

Abstract
Purpose:
Chordoma is a rare bone tumor with a high recurrence rate and limited treatment options. The aim of this study was to identify molecular subtypes of chordoma that may improve clinical management.
Experimental Design:
We conducted RNA sequencing in 48 tumors from Chinese skull-base chordoma patients and identified two major molecular subtypes. We then replicated the classification using a NanoString panel in 48 chordoma patients from North America.
Results:
Tumors in one subtype were more likely to have somatic mutations and reduced expression in chromatin remodeling genes, such as PBRM1 and SETD2, while the other subtype was characterized by the upregulation of genes in epithelial-mesenchymal transition and Sonic Hedgehog pathways. Immunohistochemical staining of top differentially expressed genes between the two subtypes in 312 Chinese chordoma patients with long-term follow-up data showed that the expression of some markers such as PTCH1 was significantly associated with survival outcomes.
Conclusions:
Our findings may improve the understanding of subtype-specific tumorigenesis of chordoma and inform clinical prognostication and targeted options.
Keywords: chordoma, molecular subtype, RNA sequencing, NanoString gene expression profiling, recurrence and survival
Introduction
Chordoma is a rare bone tumor, which is believed to originate from notochordal remnants (1) and occurs in the bones of the skull base and spine (2). Approximately one third of chordomas arise in the clivus (skull base) and patients with skull-base chordoma have the earliest average age-onset (usually less than 50 years) compared to chordomas occurring at other sites (>55 years) (3,4). Although chordomas are slow-growing, local recurrence is common especially among skull-base chordoma patients, largely due to incomplete tumor resection and difficulty in normal margin resection because of the closeness of the tumor to vital structures. Surgery with or without neoadjuvant or adjuvant radiation therapy is the first-line treatment for chordoma. Proton radiation therapy is often considered the best radiation treatment, but its availability is still limited (5). Currently, there is no clear clinical guidance on patient stratification regarding treatment such as post-surgery radiation therapy. In addition, treatment options for chordoma patients, particularly those with advanced disease, are still limited. The median survival is about 7 years; however, the clinical progression is extremely variable (6) and is likely determined by both surgical factors and tumor biology.
By morphology, chordomas are divided into classical (conventional), chondroid, poorly differentiated, and dedifferentiated. Expression of brachyury, which is a transcription factor encoded by the TBXT gene and plays a key role in notochord development, is considered as a specific diagnostic marker for chordoma (7,8). Dedifferentiated and poorly differentiated chordoma are rare subtypes and usually present with aggressive clinical behaviors (9,10), other histological subtypes did not show variation in clinical outcomes (11). A recent study identified prognostic chordoma subtypes based on DNA methylation profiles (12), suggesting that chordomas are molecularly heterogeneous. A better understanding of the molecular processes in chordoma is critically needed to develop prognostic prediction tools and better-tailored treatments. To address this question, we conducted a transcriptome analysis using RNA sequencing (RNA-Seq) in 48 chordoma tumors collected from skull-base chordoma patients in China and replicated the major findings in an independent chordoma patient population (n=48) from North America. Further, we identified several key markers defining molecular subtypes that were associated with clinical outcomes in a large set of skull-base chordoma patients (n=312) with long-term follow-up data available.
Methods
Study Populations:
We included three chordoma patient cohorts in this study. The discovery analysis (RNA-Seq cohort) included 48 patients who were diagnosed with skull-base chordoma and underwent endoscopic endonasal surgeries at the Neurosurgery Department of Beijing Tiantan Hospital, Capital Medical University, between June 2016 and May 2018. Fresh-frozen tumor samples and clinicopathological characteristics including age, tumor histologic type, tumor volume, Ki67 status, gross resection rate, pre-surgery radiation therapy (RT), post-surgery RT, recurrence, and death status were collected. The chordoma diagnosis was confirmed with brachyury staining for the majority of patients and was confirmed by morphology in combination with cytokeratin (CK) and epithelial membrane antigen (EMA) markers for the remaining patients without brachyury staining data. The survival cohort was derived from a retrospective cohort of 507 skull-base chordoma patients who were also treated at the Beijing Tiantan Hospital between January 2008 and September 2014. The current analysis included 312 patients with chordoma tumors constructed on tissue microarrays (TMAs) and clinical follow-up data collected. The study protocols were conducted in accordance with the Declaration of Helsinki and approved by the ethics committee of the Beijing Tiantan Hospital, and written informed consent was obtained for all study participants.
The replication cohort (NanoString cohort) included chordoma patients, unselected for tumor site, identified from the United States (US) and Canada who participated in an ongoing sporadic chordoma study conducted at the National Cancer Institute, which has been previously described (13). All diagnoses of chordoma were confirmed by reviewing pathologic slides or reports, medical records, or death certificates, and all study subjects were of European ancestry. The current analysis included 48 patients whose RNA was extracted from formalin-fixed paraffin-embedded (FFPE) tumors and passed all quality control measures in the expression profiling analysis using a Nanostring custom-made panel. The average age at diagnosis among these 48 patients was 50 years; the vast majority (98%) had classic chordoma histology; and 48.9% had skull-base chordoma. The study was approved by the institutional review boards at the National Institutes of Health and all participants provided written informed consent.
RNA-Seq and whole-genome sequencing (WGS) analysis:
Biospecimen collection, quality control, and tissue processing steps were described previously (14). For total RNA extraction, fresh frozen tissue sections were processed with TRIzol (Thermo, US) according to the manufacturer’s instructions. RNA was run on 1% agarose gels to check for degradation and contamination. RNA quality and quantity were assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Aligent Technologies, CA, USA). RNA samples with an integrity number (RIN) of over 6.8 were included for transcriptome library preparation and sequencing. RNA-Seq was carried out by the Novogene Corporation (Beijing, China) using Illumina Hiseq as previously described (14). Raw data in fastq format were processed by removing reads containing adapters or ploy-N and low-quality reads. Fastqc was used for quality control process (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Short reads were then mapped to hg19 genome and gene expression was further quantified as TPM (transcripts per million) using RSEM with default parameters (https://github.com/deweylab/RSEM), and log2TPM was used for statistical analyses.
WGS data based on paired tumor and blood samples was available for a subset of these patients (n=34). Sequencing and bioinformatic analyses were previously described in detail (14). Tumor purity was estimated from ESTIMATE (Estimation of STromal and Immune cells in MAlignant Tumor tissues using Expression data) (15) based on RNA expression data, and mutations and copy number alterations in the WGS analysis (14), respectively. Since purity estimated based on RNA-Seq and WGS analyses showed high correlation (Pearson Correlation Coefficient = 0.69, p=0.0004), we used RNA-based purity estimate in the downstream analysis.
Molecular classification:
Unsupervised hierarchical clustering was conducted using ConsensusClusterPlus (16), based on the 2,000 genes showing the most variable expression levels in the dataset with Euclidean distance calculated from z-statistics using the R package “Complex Heatmap” (v2.10.0). For each of the 500 resampling of subjects, we sampled 80% of the subjects and clustered them using agglomerative hierarchical clustering with Pearson correlation as the distance metric. We evaluated up to 6 clusters and chose 3 consensus clusters (k=3; CC1, CC2, and CC3) as it best fit the data. Since tumors in CC3 had lower TBXT gene expression and lower tumor purity estimated by both gene expression and WGS analyses, it is likely that this subtype is enriched with low-purity tumors. We therefore focused the subsequent analyses on CC1 and CC2 and performed differential gene expression analyses between the two subtypes. We selected 21 top-ranked differentially expressed genes (DEGs) based on p values (p <10−6) and fold changes (FC, log2FC > 1), which were included in the panel for NanoString profiling analysis of FFPE tumors in the replication dataset. Gene Set Enrichment Analysis (GSEA) was performed using DEGs ranked by scores that incorporate p values and log2FC using the GSEA package developed by the Broad Institute with the use of the Molecular Signatures Database (MSigDB v7) (17,18).
NanoString RNA profiling and DNA panel sequencing:
Genomic DNA and total RNA were extracted from FFPE tissue blocks or sections from North American chordoma patients (replication cohort) that were processed to enrich for tumor cells through macro-dissecting the tumor region. Targeted panel sequencing (gene list see Supplementary Table 1) was conducted on paired tumor and germline DNA, with an average coverage of 1,158x. Torrent Variant Caller (TVC) was used for variant calling. Variants that were QC flagged, with allele frequency >0.001 in the ExAC database, variant allele fraction <10% in tumors, and <50 total reads were removed from the analysis. Variant calls for targeted genes were checked manually through visual assessment using the Integrative Genomics Viewer (IGV).
RNA was quantitated using a ThermoScientific NanoDrop 2000 Spectrophotometer (cat. # ND-2000) and Agilent 4200 TapeStation. After assessing for low concentration or low percentages of RNA molecules > 300 nucleotides long (3%), the remaining samples were processed by the University of North Carolina Translational Genomics Laboratory using the Nanostring nCounter Platform. Samples were run on a custom codeset that included gene sets for the 21 top DEGs between CC1 and CC2 in the RNA-Seq analysis (AHR, ANGPTL4, GLI1, GULP1, HHIP, LEF1, LRRC32, ODZ2, ODZ4, OMD, PCOLCE, PDZRN4, PEG10, PKDCC, PTCH1, SLC6A20, SPON2, TMTC1, TNNC1, TP63, and TSPAN11). 48 samples passed quality check and were randomized to two batches and Stratagene Universal Human References were included to assess batch variability. Batch variability was low, with correlations between reference standards exceeding 0.99.
Tissue Microarray (TMA) and immunohistochemical staining:
TMAs were constructed from FFPE skull-base chordoma tumor donor blocks using a Tissue Array MiniCore 3 (ALPHELYS, Plaisir, France) with two 2-milimeter cores per donor block. A 4-μm thick section of each TMA was acquired using the Leica RM 2135 Rotary Microtome and sections were baked for 1 h at 65 °C followed by deparaffinization with dimethylbenzene. After being hydrated in a graded ethanol series and subjected to antigen retrieval, the sections were treated with 3% H2O2 and blocked using normal goat serum. The sections were then incubated with primary antibodies overnight at 4°C and followed by second antibody and Diaminobenzidine (DAB) treatment. Finally, the sections were dehydrated, cleared, and mounted. The following antibodies were used for IHC: anti-LEF1 (1:500, ab137872, abcam, Cambridge, Massachusetts), anti-PTCH1 (1:200, ab53715, abcam), anti-GLI1 (1:100, BS90575, bioworld, Bloomington, Illinois), anti-CLDN11 (1:100, ab53041, abcam), anti-EPHA3 (1:100, ab126261, abcam). Marker expression levels were independently assessed by two pathologists (JW and XL) and consensus calls were achieved through discussion in the case of disagreement between the two pathologists. Slightly different criteria were used for different markers to accommodate distinct patterns in staining across markers. PTCH1 expression was categorized into three groups: 0=negative staining in both nucleus and plasma, 1=positive staining in nucleus but negative or weak staining in plasma, 2=positive staining in both nucleus and plasma (strong). EPHA3 expression was assessed based on nuclear and membrane staining and was characterized into four categories: 0=negative staining, 1=overall weak staining or staining with intermediate intensity in <10% tumor cells, 2=overall intermediate staining or strong intensity in <10% tumor cells, 3=strong staining in ≥10% tumor cells. Nuclear staining was assessed for CLDN11, CLI1, and LEF1. For CLDN11, we dichotomized the staining levels (0=negative and 1=positive) because of less variable staining intensity among positive cells. For CLI1, on the other hand, the proportion of positive stained cells showed a wide range, we therefore used the quartile to categorize the staining. LEF1 was categorized into three groups: 0=negative, 1=≤10% tumor cells, and 2= >10% tumor cells. Average values were taken for the two cores from same donor blocks.
Statistical Analysis:
Wilcoxon rank or Kruskal-Wallis test was used to compare media differences in gene expression or signature contribution across tumor subtypes. Logistic regression models were used to assess the associations between risk factors/tumor features and tumor subtypes. The Kaplan–Meier curve was used to assess survival among patients, stratified by the different levels of protein expression defined by immunohistochemical staining intensity. Hazard ratios (HRs), 95% confidence intervals (CIs), and p values were obtained using multivariate Cox proportional hazards model with the adjustment of age, sex, pre- and post-surgery radiation therapy. All statistical tests in the present study were two-sided and performed using SAS version 9.4 (SAS Institute, Cary, NC, USA) or R version 3.6.3 (R Foundation for Statistical Computing, Vienna, Austria).
Data availability
The WGS and RNA-Seq data generated in this study have been deposited in the Database of Genotypes and Phenotypes (dbGaP) under Accession Code phs002301.v1.p1. The targeted sequencing and NanoString data for the European ancestry replication dataset will be released to dbGaP under accession # phs001280.v1.p1.
Results
Patient cohorts
The discovery analysis (RNA-Seq cohort) included 48 patients with skull-base chordoma who were diagnosed and treated at Beijing Tiantan Hospital, China. The detailed clinical characteristics of these patients are shown in Table 1. In brief, the mean age at initial diagnosis of chordoma among these patients was 43.6 years (range: 6–72); 64.6% were males, 54% had conventional/classical chordoma and 46% had chondroid chordoma. After an average follow-up period of 52 months, there were 30 recurrences and 9 deaths, all from chordoma (Table 1).
Table 1:
Description of the three chordoma patient cohorts included in this study
| RNAseq cohort (N= 48) | Nanostring cohort (N= 48) | Survival analysis cohort (N= 312) | ||
|---|---|---|---|---|
|
| ||||
| Variables | N (%) Mean (SD) | N (%) Mean (SD) | N (%) Mean (SD) | P-valuea |
|
| ||||
| Age (year) | 43.6 (18.7) | 50.0 (16.4) | 40.7 (14.5) | 0.0004 |
| <25 | 10 (20.8) | 5 (10.4) | 51 (16.3) | <.0001 |
| 25–50 | 15 (31.3) | 13 (27.1) | 172 (55.1) | |
| ≥50 | 23 (49.9) | 30 (62.5) | 89 (28.6) | |
| Missing | ||||
| Sex | ||||
| F | 17 (35.4) | 23 (47.9) | 137 (43.9) | 0.4323 |
| M | 31 (64.6) | 25 (52.1) | 175 (56.1) | |
| Missing | ||||
| Tumor site | ||||
| Skull/face | 48 (100) | 23 (48.9) | 312 (100) | <.0001 |
| Pelvic/sacrum | 0 (0) | 12 (25.5) | 0 (0) | |
| Vertebral | 0 (0) | 12 (25.6) | 0 (0) | |
| Missing | 1 | |||
| Histological type | ||||
| Classic | 26 (54.2) | 47 (97.9) | 227 (72.7) | <.0001 |
| Chondroid | 22 (45.8) | 1 (2.1) | 82 (26.3) | |
| Poorly differentiated | 0 (0) | 0 (0) | 3 (1.0) | |
| Dedifferentiated | 0 (0) | 0 (0) | 0 (0) | |
| Missing | ||||
| Pre-surgery RTb treatment | ||||
| No | 46 (95.8) | 266 (85.3) | 0.0419 | |
| Yes | 2 (4.2) | 46 (14.7) | ||
| Missing | ||||
| Post-surgery RTb treatment | ||||
| No | 27 (56.3) | 151 (58.5) | 0.8736 | |
| Yes | 21 (43.7) | 107 (41.5) | ||
| Missing | 54 | |||
| Post-surgery RTb treatment type | ||||
| Proton | 11 (45.8) | 16 (20.0) | <.0001 | |
| Radiosurgery | 2 (8.4) | 47 (58.8) | ||
| Photon/Carbon | 11 (45.8) | 0 (0) | ||
| Intensity-modulated radiation therapy | 0 (0) | 17 (21.2) | ||
| Missing | 27 | |||
| Recurrence status | ||||
| No | 18 (37.5) | 83 (28.9) | 0.1944 | |
| Yes | 30 (62.5) | 204 (71.1) | ||
| Missing | 25 | |||
| Recurrence-free survival (month) | 22 (2–53) | 33 (0–141) | 0.0378 | |
| Death status | ||||
| No | 39 (81.2) | 158 (50.7) | <.0001 | |
| Yes | 9 (18.8) | 154 (49.3) | ||
| Missing | ||||
| Overall survival (month) | 52 (10–174) | 61 (0–140) | 0.0916 | |
Results from Chi-square, Fisher’s Exact, or ANOVA tests.
Radiation therapy
The molecular replication dataset (NanoString cohort) included 48 patients collected from chordoma patients residing in North America (the United States and Canada) unselected for tumor sites (13) with RNA extracted from FFPE tumors. The average age at diagnosis was 50 years (range 6–78) and the site distribution was 48.9% skull-base, 25.6% spinal, and 25.6% sacral (Table 1). The vast majority of patients (98%) had classic chordoma histology.
Since follow-up time was short in the Chinese RNA-Seq cohort and clinical outcome data were not collected in the North America NanoString cohort, we used another Chinese patient cohort with a large number of chordoma tumors (N=312) constructed on TMAs and long-term follow-up data to assess the clinical outcome in relation to molecular subtypes (survival cohort). All patients in the survival cohort had skull base chordoma and the distribution of patient characteristics is shown in Table 1.
Molecular classification of chordoma based on whole-transcriptome RNA-Seq analysis
We conducted unsupervised consensus clustering analysis of 48 skull-base chordoma tumors using expression of 2,000 most variable genes. The best separation was achieved by dividing the tumors into three subtypes (CC1: n = 23; CC2: n = 11; CC3; n = 14; Figure 1A). Compared with CC1 and CC2 tumors, tumors in CC3 had lower tumor purity estimated using ESTIMATE (see Method, Figure 1B) as well as lower TBXT gene expression (Figure 1C, 1D), which is commonly considered as a hallmark of chordoma, suggesting that this subtype might be enriched with low-purity tumors. We therefore focused the subsequent analyses on CC1 and CC2. The sensitivity analysis of restricting to 34 CC1 and CC2 tumors caused minimal change in classification, with only two patients moved from CC1 to CC2. Therefore, we used the original classification in all downstream analyses.
Figure 1.
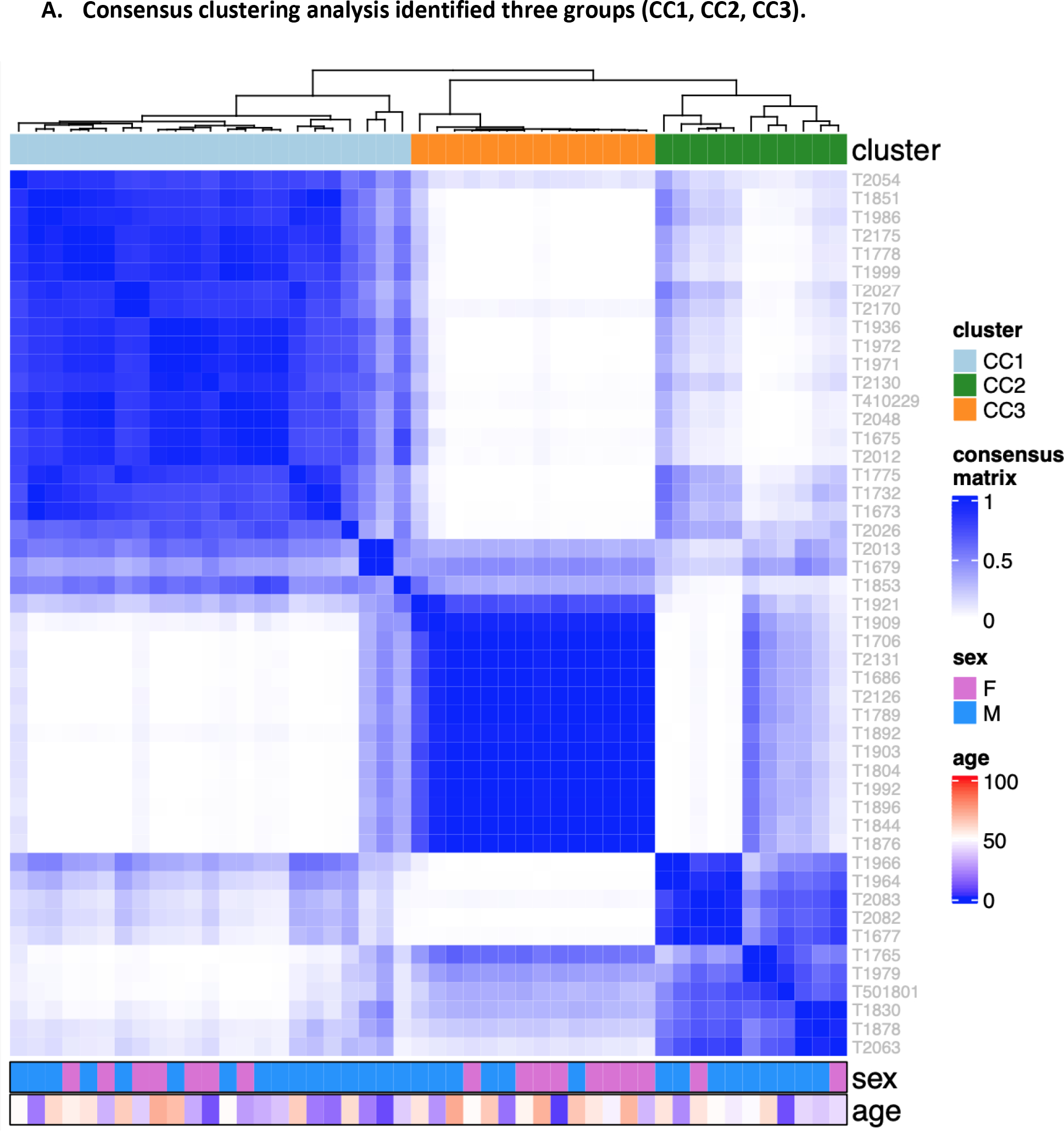
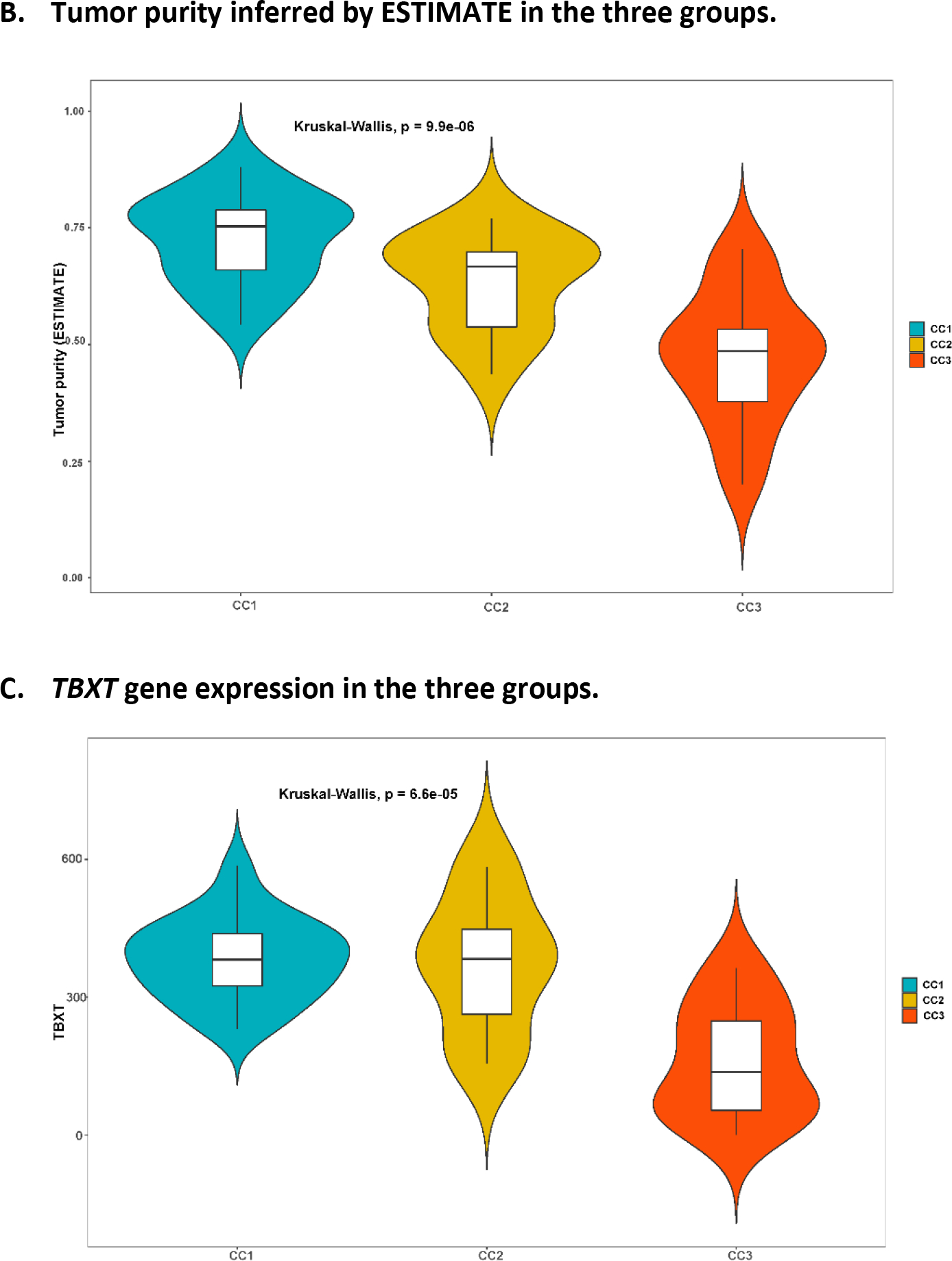
Unsupervised consensus clustering of tumors based on RNASeq data from 48 Chinese skull-base chordoma patients
CC1 and CC2 did not differ significantly in age, tumor size, or histologic subtype in the multivariable logistic regression analysis with the mutual adjustment of these variables (Supplementary Table 2), although CC2 tumors were more likely to have a higher frequency of chondroid subtype (73%) as compared to CC1 (52%). Among 22 CC1 and CC2 tumors with whole-genome sequencing (WGS) data (14), CC2 tumors were less likely to have mutations or structural variants involving PBRM1 (0 in CC2 vs. 4 in CC1) or SETD2 (0 in CC2 vs. 1 in CC1) (Supplementary Table 3), and had higher expression levels of PBRM1 (p=0.0009) and SETD2 (p=0.0012) (Supplementary Figure 1). The overall tumor mutational burden (0.43 mutations/Mb for CC1 and 0.59 mutations/Mb for CC2, p=0.33) or copy number alteration patterns(14) did not vary significantly between the two subtypes. Mutational signatures based on single-base substitution (SBS) did not show significant differences between CC1 and CC2, whereas small indel (ID) signatures demonstrated suggestive differences by subtype (Supplementary Figure 2). CC1 tumors appeared to have a higher contribution of signature A (p=0.005), which was not mapped to any COSMIC ID signatures and is composed predominantly of 2bp insertions (mainly AT and TA) at long (≥5) repeats (14). On the other hand, compared to CC1 tumors, CC2 tumors were more likely to have a higher fraction of signature E (p=0.021), which reflected combinations of COSMIC ID3, 4, 5, and 9 (14). Results based on number of mutations in these signatures showed similar patterns (Supplementary Figure 2).
Top differentially expressed genes (DEGs) between CC1 and CC2 are shown in Figure 2A, with the majority showing higher expression in CC2 than in CC1(complete list of DEGs shown in Supplementary Table 4). Top DEGs included those that play roles in embryonic development (PEG10(19), HHIP(20), PTCH1(21), EPHA3(22), LEF1(23)), neural development (ODZ2(24), SPON2(25), NGFR(26), DPYSL3(27), PPP1R9A(28), NOTCH3(29)), and bone development (OMD(30), PKDCC(31), MATN3(32), TMEM119(33), COL10A1(34)), which are biologically relevant in chordoma development. Gene set enrichment analysis (GSEA) revealed that the most significant pathways (nominal p<0.0001) these DEGs were enriched for included epithelial-mesenchymal transition (EMT), which was upregulated in CC2, and interferon alpha response and TNF alpha signaling via NFKB, which were downregulated in CC2 (Figure 2B, 2C, Supplementary Table 5).
Figure 2.
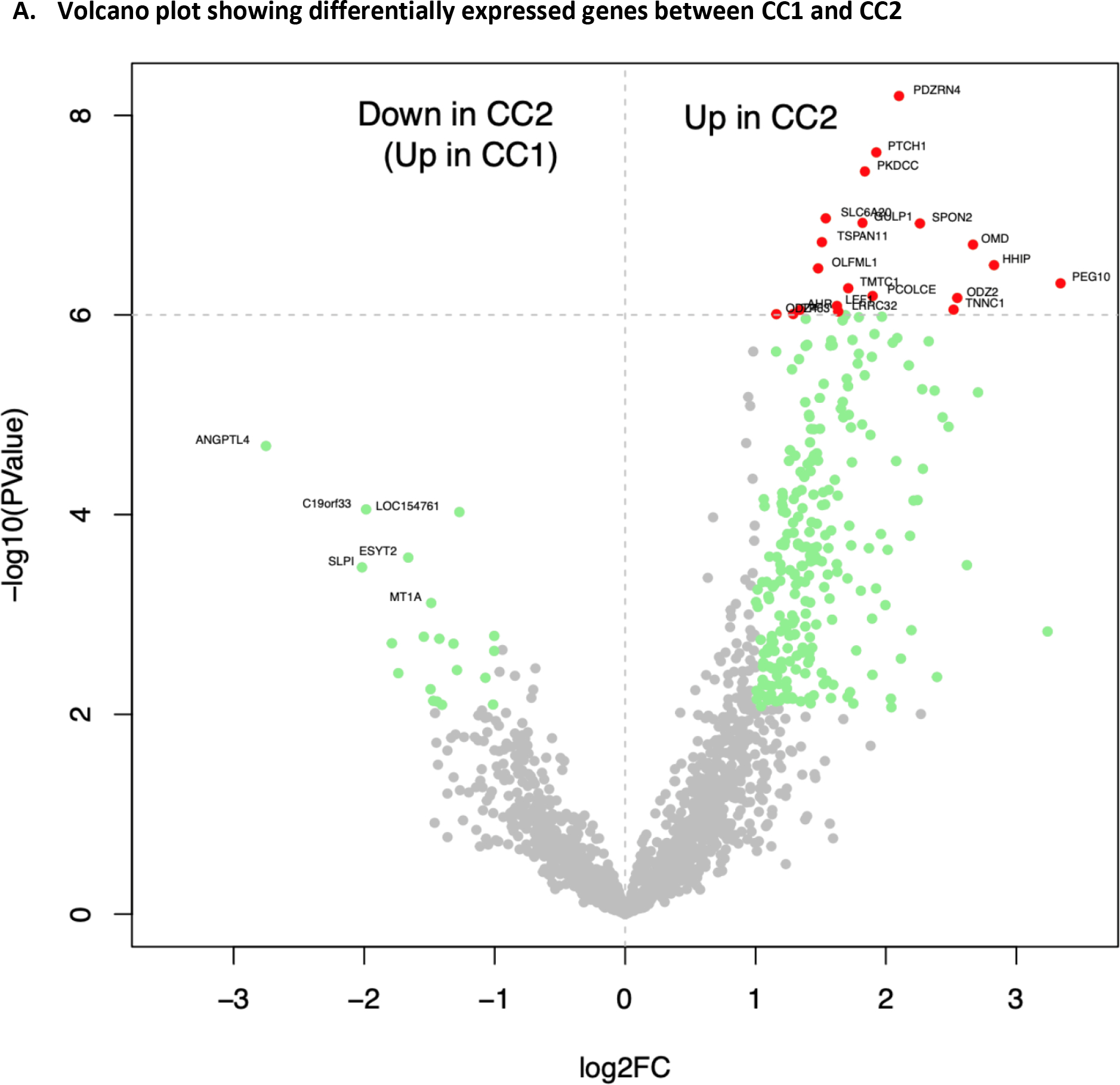
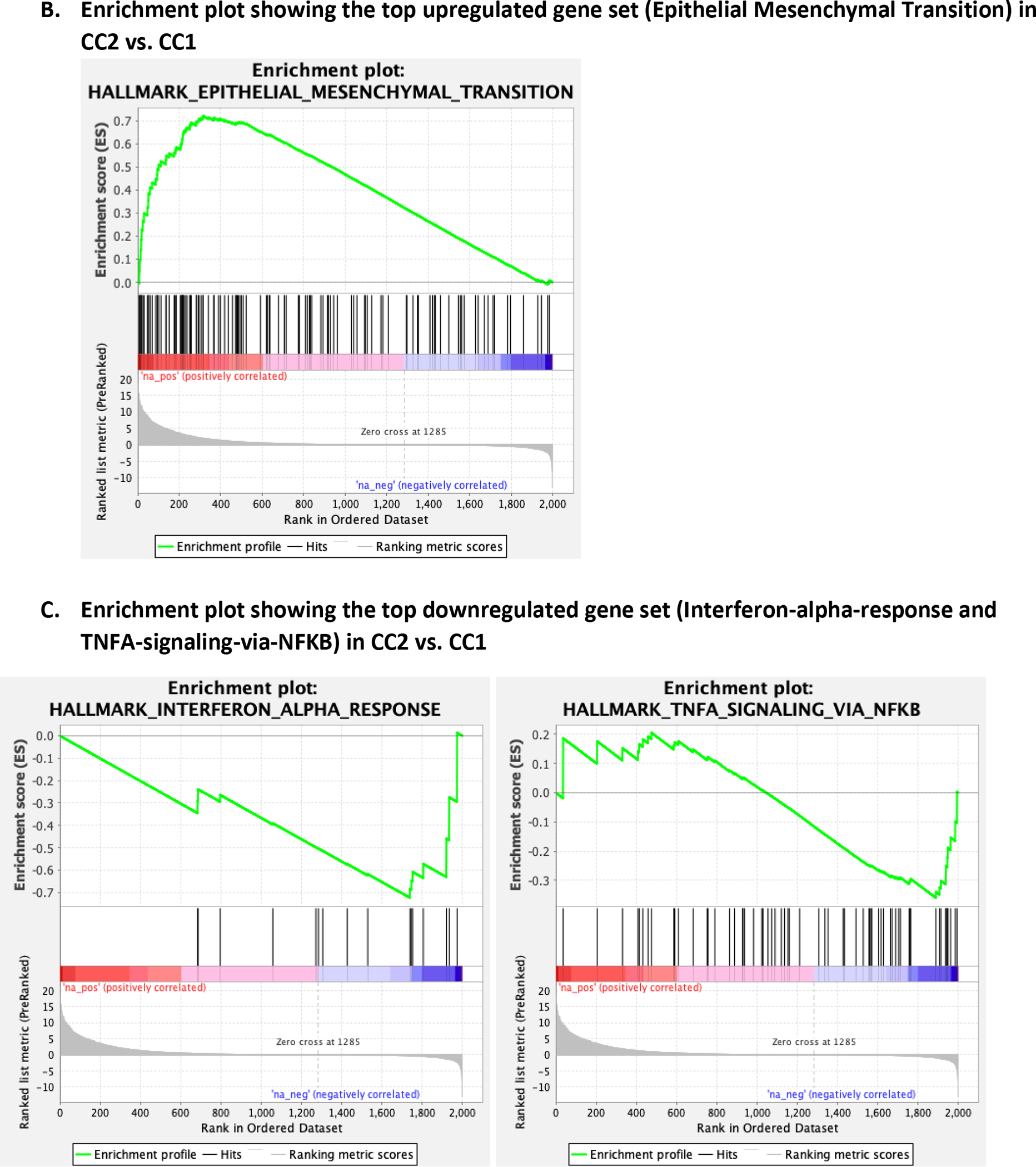
Differentially expressed genes between CC1 and CC2
Replication of the molecular classification in an independent chordoma cohort
To replicate the molecular classification, we designed a NanoString panel including 21 of the top DEGs (AHR, ANGPTL4, GLI1, GULP1, HHIP, LEF1, LRRC32, ODZ2, ODZ4, OMD, PCOLCE, PDZRN4, PEG10, PKDCC, PTCH1, SLC6A20, SPON2, TMTC1, TNNC1, TP63, and TSPAN11) from RNA-Seq and measured expression of these genes in 48 FFPE tumors collected from chordoma patients in North America (the NanoString cohort). The expression of these genes separated tumors into two main groups (NCC1 and NCC2), with tumors in NCC2 having upregulation of most genes (Figure 3), a pattern that is similar to CC2 in the RNA-Seq cohort. The distribution of sex and tumor site did not vary significantly between the two groups, although NCC2 appeared to be enriched with younger (<50 years, p=0.160) patients. Similar classifications were found in the 23 skull-base and 25 non-skull-base patients, respectively (Supplementary Figure 3).
Figure 3.
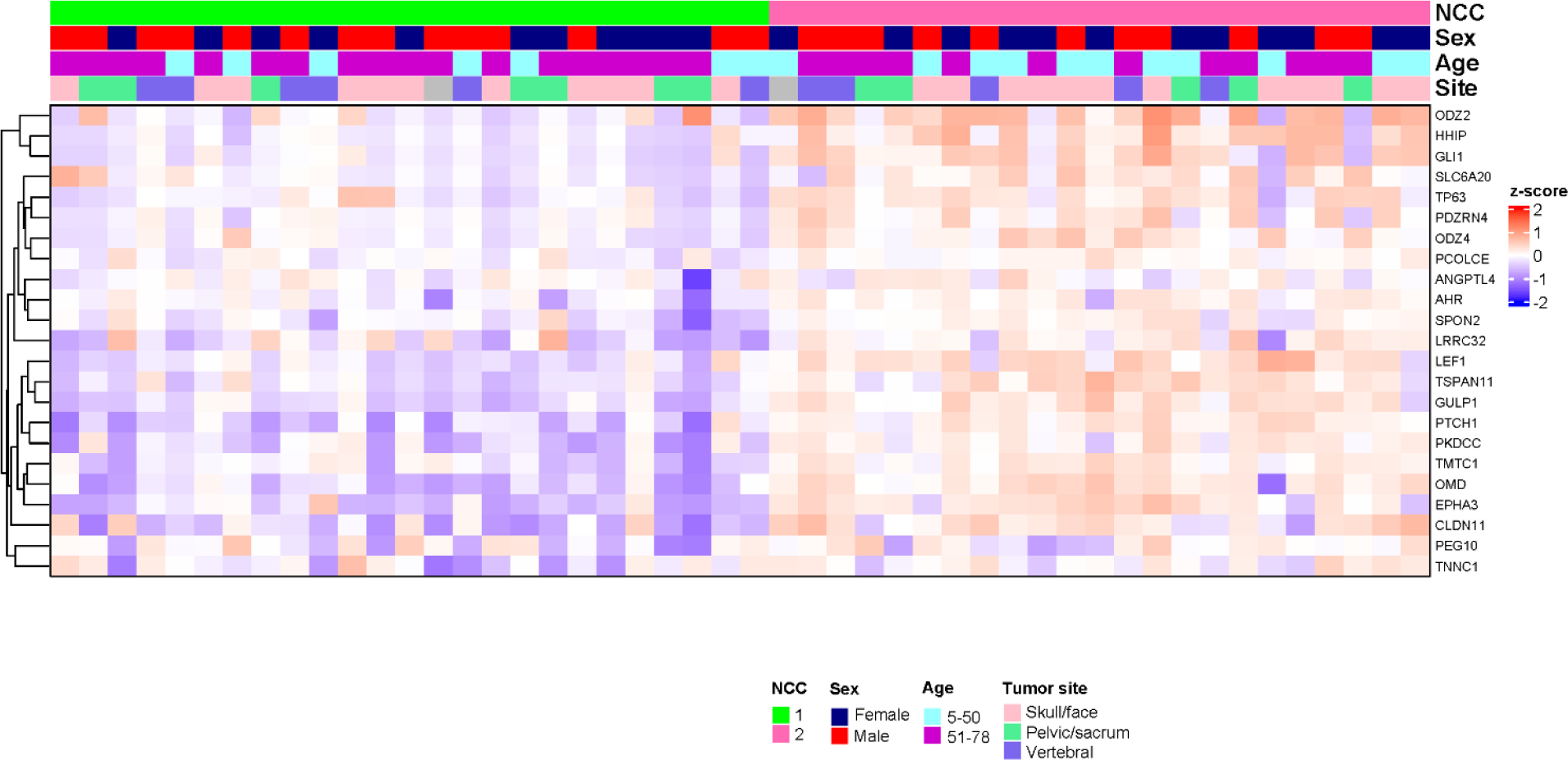
Gene expression profiling based on the NanoString panel in chordoma patients from North America
We then searched for mutations in potential chordoma driver genes in this set of tumors by designing a targeted DNA sequencing panel including genes reported in two chordoma landscape studies (Supplementary Table 1) (14,35). Consistent with results from the RNA-Seq cohort, we found that NCC2 tumors were less likely to have mutations involving PBRM1 (0 in NCC2 vs. 2 in NCC1) and SETD2 (0 in NCC2 vs. 2 in NCC1) and were more likely to have higher expression levels of these genes (PBRM1: p=0.0005, SETD2: p=0.0002, Supplementary Figure 1). In addition, among three patients carrying mutations in LYST, a potential chordoma driver gene reported by Tarpey et al.(35), all had NCC1 tumors. In contrast, among six patients carrying mutations in PIK3CA, three had NCC1 and three had NCC2 tumors, and the single TP53 mutation carrier had NCC2 tumor. These results suggest that the two major molecular subtypes may be associated with distinct sets of chordoma driver genes.
Clinical relevance of the molecular classification
Since follow-up time was short in the RNA-Seq cohort and clinical outcome data were not collected in the NanoString cohort, we examined expression levels of top DEGs in relation to clinical outcomes in the survival cohort of 312 skull base chordoma patients with an average of 5 years of follow-up time to determine the clinical relevance of the molecular subtypes. We selected five genes (PTCH1, LEF1, EPHA3, GLI1, CLDN11) that showed differential expression between the two subtypes (CC1/NCC1 and CC2/NCC2) in both RNA-Seq and NanoString analyses and had commercial antibodies available. We conducted immunohistochemical (IHC) staining of these markers on 312 tumors constructed on TMAs. Among the five markers evaluated, PTCH1 protein expression showed significant associations with both chordoma-specific survival (CSS: hazard ratio [HR] = 2.74, 95% confidence interval [CI] = 1.51 – 4.99, p=0.001, comparing strong to negative staining) and recurrence-free survival (RFS: HR = 2.52, 95% CI = 1.49 – 4.26, p=0.001, comparing strong to negative staining) (Figure 4). CLDN11 was associated with RFS (HR = 1.48, 95% CI = 1.09 – 2.01, p=0.013, comparing positive to negative staining) but not with CSS (HR = 1.26, 95% CI = 0.88 – 1.82, p=0.21, comparing positive to negative staining) (Supplementary Figure 4). The protein expression of the other three markers examined did not show significant associations with either CSS or RFS (Supplementary Figure 4).
Figure 4.
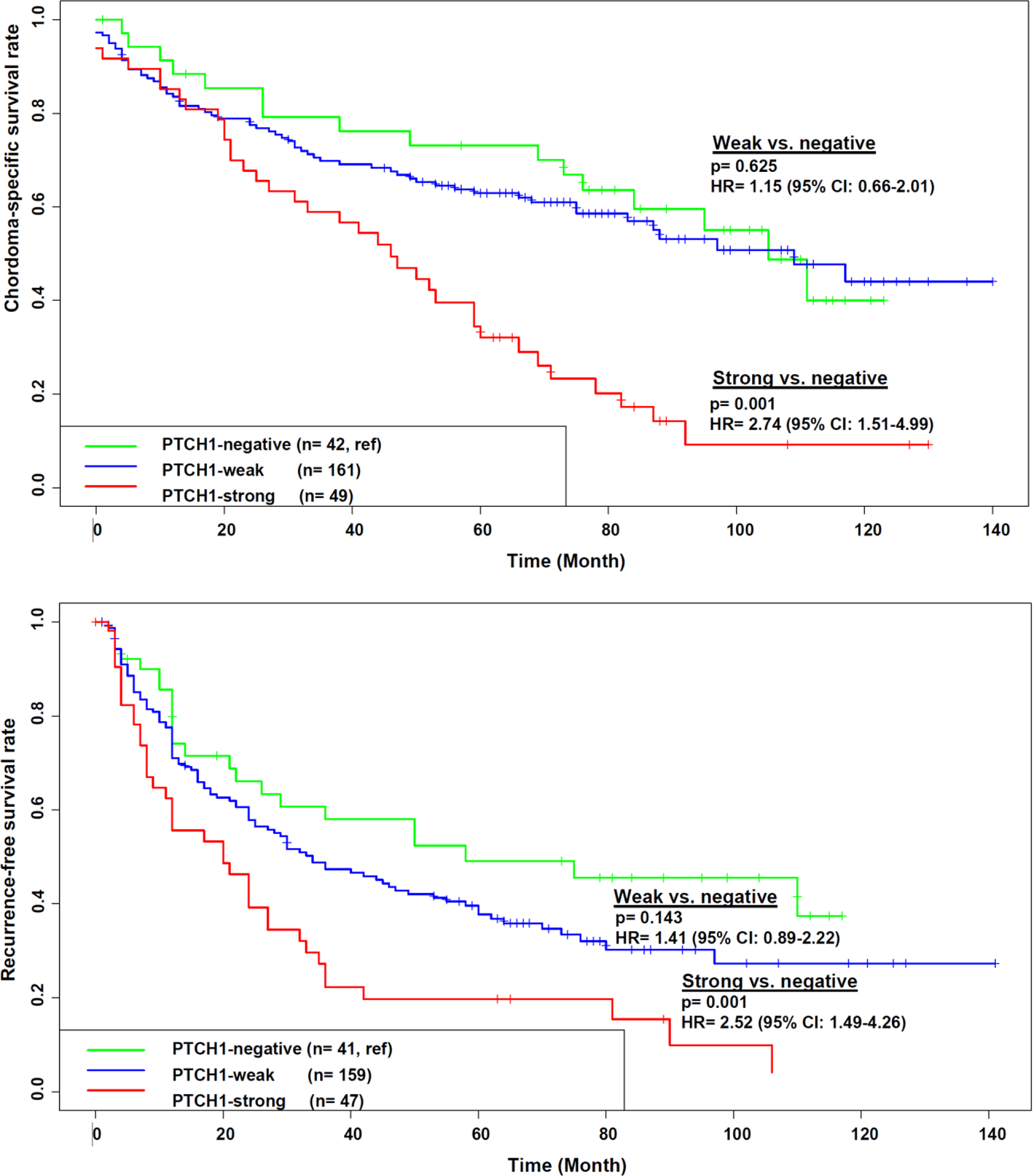
Expression of PTCH1 in relation to overall survival (upper panel) and recurrence-free survival (lower panel) in 312 Chinese chordoma patients with long-term follow-up data
Discussion
Using whole-transcriptomic analysis, we identified two major molecular subtypes of skull-base chordoma, which were distinguished by expression levels of genes involved in embryonic development, neural development, and bone development. Significant pathways enriched in the differentially expressed genes between the two subtypes included EMT, inflammatory response, angiogenesis, interferon gamma, apical junction, p53, and allograft rejection, with EMT showing the strongest enrichment. These two major subtypes were replicated in an independent chordoma cohort containing patients of different ethnic ancestries and unselected tumor locations, suggesting the robustness of the classification. DNA sequencing data from both datasets suggest that the two subtypes may be associated with distinct sets of driver genes. Further, some DEGs showed significant associations with clinical outcomes in a large chordoma cohort with long-term follow-up data, demonstrating the prognostic relevance of the molecular classification.
EMT is a process characterized by epithelial cells losing contact with their neighbors and gaining mesenchymal properties, which plays an important role in embryonic development and cancer progression (36). TBXT, which encodes a transcription factor (Brachyury) in notochord and plays an essential role in chordoma development (8,37), has been shown to promote EMT in a variety of cancers including chordoma (38). Although the two subtypes showed similar TBXT expression levels, CC2 was associated with the upregulation of EMT genes, suggesting other factors may drive the different levels of EMT between CC1 and CC2. For example, TWIST1, a transcription factor and master regulator of embryonic morphogenesis that plays an essential role in metastasis through promoting EMT (39), showed higher expression in CC2 than CC1 (p=0.046, Supplementary Figure 5). Similarly, most of the Snail family members (SNAI1 [p=0.017] and SNAI3 [p=0.098]), which are also prominent EMT inducers and are implicated in important developmental processes including neural differentiation (40), were also upregulated in CC2 tumors (Supplementary Figure 5). Since CC2 also showed significant enrichment of genes in inflammation, angiogenesis, and p53 pathways, it is possible that these processes may contribute to inducing and sustaining EMT in CC2. Since EMT is the major mechanism by which cancer cells become metastatic, targeting molecules in this pathway may present potential therapeutic applications for chordoma patients with CC2 tumors. To support this hypothesis, a recent study suggested that Twist-silenced MUG-Chor1chordoma cells were less migratory and invasive compared with negative controls (41).
Chordoma is derived from the remnants of the embryonic notochord, which plays an important role in directing vertebral column formation and segmentation (42). Sonic Hedgehog (SHH), a morphogen secreted by the embryonic notochord, directs the development of the vertebral axis when it binds to the PTCH1 receptor. SHH signaling is inactive in later embryonic stages and its persistent activation may lead to neoplastic transformation of notochordal remnants (43). Somatic mutations in PTCH1 have been identified in more than 90% of sporadic basal cell carcinoma patients but are infrequent in other tumors (44). In our patients with WGS data available, PTCH1 mutations, either germline or somatic, were not observed. However, we observed that PTCH1 and GLI1 expression was upregulated in the CC2 subtype in both discovery RNA-Seq and replication NanoString datasets, suggesting the activation of SHH signaling in CC2 tumors. In addition, we also observed the upregulation of HHIP, a member of the hedgehog-interacting protein, in CC2. HHIP acts as a vertebrate-specific inhibitor of HH signaling (45) and like PTCH1, HHIP transcription is directly induced by SHH signaling and then negatively regulates the signaling pathway(46). Thus, the upregulation of this molecule may also suggest the activation of SHH signaling in the CC2 subtype. These findings are consistent with results from a recent study by Yang et al. showing the extensive expression of SHH molecules including PTCH1 in chordoma patients and the therapeutic potential of targeting the SHH signaling pathway in treating chordoma (43). Consistent with this, we showed that increasing PTCH1 protein expression was associated with worse clinical outcomes in our survival cohort, similar to previous studies that reported higher PTCH1 expression associated with worse prognosis in breast cancer patients (47,48). On the other hand, unlike what was reported by Yang et al. (43), GLI1 protein expression was not associated with clinical outcomes in our TMA analysis (Supplementary Figure 4).
While CC2 tumors demonstrated the activation of EMT and SHH pathways, the CC1 subtype seemed to be enriched with tumors showing mutations and reduced expression in chromatin remodeling pathways, such as PBRM1 and SETD2. Although the association is based on small numbers of tumors with both molecular subtyping and DNA sequencing data, we observed similar associations in the replication dataset. SWI/SNF genes (such as PBRM1 and SMARCB1) and SETD2 have been described as potential driver genes in both skull-base and sacral chordomas (14,35). Mutations and loss of function in these genes may predict and contribute to responses to immune checkpoint inhibitors (49–51). Immune checkpoint inhibitors are a potential line of therapy in chordomas and clinical trials of agents that block the PD-1/PD-L1 pathway are ongoing in a range of cancers, some of which included chordoma patients (52).
Clinical outcomes of chordoma patients are highly variable and disease progression is likely determined by both surgical factors and tumor biology. Currently, there is no clear clinical guidance on patient stratification regarding treatment such as post-surgery RT. Previous studies have associated copy number alterations such as deletions of chromosomal regions 1p36, 9p21, 9q, and 22q (14,53) and methylation profiles (12) with clinical outcomes of chordoma patients. Our findings here further demonstrate the potential of developing molecular prognostic tools to predict patient outcomes by integrating genomic and gene expression markers. Together, these studies highlight the need and feasibility of developing molecular marker panels to predict clinical courses of chordoma patients. For patients who are predicted to have a poor prognosis, more proactive treatments should be considered such as aiming towards maximum tumor resection, application of high-dose RT and shortening the interval between RT and surgery, and early enrollment for molecular therapy trials.
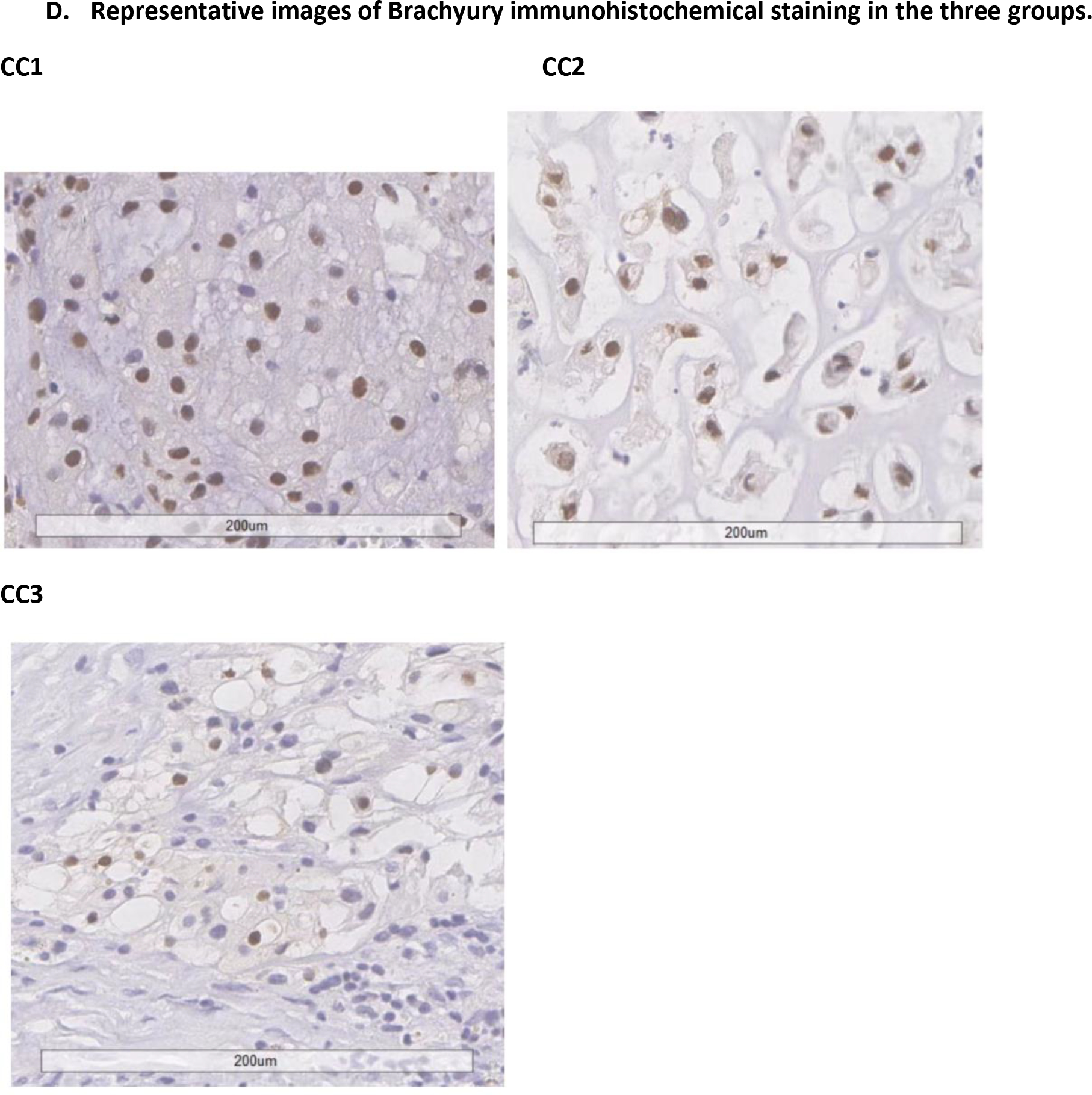
The major limitations of our study include the small sample size of the discovery dataset and lack of clinical outcome data in both discovery and replication datasets, which may have limited the discriminative power and direct assessment of clinical implications. This is particularly true for analyses involving DNA mutation data, which was based on a smaller number of patients. Future studies are needed to validate our findings and develop a set of markers to accurately discriminate subtypes regarding prognosis. In addition, in our analysis we focused on the two major subtypes with higher tumor purity, however, the third subtype we observed may not be completely driven by the low tumor purity due to sampling. Some of the tumors in this subtype may be enriched with immune and other cells in the tumor microenvironment, which may reflect a biologically distinct entity. Indeed, a recent study of epigenetic classification of chordoma tumors found that one of the subtypes they observed showed higher abundance of tumor infiltrating immune cells (12). By further examining H&E and Brachyury stained images of CC3 patients, we confirmed that samples in this subtype contained lower tumor cells, which fell into different scenarios. For example, some tumors indeed had high stromal content primarily driven by fibroblasts, endothelial cells, and inflammatory cells, while other samples were contaminated by normal adjacent tissue, such as dura mater and bone or bone marrow components. Future studies using spatial profiling analyses such as single-cell RNA-Seq are warranted to follow up this group of patients. Another potential limitation is that the Nanostring cohort had tumors from various anatomic locations and the vast majority had classic histology, while the other two datasets were exclusively skull-base and had variable histology. However, the validation of the classification across datasets indicates the robustness of the classification that is not restricted to certain ethnic groups, histology, and tumor locations. Despite the limitations, our study included several clinically well-annotated patient cohorts and replicated the molecular classification in tumors of different ancestries and profiled using different platforms. Further, using a large chordoma cohort with clinical follow-up data and tissue samples available, we demonstrated the clinical relevance of this molecular classification scheme, indicating the potential of developing key molecular markers to improve patient prognostication.
In summary, we defined at least two major molecular subtypes of chordoma using gene expression profiling data. Our findings shed light on subtype-specific molecular mechanisms underlying chordoma development and progression and may have important clinical implications in patient stratification and targeted treatments.
Supplementary Material
Supplementary Figures
Figure S1. A. PBRM1 and SETD2 expression between CC1 and CC2; B. PBRM1 and SETD2 expression between NCC1 and NCC2.
Figure S2. A. Contributions of small indel (ID) signatures (A and E) between CC1 and CC2; B. Number of mutations in small indel (ID) signatures (A and E) between CC1 and CC2.
Figure S3. Gene expression profiling based on the NanoString panel in 23 skull-base (top) and 25 non-skull-base (bottom) chordoma patients from North America.
Figure S4. Expression of selected markers in relation to overall survival (left) and recurrence-free survival (right) in Chinese chordoma patients with long-term follow-up data. A. CLDN11; B. GLI1; C. EPHA3; D. LEF1.
Figure S5. SNAI1, SNAI3, and TWIST expression in CC1 and CC2 tumors.
Supplementary Tables
Table S1. Genes included in the targeted DNA panel sequencing.
Table S2. Differences in clinical characteristics by molecular subtype.
Table S3. Distributions of mutation status in PBRM1 and SETD2 by molecular subtype in RNASeq and Nanostring datasets.
Table S4. Differentially expressed genes among molecular subtypes.
Table S5. GSEA results for up- and down-regulated gene sets comparing CC2 to CC1.
Translational relevance.
Chordoma is a rare bone tumor with a largely unclear etiology and high recurrence rate. Currently, there are no clear guidelines on patient stratification for targeted therapies. In addition, treatment options are limited for patients with advanced disease. Here, using gene expression data obtained from two independent datasets comprising of ethnically different patients, we identified at least two major molecular subtypes of chordoma that were characterized by different genomic features and biological pathways. Our findings provide a first glimpse into the subtype-specific tumorigenesis of chordoma, a tumor type with limited information on etiology and biology. Further, in a large cohort with long-term follow-up and tumor samples available, we demonstrated the association of the molecular subtypes with clinical outcomes, supporting the feasibility of developing molecular marker panels to improve patient stratification regarding prognostication and targeted treatment.
Funding:
This research was supported by Beijing Municipal Science and Technology Commission (Z171100000117002, Y Zhang), Research Special Fund for Public Welfare Industry of Health (201402008, Y Zhang), and the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Division of Cancer Epidemiology and Genetics (X Yang).
Footnotes
Conflict of Interest: The authors declare no conflict of interests.
Data availability
The WGS and RNA-Seq data generated in this study have been deposited in the Database of Genotypes and Phenotypes (dbGaP) under Accession Code phs002301.v1.p1. The targeted sequencing and NanoString data for the European ancestry replication dataset have been deposited in dbGaP under accession # phs001280.v1.p1.
References:
- 1.Salisbury JR, Deverell MH, Cookson MJ, Whimster WF. Three-dimensional reconstruction of human embryonic notochords: clue to the pathogenesis of chordoma. J Pathol 1993;171(1):59–62 doi 10.1002/path.1711710112. [DOI] [PubMed] [Google Scholar]
- 2.Boriani S, Bandiera S, Biagini R, Bacchini P, Boriani L, Cappuccio M, et al. Chordoma of the mobile spine: fifty years of experience. Spine (Phila Pa 1976) 2006;31(4):493–503 doi 10.1097/01.brs.0000200038.30869.27. [DOI] [PubMed] [Google Scholar]
- 3.Zuckerman SL, Bilsky MH, Laufer I. Chordomas of the Skull Base, Mobile Spine, and Sacrum: An Epidemiologic Investigation of Presentation, Treatment, and Survival. World Neurosurg 2018;113:e618–e27 doi 10.1016/j.wneu.2018.02.109. [DOI] [PubMed] [Google Scholar]
- 4.Parry DM, McMaster ML, Liebsch NJ, Patronas NJ, Quezado MM, Zametkin D, et al. Clinical findings in families with chordoma with and without T gene duplications and in patients with sporadic chordoma reported to the Surveillance, Epidemiology, and End Results program. J Neurosurg 2020:1–10 doi 10.3171/2020.4.JNS193505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mercado CE, Holtzman AL, Rotondo R, Rutenberg MS, Mendenhall WM. Proton therapy for skull base tumors: A review of clinical outcomes for chordomas and chondrosarcomas. Head Neck 2019;41(2):536–41 doi 10.1002/hed.25479. [DOI] [PubMed] [Google Scholar]
- 6.Jones PS, Aghi MK, Muzikansky A, Shih HA, Barker FG 2nd, Curry WT Jr. Outcomes and patterns of care in adult skull base chordomas from the Surveillance, Epidemiology, and End Results (SEER) database. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia 2014;21(9):1490–6 doi 10.1016/j.jocn.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 7.O’Donnell P, Tirabosco R, Vujovic S, Bartlett W, Briggs TW, Henderson S, et al. Diagnosing an extra-axial chordoma of the proximal tibia with the help of brachyury, a molecule required for notochordal differentiation. Skeletal Radiol 2007;36(1):59–65 doi 10.1007/s00256-006-0167-4. [DOI] [PubMed] [Google Scholar]
- 8.Vujovic S, Henderson S, Presneau N, Odell E, Jacques TS, Tirabosco R, et al. Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. The Journal of pathology 2006;209(2):157–65 doi 10.1002/path.1969. [DOI] [PubMed] [Google Scholar]
- 9.Shih AR, Cote GM, Chebib I, Choy E, DeLaney T, Deshpande V, et al. Clinicopathologic characteristics of poorly differentiated chordoma. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc 2018;31(8):1237–45 doi 10.1038/s41379-018-0002-1. [DOI] [PubMed] [Google Scholar]
- 10.Hung YP, Diaz-Perez JA, Cote GM, Wejde J, Schwab JH, Nardi V, et al. Dedifferentiated Chordoma: Clinicopathologic and Molecular Characteristics With Integrative Analysis. Am J Surg Pathol 2020;44(9):1213–23 doi 10.1097/PAS.0000000000001501. [DOI] [PubMed] [Google Scholar]
- 11.Frezza AM, Botta L, Trama A, Dei Tos AP, Stacchiotti S. Chordoma: update on disease, epidemiology, biology and medical therapies. Curr Opin Oncol 2019;31(2):114–20 doi 10.1097/CCO.0000000000000502. [DOI] [PubMed] [Google Scholar]
- 12.Zuccato JA, Patil V, Mansouri S, Liu JC, Nassiri F, Mamatjan Y, et al. DNA Methylation based prognostic subtypes of chordoma tumors in tissue and plasma. Neuro-oncology 2021. doi 10.1093/neuonc/noab235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yepes S, Shah NN, Bai J, Koka H, Li C, Gui S, et al. Rare Germline Variants in Chordoma-Related Genes and Chordoma Susceptibility. Cancers (Basel) 2021;13(11) doi 10.3390/cancers13112704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bai J, Shi J, Li C, Wang S, Zhang T, Hua X, et al. Whole genome sequencing of skull-base chordoma reveals genomic alterations associated with recurrence and chordoma-specific survival. Nat Commun 2021;12(1):757 doi 10.1038/s41467-021-21026-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nature communications 2013;4:2612 doi 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 2010;26(12):1572–3 doi 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003;34(3):267–73 doi 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 19.Clark MB, Janicke M, Gottesbuhren U, Kleffmann T, Legge M, Poole ES, et al. Mammalian gene PEG10 expresses two reading frames by high efficiency −1 frameshifting in embryonic-associated tissues. J Biol Chem 2007;282(52):37359–69 doi 10.1074/jbc.M705676200. [DOI] [PubMed] [Google Scholar]
- 20.Chuang PT, McMahon AP. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature 1999;397(6720):617–21 doi 10.1038/17611. [DOI] [PubMed] [Google Scholar]
- 21.Marigo V, Tabin CJ. Regulation of patched by sonic hedgehog in the developing neural tube. Proc Natl Acad Sci U S A 1996;93(18):9346–51 doi 10.1073/pnas.93.18.9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kudo C, Ajioka I, Hirata Y, Nakajima K. Expression profiles of EphA3 at both the RNA and protein level in the developing mammalian forebrain. J Comp Neurol 2005;487(3):255–69 doi 10.1002/cne.20551. [DOI] [PubMed] [Google Scholar]
- 23.van Genderen C, Okamura RM, Farinas I, Quo RG, Parslow TG, Bruhn L, et al. Development of several organs that require inductive epithelial-mesenchymal interactions is impaired in LEF-1-deficient mice. Genes Dev 1994;8(22):2691–703 doi 10.1101/gad.8.22.2691. [DOI] [PubMed] [Google Scholar]
- 24.Zhou XH, Brandau O, Feng K, Oohashi T, Ninomiya Y, Rauch U, et al. The murine Ten-m/Odz genes show distinct but overlapping expression patterns during development and in adult brain. Gene Expr Patterns 2003;3(4):397–405 doi 10.1016/s1567-133x(03)00087-5. [DOI] [PubMed] [Google Scholar]
- 25.Feinstein Y, Borrell V, Garcia C, Burstyn-Cohen T, Tzarfaty V, Frumkin A, et al. F-spondin and mindin: two structurally and functionally related genes expressed in the hippocampus that promote outgrowth of embryonic hippocampal neurons. Development 1999;126(16):3637–48 doi 10.1242/dev.126.16.3637. [DOI] [PubMed] [Google Scholar]
- 26.Underwood CK, Coulson EJ. The p75 neurotrophin receptor. Int J Biochem Cell Biol 2008;40(9):1664–8 doi 10.1016/j.biocel.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 27.Souopgui J, Klisch TJ, Pieler T, Henningfeld KA. Expression and regulation of Xenopus CRMP-4 in the developing nervous system. Int J Dev Biol 2007;51(4):339–43 doi 10.1387/ijdb.062235js. [DOI] [PubMed] [Google Scholar]
- 28.Nakabayashi K, Makino S, Minagawa S, Smith AC, Bamforth JS, Stanier P, et al. Genomic imprinting of PPP1R9A encoding neurabin I in skeletal muscle and extra-embryonic tissues. J Med Genet 2004;41(8):601–8 doi 10.1136/jmg.2003.014142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindsell CE, Boulter J, diSibio G, Gossler A, Weinmaster G. Expression patterns of Jagged, Delta1, Notch1, Notch2, and Notch3 genes identify ligand-receptor pairs that may function in neural development. Mol Cell Neurosci 1996;8(1):14–27 doi 10.1006/mcne.1996.0040. [DOI] [PubMed] [Google Scholar]
- 30.Lin W, Zhu X, Gao L, Mao M, Gao D, Huang Z. Osteomodulin positively regulates osteogenesis through interaction with BMP2. Cell Death Dis 2021;12(2):147 doi 10.1038/s41419-021-03404-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imuta Y, Nishioka N, Kiyonari H, Sasaki H. Short limbs, cleft palate, and delayed formation of flat proliferative chondrocytes in mice with targeted disruption of a putative protein kinase gene, Pkdcc (AW548124). Dev Dyn 2009;238(1):210–22 doi 10.1002/dvdy.21822. [DOI] [PubMed] [Google Scholar]
- 32.Ko Y, Kobbe B, Nicolae C, Miosge N, Paulsson M, Wagener R, et al. Matrilin-3 is dispensable for mouse skeletal growth and development. Mol Cell Biol 2004;24(4):1691–9 doi 10.1128/MCB.24.4.1691-1699.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mizuhashi K, Kanamoto T, Ito M, Moriishi T, Muranishi Y, Omori Y, et al. OBIF, an osteoblast induction factor, plays an essential role in bone formation in association with osteoblastogenesis. Dev Growth Differ 2012;54(4):474–80 doi 10.1111/j.1440-169X.2012.01333.x. [DOI] [PubMed] [Google Scholar]
- 34.Apte SS, Olsen BR. Characterization of the mouse type X collagen gene. Matrix 1993;13(2):165–79 doi 10.1016/s0934-8832(11)80075-2. [DOI] [PubMed] [Google Scholar]
- 35.Tarpey PS, Behjati S, Young MD, Martincorena I, Alexandrov LB, Farndon SJ, et al. The driver landscape of sporadic chordoma. Nat Commun 2017;8(1):890 doi 10.1038/s41467-017-01026-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009;139(5):871–90 doi 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 37.Yang XHR, Ng D, Alcorta DA, Liebsch NJ, Sheridan E, Li SF, et al. T (brachyury) gene duplication confers major susceptibility to familial chordoma. Nature Genetics 2009;41(11):1176–8 doi Doi 10.1038/Ng.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamilton DH, David JM, Dominguez C, Palena C. Development of Cancer Vaccines Targeting Brachyury, a Transcription Factor Associated with Tumor Epithelial-Mesenchymal Transition. Cells Tissues Organs 2017;203(2):128–38 doi 10.1159/000446495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004;117(7):927–39 doi 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, Shi J, Chai K, Ying X, Zhou BP. The Role of Snail in EMT and Tumorigenesis. Curr Cancer Drug Targets 2013;13(9):963–72 doi 10.2174/15680096113136660102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aydemir E, Kasikci E, Coskuncelebi B, Bayrak OF, Sahin F. The effect of TWIST silencing in metastatic chordoma cells. Turk J Biol 2018;42(4):279–85 doi 10.3906/biy-1801-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stemple DL. Structure and function of the notochord: an essential organ for chordate development. Development 2005;132(11):2503–12 doi 10.1242/dev.01812. [DOI] [PubMed] [Google Scholar]
- 43.Yang C, Yong L, Liang C, Li Y, Ma Y, Wei F, et al. Genetic landscape and ligand-dependent activation of sonic hedgehog-Gli1 signaling in chordomas: a novel therapeutic target. Oncogene 2020;39(24):4711–27 doi 10.1038/s41388-020-1324-2. [DOI] [PubMed] [Google Scholar]
- 44.Xu Y, Song S, Wang Z, Ajani JA. The role of hedgehog signaling in gastric cancer: molecular mechanisms, clinical potential, and perspective. Cell Commun Signal 2019;17(1):157 doi 10.1186/s12964-019-0479-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bishop B, Aricescu AR, Harlos K, O’Callaghan CA, Jones EY, Siebold C. Structural insights into hedgehog ligand sequestration by the human hedgehog-interacting protein HHIP. Nat Struct Mol Biol 2009;16(7):698–703 doi 10.1038/nsmb.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chuang PT, Kawcak T, McMahon AP. Feedback control of mammalian Hedgehog signaling by the Hedgehog-binding protein, Hip1, modulates Fgf signaling during branching morphogenesis of the lung. Genes Dev 2003;17(3):342–7 doi 10.1101/gad.1026303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia-Martinez A, Perez-Balaguer A, Ortiz-Martinez F, Pomares-Navarro E, Sanmartin E, Garcia-Escolano M, et al. Hedgehog gene expression patterns among intrinsic subtypes of breast cancer: Prognostic relevance. Pathol Res Pract 2021;223:153478 doi 10.1016/j.prp.2021.153478. [DOI] [PubMed] [Google Scholar]
- 48.Kuehn J, Espinoza-Sanchez NA, Teixeira F, Pavao MSG, Kiesel L, Gyorffy B, et al. Prognostic significance of hedgehog signaling network-related gene expression in breast cancer patients. J Cell Biochem 2021;122(5):577–97 doi 10.1002/jcb.29886. [DOI] [PubMed] [Google Scholar]
- 49.Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018;359(6377):801–6 doi 10.1126/science.aan5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu M, Zhao B, Liu M, Wu L, Li Y, Zhai Y, et al. Pan-cancer analysis of SETD2 mutation and its association with the efficacy of immunotherapy. NPJ Precis Oncol 2021;5(1):51 doi 10.1038/s41698-021-00193-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu XD, Kong W, Peterson CB, McGrail DJ, Hoang A, Zhang X, et al. PBRM1 loss defines a nonimmunogenic tumor phenotype associated with checkpoint inhibitor resistance in renal carcinoma. Nature communications 2020;11(1):2135 doi 10.1038/s41467-020-15959-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barber SM, Sadrameli SS, Lee JJ, Fridley JS, Teh BS, Oyelese AA, et al. Chordoma-Current Understanding and Modern Treatment Paradigms. J Clin Med 2021;10(5) doi 10.3390/jcm10051054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abdallah HM, Gersey ZC, Muthiah N, McDowell MM, Pearce T, Costacou T, et al. An Integrated Management Paradigm for Skull Base Chordoma Based on Clinical and Molecular Characteristics. J Neurol Surg B Skull Base 2021;82(6):601–7 doi 10.1055/s-0041-1730958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Figure S1. A. PBRM1 and SETD2 expression between CC1 and CC2; B. PBRM1 and SETD2 expression between NCC1 and NCC2.
Figure S2. A. Contributions of small indel (ID) signatures (A and E) between CC1 and CC2; B. Number of mutations in small indel (ID) signatures (A and E) between CC1 and CC2.
Figure S3. Gene expression profiling based on the NanoString panel in 23 skull-base (top) and 25 non-skull-base (bottom) chordoma patients from North America.
Figure S4. Expression of selected markers in relation to overall survival (left) and recurrence-free survival (right) in Chinese chordoma patients with long-term follow-up data. A. CLDN11; B. GLI1; C. EPHA3; D. LEF1.
Figure S5. SNAI1, SNAI3, and TWIST expression in CC1 and CC2 tumors.
Supplementary Tables
Table S1. Genes included in the targeted DNA panel sequencing.
Table S2. Differences in clinical characteristics by molecular subtype.
Table S3. Distributions of mutation status in PBRM1 and SETD2 by molecular subtype in RNASeq and Nanostring datasets.
Table S4. Differentially expressed genes among molecular subtypes.
Table S5. GSEA results for up- and down-regulated gene sets comparing CC2 to CC1.
Data Availability Statement
The WGS and RNA-Seq data generated in this study have been deposited in the Database of Genotypes and Phenotypes (dbGaP) under Accession Code phs002301.v1.p1. The targeted sequencing and NanoString data for the European ancestry replication dataset will be released to dbGaP under accession # phs001280.v1.p1.
The WGS and RNA-Seq data generated in this study have been deposited in the Database of Genotypes and Phenotypes (dbGaP) under Accession Code phs002301.v1.p1. The targeted sequencing and NanoString data for the European ancestry replication dataset have been deposited in dbGaP under accession # phs001280.v1.p1.