

Abstract
Trained immunity is characterized by long-term functional reprogramming of innate immune cells following challenge with pathogens or microbial ligands during infection or vaccination. This cellular reprogramming leads to increased responsiveness upon restimulation, and is mediated through epigenetic and metabolic modifications. In this review, we describe how molecular mechanisms underlying trained immunity, for example, induced by β-glucan or Bacille Calmette-Guérin (BCG) vaccination, can be investigated by using and integrating different layers of information including genome, epigenome, transcriptome, proteome, metabolome, microbiome, immune cell phenotyping, and function. We also describe the most commonly used experimental and computational techniques. Finally, we provide a number of examples of how a systems biology approach was applied to study trained immunity to understand interindividual variation or the complex interplay between molecular layers. In conclusion, trained immunity represents an opportunity for regulating innate immune function, and understanding the complex interplay of mechanisms that mediate trained immunity might enable us to employ it as a clinical tool in the future.
Keywords: data integration, innate immunity, multiomics, systems biology, trained immunity
Introduction
The human immune system is classically divided into the innate and the adaptive immune system. While innate immunity develops rapidly and is considered to be nonspecific, the adaptive immune system takes more time to develop, but is antigen specific and leads to the development of immunological memory. However, this dogma has been challenged, as a growing body of evidence supports the existence of innate immune memory. First of all, plants and invertebrates show enhanced protection upon reinfection, even though they lack a functional adaptive immune system [1]. In addition, certain infections and vaccinations, including Candida albicans (C. albicans) infection and the Bacille Calmette-Guérin (BCG) vaccine, are able to induce protection against unrelated infectious diseases through innate immune mechanisms [2,3]. These microbes were shown to leave an immunological imprint on innate immune cells, which leads to enhanced responsiveness, for example, increased cytokine production, upon subsequent stimulation. As shown in Fig. 1, this ability of innate immune cells, such as monocytes, to adapt after a primary challenge and exhibit improved antimicrobial activities during a secondary challenge has been termed trained immunity [3].
Figure 1.
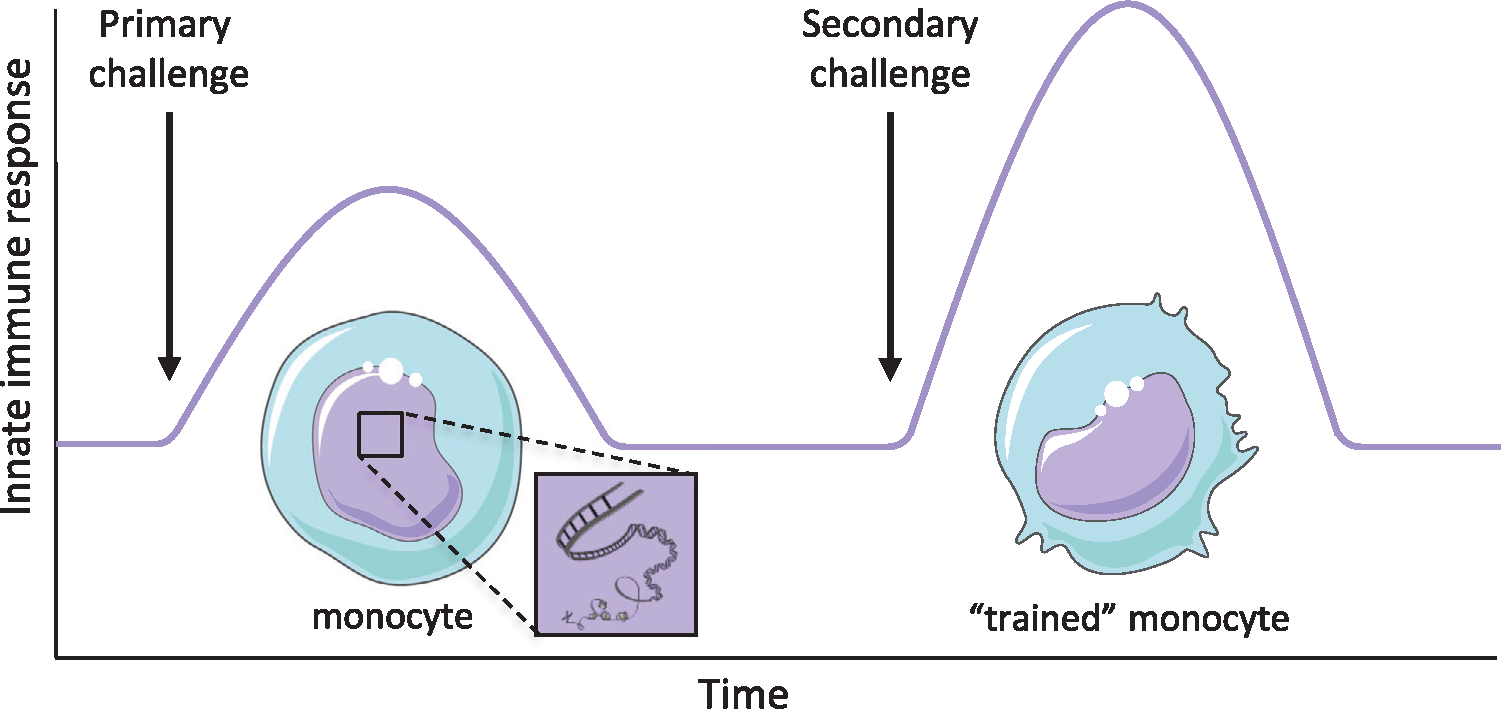
The concept of trained immunity. Innate immune cells, including monocytes, are epigenetically reprogrammed after exposure to a priming stimulus, such as BCG or β-glucan, allowing an enhanced response upon a secondary challenge when they become “trained monocytes.”
One of the well-studied inducers of trained immunity is the BCG vaccine. BCG vaccination leads to increased production of innate cytokines upon stimulation with unrelated pathogens [4], and these proinflammatory responses have been linked to increased protection against yellow fever [5] and malaria [6]. The nonspecific protection induced by BCG seems to be independent of adaptive immunity, as BCG-vaccinated severe combined immunodeficiency mice, which lack both B and T cells, are significantly better protected against infection with C. albicans compared to unvaccinated mice [4]. A cell wall component of C. albicans, β-glucan, also has the ability to induce trained immunity, and enhance cytokine responses upon restimulation [7]. Mice treated with β-glucan are also better protected against secondary infections, for example, with Staphylococcus aureus [8] and Mycobacterium tuberculosis [9]. Even sterile endogenous stimuli have been shown to induce trained immunity in humans, such as oxidized LDL and aldosterone, although the resulting proinflammatory responses after induction of trained immunity by these endogenous mediators have been linked to inflammatory diseases such as atherosclerosis [10]. It is important to note that trained immunity represents a concept of long-term adaptation of innate immune cells, and that different inducers of trained immunity, such as BCG, β-glucan, or oxidized LDL, can induce different transcriptional and functional programs. Trained immunity leading to heterologous protection might provide a tool to protect against emerging infectious diseases. For example, in the ongoing COVID-19 pandemic, BCG vaccination is currently being tested in several clinical trials to investigate its capacity to reduce susceptibility and severity of SARS-CoV-2 infection [11,12].
Underlying mechanisms of trained immunity
Trained immunity is mediated by epigenetic changes, including DNA methylation and histone modifications, which affect long-term transcriptional regulation [4,13]. The histone modifications related to trained immunity include the acquisition of histone 3 lysine 27 acetylation (H3K27ac) marks at distal enhancers (marked with histone 3 lysine 4 methylation [H3K4me1]) and the incorporation of histone 3 lysine 4 trimethylation (H3K4me3) marks at the promoters of immune-related genes [14,15]. A recent study in cell lines and human white blood cells demonstrated that these histone modifications are regulated at the level of topologically associated domains through long noncoding RNAs [16]. These epigenetic modifications are intertwined with metabolic changes. Upon BCG priming, glycolysis is increased, which is crucial for the induction of histone modifications and functional reprogramming of innate immune cells [17]. The interplay between epigenetic and metabolic reprogramming is necessary for the induction of trained immunity, as certain metabolites have a direct effect on a series of enzymes involved in epigenetic remodeling [18,19].
Trained immunity has been shown to last for at least 3 months and up to 1 year [4,20], while the heterologous protection against infections can last for at least 5 years, as BCG vaccination was associated with lower mortality in children up to 5 years of age in Uganda [21]. Considering the relatively short lifespan of innate immune cells, it is remarkable that these trained immunity effects last for such a long time. Recent studies have shown that these long-term effects are probably mediated through reprogramming of myeloid progenitor cells in the bone marrow (BM), which in turn generate myeloid cells with a trained immunity phenotype. In both mice and humans, it was observed that BCG induces transcriptional changes in hematopoietic stem and progenitor cells, which led to epigenetically modified monocytes (Fig. 2) [22,23]. Also for β-glucan, it was observed that administration led to increased myelopoiesis, which was accompanied by elevated signaling of innate immune cytokines such as IL-1β and GM-CSF [24]. In addition, considering the possibility to transmit epigenetic information through the germline, the trained immunity phenotype might even be conserved in a transgenerational manner. Indeed, maternal BCG priming has been associated with increased protection induced by BCG [25], and additional research is needed to study the impact of these transgenerational effects.
Figure 2.
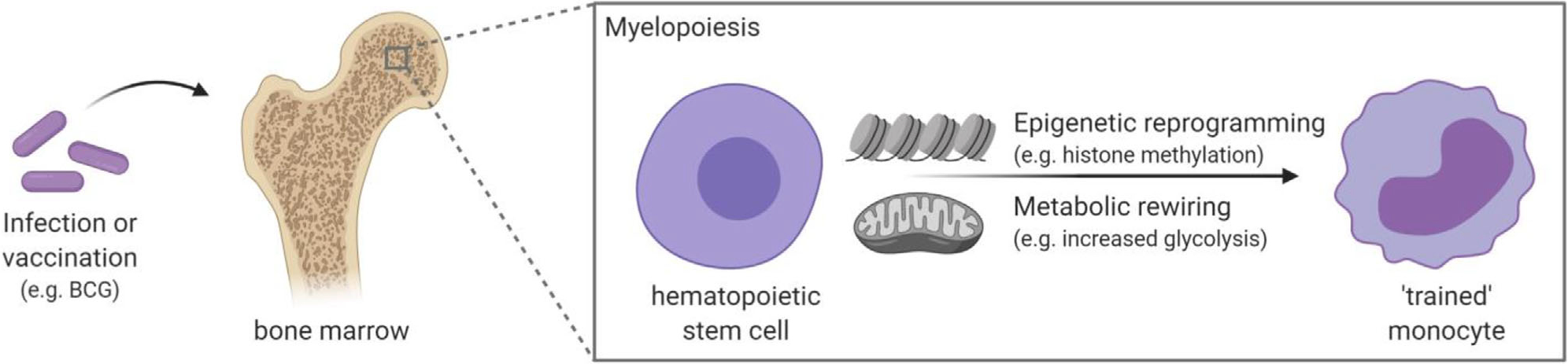
Underlying mechanism of trained immunity. Trained immunity, for example, induced by BCG vaccination, persists for at least several months due to reprogramming of myeloid progenitor cells. Epigenetic reprogramming together with metabolic rewiring mediates the transition to a trained immune cell with enhanced responsiveness upon stimulation. Created with BioRender.com.
Omics-based technologies in biological research
To be able to employ the untapped potential of trained immunity, we need to fully understand the mechanisms involved. Immune responses are the result of a complex interplay between molecular and cellular interactions. As for trained immunity, we have indeed observed the importance of epigenetic, transcriptional, and metabolic changes [26], and we need to study these different biological layers to understand their role in immune modulation. The development of high-throughput methods allows researchers to use an unbiased approach examining many potential genes or markers in relation to health and disease, rather than examining a limited number of “candidate” genes or markers. This unbiased approach can help generate novel hypotheses. When all different molecular components, such as genes, proteins, metabolites, and immunological parameters, are combined to describe an organism as a whole, we speak of systems biology (Fig. 3). With such genome-wide measurements of molecules and deep immune profiling, we have a unique opportunity to assess the interactions between molecules and reconstruct the regulatory networks, resulting in a comprehensive understanding of biological mechanisms. A systems biology approach is especially challenging in the field of immunology when we come across dozens of different cell types and many more intersecting molecular pathways and signals [27]. Nonetheless, systems biology-based methodologies are a promising approach to study trained immunity. In this next section, we describe the current technologies for genome-wide measurements of different molecular levels and the computational approaches to analyze them, which are summarized in Table 1, and integrate such data in the context of trained immunity.
Figure 3.
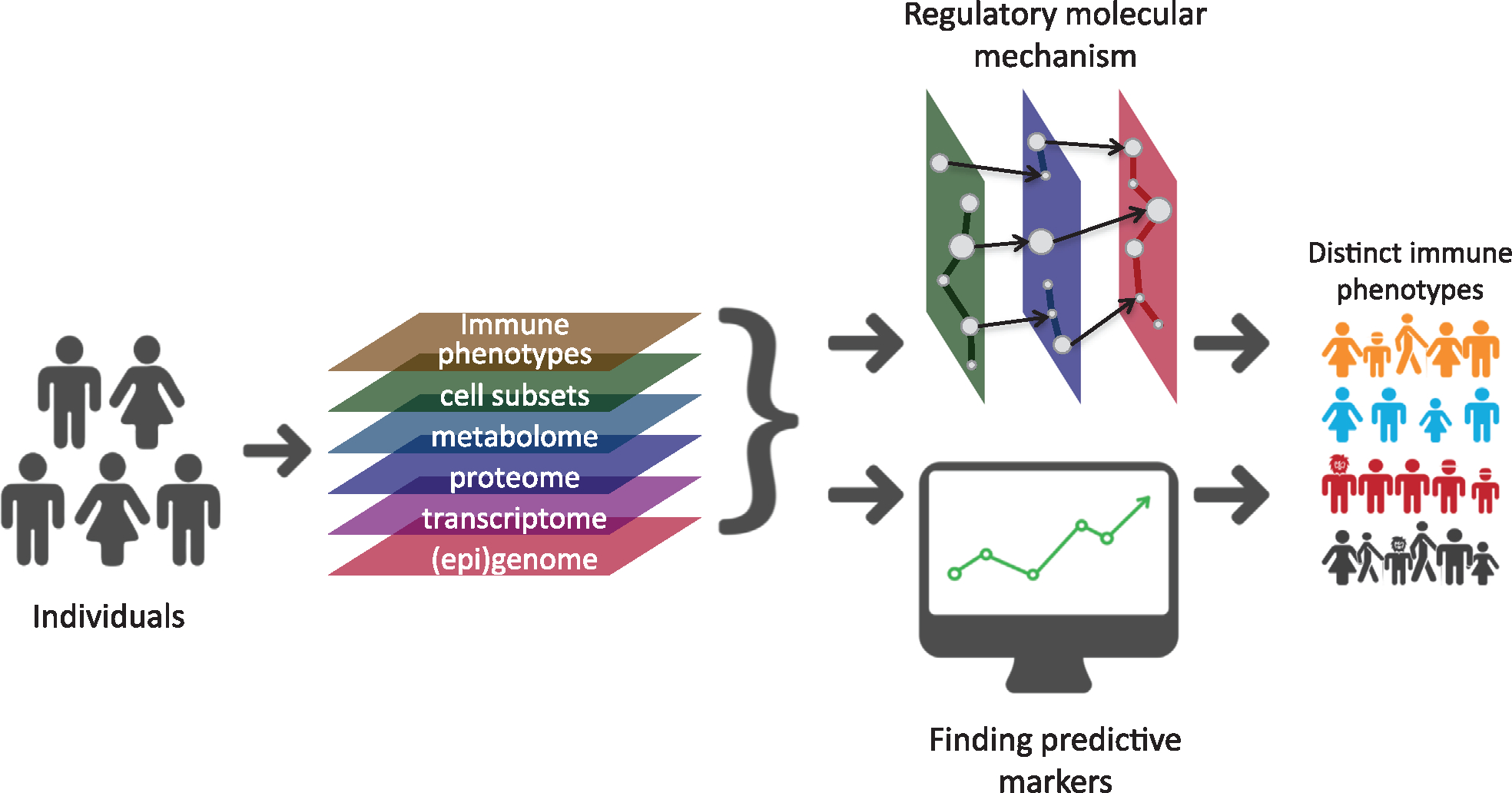
Schematic overview of systems immunology. A multiomics approach, integrating different layers of data types, can be used to understand regulatory networks of immune traits such as trained immunity or immune-related diseases. Similarly, an integrative approach can also identify biomarkers to understand interindividual variation in trained immunity or immune-related diseases.
Table 1.
Overview of different types of omics to study trained immunity
| Omics type | Technology | Reference |
|---|---|---|
|
| ||
| Genomics | DNA microarray | [31] |
| DNA-sequencing | [32] | |
| Epigenomics | DNA methylation | [38] |
| Chromatin immunoprecipitation (ChIP)-sequencing | [39] | |
| Assessment of accessible regions (e.g. using DNase-sequencing or ATAC-sequencing) | [40,41] | |
| Transcriptomics | Microarray | [51] |
| RNA-sequencing | [51] | |
| Proteomics | Mass spectrometry (MS) | [58] |
| Immunoaffinity (antibody-based) assay | [59] | |
| Metabolomics | Nuclear magnetic resonance (NMR) | [60,61] |
| Mass spectrometry (MS) | [60] | |
| Microbiome | 16S rRNA sequencing | [67] |
| Shotgun metagenomic sequencing | [68] | |
| Cell subsets | Flow cytometry | [69] |
| Mass cytometry | [70] | |
| Immune functions | Ex-vivo stimulation of immune cells | [71,79] |
| Killing assay | [90] | |
| Clinical phenotype upon infection | [74] | |
Genome assessment using DNA microarray or next-generation sequencing
The genome comprises all genetic material in an organism, which consists of protein-coding regions, noncoding regions, and mitochondrial DNA. Information on the genetic code can be generated either through genotyping, which is the process of determining specific genetic variants, or sequencing, which is a method to determine the exact sequence of nucleotides. The most commonly used technique for genotyping is microarray, which contains probes of allele-specific DNA sequences. These probes hybridize with the DNA sample when the nucleic acid sequences are complementary, which is then detected. The detection of these polymorphisms, known as single nucleotide polymorphisms (SNP), can be used to detect risk alleles for a certain disease or phenotype [28]. Using already available datasets, imputation of additional genotype variation can be performed to increase the power and the resolution with tools like Impute2 [29] or the Michigan Imputation Server [30].
Genotyping is faster and cheaper than DNA sequencing, but is limited by the amount of information that is generated [31]. The costs of sequencing decreased rapidly after the introduction of next-generation sequencing, which refer to a series of innovations including novel DNA template preparation, parallel sequencing, image capture, followed by sequence alignment, assembly and variant detection [32]. Although the sequencing costs are decreasing rapidly, whole genome sequencing is still costly for large-scale projects. In this case, SNP array-based genotyping followed by imputation is a more cost-effective strategy, even for rare variant detection [33].
By association analysis between genetic variants and immunological profiles (including trained immunity phenotypes), we can learn to understand the impact of genetic factors on immune traits and immune-related diseases [34,35]. Instead of using the traditional approach to look at specific SNPs, for example, in promotor regions of candidate genes, this genome-wide approach might reveal novel genetic associations. Linking genetic variants to immune traits can be achieved through quantitative trait loci (QTL) mapping, for example, using Plink [36] or the R package MatrixeQTL [37]. Such studies will improve our understanding of the variation in trained immunity responses, and can be used to pinpoint host factors that can be modified and targeted.
Epigenome assessment using ChIP-seq or ATAC-seq
The epigenome consists of all chemical alterations that do not change the DNA sequence, but which are added to the DNA to regulate the expression of genes in the genome. In each cell, the DNA is organized around histones to form nucleosomes, which in turn are organized into chromatin. This leaves certain genomic regions biologically active and others inactive, as these are no longer accessible for the transcription machinery. The composition and compaction of chromatin is regulated by many epigenetic mechanisms including DNA methylation, histone modifications, and noncoding RNAs. While the genetic code is for the most part static, the epigenome is not, as the chromatin structure is subject to dynamic changes.
There are various genome-wide analysis methods to understand the epigenetic structure. First of all, methylome can be assessed on a genome-wide level using microarray or bisulfite sequencing technologies. There are various experimental approaches for assessing DNA methylation including enzyme digestion, affinity enrichment or bisulfite conversion, and the selection of a profiling method depends on the specific research question [38]. Another approach is to study protein-DNA interactions using chromatin immunoprecipitation sequencing (ChIP-seq). ChIP can be used for the identification of transcription factor binding sites or the location of specific histone modifications. In combination with high-throughput sequencing analysis, this allows for an unbiased and genome-wide analysis [39]. Finally, instead of looking at specific marks, one can also study the accessible regions in the genome, for example with DNase-seq or with assay of transposase accessible chromatin (ATAC)-seq. With DNase-seq, regions in the genome that are hypersensitive to DNase I are cleaved, which are the regions that are easily accessible to the transcription machinery, after which these regions are sequenced [40]. ATAC-seq uses hyperactive Tn5 transposase to simultaneously cut and ligate adapters for sequencing of regions of increased accessibility. While other technologies require millions of cells as input material, ATAC-seq only needs a standard input of 50 000 cells [41] and can even be assessed on a single-cell level [42].
The epigenome varies across different cell types, but there are publicly available epigenetic reference datasets, for example, from the Encyclopedia of DNA Elements (ENCODE) [43] and the International Human Epigenome Consortium (IHEC) [44], of which the Roadmap Epigenomics Project is a member [45]. These datasets contain data from different cell lineages and tissues that can be used for comparison and annotation. Data on epigenetic marks can also be integrated with genotypes or transcriptomics through local or genome-wide association analysis, for example, DNA methylation QTL mapping [46] or expression quantitative trait methylation (eQTM) analysis [47], which can aid in interpreting the functionality of immune trait-associated genes.
Epigenetic changes, including the induction of histone modifications and alterations in DNA methylation levels, have a specific biological role in innate immune memory [48]. It has been observed that infection or proinflammatory stimuli affect the epigenetic landscape of innate immune cells [15,49], and these changes mediate the trained immunity responses upon a secondary challenge [14]. Differential analysis of genome-wide methylation patterns, accessible regions, and histone modifications between trained and untrained cells will reveal the regulatory mechanisms of trained immunity and identify the key epigenetic processes in this context.
Transcriptome assessment using RNA microarray or next-generation sequencing
Transcriptome refers to the collection of all RNA transcripts, both coding and noncoding. The coding RNA transcripts, also known as mRNA, are transcribed into proteins, while the noncoding RNA transcripts have other diverse functions. There are two main technologies to quantify RNA transcripts: microarray and RNA-seq. Microarrays measure the abundance of a defined set of transcripts via hybridization of RNA to complementary probes. The RNA-seq technique copies bulk RNA into cDNA transcripts which are then sequenced. These sequences are aligned to a reference genome sequence to identify which genes are transcribed, and the quantification provides the expression levels for transcribed genes. These read counts can be used for differential expression analysis, for example, using the R package DESeq2 [50]. In contrast to RNA-seq, microarray requires prior knowledge of the genomic sequence of the organism of interest to develop probes. RNA-seq methodology has constantly improved and is now the dominant transcriptomics technique used [51].
The amount of input RNA is much lower for RNA-seq technologies compared to microarray, which even allows to analyze the transcriptome on a single-cell level (scRNA-seq). This provides a higher resolution and enables us to understand individual differences on a cellular level [52]. One of the advantages of scRNA-seq over bulk RNA-seq is that changes in cell composition can also be taken into account, which can mask important changes on a bulk RNA-seq level [53]. In addition, scRNA-seq provides the opportunity to identify unique cell subpopulations. In the context of trained immunity, scRNA-seq could be used to assess cell heterogeneity after training, which could then be linked to immune function. ScRNA-seq could also be used to study hematopoietic stem and progenitor cells in the context of trained immunity, to understand the changes induced on a lineage level covered in detail in this issue by Stephenson et al. [54]. Not only the transcriptome can now be assessed on a single-cell level, but also the genome, epigenome (assessing DNA methylation patterns or using ATAC-seq), and the proteome can be studied for individual cells [55]. Furthermore, there are technologies available that are able to detect more than one type of single-cell omics in the same cell. For example, the methylome and transcriptome can be detected in parallel to explore the cellular connections between epigenetic variation and transcriptional regulation [56]. The R package Seurat version 3 presents a framework for the comprehensive integration of single-cell data [57]. An integrative approach combining these technologies will increase our understanding of different cellular modalities and their function in certain tissues.
Proteome assessment using mass spectrometry or immunoaffinity assays
Although the generation of proteins is the result of transcribing mRNA, many mechanisms, such as post-translational modifications and protein degradation, affect the correlation between transcriptome and proteome. In addition, many mRNA transcripts give rise to multiple protein products. This underlines the importance of assessing the variation in proteome in addition to transcriptome analysis.
Mass spectrometry (MS)-based technologies can be used for the systemic identification and quantification of proteins. MS is based on the principle of generating ionized analytes, separating them according to their specific m/z, and then recording the relative abundance of each ion type [58]. Another method in proteomics is to use a targeted approach by using immunoaffinity (antibody-based) assays [59]. However, in contrast to MS-based technologies, immunoaffinity assays, such as ELISA and multiplex immunoassays, are time consuming to develop per analyte, and often have a lower capacity for multiplexing.
In the context of trained immunity, the assessment of the proteome, for example, measuring cytokines and chemokines, is of great importance, as these proteins play a key role in mediating and steering the immune response. In addition, targeted proteome analyses can be used to validate the functional activity of pathways, that were identified from previous (epi)genome or transcriptome studies, and therefore, allow data integration between these different omics techniques.
Metabolome assessment using nuclear magnetic resonance or mass spectrometry
Metabolomics is the study of metabolites, which are defined as low molecular weight organic and inorganic molecules, which are the substrates, intermediates, and products of biochemical processes. These metabolites are often the result of the interaction between proteins and environmental factors, and the metabolome is therefore an important link to understand immunological processes. The diversity in metabolites leaves the complete characterization of the metabolome technically challenging and multiple strategies are often used to provide a wide coverage. Nuclear magnetic resonance (NMR) spectroscopy and MS, the latter often used in combination with liquid or gas chromatography, are the most commonly used techniques for metabolomics [60]. NMR spectroscopy is based on the principle that when a nucleus of an atom is placed in a magnetic field and exposed to a pulse of electromagnetic radiation, it will resonate at a frequency specific for that isotope. Since the magnetic field is unique to a certain compound, NMR spectroscopy can be used to identify biochemical molecules. Compared to MS-based technologies, NMR spectroscopy is less sensitive, which means the number of detectable metabolites is generally lower. On the other hand, advantages of NMR spectroscopy include minimal sample preparation, high level of experimental reproducibility, and the nondestructive nature of the technique [61]. In contrast to untargeted metabolomics, the accuracy and specificity of the measurements can be improved by using (semi-)targeted metabolomics strategies, but this requires prior knowledge of the chemical properties of analytes.
Studying the relative abundance of metabolites is a way to understand the metabolic state of a cell or organism, which is important as rewiring of cellular metabolism is essential for inducing the epigenetic changes underlying trained immunity [62]. However, the relative abundance of metabolites is also dependent on environmental factors and the gut microbiota [63]. Here, integration of transcriptome and metabolome data is a promising method for mapping metabolic networks and metabolic reactions [64].
Microbiome assessment using 16S rRNA or shotgun metagenomic sequencing
The human microbiome is the collection of microbes that live in or on our body. The gut microbiome, which is the most commonly studied part of the microbiome, has been associated with cytokine responses upon microbial stimulation [65] and immune-related diseases such as asthma, inflammatory bowel disease, and diabetes [66]. As the microbiome affects our metabolic functions and immune system, it might also be relevant for trained immunity research.
Next-generation sequencing techniques allow us to study variation in diversity and abundance of bacteria from human samples. There are two commonly used techniques for microbiome assessment: 16S sequencing and shotgun metagenomics. The 16S rRNA gene is highly conserved between different species of bacteria and archaea, but also contains species-specific variations. Therefore, the nucleotide sequence of the 16S rRNA gene can be used to identify microbial communities [67]. Another technique for microbiome assessment is shotgun metagenomic sequencing, which looks at the entire genomic content of the microbiota in an unbiased manner. Metagenomics also allows the functional characterization and de novo assembly of metagenomes [68]. However, this technique is more expensive than 16S sequencing.
Similar to studies associating microbiome composition to ex-vivo cytokine responses [65], the gut microbiome could be associated with trained immunity and vaccination responses. To this date, this has not been investigated on a population level. Further research is needed to assess the effect of microbiome on interindividual variation in trained immunity responses.
Cell subsets and surface markers assessment using flow or mass cytometry
Another layer of complexity is the large number of different cell types in the immune system. Increased resolution due to scRNA-seq led to the conclusion that individual cells within a population, which were once assumed to be homogeneous, have unique features, adding to the complexity of cellular systems. The identification and functionality of various cell types is dependent on the proteins expressed on the surface of these cells, known as cluster of differentiation (CD) markers, which can be assessed by flow cytometry using specific antibodies [69]. Since flow cytometry is dependent on the detection of fluorescent labels, one panel has a limited multiplexing capability. In contrast, mass cytometry, which is a fusion between flow cytometry and MS, offers advantages in this perspective, since it can measure up to 40+ protein parameters. Mass cytometry uses rare metal isotopes instead of fluorophores for antibodies labeling, and elemental MS is then able to discriminate isotopes of different atomic weights with high accuracy [70]. This method was commercialized and the instrument is called cytometry by time-of-flight (CyTOF). Several analytical tools have recently been developed to assist in the interpretation of mass cytometry data. These include algorithms designed to assess the global structure of a sample, the association between two molecules in single cells, or the cellular features that predict a certain immunological phenotype [70].
Due to the high dimensionality, mass cytometry can identify cells, but simultaneously assess the expression of cell-surface markers that execute critical biological processes, and even enumerate the expression of transcription factors that drive gene expression programs [70]. This allows researchers to study the behavior of individual cells in a more holistic manner, which could reveal coregulation and crosstalk between cellular programs in the context of trained immunity.
Host-pathogen interaction assessment using ex-vivo stimulation or killing assays
Finally, all these levels of data can be integrated and linked, in this context, to reveal the complex interplaying networks of molecules involved in trained immunity responses. Trained immunity is commonly assessed by measuring the immune response of innate immune cells after ex-vivo stimulation with unrelated pathogens or ligands. After stimulation, a wide variety of responses can be assessed such as cytokine production, expression of pattern recognition receptors, expression of transcription factors, and energy consumption [4,71]. Although these measurements illustrate the level of responsiveness of immune cells, we are especially interested in the ability of immune cells to clear an infection. The ability of immune cells to control outgrowth can be assessed using killing assays, which can either be studied ex vivo or in animal models [7,72]. Finally, the ultimate goal is to use this integrative approach to link these various data levels to clinical phenotypes in infectious disease cohorts or human experimental infection models [6,73,74]. Ultimately, this integrative approach might help us to understand the interindividual variation of immune traits in the context of trained immunity. Knowledge on the heterogeneity of immune traits or a patient population could lead to personalized healthcare. As an example, omics technologies are currently being applied to develop treatment for sepsis patients targeting their precise immune state [75,76]. Precision medicine could be applied in the future utilizing trained immunity to prevent infectious diseases or to improve vaccine strategies.
How to use systems biology to study trained immunity
Lessons learned about myelopoiesis
Previous studies have aimed to use a systems biology approach to get a better understanding of trained immunity. As a first example, two studies have been performed to understand the longevity of trained immunity, which is mediated through reprogramming of myeloid progenitor cells in the BM as earlier discussed. Kaufmann et al. vaccinated mice with BCG, after which they characterized the BM compartment. Using a combination of RNA-seq, ChIP-seq, ATAC-seq, and flow cytometry, they identified enhanced myelopoiesis of the hematopoietic stem cells, which in turn generated epigenetically modified macrophages. These epigenetically changed macrophages were shown to provide better protection against M. tuberculosis infection [22]. Similarly, Cirovic et al. studied the BM compartment in humans after BCG vaccination, combining RNA-seq, ATAC-seq, flow cytometry, and ex-vivo immune responses [23]. Together, these studies using an integrative approach increased our understanding of the longevity and generation of trained immunity responses in vivo.
Multiomics data integration to predict vaccine responses and immune traits
Other examples on multiomics data integration include the study from Bakker et al., which integrated 11 categories of host factors together to build a computational model which predicted stimulus-induced cytokine production [77]. In addition, Tsang et al. build predictive models of antibody responses after influenza vaccination using preperturbation cell populations [78]. Similar approaches can also be applied to study trained immunity and vaccination responses.
These previous studies integrated multiple data levels already, but a next step would be to integrate various types of data with genomic information. Currently, this has not been done in the context of trained immunity, but there are examples of studies trying to understand the variation of other immune traits using genotype data. For example, the effect of genetic heritability on cytokine production after microbial challenge was studied through QTL mapping. In this study, it was observed that genetic factors especially explained a high percentage of the variance observed for monocyte-derived cytokines (>50% of explained variance especially for IL-6 and IL1-β) [79]. In addition, heritability assessment and QTL mapping can also be applied to cell subsets instead of cytokines [80,81], which revealed that T-cell numbers are more strongly driven by genetic factors than B-cell counts [82]. A similar approach could also be used in the future to understand the variance in trained immunity immune traits. In addition, existing datasets can be used as reference to prioritize candidate genes in applying systems biology to trained immunity such as the GTEx portal with tissue-specific expression QTL data [83], the Roadmap Epigenomics [45], and the Functional Genomics Project [84].
The ultimate goal is to use multiomics to understand the protection trained immunity provides against infectious diseases, which could be studied with human experimental infection models. One example of this is the study of Arts et al., in which they vaccinated healthy controls with BCG or a placebo, after which the study participants were vaccinated with the live-attenuated yellow fever vaccine as a model for infection. Study participants who were vaccinated with BCG showed significantly lower viremia compared to the unvaccinated controls. Using a combination of data from genotyping, ChIP-seq, RNA-seq, and ex-vivo immune responses, they showed that IL-1β production is important for the induction of trained immunity and the accompanied reduction of viremia [5]. This is one example of how multiomics integration can lead to a better understanding of trained immunity and how it protects against subsequent infections.
One of the strengths of integrating multiple levels of data is an increased power to identify key regulatory molecular networks driving trained immunity. For example, results obtained from one level (i.e. genes) can be used to reduce the number of traits to test in the second level (i.e. proteins), thereby, increasing power. In addition, the shared effects observed at multiple molecular levels could provide strong evidence for real, biologically relevant traits, which in practice improves the statistical power. Lastly, the power could be further improved by using Bayesian statistics in the integrative analysis [85], where the prior knowledge from literature can be utilized.
Pitfalls of system biology approaches
There are also some pitfalls we need to acknowledge when we use systems biology to study trained immunity. One important pitfall, when it comes to designing effective omics studies, is the issue of sample size. Due to the large number of markers measured using omics technologies and the relatively small contributing effect size of individual analytes, the risk of both type 1 and 2 errors are high without a sufficient sample size for both the discovery and validation cohort. For clinical research, this is often a limitation, and a collaborative approach is often necessary to recruit enough patients. Furthermore, many multiomics studies rely on associations between markers and immune traits or patient phenotypes. Additional empirical evidence is often needed to confirm causal relationships, which can be achieved via in-vitro validation, experimental animal models, or clinical studies. In addition, with increased resolution, our definition of heterogeneity is also challenged. In the end, every individual or single cell will be unique, but not all variation is biologically or clinically relevant. Therefore, we have to be careful not to confuse noise with important functional differences. When these pitfalls are accurately addressed, systems biology provides a promising tool for studying trained immunity.
Future research
Trained immunity has the potential to have an impact on global health, as inducers of trained immunity, such as BCG and β-glucan, could be used to protect against infectious diseases. For example, it could be used to enhance host responses in (relatively) immunocompromised individuals, for example, the elderly, or to prevent postoperative infections. BCG might also provide a tool to protect against emerging infectious diseases [12]. To exemplify, in the ongoing COVID-19 pandemic, BCG vaccination is currently being tested in several clinical trials to investigate its capacity to prevent infection with the new coronavirus SARS-CoV-2 [11]. In addition to preventing infections, trained immunity might also be a useful target in other immune-mediated diseases. Local BCG instillations are being used to treat patients with bladder cancer, and trained immunity has, therefore, been proposed as a new strategy for immunotherapy in cancer [86]. Recently, nanobiologicals-inducing trained immunity were successful in suppressing tumor growth in animal models [87]. Also, other conditions characterized by defective immunity, such as immunoparalysis in sepsis, could be targeted with trained immunity [88]. Systems biology could be applied to understand interindividual variation in trained immunity responses, which could help us to improve treatment options or vaccine strategies. All things considered, trained immunity represents a powerful opportunity for regulating innate immune function to treat or prevent a variety of diseases in the future.
Over the last decade, we have gathered important new insights in the context of trained immunity. However, many important questions remain, which are summarized in Textbox 1. First of all, there is still much unknown about the durability of trained immunity responses. Future studies should investigate how long the BM alterations persist, if restimulation, for example, through BCG revaccination, could prolong these effects, and if this process is reversible. Second of all, it remains unclear why certain microbial ligands and vaccinations, such as β-glucan and BCG, are able to induce strong trained immunity responses while others are not, and what determines this ability. In addition, for many inducers of trained immunity, the level of specificity is unknown, but it will be crucial to pinpoint to which particular pathogens (viruses, bacteria, or parasites) these agents provide protection. Next, most of the research into trained immunity has focused on monocytes and NK-cells. However, recent research has revealed that neutrophils also have the ability to adapt a trained immunity phenotype after BCG vaccination [89]. Therefore, the ability of other innate or semi-innate immune cells, for example, γδ-T-cells, DCs, and stromal cells, to adapt a trained immunity phenotype should also be explored. Also, it should be further investigated how the promotion of a proinflammatory environment could possibly contribute to immune-mediated diseases, such as atherosclerosis, which could be a potential risk of trained immunity. Last, trained immunity shows high variability across individuals, possibly due to the existence of pre-existing immunity. When we understand the factors responsible for the variation in trainability, we know which individuals would benefit most from treatment, and we can aim to optimize the trained immunity inducers to generate consistent results. With this knowledge, therapeutics, such as antibodies or nanoparticles, targeting a specific epigenetic or metabolic pathway could be used to induce a more predictable trained immunity response in the future [88].
Textbox 1: Future research priorities.
Immunological questions
What is the durability of the bone marrow alterationst hat mediate trained immunity? Can the duration of these changes be prolonged by restimulation, and is the process reversible?
Which microbial or endogenous ligands are able to induce trained immunity, and how specific are these various inducers?
Which cell types and tissue-specific cells can adapt a trained immunity phenotype?
What explains the interindividual variation in trained immunity responses?
Clinical questions
Can trained immunity inducers be used to protect against infections, for example, to prevent postoperative infections, to protect the elderly or immunocompromised individuals, or in the context of emerging infection diseases?
Can trained immunity modulators be used to suppress tumor growth?
What are the possible risks of inducing trained immunity?
In conclusion, trained immunity represents an opportunity for regulating innate immune function to prevent or treat a wide variety of diseases in the future. Systems biology is a promising approach to study the complex interplay of mechanisms that mediate trained immunity and the interindividual variation in trained immunity responses. These insights might enable us to employ trained immunity as a clinical tool in the future.
Acknowledgments:
The authors thank Olivier B. Bakker for his help generating the schematic overview of systems immunology, Cheng-Jian Xu for his valuable input on multiomics techniques and Smart Servier Medical Art for generating icons for public use. BioRender.com was used to create the graphical abstract. RvC was supported by National Institute of Health for the ULTIMATE project (R01AI145781). MGN was supported by an ERC Advanced Grant (833247) and a Spinoza Grant of the Netherlands Organization for Scientific Research. YL was supported by an ERC Starting Grant (948207) and the Radboud University Medical Centre Hypatia Grant (2018) for Scientific Research.
Abbreviations:
- ATAC-seq
assay of transposase accessible chromatin-sequencing
- BCG
Bacille Calmette-Guérin
- ChIP-seq
chromatin immunoprecipitation-sequencing
- MS
mass spectrometry
- NMR
nuclear magnetic resonance
- QTL
quantitative trait loci
- scRNA-seq
single-cell RNA-sequencing
- SNP
single nucleotide polymorphism
Footnotes
Conflict of Interest: The authors declare no commercial or financial conflict of interest.
References
- 1.Netea MG and van der Meer JW Trained immunity: an ancient way of remembering. Cell Host Microbe. 2017. 21: 297–300. [DOI] [PubMed] [Google Scholar]
- 2.de Bree LCJ, Koeken V, Joosten LAB, Aaby P, Benn CS, van Crevel R and Netea MG, Non-specific effects of vaccines: current evidence and potential implications. Semin. Immunol. 2018. 39: 35–43. [DOI] [PubMed] [Google Scholar]
- 3.Netea MG, Quintin J and van der Meer JW, Trained immunity: a memory for innate host defense. Cell Host Microbe. 2011. 9: 355–361. [DOI] [PubMed] [Google Scholar]
- 4.Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Ifrim DC, Saeed S, Jacobs C et al. , Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci USA. 2012. 109: 17537–17542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arts RJW, Moorlag S, Novakovic B, Li Y, Wang SY, Oosting M, Kumar V et al. , BCG vaccination protects against experimental viral infection in humans through the induction of cytokines associated with trained immunity. Cell Host Microbe. 2018. 23: 89–100. [DOI] [PubMed] [Google Scholar]
- 6.Walk J, de Bree LCJ, Graumans W, Stoter R, van Gemert GJ, van de Vegte-Bolmer M, Teelen K et al. , Outcomes of controlled human malaria infection after BCG vaccination. Nat. Commun. 2019. 10: 874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, Jacobs L et al. , Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. 2012. 12: 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kokoshis PL, Williams DL, Cook JA and Di Luzio NR, Increased resistance to Staphylococcus aureus infection and enhancement in serum lysozyme activity by glucan. Science. 1978. 199: 1340–1342. [DOI] [PubMed] [Google Scholar]
- 9.Moorlag S, Khan N, Novakovic B, Kaufmann E, Jansen T, van Crevel R et al. , beta-glucan induces protective trained immunity against Mycobacterium tuberculosis infection: a key role for IL-1. Cell Rep. 2020. 31: 107634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riksen NP, Trained immunity and atherosclerotic cardiovascular disease. Curr. Opin. Lipidol. 2019. 30: 395–400. [DOI] [PubMed] [Google Scholar]
- 11.Curtis N, Sparrow A, Ghebreyesus TA and Netea MG, Considering BCGvaccinationtoreducetheimpactofCOVID-19.Lancet.2020.395:1545–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Netea MG, Giamarellos-Bourboulis EJ, Dominguez-Andres J, Curtis N, van Crevel R, van de Veerdonk FL et al. , Trained immunity: a tool for reducing susceptibility to and the severity of SARS-CoV-2 infection. Cell. 2020. 181: 969–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verma D, Parasa VR, Raffetseder J, Martis M, Mehta RB, Netea M and Lerm M, Anti-mycobacterial activity correlates with altered DNA methylation pattern in immune cells from BCG-vaccinated subjects. Sci. Rep. 2017. 7: 12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Heijden C, Noz MP, Joosten LAB, Netea MG, Riksen NP and Keating ST, Epigenetics and trained immunity. Antioxid Redox Signal 2018. 29: 1023–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F et al. , Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014. 345: 1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fanucchi S, Fok ET, Dalla E, Shibayama Y, Borner K, Chang EY et al. , Immune genes are primed for robust transcription by proximal long noncoding RNAs located in nuclear compartments. Nat. Genet. 2019. 51: 138–150. [DOI] [PubMed] [Google Scholar]
- 17.Arts RJW, Carvalho A, La Rocca C, Palma C, Rodrigues F, Silvestre R et al. , Immunometabolic pathways in BCG-induced trained immunity. Cell Rep. 2016. 17: 2562–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. , Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 2016. 24: 807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Netea MG, Dominguez-Andres J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E et al. , Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020. 20: 375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kleinnijenhuis J, Quintin J, Preijers F, Benn CS, Joosten LA, Jacobs C et al. , Long-lasting effects of BCG vaccination on both heterologous Th1/Th17 responses and innate trained immunity. J Innate Immun. 2014. 6: 152–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nankabirwa V, Tumwine JK, Mugaba PM, Tylleskar T, Sommerfelt H and Group P-ES, Child survival and BCG vaccination: a community based prospective cohort study in Uganda. BMC Public Health.2015.15: 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonca LE, Pacis A et al. , BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell. 2018. 172: 176–190. [DOI] [PubMed] [Google Scholar]
- 23.Cirovic B, de Bree LCJ, Groh L, Blok BA, Chan J, van der Velden W et al. , BCG vaccination in humans elicits trained immunity via the hematopoietic progenitor compartment. Cell Host Microbe. 2020. 28: 322–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T et al. , Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell. 2018. 172: 147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berendsen MLT, Oland CB, Bles P, Jensen AKG, Kofoed PE, Whittle H et al. , Maternal priming: Bacillus Calmette-Guerin (BCG) vaccine scarring in mothers enhances the survival of their child with a BCG vaccine scar. J Pediatric Infect Dis Soc. 2020. 9: 166–172. [DOI] [PubMed] [Google Scholar]
- 26.Netea MG, Dominguez-Andres J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E et al. , Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020. 20: 375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis MM, Tato CM and Furman D, Systems immunology: just getting started. Nat. Immunol. 2017. 18: 725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knight JC, Genomic modulators of the immune response. Trends Genet. 2013. 29: 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howie B, Marchini J and Stephens M, Genotype imputation with thousands of genomes. G3 (Bethesda). 2011. 1: 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A et al. , Next-generation genotype imputation service and methods. Nat. Genet. 2016. 48: 1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bumgarner R, Overview of DNA microarrays: types, applications, and their future. Curr. Protoc. Mol. Biol. 2013. Chapter 22: Unit 22 1. 10.1002/0471142727.mb2201s101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Casey G, Conti D, Haile R, Duggan D, Next generation sequencing and a new era of medicine. Gut. 2013. 62: 920–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang J, Bakshi A, Zhu Z, Hemani G, Vinkhuyzen AA, Lee SH et al. , Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat. Genet. 2015. 47: 1114–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim-Hellmuth S, Bechheim M, Putz B, Mohammadi P, Nedelec Y, Giangreco N et al. , Genetic regulatory effects modified by immune activation contribute to autoimmune disease associations. Nat. Commun. 2017. 8: 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, Oosting M, Deelen P, Ricano-Ponce I, Smeekens S, Jaeger M et al. , Inter-individual variability and genetic influences on cytokine responses to bacteria and fungi. Nat. Med. 2016. 22: 952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. , PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007. 81: 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shabalin AA, Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012. 28: 1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yong WS, Hsu FM, Chen PY, Profiling genome-wide DNA methylation. Epigenetics Chromatin. 2016. 9: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmidt D, Wilson MD, Spyrou C, Brown GD, Hadfield J and Odom DT, ChIP-seq: using high-throughput sequencing to discover protein-DNA interactions. Methods. 2009. 48: 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z et al. , High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008. 132: 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buenrostro JD, Wu B, Chang HY and Greenleaf WJ, ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015. 109: 21.29.1–21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP et al. , Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015. 523: 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davis CA, Hitz BC, Sloan CA, Chan ET, Davidson JM, Gabdank I et al. , The encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018. 46: D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bujold D, Morais DAL, Gauthier C, Cote C, Caron M, Kwan T et al. , The International Human Epigenome Consortium Data portal. Cell Syst. 2016. 3: 496–499. [DOI] [PubMed] [Google Scholar]
- 45.Roadmap Epigenomics Consortium, Kundaje A., Meuleman W, Ernst J, Bilenky M, Yen A. et al. , Integrative analysis of 111 reference human epigenomes. Nature. 2015. 518: 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu CJ, Bonder MJ, Soderhall C, Bustamante M, Baiz N, Gehring U et al. , The emerging landscape of dynamic DNA methylation in early childhood. BMC Genomics. 2017. 18: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yan Q, Forno E, Herrera-Luis E, Pino-Yanes M, Qi C, Rios R et al. , A genome-wide association study of severe asthma exacerbations in Latino children and adolescents. Eur Respir J.2020. 10.1183/13993003.02693-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun S and Barreiro LB, The epigenetically-encoded memory of the innate immune system. Curr. Opin. Immunol. 2020. 65: 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pacis A, Mailhot-Leonard F, Tailleux L, Randolph HE, Yotova V, Dumaine A et al. , Gene activation precedes DNA demethylation in response to infection in human dendritic cells. Proc Natl Acad Sci USA. 2019. 116: 6938–6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love MI, Huber W and Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014. 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lowe R, Shirley N, Bleackley M, Dolan S and Shafee T, Transcriptomics technologies. PLoS Comput. Biol. 2017. 13: e1005457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Galli E, Friebel E, Ingelfinger F, Unger S, Nunez NG and Becher B, The end of omics? High dimensional single cell analysis in precision medicine. Eur. J. Immunol. 2019. 49: 212–220. [DOI] [PubMed] [Google Scholar]
- 53.Aguirre-Gamboa R,deKlein N,diTommaso J,Claringbould A,vander Wijst MG, de Vries D et al. , Deconvolution of bulk blood eQTL effects into immune cell subpopulations. BMC Bioinformatics. 2020. 21: 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stephenson E, Webb S and Haniffa M, Multiomics uncovers developing immunological lineages in human. Eur. J. Immunol. 2021. 51: 764–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stuart T and Satija R, Integrative single-cell analysis. Nat. Rev. Genet. 2019. 20: 257–272. [DOI] [PubMed] [Google Scholar]
- 56.Hu X, Hu Y, Wu F, Leung RWT and Qin J, Integration of single-cell multi-omics for gene regulatory network inference. Comput. Struct. Biotechnol. J. 2020. 18: 1925–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, et al. , Comprehensive integration of single-cell data. Cell. 2019. 177: 1888–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aebersold R and Mann M, Mass spectrometry-based proteomics. Nature. 2003. 422: 198–207. [DOI] [PubMed] [Google Scholar]
- 59.Stoevesandt O and Taussig MJ, Affinity proteomics: the role of specific binding reagents in human proteome analysis. Expert Rev. Proteomics. 2012. 9: 401–414. [DOI] [PubMed] [Google Scholar]
- 60.Dunn WB, Broadhurst DI, Atherton HJ, Goodacre R, and Griffin JL, Systems level studies of mammalian metabolomes: the roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem. Soc. Rev. 2011. 40: 387–426. [DOI] [PubMed] [Google Scholar]
- 61.Emwas AH, Roy R, McKay RT, Tenori L, Saccenti E, Gowda GAN et al. , NMR spectroscopy for metabolomics research. Metabolites. 2019. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arts RJ, Joosten LA and Netea MG, Immunometabolic circuits in trained immunity. Semin. Immunol. 2016. 28: 425–430. [DOI] [PubMed] [Google Scholar]
- 63.Vojinovic D, Radjabzadeh D, Kurilshikov A, Amin N, Wijmenga C, Franke L et al. , Relationship between gut microbiota and circulating metabolites in population-based cohorts. Nat. Commun. 2019. 10: 5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim MK and Lun DS, Methods for integration of transcriptomic data in genome-scale metabolic models. Comput. Struct. Biotechnol. J. 2014. 11: 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schirmer M, Smeekens SP, Vlamakis H, Jaeger M, Oosting M, Franzosa EA et al. , Linking the human gut microbiome to inflammatory cytokine production capacity. Cell. 2016. 167: 1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Forbes JD, Van Domselaar G and Bernstein CN, The gut microbiota in immune-mediated inflammatory diseases. Front Microbiol.2016.7: 1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson JS, Spakowicz DJ, Hong BY, Petersen LM, Demkowicz P, Chen L et al. , Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019. 10: 5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lindgreen S, Adair KL and Gardner PP, An evaluation of the accuracy and speed of metagenome analysis tools. Sci. Rep. 2016. 6: 19233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adan A, Alizada G, Kiraz Y, Baran Y and Nalbant A, Flow cytometry: basic principles and applications. Crit. Rev. Biotechnol. 2017. 37: 163–176. [DOI] [PubMed] [Google Scholar]
- 70.Spitzer MH and Nolan GP, Mass cytometry: single cells, many features. Cell. 2016. 165: 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bekkering S, Blok BA, Joosten LA, Riksen NP, van Crevel R and Netea MG, In vitro experimental model of trained innate immunity in human primary monocytes. Clin. Vaccine Immunol. 2016. 23: 926–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Joosten SA, van Meijgaarden KE, Arend SM, Prins C, Oftung F, Korsvold GE et al. , Mycobacterial growth inhibition is associated with trained innate immunity. J. Clin. Invest. 2018. 128: 1837–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Verrall AJ, Schneider M, Alisjahbana B, Apriani L, van Laarhoven A, Koeken V et al. ,Early clearance of Mycobacterium tuberculosis is associated with increased innate immune responses. J Infect Dis. 2019. 221 8:1342–1350. [DOI] [PubMed] [Google Scholar]
- 74.Roestenberg M, Hoogerwerf MA, Ferreira DM, Mordmuller B and Yazdanbakhsh M, Experimental infection of human volunteers. Lancet Infect. Dis. 2018. 18: e312–e322. [DOI] [PubMed] [Google Scholar]
- 75.Itenov TS, Murray DD and Jensen JUS, Sepsis: Personalized Medicine Utilizing ‘Omic’ Technologies-A Paradigm Shift? Healthcare (Basel). 2018. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Karakike E and Giamarellos-Bourboulis EJ, Macrophage activation-like syndrome: a distinct entity leading to early death in sepsis. Front Immunol. 2019. 10: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bakker OB, Aguirre-Gamboa R, Sanna S, Oosting M, Smeekens SP, Jaeger M et al. , Integration of multi-omics data and deep phenotyping enables prediction of cytokine responses. Nat. Immunol. 2018. 19: 776–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tsang JS, Schwartzberg PL, Kotliarov Y, Biancotto A, Xie Z, Germain RN et al. , Global analyses of human immune variation reveal baseline predictors of postvaccination responses. Cell. 2014. 157: 499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li Y, Oosting M, Smeekens SP, Jaeger M, Aguirre-Gamboa R, Le KTT et al. , A functional genomics approach to understand variation in cytokine production in humans. Cell. 2016. 167: 1099–1110. [DOI] [PubMed] [Google Scholar]
- 80.Orru V, Steri M, Sidore C, Marongiu M, Serra V, Olla S et al. , Complex genetic signatures in immune cells underlie autoimmunity and inform therapy. Nat. Genet. 2020. 52: 1036–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brodin P, Jojic V, Gao T, Bhattacharya S, Angel CJ, Furman D, Shen-Orr S et al. , Variation in the human immune system is largely driven by non-heritable influences. Cell. 2015. 160: 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aguirre-Gamboa R, Joosten I, Urbano PCM, van der Molen RG, van Rijssen E, van Cranenbroek B, Oosting M et al. , Differential effects of environmental and genetic factors on T and B cell immune traits. Cell Rep. 2016. 17: 2474–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Carithers LJ and Moore HM, The Genotype-Tissue Expression (GTEx) project. Biopreserv Biobank. 2015. 13: 307–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Netea MG, Joosten LA, Li Y, Kumar V, Oosting M, Smeekens S, Jaeger M et al. , Understanding human immune function using the resources from the Human Functional Genomics Project. Nat. Med. 2016. 22: 831–833. [DOI] [PubMed] [Google Scholar]
- 85.Zhu J, Zhang B, Smith EN, Drees B, Brem RB, Kruglyak L, Bumgarner RE et al. , Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat. Genet. 2008. 40: 854–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lerias JR, de Sousa E, Paraschoudi G, Martins J, Condeco C, Figueiredo N et al. , Trained immunity for personalized cancer immunotherapy: current knowledge and future opportunities. Front Microbiol. 2019. 10: 2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Priem B, van Leent MMT, Teunissen AJP, Sofias AM, Mourits VP, Willemsen L, Klein EM et al. , Trained immunity-promoting nanobiologic therapy suppresses tumor growth and potentiates checkpoint inhibition. Cell. 2020. 183: 786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mulder WJM, Ochando J, Joosten LAB, Fayad ZA and Netea MG, Therapeutic targeting of trained immunity. Nat Rev Drug Discov. 2019. 18: 553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Moorlag S, Rodriguez-Rosales YA, Gillard J, Fanucchi S, Theunissen K, Novakovic B, de Boent CM et al. , BCG vaccination induces long-term functional reprogramming of human neutrophils. Cell Rep. 2020. 33: 108387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.van der Maten E, de Jonge MI, de Groot R, van der Flier M and Langereis JD, A versatile assay to determine bacterial and host factors contributing to opsonophagocytotic killing in hirudin-anticoagulated whole blood. Sci. Rep. 2017. 7: 42137. [DOI] [PMC free article] [PubMed] [Google Scholar]