

Abstract
The COVID-19 pandemic continues to pose a substantial threat to human lives and is likely to do so for years to come. Despite the availability of vaccines, searching for efficient small-molecule drugs that are widely available, including in low- and middle-income countries, is an ongoing challenge. In this work, we report the results of an open science community effort, the “Billion molecules against COVID-19 challenge”, to identify small-molecule inhibitors against SARS-CoV-2 or relevant human receptors. Participating teams used a wide variety of computational methods to screen a minimum of 1 billion virtual molecules against 6 protein targets. Overall, 31 teams participated, and they suggested a total of 639,024 molecules, which were subsequently ranked to find ‘consensus compounds’. The organizing team coordinated with various contract research organizations (CROs) and collaborating institutions to synthesize and test 878 compounds for biological activity against proteases (Nsp5, Nsp3, TMPRSS2), nucleocapsid N, RdRP (only the Nsp12 domain), and (alpha) spike protein S. Overall, 27 compounds with weak inhibition/binding were experimentally identified by binding-, cleavage-, and/or viral suppression assays and are presented here. Open science approaches such as the one presented here contribute to the knowledge base of future drug discovery efforts in finding better SARS-CoV-2 treatments.
Keywords: COVID-19, drug discovery, machine learning, SARS-CoV-2
1 |. INTRODUCTION
There is great interest in small molecule therapeutic agents for COVID-19 with high efficacy to save human lives. Even more than three years after the outbreak of the pandemic and despite the availability of vaccines [1], COVID-19 poses a threat to individuals across the world [2]. The initially-developed vaccines and boosters have so far proven protective against COVID-19, but because of multiple factors, such as new variants of the virus [2], the disease continues to pose substantial risk to life and health. Recent studies also show that reinfections act cumulatively, which is worrisome in the long term [3]. Additionally, many people cannot be vaccinated due to their medical status or refuse vaccination, and breakthrough infections occur despite vaccination. Therefore, having a small molecule therapy as an additional option or alternative is highly demanded [4]. The applicability of currently available small molecule treatments, such as nirmaltrelvir [5], baricitinib [6], remdesivir [7], and molnupiravir [8] is still restricted. For instance, the application of Paxlovid (nirmatrelvir and ritonavir) is limited due to drug-drug interactions [9], drug resistance [10–12], and rebound effects [13, 14]. In addition, molnupiravir is a mutagenic antiviral, which possibly could increase the emergence of new variants [15, 16]. Ensitrelivir has recently been developed as a small molecule antiviral specifically targeting SARS-CoV-2 [17], and has been shown to decrease viral clearance by 50 h [18]. Overall, improved pharmacological approaches are still needed.
The standard drug development process is slow compared to the time scale at which the SARS-CoV-2 virus emerged and mutates, and could easily last up to 15 years [19]. This period comprises pre-clinical phases in which large numbers of virtual or physically available molecules are considered and tested, and then clinical phases in which few molecules are validated in human trials. In early phases of the drug discovery process, computational methods have been shown to help in screening and navigating through the large chemical space [20]. Such methods should also suggest new promising ligands [21–23]. However, 90% of the molecular candidates turn out to fail later, somewhere between phase I trials and regulatory approval [24]. Therefore, using accurate computational methods to screen and filter chemical space is key to a successful and fast drug development process. With accurate computational methods, the early phases of drug discovery that usually require 3–6 years [19], might be reduced to a few weeks, after which pre-clinical studies could start [25].
The RNA genome of SARS-CoV-2 encodes 29 structural, non-structural (Nsp) and accessory proteins, which are responsible for entry and uncoating, replication, and assembly [26]. The large, multidomain transmembrane papain-like protease (Nsp3 or PLpro), the main protease (Nsp5, 3CLpro or Mpro), the RNA-dependent RNA Polymerase (RdRP or Nsp12), the nucleocapsid (N), the spike protein (S), and the human host transmembrane protease (TMPRSS2), are frequently named as potential drug targets [27–34]. Due to the frequent mutations in the spike protein S, other proteins are deemed more suitable as drug targets. Since the outbreak of the COVID-19 pandemic, there has been a quest for selective, potent, and bioavailable inhibitors of the aforementioned proteins [35–37] using a multitude of approaches, such as high-throughput screening, virtual screening, and drug repurposing.
In response to the pandemic, scientists and research groups around the world started to self-organize and work together (e.g., https://covid19-nmr.de/participants/core-team/; https://insidecorona.net/; https://app.jogl.io/; https://foldingathome.org, https://news.cnrs.fr/articles/covid-19-15-billion-compounds-to-undergo-virtual-screening); MEDIATE [38], EXSCALATE [39]). The COVID moonshot project [40–42] for example, yielded new potential inhibitors with a collaborative, crowdsourcing Open Science Discovery approach [43], now continued within the Drugs For Neglected Diseases Initiative. Here we present the results of an ad hoc crowd-sourced community initiative, the “Billion molecules against COVID-19 Challenge”, which was organized as a competition (starting May 2020) to identify inhibitors of SARS-CoV-2 proteins. Participating teams screened at least one billion molecules each using diverse computational methods. Then, the most promising drug compounds were synthesized and evaluated in wet-lab experiments. We present the computational approaches taken, biological assays performed, and the overall lessons learned from the challenge.
2 |. RESULTS
2.1 |. Set up of the community challenge
Our community effort to identify SARS-CoV-2 inhibitors was organized as a challenge, where academic and industry researchers worldwide were asked to form teams to virtually screen at least a billion small molecules each and then submit 10,000 virtual molecules as potential inhibitors for SARS-CoV-2 progression, within the timeframe May-June 2020. In response to the announcement to join, 130 teams registered, of which 31 made the submission deadline. In addition to compound lists, teams had to deliver a report outlining the methods used (see Supporting Information Section 1). Of those, 20 teams were admitted after peer-review of their reports by an ad-hoc scientific committee.
Overall, a four-step process was used during the challenge (Figure 1). The aim put forward to the teams was to find a<100 nM binder to a SARS-CoV-2 protein or human receptor of choice, which should ideally have a 100-fold reduction of live SARS-CoV-2 viral replication in whole cell assays. The teams were initially free to identify the most promising protein targets. In terms of screening databases, Zinc 15 [44], CAS (anti-virals) [45], and SweetLead [46] were suggested by the organizing team but the computational teams were free to choose other sources. The following sections will describe the four processes in detail, followed by a discussion and conclusions.
FIGURE 1.
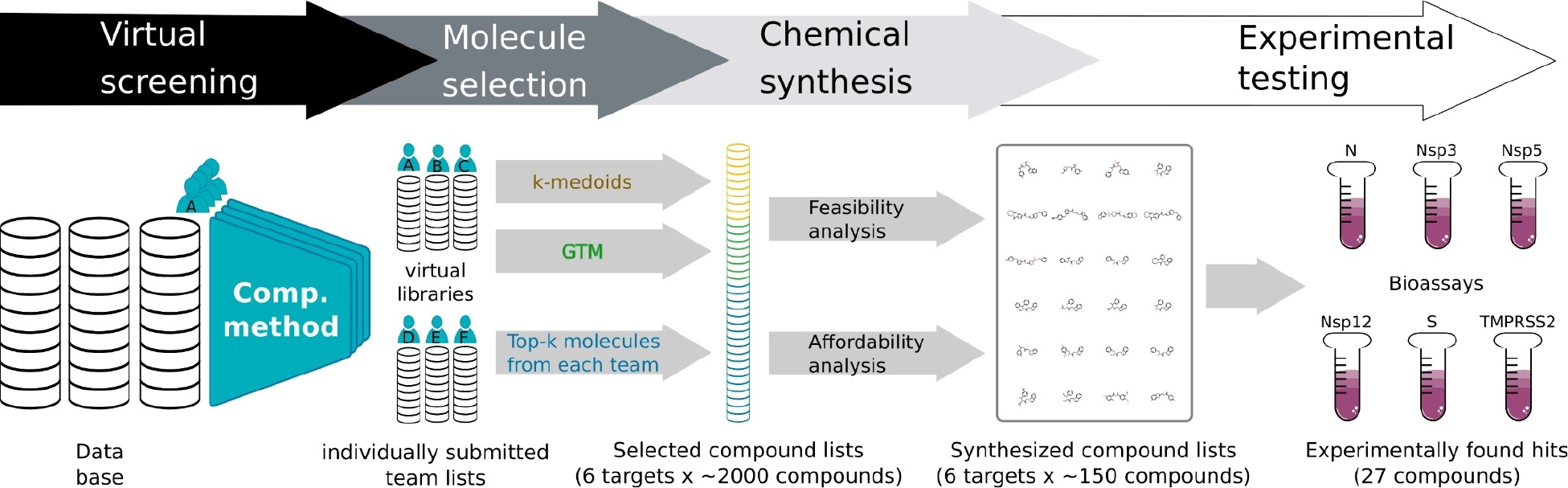
Overview of the main stages of the Billion Molecules Against COVID-19 Challenge.
2.2 |. Virtual screening using computational methods
The computational teams used a variety of machine learning [47], docking [48, 49] and hybrid approaches (Figure 2). In the group of machine learning based methods, approaches included: reinforcement learning, random forests [50], gradient boosting [51–53], kernel-based methods – e.g., Vanishing Ranking Kernels [54] – and deep learning methods – e.g., self-normalizing-networks [55], LSTMs [56], CNNs [57–61], geometric deep learning, and graph neural networks [62–64]. Also stochastic-based methods – e.g., Naive Bayes Classifier [65] and Self-Consistent Regression [66] – were used. The docking teams used different tools like GLIDE [67–69], AutoDock Vina [70, 71], QVINA2, VirtualFlow [72], Fred, Smina, Gold [73], PLANTS [74] and Data Warrior. Some teams considered molecular dynamics simulations [75]. Others combined machine learning with conventional docking approaches. This was done by a) building a pipeline in which different computational methods were stacked on top of each other – e.g., some groups used machine learning methods to make a pre-selection of the screened compounds and then used docking methods for the most promising compounds – or b) using machine learning models as a scoring function for the docking methods. Also similarity-based methods – e.g., classic similarity search [76], feature tree search [77], and the knn-algorithm [78] – and methods for dimensionality reduction – e.g., PCA, t-SNE, and GTM [79,80] – were used. Some teams added ADMET and PAINS filters to their virtual screening pipeline. On the ligand-side, multiple different molecular representations were used, e.g., SMILES, substructure-based descriptors (ECFPs), MACCs keys, continuous and data-driven molecular descriptors (CDDD) [81], MNA [82] and QNA [66] descriptors.
FIGURE 2.
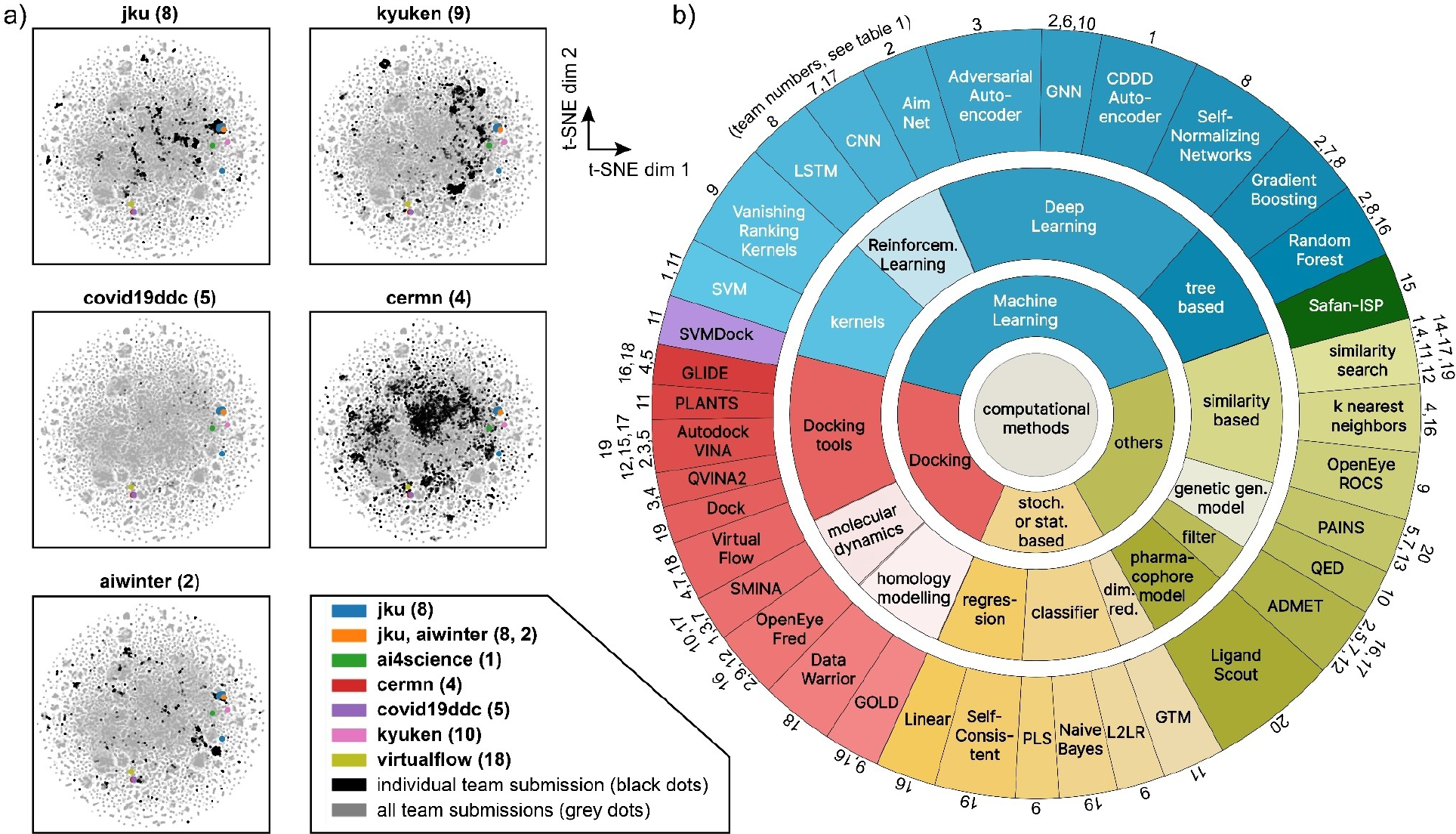
a) Scatterplots in t-SNE coordinates which show the Nsp5 experimental hits (colored dots) and the submitted compounds by the teams (black dots for single team in each panel, gray dots for all submitted compounds by all teams). For t-SNE plots for each individual team see Figure S9a,b and Supporting Information Section 3. b) Overview of computational methods used by the different teams. Numbers correspond to participating teams (see Table 1).
In terms of the hit rate, the team that ended up with the most compounds (team jku, see below) used descriptor-based deep learning methods with small molecules as inputs, thus a ligand-based approach. The self-normalizing network approach renders the models robust against domain shifts from training data to testing data. The second-ranked method by team kyuken used shallow, ligand-based, and descriptor-based machine learning methods as a first step and subsequently used structure-based approaches to refine the search. The hit compound of kyuken showed significant viral reduction in cell-based assays (see section 2.6.5 below). The third-ranked method (team aiwinter) used docking-based methods and QSAR models. For details, see Supporting Information Section 1.
2.3 |. Molecule selection and consensus ranking
A single list of molecules was made for subsequent synthesis and testing against each of the six selected SARS-CoV-2 (or host) protein targets. In total, 639,024 molecules (of which 423,466 unique ones) were submitted across all targets and teams. Many teams suggested identical compounds for the same protein target: 656 for Nsp5, 155 for Nsp3, 57 for TMPRSS2 and 54 for Nsp12.
Interestingly, 7391 compounds were suggested by multiple teams for multiple protein targets, but in 3843 cases the teams disagreed on what the target was. Also, several teams had the same identical compound on their compound list for the same target, but those duplicates were removed.
The screening capacity was estimated to be maximally 2,000 compounds for each of the 6 protein targets, considering the time and cost to synthesize compounds and perform experimental assays. ~40% of this screening capacity was reserved for testing the top-ranked molecules from each team, i.e., according to the ranking the team had determined for their own lists. The other ~60% of the screening capacity was reserved for testing consensus molecules, which are molecules that had been suggested by multiple teams or for which very similar molecules had been suggested. Two different approaches were employed to determine the set of consensus molecules: a) k-medoids clustering, and b) generative topographic mapping [79], see Supporting Information Section 2. The ‘selected molecules list’ for each of the 6 protein targets, ended up consisting of 38% top-ranked, 15% from k-medoids, and 47% from GTM (see yellow/green/blue cartoon in Figure 1). Overall, six sets of compounds for each of the protein targets were obtained amounting to 11,440 unique compounds in total.
2.4 |. Chemical synthesis of selected compounds
All compounds were synthesized by WuXi Apptec (China), based on instructions from the organizing team. 11,440 compound suggestions across 6 protein targets were provided to them. The compounds to be synthesized were selected based on 3 criteria by WuXi Apptec using proprietary methods: 1) cADME (computational absorption, distribution, metabolism, and excretion) filtering was done to arrive at compounds with molecular weight (MW) below 500 g mol−1, CLogP<5, HBA<10, HBD<5, TPSA<140, Rotatable bond<5. In addition, possible PAINS (Pan-assay interference compounds) were removed; 2) Chemical feasibility: a similarity search versus the WuXi Apptec virtual library was performed to assess feasibility (see Supporting Information Section 4.1); 3) reagent availability and cost were considered.
After the selection, 1414 compounds were selected, and synthesis was started. The synthesis period lasted from November 2020 to February 2021, and 878 compounds were delivered as 20 mM DMSO (dimethylsulfoxide) stock solution on well-plates. It was not feasible to synthesize all compounds due to delays in the delivery of starting compounds or due to practical synthetic issues (e.g., low reactivity, difficulties in purification, etc.). The compound purity was determined by LC–MS and has been reported previously [83]. Of all 878 compounds, 58 (i.e., 6.6%) had a purity below 90%, but were included in experimental assays nonetheless. The latter data set also includes information on solubility and compound chirality. Duplicate compound well-plates with DMSO stock solutions were shipped to the MIT-Broad institute (USA), Crelux GmbH (Germany), Pasteur Institute (France), and the Diamond Light Source (UK), for further experiments (see next sections).
Biases in compound selection and synthesis.
Both the methods used to obtain the list of selected compounds (from 423,466 unique ones to 12081 selected, see section 2.3) and the synthesizability of the compounds (878, see section 2.4) introduced biases. Table 1 shows that team imolecule, lci, lci, virtualflow, molecule, and cermn had the largest numbers of compounds selected for the targets N, Nsp3, Nsp5, Nsp12, S and TMPRSS2, respectively (see bold numbers). Figure S5 displays these results by the method of selection, i.e., either by GTM, k-medoids, or top-ranked. Some teams had most of their selected compounds originate from consensus selection. For example lci, cermn, kyuken, and pharmai had many compounds selected by GTM (Figure S5a,b). In contrast, other teams (e.g., covid19ddc and sarswars) had most of their selected compounds directly from their top-ranked ones. Overall, the selected compound list and the synthesized compound lists are skewed toward the top 200 positions of each team for each protein target (Figure S6). For jku, a bias was found in the number of synthesized compounds (62) versus those selected (259) likely due their chemical similarity and the fact that they can be easily synthesized (see ‘benzotriazolyl acetamide’ family in the next sections and in discussion section 3 below). Some teams had large numbers of molecules selected in the first step but none were finally synthesized. For example team belarus had 32 compounds for Nsp5 and 67 for S, but none of them were selected by WuXi Apptec since these compounds did not pass their ADME filters and/or cost/feasibility analysis. If the filtering would have been known a priori, the teams could have likely had more suitable compounds in their submitted lists thus avoiding the fact that some teams ended up with zero compounds. We could not discern a clear trend in the origin of selection of the compounds (i.e., GTM, k-medoids, or top-ranked) versus what was synthesized by WuXi Apptec in the end (Figure S7), but the percentage of GTM compounds did increase ~10% in favor of top-ranked compounds (Figure S8). We do not deem this significant, that is, the selection method did not influence which compounds were eliminated by the WuXi Apptec filtering.
TABLE 1.
Overview of selected and synthesized molecules across teams (rows) and drug targets (columns). Molecules which were selected or tested for a specific target but submitted for another target do not contribute to the team counts. For statistics which also includes molecules which were originally submitted for a different target, see Supporting Information Section 3.
| Selected molecules (section 2.3) |
Synthesized compounds (section 2.4) |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Nsp3 | Nsp5 | Nsp12 | S | TMPRSS2 | SUM | N | Nsp3 | Nsp5 | Nsp12 | S | TMPRSS2 | SUM | |
| ai4science (1) | - | - | - | - | - | 499 | 499 | - | - | - | - | - | 16 | 16 |
| aiwinter (2) | - | 64 | 88 | - | - | 63 | 215 | - | 12 | 8 | - | - | 0 | 20 |
| belarus (3) | - | - | 32 | - | 67 | - | 99 | - | - | 0 | - | 0 | - | 0 |
| cermn (4) | - | 73 | 80 | - | - | 621 | 774 | - | 7 | 3 | - | - | 49 | 59 |
| covid19ddc (5) | - | 79 | 55 | 82 | - | - | 216 | - | 6 | 5 | 10 | - | - | 21 |
| deeplab (6) | - | 60 | 69 | - | 160 | - | 289 | - | 8 | 7 | - | 11 | - | 26 |
| imolecule (7) | 1013 | 81 | 66 | 358 | 402 | 0 | 1920 | 73 | 4 | 6 | 42 | 22 | 0 | 147 |
| jku (8) | - | 86 | 259 | 57 | - | - | 402 | - | 0 | 62 | 5 | - | - | 67 |
| kyuken (9) | - | 81 | 52 | - | 424 | - | 557 | - | 15 | 0 | - | 41 | - | 56 |
| lambdazero (10) | - | - | 32 | - | - | - | 32 | - | - | 0 | - | - | - | 0 |
| lci (11) | - | 1150 | 700 | 60 | - | - | 1910 | - | 86 | 54 | 5 | - | - | 145 |
| luxscreen (12) | - | - | 73 | 323 | - | 255 | 651 | - | - | 2 | 14 | - | 5 | 21 |
| nuwave (13) | 0 | 39 | 24 | - | - | - | 63 | 0 | 0 | 0 | - | - | - | 0 |
| pharmai (14) | - | - | 42 | - | 288 | - | 330 | - | - | 0 | - | 35 | - | 35 |
| safan (15) | - | 63 | 80 | 205 | - | - | 348 | - | 2 | 17 | 15 | - | - | 34 |
| sarstroopers (16) | - | 56 | 48 | 211 | 108 | - | 423 | - | 5 | 0 | 19 | 1 | - | 25 |
| sarswars (17) | 472 | - | 85 | 298 | - | - | 855 | 8 | - | 2 | 9 | - | - | 19 |
| virtualflow (18) | 547 | 46 | 107 | 369 | 219 | 463 | 1751 | 46 | 7 | 2 | 44 | 24 | 43 | 166 |
| way2drug (19) | - | 71 | 53 | 55 | - | 97 | 276 | - | 13 | 2 | 0 | - | 21 | 36 |
| yoda (20) | - | 69 | 90 | - | 337 | - | 496 | - | 3 | 11 | - | 22 | - | 36 |
| SUM | 2032 | 2018 | 2035 | 2018 | 2005 | 1998 | 12106 | 127 | 168 | 181 | 163 | 156 | 134 | 929 * |
of 929 total team-selected compounds, there are 878 unique chemical compounds. That is, 51 identical compounds were suggested. A dash indicates the team did not submit compounds for that specific target.
2.5 |. Comparison of computational methods
Hit rate.
With the four-stage procedure described above (see Section 2.1), 27 compounds were found to have detectable biological activity (see Figure 2, Figure 3, 4, Table 2, and details in Table 3) across all SARS-CoV-2 protein targets. The experimental testing is described in the following paragraphs. Due to the multiple team submissions and the compound selection procedure some teams submitted compounds which were tested on a target which is different to the suggested one. We tackle this issue by providing a) an analysis for which these compounds are excluded (Table 1 and Table 2) and b) an analysis for which these compounds are included (Supporting Information Section 3). For a) 14 hits had been suggested by the team jku and bind to Nsp5 (see Table 2). This amounts to a hit rate of 20.9% [95% confidence interval: 11.9–32.6%] (14 actives of 67 tested) of the best team, which is followed by the teams kyuken with a hit rate of 7.1% [2.0–17.3%] (4 actives out of 56 tested) and aiwinter with a hit rate of 5.0% [0.1–24.9%] (1 active out of 20 tested). Note that three different types of assays, a) in vitro (cell-free or live cell) activity, b) biophysical binding and c) x-ray crystallography, have been used to experimentally test the compounds (see Section 2.6).
FIGURE 3.
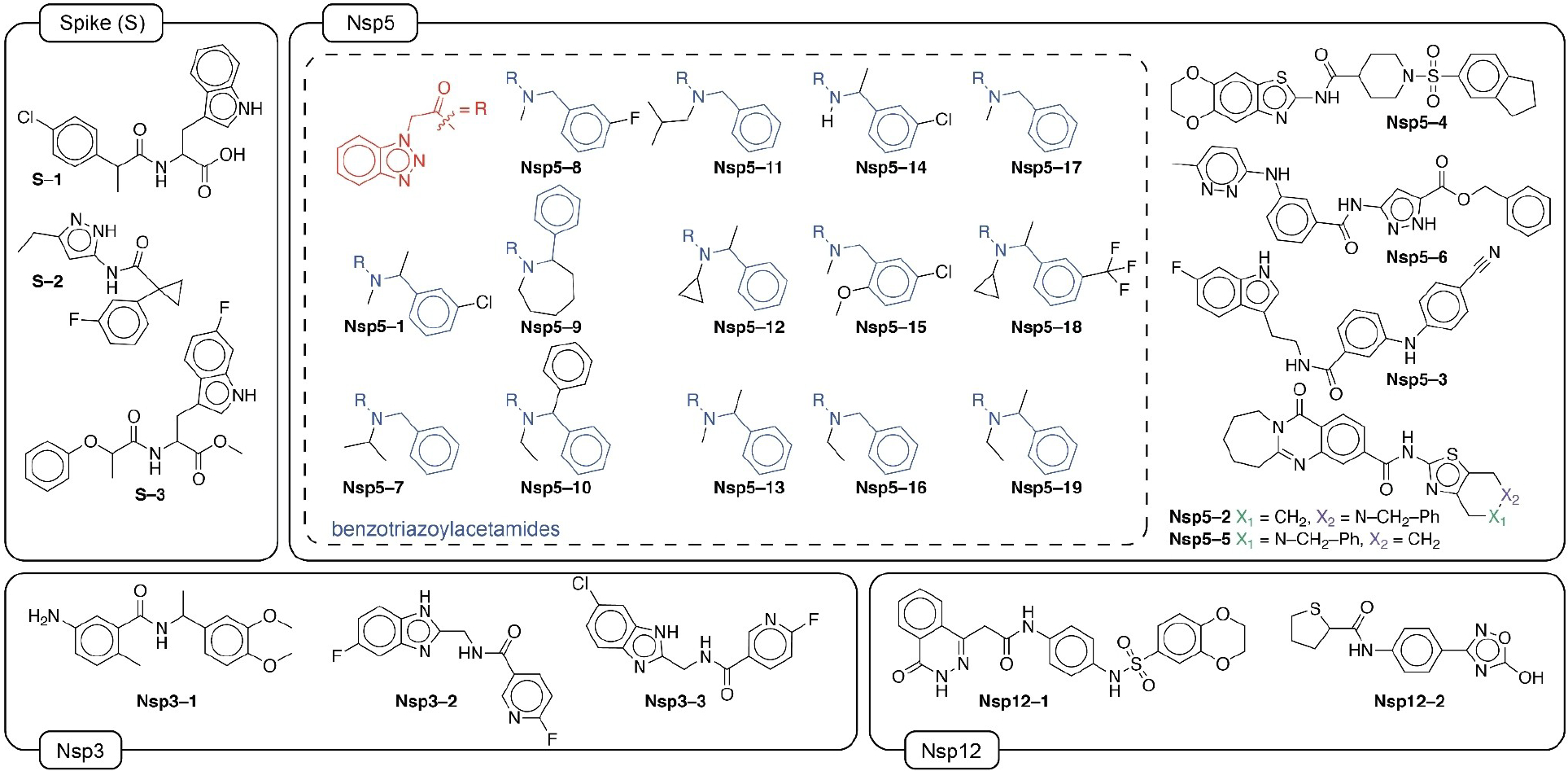
Chemical structures of 27 hit compounds that bind to one of the protein targets or have biological activity. Molecules are grouped with respect to the experimental protein target they were found to have activity, which is not always the one that was initially predicted by the teams. The benzotriazolyl acetamide family (14 compounds) of Nsp5 is shown in the dashed box.
FIGURE 4.
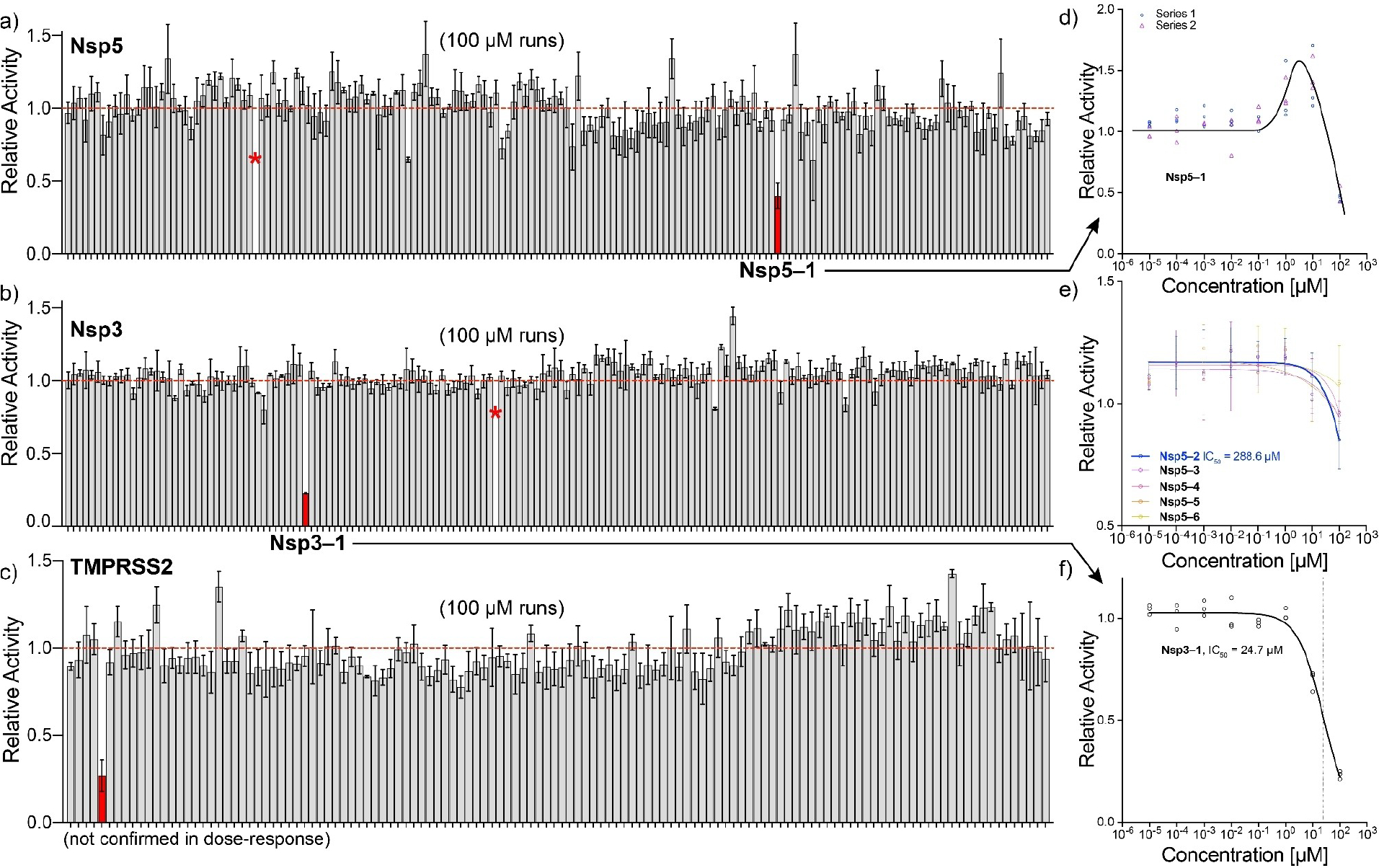
Overview of protease cleavage assays. a–c) relative activity over triplicate experiments at a fixed compound concentration of 100 μM for Nsp5, Nsp3 and TMPRSS2, respectively. Red bars show compounds that reduce cleavage (relative) activity by more than 50%. Asterisks show highly fluorescent compounds that could not be analyzed. Not all compound labels are listed for clarity. d–f) dose-response curves at different compound concentrations. Solid lines in panel e–f show fits, panel d to guide the eye.
TABLE 2.
Number of active compounds, i. e. hits, confirmed with in-vitro testing and hit-rates (ratio of active against tested compounds). The best hit-rate is marked bold. The number of tested compounds is taken from Table 1. Analogous to Table 1, hit counts just include compounds which were submitted for the tested target. For hit counts irrespective of the predicted target, see Supporting Information Section 3. Only teams with non-zero hits are listed in this table.
| Hits | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N | Nsp3 | Nsp5 | Nsp12 | S | TMPRSS2 | hits | tested | Hit rate [a] % | 95% conf-int | |
| jku (8) | - | - | 14 | 0 | - | - | 14 | 67 | 20.9 | [11.9–32.6] |
| kyuken (9) | - | 2 | 0 | - | 2 | - | 4 | 56 | 7.1 | [2.0–17.3] |
| aiwinter (2) | - | 0 | 1 | - | - | - | 1 | 20 | 5.0 | [0.1–24.9] |
| covid19ddc (5) | - | 0 | 0 | 1 | - | - | 1 | 21 | 4.8 | [0.1–23.8] |
| deeplab (6) | - | 0 | 0 | - | 1 | - | 1 | 26 | 3.8 | [0.1–19.6] |
| way2drug (19) | - | 1 | 0 | - | - | 0 | 1 | 36 | 2.8 | [0.1–14.5] |
| imolecule (7) | 0 | 0 | 0 | 1 | 0 | - | 1 | 147 | 0.7 | [0.0–3.7] |
| All teams | 0 | 3 | 14 [b] | 2 | 3 | 0 | 22 [b] | 878 | 2.5 | [1.6–3.8] |
This is the hit rate from the pooled analysis described in this paper. Some teams performed their own analysis with different results (see Supporting Information Section 3).
One hit was found by two teams.
TABLE 3.
Overview of active compounds and their labels used in the manuscript.
| Label | Teams | Predicted target | Exp. target | KD [μM] | Protease activity? | IC50 [μM] Nsp5 | X-ray structure | Viral reduction? |
|---|---|---|---|---|---|---|---|---|
| S-1 | kyuken | Ace2/S PPI | S | > 15 | - | no | ||
| S-2 | deeplab | S | S | > 172 | - | no | ||
| S-3 | kyuken | Ace2/S PPI | S | > 30 | - | no** | ||
| Nsp5–1 | jku | Nsp5 | Nsp5 | - | yes | 642±204 | yes | no** |
| Nsp5–2 | covid19ddc | Nsp12 | Nsp5 | - | yes | 40±7 | - | minor |
| Nsp5–3 | kyuken | Ace2/S PPI | Nsp5 | - | yes | 37±6 | - | Yes (IC50 9.41 μM; CC50=(19.16 μM) |
| Nsp5–4 | virtualflow | Nsp12, TMPRSS2 | Nsp5 | - | yes | 145±25 | - | minor |
| Nsp5–5 | cermn | TMPRSS2 | Nsp5 | - | yes | 59±10 | - | minor |
| Nsp5–6## | ai4science | TMPRSS2 | Nsp5 | - | yes | 68±11 | - | no |
| Nsp5–7 | jku | Nsp5 | Nsp5 | - | no | - | yes | - |
| Nsp5–8 | jku | Nsp5 | Nsp5 | - | no | - | yes | - |
| Nsp5–9 | jku | Nsp5 | Nsp5 | - | no | - | yes | - |
| Nsp5–10 | jku | Nsp5 | Nsp5 | - | no | - | yes | - |
| Nsp5–11 | jku | Nsp5 | Nsp5 | - | no | - | yes | - |
| Nsp5–12## | jku | Nsp5 | Nsp5 | - | no | - | yes | - |
| Nsp5–13## | jku | Nsp5 | Nsp5 | - | no | - | yes | - |
| Nsp5–14 | jku, aiwinter | Nsp5 | Nsp5 | - | no | - | yes | - |
| Nsp5–15 | jku | Nsp5 | Nsp5 | - | no | - | yes | - |
| Nsp5–16## | jku | Nsp5 | Nsp5 | - | no | - | yes | - |
| Nsp5–17 | jku | Nsp5 | Nsp5 | - | no | - | yes | - |
| Nsp5–18 | jku | Nsp5 | Nsp5 | - | no | - | yes | - |
| Nsp5–19 | jku | Nsp5 | Nsp5 | - | no | - | yes | - |
| Nsp3–1 | way2drug | Nsp3 | Nsp3 | 26.7# | yes | - | - | no |
| Nsp3–2 | kyuken | Nsp3 | Nsp3* | - | no | - | yes | no** |
| Nsp3–3 | kyuken | Nsp3 | Nsp3* | - | no | - | yes | - |
| Nsp12–1## | covid19ddc | Nsp12 | Nsp12 | > 39 | - | - | - | |
| Nsp12–2 | iMolecule | Nsp12 | Nsp12 | > 200 | - | - | - | |
| See section | 2.6.3/1 | 2.6.1 | 2.6.2 | 2.6.4 | 2.6.5 |
- shows that the measurement or analysis was not performed; Nsp3
indicates the Nsp3 macrodomain.
indicates an increase in viral infection in whole cell live virus assays.
X-ray structures are available via: https://fragalysis.diamond.ac.uk/or can be downloaded directly from https://github.com/hermanslab/COVID-19. Compounds are > 95% pure by HPLC analysis (see Github), except labeled compounds
that are > 90% pure.
Novelty of hits.
To evaluate the novelty of the found hits, the hit compounds are compared to prior-art molecules, which are molecules either used in filtering operations such as similarity searches or used as an active training instance for Machine Learning methods by any of the teams. The activity cut-offs for the metrics pKi, pKd, pIC50 and pChEMBL were set to 6.3. Scatterplots in t-SNE coordinates (Figure 2a and Figure S9a,b) show the relative location of the hit compounds in comparison to the prior-art compounds. Notably, compared to Nsp12 and S, Nsp3 and Nsp5 contain many prior-art molecules, due to the availability of SARS-CoV data that was assumed by the teams to be similar (in terms of binding sites) as compared to SARS-CoV-2. The hits identified by jku (14 compounds) and aiwinter (1 compound) build a cluster and overlap in the Nsp5 scatterplot. Looking in more detail we find many benzotriazolyl acetamide derivatives in the prior art data in this cluster (Figure S10). The benzotriazole family had been considered indeed for SARS-CoV in 2008 by Verschueren [84], with published protein databank structures. For secondary clusters of hits (e.g., cermn & virtualflow; lower left quadrant of Nsp5 scatter plot in Figure S10), we could not identify similar functional groups or motifs in the proximal prior art compounds. The S hits (kyuken and deeplab) and Nsp12 hit compound (imolecule) do not reside in the neighborhood of prior art compounds which is why they can be considered as highly novel (Figure S10). For targets other than Nsp5, too few hits were found to draw statistically relevant conclusions on cluster size or novelty.
2.6 |. Experimental testing of candidates
The synthesized (878) compounds were tested for their inhibitory activity or binding activity to SARS-CoV-2 targets using various assays and X-Ray crystallography. Protease cleavage assays (Nsp5, TMPRSS2, Nsp3) have been performed by the MIT-Broad Foundry to determine activity. Microscale thermophoresis (MST) assays for RdRp (Nsp12 domain), N, and S proteins have been done by Nanotemper GmbH. Details on the assays can be found in Supporting Information Section 4. In this section, we detail salient experimental issues that were encountered in the assay development, as many (especially the binding assays using MST) were not yet available or described in the literature. Initially, compound sets were only tested versus their virtually predicted protein target, but having an available chemical library, some assays were performed for all compounds (irrespective of the predicted target).
2.6.1 |. Protease cleavage assays
Protease cleavage tests were done for the compound sets of Nsp5, Nsp3, and TMPRSS2. In the assay, a peptide FRET (Förster resonance energy transfer) substrate is cleaved by the protease, which results in an increase of fluorescence intensity. The increase in fluorescence intensity over time is proportional to the rate constant of the protease, and by adding compounds at different concentrations, inhibitors can be identified. As positive controls, GC376 (IC50=9.4 ± 2.5 nM) and GRL0617 (IC50=2.8 ± 0.4 μM) were used for Nsp5 and Nsp3, respectively [85, 86] (see Supporting Information Section 4). A first brute-force screening at 100 μM showed a single compound for each of the three proteases (see red bars in Figure 4a–c). Those compounds were selected for dose-response curves, where their concentration was changed to calculate IC50 values (see Supporting Information Section 4). Nsp5-1 produced an atypical dose-response, where activity was first enhanced by ~50% and then dropped to<50% at 100 μM concentration (Figure 4d), which hampered the calculation of the IC50. Nsp3-1 showed a classical inhibition with IC50=24.7 ± 3.7 μM (Figure 4f). In addition, from cell-based Nsp5 assays (see section 2.6.2 below), 5 additional compounds were identified that did not make the<50% inhibition threshold, but were measured in dose-response using the same cleavage assay (Figure 4e). These measurements identified the IC50 of Nsp5-2 ~288 μM, whereas the remaining compounds Nsp5-3 to Nsp5-6 had much higher IC50’s that could not be determined.
2.6.2 |. Nsp5 protease cleavage assays in cells
The Pasteur Institute in Paris had previously set up a cell-based Nsp5 protease assay [87], in which cleavage of a reporter Rev-Nluc protein by Nsp5 decreases the luminescence signal. In the presence of an inhibitor, the luminescence signal is restored (see Supporting Information Section 4.3).
Here we show the data in terms of %restored activity, where no inhibition is 0% and full inhibition is 100%. GC376 was used as a control inhibitor and yielded an IC50=4.2 ± 1.0 μM. Out of all 878 compounds screened, 6 compounds had activity in the high micromolar range, while Nsp5-3 was the best inhibitor, albeit a weak one with IC50=37 ± 6 μM (see Figure 5). Interestingly, the same compound had given negligible activity in the (cell free) Nsp5 cleavage assays (see Figure 4e, purple line).
FIGURE 5.
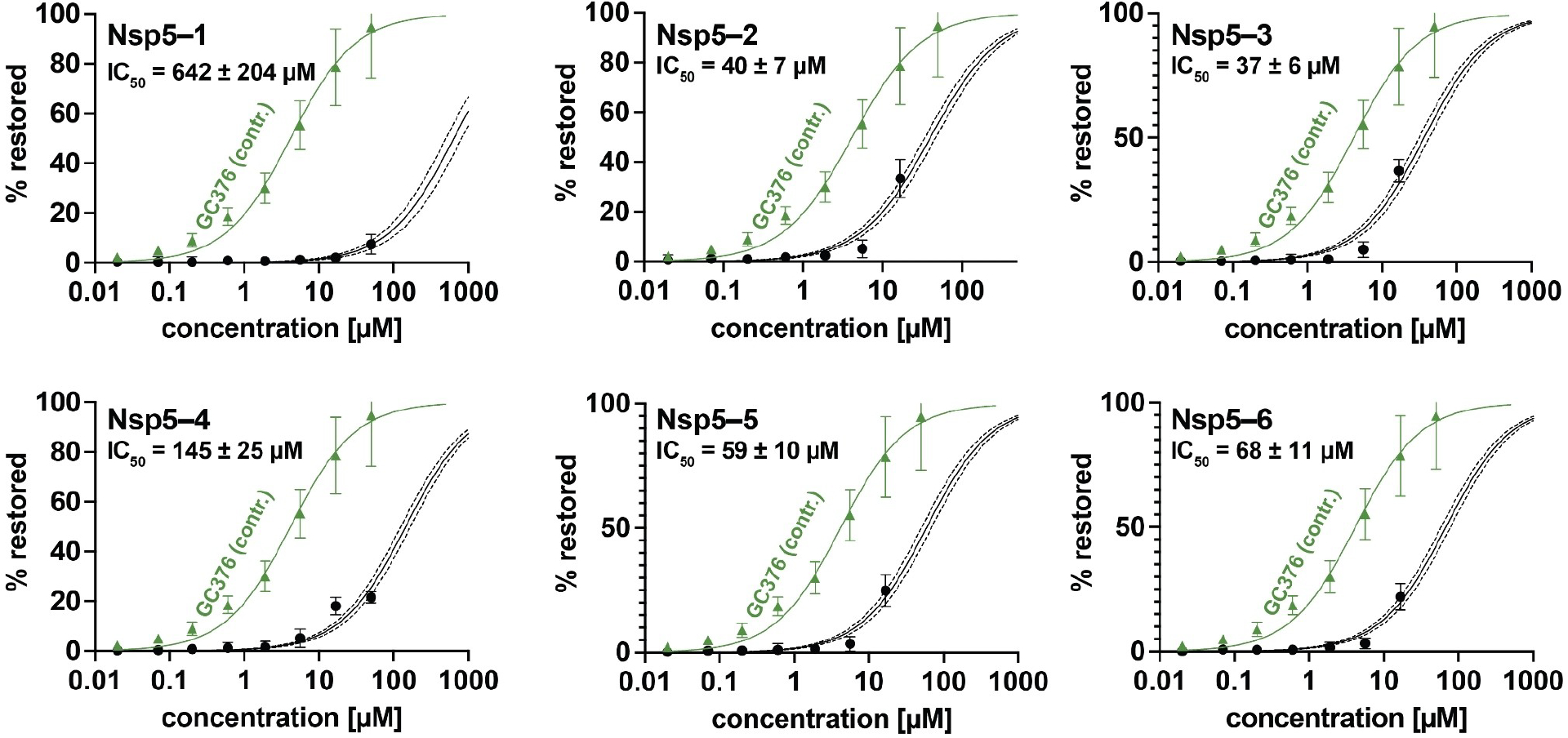
Dose-response curves of compounds in cell-based Nsp5 protease assay. IC50 values are also in Table 3 below. Solid line: curve fit result. Dashed lines: 95% confidence interval. Data are expressed as the mean ± standard deviation of 3 independent experiments each performed in triplicate. Green triangles show positive controls for inhibitor GC376 (see Supporting Information section 4.3.2). Cytotoxicity was detected above 20 μM, so higher concentrations were excluded.
2.6.3 |. Binding assays to N, RdRp (Nsp12 domain), S
Microscale thermophoresis emerged as a high-throughput label-free method to evaluate binding constants and is extensively used in the pharmaceutical industry and in CROs [88, 89]. Therefore, this method was used for the three protein targets without protease activity, i.e., S, RdRp (Nsp12 domain), and N. Various constructs of whole-length or subdomains of the targets are available from commercial sources. In this section, we will describe the assay development, the choice of positive controls (that are absolutely needed for MST), and binding outcomes.
For S, it was decided to use the stabilized trimer (R683 A, R685 A, K986P, V987P), since participating teams had also modeled trimer-specific or cryptic binding sites, other than the (classical) RBD domain. As a positive control, the natural choice was the Ace2 (human receptor) protein. Surprisingly, recombinantly expressed Ace2 did not show binding to S (stabilized trimer), we suspect due to improper folding of the construct. Fortunately, His-tagged Ace2 did provide good binding curves with a KD of 4.25 ± 1.52 nM (over 6 runs performed during the 3 days of assay measurements, see Supporting Information Section 4.2.3). This is stronger binding than previous measurements performed by Surface Plasmon Resonance [90] that showed 94.6 ± 6.5 nM for (monomeric) SARS-CoV-2-S1, but can be explained by multivalency of the trimer as shown by Kruse et al. [91]. All 152 compounds were first analyzed using 8-point dilution series between 50 nM and 100 μM concentrations, revealing 7 compounds to be potential binders. The latter 7 were measured in triplicate 12-point dilutions from 0.2 nM to 200 μM, and 3 compounds were identified as high micromolar binders: S-1, S-2 and S-3 (see Figure 6 and Table 3 above).
FIGURE 6.
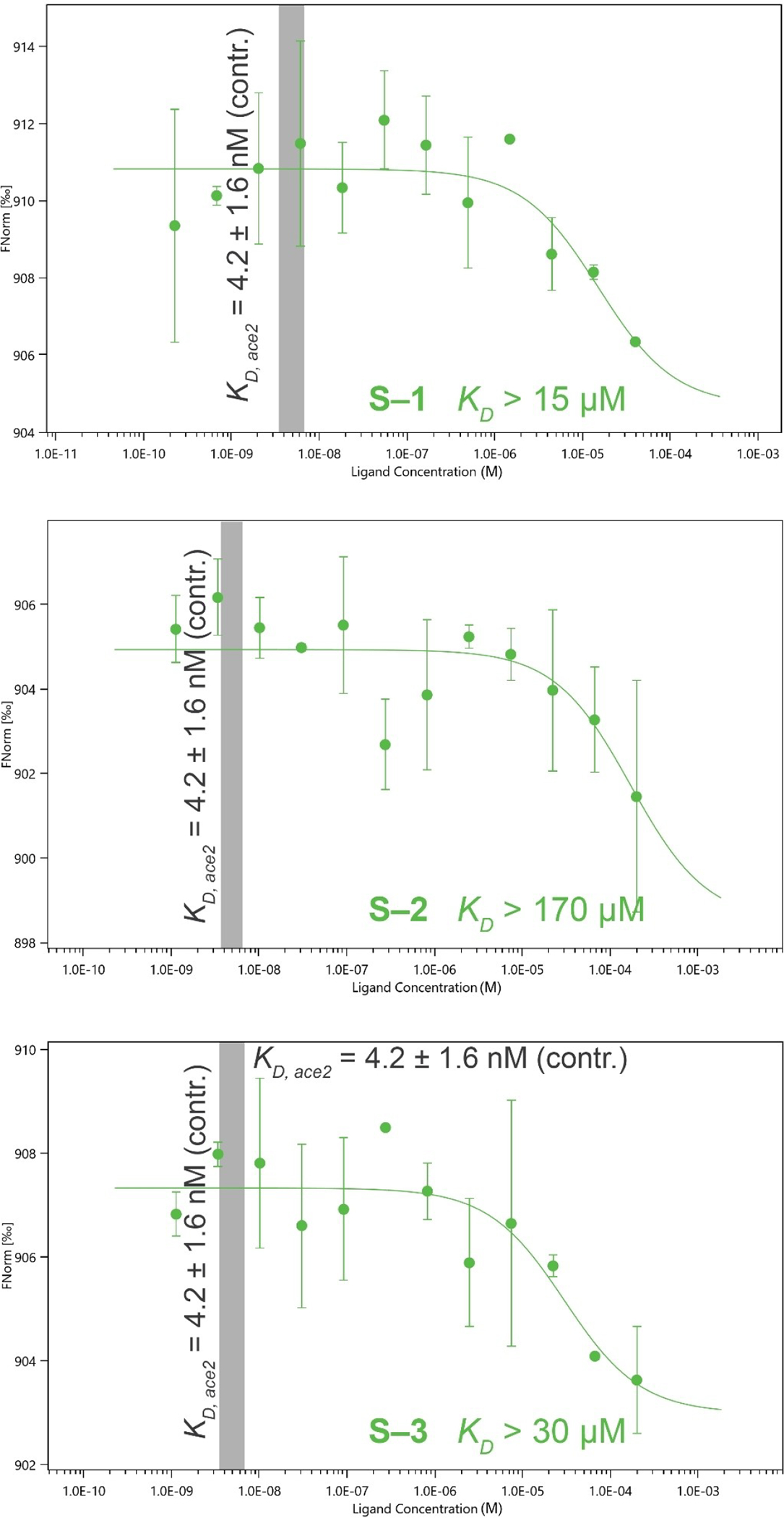
Binding curves of S compounds using Microscale thermophoresis performed in triplicate. Error bars show standard deviations. The gray region shows the KD for positive control Ace2. See Supporting Information Section 4.2 for details on assay conditions.
For RdRp, we were unable to obtain the stable trimeric complex of Nsp7/8/12 (see Supporting Information Section 4.2.1), and therefore we used only the Nsp12 subdomain. As a first control, we tried Remdesivir metabolite GS-443902, but could not detect binding. This is because the latter compound inserts itself into the RNA chain during polymerization, and therefore inhibits RdRp function, but it does not bind efficiently to Nsp12. Instead, Suramin was used as a control with a determined KD=827 ± 306 nM (over 4 triplicate measurements). Dilution (8-point) series from 0.5 nM to 250 μM were performed on 147 predicted compounds, and after pre-selection of 8 compounds and further triplicate 12-point experiments, 2 high-micromolar binders were identified: Nsp12-1 and Nsp12-2 (see Table 3 below). Three additional compounds led to Nsp12 aggregation, so no KD could be determined (Nc1nnnn1-c1cccc(c1)C(=O)NCc1cc(F)ccc1Oc1ccc(F)c-c(Cl)c1, Cc1ccc(NC(=O)c2ccc(nc2O)C2CC2)c(O)c1, and FC(F)(F)c1ccc2nnc(CNC(=O)c3ccc4 C(=O)N5CCC-CCC5=Nc4c3)n2c1).
For N, we used full-length nucleocapsid (see Supporting Information Section 4.2.2), and used nanobodies developed to bind to the N- and C-terminal domains. A total of 119 compounds were analyzed in 8-point and 12-point dilution assays between 45 nM and 100 μM. However, it was found that N would show a drop in normalized fluorescence intensity Fnorm upon the addition of 1–5% of DMSO (dimethylsulfoxide, see also Figure S12), likely due to slow polymerization and sedimentation of N over time. This made it impossible to determine KD values, and the assay development had to be abandoned.
2.6.4 |. X-ray structures
In collaboration with the Diamond light source (DLS), crystallization and X-ray diffraction experiments were carried out on Nsp5 and Nsp3 compounds. For Nsp5, 148 compounds were soaked at 2 mM and measured by synchrotron X-ray diffraction, which identified 14 potential hits all from the benzotriazolyl acetamide family: Nsp5-1 and Nsp5-7 to Nsp5-19. Comparison to the DLS database (accessible via https://fragalysis.diamond.ac.uk/viewer/react/preview/target/Mpro, use tag ‘JEDI - Benzotriazole’) showed that several other benzotriazoles had previously been identified for Nsp5. Some representative structures are shown in Figure 7 below. For Nsp3 we found two compounds that could be resolved (also shown in Figure 7).
FIGURE 7.
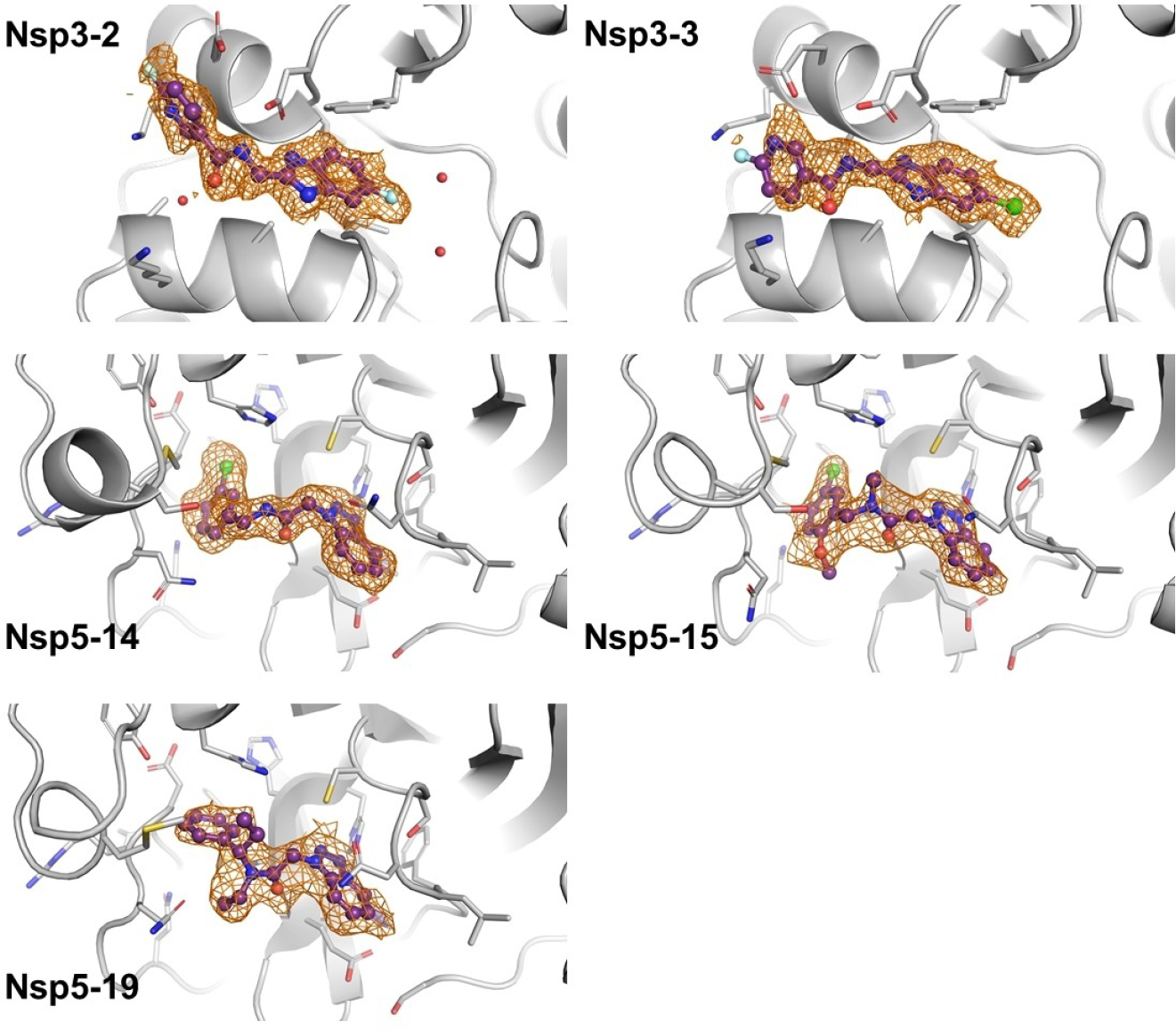
Crystal structures with examples of the Nsp5 benzotriazolyl acetamide family and Nsp3 (macrodomain) binders. The compounds are shown with purple sticks and balls and the PanDDA event map is shown as an orange mesh. PDB files can be downloaded from https://github.com/hermanslab/COVID-19.
2.6.5 |. Viral reduction assays
For a selection of compounds we performed whole-cell live-virus reduction assays using either Vero-TMPRSS2 or HeLa-ACE2 cells (see Supporting Information Section 4.3.3). In Figure 8 below, the dose-response curves of % infection and cell viability are shown. Remdesivir was used as a positive control with an IC50=347 nM (95% confidence interval CI is 161–533 nM), which is in agreement with previous reports [92]. Most of the compounds show no significant reduction of viral replication in this assay. Nsp5-3 gave significant viral reduction with IC50=9.41 μM (95% confidence interval is 5.32–19.27), but had cytotoxicity CC50=19.16 μM (95% CI is 7.191–70.01), and we cannot exclude that the latter is responsible for the viral replication reduction.
FIGURE 8.
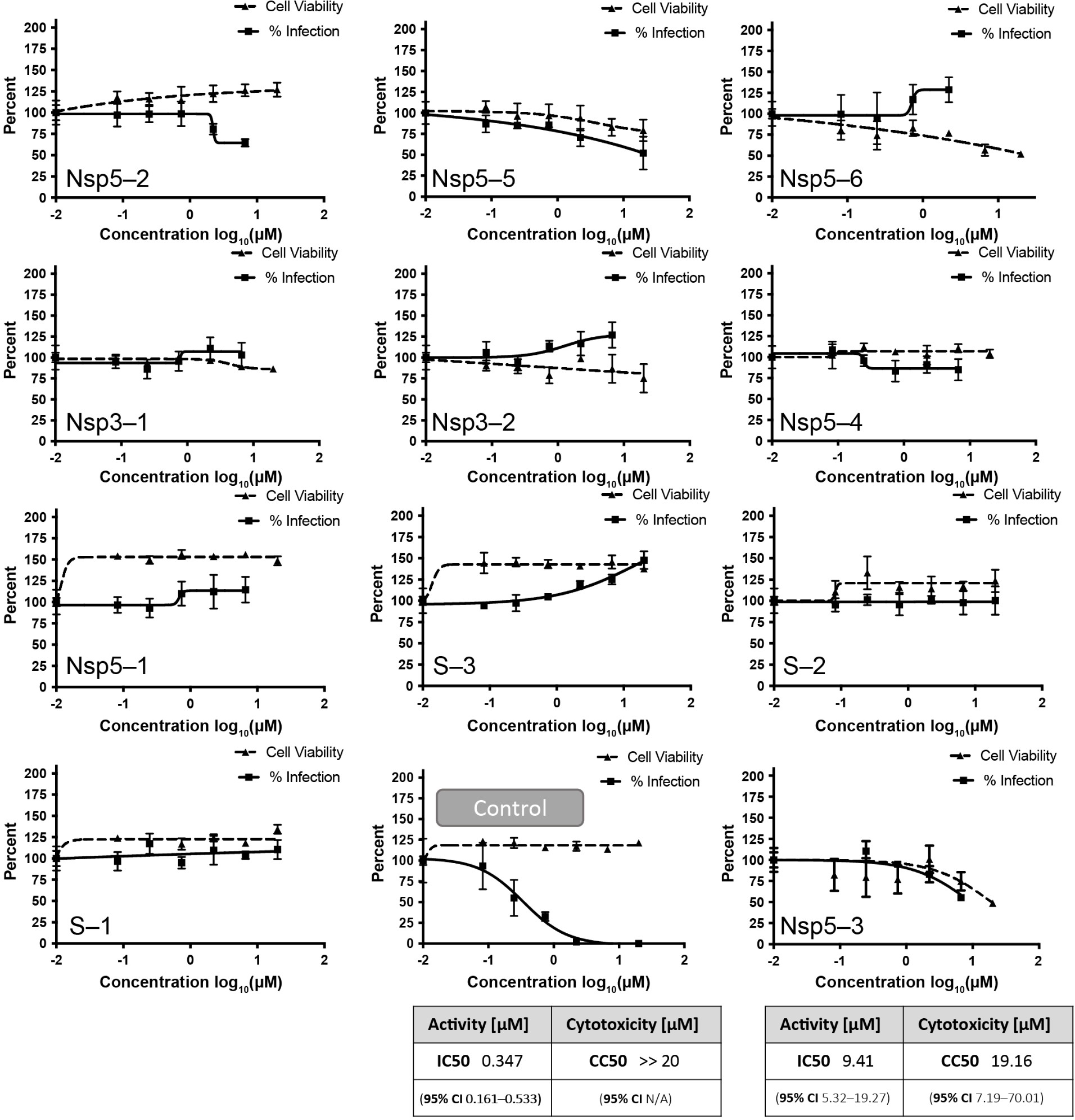
Viral reduction assays of compounds found by the teams compared to Remdesivir as the control. Error bars show standard deviations over triplicate measurements. An IC50 value could only be determined for Nsp5–3.
We have summarized the experimental findings of the previous sections in Table 3 (above). We found 6 compounds that had a quantifiable binding interaction S(3), Nsp3(1), Nsp12(2), of which only the compound for Nsp3-1 showed in vitro (cell-free) protease cleavage activity. The latter compound shows structural similarity to previously found SARS PLpro inhibitors derived from GRL-0617 [95, 96]. In live cell Nsp5 assays, 6 compounds showed weak inhibition, with the best one Nsp5-3 with IC50=37 ± 6 μM. The same compound also showed viral reduction in whole-cell live-virus reduction assays, with an IC50=9.41 μM (95% CI is 5.32–19.27), but we cannot exclude that inhibition is a side-effect of cytotoxicity. Further studies will be needed to chemically improve Nsp5-3 to increase anti-viral activity.
3 |. DISCUSSION
The COVID-19 pandemic has given an unprecedented push to scientists in academia and industry to try their hand at drug discovery. We have seen this during our “Billion molecules against COVID-19 challenge”, where even private individuals initially participated (but did not pass our internal peer-review at the report submission stage). Some novice teams were allowed to continue and submitted their compound lists, but not taking into account synthetic feasibility or ADME caused them to not have physical compounds made. We realized during the challenge that mistakes can easily be made when starting from questionable quality 3D protein structures from the Protein Databank (PDB). Fortunately, we had help from Insidecorona.net to point the teams to the best quality PDB entries for the protein targets the teams were working on. Since the challenge was organized as a winner-takes-all competition, the initial communication and sharing of results among teams was limited. The organizing team (coordinated by the last author) arranged the synthesis of compounds and all experimental studies. In hindsight, it would have been better to have a fully open communication with the teams immediately after the compound list submissions (July 2020). This would have further strengthened collaboration between protein crystallographers, computational scientists, and experimentalists. Overall, the challenge enhanced bridging of research fields, and accelerated communication (versus communication via peer-reviewed publications more traditionally).
In addition, the teams were free to choose the protein target they deemed most promising, and 6 final targets were selected by the organizing team. The experimental studies needed to validate each compound therefore took considerable effort, funding, and time (~2 years). An iterative approach on fewer targets would have likely been better and faster. With the experimental protocols in place, subsequent rounds of predicted compounds could likely be screened in<3 months, and could have served as input for additional computational rounds. Screening a library of off-the-shelf compounds, or even-better, known drugs [97] would also have accelerated things (on-demand synthesis is not as fast and costs significantly more; new molecules will require going through all clinical phases).
The computational teams chose approaches from a vast variety of different methods (see Figure 2) and therefore considered diverse orthogonal approaches. However, from today’s perspective few- (and zero-) shot methods, developed more recently, would have been an intuitive fit [98–105].
An important aspect of this challenge was its emphasis on the exploration of billions of candidate compounds for activity against the target proteins. This deviates from a more common strategy of focusing on either known drugs (e.g. DrugBank [106], DrugCentral [107]) or bio-like molecules (e.g. ChEMBL [108], SWEETLEAD [46], GEOM [109]) in that it explores a massive space of synthesizable molecules that may bear little recognized similarity to known bioactive compounds. While known drugs carry the benefit of faster path to clinical distribution, and bio-like molecules are generally perceived as being more likely to successfully translate to clinical relevance, there is reason to expect that exploration of a much larger set of candidates may yield drugs that are unlike others identified previously. For example, Lyu et al. [110] observe that billion-scale libraries are dramatically diminished for bio-like molecules relative to more focused libraries, yet still contain many experimentally-confirmed actives, as well as thousands of high-ranking molecules in docking assays. This observation justifies continued emphasis on development of methods for computationally screening billion-scale libraries. We also note that de novo generation of candidate molecules may offer a viable path to discovery.
Whereas consensus scoring has long been established in docking methods [111], extending it to other computational methods had not previously been considered until the current work. The discovered compounds have weak micromolar affinities, thus requiring further hit-to-lead development. Overall, the most potent compound Nsp5-3 found has an IC50=9.41 μM (95% CI is 5.32–19.27) in live cell assays, but with significant cytotoxicity that would need to be further addressed. The most prominent family was the benzotriazolyl acetamide family (Figure 3, Nsp5 dashed box), which has been found in other studies [112, 113] likely because several teams used ML methods starting from similar training sets, combined with the fact that benzotriazoles in general can easily be synthesized using ‘click chemistry’ [114], which is high-yielding and fast, and thus preferred by the CRO that performed the chemical synthesis. In addition, the CRO performed a proprietary synthetic feasibility and ADME screening that introduced a bias in the number of compounds that were eventually synthesized for each individual team.
In addition to the evaluation in this paper, some teams independently validated their predictions (see Supporting Information Section 3). Pharm.ai compared their top 100 predictions for Nsp5 against public data published after the competition deadline and obtained a hit rate of 17% on a highly diverse set of scaffolds. An interaction-based drug discovery screen explains known SARS-CoV-2 inhibitors and predicts new compound scaffolds [115]. The sarstrooper team experimentally tested top-ranked compounds they had submitted and found 7 compounds with IC50<10 μM (Mukherjee et al., in preparation).
Overall, we are convinced that an open communication (Open access/Open data/Open source [37]) is of the greatest importance, as previously advocated [40–42, 116]. For example, leads from the COVID Moonshot have recently been advanced by others to find a broad-spectrum nM inhibitor for SARS-CoV-2 [113]. The latter study [113], and the recent success story of Ensitrelvir (Xocava) from ultra-large computational approaches demonstrate the soundness of the approach [17]. To further accelerate the response to future pandemics, large and chemical diverse government-managed compound libraries should be readily available (such as the “Chimiothèque Nationale” [117] containing 80000 compounds and 15000 natural extracts), EU-OPENSCREEN’s unique compound collections containing over 96000 compounds [118], NCATS library containing over 10000 compounds including about 3000 drugs [119], to provide the first experimental activity/structural data, immediately and publicly shared, needed for computational researchers as a starting point.
4 |. CONCLUSIONS
Using a crowd-sourced approach, we performed the hit-finding stage of (anti-viral) drug discovery using a wide range of computational approaches that were bundled using a consensus approach. Many participating teams chose docking- or machine learning-based computational methods, for which little data was available at the start of the project (May 2020). The communication between different fields, e.g. protein crystallization, computational methods, and wet-lab experiments, was suboptimal and should be improved by direct communication and collaboration (vs. ‘communication via the scientific literature’). This would ensure that critical know-how that is easily overlooked (or not explicitly written down) in papers is efficiently transferred. Overall, the pandemic has accelerated the breaking down of silos [120] between research fields, but more is needed to act quicker to respond to future pandemics [121].
Supplementary Material
ACKNOWLEDGMENTS
Thomas Hermans led the organizing team and thanks the AXA Research Fund for financial support that was used for compound synthesis, binding studies, and protease cleavage assays that made this project possible. Please see detailed acknowledgements per author here: https://github.com/hermanslab/COVID-19/tree/main/AuthorContributions_Acknowledgements.
Funding information
AXA Research Fund
Footnotes
CONFLICT OF INTEREST STATEMENT
Nick Antonopoulos, Agamemnon Krasoulis, Vassilis Pitsikalis and Stavros Theodorakis (all members of the deeplab team) have filed non-provisional patent application PCT/EP2021/084447 in the name of Deeplab IKE relating to machine learning for efficient protein-ligand virtual screening.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The originally submitted team reports and compound lists, raw data, and scripts used to analyze the data in this manuscript are freely available: https://github.com/hermanslab/COVID-19
REFERENCES
- 1.Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, Perez JL, Pérez Marc G, Moreira ED, Zerbini C, Bailey R, Swanson KA, Roychoudhury S, Koury K, Li P, Kalina WV, Cooper D, Frenck RW, Hammitt LL, Türeci Ö, Nell H, Schaefer A, Ünal S, Tresnan DB, Mather S, Dormitzer PR, Şahin U, Jansen KU, Gruber WC, Engl N J. Med. 2020, 383, 2603–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Araf Y, Akter F, Tang Y, Fatemi R, Parvez Md. S. A., Zheng C, Hossain Md. G., J. Med. Virol. 2022, 94, 1825–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bowe B, Xie Y, Al-Aly Z, Nat. Med. 2022, 28, 2398–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Owen DR, Allerton CMN, Anderson AS, Aschenbrenner L, Avery M, Berritt S, Boras B, Cardin RD, Carlo A, Coffman KJ, Dantonio A, Di L, Eng H, Ferre R, Gajiwala KS, Gibson SA, Greasley SE, Hurst BL, Kadar EP, Kalgutkar AS, Lee JC, Lee J, Liu W, Mason SW, Noell S, Novak JJ, Obach RS, Ogilvie K, Patel NC, Pettersson M, Rai DK, Reese MR, Sammons MF, Sathish JG, Singh RSP, Steppan CM, Stewart AE, Tuttle JB, Updyke L, Verhoest PR, Wei L, Yang Q, Zhu Y, Y, Science 2021, 374, 1586–1593. [DOI] [PubMed] [Google Scholar]
- 5.Hammond J, Leister-Tebbe H, Gardner A, Abreu P, Bao W, Wisemandle W, Baniecki M, Hendrick VM, Damle B, Simón-Campos A, Pypstra R, Rusnak JM, Engl N J. Med. 2022, 386, 1397–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jorgensen SCJ, Tse CLY, Burry L, Dresser LD, Pharmacother. J. Hum. Pharmacol. Drug Ther. 2020, 40, 843–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grundeis F, Ansems K, Dahms K, Thieme V, Metzendorf MI, Skoetz N, Benstoem C, Mikolajewska A, Griesel M, Fichtner F, Stegemann M, Cochrane Database Syst. Rev. 2023, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jayk Bernal A, Gomes da Silva MM, Musungaie DB, Kovalchuk E, Gonzalez A, Delos Reyes V, Martín-Quirós A, Caraco Y, Williams-Diaz A, Brown ML, Du J, Pedley A, Assaid C, Strizki J, Grobler JA, Shamsuddin HH, Tipping R, Wan H, Paschke A, Butterton JR, Johnson MG, De Anda C, Engl N J. Med. 2022, 386, 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marzolini C, Kuritzkes DR, Marra F, Boyle A, Gibbons S, Flexner C, Pozniak A, Boffito M, Waters L, Burger D, Back DJ, Khoo S, Clin. Pharmacol. Ther. 2022, 112, 1191–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu Y, Lewandowski EM, Tan H, Zhang X, Morgan RT, Zhang X, Jacobs LMC, Butler SG, Gongora MV, Choy J, Deng X, Chen Y, Wang J, ACS Cent. Sci. 2023, 9, 1658–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ou J, Lewandowski EM, Hu Y, Lipinski AA, Aljasser A, Colon-Ascanio M, Morgan RT, Jacobs LMC, Zhang X, Bikowitz MJ, Langlais PR, Tan H, Wang J, Chen Y, Choy JS, PLoS Pathog. 2023, 19, e1011592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ip JD, Chu AW-H, Chan W-M, Leung RC-Y, Abdullah SMU, Sun Y, To KK-W, EBioMedicine 2023, 91. [Google Scholar]
- 13.Wang L, Volkow ND, Davis PB, Berger NA, Kaelber DC, Xu R, MedRxiv 2022, 2022.08.04.22278450. [Google Scholar]
- 14.Wang Y, Chen X, Xiao W, Zhao D, Feng L, Infect J 2022, 85, e134–e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nelson CW, Otto SP, Virological 2021, https://virological.org/t/mutagenic-antivirals-the-evolutionary-risk-of-low-doses/768
- 16.Swanstrom R, Schinazi RF, Science 2022, 375, 497–498. [DOI] [PubMed] [Google Scholar]
- 17.Unoh Y, Uehara S, Nakahara K, Nobori H, Yamatsu Y, Yamamoto S, Maruyama Y, Taoda Y, Kasamatsu K, Suto T, Kouki K, Nakahashi A, Kawashima S, Sanaki T, Toba S, Uemura K, Mizutare T, Ando S, Sasaki M, Orba Y, Sawa H, Sato A, Sato T, Kato T, Tachibana Y, J. Med. Chem. 2022, 65, 6499–6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mukae H, Yotsuyanagi H, Ohmagari N, Doi Y, Imamura T, Sonoyama T, Fukuhara T, Ichihashi G, Sanaki T, Baba K, Takeda Y, Tsuge Y, Uehara T, Antimicrob. Agents Chemother. 2022, 66, e00697–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes J, Rees S, Kalindjian S, Philpott K, Br. J. Pharmacol. 2011, 162, 1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reymond JL, Acc. Chem. Res. 2015, 48, 722–730. [DOI] [PubMed] [Google Scholar]
- 21.Sliwoski G, Kothiwale S, Meiler J, Lowe EW, Pharmacol. Rev. 2014, 66, 334–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneider P, Walters WP, Plowright AT, Sieroka N, Listgarten J, Goodnow RA, Fisher J, Jansen JM, Duca JS, Rush TS, Zentgraf M, Hill JE, Krutoholow E, Kohler M, Blaney J, Funatsu K, Luebkemann C, Schneider G, Nat. Rev. Drug Discovery 2020, 19, 353–364. [DOI] [PubMed] [Google Scholar]
- 23.Liu X, IJzerman AP, van GJP Westen in Artificial Neural Networks, 1st ed., (Ed.: Cartwright H), Springer US, New York, NY, 2021, pp. 139–165. [Google Scholar]
- 24.Fleming N, Nature 2018, 557, 55–57. [DOI] [PubMed] [Google Scholar]
- 25.Zhavoronkov A, Ivanenkov YA, Aliper A, Veselov MS, Aladinskiy VA, Aladinskaya AV, Terentiev VA, Polykovskiy DA, Kuznetsov MD, Asadulaev A, Volkov Y, Zholus A, Shayakhmetov RR, Zhebrak A, Minaeva LI, Zagribelnyy BA, Lee LH, Soll R, Madge D, Xing L, Guo T, Aspuru-Guzik A, Nat. Biotechnol. 2019, 37, 1038–1040. [DOI] [PubMed] [Google Scholar]
- 26.Yan W, Zheng Y, Zeng X, He B, Cheng W, Signal Transduct. Target. Ther. 2022, 7, 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang H, Xie W, Xue X, Yang K, Ma J, Liang W, Zhao Q, Zhou Z, Pei D, Ziebuhr J, Hilgenfeld R, Yuen KY, Wong L, Gao G, Chen S, Chen Z, Ma D, Bartlam M, Rao Z, PLoS Biol. 2005, 3, 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ullrich S, Nitsche C, Bioorg. Med. Chem. Lett. 2020, 30, 127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bai Z, Cao Y, Liu W, Li J, J. Viruses 2021, 13, 1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li K, Meyerholz DK, Bartlett JA, McCray PB, mBio 2021, 12, e00970–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan H, Hu Y, Jadhav P, Tan B, Wang J, J. Med. Chem. 2022, 65, 7561–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li G, Hilgenfeld R, Whitley R, De Clercq E, Nat. Rev. Drug Discovery 2023, 22, 449–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan B, Joyce R, Tan H, Hu Y, Wang J, Acc. Chem. Res. 2023, 56, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.von Delft A, Hall MD, Kwong AD, Purcell LA, Saikatendu KS, Schmitz U, Tallarico JA, Lee AA, Nat. Rev. Drug Discovery 2023, 22, 585–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKee DL, Sternberg A, Stange U, Laufer S, Naujokat C, Pharmacol. Res. 2020, 157, 104859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han Y, Duan X, Yang L, Nilsson-Payant BE, Wang P, Duan F, Tang X, Yaron TM, Zhang T, Uhl S, Bram Y, Richardson C, Zhu J, Zhao Z, Redmond D, Houghton S, Nguyen D-HT, Xu D, Wang X, Jessurun J, Borczuk A, Huang Y, Johnson JL, Liu Y, Xiang J, Wang H, Cantley LC, tenOever BR, Ho DD, Pan FC, Evans T, Chen HJ, Schwartz RE, Chen S, Nature 2021, 589, 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muratov EN, Amaro R, Andrade CH, Brown N, Ekins S, Fourches D, Isayev O, Kozakov D, Medina-Franco JL, Merz KM, Oprea TI, Poroikov V, Schneider G, Todd MH, Varnek A, Winkler DA, Zakharov AV, Cherkasov A, Tropsha A, Chem. Soc. Rev. 2021, 50, 9121–9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vistoli G, Manelfi C, Talarico C, Fava A, Warshel A, Tetko IV, Apostolov R, Ye Y, Latini C, Ficarelli F, Palermo G, Gadioli D, Vitali E, Varriale G, Pisapia V, Scaturro M, Coletti S, Gregori D, Gruffat D, Leija E, Hessenauer S, Delbianco A, Allegretti M, Beccari AR, Expert Opin. Drug Discovery 2023, 18, 821–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gadioli D, Vitali E, Ficarelli F, Latini C, Manelfi C, Talarico C, Silvano C, Cavazzoni C, Palermo G, Beccari AR, IEEE Trans. Emerg. Top. Comput. 2022, 11, 170–181. [Google Scholar]
- 40.Chodera J, Lee AA, London N, von Delft F, Nat. Chem. 2020, 12, 581–581. [DOI] [PubMed] [Google Scholar]
- 41.The COVID Moonshot Consortium, Chodera J, Lee A, London N, von Delft F, ChemRxiv. 2021. [Google Scholar]
- 42.von Delft F, Calmiano M, Chodera J, Griffen E, Lee A, London N, Matviuk T, Perry B, Robinson M, von Delft A, Nature 2021, 594, 330–332. [DOI] [PubMed] [Google Scholar]
- 43.Achdout H, Aimon A, Bar-David E, Morris GM, BioRxiv 2020. [Google Scholar]
- 44.Sterling T, Irwin JJ, J. Chem. Inf. Model. 2015, 55, 2324–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tomeo T, CAS Releases Open Access Dataset of Antiviral Chemical Compounds to Aid COVID-19 Discovery and Analysis | CAS, https://www.cas.org/resources/press-releases/open-access-covid-19-dataset.
- 46.Novick PA, Ortiz OF, Poelman J, Abdulhay AY, Pande VS, PLoS One 2013, 8, e79568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mayr A, Klambauer G, Unterthiner T, Hochreiter S, Front. Environ. Sci. 2016, 3. [Google Scholar]
- 48.Brooijmans N, Kuntz ID, Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 335–373. [DOI] [PubMed] [Google Scholar]
- 49.Brooks BR, Brooks III CL, Mackerell AD Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M, J. Comput. Chem. 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Breiman L, Mach. Learn. 2001, 45, 5–32. [Google Scholar]
- 51.Freund Y, Schapire RE, J. Comput. Syst. Sci. 1997, 55, 119–139. [Google Scholar]
- 52.Friedman J, Hastie T, Tibshirani R, Ann. Stat. 2000, 28, 337–407. [Google Scholar]
- 53.Friedman JH, Ann. Stat. 2001, 29, 1189–1232. [Google Scholar]
- 54.Berenger F, Yamanishi Y, J. Chem. Inf. Model. 2020, 60, 4376–4387. [DOI] [PubMed] [Google Scholar]
- 55.Klambauer G, Unterthiner T, Mayr A, Hochreiter S, in Advances in Neural Information Processing Systems, Vol. 30, 2017. [Google Scholar]
- 56.Hochreiter S, Schmidhuber J, Neural Comput. 1997, 9, 1735–1780. [DOI] [PubMed] [Google Scholar]
- 57.Fukushima K, Miyake S in Competition and Cooperation in Neural Nets, 1st ed (Eds.: Amari S, Arbib MA), Springer, Berlin, Heidelberg, 1982, pp. 267–285. [Google Scholar]
- 58.Lecun Y, Bottou L, Bengio Y, Haffner P, Proc. IEEE 1998, 86, 2278–2324. [Google Scholar]
- 59.LeCun Y, Bengio Y, Hinton G, Nature 2015, 521, 436–444. [DOI] [PubMed] [Google Scholar]
- 60.Schmidhuber J, Neural Netw. 2015, 61, 85–117. [DOI] [PubMed] [Google Scholar]
- 61.Bronstein MM, Bruna J, LeCun Y, Szlam A, Vandergheynst P, IEEE Signal Process. Mag. 2017, 34, 18–42. [Google Scholar]
- 62.Zhou J, Cui G, Hu S, Zhang Z, Yang C, Liu Z, Wang L, Li C, Sun M, AI Open 2020, 1, 57–81. [Google Scholar]
- 63.Wu Z, Pan S, Chen F, Long G, Zhang C, Yu PS, IEEE Trans. Neural Netw. Learn. Syst. 2021, 32, 4–24. [DOI] [PubMed] [Google Scholar]
- 64.Filimonov DA, Lagunin AA, Gloriozova TA, Rudik AV, Druzhilovskii DS, Pogodin PV, Poroikov VV, Chem. Heterocycl. Compd. 2014, 50, 444–457. [Google Scholar]
- 65.Zakharov AV, Lagunin AA, Filimonov DA, Poroikov VV, Chem. Res. Toxicol. 2012, 25, 2378–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS, J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- 67.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL, J. Med. Chem. 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- 68.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT, J. Med. Chem. 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- 69.Trott O, Olson AJ, J. Comput. Chem. 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eberhardt J, Santos-Martins D, Tillack AF, J. Chem. Inf. Model. 2021, 61, 3891–3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gorgulla C, Boeszoermenyi A, Wang Z-F, Fischer PD, Coote PW, Padmanabha Das KM, Malets YS, Radchenko DS, Moroz YS, Scott DA, Fackeldey K, Hoffmann M, Iavniuk I, Wagner G, Arthanari H, Nature 2020, 580, 663–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD, Proteins Struct. Funct. Bioinf. 2003, 52, 609–623. [DOI] [PubMed] [Google Scholar]
- 73.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD, Proteins Struct. Funct. Bioinf. 2003, 52, 609–623. [DOI] [PubMed] [Google Scholar]
- 74.Dror RO, Dirks RM, Grossman JP, Xu H, Shaw DE, Annu. Rev. Biophys. 2012, 41, 429–452. [DOI] [PubMed] [Google Scholar]
- 75.Cereto-Massagué A, Ojeda MJ, Valls C, Mulero M, Garcia-Vallvé S, Pujadas G, Methods 2015, 71, 58–63. [DOI] [PubMed] [Google Scholar]
- 76.Lessel U, Wellenzohn B, Lilienthal M, Claussen H, J. Chem. Inf. Model. 2009, 49, 270–279. [DOI] [PubMed] [Google Scholar]
- 77.Fix E, Hodges JL, Randolph Field Tex. Proj. 1951, 21–49. [Google Scholar]
- 78.Bishop CM, Svensén M, Williams CKI, Neural Comput. 1998, 10, 215–234. [Google Scholar]
- 79.Kireeva N, Baskin II, Gaspar HA, Horvath D, Marcou G, Varnek A, Mol. Inf. 2012, 31, 301–312. [DOI] [PubMed] [Google Scholar]
- 80.Winter R, Montanari F, Noé F, Clevert D-A, Chem. Sci. 2019, 10, 1692–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Filimonov D, Poroikov V, Borodina Y, Gloriozova T, J. Chem. Inf. Comput. Sci. 1999, 39, 666–670. [Google Scholar]
- 82.Le T, Hempel T, Winter R, Olsson S, Raich L, Elez K, Noé F, Narangoda C, Gokcan H, Gusev F, Zubatiuk R, Kurnikova M, Gutkin E, Bosko IP, Yushkevich A, Shuldau M, Karpenko AD, Kornoushenko YV, García-Sastre A, Furs K, Bureau R, Benabderrahmane M, Naffakh N, Cirou B, Bousquet-Melou P, Charton B, Ford B, Gil G, Epitropakis N, Krasoulis A, Pitsikalis V, Antonopoulos N, Theodorakis S, Schimunek J, Widrich M, Eghbal-zadeh H, Lee SY, Seidl P, Ruch P, Halmich C, Zhang K, Berenger F, Yamanishi Y, Brooks III CL, Kumar A, Jain M, Bengio E, Bengio Y, Marcou G, Popov P, Haupt J, Schroeder M, Kaiser F, Pugliese L, Paiardi G, Wade R, Hanke A, Goßen J, D’Arrigo G, Rossetti G, Albani S, Spyrakis F, Mukherjee G, Kokh D, Sadiq SK, Nunes-Alves A, Carloni P, Musiani F, Gianquinto E, Athanasiou C, Kovachka S, Tsengenes A-A, Joseph B, Talarico C, Manelfi C, Beccari A, Venkatraman V, Ondrechen MJ, Olson D, Copeland C, Roy A, Wheeler T, Tesseyre G, Gorgulla C, PadmanabhaDas K, Wagner G, Fackeldey K, Gruber CC, Fischer PD, Yust R, Pandita S, Wang Z-F, Veselovsky A, Poroikov V, Druzhilovskiy D, Stolbov L, Pogodin P, Sobolev B, Barnsley K, Gulotta MR, Lombino J, Simone GD, Perricone U, Mekni N, Rosa MD, Iyengar S, Watowich S, Falsafi B, Steinkellner G, Durmaz V, Cespugli M, Singh A, Gruber K, Hetmann M, Kozlovskii I, Zaretckii M, Medvedev A, Blaschitz K, Korablyov M, Allen W, Loesekrug-Pietri A, Hermans T, figshare 2021. [Google Scholar]
- 83.Verschueren KHG, Pumpor K, Anemüller S, Chen S, Mesters JR, Hilgenfeld R, Chem. Biol. 2008, 15, 597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Węglarz-Tomczak E, Tomczak JM, Talma M, Brul S, bioRxiv 2020, 2020.05.17.100768. [Google Scholar]
- 85.Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R, Science 2020, 368, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen KY, Krischuns T, Varga LO, Harigua-Souiai E, Paisant S, Zettor A, Chiaravalli J, Delpal A, Courtney D, O’Brien A, Baker SC, Decroly E, Isel C, Agou F, Jacob Y, Blondel A, Naffakh N, Antiviral Res. 2022, 201, 105272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jerabek-Willemsen M, Wienken CJ, Braun D, Baaske P, Duhr S, Assay Drug Dev. Technol. 2011, 9, 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wolff M, Mittag JJ, Herling TW, Genst ED, Dobson CM, Knowles TPJ, Braun D, Buell AK, Sci. Rep. 2016, 6, 22829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z, Lu G, Qiao C, Hu Y, Yuen K-Y, Wang Q, Zhou H, Yan J, Qi J, Cell 2020, 181, 894–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kruse M, Altattan B, Laux E-M, Grasse N, Heinig L, Möser C, Smith DM, Hölzel R, Sci. Rep. 2022, 12, 12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lin HXJ, Cho S, Meyyur Aravamudan V, Sanda HY, Palraj R, Molton JS, Venkatachalam I, Infection 2021, 49, 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kuzmič P, Sideris S, Cregar LM, Elrod KC, Rice KD, Janc JW, Anal. Biochem. 2000, 281, 62–67. [DOI] [PubMed] [Google Scholar]
- 93.Kuzmič P, Elrod KC, Cregar LM, Sideris S, Rai R, Janc JW, Anal. Biochem. 2000, 286, 45–50. [DOI] [PubMed] [Google Scholar]
- 94.Ratia K, Pegan S, Takayama J, Sleeman K, Coughlin M, Baliji S, Chaudhuri R, Fu W, Prabhakar BS, Johnson ME, Baker SC, Ghosh AK, Mesecar AD, Proc. Nat. Acad. Sci. 2008, 105, 16119–16124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Calleja DJ, Lessene G, Komander D, Front. Chem. 2022, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Belouzard S, Machelart A, Sencio V, Vausselin T, Hoffmann E, Deboosere N, Rouillé Y, Desmarets L, Séron K, Danneels A, Robil C, Belloy L, Moreau C, Piveteau C, Biela A, Vandeputte A, Heumel S, Deruyter L, Dumont J, Leroux F, Engelmann I, Alidjinou EK, Hober D, Brodin P, Beghyn T, Trottein F, Deprez B, Dubuisson J, PLoS Pathog. 2022, 18, e1010498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Altae-Tran H, Ramsundar B, Pappu AS, Pande V, ACS Cent. Sci. 2017, 3, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stanley M, Bronskill JF, Maziarz K, Misztela H, Lanini J, Segler M, Schneider N, Brockschmidt M, in Thirty-fifth Conference on Neural Information Processing Systems Datasets and Benchmarks Track (Round 2), OpenReview, 2021. [Google Scholar]
- 99.Guo Z, Zhang C, Yu W, Herr J, Wiest O, Jiang M, Chawla NV, in Proceedings of the Web Conference 2021, Association for Computing Machinery, New York, NY, 2021. [Google Scholar]
- 100.Wang Y, Abuduweili A, Yao Q, Dou D, in Advances in Neural Information Processing Systems, Vol. 34, 2021. [PMC free article] [PubMed] [Google Scholar]
- 101.Svensson E, Hoedt P-J, Hochreiter S, Klambauer G, in NeurIPS 2022 AI for Science: Progress and Promises, 2022.
- 102.Chen W, Tripp A, Hernández-Lobato JM, in The Eleventh International Conference on Learning Representations, OpenReview, 2023.
- 103.Schimunek J, Seidl P, Friedrich L, Kuhn D, Rippmann F, Hochreiter S, Klambauer G, in The Eleventh International Conference on Learning Representations, OpenReview, 2023.
- 104.Seidl P, Vall A, Hochreiter S, Klambauer G, arxiv 2023. [Google Scholar]
- 105.Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, others, Nucleic Acids Res. 2018, 46, D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ursu O, Holmes J, Knockel J, Bologa CG, Yang JJ, Mathias SL, Nelson SJ, Oprea TI, Nucleic Acids Res. 2016, gkw993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gaulton A, Hersey A, Nowotka M, Bento AP, Chambers J, Mendez D, Mutowo P, Atkinson F, Bellis LJ, Cibrián-Uhalte E, Davies M, Dedman N, Karlsson A, Magariños MP, Overington JP, Papadatos G, Smit I, Leach AR, Nucleic Acids Res. 2017, 45, D945–D954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Axelrod S, Gomez-Bombarelli R, Sci. Data 2022, 9, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lyu J, Irwin JJ, Shoichet BK, Nat. Chem. Biol. 2023, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Charifson PS, Corkery JJ, Murcko MA, Walters WP, J. Med. Chem. 1999, 42, 5100–5109. [DOI] [PubMed] [Google Scholar]
- 111.Douangamath A, Fearon D, Gehrtz P, Krojer T, Lukacik P, Owen CD, Resnick E, Strain-Damerell C, Aimon A, Ábrányi-Balogh P, Brandão-Neto J, Carbery A, Davison G, Dias A, Downes TD, Dunnett L, Fairhead M, Firth JD, Jones SP, Keeley A, Keserü GM, Klein HF, Martin MP, Noble MEM, O’Brien P, Powell A, Reddi RN, Skyner R, Snee M, Waring MJ, Wild C, London N, von Delft F, Walsh MA, Nat. Commun. 2020, 11, 5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Douangamath A, Fearon D, Gehrtz P, Krojer T, Lukacik P, Owen CD, Resnick E, Strain-Damerell C, Aimon A, Ábrányi-Balogh P, Brandão-Neto J, Carbery A, Davison G, Dias A, Downes TD, Dunnett L, Fairhead M, Firth JD, Jones SP, Keeley A, Keserü GM, Klein HF, Martin MP, Noble MEM, O’Brien P, Powell A, Reddi RN, Skyner R, Snee M, Waring MJ, Wild C, London N, von Delft F, Walsh MA, Nat. Commun. 2020, 11, 5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shi F, Waldo JP, Chen Y, Larock RC, Org. Lett. 2008, 10, 2409–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schake P, Dishnica K, Kaiser F, Leberecht C, Haupt VJ, Schroeder M, Sci. Rep. 2023, 13, 9204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Edwards A, Nature 2016, 533, S70–S70. [DOI] [PubMed] [Google Scholar]
- 116.ChemBioFrance - Infrastructure de recherche, https://chembiofrance.cn.cnrs.fr/en/.
- 117.EU-OPENSCREEN 2022: Compound Collections, https://www.eu-openscreen.eu/services/compound-collection.html.
- 118.COVID-19 OpenData Portal. National Center for Advancing Translational Sciences, https://ncats.nih.gov/expertise/covid19-open-data-portal. [Google Scholar]
- 119.Luke DA, Carothers BJ, Dhand A, Bell RA, Moreland-Russell S, Sarli CC, Evanoff BA, Clin. Transl. Oncol. 2015, 8, 143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Collins F, Adam S, Colvis C, Desrosiers E, Draghia-Akli R, Fauci A, Freire M, Gibbons G, Hall M, Hughes E, Jansen K, Kurilla M, Lane HC, Lowy D, Marks P, Menetski J, Pao W, Pérez-Stable E, Purcell L, Read S, Rutter J, Santos M, Schwetz T, Shuren J, Stenzel T, Stoffels P, Tabak L, Tountas K, Tromberg B, Wholley D, Woodcock J, Young J, Science 2023. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The originally submitted team reports and compound lists, raw data, and scripts used to analyze the data in this manuscript are freely available: https://github.com/hermanslab/COVID-19