

Summary
The duplication-triplication/inverted-duplication (DUP-TRP/INV-DUP) structure is a complex genomic rearrangement (CGR). Although it has been identified as an important pathogenic DNA mutation signature in genomic disorders and cancer genomes, its architecture remains unresolved. Here, we studied the genomic architecture of DUP-TRP/INV-DUP by investigating the DNA of 24 patients identified by array comparative genomic hybridization (aCGH) on whom we found evidence for the existence of 4 out of 4 predicted structural variant (SV) haplotypes. Using a combination of short-read genome sequencing (GS), long-read GS, optical genome mapping, and single-cell DNA template strand sequencing (strand-seq), the haplotype structure was resolved in 18 samples. The point of template switching in 4 samples was shown to be a segment of ∼2.2–5.5 kb of 100% nucleotide similarity within inverted repeat pairs. These data provide experimental evidence that inverted low-copy repeats act as recombinant substrates. This type of CGR can result in multiple conformers generating diverse SV haplotypes in susceptible dosage-sensitive loci.
Keywords: copy-number variant, break-induced replication, recombination, Xq28, inversions, segmental duplication, MECP2 duplication syndrome, Mendelian diseases, MMBIR, template switching
Graphical abstract
Highlights
-
•
Inverted triplications cause genomic disorders through alterations in gene dosage
-
•
Pairs of homologous inverted repeats generate varying structural haplotypes
-
•
Breakpoint junction mapping reveals template switches within repeats
-
•
Combining methodologies enhance the analysis of complex genomic aberrations
Analysis of 24 individuals harboring inverted triplications show surprising structural variant haplotype diversity and underlie the importance of inverted repeats acting as points of genomic instability leading to genomic disorders.
Introduction
DNA rearrangements can take many forms in a diploid genome, including deletions, duplications, inversions, and translocations, and can occur on a scale ranging from a few base pairs (bp) to several million base pairs (Mb).1 Among these diverse forms, complex genomic rearrangements (CGRs) are particularly intriguing due to their mostly unpredicted genomic architecture, potential impact on gene dosage, and the consequences for human health. CGRs represent a subset of structural variants (SVs) that involve more than one breakpoint junction in cis, often resulting in the formation of highly complex genomic structures within a chromosome.2,3,4
The duplication-triplication/inversion-duplication (DUP-TRP/INV-DUP) structure is one such CGR perturbation of genome integrity. This genomic instability can be incited by a given pair of inverted low-copy repeats (LCRs) and result from two template switches (TSs) during the process of DNA break repair.5 This recurring DNA rearrangement end product structure is increasingly recognized for its significant roles in human disease, including neurodevelopmental disorders of childhood and adult onset neurodegenerative diseases, as well as its occurrence in cancer genomes.1,5,6,7,8
Large palindromic repeat sequences have shown to be preserved by natural selection across species and point to the formation of inverted repeats in humans.9 In 2013 a genome-wide computational analysis of the GRCh37 human reference build uncovered 1,551 inverted repeats that may predispose a region to local genomic instability by generating a DUP-TRP/INV-DUP structure. That analysis predicted 1,445 dosage-sensitive genes at risk to undergo a mutational event leading to potentially pathogenic DUP-TRP/INV-DUP structures.10 Importantly, the variability in the gene copy number generated as a result of this SV and gene dosage effects (i.e., whether mapping to the duplicated or triplicated genomic interval) has been shown to influence disease severity and subsequent clinical heterogeneity.5,11,12,13,14
The DUP-TRP/INV-DUP structure has historical and clinical relevance in X-linked genomic disorders. It was originally described in the MECP2 duplication syndrome (MRXSL, MIM: 300260), a developmental disorder affecting boys that is caused by copy-number variants (CNVs) spanning the dosage-sensitive gene MECP2 at Xq28 with 100% penetrance.15 Approximately 26% of individuals with MRXSL harbor a DUP-TRP/INV-DUP in a hemizygous state mediated by inverted LCRs downstream of the gene.5,16,17 A more severe clinical phenotype is observed in patients with MECP2 triplication.5,15,18 Copy-number events not spanning the MECP2 gene but mediated by the same LCRs have also been implicated in other Xq28 duplication syndromes, sometimes with incomplete penetrance.19
Upstream of MECP2 on the X chromosome, a different pair of inverted LCRs at Xq22.2 PLP1 locus can also generate a DUP-TRP/INV-DUP structure causing Pelizaeus-Merzbacher disease (PMD, MIM: 312080). This CGR is identified in up to 20% of combined cohorts of 134 PMD subjects.20,21,22 As in MRXSL, triplications of PLP1 are associated with a more severe phenotype in patients.23 A majority of the pathogenetic effects for DUP-TRP/INV-DUP seem to be due to higher gene expression of dosage-sensitive genes (i.e., gain of function). However, loss-of-function effects of this type of variant have also been reported—for example, DUP-TRP/INV-DUP generated by LCRs within Xp21.1 disrupting exons 45–60 in the gene DMD causes Duchenne muscular dystrophy (MIM: 310200).24
Pathogenic DUP-TRP/INV-DUP structures in autosomes have been reported in multiple studies. Triplications of the gene CHRNA7 as the result of a DUP-TRP/INV-DUP structure on chromosome 15 have been associated with neuropsychiatric phenotypes and other cognitive impairments, including autism spectrum disorder and attention-deficit/hyperactivity disorder.25,26 A pair of inverted LCRs on the long arm of chromosome 7 have been shown to generate the DUP-TRP/INV-DUP structure disrupting the gene VIPR2 potentially impacting neurodevelopment and behavior.27 In addition, errors in imprinting due to template switching in the formation of DUP-TRP/INV-DUP events can underlie cases of Temple syndrome (MIM: 616222) and be associated with patients harboring multiple congenital malformations.28,29 Moreover, DUP-TRP/INV-DUP is also reported in familial genetic conditions shared by apparently unrelated families in identity-by-state inheritance. Amplifications of the gene SNCA within a DUP-TRP/INV-DUP structure at 4q22.1 has been associated as a causal factor in the progression of Parkinson disease (MIM: 168601), with duplications of the gene leading to a late onset of the disease versus triplications that lead to an early onset.30,31,32 Intriguingly, contrary to the X-linked CGRs, autosomal DUP-TRP/INV-DUP structures show lower frequency per locus, perhaps due to the smaller size of inverted repeats involved. Examples include a cohort of 27 individuals with 17p13.3 duplication syndrome, in which 10% were found to have a DUP-TRP/INV-DUP structure formed by inverted Alu elements.33
As the number of rare Mendelian disease traits and genomic disorders associated with this CGR continues to increase, our understanding of its implications in somatic cell mutagenesis and cancer genome evolution and progression is just beginning. Recent investigations into the role of structural variation in cancer genomes identified the DUP-TRP/INV-DUP structure as one of the 12 most prevalent SV mutational signatures.6 The altered copy-number state generated by the formation of such structure may lead to tumor-level selection pressures from aberrant gene dosage as well as the activation of oncogenes or inactivation of tumor suppressor genes.34,35,36 Notably, both genomic disorders and cancer genome studies provided insights into the recombinant junctions and structural haplotype possibilities that may occur.
Whether found within the constitutional or cancer genome, the DUP-TRP/INV-DUP structure seems to be formed through genomic instability triggered by a given pair of inverted repeats.5,10 The instability was proposed to result from a fork collapse during replication repaired by break-induced replication (BIR).37 The initial recombination step uses non-allelic homology provided by intrachromosomal inverted repeat substrates. DNA replication continues in the reverse direction until a second fork collapse occurs. Repair of the original strand may be accomplished by non-homologous end joining (NHEJ) or microhomology-mediated BIR (MMBIR), which resolves the second break in mitotic cells.5,28,29,38
Until recently, genomic sequencing technology limitations within large segments with high nucleotide sequence similarity stymied our ability to identify BIR breakpoints. Furthermore, we were previously unable to investigate the genomic haplotype structure of the large-size (kb or Mb) segments involved in DUP-TRP/INV-DUP events in the context of a personal genome. Here, we sought to fully resolve those CGR structures and establish the variant haplotypes utilizing multimodal experimental genomic analyses and computational tools. We also define the molecular features of the inverted repeats that serve as substrates for recurrent pathogenic BIR at a specific Xq28 locus.
Results
Inverted LCR pairs generate recurring DUP-TRP/INV-DUP patterns
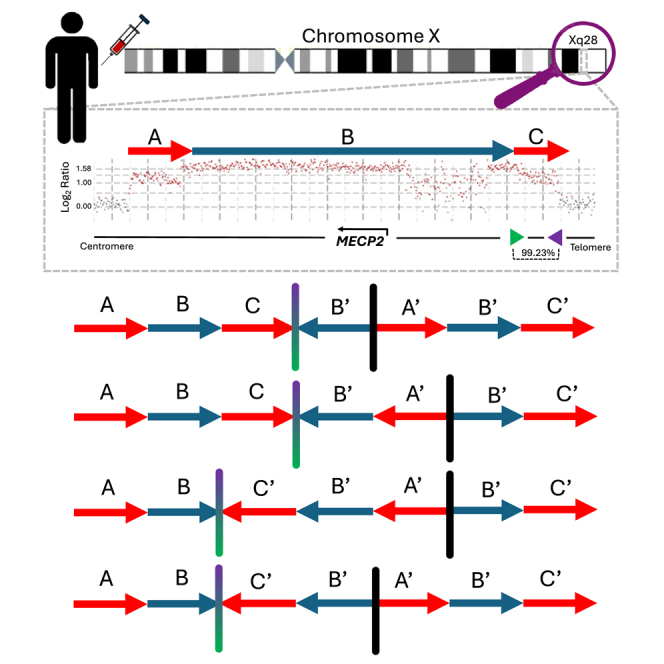
The present study includes 24 individuals who harbor a DUP-TRP/INV-DUP genomic structure as initially identified by high-resolution array comparative genomic hybridization (aCGH). Out of the 24 samples within this cohort, 23 are males with a duplication (n = 19) or triplication (n = 4) spanning the MECP2 gene causing MRXSL (Figure 1A; Table 1). There is one female subject harboring a large (approximately 7.3 Mb) DUP-TRP/INV-DUP at Xq21 without overlapping MECP2 (Figure 1B) who presented with developmental delay.
Figure 1.

Probands carrying DUP-TRP/INV-DUP genomic structure
(A) Genomic region spanning Xq28, including the MECP2 critical region, is shown with the location of selected genes and inverted repeats mediating DUP-TRP/INV-DUP formation in this cohort (43202a/43202b; 43221a/43221b [K1/K2, green and purple arrows]; 43231a/43231b [L1/L2]). The relative genomic locations of the duplication (red) and triplication (blue) are shown. Uncertainty as to the precise location of the start/end of either the duplication or triplication due to lack of probes on aCGH or low mapping quality in short-read GS within a given interval are depicted in light red and light blue, respectively.
(B) A single individual female (BAB12566) is shown with a DUP-TRP/INV-DUP within Xq21, along with the relative position of inverted LCR pairs (37696a/37696b). The naming scheme for 43221a/43221b (K1/K2) and 43231a/43231b (L1/L2) are derived from previous work detailing the DUP-TRP/INV-DUP structures at the MECP2 locus on the X chromosome.5,16,58 Genes included in this panel have an associated phenotype in OMIM.
Table 1.
Haplotype and breakpoint junction features among probands carrying DUP-TRP/INV-DUP
| Patient identifier | BH identifier | Haplotype structure | Jct1/Jct2 single molecule | MECP2 inverted (Y/N) | Junction 1 | Inverted repeat pair | Intra-/interchromosomal event | Junction 2 | Previous studies |
|---|---|---|---|---|---|---|---|---|---|
| BAB2727 | BH16106_1 | ND | ND | ND | PB (ChrX:153,613,143–153,615,342) | 43221a/43221b (K1/K2) | N/A | microhomology 2 bp + templated insertion | Carvalho et al.16 |
| BAB2769 | N/A | 3 | no | no | OGM, Sanger | 43202a/43202b | intrachromosomal | microhomology: 2 bp + deletion | Carvalho et al.5 |
| BAB2772 | N/A | 3 | yes | yes | OGM | 43221a/43221b (K1/K2) | intrachromosomal | Microhomology: 3 bp | Carvalho et al.5 |
| BAB2796 | BH16110_1 | 2 | yes | no | OGM | 43221a/43221b (K1/K2) | intrachromosomal | 2 bp insertion | Carvalho et al.5 |
| BAB2797 | N/A | ND | ND | ND | ND | 43221a/43221b (K1/K2) | intrachromosomal | 1 bp insertion | Carvalho et al.5 |
| BAB2801 | BH15649_1 | 4 | yes | yes | OGM | 43221a/43221b (K1/K2) | intrachromosomal | microhomology: 7 bp | Carvalho et al.5 |
| BAB2805 | N/A | ND | ND | yes | ND | 43221a/43221b (K1/K2) | intrachromosomal | blunt junction | Carvalho et al.5 |
| BAB3114 | BH14245_1 | 1 | no | yes | OGM, PB (ChrX:153,613,143–153,615,342) | 43221a/43221b (K1/K2) | intrachromosomal | microhomology: 2 bp | Carvalho et al.5 |
| BAB3147 | BH16111_1 | 6 | yes | no | OGM | 43221a/43221b (K1/K2) | intrachromosomal | microhomology: 2 bp | Carvalho et al.17 |
| BAB3216 | N/A | ND | ND | ND | ND | 43202a/43202b | intrachromosomal | microhomology: 4 bp + templated insertion | Carvalho et al.17 |
| BAB3255 | BH16108_1 | 1 or 3 | no | yes | OGM | 43221a/43221b (K1/K2) | intrachromosomal | microhomology: 2 bp | Carvalho et al.17 |
| BAB3274 | BH16112_1 | 1 or 3 | no | yes | OGM | 43221a/43221b (K1/K2) | intrachromosomal | microhomology: 3 bp + 9 bp + 5 bp deletions | Carvalho et al.17 |
| BAB12566 | BH13842_1 | 1 | no | N/A | OGM | 37696a/37696b | interchromosomal | microhomology: 2 bp | This study |
| BAB14392 | BH15645_1 | ND | ND | ND | ND | 43231a/43231b (L1/L2) | intrachromosomal | microhomology: 1 bp | This study |
| BAB14547 | BH15700_1 | 3 | yes | yes | OGM, PB (ChrX:153,613,143–153,615,342) | 43221a/43221b (K1/K2) | intrachromosomal | microhomology: 1 bp | This study |
| BAB14604 | BH15701_1 | 3 | yes | yes | OGM, PB (ChrX:153,613,143–153,618,666) | 43221a/43221b (K1/K2) | N/A | microhomology: 1 bp | This study |
| BAB14686 | BH15640_1 | 2 | yes | no | OGM | 43221a/43221b (K1/K2) | intrachromosomal | microhomology: 2 bp | This study |
| BAB15418 | BH16300_1 | 4 | yes | no | OGM | 43221a/43221b (K1/K2) | N/A | microhomology: 2 bp | This study |
| BAB15428 | BH16301_1 | 1 or 3 or 13 | no | yes | OGM | 43231a/43231b(L1/L2) | interchromosomal | AluY/AluSx1 at breakpoint | This study |
| BAB15702 | BH16609_1 | 6 | yes | no | OGM | 43221a/43221b (K1/K2) | intrachromosomal | microhomology: 9 bp | This study |
| BAB15705 | BH16611_1 | 2 | yes | no | OGM | 43221a/43221b (K1/K2) | intrachromosomal | microhomology: 6 bp (AluYa8/AluJo) | This study |
| BAB15740 | BH16610_1 | 2 | yes | no | OGM | 43221a/43221b (K1/K2) | intrachromosomal | microhomology: 2 bp | This study |
| BAB15789 | N/A | 2 | yes | no | OGM | 43221a/43221b (K1/K2) | N/A | microhomology: 2 bp | This study |
| BAB15420 | BH16299_2 | ND | ND | ND | ND | 43221a/43221b (K1/K2) | N/A | complexities | This study |
The majority of patients contain MECP2 in duplicated segment A/A′, except BAB3147, BAB15420 (truncated MECP2 – B/B′); BAB2801, BAB2805, BAB3114, BAB2797 (triplicated MECP2 – B/B′); BAB2769, BAB3216 – C/C′. N/A, not available; ND, not determined; OGM, optical genome mapping; PB, PacBio HiFi.
Based on customized aCGH data (hg19), the genomic rearrangements range in size from 417 kb to 7.4 Mb (from the beginning of the first duplication to the end of the second duplication) (Data S1; Table S1). The sizes of the initial duplication as well as triplication are variable, ranging from 18.5 kb (BAB2797) to 1.23 Mb (BAB15705) for the duplication region and 8.2 kb (BAB3216) to 7 Mb for the triplicated region (BAB12566). The size of the duplication and triplication events are dependent on the location of the second TS forming junction 2. The size of the second duplication is dependent on the distance between the two initiating LCRs within a given genomic loci. In contrast, the size of the second duplication tends to be constant for the same loci since that CNV is often mediated by inverted repeats. In this cohort, the second duplication varies from 16 to 575 kb, but 18 out of 24 CGRs show the same duplicated segment of 47 kb.
In this cohort, four different pairs of inverted repeats were identified to initiate the formation of the CGR event; three pairs are located at Xq28 (43202a/43202b; 43221a/43221b [K1/K2]; and 43231a/43231b [L1/L2]), whereas the fourth pair is located at Xq22.1 (37696a/37696b). The size of each inverted repeat pair included in this study as well as the distance between the pairs varied. The smallest pair (43202a/43202b) was 926 and 917 bp in length, with 98.12% similarity, separated by 317,810 bp. The next smallest (43221a/43221b [K1/K2]) was 11,455 and 11,446 bp in size, with 99.23% similarity, separated by 37,614 bp. The next largest pair (43231a/43231b [L1/L2]) had both repeats approximately 35,968 bp in size and 99.92% similarity, with 21,624 bp separating the two. The largest repeat pair identified in this cohort (37696a/37696b) was 140,562 bp and 140,621 bp in size, with a distance of 10,767 bp apart with 99.89% similarity (Table S2).
The breakpoint junction alignments for junction 2 in the structure for each sample were determined through either short-read genome sequencing (GS), long-read PacBio HiFi sequencing, traditional Sanger dideoxy sequencing, or a combination of methods. Out of a total of 24 samples, 14 samples showed a 1- to 9-bp microhomology at the breakpoint junction, one showing 7 bp of microhomeology, one with a blunt junction, two showed a 1- to 2-bp insertion, one sample displayed an Alu/Alu fusion at the breakpoint, and five displayed additional complexities such as templated insertions or microhomology-mediated deletions (Table 1). BAB15428 showed a chimeric fusion of AluY and AluSx1; these Alu repetitive elements were present in an inverted orientation on the reference genome and share 83% nucleotide sequence similarity. Although we have not obtained the breakpoint junction at the nucleotide level for this junction, we hypothesize that it is an Alu-Alu mediated event.39 Junction 2 in BAB15705 was found to be mediated by AluYa8 and AluJo, which were also in an inverted orientation and shared 36% sequence similarity when aligned to each other using the NCBI BLAST tool.5,16,17,40
Modeling SV haplotype conformers of mutational events
The previous identification of triplications being inverted and embedded within duplicated sequences provided evidence for two breakpoint junctions that occur in cis, forming the DUP-TRP/INV-DUP structure, with inverted LCRs acting as a recombinant substrate through BIR to generate this type of structure.5 The model for the formation of each haplotype conformer is predicated on which inverted LCR was used to generate the event and the distance and location that the template then switches back to the reference strand, continuing through replication and resolved by extended replication, repair by NHEJ, or the formation of half-crossover.6
We developed a prediction model based on an LCR pair within the MECP2 locus 43221a (K1) and 43221b (K2) (Data S2). This model is based on experimental interpretation and inferences from previous studies5 and expanded based on the results we obtained here using GS and optical genome mapping (OGM) approaches, which enable the incorporation of the diversity of observed haplotypes. It can be used to infer whether the TS occurred from the first LCR (K1) to the second LCR (K2) on the sister chromatid via homologous recombination and re-initiation of the replication fork to resume replication in the opposite orientation (haplotype conformer 1 and 2) or a TS from the second LCR (K2) to the first LCR (K1) on the opposite sister chromatid (haplotype conformers 3 and 4) (Figure 2). Both form a chimeric LCR (i.e., recombinant representing junction 1) and a recurrent duplication (DUP2) spanning the genomic segment in between the inverted repeats. Junction 2 results from a second TS triggered by double-stranded break or replication fork stalling/collapse, which will produce the inverted triplication segment and DUP1. The size of the inverted triplication and DUP1 depends on the location of the second TS. The linearized final structure thus allows inferences as to the temporal replication fork jumps (i.e., iterative TS of the progressing replication fork) forming junctions 1 and 2 with the formation of a chimeric LCR. The same prediction model can be inferred for all inverted repeats detailed in this study (43202a, 43202b; 43221a/43221b [K1/K2]; and 43231a/43231b [L1/L2], 37696a, and 37696b), as well as other pairs that generate additional DUP-TRP/INV-DUP events at other positions (Figure S1).
Figure 2.

Predictive model for DUP-TRP/INV-DUP formation
At least 4 haplotype sub-structures can be derived from rearrangement involving a pair of inverted LCRs. This figure depicts the LCRs K1 and K2 (green and purple arrowheads) within the MECP2 locus used as substrates during an intrachromosomal event. The same model can be applied to other DUP-TRP/INV-DUPs formed through inverted LCRs pairs nearby dosage-sensitive genes.59 The formation of the DUP-TRP/INV-DUP event may start due to a replication fork stall and collapse at or nearby the LCR (K1), denoted as a green arrowhead. Homology drives strand invasion at the inverted LCR (K2′) on the opposite strand (denoted in purple), producing junction 1. DNA replication continues in the opposite direction until a second replication fork collapse and repair on the original strand through either MMBIR or NHEJ resolves the second junction. The 4 conformer possibilities shown here are determined by the replication fork collapsing and jumping (TS denoted by dashed black arrows) from either K1 to K2′ or K2 to K1′.
Structural variant haplotype conformers within DUP-TRP/INV-DUP events
In samples for which cell lines or whole blood was previously frozen and available, we utilized ultra-high-molecular-weight DNA and OGM to phase genomic fragments in the context of the larger structure and the diploid genome through the visualization of single DNA molecules containing genomic segments in the structure (Figure 3). Out of 19 samples on which OGM was performed, we could phase the DUP-TRP-DUP CGR into four distinct and predicted substructures that are possible through two TSs (Figure 2). The four identified conformers are (1) the initial duplication, triplication, and final duplication, all in an inverted orientation (haplotype structure 1) (BAB3114 and BAB12566); (2) the triplication and final duplication in an inverted orientation (haplotype structure 2) (BAB2796, BAB14686, BAB15705, BAB15740, and BAB15789); (3) the triplication and initial duplication in an inverted orientation (haplotype structure 3) (BAB2772, BAB14547, BAB14604, and BAB2769) (Figure 3); and finally, (4) just the triplication in an inverted orientation (haplotype structure 4) (BAB2801 and BAB15418) (Figure 3; Table 1). Two samples (BAB3147 and BAB15702) were found to harbor an additional structure (haplotype structure 6), which is formed through the same mechanism as haplotype structure 2 but leads to an appearance of an inversion of only the triplication due to a potential ancestral inversion of the segment C relative to reference (Data S1 and S2). For the LCR K1 and K2, a polymorphic inversion is known to be present in approximately 18% of the population of European descent.41
Figure 3.

Haplotype resolution using OGM
Structural haplotype determination and conformer configuration was established based upon single-molecule support through junction 1 and junction 2 in cis. Two samples in the cohort are highlighted, BAB14604 (left) and BAB15418 (right).
(A) ArrayCGH plots for each sample show a similarly sized DUP-TRP-DUP event both mediated by inverted LCRs (shown as green and purple arrows) downstream of MECP2 (black rectangle).
(B) OGM reference (green rectangle) shows in silico motifs throughout the MECP2 locus. The red arrows correspond to the duplicated segments, whereas the blue arrow corresponds to the triplicated segments. The length of the CNVs is proportional to the aCGH CNV.
(C) OGM de novo assembly from proband samples are shown in blue rectangles. Sequence motifs aligned to the reference shown as connecting gray lines enable restriction fragment genome mapping and pattern recognition. Red and blue arrows are overlayed to represent the position and orientation of each amplified genomic fragment within the DUP-TRP-DUP structure. The connection points forming junctions 1 and 2 are shown as black vertical dashed lines/bars.
(D) Single DNA molecules that span both junctions 1 and 2 are highlighted in blue, confirming that both junctions are present in cis.
(E) Hypothesized resolved haplotypes based on CNV and in cis junction analysis. Although both samples show nearly identical aCGH patterns, BAB14604 has conformer haplotype 3 and BAB15418 shows conformer haplotype 4.
The two breakpoint junctions (Jct1 and Jct2) have the same nucleotide sequencing at the connection point in all sub-haplotype structures for the same individual; however, the orientations of genomic fragments in the structure differ between distinct haplotype conformers when the structure is visualized in a linear fashion (Figure 2). Single DNA molecule resolution in cis through both breakpoint junctions 1 and 2 (Figure 3D) enables an interpretation of each individual haplotype structure, given the rearrangements occur in a male on the X chromosome and not on an autosome. For samples for which we did not have a single DNA molecule that spans both junctions 1 and 2 (BAB3255, BAB3274, and BAB15428), we could not definitively refine the structure to a single haplotype, but we could refine it to either haplotype 1 or 3 or 13 (Data S1 and S2).
Within DUP-TRP/INV-DUP, all samples with triplications encompassing the entire MECP2 gene have MECP2, an inverted orientation (BAB3114, BAB2805, and BAB2801). Additionally, haplotype structures 1 and 3 have the initial duplication (including the MECP2 gene) in an inverted orientation on the amplified genomic fragment (BAB15428, BAB14604, BAB14547, BAB3274, BAB3255, and BAB2772) (Table 1). The remainder of the samples with an identified haplotype structure include the amplified copy of MECP2 that appears to be present in the structure in a proposed haploid human genome reference orientation.
Long-read sequencing facilitated breakpoint mapping within inverted repeats
PacBio HiFi facilitated the ability to generate highly accurate reads though repetitive sequences that were not possible using previous short-read technologies due to low mapping quality within a given region. For the MECP2 region, LCRs K1 and K2 are approximately 11 kb in length and 99.23% similar (Hg19) (Table S2). The CGR generates a hybrid K1/K2 resulting from copy-number event in addition to extra copies of either K1 or K2. All the long-reads that span the LCRs are mapped to the reference, but we can refine the exact reads that map to the K1/K2 hybrid as they will present soft-clipping junctions flanking the genomic border of the LCRs (Figure 4). This approach enabled the re-mapping of the hybrid reads, which revealed the recombinant breakpoint junctions within the LCRs in four samples (BAB2727, BAB3114, BAB14547, and BAB14604). These reads indicate the connection of LCR K1 and K2 forming Jct1 within the DUP-TRP/INV-DUP structure that as a result form a recombinant or chimeric LCR.
Figure 4.

Refined position of TS within LCR K1/K2
Identification of PSVs through inverted LCRs allows for a determination of the relative position of the breakpoint junction within the inverted LCRs for BAB2727.
(A) aCGH showing a DUP-TRP-DUP structure, with MECP2 locus highlighted and magnified. The inverted LCRs K1 and K2 (shown as green and purple arrows) are located flanking the terminal/3′ end duplication in the structure.
(B) Positions of K1 and K2 are shown with representative HiFi data below, highlighting sequence reads that span the region. Ancestral reads denote HiFi reads are uniquely aligned with LCR (e.g., reads 2, 4, and 5). Breakpoint reads denote HiFi reads that begin in unique sequence and show soft clipping as they exit the LCR (e.g., reads 1 and 3). PSVs are visualized in LCR K2 with the green (A nucleotide) and red (T nucleotide) positions (ChrX:153,615,342 and ChrX:153,615,645, respectively) that are found breakpoint reads in K2 and are present within points of homology in K1.
(C) Linearized structure showing the reads found within each position. The chimeric K1/K2 shows the positioning of PSVs used to refine the position of Jct1.
(D) Percentage of uniquely aligned base in slide window of 20 bp (i.e., sequence similarities were shown as a heatmap). A “hot” color, orange, denotes a 100% match, while a “cold” color, purple, denotes reduced similarity. The position of the PSV can be used to estimate the distance the replication fork proceeds before the TS to K2 occurred. Samples in this cohort could be narrowed to a 2.2- or 5.5-kb region.
Moreover, the high accuracy rate of PacBio HiFi sequencing allows the identification of single nucleotide changes even within highly similar sequences (i.e., paralogous sequence variants [PSVs]).42,43 Single nucleotide variation between the LCRs enables one to refine the point from where BIR uses homology to switch to its LCR pair (i.e., the recombinant join point) (Figure 4). We could further refine the “crossover uncertainty” to approximately 5.5 kb in one sample (BAB14604) and 2.2 kb in three samples (BAB14547, BAB2727, and BAB3114) (Figure 4D). The uncertainty range is based on the presence of informative PSVs (i.e., SNPs that are present in K2 but map to the same reference location in K1 or vice versa). If no informative SNPs were present within the breakpoint spanning read, the uncertainty range as to where the homology-driven TS occurred cannot be determined within a given individual sequencing read.
CRISPR-Cas9 enrichment (ONT) and strand-seq orthogonally validate fusion junction formation and haplotype structure
CRISPR-Cas9 enrichment and subsequent Oxford Nanopore Technologies (ONT) sequencing for the MECP2 critical region was performed on three individuals in family BH14245, including BAB3114 (proband), BAB3115 (carrier mother), and BAB3121 (maternal grandfather). Targeted nanopore sequencing and Cas9-guided adapter ligation on ultra-high-molecular-weight extracted DNA allowed for sequencing within the MECP2 region through the LCRs K1 and K2 as well as through duplication-triplication-duplication event with single DNA molecule resolution of the structure. ONT long-read sequencing through both K1 and K2 LCRs orthogonally validated the informative PSVs that were detected within K2 at position ChrX:153,615,342 and ChrX:153,615,645 on reads that span the chimeric LCR and that are present within the same position on K1 as independently visualized though HiFi sequencing data (Data S3).
Additionally, the presence of a large single 530-kb read that spanned (in a single molecule) the duplication and triplication regions enabled refinement of the haplotype structure in the context of the larger CGR (Data S3). This method allowed for an additional orthogonal confirmation of the haplotype structure that was observed in the OGM analysis of the same sample (BAB3114) (haplotype conformer 1).
The implementation of single-cell DNA template strand sequencing (strand-seq) for samples BAB3114 and BAB14547 provided an orthogonal confirmation of the haplotype structures 1 and 3, respectively. Strand-seq was particularly important to validate the haplotype of BAB3114 because there were no molecules spanning both junctions 1 and 2 in the optical mapping data for BAB3114 (Data S4).
Discussion
We studied 24 individuals harboring a DUP-TRP/INV-DUP structure mediated by inverted repeats, including three sets that reside at the MECP2 critical region at Xq28 and an additional pair at Xq21. The size of the genomic fragments as well as the fact that breakpoint junctions may occur within repetitive regions of the genome previously obfuscated resolution of the SV haplotypes. Utilizing data from high-resolution aCGH as well as short- and long-read GS (ONT and PacBio HiFi), OGM and strand-seq enabled elucidation of the recombinant events within each SV haplotype and visualization of the individual conformers (Tables 1 and S3).
The formation of the DUP-TRP/INV-DUP structure was hypothesized to occur by a combination of BIR and MMBIR or NHEJ using inverted repeats as the recombinant substrate during the process of generating breakpoint junctions 1 and 2.5 A combination of Southern blot and Sanger dideoxy sequencing of these breakpoint junctions revealed the inverted orientation of the triplication and its connections to flanking duplications.5 Partly due to technical limitations, previously applied methodologies did not identify SV haplotype differences specifically concerning the order and position of which the copy-number segments are assembled in the derivative genome of individuals harboring DUP-TRP/INV-DUP.5,21,28,29 Therefore, although the haplotype diversity was recently predicted,6 the haplotype conformers cannot be distinguished by nucleotide breakpoint junction analysis alone, requiring ultra-long molecule methodologies such as OGM or CRISPR-Cas9-targeted ONT.
Within this study, all four hypothesized SV haplotypes were detected (Figure 2) in unrelated families with MRXSL in addition to new ones (haplotype 6) (Table 1). The utilization of OGM together with long-read ONT and HiFi sequencing data allowed for (1) identification of the relative orientation and order of each genomic fragment and (2) breakpoint junction sequence to the bp level resolution, including within large inverted repeats. Specifically, the initial LCR used to mediate the rearrangement is now defined, and the recombinant region within that LCR is delineated. The formation of this genomic aberration may lead to pathogenic alleles due to increased gene expression of dosage-sensitive genes, gene interruption, or gene fusion44 (Figure S2). Identification of these SV haplotype structures presents a previously unknown level of complexity to SV mutagenesis.
Based on the experimental data provided by OGM, long-read GS, and CRISPR-Cas9-targeted sequencing for this cohort, we developed a predictive model that can be applied to susceptible loci in the genome.10 The implementation of Strand-Seq allowed for the refinement of haplotype structure 1 in BAB3114. In this case, the molecule size limitations in OGM data restricted our ability to resolve the haplotype from one data source alone. Additional derivations of these four observed haplotypes can occur due to the presence of inversion alleles in the ancestral X chromosome, as observed in BAB3147 and BAB15702 (Data S2). Moreover, haplotype diversity can also occur due to interchromosomal events (as opposed to intrachromosomal events) such as those in BAB12566 and BAB15428 (Table 1; Figure S3). Of note, BAB15428 is the first individual reported to carry an interchromosomal DUP-TRP/INV-DUP structure in a male MRXSL cohort, suggesting a contribution from two X chromosomes (Figure S3). Interestingly, the inverted repeats that mediate this event (43231a/43231b [L1/L2]) differ from the majority of DUP-TRP/INV-DUP in the MRXSL cohort, which often involve LCRs K1 and K2. This result possibly indicates a gender preference for certain inverted repeats in BIR.
We elucidated four pairs of inverted repeats that act as recombinant substrates for the formation of this genomic event. Shared nucleotide similarity ranges from 98.12% to 99.92%, whereas there are significant differences in size and distance separating them (Table S1). Most of the samples within this cohort (19/24) have CGRs mapping to the LCR pairs 43221a/43221b (K1/K2) downstream of the MECP2 locus. The K1/K2 pair has the third largest separation distance (37,614 bp) and is the second-smallest sized LCR pair (∼11.5 kb). The average size of the duplication 1 was 380,900 bp, with a median size of 320,848 bp. The distance from MECP2 for the first LCR pair in 43221a/43221b (K1/K2) is 201,074 bp, while the distance from MECP2 to the LCR pair 43231a/43231b (L1/L2) is 420,500 bp, which is 99,652 bp larger than the median size of the initial duplication. It is possible that an unstable replication fork is generated as a result of BIR within an inverted LCR moving in a reverse direction (generating the initial duplication).45 The preference for 43221a/43221b (K1/K2) may represent an ascertainment bias within our cohort due to the distance the pairs sit from a dosage-sensitive gene, in this case MECP2. Other inverted pairs on the X chromosome are known to mediate the same type of CGRs. For instance, the inverted LCR pairs A1a/A1b (38209a/38209b), downstream of the gene PLP1 at Xq22, form DUP-TRP/INV-DUP structures in PMD. A1a/A1b are 20,349 and 20,353 bp in size and share 99.27% sequence similarity, with a distance of 60,043-bp apart. Moreover, inverted repetitive elements such as Alu with shared nucleotide similarity as low as 85% (e.g., AluSg/AluSg, AluSx3/AluSz) have also been identified as mediators of the CGR event as seen in 17p13.3.33 However, contrary to K1/K2, L1/L2 at Xq28, or A1a/A1b at Xq22 responsible for multiple independent events involving those loci, 17p13.3 Alus have not been reported in more than a single DUP-TRP/INV-DUP event. In aggregate, these data support a combined role of inverted repeat size (>10 kb), shared nucleotide similarity (>98%), and proximity (<100 kb) in the recurrent formation of this type of CGR.
The region of uncertainty in which the recombination crossover takes place within the first or second LCR in 43221a/43221b (K1/K2) could be narrowed to 2.2–5.5 kb thanks to the use of long-read sequencing. The segment defined in four crossovers shares 100% sequence similarity between the LCRs K1 and K2, supporting the hypothesis that large stretches of homology are optimum substrates for non-allelic recombination. While this length and sequence similarity is comparable to those that generate rearrangements through non-allelic homologous recombination,46,47 the recombination formed within the LCR generate inversions accompanied by CNVs that are further resolved by a second mechanism (MMBIR or NHEJ), which are consistent with an unstable BIR event.48
Recently, new studies have proposed an alternative model for the origin of DUP-TRP/INV-DUP events. Martin et al. postulate a process called origin-dependent inverted repeat amplification (ODIRA) that involves template switching between leading and lagging strands, where the leading strand at a replication fork switches to the lagging strand template at short, interrupted inverted repeats.49,50 This event then forms an unstable full triplication with an extrachromosomal intermediate step, which is further processed to the formation of a DUP-TRP/INV-DUP structure. While we have investigated numerous individuals carrying DUP-TRP/INV-DUP affecting diverse genomic loci, including de novo structures, we have not observed full triplications with similar features proposed as part of the initial step in the ODIRA process. Additionally, the inverted repeats we detail in human genomic disorders (Table S2) are too far apart to mediate TSs within the same replication fork as proposed in the ODIRA model. While our data are not definitively conclusive, they do provide experimental evidence through phasing of SNVs directly within LCR regions to support that BIR is a driving mechanism forming DUP-TRP/INV-DUP events.
In summary, OGM and long-read GS approaches facilitated phasing and assembly of a clinically relevant recurrent SV structure—the DUP-TRP/INV-DUP CGR at Xq28 (Figure 5). Furthermore, this work provides insights into BIR, a molecular mechanism contributing to the formation of inversion alleles and CGRs in genomic disorders. The ability to resolve SV haplotypes and to determine gene structure perturbations will guide our understanding of neomorphic alleles and gene fusion formation. Complex structural variation may have pathogenic consequences, as well as beneficial clinical ramifications51 and the potential of driving genome evolution.52,53
Figure 5.

Multipronged approach resolving DUP-TRP/INV-DUP events
Sample BAB3114, including the methodology used to fully resolve the SV haplotype and breakpoint junctions 1 and 2.
(A) aCGH showing a DUP-TRP-DUP structure at Xq28, including the MECP2 gene and LCRs K1 (green arrow) and K2 (purple arrow).
(B) Illumina short-read GS showing the read depth for the region as visualized in the VizCNV plotting program.60
(C) Red arrows denote the regions of copy-number change as seen in the short-read sequencing in the Integrative Genomics Viewer. Of note, soft clipping can be seen in the regions of unique sequence (left) versus the unmapped reads at the region with K1 and K2 due to sequence similarity of the region.
(D) PacBio HiFi data show the reads that include the breakpoint region (shown as soft clipping) within both junction 1 and junction 2.
(E) CRISPR-Cas9-targeted ONT facilitated ultra-long molecule (>500 kb) sequencing to capture the haplotype structure within a single DNA molecule.
(F) Bionano OGM shows orientation and connection points of amplified genomic fragments forming junctions 1 and 2 within the structure.
(G) Strand-seq data showing the points of breakpoint (purple peaks) with the inverted genomic sequence between.
(H) Resolved haplotype structure 1 for BAB3114 shows the triplication and initial duplication in an inverted orientation.
(I) Junction 1 shows a heatmap of K1/K2 similarity. The point of fork stall/collapse and strand invasion to the inverted LCR occurs within a 2.2-kb stretch of the LCR K1/K2 (as shown with the red arrow). Junction 2 can be determined to nucleotide-level resolution and shows a 2-bp microhomology.
Untangling complex genomic events including the DUP-TRP/INV-DUP enables better comprehension of the underlying molecular basis of Mendelian disease, but it also enlightens the mechanisms leading to genomic instability and provides insights into cancer mutagenesis and the evolution of genes and genomes. Drastic and rapid changes to the genome caused by complex structural variation such as that observed herein have the effect of generating changes beyond simple Watson-Crick single base pair “editing.”54 Through the generation of CGRs, large portions of the genome are moved, reordered, inverted, and connected in ways not previously seen, driving new and unknown possible outcomes and involving previously cryptic genomic complexities.55,56,57 The subsequent gene expression and clinical effect(s) of such genomic perturbations must be further investigated.
Limitations of the study
This work has been performed on a disease-specific cohort carrying ultra-rare pathogenic SVs and was not extrapolated to other human populations. The relatively small sample size and inclusion of individuals with aberrations only occurring on the X chromosome limit our ability to expand our conclusion to similar events involving autosomes, which requires additional studies. Lastly, the technological approaches applied here may have limited application to investigate nucleotide-level resolution of breakpoints within inverted pairs of LCRs smaller than ∼15 kb.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Genomic DNA Extracted from BAB2727 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB2769 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB2772 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB2796 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB2797 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB2801 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB2805 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB3114 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB3147 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB3216 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB3255 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB3274 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB12566 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB14392 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB14547 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB14604 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB14686 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB15418 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB15428 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB15702 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB15705 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB15740 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB15789 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Genomic DNA Extracted from BAB15420 | Baylor College of Medicine/Pacific Northwest Research Institute Lupski Lab/Carvalho Lab |
N/A |
| Deposited Data | ||
| Microarray Data | This Paper, Carvalho et al.5,16 | GEO: GSE49440, GSE49446, GSE250451 |
| Oxford Nanopore Datasets | This Paper, Smolka and Paulin et al.67 | SRA: PRJNA953021 |
| Short-Read GS | This Paper | dbGAP: Phs002999.v2.p1 |
| Software and Algorithms | ||
| Megalodon | N/A | https://github.com/nanoporetech/megalodon |
| PRINCESS | Mahmoud et al.64 | https://github.com/MeHelmy/princess |
| Minimap2 | Li et al.65 | https://github.com/lh3/minimap2 |
| Sniffles | Sedlazeck et al.66 | https://github.com/fritzsedlazeck/Sniffles |
| Clair3 | Luo et al.68 | https://github.com/HKU-BAL/Clair3 |
| VizCNV | Du et al.60 | https://github.com/BCM-Lupskilab/VizCNV |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Claudia M. B. Carvalho, PhD. Email: ccarvalho@pnri.org.
Materials availability
This study did not generate new unique reagents.
Data and code availability
Microarray data generated in previous studies17 are available through the gene expression omnibus (GEO) under accessions GEO: GSE49440, GSE49446; new microarray data within this study is available under accession GEO: GSE250451. Oxford nanopore datasets are available within SRA BioProject ID SRA: PRJNA953021. Samples that had GS and consented for broad data sharing are available under dbGAP: phs002999.v2.p1. Optical genome mapping data is available upon request to the authors. All original code is available in this paper’s key resource table.
Experimental model and study participants
Study participants (N = 24) (Table 1) included 23 males and 1 female were consented according to the Institutional Review Board for Human Subject Research at Baylor College of Medicine approved protocols: H-29697, H-20268, and H-47127/Pacific Northwest Research Institute WIRB #20202158. Whole blood samples (3-10mL) were collected via peripheral venous blood draw in Ethylenediaminetetraacetic acid (EDTA) and Acid Citrate Dextrose (ACD) vacutainer tubes from patients diagnosed with MECP2 Duplication Syndrome. Ancestry and race demographics were not captured as part of the consenting process. All study participants have consented for publication.
Method details
Array comparative genomic hybridization
To evaluate copy-number changes in chromosomes X and Y, we designed a custom 4 × 180K tiling-path oligonucleotide microarray spanning the entirety of X and Y, including the MECP2 region on Xq28 (Hg19). The custom 4 × 180K Agilent Technologies microarray (AMADID #086099) was designed using the Agilent Sure Design Website version 6.9.1.1 (https://earray.chem.agilent.com/suredesign/) on NCBI Build 37. We selected 143,860 probes interrogating chrX: 1–155,270,560 for a median probe spacing of 797 bp and 23,912 probes covering chrY: 1–59,373,566 for a median probe spacing of 425 bp. Arrays were run according to the manufacturer’s protocol (Agilent Oligonucleotide Array-Based CGH for Genomic DNA Analysis, version 7.2, Agilent Technologies) with modifications5 on probands, mothers, and in select cases, fathers and maternal grandparents (if available) to determine inherited vs. de novo rearrangements. Arrays were performed on DNA of sex-matched controls from Coriell, NA1550 and NA10851. 1.2 μg of DNA was digested with the restriction enzymes AluI and RsaI at 37°C for 2 h. Digested DNA was labeled with Cy5 and Cy3 using the BioPrime Array CGH Genomic Labeling kit at 37°C for 2 h with the labeling efficiency determined afterward using a nanodrop. Fluorescently labeled sample and sex-matched control DNA were combined with 5 μg of human Cot-1 DNA. The mixture was placed at 95°C for 5 min with the Agilent 10X blocking agent and 2X Agilent hybridization buffer then incubated at 37°C for 30 min. The mixture was placed on the appropriate array and placed in a revolving hybridization chamber at 65°C for 40 h. After hybridization the arrays were washed with Agilent OligoCGH Wash buffers 1 and 2. Slides were scanned using the Agilent SureScan Microarray Scanner and resulting image processed using the Agilent feature extraction software.
Coordinates for each CNV observed along those chromosomes were annotated using the Agilent Genomic Workbench software. The genomic context where breakpoint junctions occur were investigated using UCSC Genome Browser GRCh37/hg19 Assembly (http://genome.ucsc.edu)61 for information about the presence of repeats, low-copy repeats, and genes or pseudogenes. To identity inverted and direct repeats that may mediate DUP-TRP/INV-DUP events, we mapped the breakpoints to genome-wide maps of highly-similar intrachromosomal repeats.59,62
Short-read genome sequencing
Whole genome sequencing was performed at the Human Genome sequencing center (HGSC) at Baylor College of Medicine. Following sample QC, libraries were prepared with KAPA Hyper reagents and sequenced using the Illumina Novaseq 6000 to generate 150 bp paired-end sequence reads for all samples in a format of multiplexed pools to generate an average of 30X coverage. Post-sequencing data analysis was performed using the HGSC HgV analysis pipeline, which executed base calling, mapping (BWA-mem) to the reference genome (Hg19), merging, variant calling (xAtlas), post-processing, annotation and QC metric collection for all sequencing events. To ensure sample identify and integrity the Fluidigm SNPtrace method for rapidly genotyping 96 SNP sites was employed to verify gender prior to sequencing and to detect contamination. Using this assay sample identity was verified using the Error Rate In Sequencing (ERIS) software developed at the HGSC. A subset of samples (N = 2) had sequencing performed at the National Genomics Infrastructure (NGI), in Stockholm, Sweden using an Illumina 30X PCR-free paired-end (PE) approach.63
Optical genome mapping
Ultra-high molecular weight (UHMW) DNA was isolated from frozen EDTA blood or cryopreserved cells following manufacturer instructions (documents 30246 rev F, 30268 rev D). In short, the frozen samples were first thawed in a 37°C water bath. Then blood and cell samples were counted using a either a HemoCue WBC System (HemoCue AB) or hemocytometer, respectively. A volume containing 1.5 million cells was pelleted via centrifugation at 2,200x g for 5 min. The pellets were resuspended in a DNA stabilization buffer and treated with proteinase K in lysis and binding buffer. Cryopreserved cell samples were also treated with RNAse A at this step. After proteinase K digestion, samples were treated with PMSF, bound to a nanobind disk, washed, and eluted. DNA extracts were homogenized via end-over-end rotation and incubated at room temperature overnight before fluorescent labeling.
For each sample, 750 ng of DNA was labeled at the recognition site CTTAAG using Direct Labeling Enzyme 1 (DLE-1) and counter-stained following manufacturer instructions (document 30206 Rev F). Labeled DNA was imaged on a Saphyr Gen2 platform, collecting 400X-1500X effective coverage for each dataset. De novo assembly and structural variant calling was performed using Solve version 3.7 as described by the Bionano Solve Theory of Operation: Structural Variant Calling (Document 30110 Rev J). To reduce the computation time for de novo assemblies, each dataset was down-sampled to 250X effective coverage by filtering for the longest molecules of each dataset.
Structural variants (SV) were called against the human reference genome Hg19. Using the Variant Annotation Pipeline, SV calls were annotated and compared to the Bionano control sample database, which contains >600,000 SV calls from >150 phenotypically normal individuals from >26 populations. See the Bionano Solve Theory of Operation: Variant Annotation Pipeline (document 30190 revision H) for more details. The raw molecules contained in each contig present in the OGM data were interrogated to identify molecules that contained both breakpoint junctions in cis. The pattern of sequence motifs for the region allowed for interpretation of each genomic fragment in context of the larger structure to identify the haplotype differences for each individual.
Pacific Biosciences (PacBio HiFi)
Whole genome sequencing was performed at the HGSC at Baylor College of Medicine using long reads from the Pacific Biosciences sequencing platform. After DNA quality was assessed using Qubit and pulsed-field gel electrophoresis (PFGE), 15ug genomic DNA was used to construct a library using the SMRTbell Express Template Preparation Kit 2.0 with an average fragment length of 15 kb. Using the PacBio Sequel II instrument, two SMRTcells were sequenced per library for an average of 43Gb of HiFi reads per sample with an average coverage of 15-20x.
Oxford Nanopore (Promethion)
Long read whole genome sequencing data was also generated using the Oxford Nanopore Technologies sequencing platform. After DNA quality was assessed using Qubit and pulsed-field gel electrophoresis (PFGE), A library was constructed with 15ug input genomic DNA using the SQK-LSK110 ligation sequencing kit with an average fragment length of 15Kb. Using the Oxford Nanopore Technologies Promethion instrument, one flowcell was sequenced per library with an average yield of 90Gb per sample. Basecalling was performed using Guppy version 4.3.4+ecb2805 and methylation analysis with Megalodon version 2.3.1 [https://github.com/nanoporetech/megalodon] using the default parameters of the program.
Additional data processing and analysis of long read sequencing data
Using PRINCESS version 2.0 64, a workflow for long read sequence analysis, reads were aligned to GRCh37 and phased variant calls were generated for SVs and SNVs. Briefly, PRINCESS will start by aligning reads using the appropriate parameters based on the type of sequencing technology using Minimap2 version 2.2465 followed by calling SVs using Sniffles version 2.0.566,67 and will identify SNVs and indels using Clair3 version 0.1.11.68 Finally, PacBio HiFi data was processed using the same methods using PRINCESS with the read-option set to CCS (--ReadType ccs). For SVs from both sequencing platforms, variants were filtered based on read support to require a maximum ∼25k SVs per sample.
Additionally, to determine reads that contained a PSV within either LCRs K1 and K2, reads were manually inspected within IGV to determine reads that were “ancestral” that is reads that started within unique sequence, spanned the LCR and ended within unique sequence within the LCR and were not part of the chimeric K1/K2 junction. These reads were then extracted and aligned in the Geneious software suite to the corresponding reference position in Hg19. We then determined reads that were part of the chimeric junction my visualizing reads that started within unique sequence outside of the LCR (and were thus “anchored”) in unique sequence and showed soft-clipping as they exited each LCR indicating that read spanned the breakpoint junction sequence. That read was then extracted and aligned to the “ancestorial read” in Geneious to determine PSVs that were present in either K1 or K2 to determine the relative point of template switch forming junction 1.
Nanopore Cas9 enrichment and sequencing for BH14245 family
Patient derived immortalized lymphoblastoid cell lines were cultured in RPMI-1640 media (ATCC) supplemented with 10% FBS (ATCC) and 1% penicillin/streptomycin/amphotericin B (Thermo Fisher). Cells were maintained at 37°C in 5% CO2. Genomic DNA Extraction and Purification: Genomic DNA was extracted from 5 M cells using the Gentra Puregene Cell Kit (Qiagen) following the manufacturer’s instructions. Extracted DNA was further purified by isopropanol precipitation. For Adaptive Sampling experiments, DNA was sheared to approximately 20 kb using g-tube (Covaris). Ultra-high molecular weight (UHMW) DNA was purified using the Nanobind CBB Big DNA Kit (Circulomics) following the manufacturer’s instructions, eluted into 150 μL Circulomics EB containing 0.02% Triton X-100, and equilibrated overnight at room temperature. DNA was quantified using the Qubit fluorometer (Thermo Fisher). Adaptive Sampling: DNA was prepared for sequencing using the Ligation Sequencing Kit (Oxford Nanopore Technologies, catalog no. SQK-LSK109) and sequenced using the GridION sequencer (ONT) with readfish1 integration (minKNOW 20.10.6) or using MinKNOW Adaptive Sampling2 (minKNOW 19.16.6, guppy 3.4.5) with a target region of interest defined as chrX:141,000,000-156,000,000 in the GRCh38 reference. Cas9 Sequencing: Guide RNAs were designed and ordered using the Custom Alt-R CRISPR-Cas9 guide RNA design tool (Integrated DNA Technologies) with a 1 kb reference input fasta target region from the human GRCh38 genome. Cas9 sequencing libraries were prepared using the Cas9 Sequencing Kit (Oxford Nanopore Technologies, catalog no. SQK-CS9109) with modifications as described.3 Ultra-high molecular weight (UHMW) Cas9 sequencing libraries were prepared with the following additional modifications: adapter-ligated libraries were purified via Nanobind disk (Circulomics) with precipitation in NAF10 buffer (Circulomics); Nanobind disks were washed three times with magnetic separation in Long Fragment Buffer (Oxford Nanopore Technologies, catalog no. LFB); final elutions were carried out at room temperature overnight with 60 μL or 120 μL Elution Buffer for MinION or PromethION libraries, respectively. Samples were sequenced using the MinION, GridION, or PromethION sequencer (Oxford Nanopore Technologies) using R9.4.1 flow cells. Final libraries were combined with 30 μL or 120 μL of Sequencing Buffer for MinION or PromethION flow cells, respectively, and equilibrated for 30 min at room temperature prior to loading with a wide-bore pipette tip. Flow cells were flushed and reloaded as needed using the Flow Cell Wash Kit (Oxford Nanopore Technologies, catalog no. EXP-WSH004). Analysis: Due to each genome in this family containing a different mecp2 locus with different expected ploidy, each genome was examined using a different method. Adaptive Sampling reads for BAB3121 (flowcell: FAO74863) were aligned to GRCh38 with minimap2 (version 2.17), and SNVs were detected using the medaka_variant wrapper (version 1.0.3). Cas9 targeted reads for BAB3121 (flow cell: FAN49258) were analyzed using an identical workflow. BAB3114 was sequenced using either a pair of Cas9 targets that flank the region of interest (flowcell: PAG08429) or a single Cas9 target next to the region of interest (flowcell: FAO31820). Flanking target Cas9 reads were aligned with minimap2, and SNVs were detected with medaka using medaka’s default diploid method. Single-target Cas9 reads (flowcell: FAO31820) were aligned with minimap2 and then inspected manually for ultra-long reads.
BAB3115 was sequenced using two separate single-target Cas9 guide RNAs (flowcells: FAN40573 and PAG08038) in order to preferentially enrich for either the two original copies of FLNA, or the additional copy created in the rearrangement, which we call FLNA’. Reads for the two original FLNA copies were enriched by targeting five Cas9 guide RNAs to the RPL10 gene, which is only found adjacent to these two copies of FLNA. Reads were aligned with minimap2, and SNVs were called using medaka_variant. To enrich for FLNA’ reads, three Cas9 guide RNAs targeting TKTL-1 were used, which flanks FLNA’ on both sides but is only on one side of the original FLNA gene copies. Reads were filtered to include reads producing either primary alignments or any supplementary alignments ending within 50 bp of the telomeric end of the FLNA flanking repeat (chrX:154,384,868-154,396,222 in GRCh38). All alignments from these reads were then subjected to variant calling with medaka before final analysis validation was performed.
Oxford Nanopore (Minion)
In house nanopore sequencing used a minion R.10.4.1 flow cell, with the V14 ligation sequencing kit (LSK114) following the manufacturer’s directions with modifications. DNA was sheared to an N50 of 10 kb using a g-tube (Covaris), 2 μg of DNA was sheared by centrifugation at 5500 rpm in an Eppendorf 5424r centrifuge two times for 1 min each. Shearing was confirmed by visualization on a 1% agarose gel. DNA ends were repaired using the NEBNext® Companion Module for Oxford Nanopore Technologies® Ligation Sequencing (NEB cat# E7180S) following the manufactures directions. DNA was purified by AMPure magnetic beads. Sequencing adapters ligation was carried out using NEB Quick Ligase (NEB cat #7180S) and Oxford Nanopore’s ligation buffer. Following purification with AMPure magnetic beads. Fifteen femtomoles of library were loaded onto the R.10.4.1 flow cell following priming. Post run base calling used guppy 6.0.1. Reads were mapped with minimap2 to the hg19 reference genome.
Read-depth and B-allele frequency analysis of short-read GS via VizCNV platform
The depth of sequencing coverage was computed using mosdepth (version 0.3.4)69 and subsequently visualized using our custom visualization tool, VizCNV (https://github.com/BCM-Lupskilab/VizCNV).60 This tool enables the plotting of normalized read depth for the individuals’ sequencing data, which facilitates manual assessment of CNVs exceeding 3 kilobases in size as well as the determination of B-allele frequency for a given genomic range. Analysis of B-allele frequency was performed on cases with available parental samples to determine if the CGR was formed from an intrachromosomal or interchromosomal event. An intrachromosomal event would have an expected B-allele frequency within a specified copy-number gain of 0 and 1 since there are no other contributions of a specified SNP position involved. Alternatively, in an interchromosomal event involving two X chromosomes, the expected B-allele frequency within a duplication and hemizygous triplication would be: 0, 0.33, 0.66, 1. An interchromosomal event for a triplication in a female would be 0, 0.25, 0.5, 0.75, 1.28,29
Strand-Seq
Strand-Seq data generation and data processing
Strand-Seq data were generated at the European Molecular Biology Laboratory using a modification of the OP-Strand-Seq library preparation protocol.70 Briefly, lymphoblastoid cell lines derived from two patients (BAB3114, BAB14547) were first cultured in RPMI media and subjected to 40μM BrdU treatment for 18h and 24h. The cells were then lysed to release the nuclei and nuclei were digested with RNase and MNase as described in the original protocol. Cells were fixed with formaldehyde, and the crosslinked nuclei were stained with Hoechst to reveal the population of cells that had incorporated BrdU for a single cell division. The population of once-divided cell nuclei was used to sort single nuclei into 96 well plates using fluorescence-activated cell sorting (FACS). The individual nuclei were processed to produce a sequencing library using a robotic liquid handler. In brief, nuclei were first de-crosslinked, protease-digested, fragmented DNA ends “polished” and Illumina adapters ligated as described in the original OP protocol with the necessary volumetric adjustments according to the starting volume. A necessary deviation from the protocol in our hands was to introduce a bead-based clean up after adapter ligation to remove adapter dimers prior to PCR amplification. This clean up was done at a 0.8x bead: DNA ratio. The adapter dimer free DNA was then exposed to Hoechst and UV light to ablate the BrdU-substituted strands. Finally, the libraries were PCR amplified for 15x cycles and simultaneously barcoded using a dual indexing strategy with iTru adapters. Amplified libraries were again subjected to a bead-based clean up at a 0.8x bead ratio and pooled for size selection. Final, size-selected libraries were subjected to deep sequencing on the Illumina NextSeq500 platform (MID-mode, 75 bp paired-end protocol). The resulting raw read files were aligned to the GRCh38 reference assembly (GCA_000001405.15) using BWA aligner (version 0.7.17). Low-quality libraries were automatically flagged using ASHLEYS (version 1.0),71 resulting in 41/96 (43%) and 52/96 (54%) viable, high-quality single-cell libraries for BAB14547 and BAB3114, respectively.
Strand-Seq data analysis
To detect SV breakpoint candidates, we initially flagged genomic regions which displayed a switch in read directionality, suggestive of inversion- or inverted duplication breakpoints, using the breakpointR tool (version 0.99.0) with default settings.72 Using this procedure, we generated breakpoint estimates from each individual cell, with confidence intervals between 10 kb (default minimum resolution) and >100 kb for poorly covered regions. Under the assumption of clonality, we merged breakpoint estimates of cells from the same sample and extracted ‘peak’ regions in which at least 75% of cells predicted a breakpoint, yielding high-confidence consensus breakpoint regions of typically 10 kb size. Genotypes for all regions were subsequently obtained using the ArbiGent tool,73 which estimates regional genotypes based on a directionality-specific read depth model and integrates this information across cells. Lastly, to confirm the obtained breakpoints and genotypes over long-range haplotype stretches visually, we generated a pseudo-bulk data track for each sample, which is conceptually similar to Strand-Seq based ‘composite files’ described previously.74 In these tracks, we combined the reads from all cells and synchronised their read directionality in a way that ‘reference’ and ‘inverse’ orientation are encoded by reads mapping on the ‘W’ and ‘C’ strands, respectively. All previously obtained breakpoint regions and genotypes could be confirmed after visualising this pseudo-bulk track in the UCSC browser.
Acknowledgments
This work was supported by the US National Institute of General Medical Sciences (NIGMS) R01 GM132589 (to C.M.B.C.) and in part by the Swedish Brain Foundation (FO2020-0351, to A.L.), the US National Institute of Neurological Disorders and Stroke (NINDS) (R35 NS105078, to J.R.L.), the National Human Genome Research Institute/National Heart, Lung, and Blood Institute (UM1HG006542, to the Baylor-Hopkins Center for Mendelian Genomics), and IDDRC grant no. 1U54 HD083092 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD). Work at the European Molecular Biology Laboratory was provided by the European Council (ERC Consolidator grant no. 773026, to J.O.K.). D.P. is supported by the International Rett Syndrome Foundation (IRSF grant 3701-1), the Rett Syndrome Research Trust, the Doris Duke Charitable Foundation (2023-0235), and NINDS (1K23 NS125126-01A1). The project described was supported in part by the Clinical Translational Core at Baylor College of Medicine, which is supported by IDDRC grant no. P50103555, from the NICHD. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NICHD or the NIH.
Author contributions
C.M.G. performed the laboratory experiments, interpreted and analyzed the data, and wrote the manuscript. J.D.B., M.G., K.P., J.M.F., and W.H. performed the laboratory experiments as well as analyzed and interpreted data. J.O.K., S.N.J., D.M.M., R.A.G., A.L., and J.R.L. interpreted and/or analyzed the data. M.Y.L., E.B., P.H., J.P.H., S.V.B., H.D., M.G.M., M.M., L.F.P., M.P., E.H., S.J., and F.J.S. performed the bioinformatics analyses and interpretation. D.P. provided the patient samples as well as clinical interpretation. C.M.B.C. conceptualized the study, analyzed and interpreted the data, and contributed to writing the manuscript. All authors have read, edited, and approved the final manuscript.
Declaration of interests
Baylor College of Medicine and Miraca Holdings have formed a joint venture with shared ownership and governance of BG, which performs clinical microarray analysis, clinical ES, and clinical biochemical studies. J.R.L. serves on the scientific advisory board of the BG. J.R.L. has stock ownership in 23andMe, is a paid consultant for Genomics International, and is a co-inventor on multiple US and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, genomic disorders, and bacterial genomic fingerprinting. E.H. and S.J. are employees of ONT and shareholders and/or share option holders of ONT. D.P. provides consulting services for Ionis Pharmaceuticals. F.J.S. receives research support from Genetech, Illumina, Pacbio, and ONT.
Published: June 21, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2024.100590.
Supplemental information
References
- 1.Carvalho C.M.B., Lupski J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016;17:224–238. doi: 10.1038/nrg.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pellestor F., Anahory T., Lefort G., Puechberty J., Liehr T., Hédon B., Sarda P. Complex chromosomal rearrangements: origin and meiotic behavior. Hum. Reprod. Update. 2011;17:476–494. doi: 10.1093/humupd/dmr010. [DOI] [PubMed] [Google Scholar]
- 3.Liu P., Carvalho C.M.B., Hastings P.J., Lupski J.R. Mechanisms for recurrent and complex human genomic rearrangements. Curr. Opin. Genet. Dev. 2012;22:211–220. doi: 10.1016/j.gde.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schuy J., Grochowski C.M., Carvalho C.M.B., Lindstrand A. Complex genomic rearrangements: an underestimated cause of rare diseases. Trends Genet. 2022;38:1134–1146. doi: 10.1016/j.tig.2022.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carvalho C.M.B., Ramocki M.B., Pehlivan D., Franco L.M., Gonzaga-Jauregui C., Fang P., McCall A., Pivnick E.K., Hines-Dowell S., Seaver L.H., et al. Inverted genomic segments and complex triplication rearrangements are mediated by inverted repeats in the human genome. Nat. Genet. 2011;43:1074–1081. doi: 10.1038/ng.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Y., Roberts N.D., Wala J.A., Shapira O., Schumacher S.E., Kumar K., Khurana E., Waszak S., Korbel J.O., Haber J.E., et al. Patterns of somatic structural variation in human cancer genomes. Nature. 2020;578:112–121. doi: 10.1038/s41586-019-1913-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newman S., Hermetz K.E., Weckselblatt B., Rudd M.K. Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am. J. Hum. Genet. 2015;96:208–220. doi: 10.1016/j.ajhg.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lupski J.R. Clan genomics: From OMIM phenotypic traits to genes and biology. Am. J. Med. Genet. 2021;185:3294–3313. doi: 10.1002/ajmg.a.62434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson E.K., Bellott D.W., Cho T.-J., Skaletsky H., Hughes J.F., Pyntikova T., Page D.C. Large palindromes on the primate X Chromosome are preserved by natural selection. Genome Res. 2021;31:1337–1352. doi: 10.1101/gr.275188.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dittwald P., Gambin T., Gonzaga-Jauregui C., Carvalho C.M.B., Lupski J.R., Stankiewicz P., Gambin A. Inverted low-copy repeats and genome instability--a genome-wide analysis. Hum. Mutat. 2013;34:210–220. doi: 10.1002/humu.22217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cook E.H., Jr., Scherer S.W. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–923. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- 12.Bodkin J.A., Coleman M.J., Godfrey L.J., Carvalho C.M.B., Morgan C.J., Suckow R.F., Anderson T., Öngür D., Kaufman M.J., Lewandowski K.E., et al. Targeted Treatment of Individuals With Psychosis Carrying a Copy Number Variant Containing a Genomic Triplication of the Glycine Decarboxylase Gene. Biol. Psychiatry. 2019;86:523–535. doi: 10.1016/j.biopsych.2019.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malhotra D., Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223–1241. doi: 10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang F., Khajavi M., Connolly A.M., Towne C.F., Batish S.D., Lupski J.R. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 2009;41:849–853. doi: 10.1038/ng.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramocki M.B., Tavyev Y.J., Peters S.U. The MECP2 duplication syndrome. Am. J. Med. Genet. 2010;A.152A:1079–1088. doi: 10.1002/ajmg.a.33184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carvalho C.M.B., Zhang F., Liu P., Patel A., Sahoo T., Bacino C.A., Shaw C., Peacock S., Pursley A., Tavyev Y.J., et al. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum. Mol. Genet. 2009;18:2188–2203. doi: 10.1093/hmg/ddp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carvalho C.M.B., Pehlivan D., Ramocki M.B., Fang P., Alleva B., Franco L.M., Belmont J.W., Hastings P.J., Lupski J.R. Replicative mechanisms for CNV formation are error prone. Nat. Genet. 2013;45:1319–1326. doi: 10.1038/ng.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.del Gaudio D., Fang P., Scaglia F., Ward P.A., Craigen W.J., Glaze D.G., Neul J.L., Patel A., Lee J.A., Irons M., et al. Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet. Med. 2006;8:784–792. doi: 10.1097/01.gim.0000250502.28516.3c. [DOI] [PubMed] [Google Scholar]
- 19.Leffler M., Christie L., Hackett A., Bennetts B., Goel H., Amor D.J., Peters G.B., Field M., Dudding-Byth T. Further delineation of dosage-sensitive K/L mediated Xq28 duplication syndrome includes incomplete penetrance. Clin. Genet. 2023;103:681–687. doi: 10.1111/cge.14303. [DOI] [PubMed] [Google Scholar]
- 20.Bahrambeigi V., Song X., Sperle K., Beck C.R., Hijazi H., Grochowski C.M., Gu S., Seeman P., Woodward K.J., Carvalho C.M.B., et al. Distinct patterns of complex rearrangements and a mutational signature of microhomeology are frequently observed in PLP1 copy number gain structural variants. Genome Med. 2019;11:80. doi: 10.1186/s13073-019-0676-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beck C.R., Carvalho C.M.B., Banser L., Gambin T., Stubbolo D., Yuan B., Sperle K., McCahan S.M., Henneke M., Seeman P., et al. Complex genomic rearrangements at the PLP1 locus include triplication and quadruplication. PLoS Genet. 2015;11 doi: 10.1371/journal.pgen.1005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L., Wang J., Zhang C., Li D., Carvalho C.M.B., Ji H., Xiao J., Wu Y., Zhou W., Wang H., et al. Efficient CNV breakpoint analysis reveals unexpected structural complexity and correlation of dosage-sensitive genes with clinical severity in genomic disorders. Hum. Mol. Genet. 2017;26:1927–1941. doi: 10.1093/hmg/ddx102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimojima K., Mano T., Kashiwagi M., Tanabe T., Sugawara M., Okamoto N., Arai H., Yamamoto T. Pelizaeus-Merzbacher disease caused by a duplication-inverted triplication-duplication in chromosomal segments including the PLP1 region. Eur. J. Med. Genet. 2012;55:400–403. doi: 10.1016/j.ejmg.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 24.Ishmukhametova A., Chen J.-M., Bernard R., de Massy B., Baudat F., Boyer A., Méchin D., Thorel D., Chabrol B., Vincent M.-C., et al. Dissecting the structure and mechanism of a complex duplication-triplication rearrangement in the DMD gene. Hum. Mutat. 2013;34:1080–1084. doi: 10.1002/humu.22353. [DOI] [PubMed] [Google Scholar]
- 25.Soler-Alfonso C., Carvalho C.M.B., Ge J., Roney E.K., Bader P.I., Kolodziejska K.E., Miller R.M., Lupski J.R., Stankiewicz P., Cheung S.W., et al. CHRNA7 triplication associated with cognitive impairment and neuropsychiatric phenotypes in a three-generation pedigree. Eur. J. Hum. Genet. 2014;22:1071–1076. doi: 10.1038/ejhg.2013.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gillentine M.A., Lozoya R., Yin J., Grochowski C.M., White J.J., Schaaf C.P., Calarge C.A. CHRNA7 copy number gains are enriched in adolescents with major depressive and anxiety disorders. J. Affect. Disord. 2018;239:247–252. doi: 10.1016/j.jad.2018.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beri S., Bonaglia M.C., Giorda R. Low-copy repeats at the human VIPR2 gene predispose to recurrent and nonrecurrent rearrangements. Eur. J. Hum. Genet. 2013;21:757–761. doi: 10.1038/ejhg.2012.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carvalho C.M.B., Pfundt R., King D.A., Lindsay S.J., Zuccherato L.W., Macville M.V.E., Liu P., Johnson D., Stankiewicz P., Brown C.W., et al. Absence of heterozygosity due to template switching during replicative rearrangements. Am. J. Hum. Genet. 2015;96:555–564. doi: 10.1016/j.ajhg.2015.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carvalho C.M.B., Coban-Akdemir Z., Hijazi H., Yuan B., Pendleton M., Harrington E., Beaulaurier J., Juul S., Turner D.J., Kanchi R.S., et al. Interchromosomal template-switching as a novel molecular mechanism for imprinting perturbations associated with Temple syndrome. Genome Med. 2019;11:25. doi: 10.1186/s13073-019-0633-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robak L.A., Du R., Yuan B., Gu S., Alfradique-Dunham I., Kondapalli V., Hinojosa E., Stillwell A., Young E., Zhang C., et al. Integrated sequencing and array comparative genomic hybridization in familial Parkinson disease. Neurol. Genet. 2020;6 doi: 10.1212/NXG.0000000000000498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zafar F., Valappil R.A., Kim S., Johansen K.K., Chang A.L.S., Tetrud J.W., Eis P.S., Hatchwell E., Langston J.W., Dickson D.W., Schüle B. Genetic fine-mapping of the Iowan SNCA gene triplication in a patient with Parkinson’s disease. NPJ Parkinsons Dis. 2018;4:18. doi: 10.1038/s41531-018-0054-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fuchs J., Nilsson C., Kachergus J., Munz M., Larsson E.-M., Schüle B., Langston J.W., Middleton F.A., Ross O.A., Hulihan M., et al. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology. 2007;68:916–922. doi: 10.1212/01.wnl.0000254458.17630.c5. [DOI] [PubMed] [Google Scholar]
- 33.Gu S., Yuan B., Campbell I.M., Beck C.R., Carvalho C.M.B., Nagamani S.C.S., Erez A., Patel A., Bacino C.A., Shaw C.A., et al. Alu-mediated diverse and complex pathogenic copy-number variants within human chromosome 17 at p13.3. Hum. Mol. Genet. 2015;24:4061–4077. doi: 10.1093/hmg/ddv146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beroukhim R., Mermel C.H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J.S., Dobson J., Urashima M., et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bignell G.R., Greenman C.D., Davies H., Butler A.P., Edkins S., Andrews J.M., Buck G., Chen L., Beare D., Latimer C., et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463:893–898. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Futreal P.A., Coin L., Marshall M., Down T., Hubbard T., Wooster R., Rahman N., Stratton M.R. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramakrishnan S., Kockler Z., Evans R., Downing B.D., Malkova A. Single-strand annealing between inverted DNA repeats: Pathway choice, participating proteins, and genome destabilizing consequences. PLoS Genet. 2018;14 doi: 10.1371/journal.pgen.1007543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weckselblatt B., Rudd M.K. Human Structural Variation: Mechanisms of Chromosome Rearrangements. Trends Genet. 2015;31:587–599. doi: 10.1016/j.tig.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song X., Beck C.R., Du R., Campbell I.M., Coban-Akdemir Z., Gu S., Breman A.M., Stankiewicz P., Ira G., Shaw C.A., Lupski J.R. Predicting human genes susceptible to genomic instability associated with Alu/Alu-mediated rearrangements. Genome Res. 2018;28:1228–1242. doi: 10.1101/gr.229401.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 41.Small K., Iber J., Warren S.T. Emerin deletion reveals a common X-chromosome inversion mediated by inverted repeats. Nat. Genet. 1997;16:96–99. doi: 10.1038/ng0597-96. [DOI] [PubMed] [Google Scholar]
- 42.Lang D., Zhang S., Ren P., Liang F., Sun Z., Meng G., Tan Y., Li X., Lai Q., Han L., et al. Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of Pacific Biosciences Sequel II system and ultralong reads of Oxford Nanopore. GigaScience. 2020;9 doi: 10.1093/gigascience/giaa123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lindsay S.J., Khajavi M., Lupski J.R., Hurles M.E. A chromosomal rearrangement hotspot can be identified from population genetic variation and is coincident with a hotspot for allelic recombination. Am. J. Hum. Genet. 2006;79:890–902. doi: 10.1086/508709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuccherato L.W., Alleva B., Whiters M.A., Carvalho C.M.B., Lupski J.R. Chimeric transcripts resulting from complex duplications in chromosome Xq28. Hum. Genet. 2016;135:253–256. doi: 10.1007/s00439-015-1614-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malkova A., Ira G. Break-induced replication: functions and molecular mechanism. Curr. Opin. Genet. Dev. 2013;23:271–279. doi: 10.1016/j.gde.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu P., Lacaria M., Zhang F., Withers M., Hastings P.J., Lupski J.R. Frequency of nonallelic homologous recombination is correlated with length of homology: evidence that ectopic synapsis precedes ectopic crossing-over. Am. J. Hum. Genet. 2011;89:580–588. doi: 10.1016/j.ajhg.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reiter L.T., Hastings P.J., Nelis E., De Jonghe P., Van Broeckhoven C., Lupski J.R. Human meiotic recombination products revealed by sequencing a hotspot for homologous strand exchange in multiple HNPP deletion patients. Am. J. Hum. Genet. 1998;62:1023–1033. doi: 10.1086/301827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sakofsky C.J., Ayyar S., Deem A.K., Chung W.-H., Ira G., Malkova A. Translesion Polymerases Drive Microhomology-Mediated Break-Induced Replication Leading to Complex Chromosomal Rearrangements. Mol. Cell. 2015;60:860–872. doi: 10.1016/j.molcel.2015.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin R., Espinoza C.Y., Large C.R.L., Rosswork J., Van Bruinisse C., Miller A.W., Sanchez J.C., Miller M., Paskvan S., Alvino G.M., et al. Template switching between the leading and lagging strands at replication forks generates inverted copy number variants through hairpin-capped extrachromosomal DNA. PLoS Genet. 2024;20 doi: 10.1371/journal.pgen.1010850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brewer B.J., Dunham M.J., Raghuraman M.K. A unifying model that explains the origins of human inverted copy number variants. PLoS Genet. 2024;20 doi: 10.1371/journal.pgen.1011091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McDermott D.H., Gao J.-L., Liu Q., Siwicki M., Martens C., Jacobs P., Velez D., Yim E., Bryke C.R., Hsu N., et al. Chromothriptic cure of WHIM syndrome. Cell. 2015;160:686–699. doi: 10.1016/j.cell.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Joron M., Frezal L., Jones R.T., Chamberlain N.L., Lee S.F., Haag C.R., Whibley A., Becuwe M., Baxter S.W., Ferguson L., et al. Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature. 2011;477:203–206. doi: 10.1038/nature10341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taylor J.S., Van de Peer Y., Meyer A. Genome duplication, divergent resolution and speciation. Trends Genet. 2001;17:299–301. doi: 10.1016/s0168-9525(01)02318-6. [DOI] [PubMed] [Google Scholar]
- 54.Fuller Z.L., Koury S.A., Phadnis N., Schaeffer S.W. How chromosomal rearrangements shape adaptation and speciation: Case studies in Drosophila pseudoobscura and its sibling species Drosophila persimilis. Mol. Ecol. 2019;28:1283–1301. doi: 10.1111/mec.14923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu P., Erez A., Nagamani S.C.S., Bi W., Carvalho C.M.B., Simmons A.D., Wiszniewska J., Fang P., Eng P.A., Cooper M.L., et al. Copy number gain at Xp22.31 includes complex duplication rearrangements and recurrent triplications. Hum. Mol. Genet. 2011;20:1975–1988. doi: 10.1093/hmg/ddr078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Voet T., Vermeesch J.R. Mutational processes shaping the genome in early human embryos. Cell. 2017;168:751–753. doi: 10.1016/j.cell.2017.02.008. [DOI] [PubMed] [Google Scholar]
- 57.Pettersson M., Grochowski C.M., Wincent J., Eisfeldt J., Breman A.M., Cheung S.W., Krepischi A.C.V., Rosenberg C., Lupski J.R., Ottosson J., et al. Cytogenetically visible inversions are formed by multiple molecular mechanisms. Hum. Mutat. 2020;41:1979–1998. doi: 10.1002/humu.24106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee J.A., Lupski J.R. Genomic rearrangements and gene copy-number alterations as a cause of nervous system disorders. Neuron. 2006;52:103–121. doi: 10.1016/j.neuron.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 59.Fernandez-Luna L., Aguilar-Perez C., Grochowski C.M., Mehaffey M., Carvalho C.M., Gonzaga-Jauregui C. Genome-wide maps of highly-similar intrachromosomal repeats that mediate ectopic recombination in three human genome assemblies. bioRxiv. 2024 doi: 10.1101/2024.01.29.577884. Preprint at. [DOI] [Google Scholar]
- 60.Du H., Jolly A., Grochowski C.M., Yuan B., Dawood M., Jhangiani S.N., Li H., Muzny D., Fatih J.M., Coban-Akdemir Z., et al. The multiple de novo copy number variant (MdnCNV) phenomenon presents with peri-zygotic DNA mutational signatures and multilocus pathogenic variation. Genome Med. 2022;14:122. doi: 10.1186/s13073-022-01123-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hijazi H., Coelho F.S., Gonzaga-Jauregui C., Bernardini L., Mar S.S., Manning M.A., Hanson-Kahn A., Naidu S., Srivastava S., Lee J.A., et al. Xq22 deletions and correlation with distinct neurological disease traits in females: Further evidence for a contiguous gene syndrome. Hum. Mutat. 2020;41:150–168. doi: 10.1002/humu.23902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nilsson D., Pettersson M., Gustavsson P., Förster A., Hofmeister W., Wincent J., Zachariadis V., Anderlid B.-M., Nordgren A., Mäkitie O., et al. Whole-Genome Sequencing of Cytogenetically Balanced Chromosome Translocations Identifies Potentially Pathological Gene Disruptions and Highlights the Importance of Microhomology in the Mechanism of Formation. Hum. Mutat. 2017;38:180–192. doi: 10.1002/humu.23146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mahmoud M., Doddapaneni H., Timp W., Sedlazeck F.J. PRINCESS: comprehensive detection of haplotype resolved SNVs, SVs, and methylation. Genome Biol. 2021;22:268. doi: 10.1186/s13059-021-02486-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sedlazeck F.J., Rescheneder P., Smolka M., Fang H., Nattestad M., von Haeseler A., Schatz M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods. 2018;15:461–468. doi: 10.1038/s41592-018-0001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smolka M., Paulin L.F., Grochowski C.M., Horner D.W., Mahmoud M., Behera S., Kalef-Ezra E., Gandhi M., Hong K., Pehlivan D., et al. Detection of mosaic and population-level structural variants with Sniffles2. Nat. Biotechnol. 2024 doi: 10.1038/s41587-023-02024-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Luo R., Wong C.-L., Wong Y.-S., Tang C.-I., Liu C.-M., Leung C.-M., Lam T.-W. Exploring the limit of using a deep neural network on pileup data for germline variant calling. Nat. Mach. Intell. 2020;2:220–227. [Google Scholar]
- 69.Pedersen B.S., Quinlan A.R. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics. 2018;34:867–868. doi: 10.1093/bioinformatics/btx699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hanlon V.C.T., Chan D.D., Hamadeh Z., Wang Y., Mattsson C.-A., Spierings D.C.J., Coope R.J.N., Lansdorp P.M. Construction of Strand-seq libraries in open nanoliter arrays. Cell Rep. Methods. 2022;2 doi: 10.1016/j.crmeth.2021.100150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gros C., Sanders A.D., Korbel J.O., Marschall T., Ebert P. ASHLEYS: automated quality control for single-cell Strand-seq data. Bioinformatics. 2021;37:3356–3357. doi: 10.1093/bioinformatics/btab221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Porubsky D., Sanders A.D., Taudt A., Colomé-Tatché M., Lansdorp P.M., Guryev V. breakpointR: an R/Bioconductor package to localize strand state changes in Strand-seq data. Bioinformatics. 2020;36:1260–1261. doi: 10.1093/bioinformatics/btz681. [DOI] [PubMed] [Google Scholar]
- 73.Porubsky D., Höps W., Ashraf H., Hsieh P., Rodriguez-Martin B., Yilmaz F., Ebler J., Hallast P., Maria Maggiolini F.A., Harvey W.T., et al. Recurrent inversion polymorphisms in humans associate with genetic instability and genomic disorders. Cell. 2022;185:1986–2005. doi: 10.1016/j.cell.2022.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chaisson M.J.P., Sanders A.D., Zhao X., Malhotra A., Porubsky D., Rausch T., Gardner E.J., Rodriguez O.L., Guo L., Collins R.L., et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019;10:1784. doi: 10.1038/s41467-018-08148-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microarray data generated in previous studies17 are available through the gene expression omnibus (GEO) under accessions GEO: GSE49440, GSE49446; new microarray data within this study is available under accession GEO: GSE250451. Oxford nanopore datasets are available within SRA BioProject ID SRA: PRJNA953021. Samples that had GS and consented for broad data sharing are available under dbGAP: phs002999.v2.p1. Optical genome mapping data is available upon request to the authors. All original code is available in this paper’s key resource table.