

Abstract
Pharmacogenomic Polygenic Risk Scores (PRS) have emerged as a tool to address the polygenic nature of pharmacogenetic phenotypes, increasing the potential to predict drug response. Most pharmacogenomic PRS have been extrapolated from disease‐associated variants identified by genome wide association studies (GWAS), although some have begun to utilize genetic variants from pharmacogenomic GWAS. As pharmacogenomic PRS hold the promise of enabling precision medicine, including stratified treatment approaches, it is important to assess the opportunities and challenges presented by the current data. This assessment will help determine how pharmacogenomic PRS can be advanced and transitioned into clinical use. In this review, we present a summary of recent evidence, evaluate the current status, and identify several challenges that have impeded the progress of pharmacogenomic PRS. These challenges include the reliance on extrapolations from disease genetics and limitations inherent to pharmacogenomics research such as low sample sizes, phenotyping inconsistencies, among others. We finally propose recommendations to overcome the challenges and facilitate the clinical implementation. These recommendations include standardizing methodologies for phenotyping, enhancing collaborative efforts, developing new statistical methods to capitalize on drug‐specific genetic associations for PRS construction. Additional recommendations include enhancing the infrastructure that can integrate genomic data with clinical predictors, along with implementing user‐friendly clinical decision tools, and patient education. Ethical and regulatory considerations should address issues related to patient privacy, informed consent and safe use of PRS. Despite these challenges, ongoing research and large‐scale collaboration is likely to advance the field and realize the potential of pharmacogenomic PRS.
INTRODUCTION
The field of pharmacogenomics was driven by the observation that individuals varied widely with respect to the pharmacodynamic and pharmacokinetic measures including drug response and risk of adverse reactions. Early studies, typically conducted on small populations and examining extreme phenotypes identified genetic variants of large effect sizes such as those impacting the cytochrome P450 and TPMT enzymes that contributed to some of this variability. 1 However, newer genetic methods have demonstrated that drug response phenotypes are highly polygenic, and driven by a large numbers of common variants of modest or small effect sizes. 2
Historically, candidate gene studies and genome‐wide association studies (GWAS) have served as the cornerstone of genetic research, including pharmacogenomic studies. However, both approaches are complicated by a lack of efficiency in handling the multi‐locus involvement typical of drug response phenotypes. Thus, the field of pharmacogenomics has evolved from trying to predict drug responses using simple single‐variant predictors to the use of polygenic instruments such as Polygenic Risk Score (PRS), which more fully capture the polygenic variability underlying drug responses. 3 Other efforts to account for the genetic underpinnings of complex phenotypes led to the development of recent approaches, such as Transcriptome‐Wide Association Studies (TWAS), Phenome‐Wide Association Studies (PheWAS), and Mendelian Randomization, among others. 4 , 5
PRS arose as a potential solution for addressing the polygenicity inherent to pharmacogenomic phenotypes. PRS amalgamate the cumulative effects of multiple genetic variants associated with a particular trait or pharmacogenomics phenotype, such as drug efficacy or adverse events. In the context of pharmacogenomics, PRS offer a means to combine the impact of a multitude of genetic variants across many genes and genomic regions, 6 and thus PRS may capture a larger portion of the genetic liability of drug responses, 7 potentially improving risk prediction, as compared to single variant predictors.
PRS have emerged as a promising tool for disease risk prediction and patients' stratification. In recent years, many PRS have been developed for the prediction of complex diseases such as coronary artery, diabetes, cancer, among others. 8 , 9 , 10 , 11 The progress in disease PRS was largely facilitated by large scale, collaborative consortia of disease phenotypes. Moreover, many publications have provided recommendations on how to facilitate and enhance the transition of these PRS to the clinics. 12 While the research on the pharmacogenomic PRS has also grown, it has lagged behind disease PRS. Enhancing research in pharmacogenomic PRS and addressing its current challenges can facilitate the clinical utilization, which can advance personalized medicine, through treatment stratification and drug response prediction.
Herein, we aim to review the recent landscape of pharmacogenomic PRS, describing the current approaches and most recent methods, shedding light on their limitations and impediments to clinical implementation. We finally propose a set of recommendations to address these challenges and facilitate a widespread utilization of pharmacogenomic PRS. This review provides summaries, rather than in‐depth reviews of relevant studies, as extensive reviews of the addressed studies have been published in recent systematic reviews. 6 , 13 , 14
CURRENT APPROACHES FOR CONSTRUCTING PRS
PRS measure the burden of additive genetic variants associated with a disease phenotype or an outcome that an individual has. 15 For a given SNP, an individual can have 0, 1, or 2 copies of the risk associated allele. Initially, PRS represented a tally (i.e., an unweighted sum) of the number of risk alleles across all variants that an individual carried. 16 This approach relied on the simple assumption that each variant has an equivalent contribution to disease risk, although risks associated with a given variant can vary widely. Thus, weighted PRS have now largely supplanted unweighted risk scores. These PRS are a weighted sum of risk alleles that an individual carries, with the effect size (often measured as the log of the odds‐ratio) of the risk allele typically used as the weight. 17 A weighted PRS is a continuous variable and, across a population, PRS will generally demonstrate a normal distribution. An important consideration is which variants should be included in a PRS. Ideally, SNPs and weights are derived from a single GWAS, as this ensures that the relative weights of the SNPs are comparable. Initially, PRS were restricted to those SNPs that met a genome‐wide significance threshold (typically a p‐value <5 × 10−8). However, Purcell et al. demonstrated that inclusion of SNPs with lower statistical significance in the PRS improved predictive performance. 17 Hence, PRS typically include large numbers of SNPs that have a lower level of significance.
Among the numerous approaches used to select SNPs for inclusion in the PRS, all include methods to account for correlation (or linkage‐disequilibrium) among the large numbers of SNPs. The earliest and simplest strategy was a pruning‐and‐thresholding (or “clumping”) approach. 17 With this iterative strategy, the SNP with the lowest p‐value in the GWAS is selected. Then, all SNPs in linkage‐disequilibrium (LD) with that SNP are removed from the GWAS. The process is repeated until all independent SNPs below a specified threshold are selected. Alternatives to this pruning‐and‐thresholding approach employ various statistical methodologies and often seek to retain a larger number of SNPs for inclusion in the risk score. Bayesian approaches empirically determine a distribution of effect sizes among SNPs and then reweight SNP effect sizes to conform to the distributional expectations. 18 , 19 , 20 , 21 , 22 , 23 Other approaches use LASSO or ridge regression to adjust SNP effect sizes according to different distributional assumptions. 24 As GWAS studies have grown larger, the differences in predictive performance of PRS developed using differing methods has become relatively modest, though some methods may perform better for particular underlying genetic architectures. 25
Most of the existing pharmacogenomic PRS studies use variants from disease genetics, a method that has its limitations, since it assumes that these variants represent the drug's effects. This approach assumes a constant ratio between the genetic main effect and its interaction effect with the drug(s), which may not always hold true (discussed in details in the “ Brief review of recent PRS Pharmacogenomics research section,” below). 3 Further, the majority of the pharmacogenomic PRS studies employ the clumping‐and‐thresholding method to create PRS. Researchers have begun to investigate newer statistical methods for constructing pharmacogenomic PRS using Bayesian modeling that account for both the disease and drug effects. Of note, the recent methods developed by a single group of investigators have focused on overcoming some of the challenges of pharmacogenomic PRS, including the reliance on disease variants. Table 1 summarizes the benefits and limitations of the Bayesian‐based techniques, including the novel approaches.
TABLE 1.
A summary of recently developed methods for constructing pharmacogenomic PRS.
| Name | Premise | Benefits | Limitations |
|---|---|---|---|
| Bayesian modeling (e.g., PRS‐CS) 20 , 49 | Bayesian shrinkage approach that uses global and local scale mixtures of normal prior distributions |
|
|
| Novel pharmacogenomics PRS | |||
| PRS‐PGx‐Bayes 3 | Bayesian based |
|
|
| mtPRS‐PCA 26 | Integrates multiple PRS from traits, weighted by the PCA derived from genetic correlation matrix |
|
|
| PRS‐PGx‐Bayesx 27 | Bayesian method that builds on PRS‐PGx‐Bayes accounting for disease‐related and drug–response‐related effects of genetic variants |
|
|
Zhai et al. 3 proposed a novel method (PRS‐PGx‐Bayes) to incorporate the genotypic main effect of the disease and the “genotype × treatment interaction” on drug response. They applied this method to predict the LDL‐lowering impact of ezetimibe and simvastatin compared to the simvastatin only arm using the IMPROVE‐IT clinical trial data, and demonstrated superiority of this method over other existing methods in terms of prediction accuracy. 3 Zhai et al. developed a framework for integrating multiple correlated traits for developing pharmacogenomic PRS. They proposed multiple integrative approaches in this paper including the mtPRS‐PCA. The mtPRS‐PCA method applies Principal Component Analysis (PCA) to a genetic correlation matrix of the traits, and uses the top PCAs as weights, followed by combining the PRS of each trait to create a final PRS. 26 In the paper, Zhai et al. applied mtPRS‐PCA and other methods to an anacetrapib GWAS study to predict the effect of the medication on LDL lowering. In their analysis, they included traits that are genetically correlated and used the summary statistics from the UK Biobank GWAS such as those for HDL, LDL, triglycerides, apolipoprotein B, apolipoprotein A1, and lipoprotein (a). The investigators showed that this method improved both the prediction and risk stratification compared to other PRS methods. 26 The same investigators proposed a newer approach for pharmacogenomic PRS to address three challenges in the development of pharmacogenomic PRS, which are: (1) the lack of systematic approaches to choosing the summary statistics (i.e., those for the disease, pharmacogenomics, or both) for PRS construction; (2) the Eurocentric or trans‐ethnic bias in cross‐population PRS prediction; and (3) the small sample sizes, low power, and complex PRS modeling in pharmacogenomics GWAS. 27 This paper developed a new approach using a Bayesian regression method called PRS‐PGx‐Bayesx, which is an extension of PRS‐PGx‐Bayes. The method uses pharmacogenomic GWAS; however, they replace the effect size estimates of the pharmacogenomic summary statistics with those from disease GWAS summary statistics. 27 This method has the potential to improve the accuracy and power of PRS through leveraging the increased power of disease GWAS owing to the large number, and the specific predictive power of pharmacogenomic variants. A schematic figure of how pharmacogenomic PRS are constructed or proposed to be constructed is displayed (Figure 1). We refer the readers to the recent paper by Zhai et al., 27 for the additional technical details of these methods.
FIGURE 1.
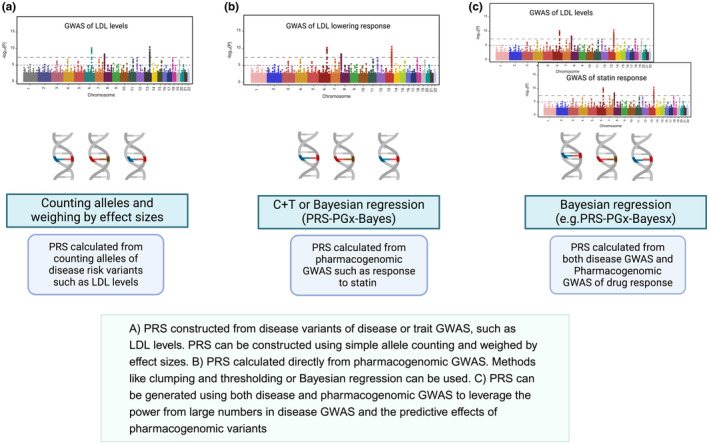
Illustration of select approaches for building pharmacogenomic PRS.
Various metrics are used to assess the predictive performance of PRS, including the Area Under the Receiver Operating Characteristic‐Curve (AUC‐ROC), pseudo‐R 2, and the odds ratios (OR) per PRS quantiles. The AUC derived from the ROC curve (plotting true‐positive rate against false‐positive rate) measures the PRS's ability to discriminate between cases and controls. An AUC of 0.5 indicates a discrimination that is no better than a random chance, while an AUC of 1 indicates a perfect discriminatory power. 28 Pseudo‐R 2 statistics such as Nagelkerke R 2 estimates the proportion of variance explained by PRS. 15 In the event that the case and control prevalence in a study differs from the actual population prevalence, alternative pseudo‐R 2 measures such as Lee R 2 are recommended to avoid bias in the estimate 15 The OR plot is a visual representation that is commonly reported to quantify the association between PRS and the phenotype. It shows the likelihood of being a case across different PRS percentiles (e.g., the OR of developing a side effect comparing patients in the 90th PRS percentile to those in the 10th percentile).
BRIEF REVIEW OF RECENT PRS PHARMACOGENOMICS RESEARCH AND ITS ASSOCIATED LIMITATIONS AND CHALLENGES
Pharmacogenomic studies have used two main approaches for PRS: the first approach relies on using genetic variants associated with disease or traits, while the second approach uses genetic variants associated with drug response, as shown in Table 2. In the next section, we provide a selective review of the recent studies (within the past 5–10 years) that fall under these two approaches, describing the advantages and challenges. More extensive reviews of the pharmacogenomics PRS studies were recently published. 6 , 14
TABLE 2.
A selected list of studies that used PRS using: (1) extrapolation method from disease variants, or (2) direct pharmacogenomics variants of drug response.
| Study | Disease/trait | Medication/medication class | Outcome measure (e.g., treatment stratification, response, dose) | Method | Number of variants, if reported | Ancestry of patients in the study | Prediction metrics (e.g., R 2), if reported |
|---|---|---|---|---|---|---|---|
| PRS Extrapolated from disease or trait GWAS | |||||||
| Natarajan et al. 32 | Atherosclerosis/coronary artery disease | Statin | Treatment stratification (i.e., risk reduction with statin versus placebo per PRS strata) | Risk allele counting weighed by effect size | 57 | European | NA |
| Oni‐orisan et al. 33 | Coronary heart disease | Statin | Risk reduction with statin versus nonstatin per PRS strata | Risk allele counting weighed by effect size | 164 | European and African American | NA |
| Kappel et al. 37 | Schizophrenia | Clozapine | Clozapine dose | Bayesian regression and continuous shrinkage 20 | European | 32% | |
| Guo et al. 38 | Schizophrenia | Antipsychotics | Treatment response (reduction of PANSS) | Clumping + thresholding (PRSice‐2 68 ) | Han Chinese | 48% for both clinical and PRS | |
| PRS constructed from pharmacogenomic variants | |||||||
| Lanfear et al. 41 | Heart failure | β‐blockers | Survival benefit (β‐blockers*PRS interaction) | Risk allele counting weighed by effect size | 44 | European | NA |
| Pardiñas et al. 48 | Schizophrenia | Clozapine | Clozapine metabolic ratio | Clumping + thresholding (PRSice‐2 68 ) | 8 | Multi‐ancestry (European, African+Asian) | 7.2% |
| Amare AT et al. 49 | Bipolar | Lithium | Lithium response | Bayesian regression and continuous shrinkage 20 | European | 2.6% | |
| De Pieri et al. 50 | Schizophrenia, bipolar | Antipsychotics | Treatment response | Clumping + thresholding (PRSice‐2 68 ) | 11 | European | PPV:64%, NPV:57%, sensitivity 63%, Specificity: 58% |
PRS USING VARIANTS ASSOCIATED WITH DISEASES
Most existing studies have developed PRS using variants identified by GWAS of common diseases (e.g., cardiovascular and psychiatric diseases) to assess disease‐related drug response. This approach leveraged the existing PRS, developed from large‐scale GWAS data. For example, PRS derived from coronary artery disease (CAD) GWAS improved CAD prediction and demonstrated a value in identifying patients likely to receive clinical benefits from interventions. 10 , 29 , 30 CAD PRS have since been tested for allocating of lipid lowering treatment, including statins, and more recently PCSK9 inhibitors. In the statin prevention clinical trials, individuals with a high CAD PRS (top quintile) had a greater absolute and relative CAD risk reduction compared to other PRS strata (quintiles 2 to 4), despite a similar low‐density lipoprotein cholesterol (LDL‐C) lowering among individuals of different CAD genetic risk strata. 31 , 32 A recent study by Oni‐Orisan et al. investigated if a PRS for coronary heart disease (CHD), constructed from 164 variants of the CARDIoGRAMplusC4D GWAS could stratify patients based on the benefits derived from statin therapy in a large, diverse cohort of primary prevention patients. 33 They found that a high CHD PRS value (top 20%) was associated with a greater relative risk reduction from statins (59%), compared to a smaller relative risk reduction from statins (33%) among patients with lower PRS values (quintiles 2–4). 33 Similar to the statin trials, a greater absolute risk and relative risk reduction in cardiovascular events from alirocumab, a PCSK9 inhibitor, was observed among patients with high PRS. 34 Likewise, a CAD PRS was evaluated with evolocumab, another PCSK9 inhibitor among patients with a history of cardiovascular disease. 35 In this study by Marston et al., patients in the highest PRS category had 31% and 4% relative and absolute risk reductions from evolocumab, respectively, compared to placebo. While these studies together suggested that PRS of CAD could be utilized to stratify patients with high PRS for statins or PCSK9 inhibitors, for early prevention effort, most of the studies have not assessed the interaction of PRS and treatment benefit, which would be important to ascertain whether relative risk reductions vary across PRS strata with respect to medications.
Evaluating pharmacogenomic PRS using variants extrapolated from other traits has been prolific in the field of psychiatry, with the majority of these studies evaluating the use of PRS in predicting treatment efficacy, and fewer evaluating the association with doses and treatment related adverse events. Unlike the case with CAD PRS in stratifying patients benefiting from lipid lowering treatment, the use of PRS in psychiatric diseases has shown less consistent results. For example, several schizophrenia PRS were associated with high clozapine dose and poor treatment outcomes in patients with severe schizophrenia. 36 , 37 , 38 In contrast, no association was reported between schizophrenia PRS and treatment resistant schizophrenia. 39 Similarly, a systematic review of 26 PRS for antipsychotic or antidepressant treatment outcomes reported associations for less than half of the studies. 6
PRS USING VARIANTS ASSOCIATED WITH DRUG RESPONSE
Emerging data suggest that pharmacogenomics derived PRS (i.e., PRS derived from genetic associations of drug responses) may offer better prediction of response to medications. The first PRS were developed in the context of cardiovascular medications, with more recent studies in the context of psychiatric diseases. (Table 1). In 2020, Lewis et al. constructed a pharmacogenomics score for clopidogrel using allele counting, weighted on effect size of 31 candidate genetic variants related to platelet reactivity. The score aimed to predict major cardiovascular events in clopidogrel‐treated patients, while accounting for other co‐morbidities including age, sex, participating site, body mass index (BMI), diabetes, proton pump inhibitor use, and smoking. 40 While no single variant was associated with cardiovascular events in the replication cohort, the higher PRS (i.e., patients with eight or more risk alleles for platelet reactivity) were significantly associated with higher risk of cardiovascular events (Odds ratio (OR) = 1.78, 95% CI: 1.14–2.76, p = 0.01) or cardiovascular‐related death (OR = 4.39, 95% CI: 1.35–14.27, p = 0.01), compared to patients with six or less risk alleles, suggesting that patients with high PRS are at high risk of clinical outcomes with clopidogrel and can be offered an alternative treatment. 40 Lanfear et al. derived a PRS of 44 SNPs from GWAS of β‐blocker related survival within Henry Ford Heart Failure Pharmacogenomic Registry (HFPGR) and tested it in additional independent cohorts (HF‐ACTION and TIME‐CHF). 41 These 44 SNPs were selected from a GWAS that evaluated how the genetic variants interact with β‐blocker treatment (SNP x treatment interaction), to predict survival benefits in patients with reduced ejection fraction heart failure. This GWAS analysis adjusted for the Meta‐Analysis Global Group in Chronic Heart Failure (MAGGIC) score, a predictor of death or hospitalization in patients with heart failure and a β‐blocker propensity score to account for potential confounding by indication or treatment bias. The results showed that β‐blocker exposure was associated with a non‐significant decrease in mortality risk among patients with low PRS (≤30th percentile) in the HFPGR and HF‐ACTION cohorts, and a significantly lower risk in the TIME‐CHF cohort (p = 0.049). In the total validation set of the meta‐analysis of the three cohorts, the β‐blocker significantly lowered mortality (Hazard Ratio (HR): 0.19, 95% CI: 0.06–0.64, p = 0.008) in the low PRS, while no significant association was found in the high PRS group (HR: 0.84, 95% CI: 0.53–1.30, p = 0.45). The interaction analysis was significant for the effect of β‐blocker exposure between low and high PRS groups (p = 0.02). 41 The PRS was further validated in a separate cohort within the UK Biobank. 42 In the overall study (N = 7141), the PRS was not significantly associated with mortality (p = 0.92). Similar to the first paper by Lanfear et al., 42 patients in the low PRS category had a trending significant survival benefit with β‐blocker treatment (HR = 0.57, 95% CI: 0.33–1.00, p = 0.052), compared to these in the high PRS (HR = 0.90, 95% CI: 0.70–1.15, p = 0.4), with a statistically significant interaction between the PRS and β‐blocker exposure (p = 0.04).
In psychiatry, a number of studies have examined the association of PRS developed from pharmacogenomic association with treatment outcomes for several classes including antidepressants, antipsychotics, and lithium, among others. A 2023 study investigated the association of pharmacogenomic PRS with remission of psychotic depression treated with sertraline and olanzapine. 43 The investigators used a PRS generated from GWAS of response to antidepressants including SSRIs (e.g., citalopram and sertraline) and norepinephrine reuptake inhibitor (NRI, e.g., reboxetine), and tested its association with remission, adjusting for age, sex, baseline depression and delusion severity, and principal components of ancestry (PCA). 43 , 44 It was shown that the PRS was associated with remission (OR: 1.95, 95% CI: 1.20–3.17; p = 0.007). 43 Another study evaluated the PRS for antidepressant response with its association with functional brain network from EEG data, after controlling for age and five principal components. 45 The PRS was shown to be associated with the functional network, which is a predictive of treatment response. 45 Given that ketamine and scopolamine are thought to similarly activate synaptic plasticity, 46 a PRS developed using GWAS data of ketamine response was evaluated for the association with scopolamine response. 46 , 47 The PRS association was not significant for the scopolamine response, likely due to a limited sample size of 37 patients. 47
A number of studies investigated the association of pharmacogenomic PRS for antipsychotics and lithium. A recent study created a PRS from eight pharmacogenomics associations with clozapine metabolism. The PRS was associated with clozapine levels, its metabolite, and the metabolic ratio, after adjusting for sex, age, age, 2 principal components, genetic ancestry probabilities, the clozapine dose, and the time between dosing and sampling. This PRS explained 7% of the variance in clozapine metabolic ratio in a diverse cohort with multiple genetic ancestry. 48 A pharmacogenomic PRS for lithium response was recently constructed in the ConLi + Gen consortium using a Bayesian regression and continuous shrinkage methods, including SNPs from the lithium GWAS summary statistics. 49 The study assessed treatment response to lithium and determined the contribution of PRS to the variability in lithium response, using R‐squared (R 2). The difference in R 2 was calculated for a model that included PRS and covariates (age, sex, and principal components of ancestry), and another model with covariates only. Patients in the highest PRS decile (10th decile) were 3.47 times more likely to respond to lithium (95% CI: 2.22–5.47), compared to patients in the second PRS decile (OR: 1.95, 95% CI: 1.02–2.49, p = 6.4 × 10−9). 49 The difference in R 2 was reported to be 2.6%. 49 Most recently in 2024, a PRS was constructed from 11 variants of GWAS of antipsychotic response. 50 The PRS was tested for its association with response to antipsychotic medications in real world clinical setting, adjusting for age and five principal components. 50 The PRS was associated with response to antipsychotic treatment, regardless of the diagnosis (OR = 1.14, 95% CI: 1.03–1.26, p = 0.01), with a positive predictive value of 64%, negative predictive value of 57%, specificity of 58% and sensitivity of 63%. 50
As stated earlier in this paper, the observed inconsistency in the association of CAD PRS and some of the psychiatry suggests that the effectiveness of PRS in stratifying patients may depend on the specific disease and the medications. While the current studies in CAD and those in psychiatry show promise for PRS in predicting treatment response or patient stratification based on the PRS strata, there are no current data on whether PRS can guide the selection of treatment based on the underlying diagnosis, an area that is worth investigating in future studies.
CURRENT CHALLENGES AND LIMITATIONS OF THE PHARMACOGENOMIC PRS
It is notable that the research in developing disease PRS has advanced in recent years, at a much faster pace than in pharmacogenomic PRS. As of May 2024, a PubMed search using the keywords “Polygenic Risk Score” and “Disease” yielded over 10,477 publications, whereas the same search with “Pharmacogenomics” returns only 322 papers, with many papers focusing on psychiatry and less on cardiovascular and chemotherapy. 14 , 32 , 43 , 48 , 51 , 52 , 53 , 54 , 55 , 56 , 57 The difference in the number of publications suggests potential challenges that may be impeding the advancement of pharmacogenomics PRS, which will be highlighted in this section. Additionally, the landscape of pharmacogenomic PRS show that the majority of existing studies relied on a shortcut of constructing these PRS from disease variants, while fewer studies currently evaluated PRS directly from genetic variants of drug response.
For the pharmacogenomic PRS to advance, the current pharmacogenomics studies need to address some limitations that are inherent to pharmacogenomics, particularly with respect to measuring drug response and standardizing the phenotype definitions. Additionally, accurate phenotyping requires extensive data collection on medication adherence, dosage, and clinical outcomes in relation to medication start times. Further, although smaller sample sizes may theoretically be sufficient for pharmacogenomics studies given the larger effect sizes of pharmacogenomic variants compared to disease variants, the inconsistency and complexity of drug response phenotyping may necessitate large sample sizes to overcome the noise and variability of the phenotype. This power is not often available in pharmacogenetics studies. While resources such as large biobanks and disease consortia are available in disease genetics, such comparable resources are relatively nascent for pharmacogenomics phenotypes.
As discussed earlier, most of the studies extrapolate from existing disease variants to build pharmacogenomic PRS. A recent systematic review by Zhai et al. 27 found that 82% of the pharmacogenomic PRS studies used variants from disease GWAS. This approach of extrapolating from disease GWAS may not be an optimal approach for pharmacogenomic PRS due to a common, but not always accurate, assumption that disease‐related variants directly impact the effects of drugs. While there may be some genetic overlap between variants associated with disease predisposition and medication response, this relationship does not necessarily hold true for all medications and phenotypes. Additionally, drug response phenotypes, similar to complex disease traits, are polygenic and are also influenced by environmental factors. Therefore, environmental effects such as gene–drug interactions, which are important for drug response phenotypes, may not be accounted for when only relying on variants from disease genetics. Indeed, current studies rarely evaluate how PRS interact with medications in different PRS risk groups (high vs. low PRS), as done in the study by Lanfear et al. 41 Further, building pharmacogenomics PRS from pharmacogenomic variants have relied on candidate gene variants rather than using pharmacogenomic GWAS variants. 13 Selecting candidate gene variants to build PRS will often miss key pharmacogenomics variants, leading to a less accurate PRS. Together, these factors may explain some of the inconsistency observed among studies of pharmacogenomic PRS, with respect to the association with drug response phenotypes, across different medications and diseases, as was described. 6
A critical gap in pharmacogenomic PRS research is the need to evaluate the added value of PRS besides the established clinical predictors, including independent validation of existing pharmacogenomics PRS. While many studies found a significant association between the PRS and the drug response, less than half of the studies compare the PRS model to clinical predictors. 13 Additionally, many of these studies include some sort of internal validation, and do not validate in external cohorts, which can impact the generalizability of the developed pharmacogenomics PRS. 13 Siemens et al. found in his review that only a small percentage (4.5%) of the evaluated studies focused on validating existing PRS, reflecting a tendency of researchers toward building new PRS rather than replicating the existing ones. 6 , 13 A similar observation was reported in another review. 6 Compounding the lack of validation of pharmacogenomics PRS, other key challenges that hinder comparisons across studies include: (1) low reporting of prediction metrics, (2) variability in the reported metrics (e.g., AUC, C‐statistics, pseudo‐R, 2 positive predictive value (PPV), and negative predictive value (NPV)); and (3) the lack of guidance on standardized cutoffs for relative or absolute risk reduction estimation. 58 As discussed earlier in “Current Approaches for construction of pharmacogenomic PRS,” some studies report metrics like AUC, C‐statistics, pseudo‐R, 2 and rank‐based risk classifications such as odds ratios per percentile, which while informative, may not directly translate to individual level predictions as they do not often account for the baseline risk in the untreated population. 28 Absolute risk metrics, such as PPV and NPV, which define the proportions of patients who truly have or do not have the drug response phenotype among those who test positive or negative, respectively, offer more clinical utility, but are less commonly reported. 28 Further, there are insufficient data to show the added value of PRS for predicting drug response beyond what can be predicted by the clinical and demographic factors (e.g., age, sex, renal function, or hepatic function). A recent study that included PRS and other clinical predictors was able to explain 48% of variability in the response to antipsychotics. 38 For future clinical implementation, it is necessary to develop and validate integrated models that incorporate both pharmacogenomic PRS and clinical factors, similar to some of the recent disease PRS papers. 10
ARE WE READY FOR PRIME TIME? IF NOT NOW, WHEN?
Pharmacogenomics PRS are poised to gain prominence. Compared to disease PRS, pharmacogenomics PRS for most drugs are less likely to encounter concerns of needing extra diagnostic tests or scans, potentially reducing patient burden and streamlining the medication decision‐making process. Additionally, the large effect sizes typically observed for PRS, may translate to a better prediction of drug response. 30 The current evidence suggests that the utility of pharmacogenomic PRS will likely be in stratifying patients for drug treatments, such as initiating PCSK9 inhibitors. A broader utility of pharmacogenomic PRS to guide treatment selection based on diagnosis requires further research.
While there has been steady advancement in the field of pharmacogenomic PRS, the current evidence suggests that the time for widespread adoption is yet to come. To effectively evaluate pharmacogenomic PRS use in precision medicine and facilitate their widespread adoption, we propose recommendations (Figure 2) in three categories aimed at mitigating the existing limitations and challenges: (1) enhance the research of developing pharmacogenomic PRS; (2) facilitate the clinical implementation via enhancing the infrastructure; and (3) address the ethical and regulatory issues.
FIGURE 2.
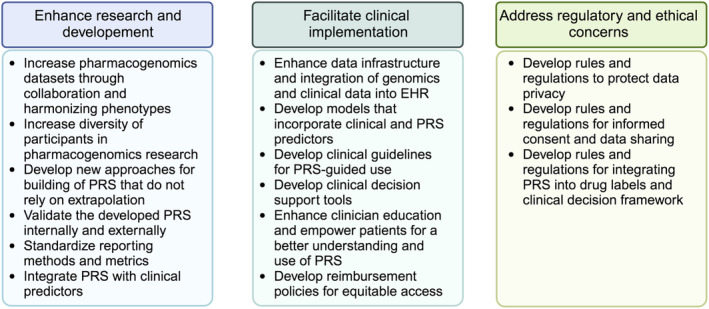
Recommendations to advance the utilization of the pharmacogenomics PRS.
Pharmacogenomic phenotypes represent an intricate interplay of multiple genetic variants and, therefore, pharmacogenomic PRS development will require the collection of large‐scale, high‐quality, standardized genomic data from diverse populations. Given the dynamic nature of drug responses, longitudinal data collection and analysis are essential to continuously refine and update PRS. Further, establishing standardized definitions for measuring drug response phenotypes, coupled with enhanced data collection efforts to capture information on medication dates, dosage, and clinical outcomes, are essential. Researchers should engage in collaborative efforts to harmonize phenotypes, increase sample sizes, and develop statistical methods that transition away from reliance on candidate gene variants and /or extrapolation from disease GWAS associations. Newer methods should explore integrating GWAS variants from disease traits and drug response, incorporating newer Bayesian regression methods, similar to the recently proposed. 3 , 26 The newly developed methods should identify the relevant variants to be included in PRS, rather than an arbitrary selection of pharmacogenomic variants based on genome‐wide significance thresholds, with ambiguity toward the independent SNPs, ultimately leading to inaccurate PRS.
The existing PRS methods generally have limited portability across‐ and sometimes even within‐ancestries. 59 , 60 This effect can be due to ancestral differences in the frequencies of alleles, patterns of linkage disequilibrium, or effect sizes associated with a variant. Moreover, the existing PRS are Eurocentric as the largest GWAS are typically comprised largely of individuals of European ancestry, and therefore the application of PRS has the potential to create systematic disparities. 61 While SNP selection approaches to mitigate this problem are under development, 62 the underlying issue is likely best addressed by expanded inclusion of other ancestral populations in genetic studies. 63 Forming large consortia for pharmacogenomic PRS development that include diverse genetic ancestry, similar to existing disease PRS consortia like the Polygenic RIsk MEthods in Diverse populations (PRIMED), can address concerns of widening disparity if pharmacogenomic PRS continue to be developed in European ancestry.
Prior to reaching the clinical implementation stage, pharmacogenomic PRS will require validation to ensure their accuracy and reproducibility across independent datasets. Given the limited clinical evidence linking numerous genetic variants to drug responses, replicating and/or validating PRS across diverse datasets merits particular attention. Furthermore, standardizing prediction metrics and reporting practices is essential to enable comparison across studies, including both internal and external validation. Finally, prioritizing and assessing the integration of pharmacogenomic PRS with clinical predictors, similar to clinical risk tools, 64 should be done to discern the contribution of pharmacogenomic PRS and justify their clinical use.
The clinical use of pharmacogenomic PRS requires overcoming technical challenges and enhancing the infrastructure and clinician support. The voluminous nature of genomic data necessitates secure storage solutions, which is accompanied with high costs. Additionally, integrating the genomic data and other clinical data within EHR is essential for obtaining a comprehensive clinical view of patients' health. All of these create financial constraints, particularly in resource‐limited settings. Moreover, PRS generate numerical scores that can pose challenges for clinicians with regards to the interpretation and practical application. Therefore, there is an urgent need for the development of user‐friendly tools to facilitate a seamless incorporation of PRS into clinical practice similar to the efforts led by the eMERGE network. 65 Clinical guidelines, such as those developed by the Clinical Pharmacogenetics Implementation Consortium (CPIC) and others, will become essential for interpreting PRS results and guiding treatment decisions. The creation of these guidelines will need to be developed in coordination with training for healthcare professionals to effectively understand and utilize PRS. Further, initiatives to address health literacy and empower patients to act upon their PRS information, will need to take place. Patients must be educated that PRS is a risk prediction and not a diagnostic tool. 66 , 67 Simultaneously, a rigorous evaluation of the cost‐effectiveness of PRS implementation in clinical practice is critical to ensure sustainable and equitable access of PRS across diverse populations, alongside with the development of reimbursement policies that incentivize the adoption of PRS‐guided treatments.
Finally, a regulatory framework must be established if PRS are to be incorporated into drug labeling and clinical decision processes. Policies and procedures are essential to ensure that patients understand the implications of PRS generation. Laws and regulations are needed to protect patients' privacy and prevent genomic data breaches, including strong data security measures. The regulatory framework should prevent against discrimination based on a patients' PRS, allow equitable access, and prevent harms from any misuse of PRS. Further, regulatory guidance should be in place to promote the standardization of PRS development and validation. The regulatory framework should be flexible to accommodate scientific discoveries, dynamic changes in PRS and address arising challenges.
SUMMARY AND CONCLUSION
Overcoming the current limitations of the existing pharmacogenomic PRS research presents the next logical step before clinical implementation. The adoption of pharmacogenomic PRS in the clinics is a complex process, and will require a comprehensive approach that addresses infrastructure, clinical interpretation and use, and ethical and regulatory considerations. This process will likely be gradual, requiring ongoing collaboration among researchers from academic and industry, implementers of pharmacogenomics, healthcare professionals, regulators, and policymakers. Together, they can ensure robust development and rigorous evaluation of pharmacogenomics PRS, followed by a safe and effective implementation into clinical care. Ongoing research and large‐scale collaboration are crucial to overcoming existing obstacles and bring PRS to fruition.
FUNDING INFORMATION
No funding was received for this work.
CONFLICT OF INTEREST STATEMENT
The authors declare no competing interests for this work.
Singh S, Stocco G, Theken KN, et al. Pharmacogenomics polygenic risk score: Ready or not for prime time? Clin Transl Sci. 2024;17:e13893. doi: 10.1111/cts.13893
Sonal Singh and Nihal El Rouby contributed equally.
Disclaimer: As an Associate Editor for Clinical and Translational Science, Sonal Singh was not involved in the review or decision process for this paper.
Nihal El Rouby and Jonathan D. Mosley shared corresponding author.
Contributor Information
Jonathan D. Mosley, Email: jonathan.d.mosley@vumc.org.
Nihal El Rouby, Email: elroubnl@ucmail.uc.edu.
REFERENCES
- 1. Liu C, Yang W, Pei D, et al. Genomewide approach validates thiopurine methyltransferase activity is a monogenic pharmacogenomic trait. Clinical Pharmacology and Therapeutics. 2017;101(3):373‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Muhammad A, Aka IT, Birdwell KA, et al. Genome‐wide approach to measure variant‐based heritability of drug outcome phenotypes. Clinical Pharmacology and Therapeutics. 2021;110(3):714‐722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhai S, Zhang H, Mehrotra DV, Shen J. Pharmacogenomics polygenic risk score for drug response prediction using PRS‐PGx methods. Nature Communications. 2022;13(1):5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Krebs K, Milani L. Harnessing the power of electronic health records and genomics for drug discovery. Annual Review of Pharmacology and Toxicology. 2023;63:65‐76. [DOI] [PubMed] [Google Scholar]
- 5. Li B, Ritchie MD. From GWAS to gene: transcriptome‐wide association studies and other methods to functionally understand GWAS discoveries. Frontiers in Genetics. 2021;12:713230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Johnson D, Wilke MAP, Lyle SM, et al. A systematic review and analysis of the use of polygenic scores in pharmacogenomics. Clinical Pharmacology and Therapeutics. 2022;111(4):919‐930. [DOI] [PubMed] [Google Scholar]
- 7. O'Sullivan JW, Raghavan S, Marquez‐Luna C, et al. Polygenic risk scores for cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2022;146(8):e93‐e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu X, Littlejohns TJ, Bešević J, et al. Incorporating polygenic risk into the Leicester risk assessment score for 10‐year risk prediction of type 2 diabetes. Diabetes and Metabolic Syndrome: Clinical Research and Reviews. 2024;18(4):102996. [DOI] [PubMed] [Google Scholar]
- 9. Iribarren C, Lu M, Elosua R, et al. Polygenic risk and incident coronary heart disease in a large multiethnic cohort. Am J Prev Cardiol. 2024;18:100661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Patel AP, Wang M, Ruan Y, et al. A multi‐ancestry polygenic risk score improves risk prediction for coronary artery disease. Nature Medicine. 2023;29(7):1793‐1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lennon NJ, Kottyan LC, Kachulis C, et al. Selection, optimization and validation of ten chronic disease polygenic risk scores for clinical implementation in diverse US populations. Nature Medicine. 2024;30(2):480‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Slunecka JL, van der Zee MD, Beck JJ, et al. Implementation and implications for polygenic risk scores in healthcare. Human Genomics. 2021;15(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Siemens A, Anderson SJ, Rassekh SR, Ross CJD, Carleton BC. A systematic review of polygenic models for predicting drug outcomes. J Pers Med. 2022;12(9):1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cross B, Turner R, Pirmohamed M. Polygenic risk scores: an overview from bench to bedside for personalised medicine. Frontiers in Genetics. 2022;13:1000667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choi SW, Mak TS, O'Reilly PF. Tutorial: a guide to performing polygenic risk score analyses. Nature Protocols. 2020;15(9):2759‐2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burgess S, Thompson SG. Use of allele scores as instrumental variables for Mendelian randomization. International Journal of Epidemiology. 2013;42(4):1134‐1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Purcell SM, Wray NR, Stone JL, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vilhjálmsson BJ, Yang J, Finucane HK, et al. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. American Journal of Human Genetics. 2015;97(4):576‐592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Newcombe PJ, Nelson CP, Samani NJ, Dudbridge F. A flexible and parallelizable approach to genome‐wide polygenic risk scores. Genetic Epidemiology. 2019;43(7):730‐741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ge T, Chen CY, Ni Y, Feng YA, Smoller JW. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nature Communications. 2019;10(1):1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lloyd‐Jones LR, Zeng J, Sidorenko J, et al. Improved polygenic prediction by Bayesian multiple regression on summary statistics. Nature Communications. 2019;10(1):5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Privé F, Arbel J, Vilhjálmsson BJ. LDpred2: better, faster, stronger. Bioinformatics. 2021;36(22–23):5424‐5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang S, Zhou X. Accurate and scalable construction of polygenic scores in large biobank data sets. American Journal of Human Genetics. 2020;106(5):679‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mak TSH, Porsch RM, Choi SW, Zhou X, Sham PC. Polygenic scores via penalized regression on summary statistics. Genetic Epidemiology. 2017;41(6):469‐480. [DOI] [PubMed] [Google Scholar]
- 25. Ma Y, Zhou X. Genetic prediction of complex traits with polygenic scores: a statistical review. Trends in Genetics. 2021;37(11):995‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhai S, Guo B, Wu B, Mehrotra DV, Shen J. Integrating multiple traits for improving polygenic risk prediction in disease and pharmacogenomics GWAS. Briefings in Bioinformatics. 2023;24(4):1–11. [DOI] [PubMed] [Google Scholar]
- 27. Zhai S, Mehrotra DV, Shen J. Applying polygenic risk score methods to pharmacogenomics GWAS: challenges and opportunities. Briefings in Bioinformatics. 2023;25(1):bbad470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kachuri L, Chatterjee N, Hirbo J, et al. Principles and methods for transferring polygenic risk scores across global populations. Nature Reviews. Genetics. 2024;25(1):8‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Klarin D, Natarajan P. Clinical utility of polygenic risk scores for coronary artery disease. Nature Reviews. Cardiology. 2022;19(5):291‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Khera AV, Chaffin M, Aragam KG, et al. Genome‐wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nature Genetics. 2018;50(9):1219‐1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mega JL, Stitziel NO, Smith JG, et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet. 2015;385(9984):2264‐2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Natarajan P, Young R, Stitziel NO, et al. Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation. 2017;135(22):2091‐2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oni‐Orisan A, Haldar T, Cayabyab MAS, et al. Polygenic risk score and statin relative risk reduction for primary prevention of myocardial infarction in a real‐world population. Clinical Pharmacology and Therapeutics. 2022;112(5):1070‐1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Damask A, Steg PG, Schwartz GG, et al. Patients with high genome‐wide polygenic risk scores for coronary artery disease May receive greater clinical benefit from Alirocumab treatment in the ODYSSEY OUTCOMES trial. Circulation. 2020;141(8):624‐636. [DOI] [PubMed] [Google Scholar]
- 35. Marston NA, Kamanu FK, Nordio F, et al. Predicting benefit from Evolocumab therapy in patients with atherosclerotic disease using a genetic risk score: results from the FOURIER trial. Circulation. 2020;141(8):616‐623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Frank J, Lang M, Witt SH, et al. Identification of increased genetic risk scores for schizophrenia in treatment‐resistant patients. Molecular Psychiatry. 2015;20(2):150‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kappel DB, Legge SE, Hubbard L, et al. Genomic stratification of clozapine prescription patterns using schizophrenia polygenic scores. Biological Psychiatry. 2023;93(2):149‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guo LK, Su Y, Zhang YY, et al. Prediction of treatment response to antipsychotic drugs for precision medicine approach to schizophrenia: randomized trials and multiomics analysis. Military Medical Research. 2023;10(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wimberley T, Gasse C, Meier SM, Agerbo E, MacCabe JH, Horsdal HT. Polygenic risk score for schizophrenia and treatment‐resistant schizophrenia. Schizophrenia Bulletin. 2017;43(5):1064‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lewis JP, Backman JD, Reny JL, et al. Pharmacogenomic polygenic response score predicts ischaemic events and cardiovascular mortality in clopidogrel‐treated patients. European Heart Journal ‐ Cardiovascular Pharmacotherapy. 2020;6(4):203‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lanfear DE, Luzum JA, She R, et al. Polygenic score for β‐blocker survival benefit in European ancestry patients with reduced ejection fraction heart failure. Circulation. Heart Failure. 2020;13(12):e007012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lanfear DE, Luzum JA, She R, et al. Validation of a polygenic score for Beta‐blocker survival benefit in patients with heart failure using the United Kingdom biobank. Circ Genom Precis Med. 2023;16(2):e003835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Men X, Marshe V, Elsheikh SS, et al. Genomic investigation of remission and relapse of psychotic depression treated with sertraline plus olanzapine: the STOP‐PD II study. Neuropsychobiology. 2023;82(3):168‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pain O, Hodgson K, Trubetskoy V, et al. Identifying the common genetic basis of antidepressant response. Biol Psychiatry Glob Open Sci. 2022;2(2):115‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Meijs H, Prentice A, Lin BD, et al. A polygenic‐informed approach to a predictive EEG signature empowers antidepressant treatment prediction: a proof‐of‐concept study. European Neuropsychopharmacology. 2022;62:49‐60. [DOI] [PubMed] [Google Scholar]
- 46. Duman RS, Aghajanian GK, Sanacora G, Krystal JH. Synaptic plasticity and depression: new insights from stress and rapid‐acting antidepressants. Nature Medicine. 2016;22(3):238‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guo W, Machado‐Vieira R, Mathew S, et al. Exploratory genome‐wide association analysis of response to ketamine and a polygenic analysis of response to scopolamine in depression. Translational Psychiatry. 2018;8(1):280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pardiñas AF, Kappel DB, Roberts M, et al. Pharmacokinetics and pharmacogenomics of clozapine in an ancestrally diverse sample: a longitudinal analysis and genome‐wide association study using UK clinical monitoring data. Lancet Psychiatry. 2023;10(3):209‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Amare AT, Thalamuthu A, Schubert KO, et al. Association of polygenic score and the involvement of cholinergic and glutamatergic pathways with lithium treatment response in patients with bipolar disorder. Molecular Psychiatry. 2023;28:5251‐5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. De Pieri M, Ferrari M, Pistis G, et al. Prediction of antipsychotics efficacy based on a polygenic risk score: a real‐world cohort study. Frontiers in Pharmacology. 2024;15:1274442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pardiñas AF, Smart SE, Willcocks IR, et al. Interaction testing and polygenic risk scoring to estimate the Association of Common Genetic Variants with Treatment Resistance in schizophrenia. JAMA Psychiatry. 2022;79(3):260‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Laaboub N, Gholam M, Sibailly G, et al. Associations between high plasma Methylxanthine levels, sleep disorders and polygenic risk scores of caffeine consumption or sleep duration in a Swiss psychiatric cohort. Frontiers in Psychiatry. 2021;12:756403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Campos AI, Ngo TT, Medland SE, et al. Genetic risk for chronic pain is associated with lower antidepressant effectiveness: converging evidence for a depression subtype. The Australian and New Zealand Journal of Psychiatry. 2022;56(9):1177‐1186. [DOI] [PubMed] [Google Scholar]
- 54. Zwicker A, Fabbri C, Rietschel M, et al. Genetic disposition to inflammation and response to antidepressants in major depressive disorder. Journal of Psychiatric Research. 2018;105:17‐22. [DOI] [PubMed] [Google Scholar]
- 55. Biernacka JM, Coombes BJ, Batzler A, et al. Genetic contributions to alcohol use disorder treatment outcomes: a genome‐wide pharmacogenomics study. Neuropsychopharmacology. 2021;46(12):2132‐2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Werner MCF, Wirgenes KV, Haram M, et al. Indicated association between polygenic risk score and treatment‐resistance in a naturalistic sample of patients with schizophrenia spectrum disorders. Schizophrenia Research. 2020;218:55‐62. [DOI] [PubMed] [Google Scholar]
- 57. Duconge J, Santiago E, Hernandez‐Suarez DF, et al. Pharmacogenomic polygenic risk score for clopidogrel responsiveness among Caribbean Hispanics: a candidate gene approach. Clinical and Translational Science. 2021;14(6):2254‐2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Simona A, Song W, Bates DW, Samer CF. Polygenic risk scores in pharmacogenomics: opportunities and challenges—a mini review. Frontiers in Genetics. 2023;14:1217049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Martin AR, Gignoux CR, Walters RK, et al. Human demographic history impacts genetic risk prediction across diverse populations. American Journal of Human Genetics. 2017;100(4):635‐649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mostafavi H, Harpak A, Agarwal I, Conley D, Pritchard JK, Przeworski M. Variable prediction accuracy of polygenic scores within an ancestry group. eLife. 2020;9:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nature Genetics. 2019;51(4):584‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ruan Y, Lin YF, Feng YA, et al. Improving polygenic prediction in ancestrally diverse populations. Nature Genetics. 2022;54(5):573‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Novembre J, Stein C, Asgari S, et al. Addressing the challenges of polygenic scores in human genetic research. American Journal of Human Genetics. 2022;109(12):2095‐2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fuat A, Adlen E, Monane M, et al. A polygenic risk score added to a QRISK®2 cardiovascular disease risk calculator demonstrated robust clinical acceptance and clinical utility in the primary care setting. European Journal of Preventive Cardiology. 2024;31:716‐722. [DOI] [PubMed] [Google Scholar]
- 65. Linder JE, Allworth A, Bland HT, et al. Returning integrated genomic risk and clinical recommendations: the eMERGE study. Genetics in Medicine. 2023;25(4):100006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Abu‐El‐Haija A, Reddi HV, Wand H, et al. The clinical application of polygenic risk scores: a points to consider statement of the American College of Medical Genetics and Genomics (ACMG). Genetics in Medicine. 2023;25(5):100803. [DOI] [PubMed] [Google Scholar]
- 67. Alliance PRSTFotICD . Responsible use of polygenic risk scores in the clinic: potential benefits, risks and gaps. Nature Medicine. 2021;27(11):1876‐1884. [DOI] [PubMed] [Google Scholar]
- 68. Choi SW, O'Reilly PF. PRSice‐2: polygenic risk score software for biobank‐scale data. GigaScience. 2019;8(7):giz082. [DOI] [PMC free article] [PubMed] [Google Scholar]