

Abstract
Modern histologic imaging platforms coupled with machine learning methods have provided new opportunities to map the spatial distribution of immune cells in the tumor microenvironment. However, there exists no standardized method for describing or analyzing spatial immune cell data, and most reported spatial analyses are rudimentary. In this review, we provide an overview of two approaches for reporting and analyzing spatial data (raster versus vector-based). We then provide a compendium of spatial immune cell metrics that have been reported in the literature, summarizing prognostic associations in the context of a variety of cancers. We conclude by discussing two well-described clinical biomarkers, the breast cancer stromal tumor infiltrating lymphocytes score and the colon cancer Immunoscore, and describe investigative opportunities to improve clinical utility of these spatial biomarkers.
Keywords: spatial statistics, tumor infiltrating lymphocytes, TIL, sTIL score, Immunoscore, multispectral immunofluorescence, spatial heterogeneity
Introduction
With advances in technology, it is now possible to visualize the spatial interface between tumors and host immunity on the level of individual cells, thus generating vast spatial datasets that may contain clinically important prognostic and predictive information. However, this capability is accompanied by the challenge of developing rigorous and reproducible statistical methods for reporting and interpreting spatial immune cell (IC) data. Currently there is no standardized approach; most published analyses only scratch the surface of the rich information hidden within the spatial orientation of ICs. Here, we summarize published spatial IC analysis methods and future directions. The review is organized into three sections. First, we summarize the status of histologic imaging technology, including machine learning (ML) approaches to identify IC cancer cell spatial locations and their attributes, such as cellular phenotype. We also introduce the basic data structure, terminology, and analytic methods for spatial datasets. Second, we summarize published research in the field, providing examples of different types of spatial metrics across categories including IC density, heterogeneity, clustering, colocalization, interfacing, and tissue segmentation. Finally, we conclude by discussing the spatial analytic methods of two leading IC prognostic biomarkers, the breast cancer stromal tumor infiltrating lymphocytes (sTIL) score [1] and the colon cancer Immunoscore [2], and propose how advanced spatial analysis methods could be applied to create new spatial biomarkers.
Data acquisition: technologies, ML, and spatial data structure
Here, we provide a summary of imaging technologies, methods of acquiring spatial data from images, and the organizational structure of spatial data.
Imaging technologies
Modern imaging technologies have permitted the acquisition of spatial datasets of ICs in cancer. The most common platforms employ high-resolution microscopy to evaluate the expression of multiple cell markers from a single tumor slide. A shared feature of these platforms (which include multiplex immunohistochemistry [mIHC] and immunofluorescence [mIF]) is the use of multiple antibodies that bind to and label specific cellular proteins, to visualize various cell types and identify their precise two-dimensional geographic location in relation to cancer cells. Analysis of multiple markers on a single slide ensures that spatial relationships of cell types are preserved, whereas single-marker analyses on sequential slides may be less accurate for characterizing spatial relationships between cell types. Great care must be taken to standardize and validate the assay workflow, which includes steps such as antibody/reagent selection, antibody staining order, sample storage, processing, and staining, and quality control steps employing positive and negative controls to ensure consistency. Best practices are summarized in an expert consensus statement by the Society for Immunotherapy of Cancer (SITC) [3]. In addition to mIF and mIHC, nascent technologies permit extensive characterization of protein or RNA differential expression on the level of individual cells, or within spatially defined subregions of the tumor. Such technological platforms include mass ion beam imaging, imaging mass cytometry, digital spatial profiling, or spatial single-cell RNA sequencing, which are summarized elsewhere [3–5]. Of note, older platforms, such as single-stain chromogenic IHC or H&E can still provide a wealth of spatial information, which can be leveraged using imaging software such as QuPath [6,7].
ML to extract spatial IC data from images
A shared and crucial step across all imaging platforms is to extract spatial data from raw images. High-resolution microscopy images are large datasets comprised of many pixel locations, each with associated intensity or color/hue values. Automated methods using ML are helpful in extracting spatial features from the imaging data that summarize the spatial distribution of cell and tissue types. Additionally, computational platforms are used to spectrally unmix multiplexed imaging data to accurately identify the spatial coordinates of the different biomarkers. Using various ML-based approaches, images can be used to generate data features such as tissue type (i.e. stroma, necrosis, fat, microvessels, cancer cell nest, blank/acellular) [8], region-specific features (such as the invasive margin [IM] of the tumor), and cells (annotated by location and phenotype). ML methods can be either fully automated, such as with the Definiens imaging analysis platform for the colon cancer Immunoscore [2] (Developer XD, Definiens, Munich, Germany), or they can depend on user supervision to refine the classification of tissues or cells, as with the mIF InForm software package (Akoya Biosciences, Marlborough, MA, USA) [9,10]. A separate summary statement has been published that describes ML and computational methods for acquiring spatial IC data [11].
Structure and analysis of spatial IC datasets
Before the advent of these technologies, spatial IC data were reported descriptively, visually, or in the form of whole-slide summary metrics, such as cell count, intensity, density, or other textural features. These metrics are poor at characterizing heterogeneity of ICs across the span of the tumor. It is now possible to quantify IC densities precisely across multiple geographic subregions of the tumor. In the context of mIF/mlHC, this is often conducted across high-power microscopy fields (HPFs), determined by the field of view of the microscope lens [3,12]. When ICs are interrogated across multiple subregions, characteristics of subregions within a tumor can be compared. With these data, additional information, such as ‘hotspot zones’ or inter-region variability can be reported (described further below).
When a tumor is repeatedly sampled across HPFs, it is important to consider the underlying geographic interdependencies across the various HPFs. Summary statistics, such as mean IC density across multiple HPFs, can be confounded if sampling of subregions is non-uniform and/or if an insufficient proportion of the tumor is sampled. To build robust analytic pipelines using HPFs, it will be important to determine the number of HPFs required to accurately characterize the tumor and to ensure that the sampling distribution is not spatially biased toward certain tissue architectures. It was recently shown in the context of a breast cancer cytokine-based immunotherapy trial that at least 15 HPFs per patient were required to overcome the potential confounding effect of spatial heterogeneity and accurately characterize changes in IC density related to the cytokine treatment [12], This trial also illustrated how statistical models, such as hierarchical linear modeling, could be leveraged to better capture intratumoral heterogeneity and estimate IC density [12]. In a melanoma cohort it was demonstrated how HPFs could be tiled to cover the entirety of the tumor space and overlapped by 20% to permit correction of errors on the periphery of HPF images due to illumination and lens effects [13]. An alternative to tiling HPFs is to utilize whole-slide imaging platforms, such as the mIF Polaris (Akoya Biosciences); however, these platforms are still in development and the imaging can be time-consuming.
To fully utilize spatial data, observations must be annotated by their exact spatial/geographic position, so that both spatial interdependencies and heterogeneity can be fully explored. There are two distinct approaches for analyzing spatial data, raster analysis and vector analysis, each of which can be used in different contexts of spatial IC analysis (illustrated in Figure 1A–D). Vector-based datasets can be converted to raster-based and vice versa. However, when converting raster to vector, the resolution depends on the size of the raster cells.
Figure 1.
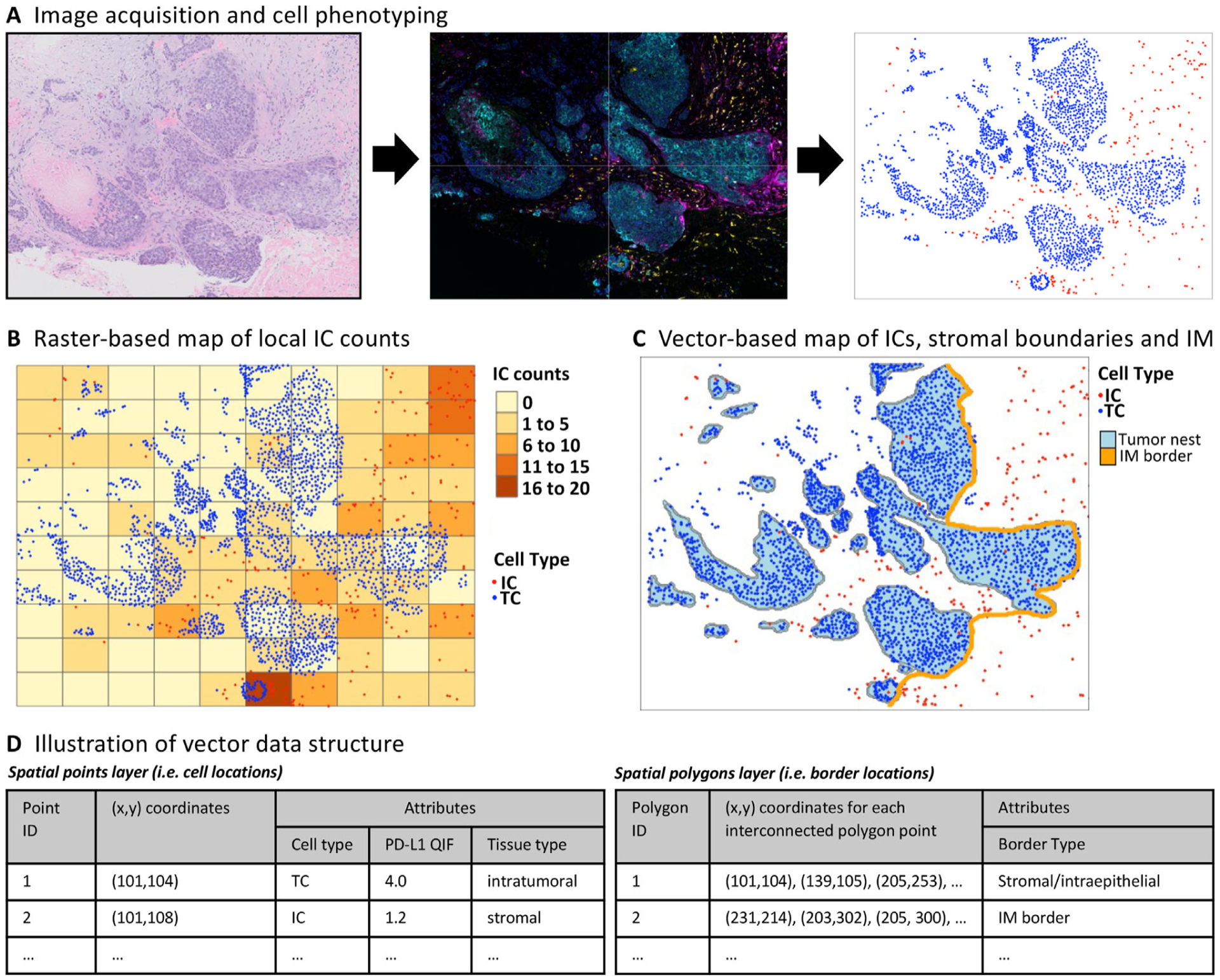
Raster versus vector spatial data structure. (A) An example of a TNBC specimen imaged by H&E, with high-resolution multi-color images obtained using mIF (Vectra platform). High-resolution images are used to obtain cell coordinates and phenotypes. (B) In raster-based spatial analysis, the tumor is divided into small subregions (usually by a rectangular grid) and spatial metrics are calculated independently across each subregion, allowing for analysis of spatial metrics such as average cell count, deviation/skewness, and hotspot analysis. (C and D) In vector-based spatial analysis, cells are annotated by their phenotype, (X,Y) geographic location, and other attributes, such as PD-L1 expression. These data can then be analyzed using statistical software to calculate a variety of metrics.
With raster-based datasets, spatial analyses are accomplished by partitioning the evaluable space by a grid of subregions or raster cells, each annotated by their geographic location. For each raster cell, measurements pertaining to IC infiltration (such as IC density, programmed cell death ligand 1 [PD-L1] intensity, percentage tumor/stroma) are individually estimated, thus generating a mosaic of estimations across the entirety of the tumor space (Figure 1B). This method allows for the identification of hotspot zones, investigation of spatial relationships between ICs and locoregional anatomic structures, and evaluation of the heterogeneity of IC characteristics.
One important factor affecting raster analyses is the partitioning scale/grid size: if the raster cell size is too large, the resolution is poor for detecting locoregional variations (such as hotspots or variations in IC density); whereas if the raster cell size is too small, estimates of IC density become volatile due to inadequate sampling space within each cell. In a recent breast cancer prognostic evaluation, raster cell size affected the overall prognostic performance of IC hotspot analysis [14]. There are several proposed methods for optimizing raster cell size. One is to select a size that reflects underlying biological considerations, such as the span of cell-cell paracrine signaling [15]. An alternative to rectangular grids is provided by Voronoi tessellation [6], which can be used to subdivide the tumor space into uneven polygons that maximize grouping of cells with their nearest neighbors, and could be more flexible to adapt to underlying anatomic structures [7,16]. Superpixels is another method that segments tumors into raster cells according to similarities in adjacent pixel colors or other features [17,18]. Finally, approaches can be implemented that analyze tumors on different scales (multiscaling) or to optimize raster cell size as a hyperparameter, as it is effectively changing the resolution and accuracy of the statistical modeling [19].
Vector-based datasets are comprised of a set of unique observations corresponding to each visualized cell across the tumor space, with measured attributes such as cell type (cancer versus immune versus stromal cell) or protein expression (such as PD-L1 quantitative intensity by mIF). Each observation/cell is spatially annotated by its point location (x, y coordinates in two-dimensional space; Figure 1C,D) [20]. Locations of anatomic boundaries within the tumor, such as the IM or cancer cell/stromal interface, are defined as a series of (x,y) spatial points, that when connected linearly, create either a line or a polygon in two-dimensional space (Figure 1C,D). With these multi-featured vector datasets, software packages (such as Python or Simple Features sf package in R [20]) can efficiently calculate distances to the IM for each IC, calculate locoregional IC densities or PD-L1 expression, or label ICs according to the location within different tissue compartments (such as intraepithelial versus stromal). Another benefit of vector datasets is the ability to convert the data into detailed spatial maps that graphically illustrate the interrelationships between cancer cells and ICs (illustrated in Figure 1C).
Analysis of spatial data: overview of published research
Here we provide an overview of spatial IC analyses that have been conducted across a variety of cancer contexts and summarize their potential clinical prognostic and/or predictive utility. When possible, we describe these metrics using the terminology and framework of raster versus vector spatial analyses. We also describe the metrics as either global (i.e. summarized across all spatial subunits) or local (i.e. metrics that are reported repeatedly across spatial subunits). These metrics are summarized in Table 1.
Table 1.
Examples of spatial metrics and definitions.
| Metric/references | Local versus global* | Raster versus vector | Description |
|---|---|---|---|
| Count/density metrics | |||
| Cell count [21–23] | Either | Raster | Count of cells within a given analysis area |
| Cell density [21–23] | Either | Raster | Count of cells, normalized for area |
| Density ratio [24–26] | Either | Raster | Ratio of two cellular densities |
| Radial density [27] | Either | Vector | Density of cells within a given radius of a given geographic location |
| Kernel density [28] | Local | Vector | Density of cells in an area, smoothed for densities in surrounding area |
| Heterogeneity metrics | |||
| Density variance [29] | Global | Raster | Variation in densities across subregions of a tumor |
| Coefficient of variation [30] | Global | Raster | Variance, normalized for overall cell density |
| Quartile coefficient of dispersion [30] | Global | raster | Alternate measure of heterogeneity, less influenced by outlier regions |
| Skewness [31] | Global | Raster | Global measure of degree of outliers in a sample |
| Shannon entropy [6] | Global | Raster | Global measure of heterogeneity/randomness across subregions |
| Clustering metrics | |||
| Quadrat analysis [6] | Global | Raster | Variance/mean ratio of densities across subregions, scores >1 indicate clustering |
| Moran’s I statistic [20] | Global | Raster | Strength of correlation of cell densities with those of neighboring subregions |
| Ripley’s K function [32] | Global | Vector | Metric providing clustering strength at different cell–cell distances |
| Getis-Ord general G-statistic [20] | Global | Raster | Generalized metric summarizing degree of clustering across neighboring subregions |
| Hotspot enumeration [33] | Global | Raster | Quantification of numbers of hotspots in a sample |
| Hotspot fractional area [33] | Global | Raster | Proportion of tumor that meets hotspot criteria |
| Hotspot density [2,13,29,34] | Global | Raster | Average density within hotspot regions (various definitions of hotspots can be used) |
| Getis-Ord local Gi [20] | Local | Raster | Hypothesis test to evaluate presence/absence of hotspot (p value for each subregion) |
| Local Moran’s l [20] | Local | Raster | Application of Moran’s I statistic to identify subregions likely to be hotspots |
| Cluster morphometric analysis [6,35] | Local | Vector | Evaluation of shape and other geometric characteristics of IC clusters |
| Colocalization metrics | |||
| Morisita-Horn Index [7,16] | Global | Raster | Global measure of degree of localization of two cell species |
| G-cross function AUC [36] | Global | Vector | Estimation of likelihood of two cell types to be adjacent, across various distances |
| Nearest neighbor [27] | Global | Vector | Average nearest pairwise distance of two cell types |
| Interface metrics | |||
| Band density [6,37] | Global | Either | Cell densities measured at intervals of distances from IM |
| Band density ratio [37] | Global | Either | Ratio of band densities (measure of dependency of cell density with IM) |
| Fractal dimension [7,34,38] | Global | Vector | Measure of complexity of tumor/stromal border |
| Density gradient [39] | Global | Either | Rate of change of cell density, by distance from tumor IM |
| Segmentation metrics | |||
| Intratumoral density ratio [28] | Global | Raster | Ratio of cellular densities, comparing stromal versus intraepithelial |
| Area ratios [20] | Global | Vector | Ratio of stromal area to intraepithelial area (or other areas of interest) |
Local refers to analyses conducted across various tumor subregions; global refers to analyses that summarize the whole tumor.
IC counts/densities
The most basic spatial metric is IC count or density. Cell count is defined as the number of detected cells within a given spatial unit (i.e. HPF), whereas density is the cell count normalized by the two-dimensional area of the spatial unit. Cell counts can be influenced dramatically by tissue composition, which may be an admixture of cancer cells, non-cancerous tissue (stroma, adipocytes, vessels/lymphatics), non-viable tissue (necrosis or artifacts), or empty space. Cell densities are preferred over counts when comparing IC infiltration across regions with a non-uniform evaluable tumor area. Densities of certain cell types have been shown across multiple studies to be prognostic of outcome or predictive of response to immunotherapy or chemotherapy. For example, CD8+ cell density within either the tumor compartment or the IM was shown to predict melanoma response to treatment with anti-PD-1 (pembrolizumab) [21]. In early-stage triple-negative breast cancer (TNBC), the TIL density within the stromal compartment in an H&E slide is prognostic of outcome [40], predictive of neoadjuvant chemotherapy response [22], and predictive of anti-PD-1 response [23]. Interestingly, in a study on TNBC correlating TIL density assessed by AI in different tissue compartments (intratumoral, within the tumor stroma, at the tumor periphery, etc.) with patient outcome, it was shown that a similar prognostic value could be observed independent of the measurement region, indicating that for these tumors, TIL assessment may be less sensitive to variations between methods [41].
Once cell counts or densities are determined, derivative metrics, such as ratios, can be calculated. One example is the PD-L1 combined positive score (CPS), which uses single-color IHC labeling of PD-L1 to identify patients who may benefit from anti-PD-1/PD-L1 immunotherapy. The CPS is a ratio calculated as the PD-L1+ cell count (ICs or cancer cells) within the tumor region, divided by cancer cell count, on a single slide [24–26]. The PD-L1 CPS can predict the response to PD-1 immunotherapy (pembrolizumab) in some disease contexts (such as with chemotherapy in first-line metastatic TNBC) [25], but fails to predict the response in other disease contexts (such as in stage II/III TNBC) [42]. One downside of the CPS method is that the score is capped at 100 and, therefore, has a more limited dynamic range for describing PD-L1 expression compared with other methods. Another downside is that the inclusion of cancer cells in the denominator makes the score particularly sensitive to variations in the ratio of cancer cells and other cells. In another example, a high ratio of cytotoxic CD8+ T cells to CD68+ macrophages within primary melanoma was found to be associated with distant recurrence-free survival [43].
IC densities and ratios can also be analyzed across tumor subregions as a local metric, using either raster-based or vector-based methods. As used in the colon cancer Immunoscore (discussed below), the Developer XD software (Definiens) can be used to estimate IC density across rectangular raster cells and to generate novel metrics, such as hotspot density, defined as the mean IC density across the three raster grid cells with the highest-ranked IC density. The hotspot density was shown to be prognostic in patients with metastatic colon cancer [2,29]. With vector datasets, IC densities across anatomically defined subregions (such as stromal versus intraepithelial) can be efficiently calculated using statistical software (such as the sf or sp packages in R) [39].
Kernel density smoothing or estimation is another statistical method of estimating IC density across spatial subregions of a tumor. In kernel density estimation, vector datasets are used to estimate IC density as the sum of kernel functions centered in the IC locations. With this approach, a smoothed estimation of IC density can be calculated for each (x,y) coordinate within the tissue. This method was employed to create topographical maps of lymphocyte densities across breast tumors [28]. The method was also used to characterize lymphocytes according to the density of cancer cells within their locoregional environment. Lymphocytes were categorized into three spatial subtypes: intratumoral, adjacent to tumor, and distant to tumor [28]. The ratio of intratumoral lymphocytes to tumor cells, designated the intratumoral lymphocyte ratio, was prognostic of clinical outcome, independent of other clinical factors.
Metrics of spatial heterogeneity
Cell counts, densities, and ratios define IC patterns based upon the central tendency of immune infiltrates within a given tumor region. However, the underlying spatial distribution of IC across tumor subregions may provide additional prognostic/predictive information. Using raster analysis, once IC densities are estimated across raster cells/subregions of the tumor, spatial heterogeneity summary metrics can be calculated. One common metric is the standard deviation (SD), which indicates how much subregion IC densities differ from the mean IC density. In an analysis of metastatic colon cancer specimens, the SD of CD8+ cell density was shown to associate with inferior disease-free survival and overall survival [29]. SDs are influenced heavily by the underlying mean IC density (with densely infiltrated tumors having a greater tendency to exhibit higher SDs). An alternative statistic is the coefficient of variation (CV), which is the ratio of the SD to the mean. These statistics may also be influenced by the size of the subregion.
Another metric of heterogeneity is skewness, which is a measure of the asymmetry of distributions and can be calculated as the difference between the mean and the median, normalized by the SD (non-parametric skew). A positive skewness value indicates the occurrence of high-value outliers. In an analysis of tissue microarrays of 998 early-stage breast cancer specimens, high skewness of the lymphocyte/cancer cell ratio (indicating the presence of subregions with unusually high ratios) was prognostic for improved disease-free survival in HER2-positive breast cancer [31]. Shannon’s entropy metric provides a measure of randomness across the tumor subregions, with higher scores indicating a higher degree of heterogeneity of IC density. In a recent analysis of TNBC specimens, Shannon’s entropy was shown to be higher in tumor subregions adjacent to the IM of the tumor [6]. Other useful measures of species richness borrowed from the ecology field may be useful for describing heterogeneity, including the Gini Simpson, inverse Simpson, and hill numbers.
Cluster/hotspot analysis
Cluster analysis aims to define IC location along a spectrum that varies from clustered to dispersed. Cell type ‘clusters’ indicate that the cells of interest are more likely to be colocalized with one another, whereas ‘dispersed’ indicates that cells are repelling one another. Following this paradigm, statistical testing can be employed to evaluate whether the ICs are clustered or uniformly dispersed versus the alternative hypothesis that ICs are spatially random (i.e. neither clustered nor uniformly dispersed). Because IC clusters may reflect underlying biological immune engagement, there is tremendous interest to identify tumor subregions with abundant IC density (‘hotspots’) versus areas with IC paucity (‘coldspots’).
Numerous hotspots/clusters analysis approaches have been reported in the literature. Visual characterization of H&E breast cancer TIL hotspots and coldspots (as binary ‘absent or present’ variables) were shown to predict progression-free survival, independent of clinical risk factors, in a cohort from the Cancer Genome Atlas and Carolina Breast Cancer Study [44]. A commonly adopted quantitative method for analyzing hotspots is to rasterize the tumor area, rank-order the subregions by IC density, and define hotspots as either a fixed number, fixed percentile, or a fixed SD cut-off of highest-density subregions (Figure 2A) [13,29,34]. In a series of 53 melanoma patients treated with pembrolizumab, it was concluded that a hotspot definition of IC densities of 30% of the highest-ranking HPFs resulted in the best prediction of the clinical response to pembrolizumab and outperformed conventional assessments of mean IC density [13]. In another study of metastatic colon cancer using the Definiens platform, hotspot density was defined as the density within the three most densely infiltrated raster cells within the tumor area [29]. Using this definition, patients with high hotspot density tumors had improved survival.
Figure 2.
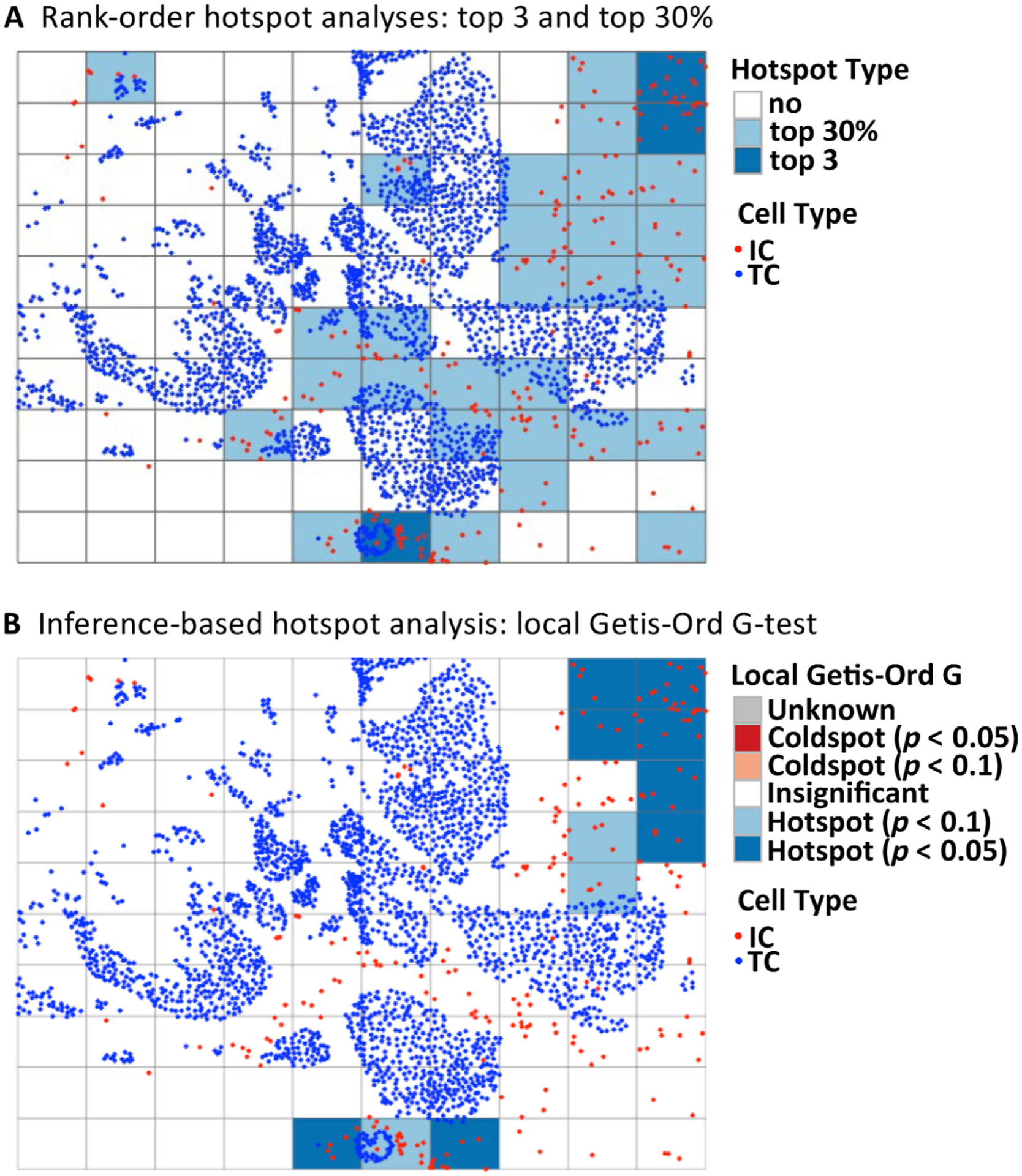
Illustration of various hotspot metrics. Various methods of calculating IC hotspots have been described in the literature, and include methods based upon rank-ordering of IC density across subregions, or based upon inferential testing. (A) Rank-order-based approaches, which define hotspots as either ‘top 3’ (the three most densely infiltrated subregions) or ‘top 30%’ (the top 30% most densely infiltrated subregions). (B) The Getis-Ord Gi* method, which uses inferential statistical testing to estimate p values indicating the likelihood of each subregion being a hotspot or a coldspot. The Getis-Ord test statistic follows a normal distribution and can be thought of as a measure of local IC density in neighboring subregions, relative to overall IC density. IC, immune cell; TC, tumor cell.
One limitation of rank-order hotspot analysis is that it can be influenced by tumor size: larger tumors that have more extensive spatial sampling will have a higher inherent likelihood of having hotspots with greater IC density. This observation was noted in the colon cancer hotspot analysis [45]. Another limitation is that it rests upon the presumption that all tumors have hotspots, whereas it is biologically possible that IC hotspots occur at different frequencies across tumors. As an alternative, the Getis-Ord Gi* is a probabilistic approach for defining hotspots and coldspots within a tumor (Figure 2B). The approach involves defining neighbors for each tumor subregion (i.e. usually defined as immediately adjacent subregions) and conducting a formal hypothesis test using these data to estimate a likelihood that any given neighborhood is a hotspot, given the totality of the spatial data. With this approach, not all tumors will have hotspots, and derivative global metrics describing the relative proportion of hotspots (as a fraction of total tumor area) can be calculated. In a breast cancer dataset, fractional area of cancer/IC colocalization hotspots were found to be independently prognostic for clinical outcome in estrogen receptor-negative breast cancer [14] and estrogen receptor-positive breast cancer [33]. This metric was only weakly correlated with lymphocyte density, indicating that the two metrics may be useful in conjunction for stratifying tumors by immune response. Figure 2 illustrates the top 3, top 30%, and Getis-Ord Gi* approaches for defining hotspots in a breast cancer specimen. In this example, the mean IC hotspot density varies substantially depending on the method (top 3: 3.6 × l0−3/μm2; top 30%: 1.6 × 10−4/μm2; Getis-Ord p < 0.1: 2.2 × 10−4/μm2; Getis-Ord p < 0.05: 1.8 × 10−4/μm2).
Once clusters are defined, they can be further characterized according to features such as size (e.g. number of cells, area, maximum diameter of perimeter), shape (e.g. convexity, circularity, eccentricity, dimensionality of the perimeter), or IC phenotype/composition [6]. Clusters could also be characterized by their spatial relationships to other neighboring clusters while using different distance metrics like Euclidean norm, correlation, or nearest neighbors [35]. In a study of lung cancer specimens, cluster analysis was used in conjunction with supervised ML to generate a spatial score called the spaTIL score, which was shown to be prognostic of recurrence-free survival in non-small cell lung cancer (NSCLC) [35].
There are numerous hotspot/clustering metrics employed in the literature (some listed in Table 1), highlighting the lack of standardization in the field. Efforts should be made by investigators to justify their selection of hotspot method and to conduct sensitivity analyses to evaluate the impact of the hotspot method on their conclusions.
Metrics of cellular colocalization
Vector analysis of spatial IC data allows for efficient calculation of pairwise distances between individual cells, which is particularly useful for characterizing cell–cell interactions. One metric for describing colocalization of cells is the nearest neighbor distance metric. The nearest neighbor distance is calculated for each IC in the specimen and is defined as the shortest pairwise Euclidean distance between two cell types of interest. Similarly, with vector datasets, one can calculate the locoregional density of cells found within a specified radius of each cell. This method was employed to explore the relationship between clinical outcome and T-regulatory (Treg) suppressor cells in oral squamous cell carcinoma [27]. The authors posited that the density of FoxP3+ T-cells within a 30-μm radius of CD8+ effector cells (a biologically plausible distance for cell–cell interactions) could serve as a metric for Treg suppressor engagement with effector T-cells. They found that high Treg/CD8+ radial densities were associated with inferior overall survival, whereas the simple density ratio of Tregs to CD8 cells was not prognostic.
Using the G and cross-G function method, with vector datasets one can calculate the likelihood that ICs colocalize with themselves or other cell types at specified distances [36]. The likelihoods are plotted as a curve, with the x axis representing distance and the y axis representing the cumulative probability density of the nearest IC being within that distance (Figure 3A–D). This provides a graphical representation of colocalization (blue line) compared with what would be expected by random chance (calculated as a Poisson-distributed homogenous point process, red line). For example, in Figure 3C, for cancer cells the red line exceeds the blue line between distances of 0 and 13 pixels, suggesting that cancer cells tend to repel each other at these distances, whereas at distances >13 pixels, the cancer cells tend to group together. In Figure 3D, the red line exceeds the blue line across all distances, suggesting that cancer cells and ICs do not colocalize. The area under the curve (AUC) can be calculated to provide a global metric of colocalization of two cell types within a certain proximity range (e.g. 50 pixels, as illustrated in Figure 3D). With this approach, in a NSCLC cohort, AUC scores of cross-G functions between Treg or CD8+ ICs to cancer cells were prognostic for survival [36]. As a raster-based alternative to the G function, Ripley’s K function quantifies colocalization across specified distances by comparing IC densities across neighboring raster cells. In an analysis of H&E breast cancer specimens, extremes in the K function for stromal cells were associated with improved prognosis [32].
Figure 3.
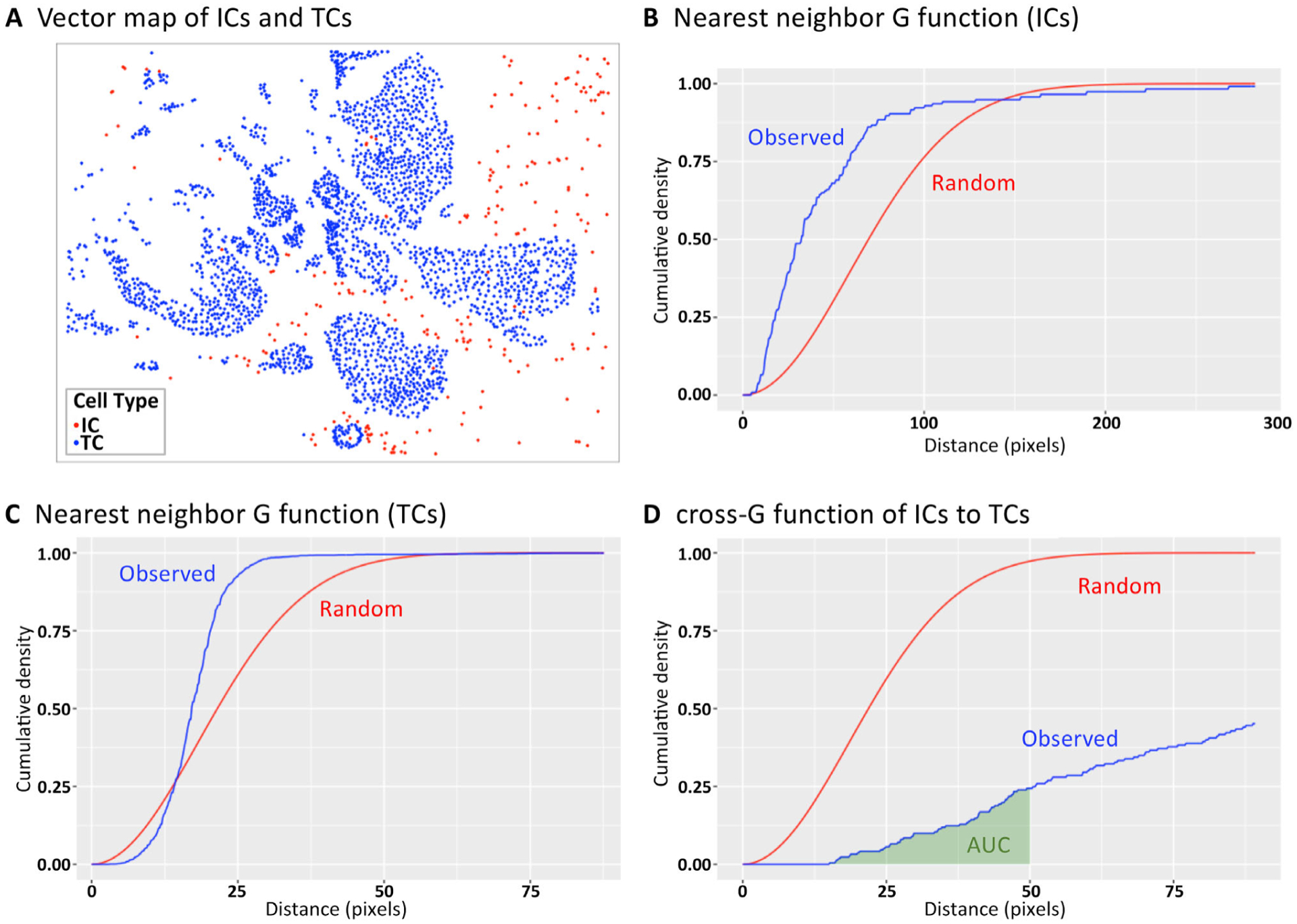
G and cross-G function for describing cellular colocalization. (A) The (X,Y) locations of ICs in relation to cancer cells of an early-stage breast cancer specimen. Colocalization of cells at specified distances can be illustrated using (B and C) the G function (colocalization of the same cell type) and (D) the cross-G function (colocalization of two distinct cell types). The blue lines illustrate the observed colocalization patterns of the sample, whereas the red lines illustrate the expected colocalization under the assumption of randomness/homogeneous point pattern. In (D), the AUC is illustrated in green and is used to provide a global metric of colocalization of two cell types within a certain proximity range (<50 pixels in this example). TC, tumor cell.
Another proposed colocalization metric borrowed from the ecology literature is the Morisita-Horn index. The method is raster-based and generates a scaled score between 0 and 1 based on the calculated ratios of cell densities for two cell types within each raster cell [7], with scores close to 1 indicating colocalization and scores close to 0 indicating spatial independence of the two cell types. In a breast cancer cohort, higher Morisita-Horn indices between ICs and cancer cells were prognostic for disease-free survival, particularly in HER2-positive breast cancer. This metric outperformed IC density or hotspot analyses [16]. Numerous alternative methods for quantifying colocalization patterns have been reported, again highlighting the lack of standardization in the field and the need for investigators to justify their selection of test [6].
Using multiplex assays, recurrent structures of complex cell–cell spatial interactions can be uncovered within the tumor microenvironment. Using a spatial database of 458 breast tumors, hierarchical clustering methods were used to uncover 10 cell–cell interaction profiles, defined by unique clustering patterns of mixtures of cell types [46]. Examples of profiles include a tertiary lymphoid structure-like profile and a regulatory-cell profile that included Tregs combined with cells with upregulated checkpoint proteins such as PD-1. Classification of tumors according to the presence/absence of these structures was found to predict long-term survival [46]. In another study, a graph neural-network ML approach was used to analyze patterns of cell–cell distances and predict long-term survival in a cohort of gastric carcinomas [47].
Segmentation and interface metrics
Segmentation of a tumor into biologically distinct tissue compartments (such as epithelial nest versus stroma) is a crucial component of spatial IC analysis. The segmentation process can be either manual (e.g. via visual pathologist inspection, as exemplified in the breast cancer sTIL score, or by using pan-cytokeratin stains to identify epithelial populations) [1,48], semi-automated using supervised ML (e.g. using the InForm package for mIF or QuPath) [9,12,49], or fully automated with novel ML platforms [8,50]. In the breast cancer sTIL score, ICs are separately scored across stromal versus intraepithelial compartments, providing a powerful prognostic tool (described below) [1,40].
In many epithelial cancers, such as invasive ductal carcinoma of the breast, the epithelial/stromal boundary is well-defined and can be further characterized according to the distributions of cellular phenotypes at various distances from the boundary or by the geometric shape of the boundary. Using multiplex staining it has been shown that certain cell phenotypes (such as myofibroblasts) are enriched at the stromal boundary of breast cancers, whereas other phenotypes (including B-cells) are depleted [46]. With fractal dimension analysis, the geometric complexity of the interface is quantified (with a simple line receiving the lowest score and jagged interfaces receiving higher scores) [7,34]. The epithelial/stromal fractal dimension may reflect differences in underlying tumor biology. For example, a high fractal dimension in breast cancer is associated with higher cancer cell proliferation indices (measured by Ki67 labeling) [38]. Because a high fractal dimension results in increased stroma/cancer cell surface area, it has been proposed that the fractal dimension could modulate suppressive immune reactions between stromal fibroblasts that reside at the interface [34]. Supporting this hypothesis, the fractal dimension of NSCLC specimens was higher in immune cold regions compared with immune hot regions [34]. Fractal dimension analysis of fibers of the extracellular matrix, and other features of matrix fibers, can be quantified with image analysis software, and may contain prognostic information [51].
In breast, colon, and other cancers, ICs congregate in greater density at the IM of the tumor relative to the tumor center [2,37] and thus the margin could influence overall IC density within a specimen. This is especially relevant for the formation of tertiary lymphoid structures that form IC ‘hotspots’ found preferentially near the IM and provide relevant insights into activation of the immune system [52,53]. Biologically, differences at the IM could be related to physical impediments such as desmoplastic/fibrous stroma or interstitial pressure, versus chemical barriers related to secreted factors such as chemokines [37,54]. The IM is most commonly identified by pathologist visual annotation [6,37]; however, ML approaches are also being developed to automate this process [2,8]. In spatial analyses, the IM is often defined as a subregion of tissue spanning a fixed distance from the invasive edge. The colon cancer Immunoscore uses 360 μm, but other cut-offs have been proposed, including 500 or 100 μm [6,37,55]. Once these regions are defined, metrics can be calculated, such as the IM cell density [2] or ratios of densities [37]. These metrics can also be used to categorize tumors according to the degree of infiltration at the IM tumor center (i.e. ‘immune desert’, which describes low IC density at both, versus ‘immune excluded’, which describes low IC density in the tumor center but higher IC density at the IM surrounding the tumor). Using vector datasets, more sophisticated metrics, such as density gradients, can be calculated [39]. One proposed method is to partition the tumor area into bands defined by Euclidean distance from the IM and to estimate IC densities and 95% CIs of density across each distance range [6,56].
Case discussions: the breast cancer sTIL score and colon cancer Immunoscore
Here, from a spatial data perspective, we summarize two leading IC clinical biomarkers, breast cancer sTIL score and the colon cancer Immunoscore, and describe future enhancements that could be achieved by applying advanced spatial analysis techniques, such as those described above.
sTIL score in breast cancer
The breast cancer sTIL score is a visual estimation of the mean proportion of stromal area occupied by lymphocytes from a single H&E slide. Based upon early observations of the prognostic significance of sTILs [22], the International Immuno-Oncology Working Group (TIL working group or TIL-WG) was created to standardize and validate a method of scoring sTILs [1], which has since been shown in multiple datasets to be prognostic [40,57,58] and predictive of chemotherapy ± immunotherapy response [23,59]. These efforts have contributed to the endorsement of the sTIL score for clinical use in early-stage breast cancer by the St. Gallen International Consensus Guidelines [60]. Because the TIL-WG sTIL score relies on visual estimation, it exhibits only modest interobserver scoring concordance [61]. The RING studies demonstrated a modest concordance score (intraclass correlation of 0.70 by untrained pathologists [95% CI 0.62–0.78]), which was improved to 0.89 (0.85–0.92) with the incorporation of training, additional safeguards including visual controls/scales, and the requirement for sub-sampling or the evaluation of at least three regions of tumor. An intraclass correlation greater than 0.8 is generally considered to be sufficient for adoption in clinical practices. Many of these safeguards are addressed in a continuing medical education (CME)-accredited FDA course on TILs available on the TIL-WG website [62]. A root-cause analysis identified the following recurring contributors to discordance: intratumoral heterogeneity of sTIL density, poor-quality staining, misclassification of cell type or cell segment, and insufficient spatial sampling (in biopsies with minimal evaluable tumor area).
Ongoing efforts are underway to evaluate whether novel imaging platforms or spatial analyses could produce spatial metrics that perform similarly or superior to the H&E sTIL score for clinical prognosis/prediction (illustrated and summarized in Figure 4) [3,11,63–65]. These can be summarized across several discrete steps of the sTILs scoring process: (1) segmentation; (2) subregion sampling and density estimation; and (3) calculation of an overall sTIL score. In the segmentation step, a tumor must be accurately separated into stromal and non-stromal compartments, and cells must be accurately categorized as lymphocytes versus other cells. This step was identified as a frequent source of discordance in the RING studies, with pathologists erroneously categorizing intraepithelial TILs as sTILs, or categorizing neutrophils, histocytes, and apoptotic cells as sTILs [66]. Proposed methods for improving segmentation involve the use of automated image analysis/ML methods that can more accurately classify tissues and cell types (summarized in [11]) or the use of mIF/mIHC to enable more precise segmentation through the use of cytokeratin staining of cancer cell-containing regions, as well as multiple cellular markers to discern lymphocytes from non-lymphocytes [12,63].
Figure 4.
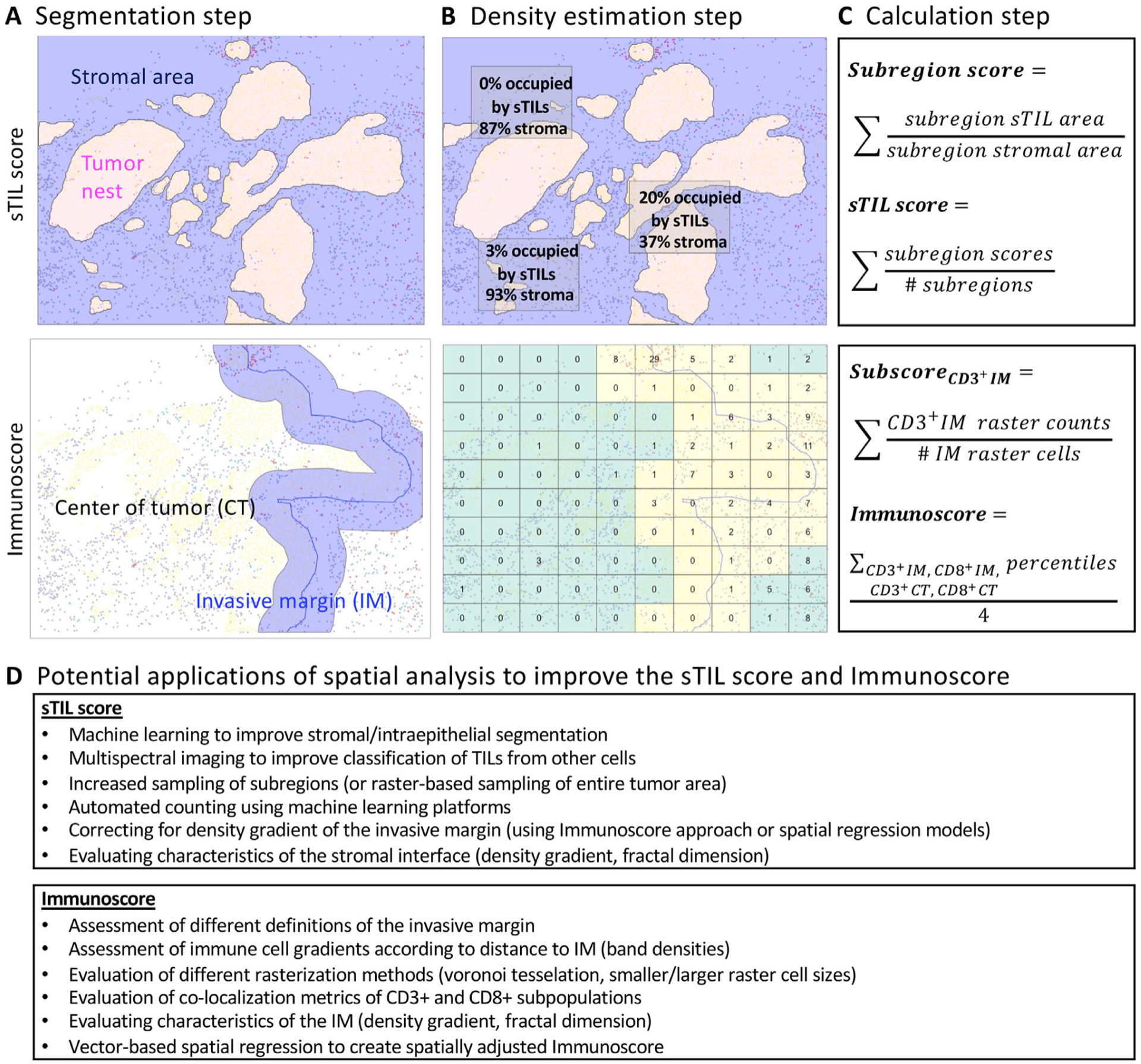
sTILs score and Immunoscore: methodology and opportunities for spatial applications. (A) Segmentation step: for the sTILs score, the intraepithelial versus stromal tumor compartment are visually determined by a pathologist, whereas for Immunoscore, the IM versus tumor center is determined using an automated ML platform. (B) Sampling and density estimation steps: for the sTIL score, several representative subregions are visually selected and sTIL counts are estimated across each subregion, whereas for Immunoscore, the entirety of the tumor area is divided by a rectangular raster grid, and CD3+ and CD8+ ICs are counted for each raster cell. (C) Calculation step: for the sTIL score, the arithmetic mean of sTIL densities for each subregion is calculated, whereas for Immunoscore, the arithmetic mean of cohort-level percentile scores across the four cellular compartments is calculated (CD3+ IM, CD3+ TC, CD8+ IM, CD8+ TC). (D) Advanced ML, histologic imaging, and spatial analytic approaches can be applied to the sTIL score and Immunoscore to potentially improve predictive/prognostic utility.
In the subregion sampling step, pathologists must sample enough subregions of the tumor space to reflect the entirety of the specimen. In the RING studies, interobserver concordance improved substantially when pathologists were explicitly required to evaluate at least three tumor subregions [66], thus raising the hypothesis that pathologists, when unassisted, may not visually assess enough areas of the tumor. In this regard, automated imaging and/or multispectral imaging would provide a clear advantage over H&E visual sTIL estimation. ML-based software platforms could permit efficient sampling across the entirety of the tumor slide or even provide whole-slide analysis. However, further work is necessary to optimize the accuracy of these platforms and to evaluate their prognostic/predictive utility in comparison with the standard visual TIL-WG method [11]. Alternatively, high-resolution multispectral imaging permits sampling of many HPFs across a specimen. In a recent evaluation of mIF in breast cancer, using Monte Carlo simulation studies, it was shown that at least 15 HPFs per patient would be required to adequately characterize changes in sTILs in the context of an immunotherapy clinical trial [12]. Before mIF can be used clinically, significant progress must be made to standardize the mIF/mIHC workflow across institutions and validate the assay [3,67].
When TIL densities are estimated for each subregion, pathologists must accurately visually estimate the proportion of stromal area occupied by sTILs within a given subregion. This was a frequent cause of interobserver discordance. However, it was improved in the second RING study, when pathologists were provided standardized images of various sTIL scores for visual comparison [66]. Nonetheless, this step can be improved tremendously with automated analysis and/or mIF, which has adequate resolution to localize individual TILs, their precise geometric location in relation to the stromal compartment, and to compute the overall stromal area for each subregion.
The last step is calculation of overall sTIL density, which is where spatial data analysis can provide the largest gains for accuracy and precision. In the RING studies, pathologists were instructed to report the arithmetic mean of sTIL scores across the various tumor subregions. However, from a spatial analytic perspective, this method relies on a simplistic assumption that IC density is homogenous, when in fact biologically the IC density can vary tremendously across various subregions of the tumor, and can be influenced by spatial interdependencies (i.e. spatial autocorrelation) as well as anatomic features (such as the IM, which exhibits inherently higher IC density relative to the tumor center). Using vector-based datasets that include (x, y) coordinates of ICs as well as anatomic landmarks (such as the IM), spatial regression methods (such as inhomogeneous Poisson process models) [20] could be used to generate adjusted sTIL scores that may more accurately reflect the intrinsic immunogenicity of the tumor. With inhomogeneous Poisson process models, IC densities are estimated repeatedly across the tumor sample (using kernel density smoothing, radial densities, or other methods). Furthermore, for each observation, spatial covariates such as proximity to the IM can be estimated and included as parameters in a regression model, thus allowing for a spatially adjusted estimation of IC density. Other variables of interest could also be included in the model, such as local PD-L1 protein expression or treatment exposure [20].
Whole-slide TIL density metrics have also been shown to be prognostic in melanoma, and there are ongoing efforts to standardize and automate scoring. Recently, automated TIL density estimation using ML classifiers (NN192 or QuIP TIL CNN) has been shown across multiple datasets to improve prognostication compared with the historical Clark grading system for TILs [68–71]. A detailed analysis of the potential pitfalls of ML assessment is provided in a companion article [72].
The colon cancer Immunoscore
Immunoscore was developed as a prognostic biomarker in the context of early-stage colon cancer. It differs from the breast cancer sTIL score in several regards. First, it is based upon quantification of T-cell populations using IHC staining of CD3 and CD8. The selection of these markers was based upon expert consensus, genomic studies illustrating the prognostic value of Thl-type gene signatures (that would correlate with CD3 and CDS infiltration and initially also on T-cell activation reflected by granzyme B expression) [73], and the relative stability and quality of staining of these antigens. Second, whereas the H&E sTIL score aims to characterize the mean cell density across the entirety of the tumor stroma, Immunoscore addresses spatial heterogeneity by segmenting and quantifying IC densities within two compartments, the tumor center and the IM, with the margin defined as a region surrounding the border between normal cells and cancerous cells. Analysis of these two distinct compartments was shown in a large cohort to better predict colon cancer outcomes, compared with single-region analysis. Third, Immunoscore relies on image analysis software (Immunoscore Analyzer, HalioDx), and the coding and detailed methods are not available publicly.
Extensive collaborative efforts by a SITC consortium have been undertaken to clinically validate Immunoscore as a prognostic biomarker for colon cancer [2]. In this study, Immunoscore was found to be prognostic across a training set, a validation set, and an external validation set. Consistency of staining was established through the use of a biomarker reference center that disseminated an optimized immunostaining protocol and control specimens. Cut-offs for high versus low Immunoscore were defined using the training set. Cut-offs of 25% and 70% were selected to create a three-tier Immunoscore (low versus intermediate versus high). A cut-off of 25% was selected for a two-tier Immunoscore (low versus high). Using this methodology, Immunoscore was proven across training and two validation cohorts to be highly prognostic of disease-free and overall survival, independent of other clinical factors. Recently, Immunoscore was also shown to have prognostic significance in the context of metastatic colorectal carcinoma [29].
Reframed using spatial metrics, the Immunoscore method includes the following steps: (1) segmentation of the tumor into IM versus tumor center; (2) rasterization of each compartment and estimation of IC densities in each raster cell; (3) calculation of mean IC densities for each compartment and calculation of a composite Immunoscore. Like the breast cancer sTIL score, Immunoscore could be enhanced by leveraging novel spatial analytic techniques across each of these steps. In the segmentation step, tumor regions are segmented into one of two tissue types, IM or tumor center. This is conducted by defining an IM buffer zone as 360 pm spanning each side of the border separating tumor from normal tissue. This approach is reasonable, albeit arbitrarily defined, but there exist alternative methods for adjusting spatial heterogeneity in relation to the IM. Across multiple tumor types, ICs have been shown to exhibit a density gradient according to distance from the margin. To capture the data inherent to the density gradient, IC and IM spatial locations could be collected in vector dataset format, allowing for computation of densities across regions spanning various distances from the margin. These densities could then be assessed for prognostic significance using regression modeling. One caveat of Immunoscore is that the IM may not be adequately sampled in every biopsy.
In the rasterization step, the tumor is divided into rectangular subregions. Each subregion is labeled as margin or center, and the IC densities are estimated within each subregion. Because raster grid size or shape (e.g. rectangular, hexagonal, Voronoi polygon) has been previously shown to influence overall IC estimation and prognostic outcome, in the context of the SITC Immunoscore validation project [2], it would be of interest to use this dataset to conduct a sensitivity analysis that illustrates the impact of rasterization method on Immunoscore. These data may be helpful for improving the performance of Immunoscore but may also enhance other investigations in the field of spatial IC analysis.
In the calculation step, raster cell IC densities are used to calculate the sample’s overall Immunoscore. First, for each cell type (CD3 and CD8) and compartment (IM and tumor center), the mean IC density across all raster cells is computed. The mean is then converted into a percentile score by comparing against scores across the entire cohort of samples. Finally, the percentiles across the four cell types (CD3/margin, CD3/center, CD8/margin, CD8/center) are averaged, creating a single Immunoscore for each patient, which is prognostic of survival. Using the clinically annotated SITC Immunoscore dataset, alternative spatial metrics could be developed and tested for clinical utility. Examples would be to explore the prognostic significance of colocalization metrics of ICs with tumor cells (as summarized above), to evaluate the significance of the shape of the IM (i.e. fractal dimension, as described above), or to evaluate the prognostic impact of IC hotspots/coldspots (as described above). A ML approach could be adopted to identify a unique combination of spatial metrics that confers the optimal prognostic utility.
Conclusion
Modern histologic imaging combined with ML has provided the opportunity to evaluate not only the overall IC density of tumors, but also the degree of heterogeneity of IC infiltration, the absence/presence of IC clustering, patterns of cell–cell colocalization, the complexity of the tumor/stromal interface, and the gradient of IC density in relation to the IM of the tumor. Two complementary analytic approaches, raster-based and vector-based, can be leveraged to generate spatial biomarkers that offer a more fine-grained picture of tissue architecture. An important future step is to amalgamate spatial and transcriptomic data using next-generation spatial sequencing technologies, which provides us with the challenging opportunity to analyze all cell types of a tissue in their spatial context. Although most of the available technologies focus on analysis of two-dimensional images, in the future, strategies may be developed to analyze TIL patterns in three-dimensional space. Additionally, sustainable strategies for cohort generation, data sharing, and dimensionality reduction methods to decrease data storage requirements are becoming more crucial than ever.
In this nascent field, a key deficiency is the lack of an investigative path to establish that novel spatial biomarkers can improve clinical prediction/prognosis beyond that of existing biomarkers. With the advent of cloud computing and with increasing emphasis on data sharing by regulatory agencies and editorial boards, in the future it may be possible to leverage shared databases that contain digitized imaging files and outcomes data from pivotal clinical trial datasets. Such databases could provide necessary standardization, quality control, and sample sizes to permit the validation of clinical utility of a novel spatial biomarker. Efforts should be made by industry, consortia, investigators, and other agencies to contribute to these databases. Oversight by a committee may be required to ensure that the data are used efficiently, without bias, and without compromising patient privacy. An additional validation step for a novel biomarker is external quality assessment, whereby multiple laboratories replicate and confirm findings. In the context of mIF/mIHC-based biomarkers, external validation might be achieved via multi-institutional consortia sponsored by the National Institutes of Health, SITC, and other organizations.
Acknowledgements
This work was made possible through the donations of Walter Bowen (Portland, OR, USA) and others. The authors thank Torsten Nielsen, MD/PhD (Professor of Pathology & Laboratory Medicine, University of British Columbia) for critical revision of the manuscript. Acknowledgements of funds supporting co-authors are as follows: GB: funded by the Gilead Breast Cancer Research Grant 2023. SV: supported by ‘Interne Fondsen KU Leuven/Internal Funds KU Leuven. BA: the Swedish Society for Medical Research (Svenska Saäskapet för Medicinsk Forskning) postdoctoral grant, Swedish Breast Cancer Association (Bröstcancerförbundet) research grant 2021. GC: the Peer Reviewed Cancer Research Program (award W81XWH-21-1-0160) from the US Department of Defense and the Mayo Clinic Breast Cancer SPORE grant P50 CA116201 from the National Institutes of Health (NIH). CF-M: Horizon 2020 European Union Research and Innovation Programme under the Marie Sklodowska Curie Grant agreement no. 860627 (CLARIFY Project). SBF: NHMRC GNT1193630. WMG: Higher Education Authority, Department of Further and Higher Education, Research, Innovation and Science and the Shared Island Fund (AICRIstart: A Foundation Stone for the All-Island Cancer Research Institute [AICRI]: Building Critical Mass in Precision Cancer Medicine: https://www.aicri.org/aicristart): Irish Cancer Society (Collaborative Cancer Research Centre BREAST-PREDICT; CCRC13GAL; https://www.breastpredict.com), the Science Foundation Ireland Investigator Programme (OPTi-PREDICT; 15/IA/3104), the Science Foundation Ireland Strategic Partnership Programme (Precision Oncology Ireland; 18/SPP/3522; https://www.precisiononcology.ie). SG: partially supported by National Institutes of Health (NIH) grant CA224319, DK124165, CA263705, and CA196521. AG: Breast Cancer Now (and their legacy charity Breakthrough Breast Cancer) and Cancer Research UK (CRUK/07/012, KCL-BCN-Q3). TRK: Japan Society for the Promotion of Science (JSPS) KAKENHI (21K06909). UK: Horizon 2020 European Union Research and Innovation Programme under the Marie Sklodowska Curie grant agreement no. 860627 (CLARIFY Project). JKL: part supported by NIH (R37 CA225655). AM: National Cancer Institute award numbers R01CA268287A1, U01CA269181, R0CA26820701A1, R01CA249992-01A1, R01CA202752-01A1, RO 1CA208236-01A1, R01CA216579-01A1, R01CA220581-01A1, R01CA257612-01A1,1U01CA239055-01, 1U01CA248226-01, 1U54CA254566-01, National Heart, Lung and Blood Institute 1R01HL15127701A1, R01HL15807101A1, National Institute of Biomedical Imaging and Bioengineering 1R43EB028736-01, VA Merit Review Award IBX004121A from the US Department of Veterans Affairs Biomedical Laboratory Research and Development Service the Office of the Assistant Secretary of Defense for Health Affairs, through the Department of Defense Breast Cancer Research Program (W81XWH-19-1-0668), the Prostate Cancer Research Program (W81XWH-20-1-0851), the Lung Cancer Research Program (W81XWH-18-1-0440, W81XWH-20-1-0595), the Peer Reviewed Cancer Research Program (W81XWH-18-1-0404, W81XWH-21-1-0345, W81XWH-21-1-0160), the Kidney Precision Medicine Project (KPMP) Glue Grant and sponsored research agreements from Bristol Myers-Squibb, Boehringer-Ingelheim, Eli-Lilly, and AstraZeneca. SKM: Kay Pogue-Geile, Director of Molecular Profiling at NSABP for her constant support and encouragement, Roberto Salgado, for getting me initiated into the wonderful subject of immuno-oncology and its possibilities. FuAAM: EPSRC EP/W02909X/1 and PathLAKE consortium. FP-L: Fondation ARC, La Ligue Contre le Cancer. RDP: the Melbourne Research Scholarship and a scholarship from the Peter MacCallum Cancer Centre. JSR-F: part by the Breast Cancer Research Foundation, a Susan G Komen Leadership Grant, and the NIH/NCI P50 CA247749 01 grant. JS: National Cancer Institute grant UH3CA225021, U24CA215109. ST: ‘Interne Fondsen KU Leuven/Internal Funds KU Leuven. JT: institutional grants of the Dutch Cancer Society and the Dutch Ministry of Health, Welfare and Sport. EAT: Breast Cancer Research Foundation grant 22-161. GEV: Breast Cancer Now (and their legacy charity Breakthrough Breast Cancer) and Cancer Research UK (CRUK/07/012, KCL-BCN-Q3). TW: S French Government Under Management of Agence Nationale de la Recherche as part of the ‘Investissements d’avenir’ program, reference ANR-19-P3IA-0001 (PRAIRIE 3IA Institute) and Q-Life (ANR-17-CONV-0005). HYW: in part by the NIH/NCI P50 CA247749 01 grant. YY: Cancer Research UK Career Establishment Award (CRUK C4598 2/A21808). PS: National Health and Medical Research Council, Australia. SL: National Breast Cancer Foundation of Australia (NBCF) (APP ID: EC-17-001), the Breast Cancer Research Foundation, New York (BCRF [APP ID: BCRF-21-102]), and a National Health and Medical Council of Australia (NHMRC) Investigator Grant (APP ID: 1162318). RS: Breast Cancer Research Foundation (BCRF, grant no. 17-194).
Conflict of interest statement:
DBP is a member of the Speaker’s Bureau for Genentech, Novartis, Qinical Care Options, and Oncocyte: receives research support from WindMIL Brooklyn immunotherapeutics, Merck Bristol-Myers Squibb, and IMV, consults for Merck Biotheranostics, Puma, Gilead, Lilly, Sanofi, NGM Bio, Sanford Burnham Prebys, and AstraZeneca. GB receives speaker’s fees from MSD and Novartis and is on the advisory boards of Roche and MSD, is a consultant for MSD, Novartis and Roche, receives travel and conference support from Roche, MSD, and Gilead. JT is an employee ofVisiopharm A/S. ZK has a paid advisory role for Eli Lilly and AstraZeneca Canada. KRB is on the Scientific Advisor Board for CDI Labs and receives research funding from Carevive. FC is the Chair of the Scientific and Medical Advisory Board of TRIBVN Healthcare, France, and has received advisory board fees from TRIBVN Healthcare, France in the last 5 years, and is a shareholder of Aiosyn BV, the Netherlands. LAC participates in the Tempus Algorithm Advisors program. AC is a contracted researcher for Oncoinvent AS and Novocure and a consultant for Sotio a.s. and Epics Therapeutics SA. ME is part of the Egyptian missions sector. SE is an employee of BMS. SF is on the expert advisory panel for the AXDEV Group. WMG is the co-founder, shareholder, and part-time Chief Scientific Officer of OncoAssure Limited, a shareholder in Deciphex, and a member of the Scientific Advisory Board of Carrick Therapeutics. JMG is an employee and stockholder of Rochel/Genentech. SC receives research funding from Regeneron Pharmaceuticals, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Genentech, EMD Serono, Pfizer, and Takeda, unrelated to the current work; is a named co-inventor on an issued patent for multiplex immunohistochemistry to characterize tumors and treatment responses. The technology is filed through icahn School of Medicine at Mount Sinai (ISMMS) and is currently unlicensed. NH has a patent on a technology to measure immune infiltration in cancer to predict treatment outcome (W02012038068A2). MGH is a consultant for PaigeAI, VolastraTx, and an advisor for PathPresenter. JH receives speaker’s honoraria or advisory board remunerations from Roche, Novartis, AstraZeneca, Eli Lilly, and MSD and is co-founder and shareholder of Stratipath AB. AIH received a research fund from Visiopharm A/S. KK is an employee and stockholder of Roche. AK received an honorarium from Roche, MSD, and Pfizer and is a member of the Advisory Board of Pfizer. A-VL received institutional grants from AstraZeneca and personal grants from AstraZeneca (travel and honorarium from advisory board), MSD (honorarium from advisory board), and Daiichi Sankyo (travel). XL is an advisor for Eli Lilly Company and Cancer Expert Now, and received research funding from Champions Oncology. AM is an equity holder in Picture Health, Elucid Bioimaging, and Inspirata Inc, on the advisory board of Picture Health, Aiforia Inc, and SimBioSys, a consultant for SimBioSys, has sponsored research agreements with AstraZeneca, Boehringer-lngelheim, Eli-Lilly, and Bristol Myers-Squibb, has technology licensed to Picture Health and EJucid Bioimaging, involvement in three different R01 grants with Inspirata Inc. DKM consults for AstraZeneca, Lilly USA LLC, Hologic and sponsored research for Merck and Agendia. SM is a Scientific Committee Study member for Roche and a data and safety monitoring member of clinical trials for Sensorion, Biophytis, Servier, IQVIA, Yuhan, and Kedrion. FuAAM receives research studentship funding from GSK DAM received speaker’s fees from AstraZeneca, Eli Lilly, and Takeda, and consultancy fees from AstraZeneca, Thermo Fisher, Takeda, Amgen, Janssen, MIM Software, Bristol-Myers Squibb, and Eli Lilly, and has received educational support from Takeda and Amgen. FP-L has personal financial interests in AbbVie, Agendia, Amgen, Astellas, AstraZeneca, Bayer, BMS, Daiichi-Sankyo, Eisai, Exact Science, GSK lliumina, Incyte, Janssen, Lilly, MERCK Lifa, Merck-MSD, Myriad, Novartis, Pfizer, Pierre-Fabre, Roche, Sanofi, Seagen, Takeda, Veracyte, and Servier, and has institutional financial interests in AstraZeneca, Bayer, BMS, MSD, Myriad, Roche, and Veracyte, has congress invitations from AbbVie, Amgen, AstraZeneca, Bayer, BMS, Gilead, MSD, Novartis, Roche, Lilly, and Pfizer. NMR is a Co-Founder, Director, and CSO of Histofy Ltd, UK JSR-F is an Associate Editor of The Journal of Pathology; he receives personal/consultancy fees from Goldman Sachs, Bain Capital, REPARE Therapeutics, Saga Diagnostics, and PaigeAI, is a member of the scientific advisory boards of VolitionRx, REPARE Therapeutics, and PaigeAI, a member of the Board of Directors ofGrupo Oncoclinicas, and ad hoc membership of the scientific advisory boards of AstraZeneca, Merck, Daiichi Sankyo, Roche Tissue Diagnostics, and Personalis, outside the scope of this study. AS is on Advisory Board/Speaker’s Bureau of Aignostics, Astra Zeneca, Bayer, BMS, Eli Lilly, lliumina, Incyte, Janssen, MSD, Novartis, Pfizer, Roche, Seagen, Takeda, and Thermo Fisher, receives grants from Bayer, BMS, Chugai, and Incyte. TT is an employee of Tempus Labs. JT is a shareholder of EliogonAI BV. TT receives speaker’s fees from Pfizer. JvdL is a member of the advisory boards of Philips, the Netherlands and ContextVision, Sweden, and has received research funding from Philips, the Netherlands, ContextVision, Sweden, and Sectra, Sweden in the last 5 years, is CSO and shareholder of Aiosyn BV, the Netherlands. TW collaborates with TRIBUN Health on automatic grading of biopsies for head and neck cancer, has a patent on the prediction of homologous recombination deficiency (HRD) in breast cancer. YW is an employee of CellCarta. HYW is part of the advisory faculty of AstraZeneca. YY is a speaker/consultant for Roche and Merck JMSB consults for Biotheranostics, Inc., Rna Diagnostics Inc., AstraZeneca, Cerca Biotech, is on the Scientific Advisory Board for Medcomxchange Communications Inc., receives honoraria from Medcomxchange Communications Inc, research funding from ThermoFisher Scientific, Genoptix, Agendia, Nanostring, Technologies, Inc, Stratifyer Gmbh, Biotheranostics, Inc, Exact Sciences, has applied for patents: Cin4 Predicts Benefit From Anthracycline, National Phase Application, (Canada, 11 January 2017), Systems, Devices And Methods For Constructing And Using A Biomarker, National Phase Application, 15/328, 108 (United States, 23 January 2017); 15824751.0 (Europe, 12 January 2017); (Canada, 12 January 2017), Targeting The Histone Pathway To Detect And Overcome Anthracycline Resistance (Ip Title), National Phase Application, Pct/Ca2016/000247, Patent Application #: 3000858 (Canada - Patent Application Date: 4 April 2018), Immune Gene Signature Predicts Anthracydine Benefit Pct (International Application), Pct/Ca2016/000305, Filing Date: 7 December 2016, Gene Signature Of Residual Risk Following Endocrine Treatment In Early Breast Cancer (Patent Title), National Phase Application, Patent Application Number 3007118 (Canada - Patent Application Date: 1 June 2018); 15/781,939 (USA - 6 June 2018), A Molecular Classifier For Personalized Risk Stratification For Patients With Prostate Cancer (Invention Title), Pet International Application No.: Pct/Ca2021/050837, International Filing Date: 18 June 2021; has patents granted for: Cin4 Predicts Benefit From Anthracycline (Invention Title), National Phase Application, Patent Number: 11214836 (USA, Date: 4 January 2022); 3169815 (Europe, 23 December 2020), Targeting The Histone Pathway To Detect And Overcome Anthracycline Resistance (Ip Title), National Phase Application, Patent Number. 2016800728463 (PR China, Date: 27 August 2021); 11,015.226 (USA, 25 May 2021); 3359508 (Europe, 9 September 2020), Gene Signature Of Residual Risk Following Endocrine Treatment In Early Breast Cancer (Patent Title), National Phase Application, Patent Number. 2016368696 (Australia, 10 March 2022); 2016800813945 (PR China, 18 March 2022); 3387168 (Europe, 12 May 2022); 7043404 (japan, 18 March 2022), Invention Disclosure: Disclosure Name: A Molecular Classifier For Personalized Risk Stratification For Patients With Prostate Cancer, Date: 21/08/2019. PS is a consultant (uncompensated) to Roche-Genentech. SL receives research funding to her institution from Novartis, Bristol-Meyers Squibb, Merck Puma Biotechnology, Eli Lilly, Nektar Therapeutics, Astra Zeneca, Roche-Genentech, and Seattle Genetics. SLoi has acted as consultant (not compensated) to Seattle Genetics, Novartis, Bristol-Meyers Squibb, Merck, AstraZeneca, Eli Lilly, Pfizer, and Roche-Genentech and has acted as a consultant (paid to her institution) to Aduro Biotech, Novartis, GlaxoSmithKline, Roche-Genentech, Astra Zeneca, Silverback Therapeutics, Gl Therapeutics, PUMA Biotechnologies, Pfizer, Gilead Therapeutics, Seattle Genetics, Daiichi-Sankyo, Amunix, Tallac Therapeutics, Eli Lilly, and Bristol-Meyers Squibb. RS receives non-financial support from Merck and Bristol Myers Squibb, research support from Merck, Puma Biotechnology and Roche, and personal fees from Roche, Bristol Myers Squibb, and Exact Sciences for advisory boards. GA, is an employee of Merck.
References
- 1.Salgado R, Denkert C, Demaria S, et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: recommendations by an International TILs Working Group 2014. Ann Oncol 2015; 26: 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pagès F, Mlecnik B, Marliot F, et al. International validation of the consensus immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 2018; 391: 2128–2139. [DOI] [PubMed] [Google Scholar]
- 3.Taube JM, Akturk G, Angelo M, et al. The Society for Immunotherapy of Cancer statement on best practices for multiplex immunohistochemistry (IHC) and immunofluorescence (IF) staining and validation. J Immunother Cancer 2020; 8: e000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tien TZ, Lee J, Lim JCT, et al. Delineating the breast cancer immune microenvironment in the era of multiplex immunohistochemistry/immunofluorescence (mIHC/IF). Histopathology 2021; 79: 139–159. [DOI] [PubMed] [Google Scholar]
- 5.Toki MI, Merritt CR, Wong PF, et al. High-Plex predictive marker discovery for melanoma immunotherapy-treated patients using digital spatial profiling. Clin Cancer Res 2019; 25: 5503–5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mi H, Gong C, Sulam J, et al. Digital pathology analysis quantifies spatial heterogeneity of CD3, CD4, CD8, CD20, and FoxP3 immune markers in triple-negative breast cancer. Front Physiol 2020; 11: 583333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan Y Spatial heterogeneity in the tumor microenvironment. Cold Spring Harb Perspect Med 2016; 6: a026583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zormpas-Petridis K, Noguera R, Ivankovic DK, et al. SuperHistopath: a deep learning pipeline for mapping tumor heterogeneity on low-resolution whole-slide digital histopathology images. Front Oncol 2020; 10: 586292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.inForm User Manual Version 2.4.2 [Accessed 27 December 2019]. Available from: https://research.pathology.wisc.edu/wp-content/uploads/sites/510/2018/12/inFormUserManual_2_4_2_rev0.pdf.
- 10.Stack EC, Wang C, Roman KA, et al. Multiplexed immunohistochemistry, imaging, and quantitation: a review, with an assessment of tyramide signal amplification, multispectral imaging and multiplex analysis. Methods 2014; 70: 46–58. [DOI] [PubMed] [Google Scholar]
- 11.Amgad M, Stovgaard ES, Balslev E, et al. Report on computational assessment of tumor infiltrating lymphocytes from the International Immuno-oncology Biomarker Working Group. NPJ Breast Cancer 2020; 6: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanchez K, Kim I, Chun B, et al. Multiplex immunofluorescence to measure dynamic changes in tumor-infiltrating lymphocytes and PD-L1 in early-stage breast cancer. Breast Cancer Res 2021; 23: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berry S, Giraldo NA, Green BF, et al. Analysis of multispectral imaging with the AstroPath platform informs efficacy of PD-1 blockade. Science 2021; 372: eaba2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nawaz S, Heindl A, Koelble K, et al. Beyond immune density: critical role of spatial heterogeneity in estrogen receptor-negative breast cancer. Mod Pathol 2015; 28: 766–777. [DOI] [PubMed] [Google Scholar]
- 15.Francis K, Palsson BO. Effective intercellular communication distances are determined by the relative time constants for cyto/chemokine secretion and diffusion. Proc Natl Acad Sci USA 1997; 94: 12258–12262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maley CC, Koelble K, Natrajan R, et al. An ecological measure of immune-cancer colocalization as a prognostic factor for breast cancer. Breast Cancer Res 2015; 17: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilm F, Benz M, Bruns V, et al. Fast whole-slide cartography in colon cancer histology using superpixels and CNN classification. J Med Imaging (Bellingham) 2022; 9: 027501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turkki R, Linder N, Kovanen PE, et al. Antibody-supervised deep learning for quantification of tumor-infiltrating immune cells in hema-toxylin and eosin stained breast cancer samples. J Pathol Inform 2016; 7: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verghese G, Li M, Liu F, et al. Multiscale deep learning framework captures systemic immune features in lymph nodes predictive of triple negative breast cancer outcome in large-scale studies. J Pathol 2023; 260: 376–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bivand RS, Pebesma E, Gomez-Rubio V. Applied Spatial Data Analysis with R. Springer: New York, NY, 2013. [Google Scholar]
- 21.Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515: 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denkert C, Loibl S, Noske A, et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol 2010; 28: 105–113. [DOI] [PubMed] [Google Scholar]
- 23.Loi S, Winer E, Lipatov O, et al. Abstract PD5–03: relationship between tumor-infiltrating lymphocytes (TILs) and outcomes in the KEYNOTE-119 study of pembrolizumab vs chemotherapy for previously treated metastatic triple-negative breast cancer (mTNBC). Cancer Res 2020; 80: PD5–03-PD05–03. [Google Scholar]
- 24.Gonzalez-Ericsson PI, Stovgaard ES, Sua LF, et al. The path to a better biomarker: application of a risk management framework for the implementation of PD-L1 and TILs as immuno-oncology biomarkers in breast cancer clinical trials and daily practice. J Pathol 2020; 250: 667–684. [DOI] [PubMed] [Google Scholar]
- 25.Cortes J, Cescon DW, Rugo HS, et al. KEYNOTE-355: randomized, double-blind, phase III study of pembrolizumab + chemotherapy versus placebo + chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer. In ASCO Annual Meeting. Chicago, IL, 2020. [DOI] [PubMed] [Google Scholar]
- 26.Scott M, Scorer P, Barker C, et al. Comparison of patient populations identified by different PD-L1 assays in in triple-negative breast cancer (TNBC). Ann Oncol 2019; 30: iii4. [Google Scholar]
- 27.Feng Z, Bethmann D, Kappler M, et al. Multiparametric immune profiling in HPV-oral squamous cell cancer. JCI Insight 2017; 2: e93652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan Y Modelling the spatial heterogeneity and molecular correlates of lymphocytic infiltration in triple-negative breast cancer. J R Soc Interface 2015; 12: 20141153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van den Eynde M, Mlecnik B, Bindea G, et al. The link between the multiverse of immune microenvironments in metastases and the survival of colorectal cancer patients. Cancer Cell 2018; 34: el013. [DOI] [PubMed] [Google Scholar]
- 30.Etzioni R Statistics for Health Data Science: An Organic Approach. Springer: Cham, Switzerland, 2021. [Google Scholar]
- 31.Khan AM, Yuan Y. Biopsy variability of lymphocytic infiltration in breast cancer subtypes and the ImmunoSkew score. Sci Rep 2016; 6: 36231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan Y, Failmezger H, Rueda OM, et al. Quantitative image analysis of cellular heterogeneity in breast tumors complements genomic profiling. Sci Transl Med 2012; 4: 157ral43. [DOI] [PubMed] [Google Scholar]
- 33.Heindl A, Sestak I, Naidoo K, et al. Relevance of spatial heterogeneity of immune infiltration for predicting risk of recurrence after endocrine therapy of ER+ breast cancer. J Natl Cancer Inst 2018; 110: 10.1093/jnci/djxl37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.AbdulJabbar K, Raza SEA, Rosenthal R, et al. Geospatial immune variability illuminates differential evolution of lung adenocarcinoma. Nat Med 2020; 26: 1054–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corredor G, Wang X, Zhou Y, et al. Spatial architecture and arrangement of tumor-infiltrating lymphocytes for predicting likelihood of recurrence in early-stage non-small cell lung cancer. Clin Cancer Res 2019; 25: 1526–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barua S, Fang P, Sharma A, et al. Spatial interaction of tumor cells and regulatory T cells correlates with survival in non-small cell lung cancer. Lung Cancer 2018; 117: 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X, Gruosso T, Zuo D, et al. Infiltration of CD8+ T cells into tumor cell clusters in triple-negative breast cancer. Proc Natl Acad Sci USA 2019; 116: 3678–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bose P, Brockton NT, Guggisberg K, et al. Fractal analysis of nuclear histology integrates tumor and stromal features into a single prognostic factor of the oral cancer microenvironment. BMC Cancer 2015; 15: 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rasmusson A, Zilenaite D, Nestarenkaite A, et al. Immunogradient indicators for antitumor response assessment by automated tumor-stroma interface zone detection. Am J Pathol 2020; 190: 1309–1322. [DOI] [PubMed] [Google Scholar]
- 40.Loi S, Drubay D, Adams S, et al. Tumor-infiltrating lymphocytes and prognosis: a pooled individual patient analysis of early-stage triple-negative breast cancers. J Clin Oncol 2019; 37: 559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balkenhol MC, Ciompi F, Swiderska-Chadaj Z, et al. Optimized tumour infiltrating lymphocyte assessment for triple negative breast cancer prognostics. Breast 2021; 56: 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmid P, Cortes J, Pusztai L, et al. Pembrolizumab for early triple-negative breast cancer. N Engl J Med 2020; 382: 810–821. [DOI] [PubMed] [Google Scholar]
- 43.Gartrell-Corrado RD, Chen AX, Rizk EM, et al. Linking transcriptomic and imaging data defines features of a favorable tumor immune microenvironment and identifies a combination biomarker for primary melanoma. Cancer Res 2020; 80: 1078–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fassler DJ, Torre-Healy LA, Gupta R, et al. Spatial characterization of tumor-infiltrating lymphocytes and breast cancer progression. Cancer 2022; 14: 2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Apetoh L, Smyth MJ, Drake CG, et al. Consensus nomenclature for CD8+ T cell phenotypes in cancer. Onco Targets Ther 2015; 4: e998538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Danenberg E, Bardwell H, Zanotelli VRT, et al. Breast tumor microenvironment structures are associated with genomic features and clinical outcome. Nat Genet 2022; 54: 660–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, Wang YG, Hu C, et al. Cell graph neural networks enable the precise prediction of patient survival in gastric cancer. NPJ Precis Oncol 2022; 6: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Powles T, Kockx M, Rodriguez-Vida A, et al. Clinical efficacy and biomarker analysis of neoadjuvant atezolizumab in operable urothelial carcinoma in the ABACUS trial. Nat Med 2019; 25: 1706–1714. [DOI] [PubMed] [Google Scholar]
- 49.Berben L, Wildiers H, Marcelis L, et al. Computerised scoring protocol for identification and quantification of different immune cell populations in breast tumour regions by the use of QuPath software. Histopathology 2020; 77: 79–91. [DOI] [PubMed] [Google Scholar]
- 50.Ho DJ, Yarlagadda DVK, D’Alfonso TM, et al. Deep multi-magnification networks for multi-class breast cancer image segmentation. Comput Med Imaging Graph 2021; 88: 101866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wershof E, Park D, Barry DJ, et al. A FIJI macro for quantifying pattern in extracellular matrix. Life Sci Alliance 2021; 4: e202000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vanhersecke L, Bougouin A, Crombe A, et al. Standardized pathology screening of mature tertiary lymphoid structures in cancers. Lab Invest 2023; 103: 100063. [DOI] [PubMed] [Google Scholar]
- 53.Vanhersecke L, Brunet M, Guégan JP, et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat Cancer 2021; 2: 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Halama N, Zoemig I, Berthel A, et al. Tumoral immune cell exploitation in colorectal cancer metastases can be targeted effectively by anti-CCR5 therapy in cancer patients. Cancer Cell 2016; 29: 587–601. [DOI] [PubMed] [Google Scholar]
- 55.Halama N, Michel S, Kloor M, et al. Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer Res 2011;71:5670–5677. [DOI] [PubMed] [Google Scholar]
- 56.Berthel A, Zoernig I, Valous NA, et al. Detailed resolution analysis reveals spatial T cell heterogeneity in the invasive margin of colorectal cancer liver metastases associated with improved survival. Oncoimmunology 2017; 6: el286436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Denkert C, von Minckwitz G, Darb-Esfahani S, et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol 2018; 19: 40–50. [DOI] [PubMed] [Google Scholar]
- 58.Stanton SE, Disis ML. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J Immunother Cancer 2016; 4: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Emens LA, Cruz C, Eder JP, et al. Long-term clinical outcomes and biomarker analyses of atezolizumab therapy for patients with metastatic triple-negative breast cancer: a phase 1 study. JAMA Oncol 2019; 5: 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burstein HJ, Curigliano G, Loibl S, et al. Estimating the benefits of therapy for early-stage breast cancer: the St. Gallen international consensus guidelines for the primary therapy of early breast cancer 2019. Ann Oncol 2019; 30: 1541–1557. [DOI] [PubMed] [Google Scholar]
- 61.Swisher SK, Wu Y, Castaneda CA, et al. Interobserver agreement between pathologists assessing tumor-infiltrating lymphocytes (TILs) in breast cancer using methodology proposed by the International TILs Working Group. Ann Surg Oncol 2016; 23: 2242–2248. [DOI] [PubMed] [Google Scholar]
- 62.TILs in breast cancer [Accessed 4 August 2020]. Available from: https://www.tilsinbreastcancer.org/.
- 63.Brown JR, Wimberly H, Lannin DR, et al. Multiplexed quantitative analysis of CD3, CD8, and CD20 predicts response to neoadjuvant chemotherapy in breast cancer. Clin Cancer Res 2014; 20: 5995–6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yaghoobi V, Martinez-Morilla S, Liu Y, et al. Advances in quantitative immunohistochemistry and their contribution to breast cancer. Expert Rev Mol Diagn 2020; 20: 509–522. [DOI] [PubMed] [Google Scholar]
- 65.Sun P, He J, Chao X, et al. A computational tumor-infiltrating lymphocyte assessment method comparable with visual reporting guidelines for triple-negative breast cancer. EBioMedicine 2021; 70: 103492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kos Z, Roblin E, Kim RS, et al. Pitfalls in assessing stromal tumor infiltrating lymphocytes (sTILs) in breast cancer. NPJ Breast Cancer 2020; 6: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen HX, Song M, Maecker HT, et al. Network for biomarker immunoprofiling for cancer immunotherapy: cancer immune monitoring and analysis centers and cancer immunologic data commons (CIMAC-CIDC). Clin Cancer Res 2021; 27: 5038–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chou M, Illa-Bochaca I, Minxi B, et al. Optimization of an automated tumor-infiltrating lymphocyte algorithm for improved prognostication in primary melanoma. Mod Pathol 2021; 34: 562–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Acs B, Ahmed FS, Gupta S, et al. An open source automated tumor infiltrating lymphocyte algorithm for prognosis in melanoma. Nat Commun 2019; 10: 5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saldanha G, Flatman K, Teo KW, et al. A novel numerical scoring system for melanoma tumor-infiltrating lymphocytes has better prognostic value than standard scoring. Am J Surg Pathol 2017; 41: 906–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moore MR, Friesner ID, Rizk EM, et al. Automated digital TIL analysis (ADTA) adds prognostic value to standard assessment of depth and ulceration in primary melanoma. Sci Rep 2021; 11: 2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thagaard J, Broeckx G, Page DB, et al. Pitfalls in machine learning-based assessment of tumor-infiltrating lymphocytes in breast cancer: a report of the International Immuno-oncology Biomarker Working Group. J Pathol 2023; 250: 498–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313: 1960–1964. [DOI] [PubMed] [Google Scholar]