cGAMP; (6) degarelix plus FR-β-antibody coated LNPs containing active cGAMP; (7) degarelix plus control rat IgG-antibody coated LNPs containing inactive 2’3-cGAMP; (8) degarelix plus control rat IgG-antibody coated LNPs containing active 2’3-cGAMP and (9) degarelix plus LNPs with no antibody coating but containing active 2’3-cGAMP.
A further two groups of mice were also included—to examine the role of CD8+T cells in mediating the effects of FR-β-antibody coated LNPs containing active cGAMP on the onset of CRPC after ADT. For these, mice were administered i.p. injections of 200 µg of anti-CD8 antibody (or an isotype control IgG, both supplied by BioXcell). These started 2 days before inoculation with Myc-CaP-LUC cells, and then proceded every 4 days throughout the course of tumor growth.
Mice were monitored daily for tumor growth using luminometry and well-being (including body weight) every 2–3 days. Tumors were snap frozen prior to being embedded in OCT compound for frozen sectioning.
Flow cytometry
Myc-CaP cells were detached from 6-well plates using trypsin/EDTA, washed and resuspended in the following primary antibodies: a rat monoclonal (IgG2a) anti-mouse FR-β (BioLegend,1:100) or a sheep polyclonal anti-mouse FR-α (R&D Systems, 1:100) for 45–60 min. Cells were then washed twice and resuspended in FACS buffer. Flow cytometry was performed using the BD LSR II flow cytometer and data processed using FlowJo Software.
Statistical analysis
All data shown are means±SEMs. Dots on jitter plots represent values for individual tumors. All data were analyzed using GraphPad Prism V.8.02 software. Data analysis was conducted blind and statistical analysis performed using the Mann-Whitney U-test with p values of <0.05 considered to be significant.
Results
ADT induces PV accumulation of protumoral TAMs in mouse and human prostate tumors
The effects of ADT on the distribution and phenoptype of TAMs were investigated just prior to CRPC in the immunocompetent Myc-CaP model. It caused an initial reduction in tumor burden within the first 7 days of treatment but then started to regrow (ie, acquire CRPC) 7–10 days post-ADT treatment (figure 1A). Sections from tumors harvested on day 17 from both control and ADT-treated mice were coimmunofluorescently stained for the macrophage marker F4/80 and the endothelial cell marker CD31. While both PV and non-PV F4/80+TAMs were increased by ADT relative to the control group (figure 1B,C), a significantly greater increase was seen in PV TAMs. Elevated PV F4/80+TAMs were also observed prior to the acquisition of CRPC in a Pten-deficient transgenic mouse model of prostate cancer (PBiCre+/−;Ptenfl/fl)23 2 weeks postsurgical castration, compared with sham castration (online supplemental file 4).
Figure 1. ADT stimulates the PV accumulation of FR-β+ TAMs in mouse (Myc-CaP) and localized, human prostate tumors. (A) Two phases of tumor response to the LHRH antagonist, degarelix, in Myc-CaP tumors: an initial, hormone sensitive (HS) period of tumor growth inhibition followed by the start of castration resistance (CR) when tumors start to regrow. In Myc-CaP tumors (B–E), immunofluorescence staining shows that the density of both PV F4/80+TAMs (B, C)—and the FR-β+ subset of these cells (D, E) increased by the start of CR in ADT-treated tumors. Similar changes occurred in non-PV tumor areas but to a lesser extent than in PV areas. The proportion of F4/80+TAMs expressing FR-β also increased in PV and non-PV areas at this time. Immunofluorescence staining of matched human prostate tumors (F, H) sampled before and after ADT showed that ADT increased the PV density of PV CD68+ tumors (G) and the CD68+TAM subset expressing FR-β (I, left panel). The proportion of CD68+TAMs expressing FR-β rose in PV and non-PV tumor areas after ADT (I, right panel). (NPV=non-PV). Data are presented as means±SEMs. All fluorescence images are of ADT-treated tumors (except the top one in panel B). *p<0.05, **p<0.01, ***p<0.001. Magnification bars=50 µm. ADT, androgen deprivation therapy; LHRH, Luteinising hormone-releasing hormone; PV, perivascular; TAM, tumor-associated macrophage.

To determine if ADT also alters macrophage phenotype, we interrogated the phenotype of F4/80+TAMs in PV and non-PV areas post-ADT using a panel of well-characterized markers of protumoral macrophages, including folate receptor beta (FR-β), CD169 (SIGLEC1), V-domain immunoglobulin suppressor of T cell activation (VISTA) and MRC1 (CD206), (figure 1D,E and online supplemental file 2).27,30 Strikingly, the density of F4/80+ TAMs coexpressing each of the tumor-promoting cell surface macrophage markers analyzed in PV regions were significantly upregulated on ADT relative to the control. An increase in TAMs expressing these markers was also observed in non-PV regions but was significantly lower relative to than in PV regions (figure 1D,E and online supplemental file 2). Collectively, these findings indicate that ADT promotes the accumulation of PV protumor TAMs.
Given that both the densities and proportions of PV F4/80+TAMs expressing FR-β, CD169, VISTA or MRC1 were virtually identical (online supplemental file 2), we investigated whether the same PV TAMs coexpressed these markers. Multiplex immunofluorescence staining for F4/80, FR-β, CD169 and VISTA confirmed that these markers were expressed by the same TAM subset. This showed that ADT treatment induces a subset of TAMs with a distinct protumoral phenotype in Myc-CaP tumors at the onset of CRPC, with significantly higher frequency in PV areas (online supplemental file 3).
To establish if the observed induction of protumoral PV TAMs was specific to androgen deprivation in the Myc-CaP model, immunofluorescence staining was performed to colocalize F4/80 and MRC1 in primary prostate tumors from PBiCre+;Ptenfl/fl mice, 2 weeks postsurgical castration. A similar increase in the PV density of MRC1+F4/80+TAMs was observed (online supplemental file 4). PV F4/80+TAMs also expressed FR-β following castration (online supplemental file 4).
To investigate the clinical relevance of the above findings, we first examined matching human prostate tumor specimens collected before and after ADT for the human macrophage marker, CD68 and CD31. Immunofluorescence analysis revealed that while the density of CD68+TAMs was significantly higher in PV than non-PV areas before and after ADT, their PV density increased further after ADT (figure 1F,G).
The density of PV and non-PV FR-β+CD68+TAMs was significantly higher after ADT than before, but this effect of ADT was significantly greater in PV areas (figure 1H&I).
We then investigated whether CD68+TAM distribution correlated with tumor responses to ADT by dividing patients who received this treatment for 6 months into those that showed increased tumor growth during ADT (ie, were “NRs”) or did not grow (ie, were responders, “Rs”) (online supplemental file 5). There were no differences in CD68+TAMs between Non-Rs and Rs, before and after ADT. While the FR-β+ subset of CD68+TAMs showed a similar PV location to TAMs labeled for CD68 alone (ie, both before and after ADT), the density of PV FR-β+CD68+ TAMs was significantly higher for NRs than R’s before ADT. There was a non-significant (p=0.07) trend for PV FR-β+CD68+ TAMs to also be higher in NRs than Rs after ADT (online supplemental file 5). The non-PV density of these TAMs was also higher in NRs than Rs before ADT but this was significantly lower than in PV areas (online supplemental file 5). These data confirm the abundance of protumoral (FR-β+) TAMs in PV areas of human prostate tumors after ADT, especially those entering CRPC (ie, NRs).
ADT alters the activation status of various immune effectors in PV areas of tumors
Given that protumoral TAMs were observed to increase in PV areas on ADT, we reasoned that this might lead to (or coincide with) changes in effectors cells with cytotoxic potential (CD4+and CD8+ T cells, and NK cells). To address this, we assessed the density and activation status of these immune effector cells in PV and non-PV areas of control and ADT-treated Myc-CaP tumors
We show that CD8+T cell density dramatically increases significantly in PV areas after ADT (this was also observed in non-PV regions although in a significantly smaller CD8+T cell population) (figure 2A). Interestingly, our analysis of PD-1-expression by CD8+T cells revealed that the majority (70%) of CD8+T cells accumulating at PV regions on ADT lacked this activation marker and so were antigen-naïve (figure 2B).
Figure 2. ADT stimulates the PV accumulation of PD-1-CD8+T cells in mouse (Myc-CaP) (A, B) and human (C, D) prostate tumors. (A, B). (A) Representative fluorescence images showing the presence of mainly PD-1- CD8+T cells in PV areas of ADT-treated Myc-CaP (A) and human (C) prostate tumors (left panels in both). (A, C) Yellow arrows=PD-1+CD8+T cells, orange arrows=PD-1-CD8+T cells. ADT stimulates the PV accumulation of CD8+T cells (A, C, right panels), which are mainly PD-1- (B, D left panels). The majority of CD8+T cells lack expression of PD-1 across tumors, which increases further after ADT (B, D, right panels). (NPV=non-PV). Data are presented as means±SEMs. Fluorescence images in A, C are from ADT-treated tumors. *p<0.05, **p<0.01, ***p<0.001. Magnification bars=20 µm. ADT, androgen deprivation therapy; PV, perivascular.
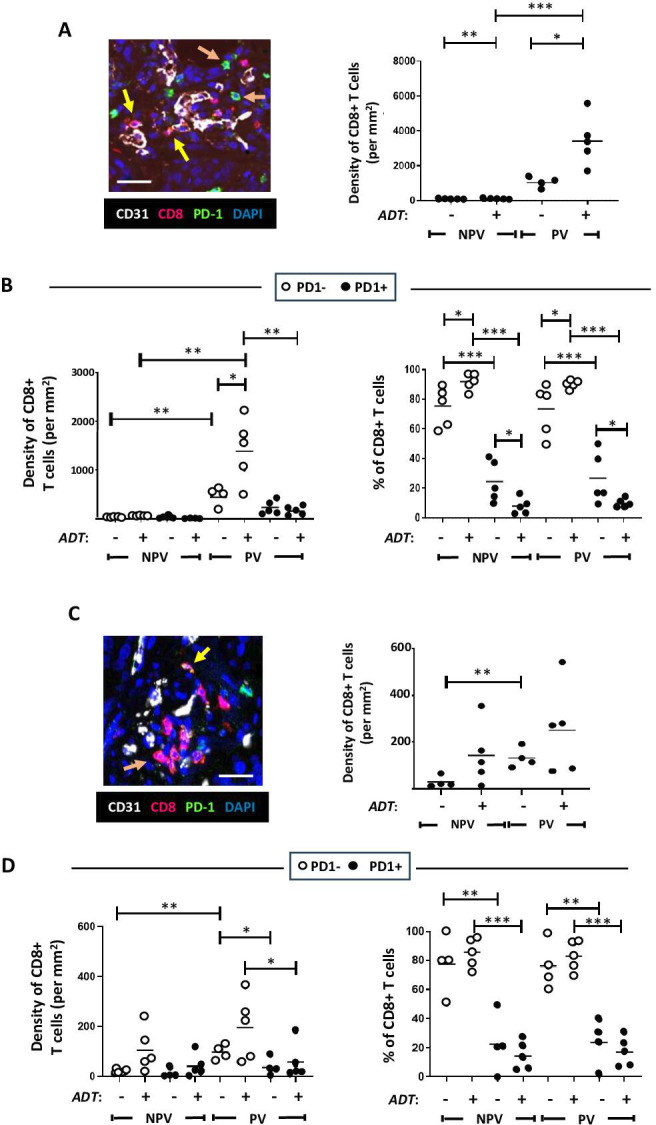
While CD8+T cells tended toward a higher density in PV than non-PV areas of both untreated and ADT-treated tumors (figure 2C, right panel), a significant increase in PD-1 negative CD8+T cells was evident in PV (but not non-PV) areas after ADT (figure 2D), resembling the Myc-CaP model. Together, these data indicate that ADT causes naive CD8+T cells to accumulate in PV areas.
Analysis of CD4+T cells in Myc-CaP prostate tumors showed that CD4+T cells also principally accumulate in PV areas of both control and ADT-treated tumors (online supplemental figure 5A). However, in both PV and non-PV areas, the density and proportion of CD4+T cells expressing PD-1 (approximately 50% in all groups) did not change during ADT (online supplemental figure 5B). So, ADT did not alter the distribution or activation status of CD4+T cells in Myc-CaP tumors.
NK cells were identified using the antibody, NK1.1, and their activation status investigated using CD69, an established marker of activation in NK cells (online supplemental figure 5C,D). NK cells were more frequent in PV than non-PV areas of both control and ADT-treated Myc-CaP tumors (online supplemental file 5C, right panel) and were almost all CD69 positive (ie, active). A similar trend was observed in non-PV areas, however, the density of NK cells was significantly lower in these regions. Of note, differences in the PV density of CD69+NK cells in ADT-treated versus control tumors failed to reach significance (p=0.056) (online supplemental figure 5D).
FR-β-targeted LNPs selectively target the STING agonist, cGAMP, to PV TAMs in ADT-treated Myc-CaP tumors, and delay CRPC
To take advantage of these immune-activating functions of the cGAMP-STING signaling pathway,18 19 we generated LNPs containing cGAMP and targeted these to PV TAMs in ADT-treated tumors by coating them with an antibody to FR-β. The two main aims of this part of the study were to investigate whether FR-β-targeted LNPs could selectively deliver cGAMP to PV TAMs in ADT-treated tumors and stimulate them to express IFNβ, and whether this would stimulate antitumor immunity and delay the onset of CRPC.
As a prelude to the LNP in vivo experiment, we first confirmed that the FR-β antibody used to coat LNPs did not bind to a related molecule, FR-α (known to be expressed by Myc-CaP cells). Flow cytometry and immunofluorescence staining using a specific FR-α antibody alongside our FR-β antibody confirmed that Myc-CaP cells express FR-α but not FR-β, and that the opposite was the case for TAMs in Myc-CaP tumors (online supplemental figure 6B,C).
Multiple, novel formulations of LNPs were synthesized containing either an active or inactive form of cGAMP and coated with either an anti-mouse FR-β antibody or a control IgG (figure 3A; see method for LNP synthesis in online supplemental file 7).
Figure 3. LNPs coated with FR-β-antibody target the STING agonist, cGAMP, to PV TAMs and delay CR in ADT-treated Myc-CaP tumors. (A) Design of LNPs used in vivo. The Fc regions of either a FR-β antibody or a control IgG were attached to LNPs containing either an active cGAMP or an inactive version of this (“cGAMP Ctrl”). (B) Tumor growth in mice administered either PBS or ADT alone, or these followed by administration every 2 days of the various forms of LNP listed. (C and D) Various key groups have been selected from (B) and shown separately (for clarity). (C) Tumor growth curves in response to PBS alone (control) versus a single dose of ADT or (D) PBS alone (control), ADT alone, ADT plus FR-β antibody-coated LNPs containing either cGAMP or cGAMP Ctrl. (E) Effect of in vivo administration of (i) an antibody against CD8 or (ii) an isotype-matched control IgG2b on tumor-infiltrating CD8+T cells after ADT plus FR-β antibody-coated LNPs containing cGAMP (magnification bar=50 µm). (iii) Tumor growth curves showing the effect of depleting CD8+T cells on tumor responses to ADT plus by FR-β antibody-coated LNPs containing cGAMP. Data are presented as means±SEMs. *p<0.001 (comparing tumor sizes at sacrifice). ADT, androgen deprivation therapy; LNP, lipid nanoparticle; TAM, tumor-associated macrophage.

Mice-bearing orthotopic Myc-CaP tumors were then administered either PBS alone (the vehicle for ADT) or a single dose of ADT alone on day seven after implantation (“ADT” group). Two days later, separate groups of control or ADT-treated mice were administered the various forms of LNPs listed in figure 3B (see box). Controls included LNPs coated with either no antibody or a control rat IgG instead of the FR-β antibody, and others containing an inactive form of cGAMP (“cGAMP Ctrl”) rather than active cGAMP. LNP injections were continued every 2 days until day 22, and tumor growth was assessed at regular intervals. None of the LNP groups appeared to have deleterious effects on the mice in terms of their eating/drinking behavior, overall health and body weight (online supplemental file 8).
The effects of these various treatments on tumor growth are shown in figure 3B (and selected groups are shown separately to facilitate comparisons in figure 3C–F). A single dose of ADT was shown to have a similar effect on the growth of Myc-CaP tumors as two ADT doses, indicating the onset of CRPC within 7 days of ADT in this model (figures1A3C). The various control LNP groups showed no effect on tumor growth in the presence or absence of ADT (online supplemental file 7). In contrast, LNPs coated with FR-β antibody and containing active cGAMP significantly delayed the onset of CRPC after ADT (figure 3D). This effect was found to be dependent on CD8+T cells as it was abolished by antibody depletion of these effectors in tumors. Alternatively, an isotype-matched control IgG had no effect on the onset of CRPC after these LNPs (figure 3E).
Immunofluorescence staining showed that LNPs coated with FR-β antibody and containing active cGAMP (“LNPs(E)”) were taken up mainly by PV F4/80+TAMs rather than non-PV TAMs or F4/80 cells in PV or non-PV areas (figure 4A). PV TAMs bearing LNPs were FR-β+ (figure 4B–D) and FR-β antibody-coating of LNPs was essential for their uptake by PV TAMs. Only<5% of PV F4/80+cells took up LNPs when they had either a control IgG or no IgG on their surface. This is supported by the fact that neither of these two LNP groups (with or without cGAMP) delayed the start of CRPC (figure 3B and online supplemental file 7).
Figure 4. Selective delivery of cGAMP to PV FR-β+ TAMs in Myc-CaP tumors results in STING activation and upregulation of IFNβ. Following the administration of ADT plus LNPs: (A). The proportion of cells in PV and non-PV areas bearing LNPs that were F4/80− vs F4/80+. (B) Fluorescently labeled LNPs colocalized with PV FR-β+F4/80+TAMs. (C, D) FR-β+F4/80+TAMs bearing LNPs were only present in PV areas. (E) When LNPs bearing active cGAMP (LNPs(E)) were administered, the expression of active phosphorylated STING (P-STING) could be detected in LNP+FR-β+F4/80+TAMs. This was accompanied by a significant increase in IFNβ detection PV LNP+F4/80+TAMs (ie, in the LNPs(E) group) (E). In this group, IFNβ detection was only detectable in F4/80+TAMs in PV not NPV areas, (F) but often extended beyond LNP+cells, indicating its possible release and uptake by other cells in tumors (G). This did not occur when mice were injected with LNPs bearing inactive cGAMP. (NPV=non-PV). Data are presented as means±SEMs. *p<0.05, **p<0.01, ***p<0.001. Magnification bars=50 µm. ADT, androgen deprivation therapy; LNP, lipid nanoparticle.

Immunofluorescence analysis revealed that LNP+FR-β+F4/80+TAMs display p-STING in response to LNPs(E) treatment (figure 4E). This was not seen with either FR-β+ antibody-coated LNPs containing inactive cGAMP (“LNPs(C)”) or LNPs coated with control IgG (data are not shown), indicating that the cGAMP-STING pathway was successfully activated only by LNPs(E) treatment.
The effect of cGAMP activation of STING on expression of IFNβ by PV FR-β+ TAMs was then demonstrated. The density of LNP+TAMs expressing immunoreactive IFNβ was significantly higher in PV than non-PV areas of LNPs(E)-treated tumors. Indeed, very few IFNβ+LNP+ TAMswere present in non-PV areas of LNPs(E)-treated tumors (figure 4F). Interestingly, IFNβ was detected beyond PV TAMs indicating the release of this cytokine by PV FR-β+ TAMs and subsequent uptake by neighboring cells (figure 4G). This accords well with the finding that many cell types in tumors express receptors for type I IFNs.31 This increase in tumor IFNβ levels after LNPs(E) treatment was not observed with FR-β antibody-coated LNPs bearing the inactive form of cGAMP (“LNPs(C)”) (figure 4H,I), illustrating that LNP(E) treatment is specific and reliant on cGAMP-STING pathway activity.
We then examined the effect of elevated IFNβ (a known immunostimulant) on the density, distribution and activation status of CD8+T cells, CD4+T cells and NK cells. As shown in figure 2A (after 2 doses of ADT), a single dose of ADT alone in the LNP experiment resulted in a significant increase in PV CD8+T cells. Neither the coadministration of FR-β-antibody coated LNPs with LNPs(E) nor LNPs(C) with ADT altered this ADT-induced PV accumulation of CD8+T cells (figure 5A,B). However, ADT plus LNPs(E) increased the density and proportion of PV PD-1+CD8+ T cells (ie, reversed the induction by ADT alone of PV PD-1-CD8+T cells—see figure 2B). This induction of active (PD-1+) CD8+T cells by LNPs(E) occurred only in PV areas of ADT-treated tumors and did not occur with LNPs(C) (figure 5C). Finally, we immunostained sections with antibodies against PD-1, LAG3 (a marker of T cell exhaustion) and CD8 to investigate the functional status of PV CD8+T cells after LNP treatment. Fully active T cells were described as CD8+PD-1+LAG3 and exhausted ones as CD8+PD-1+LAG3+. Figure 5F shows that both PV and non-PV PD-1+CD8+ T cells in the “ADT plus LNPs(E)” group were predominantly LAG3- (ie, fully active).
Figure 5. Selective delivery of cGAMP to PV FR-β+ TAMs in Myc-CaP tumors reverses the effect of ADT on the activation status of CD8+T cells in Myc-CaP tumors. (A) Representative fluorescence image showing CD8+T cells in a vascularized area of a tumor treated with ADT plus FR-β antibody-coated LNPs containing active cGAMP (“LNPs(E)”). (B) ADT increased the PV accumulation of CD8+T cells (a change that was unaffected by coadministration with FR-β antibody-coated LNPs containing either inactive cGAMP (“LNPs(C)”) or LNPs(E)). (C) Representative fluorescence image showing colocalization of PD-1 and CD8 in a vascularized area of a tumor treated with ADT plus LNPs(E). (D, E) ADT administered with LNPs(E) reversed the PV accumulation of inactive (PD-1-) CD8+T cells induced by ADT alone, and led to an increase in both the PV density (D) and proportion (E) of CD8+T cells expressing PD-1. (F) The majority of PD-1+CD8+ T cells did not express the exhaustion marker, LAG3 following treatment with ADT plus LNPs(E). (NPV=non-PV). Data are presented as means±SEMs. *p<0.05, **p<0.01, ***p<0.001. Magnification bars=50 µm. ADT, androgen deprivation therapy; LNP, lipid nanoparticle.

Figure 6A,B shows that CD4+T cells are mainly PV in Myc-CaP tumors and remain so after ADT. There was a non-significant trend toward this PV accumulation of CD4+T cells being increased further by the administration LNPs(E), but not LNPs(C) with ADT. Neither form of LNP altered the proportion of CD4+T cells expressing the activation marker, PD-1, in PV areas but the density of PV PD-1+CD4+ T cells was significantly increased during ADT by LNPs(E), but not LNPs(C) (figure 6D,E).
Figure 6. Selective delivery of cGAMP to PV FR-β+ TAMs in Myc-CaP tumors increased the PV accumulation of PD-1+CD4+ T cells in Myc-CaP tumors. (A) Representative fluorescence image showing CD4+T cells in a vascularized area of a tumor treated with ADT plus FR-β antibody-coated LNPs containing active cGAMP (“LNPs(E)”). (B) CD4+T cells were mainly PV in PBS-treated tumors and this was unaffected by ADT alone or coadministration of ADT with FR-β antibody-coated LNPs containing either inactive cGAMP (“LNPs(C)”) or LNPs(E). (C) Representative fluorescence image showing colocalization of PD-1 and CD4 in a vascularized area of a tumor treated with ADT plus FR-β antibody-coated LNPs containing active cGAMP. (D, E) ADT administered with LNPs(E) resulted in the PV accumulation of PD-1+CD4+ T cells. The proportion of CD4+T cells expressing PD-1 remained unaltered by this treatment. (NPV=non-PV). Data are presented as means±SEMs. *p<0.05, ***p<0.001. Magnification bars=50 µm. ADT, androgen deprivation therapy; LNP, androgen deprivation therapy.

As mentioned previously, the density of NK cells is higher in PV than non-PV areas of both control and ADT-treated Myc-CaP tumors, and they mainly express CD69 in this location (online supplemental file 6). This was not altered when LNPs(E) or LNPs(C) were administered with ADT (figure 7), but the trend toward a lower density of activated PV NK cells after ADT alone (online supplemental file 6, left panel) was reversed by LNPs(E) but not LNPs(C) (figure 7D).
Figure 7. Selective delivery of cGAMP to PV FR-β+ TAMs in Myc-CaP tumors increases the PV density of active NK cells in Myc-CaP tumors. (A) Representative fluorescence image showing NK1.1+ NK cells in a vascularized area of a tumor treated with ADT plus FR-β antibody-coated LNPs containing active cGAMP (“LNPs (E)”). (B) NK cells were mainly PV in PBS-treated tumors and this was unaffected by ADT alone. Coadministration of ADT with LNPs (E) increased both the non-PV and PV density of NK cells compared with ADT alone or ADT plus FR-β antibody-coated LNPs containing inactive cGAMP (“LNPs (C)”). (C) Representative fluorescence image showing colocalization of CD69 and NK1.1 in a vascularized area of a tumor treated with ADT plus LNPs (E). (D, E) ADT administered with LNPs(E) increased the PV density of CD69+ (ie, activated) NK cells compared with ADT alone or ADT+LNPs (C). The majority of PV NK cells expressed CD69 in PBS-treated tumors. This did not change after ADT, with or without LNPs. (NPV=non-PV). Data are presented as means±SEMs. *p<0.05, **p<0.01,***p<0.001. Magnification bars=20 µm. ADT, androgen deprivation therapy; LNP, androgen deprivation therapy.

The above findings indicate that when LNPs containing cGAMP are targeted to PV FR-β+ TAMs, it results in their STING activation and increased IFNβ release. While this activated a number of immune effectors in the PV niche, the resultant delay in the onset of CRPC after ADT was mediated mainly by CD8+T cells.
Finally, it was important to confirm that LNPs(E) do not target FR-β+macrophages residing in healthy tissues. We, therefore, examined the effects of LNP exposure on a tissue known to contain FR-β+ macrophages the liver (online supplemental file 9). FR-β was detected in>80% of F4/80+Kupffer cells (online supplemental file 9). Despite this, only 20% of these cells took up LNPs or expressed IFNβ (online supplemental file 9). When the extent of IFNβ immunoreactivity by all cells was examined in the liver, only 15%–20% of all nucleated cells contained this cytokine (online supplemental file 9). This matched the proportion of cells in the liver found to be FR-β+ F4/80+Kupffer cells (online supplemental file 9) and indicated that little, if any, IFNβ was taken up by other cell types in the liver.
Discussion
ADT is a mainstay treatment for prostate cancer but the development of CRPC limits its long-term efficacy in both the neoadjuvant and adjuvant settings.1 32 In the current study, we show that ADT induces the PV accumulation of protumoral (FR-β+MRC1+CD169+VISTA+) TAMs in both human and mouse prostate tumors prior to the onset of CRPC. Interestingly, TAMs with a similar phenotype accumulate in PV areas of mouse tumors during chemotherapy and promote tumor relapse.31 33 34 Further studies are now needed to investigate a possible causal link between such PV cells and tumor regrowth in CRPC.
We also show that ADT causes naïve (PD-1-) CD8+T cells to gather in PV sites of prostate tumors suggesting their selective recruitment and/or suppression/retention in these areas. PV TAMs are known to be immunosuppressive27 31 33 so they may inhibit neighboring T cells. Indeed, a number of recent studies suggest this may be the case. For example, Bao et al35 revealed a close cell-to-cell interaction between a subset of FR-β+MRC1+ TAMs and inactive (mainly PD-1-) CD8+T cells in human colorectal carcinomas. Furthermore, depletion of FR-β+ TAMs expressing a transcriptional profile typical of PV TAMs (ie, mrc1, Lyve1, Hmox1,Il10 and stab1) in a mouse model of ovarian cancer increased the frequency and activation of ascitic CD8+T cells.36
VISTA expression by PV TAMs in ADT-treated tumors is likely to contribute to the inactive status of PV CD8+T cells as it is known to bind to receptors/ligands on T cells and suppress their functions.37 38 This could include their activation by dendritic cells–cells known to be abundant in PV tumor areas of mouse tumors.39 The coexpression of CD169 by PV TAMs also suggests an immunosuppressive phenotype. A recent study revealed that CD169+TAMs colocalize with T cells (along with regulatory T cells) in human breast tumors, and suppress T cells and NK cells, possibly via the release of PGE2, ROS and IL-1.40 It is not known whether CD169+PV TAMs do so in ADT-treated tumors and whether this contributes to the resistance of primary tumors to ADT. Interestingly, a recent study showed that CD169+macrophages promote resistance to the anti-androgen, enzalutamide (an AR antagonist) in mouse (prostate) bone metastases.41
The increase in PV naïve CD8+T cells seen after ADT in our studies could also have been due, in part, to thymus enlargement, a known side effect of ADT.42 This enhances thymic release of naïve T cells into the circulation,43 which could lead to more then being recruited into tumors. However, thymic release of naïve CD4+T cells also occurs during ADT,44 but these cells were not increased in ADT-treated mouse tumors, suggesting that other mechanisms are involved in the selective increase in naïve CD8+T cells in PV areas.
The main aim of our study was to see whether “re-educating” PV TAMs to enable them to release a potent immunostimulant, IFNβ, reverses the suppressive effects of ADT on tumor-infiltrating T cells. To do this, LNPs coated with an antibody to FR-β were used to deliver the STING agonist, cGAMP, to PV TAMs. In mouse prostate tumors, these LNPs were selectively taken up by PV TAMs after ADT, where they released cGAMP, activated STING signaling, and released IFNβ. The PV niche is an ideal location for this targeted form of immunotherapy as IFNβ has been shown to stimulate the functions of a number of immune cell types present in this site in tumors. For example, it stimulates antigen presentation by DCs to naïve T cells,45 as well as the proliferation and effector functions of T cells and NK cells.46 Type I IFNs can also decrease the immunosuppressive functions of MDSCs and Tregs and promote the expression of antitumor phenotype of TAMs.47 So, although the antitumor effects of our LNP therapy could be multifaceted in ADT-treated tumors, our CD8 depletion study showed that these cells play an essential role as mediators of our LNP approach. IFNβ is known to stimulate the release of various potent chemokines for T cells (eg, CXCL9, 10 and 11).48,50 The latter are highly likely to have been involved in the recruitment and activation of CD8+T cells seen in tumors after LNP-induction of tumor IFNβ levels.
To date, clinical trials using such agents (ie, immune checkpoint inhibitors, vaccines and CAR-T cells) have shown limited efficacy in prostate cancer.51 Our studies suggest that such targeted LNPs could be used to enhance the efficacy of immunotherapies that require functional CD8+T cells in this disease.
The FR-β-targeted delivery of cGAMP in LNPs to PV TAMs—and downstream IFNβ—could help circumvent the serious side effects recorded when non-encapsulated cGAMP or type I IFNs are injected into the systemic circulation. Indeed, no deleterious effects were seen in the livers of mice injected with cGAMP-containing LNPs despite the presence of FR-β-expressing Kupffer cells. The presence of LNPs and expression of IFNβ were detectable in only a small subset (20%) of these cells with no signs of IFNβ uptake by other cell types. This accords well with the finding that the expression of receptors for type I IFNs like IFNβ (IFNARs1 and 2) is negligible in the liver.52
Taken together, our studies show that LNP delivery of cGAMP to PV TAMs in ADT-treated prostate tumors increases antitumor immunity and delays CR. If reproduced in patients with prostate cancer, it could extend their treatment window for ADT and limit the metastatic spead of their disease.
supplementary material
Acknowledgements
We thank Dr Mukund Seshadri at the Roswell Park Comprehensive Cancer Center, USA, for the kind gift of the Myc-CaP cells stably transfected with luciferase.
Footnotes
Funding: This project was funded by a grant from Prostate Cancer-UK to CL and JB (RIA16-ST2-022). HP is supported by a CRUK Career Development award (#A27894).
Provenance and peer review: Not commissioned; externally peer reviewed.
Patient consent for publication: Not applicable.
Ethics approval: This study involves human participants and the prostate cancer samples provided by the Institute of Cancer Research and Royal Marsden Hospital in London: patients provided written, informed consent, and the ethics committee was the Royal Marsden Hospital Ethics Review Committee (reference no. 04/Q0801/60). For the second set of prostate cancer samples (provided by the Dana Farber Cancer Centre, USA), the name of the ethics committee was the Dana-Farber Cancer Center IRB (reference no. 01-045). Participants gave informed consent to participate in the study before taking part.
Data availability free text: Further information and requests for data and/or resources/reagents should be directed to—and will be fulfilled by—the lead contact, CL (claire.lewis@sheffield.ac.uk).
Contributor Information
Haider Al-janabi, Email: h.h.al-janabi@sheffield.ac.uk.
Katy Moyes, Email: k.moyes@sheffield.ac.uk.
Richard Allen, Email: r.j.allen@sheffield.ac.uk.
Matthew Fisher, Email: Matthew.fisher@sheffield.ac.uk.
Mateus Crespo, Email: mateus.crespo@icr.ac.uk.
Bora Gurel, Email: bora.gurel@icr.ac.uk.
Pasquale Rescigno, Email: Pasquale.Rescigno@icr.ac.uk.
Johann de Bono, Email: johann.debono@icr.ac.uk.
Harry Nunns, Email: harry.nunns@neogenomics.com.
Christopher Bailey, Email: info@lipexogen.com.
Anna Junker-Jensen, Email: anna.juncker-jensen@neogenomics.com.
Munitta Muthana, Email: munitta.muthana@sheffield.ac.uk.
Wayne A Phillips, Email: wayne.phillips@petermac.org.
Helen B Pearson, Email: pearsonh2@cardiff.ac.uk.
Mary-Ellen Taplin, Email: Mary_Taplin@dfci.harvard.edu.
Janet E Brown, Email: j.e.brown@sheffield.ac.uk.
Claire E Lewis, Email: Claire.lewis@sheffield.ac.uk.
Data availability statement
Data are available on reasonable request.
References
- 1.Jamroze A, Chatta G, Tang DG. Androgen receptor (AR) heterogeneity in prostate cancer and therapy resistance. Cancer Lett. 2021;518:1–9. doi: 10.1016/j.canlet.2021.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang Y, Jiang X, Liang X, et al. Molecular and cellular mechanisms of castration resistant prostate cancer (review) Oncol Lett. 2018;15:6063–76. doi: 10.3892/ol.2018.8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mansinho A, Macedo D, Fernandes I, et al. Castration-resistant prostate cancer: mechanisms, targets and treatment. Adv Exp Med Biol. 2018;1096:117–33. doi: 10.1007/978-3-319-99286-0_7. [DOI] [PubMed] [Google Scholar]
- 4.Cattrini C, Zanardi E, Vallome G, et al. Targeting androgen-independent pathways: new chances for patients with prostate cancer? Crit Rev Oncol Hematol. 2017;118:42–53. doi: 10.1016/j.critrevonc.2017.08.009. [DOI] [PubMed] [Google Scholar]
- 5.Shen Y-C, Ghasemzadeh A, Kochel CM, et al. Combining intratumoral treg depletion with androgen deprivation therapy (ADT): preclinical activity in the myc-cap model. Prostate Cancer Prostatic Dis. 2018;21:113–25. doi: 10.1038/s41391-017-0013-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Obradovic AZ, Dallos MC, Zahurak ML, et al. T-cell infiltration and adaptive treg resistance in response to androgen deprivation with or without vaccination in localized prostate cancer. Clin Cancer Res. 2020;26:3182–92. doi: 10.1158/1078-0432.CCR-19-3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mercader M, Bodner BK, Moser MT, et al. T cell infiltration of the prostate induced by androgen withdrawal in patients with prostate cancer. Proc Natl Acad Sci USA. 2001;98:14565–70. doi: 10.1073/pnas.251140998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gannon PO, Poisson AO, Delvoye N, et al. Characterization of the intra-prostatic immune cell infiltration in androgen-deprived prostate cancer patients. J Immunol Methods. 2009;348:9–17. doi: 10.1016/j.jim.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Pu Y, Xu M, Liang Y, et al. Androgen receptor antagonists compromise T cell response against prostate cancer leading to early tumor relapse. Sci Transl Med. 2016;8:333ra47. doi: 10.1126/scitranslmed.aad5659. [DOI] [PubMed] [Google Scholar]
- 10.Sommer U, Ebersbach C, Beier A-MK, et al. Influence of androgen deprivation therapy on the PD-L1 expression and immune activity in prostate cancer tissue. Front Mol Biosci. 2022;9:878353. doi: 10.3389/fmolb.2022.878353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qin C, Wang J, Du Y, et al. Immunosuppressive environment in response to androgen deprivation treatment in prostate cancer. Front Endocrinol (Lausanne) 2022;13:1055826. doi: 10.3389/fendo.2022.1055826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang S, Moore ML, Grayson JM, et al. Increased CD8+ T-cell function following castration and immunization is countered by parallel expansion of regulatory T cells. Cancer Res. 2012;72:1975–85. doi: 10.1158/0008-5472.CAN-11-2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalina JL, Neilson DS, Comber AP, et al. Immune modulation by androgen deprivation and radiation therapy: implications for prostate cancer immunotherapy. Cancers (Basel) 2017;9:13. doi: 10.3390/cancers9020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Escamilla J, Schokrpur S, Liu C, et al. CSF1 receptor targeting in prostate cancer reverses macrophage-mediated resistance to androgen blockade therapy. Cancer Res. 2015;75:950–62. doi: 10.1158/0008-5472.CAN-14-0992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amouzegar A, Chelvanambi M, Filderman J, et al. STING agonists as cancer therapeutics. Cancers (Basel) 2021;13:2695. doi: 10.3390/cancers13112695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hines JB, Kacew AJ, Sweis RF. The development of STING agonists and emerging results as a cancer immunotherapy. Curr Oncol Rep. 2023;25:189–99. doi: 10.1007/s11912-023-01361-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan X, Zhang W, Guo H, et al. Strategies involving STING pathway activation for cancer immunotherapy: mechanism and agonists. Biochem Pharmacol. 2023;213:S0006-2952(23)00187-9. doi: 10.1016/j.bcp.2023.115596. [DOI] [PubMed] [Google Scholar]
- 18.Luo K, Li N, Ye W, et al. Activation of stimulation of interferon genes (STING) signal and cancer immunotherapy. Molecules. 2022;27:4638. doi: 10.3390/molecules27144638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuka M, De Giovanni M, Iannacone M. The role of type I interferons in CD4+ T cell differentiation. Immunol Lett. 2019;215:19–23. doi: 10.1016/j.imlet.2019.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Müller L, Aigner P, Stoiber D. Type I interferons and natural killer cell regulation in cancer. Front Immunol. 2013;8:304. doi: 10.3389/fimmu.2017.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Motedayen Aval L, Pease JE, Sharma R, et al. Challenges and opportunities in the clinical development of STING agonists for cancer immunotherapy. JCM. 2020;9:3323. doi: 10.3390/jcm9103323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang C, Shao N, Huang Y, et al. Overcoming challenges in the delivery of STING agonists for cancer immunotherapy: a comprehensive review of strategies and future perspectives. Mater Today Bio . 2023;23:100839. doi: 10.1016/j.mtbio.2023.100839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watson PA, Ellwood-Yen K, King JC, et al. Context-dependent hormone-refractory progression revealed through characterization of a novel murine prostate cancer cell line. Cancer Res. 2005;65:11565–71. doi: 10.1158/0008-5472.CAN-05-3441. [DOI] [PubMed] [Google Scholar]
- 24.Pearson HB, Li J, Meniel VS, et al. Identification of pik3ca mutation as a genetic driver of prostate cancer that cooperates with pten loss to accelerate progression and castration-resistant growth. Cancer Discov. 2020;8:764–79. doi: 10.1158/2159-8290.CD-17-0867. [DOI] [PubMed] [Google Scholar]
- 25.Moamin MR, Allen R, Woods SL, et al. Changes in the immune landscape of TNBC after neoadjuvant chemotherapy: correlation with relapse. Front Immunol. 2023;14:1291643. doi: 10.3389/fimmu.2023.1291643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swart LE, Koekman CA, Seinen CW, et al. A robust post-insertion method for the preparation of targeted sirna lnps. Int J Pharm. 2022;620:S0378-5173(22)00296-4. doi: 10.1016/j.ijpharm.2022.121741. [DOI] [PubMed] [Google Scholar]
- 27.Sharma A, Seow JJW, Dutertre C-A, et al. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell. 2020;183:377–94. doi: 10.1016/j.cell.2020.08.040. [DOI] [PubMed] [Google Scholar]
- 28.Jing W, Guo X, Wang G, et al. Breast cancer cells promote CD169+ macrophage-associated immunosuppression through JAK2-mediated PD-L1 upregulation on macrophages. Int Immunopharmacol. 2020;78:S1567-5769(19)31583-8. doi: 10.1016/j.intimp.2019.106012. [DOI] [PubMed] [Google Scholar]
- 29.Yuan L, Tatineni J, Mahoney KM, et al. VISTA: a mediator of quiescence and a promising target in cancer immunotherapy. Trends Immunol. 2021;42:209–27. doi: 10.1016/j.it.2020.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larionova I, Tuguzbaeva G, Ponomaryova A, et al. Tumor-associated macrophages in human breast, colorectal, lung, ovarian and prostate cancers. Front Oncol. 2020;10:566511. doi: 10.3389/fonc.2020.566511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hughes R, Qian B-Z, Rowan C, et al. Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res. 2015;75:3479–91. doi: 10.1158/0008-5472.CAN-14-3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu EM, Aragon-Ching JB. Advances with androgen deprivation therapy for prostate cancer. Expert Opin Pharmacother. 2022;23:1015–33. doi: 10.1080/14656566.2022.2033210. [DOI] [PubMed] [Google Scholar]
- 33.Anstee JE, Feehan KT, Opzoomer JW, et al. LYVE-1+ macrophages form a collaborative CCR5-dependent perivascular niche that influences chemotherapy responses in murine breast cancer. Dev Cell. 2023;58:1548–61. doi: 10.1016/j.devcel.2023.06.006. [DOI] [PubMed] [Google Scholar]
- 34.Coffelt SB, Chen Y-Y, Muthana M, et al. Angiopoietin 2 stimulates TIE2-expressing monocytes to suppress T cell activation and to promote regulatory T cell expansion. J Immunol. 2011;186:4183–90. doi: 10.4049/jimmunol.1002802. [DOI] [PubMed] [Google Scholar]
- 35.Bao X, Li Q, Chen D, et al. A multiomics analysis-assisted deep learning model identifies a macrophage-oriented module as a potential therapeutic target in colorectal cancer. Cell Rep Med. 2024;5:101399. doi: 10.1016/j.xcrm.2024.101399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez-Garcia A, Lynn RC, Poussin M, et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat Commun. 2021;12:877. doi: 10.1038/s41467-021-20893-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnston RJ, Su LJ, Pinckney J, et al. VISTA is an acidic ph-selective ligand for PSGL-1. Nature New Biol. 2019;574:565–70. doi: 10.1038/s41586-019-1674-5. [DOI] [PubMed] [Google Scholar]
- 38.Schuette V, Embgenbroich M, Ulas T, et al. Mannose receptor induces T-cell tolerance via inhibition of CD45 and up-regulation of CTLA-4. Proc Natl Acad Sci U S A. 2016;113:10649–54. doi: 10.1073/pnas.1605885113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stoltzfus CR, Sivakumar R, Kunz L, et al. Multi-parameter quantitative imaging of tumor microenvironments reveals perivascular immune niches associated with anti-tumor immunity. Front Immunol. 2021;12:726492. doi: 10.3389/fimmu.2021.726492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gunnarsdottir FB, Briem O, Lindgren AY, et al. Breast cancer associated CD169+ macrophages possess broad immunosuppressive functions but enhance antibody secretion by activated B cells. Front Immunol. 2023;14:1180209. doi: 10.3389/fimmu.2023.1180209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li X-F, Selli C, Zhou H-L, et al. Macrophages promote anti-androgen resistance in prostate cancer bone disease. J Exp Med. 2023;220:e20221007. doi: 10.1084/jem.20221007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Polesso F, Caruso B, Hammond SA, et al. Restored thymic output after androgen blockade participates in antitumor immunity. J Immunol. 2023;210:496–503. doi: 10.4049/jimmunol.2200696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lai J-J, Lai K-P, Zeng W, et al. Androgen receptor influences on body defense system via modulation of innate and adaptive immune systems: lessons from conditional AR knockout mice. Am J Pathol. 2012;181:1504–12. doi: 10.1016/j.ajpath.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morse MD, McNeel DG. Prostate cancer patients on androgen deprivation therapy develop persistent changes in adaptive immune responses. Hum Immunol. 2010;71:496–504. doi: 10.1016/j.humimm.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cucak H, Yrlid U, Reizis B, et al. Type I interferon signaling in dendritic cells stimulates the development of lymph-node-resident T follicular helper cells. Immunity. 2009;31:491–501. doi: 10.1016/j.immuni.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 46.Kolumam GA, Thomas S, Thompson LJ, et al. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202:637–50. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borden EC. Interferons α and β in cancer: therapeutic opportunities from new insights. Nat Rev Drug Discov. 2019;18:219–34. doi: 10.1038/s41573-018-0011-2. [DOI] [PubMed] [Google Scholar]
- 48.Henig N, Avidan N, Mandel I, et al. Interferon-beta induces distinct gene expression response patterns in human monocytes versus T cells. PLoS One. 2013;8:e62366. doi: 10.1371/journal.pone.0062366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buttmann M, Berberich-Siebelt F, Serfling E, et al. Interferon-beta is a potent inducer of interferon regulatory factor-1/2-dependent IP-10/CXCL10 expression in primary human endothelial cells. J Vasc Res. 2007;44:51–60. doi: 10.1159/000097977. [DOI] [PubMed] [Google Scholar]
- 50.Coelho LFL, Magno de Freitas Almeida G, Mennechet FJD, et al. Interferon-alpha and -beta differentially regulate osteoclastogenesis: role of differential induction of chemokine CXCL11 expression. Proc Natl Acad Sci U S A. 2005;102:11917–22. doi: 10.1073/pnas.0502188102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rehman LU, Nisar MH, Fatima W, et al. Immunotherapy for prostate cancer: a current systematic review and patient centric perspectives. J Clin Med. 2023;12:1446. doi: 10.3390/jcm12041446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tochizawa S, Muraguchi M, Ohmoto Y, et al. Functional expression of human type I interferon receptors in the mouse liver. Biochem Biophys Res Commun. 2006;346:61–6. doi: 10.1016/j.bbrc.2006.05.081. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available on reasonable request.