

Abstract
Background
Identifying patients with COVID‐19 disease who will deteriorate can be useful to assess whether they should receive intensive care, or whether they can be treated in a less intensive way or through outpatient care. In clinical care, routine laboratory markers, such as C‐reactive protein, are used to assess a person's health status.
Objectives
To assess the accuracy of routine blood‐based laboratory tests to predict mortality and deterioration to severe or critical (from mild or moderate) COVID‐19 in people with SARS‐CoV‐2.
Search methods
On 25 August 2022, we searched the Cochrane COVID‐19 Study Register, encompassing searches of various databases such as MEDLINE via PubMed, CENTRAL, Embase, medRxiv, and ClinicalTrials.gov. We did not apply any language restrictions.
Selection criteria
We included studies of all designs that produced estimates of prognostic accuracy in participants who presented to outpatient services, or were admitted to general hospital wards with confirmed SARS‐CoV‐2 infection, and studies that were based on serum banks of samples from people. All routine blood‐based laboratory tests performed during the first encounter were included. We included any reference standard used to define deterioration to severe or critical disease that was provided by the authors.
Data collection and analysis
Two review authors independently extracted data from each included study, and independently assessed the methodological quality using the Quality Assessment of Prognostic Accuracy Studies tool. As studies reported different thresholds for the same test, we used the Hierarchical Summary Receiver Operator Curve model for meta‐analyses to estimate summary curves in SAS 9.4. We estimated the sensitivity at points on the SROC curves that corresponded to the median and interquartile range boundaries of specificities in the included studies. Direct and indirect comparisons were exclusively conducted for biomarkers with an estimated sensitivity and 95% CI of ≥ 50% at a specificity of ≥ 50%. The relative diagnostic odds ratio was calculated as a summary of the relative accuracy of these biomarkers.
Main results
We identified a total of 64 studies, including 71,170 participants, of which 8169 participants died, and 4031 participants deteriorated to severe/critical condition. The studies assessed 53 different laboratory tests. For some tests, both increases and decreases relative to the normal range were included. There was important heterogeneity between tests and their cut‐off values. None of the included studies had a low risk of bias or low concern for applicability for all domains. None of the tests included in this review demonstrated high sensitivity or specificity, or both. The five tests with summary sensitivity and specificity above 50% were: C‐reactive protein increase, neutrophil‐to‐lymphocyte ratio increase, lymphocyte count decrease, d‐dimer increase, and lactate dehydrogenase increase.
Inflammation
For mortality, summary sensitivity of a C‐reactive protein increase was 76% (95% CI 73% to 79%) at median specificity, 59% (low‐certainty evidence). For deterioration, summary sensitivity was 78% (95% CI 67% to 86%) at median specificity, 72% (very low‐certainty evidence). For the combined outcome of mortality or deterioration, or both, summary sensitivity was 70% (95% CI 49% to 85%) at median specificity, 60% (very low‐certainty evidence). For mortality, summary sensitivity of an increase in neutrophil‐to‐lymphocyte ratio was 69% (95% CI 66% to 72%) at median specificity, 63% (very low‐certainty evidence). For deterioration, summary sensitivity was 75% (95% CI 59% to 87%) at median specificity, 71% (very low‐certainty evidence). For mortality, summary sensitivity of a decrease in lymphocyte count was 67% (95% CI 56% to 77%) at median specificity, 61% (very low‐certainty evidence). For deterioration, summary sensitivity of a decrease in lymphocyte count was 69% (95% CI 60% to 76%) at median specificity, 67% (very low‐certainty evidence). For the combined outcome, summary sensitivity was 83% (95% CI 67% to 92%) at median specificity, 29% (very low‐certainty evidence). For mortality, summary sensitivity of a lactate dehydrogenase increase was 82% (95% CI 66% to 91%) at median specificity, 60% (very low‐certainty evidence). For deterioration, summary sensitivity of a lactate dehydrogenase increase was 79% (95% CI 76% to 82%) at median specificity, 66% (low‐certainty evidence). For the combined outcome, summary sensitivity was 69% (95% CI 51% to 82%) at median specificity, 62% (very low‐certainty evidence).
Hypercoagulability
For mortality, summary sensitivity of a d‐dimer increase was 70% (95% CI 64% to 76%) at median specificity of 56% (very low‐certainty evidence). For deterioration, summary sensitivity was 65% (95% CI 56% to 74%) at median specificity of 63% (very low‐certainty evidence). For the combined outcome, summary sensitivity was 65% (95% CI 52% to 76%) at median specificity of 54% (very low‐certainty evidence).
To predict mortality, neutrophil‐to‐lymphocyte ratio increase had higher accuracy compared to d‐dimer increase (RDOR (diagnostic Odds Ratio) 2.05, 95% CI 1.30 to 3.24), C‐reactive protein increase (RDOR 2.64, 95% CI 2.09 to 3.33), and lymphocyte count decrease (RDOR 2.63, 95% CI 1.55 to 4.46). D‐dimer increase had higher accuracy compared to lymphocyte count decrease (RDOR 1.49, 95% CI 1.23 to 1.80), C‐reactive protein increase (RDOR 1.31, 95% CI 1.03 to 1.65), and lactate dehydrogenase increase (RDOR 1.42, 95% CI 1.05 to 1.90). Additionally, lactate dehydrogenase increase had higher accuracy compared to lymphocyte count decrease (RDOR 1.30, 95% CI 1.13 to 1.49). To predict deterioration to severe disease, C‐reactive protein increase had higher accuracy compared to d‐dimer increase (RDOR 1.76, 95% CI 1.25 to 2.50). The neutrophil‐to‐lymphocyte ratio increase had higher accuracy compared to d‐dimer increase (RDOR 2.77, 95% CI 1.58 to 4.84). Lastly, lymphocyte count decrease had higher accuracy compared to d‐dimer increase (RDOR 2.10, 95% CI 1.44 to 3.07) and lactate dehydrogenase increase (RDOR 2.22, 95% CI 1.52 to 3.26).
Authors' conclusions
Laboratory tests, associated with hypercoagulability and hyperinflammatory response, were better at predicting severe disease and mortality in patients with SARS‐CoV‐2 compared to other laboratory tests. However, to safely rule out severe disease, tests should have high sensitivity (> 90%), and none of the identified laboratory tests met this criterion. In clinical practice, a more comprehensive assessment of a patient's health status is usually required by, for example, incorporating these laboratory tests into clinical prediction rules together with clinical symptoms, radiological findings, and patient's characteristics.
Keywords: Humans, Bias, Biomarkers, Biomarkers/blood, C-Reactive Protein, C-Reactive Protein/analysis, Clinical Deterioration, COVID-19, COVID-19/blood, COVID-19/diagnosis, COVID-19/mortality, COVID-19 Testing, COVID-19 Testing/methods, Pandemics, Prognosis, SARS-CoV-2, Sensitivity and Specificity, Severity of Illness Index
Plain language summary
How accurate are routine laboratory tests in predicting mortality and deterioration to severe or critical COVID‐19 in people with SARS‐CoV‐2?
What are routine laboratory tests? Routine laboratory tests are a set of commonly performed blood tests that provide information about a patient's health status. These tests can be used to identify disease or monitor health.
What did we want to find out? It is important to identify patients, presenting at a doctor's appointment at an outpatient service or at the emergency department who are at high risk of developing severe COVID‐19 disease or dying. It can help clinicians in deciding if the patients need hospitalisation. We wanted to know if routine laboratory tests were sufficiently accurate to predict mortality and deterioration in patients with confirmed SARS‐CoV‐2.
What did we do? We searched for studies that assessed how well routine laboratory tests predict mortality and deterioration in patients with confirmed SARS‐CoV‐2. We included studies of any design and set anywhere in the world. Patients of any age or sex were included.
What we found We found 64 studies that looked at 53 different routine laboratory tests. These studies assessed how well these tests could predict mortality, deterioration, or both. A total of 71,170 patients were included, of which 8169 (11.5%) patients died, and 4031 (5.7%) patients deteriorated to severe/critical disease. Adult patients were included in 31 studies, two studies reported on patients more than 60 years, two studies included a mix of children and adults, and one study included only children. Most studies were done in China, followed by Spain and Italy. All studies took place in hospitals.
'Sensitivity' and 'specificity' are often used to report the performance of tests. Sensitivity is the proportion of patients with the outcome (= mortality or deterioration) that are correctly detected by the test, and specificity is the proportion of people without the outcome that are correctly detected by the test. The closer the sensitivity and specificity of a test are to 100%, the more accurate the test is. To safely rule out patients who will not die or deteriorate, a high sensitivity of more than 90% is necessary. When four or more studies assessed the same tests, we pooled the data and analysed them together. We did not find any tests that were accurate enough to safely rule out a severe outcome, such as deterioration or death. We found five tests with both sensitivity and specificity exceeding 50%. Four of these laboratory tests indicate important inflammation in a SARS‐CoV‐2 infection. These four tests are C‐reactive protein, neutrophil‐to‐lymphocyte ratio, lymphocyte count, and lactate dehydrogenase. The fifth test, d‐dimer, reflects a state of increased blood clotting in a SARS‐CoV‐2 infection.
How reliable are the results? We have low confidence in the evidence of this review, because there were important differences between the included studies, and it was, therefore, difficult to compare them. Sensitivity and specificity depend on where the cut‐off point is made between positive (indicative of disease) and negative (disease‐free). For some studies, the authors decided on the cut‐off value (for a test) before doing the test (less likely to create bias) and in others, they chose the cut‐off value after analysis of the test (more likely to be biassed).
Who do the results of this review apply to? Routine laboratory tests can be performed at a doctor's appointment or at the emergency department. However, the included studies only assessed patients presenting to the hospital. We included patients with confirmed SARS‐CoV‐2 infection. Only one study reported on vaccinated patients, and we could not assess the effect of different SARS‐CoV‐2 variants of concern. Therefore, our results might not be representative for vaccinated patients or different variants of concern.
What does this mean? These routine laboratory tests, linked to inflammation and blood clotting in patients with COVID‐19 disease, can be used for risk stratification to assess a patient. However, none of these tests performed well enough to safely rule out progression to severe or deadly disease. These tests might serve to assess the overall health status of the patient. To predict deterioration or mortality, a more comprehensive assessment, including clinical symptoms, radiological findings and patient's characteristics, may be considered.
How up‐to‐date is this review? We searched for all COVID‐19 studies up to 25 August 2022.
Summary of findings
Summary of findings 1. Routine laboratory tests for COVID‐19: single tests (including markers of hyperinflammation).
|
Population: people who present to outpatient services, or are admitted to general hospital wards with confirmed SARS‐CoV‐2 infection. Index test: all routine blood‐based laboratory tests, including blood, plasma, and serum biomarkers, performed at the first assessment of the person as part of the initial routine diagnostic workup (e.g. during admission to hospital). The tests were classified into the following groups: markers of inflammation; complete blood count; liver function tests; biochemistry; coagulation markers; kidney function tests; cardiac markers; other tests. Target condition:
Reference standard: we expected the definitions for mild, moderate, severe, and critical COVID‐19 to vary between publications, and to be poorly reported. Therefore, we included any reference standard used to define severity that was provided by the authors, and documented the definitions. | |||||||
| Test | Target condition | Number of studies (number of cases/number of non‐cases) | % Median prevalence (IQR) |
% Specificity
Q11
Median1 Q31 |
% Summary sensitivity corresponding with fixed specificity (95% CI) | Certainty of the evidence | Interpretation of the results |
| C‐reactive protein increase | Mortality | 14 studies (1977/9834) | 17 (14 to 31) | 27 | 90 (86 to 92) | Lowa,d | C‐reactive protein is an acute‐phase protein that increases in response to inflammation. Most cases will have an increase in CRP, although many non‐cases will also show a rise in CRP levels. Therefore, it is neither suitable for ruling in or ruling out. |
| 59 | 76 (73 to 79) | ||||||
| 76 | 63 (57 to 69) | ||||||
| Deterioration | 18 studies (1221/4819) | 22 (13 to 34) | 62 | 85 (75 to 91) | Very lowa,c,d | CRP increase is one of the more sensitive tests for deterioration. However, the test by itself cannot safely rule in or rule out cases. | |
| 72 | 78 (67 to 86) | ||||||
| 81 | 67 (53 to 78) | ||||||
| Combined2 | 4 studies (224/1618) | 21 (14 to 26) | 54 | 78 (55 to 91) | Very lowa,b,c,d | A rise in CRP levels will be seen in many cases, although non‐cases will also show a rise in CRP levels. Therefore, it is neither suitable for ruling in or ruling out. | |
| 60 | 70 (49 to 85) | ||||||
| 64 | 64 (39 to 83) | ||||||
| Procalcitonin increase | Mortality | 9 studies (1655/8045) | 18 (15 to 27) | 82 | 54 (51 to 57) | Lowa,d | Procalcitonin levels are often elevated in bacterial infections. In COVID‐19, which is caused by a virus, increased procalcitonin levels may be associated with bacterial co‐infection. Many non‐cases will not have a procalcitonin increase. However, only half of cases or fewer will have a rise in procalcitonin. |
| 84 | 52 (49 to 55) | ||||||
| 93 | 38 (31 to 46) | ||||||
| Deterioration | 7 studies (718/2636) | 19 (14 to 37) | 86 | 40 (25 to 58) | Very lowa,c,d | Many cases will be missed at any cut‐off value. | |
| 93 | 25 (17 to 35) | ||||||
| 96 | 16 (8 to 29) | ||||||
| Ferritin increase | Mortality | 9 studies (1216/5288) | 15 (12 to 27) | 35 | 88 (65 to 97) | Very lowa,c,d | Ferritin is a known inflammatory marker. Many cases will have a rise in ferritin levels. However, the test by itself cannot safely rule in or rule out cases. |
| 64 | 64 (40 to 83) | ||||||
| 79 | 42 (17 to 73) | ||||||
| Deterioration | 5 studies (446/1711) | 20 (15 to 37) | 74 | 58 (38 to 76) | Very lowa,b,c,d | A little over half of cases will have a rise in ferritin. | |
| 75 | 57 (37 to 74) | ||||||
| 77 | 53 (35 to 71) | ||||||
| White blood cell count increase | Mortality | 10 studies (1516/12352) | 19 (13 to 39) | 61 | 38 (35 to 40) | Very lowa,c,d | The white blood cell count increases in inflammation. Many non‐cases will not have a rise in white blood cell count. However, many patients will be missed. |
| 82 | 37 (34 to 40) | ||||||
| 89 | 36 (33 to 39) | ||||||
| Deterioration | 13 studies (1088/8218) | 9 (13 to 28) | 75 | 40 (35 to 44) | Lowa,d | Many cases will be missed at any cut‐off value. | |
| 89 | 32 (28 to 36) | ||||||
| 94 | 27 (22 to 32) | ||||||
| White blood cell count decrease | Mortality | 5 studies (500/8714) | 4 (3 to 12) | 68 | 9 (6 to 14) | Lowa,d | Low white blood cell count can indicate an immune system problem. Many cases with COVID‐19 will be missed at any cut‐off value. |
| 83 | 9 (6 to 14) | ||||||
| 87 | 9 (6 to 14) | ||||||
| Deterioration | 6 studies (412/5907) | 21 (15 to 23) | 83 | 15 (6 to 33) | Very lowa,c,d | Many non‐cases will not have a white blood cell count decrease. However, many cases will be missed at any cut‐off level. | |
| 85 | 12 (5 to 26) | ||||||
| 88 | 8 (/) | ||||||
| Neutrophil count increase | Deterioration | 5 studies (232/1338) | 16 (13 to 19) | 88 | 62 (46 to 75) | Very lowa,c,d | Infections can increase the number of neutrophils in the blood. Many non‐cases will not have a rise in the neutrophil count. However, half of cases will be missed. |
| 89 | 49 (37 to 62) | ||||||
| 93 | 41 (28 to 55) | ||||||
| Lymphocyte count increase | Deterioration | 4 studies (331/5716) | 16 (11 to 20) | 99 | 7 (0 to 83) | Lowa,c | Lymphocyte blood levels can increase when dealing with infection or inflammatory conditions. |
| 99 | 7 (0 to 83) | ||||||
| 99 | 7 (0 to 83) | ||||||
| Lymphocyte count decrease | Mortality | 13 studies (3241/21,207) | 17 (12 to 34) | 48 | 79 (68 to 87) | Very lowa,c,d | Viral infections can decrease the number of lymphocytes. Lymphocyte count decrease is one of the more sensitive tests for predicting mortality. However, the test by itself cannot safely rule in or rule out cases. |
| 61 | 67 (56 to 77) | ||||||
| 70 | 56 (43 to 68) | ||||||
| Deterioration | 14 studies (1101/8698) | 20 (13 to 24) | 61 | 74 (64 to 81) | Very lowa,c,d | Lymphocyte count decrease is one of the more sensitive tests for predicting deterioration to severe disease. However, the test by itself cannot safely rule in or rule out cases. | |
| 67 | 69 (60 to 76) | ||||||
| 74 | 62 (52 to 71) | ||||||
| Combined | 4 studies (326/1516) | 22 (15 to 26) | 20 | 87 (66 to 96) | Very lowa,b,c,d | Many non‐cases will also have a decrease in lymphocyte count. | |
| 29 | 83 (67 to 92) | ||||||
| 46 | 76 (58 to 88) | ||||||
| Neutrophil‐to‐lymphocyte ratio increase | Mortality | 11 studies (1369/6092) | 15 (13 to 22) | 54 | 73 (69 to 77) | Very lowa,b,d | NLR increase can be an indicator of a severe infection. NLR increase was identified as one of the more sensitive tests for predicting mortality. However, the test by itself cannot safely rule in or rule out cases. |
| 63 | 69 (66 to 72) | ||||||
| 78 | 59 (52 to 66) | ||||||
| Deterioration | 9 studies (673/2034) | 29 (16 to 37) | 64 | 82 (65 to 91) | Very lowa,b,d | NLR increase is one of the more sensitive tests for predicting deterioration to severe disease. However, the test by itself cannot safely rule in or rule out cases. | |
| 71 | 75 (59 to 87) | ||||||
| 82 | 59 (38 to 77) | ||||||
| Platelets increase | Deterioration | 6 studies (703/6831) | 15 (14 to 18) | 93 | 15 (12 to 20) | Lowa,d | Platelet count can increase in response to SARS‐CoV‐2 infection. Many cases will be missed. |
| 96 | 10 (7 to 15) | ||||||
| 98 | 6 (5 to 8) | ||||||
| Platelets decrease | Mortality | 12 studies (1552/13,025) | 15 (8 to 32) | 79 | 32 (26 to 39) | Lowa,d | Viruses can trigger a decrease in platelet production. Many cases will be missed. |
| 88 | 21 (17 to 26) | ||||||
| 90 | 19 (14 to 26) | ||||||
| Deterioration | 12 studies (883/7640) | 19 (7 to 37) | 77 | 47 (27 to 68) | Very lowa,c,d | Many cases will be missed. | |
| 83 | 37 (22 to 54) | ||||||
| 90 | 22 (12 to 38) | ||||||
| Combined | 4 studies (335/1484) | 25 (20 to 26) | 63 | 52 (28 to 75) | Very lowa,c,d | Many cases will be missed. | |
| 73 | 41 (21 to 64) | ||||||
| 86 | 24 (1 to 88) | ||||||
| Haemoglobin decrease | Mortality | 5 studies (988/9115) | 17 (14 to 34) | 41 | 52 (43 to 61) | Very lowa,c,d | Haemoglobin decrease is called anaemia, and can be caused by infection. Many cases will be missed. |
| 60 | 45 (40 to 51) | ||||||
| 75 | 39 (34 to 45) | ||||||
| Deterioration | 5 studies (585/6455) | 16 (13 to 19) | 77 | 47 (5 to 94) | Very lowa,c,d | Many cases will be missed. | |
| 77 | 47 (5 to 94) | ||||||
| 88 | 0 (0 to 99) | ||||||
| Eosinophil count decrease | Mortality | 4 studies (1943/8997) | 20 (17 to 24) | 13 | 91 (14 to 100) | Very lowa,c,d | Eosinophil count decrease can be present in patients with severe COVID‐19 disease. Most cases will have a decrease in the eosinophil count, although many non‐cases will also show a rise in levels. |
| 25 | 75 (5 to 99) | ||||||
| 36 | 57 (1 to 99) | ||||||
| Alanine aminotransferase increase | Mortality | 7 studies (2658/13609) | 24 (15 to 45) | 57 | 60 (37 to 79) | Very lowa,c,d | Alanine aminotransferase increases because of liver cell damage. Many cases will be missed. |
| 65 | 45 (29 to 61) | ||||||
| 73 | 30 (19 to 44) | ||||||
| Deterioration | 11 studies (474/2111) | 16 (12 to 23) | 75 | 56 (36 to 74) | Very lowa,c,d | Many cases will be missed. | |
| 81 | 47 (33 to 62) | ||||||
| 88 | 34 (26 to 43) | ||||||
| Aspartate aminotransferase increase | Mortality | 6 studies (2468/13,354) | 20 (15 to 40) | 51 | 69 (41 to 87) | Very lowa,c,d | Aspartate aminotransferase indicates liver cell damage. A little over half of cases will have a rise in aspartate aminotransferase increase. |
| 64 | 58 (36 to 77) | ||||||
| 80 | 40 (18 to 66) | ||||||
| Deterioration | 11 studies (490/2256) | 15 (12 to 22) | 71 | 60 (47 to 71) | Very lowa,c,d | Many cases will be missed. | |
| 81 | 48 (36 to 60) | ||||||
| 86 | 40 (28 to 54) | ||||||
| Total bilirubin increase | Deterioration | 5 studies (306/1244) | 13 (12 to 19) | 74 | 40 (16 to 70) | Very lowa,c,d | Total bilirubin is a waste product produced during the normal process of haemoglobin breakdown, primarily in the liver. Infection can affect overall liver function, including the processing of bilirubin. Many cases will be missed. |
| 90 | 16 (6 to 35) | ||||||
| 91 | 14 (5 to 35) | ||||||
| Potassium decrease | Deterioration | 4 studies (255/870) | 18 (11 to 35) | 69 | 64 (11 to 96) | Very lowa,c,d | COVID‐19 disease can present with a decrease in potassium levels in the blood, known as hypokalemia. Many cases will be missed. |
| 76 | 51 (9 to 91) | ||||||
| 83 | 35 (5 to 84) | ||||||
| Sodium decrease | Deterioration | 4 studies (397/1352) | 18 (11 to 35) | 52 | 75 (14 to 98) | Very lowa,c,d | Sodium decrease, known as hyponatremia, has been reported in patients with COVID‐19. Many cases will have a decrease in sodium. However, the test by itself cannot safely rule in or rule out cases. |
| 59 | 67 (13 to 97) | ||||||
| 69 | 53 (8 to 93) | ||||||
GRADE Working Group grades of evidence High certainty: we believe strongly that the true effect lies close to the estimated effect. Moderate certainty: we have moderate confidence in the estimated effect; the true effect is likely to be close to the estimate of the effect, but possibly is substantially different. Low certainty: we have limited confidence in the effect estimate; the true effect could differ significantly from the estimated effect. Very low certainty: there is very little confidence in the effect estimate; it's highly likely that the actual effect is substantially different from the estimated effect.
Abbreviations: CI: confidence interval, IQR: interquartile range, Q1: first quartile, Q3: third quartile.
1We used the first quartile, median and third quartile specificity of all specificities of the included studies to estimate the corresponding sensitivity estimates from the HSROC model. 2Combined target condition of death due to any cause or deterioration, or both, from mild or moderate to severe or critical COVID‐19 cases.
We started at a high certainty of the evidence.
aWe downgraded the evidence by one level for risk of bias when at least half of the studies had high risk of bias on one or more domains. bWe downgraded the evidence by one level for indirectness when at least half of the studies in the meta‐analyses had high concerns regarding applicability on at least one domain. cWe downgraded the evidence by one level for imprecision when fewer people with the target condition were included than would have been needed to achieve the sensitivity estimates listed with a width of the CI of at most 10% points. dWe downgraded the evidence by one level for inconsistency when study estimates differed more than 20% points from each other. Publication bias was not considered to be a problem.
Summary of findings 2. Routine laboratory tests for COVID‐19: single tests (including markers of hypercoagulability).
|
Population: people who present to outpatient services, or are admitted to general hospital wards with confirmed SARS‐CoV‐2 infection. Index test: all routine blood‐based laboratory tests, including blood, plasma, and serum biomarkers, performed at the first assessment of the person as part of the initial routine diagnostic workup (e.g. during admission to hospital). The tests were classified into the following groups: markers of inflammation; complete blood count; liver function tests; biochemistry; coagulation markers; kidney function tests; cardiac markers; other tests. Target condition:
Reference standard: we expected the definitions for mild, moderate, severe, and critical COVID‐19 to vary between publications, and to be poorly reported. Therefore, we included any reference standard used to define severity that was provided by the authors, and documented the definitions. | |||||||
| Test | Target condition | Number of studies (number of cases/number of non‐cases) | % Median prevalence (IQR) |
% Specificity
Q11
Median1 Q31 |
% Summary sensitivity corresponding with fixed specificity (95% CI) | Certainty of the evidence | Interpretation of the results |
| Activated partial thromboplastin time increase | Deterioration | 5 studies (255/1328) | 19 (13 to 46) | 73 | 41 (28 to 56) | Very lowa,c,d | Activated partial thromboplastin time characterises coagulation of the blood. When it is increased, it means it takes longer for the blood to form a clot. Many cases will be missed at any cut‐off level. |
| 80 | 34 (23 to 48) | ||||||
| 90 | 22 (12 to 37) | ||||||
| Prothrombin time increase | Deterioration | 4 studies (298/725) | 31 (18 to 43) | 77 | 37 (18 to 60) | Very lowa,c,d | When prothrombin time is increased, it takes longer for the blood to form a clot. Many cases will be missed at any cut‐off level. |
| 92 | 13 (4 to 34) | ||||||
| 95 | 8 (2 to 28) | ||||||
| D‐dimer increase | Mortality | 14 studies (5565/28,556) | 15 (12 to 29) | 48 | 77 (71 to 83) | Very lowa,c,d | Increased D‐dimer can be associated with thromboembolic complications. D‐dimer increase is one of the more sensitive tests to predict mortality. However, the test by itself cannot safely rule in or rule out cases. |
| 56 | 70 (64 to 76) | ||||||
| 63 | 62 (56 to 69) | ||||||
| Deterioration | 10 studies (651/1970) | 25 (15 to 41) | 46 | 80 (69 to 88) | Very lowa,c,d | D‐dimer increase is one of the more sensitive tests to predict deterioration to severe disease. However, the test by itself cannot safely rule in or rule out cases. | |
| 63 | 65 (56 to 74) | ||||||
| 71 | 56 (44 to 67) | ||||||
| Combined2 | 4 studies (326 cases/1516) | 20 (16 to 24) | 52 | 67 (55 to 76) | Very lowa,b,c | Many non‐cases will also have an increase in D‐dimer level. | |
| 54 | 65 (52 to 76) | ||||||
| 57 | 62 (46 to 76) | ||||||
| Fibrinogen increase | Deterioration | 5 studies (337/1716) | 19 (16 to 46) | 22 | 90 (79 to 95) | Very lowa,b,c,d | Fibrinogen can increase in response to infection or inflammation. Many cases will have an increase in fibrinogen. However, half of non‐cases will also have a fibrinogen increase. |
| 49 | 77 (66 to 86) | ||||||
| 78 | 56 (40 to 71) | ||||||
| Serum creatinine increase | Mortality | 7 studies (2705/13,807) | 22 (15 to 36) | 73 | 43 (40 to 45) | Lowa,d | Serum creatinine can be a sign of poor kidney function. Many cases will be missed. |
| 79 | 43 (40 to 45) | ||||||
| 94 | 42 (40 to 45) | ||||||
| Deterioration | 6 studies (291/1307) | 12 (8 to 20) | 93 | 19 (1 to 81) | Very lowa,c,d | Many cases will be missed. | |
| 97 | 9 (0 to 67) | ||||||
| 97 | 9 (0 to 67) | ||||||
| Blood urea nitrogen increase | Deterioration | 5 studies (387/1149) | 19 (13 to 22) | 90 | 32 (23 to 42) | Very lowa,c,d | Blood urea nitrogen increase may indicate impaired kidney function. Many cases will be missed. |
| 94 | 27 (16 to 42) | ||||||
| 96 | 23 (12 to 40) | ||||||
| Troponin I increase | Mortality | 4 studies (805/2745) | 27 (24 to 33) | 57 | 84 (65 to 93) | Very lowa,c,d | Troponin I increase indicates myocardial injury. Troponin I increase is one of the more sensitive tests to predict mortality. However, the test by itself cannot safely rule in or rule out cases. |
| 78 | 64 (47 to 78) | ||||||
| 88 | 42 (28 to 58) | ||||||
| Troponin increase | Deterioration | 4 studies (387/2133) | 16 (13 to 24) | 75 | 34 (22 to 50) | Very lowa,c,d | Troponin increase indicates myocardial injury. Many cases will be missed. |
| 76 | 34 (22 to 49) | ||||||
| 83 | 30 (18 to 45) | ||||||
| Lactate dehydrogenase increase | Mortality | 10 studies (2786/14,208) | 21 (11 to 38) | 19 | 91 (80 to 96) | Very lowa,c,d | High lactate dehydrogenase can be indicative of tissue damage or inflammation. LDH increase is one of the more sensitive tests to predict mortality. However, the test by itself cannot safely rule in or rule out cases. |
| 60 | 82 (66 to 91) | ||||||
| 80 | 74 (53 to 88) | ||||||
| Deterioration | 12 studies (780/2838) | 21 (13 to 25) | 51 | 80 (76 to 83) | Lowa,d | LDH increase is one of the more sensitive tests to predict deterioration to severe disease. However, the test by itself cannot safely rule in or rule out cases. | |
| 66 | 79 (76 to 82) | ||||||
| 78 | 79 (75 to 81) | ||||||
| Combined | 4 studies (335/1484) | 22 (18 to 25) | 57 | 72 (52 to 86) | Very lowa,c,d | LDH increase is one of the more sensitive tests to predict cases. However, the test by itself cannot safely rule in or rule out cases. | |
| 62 | 69 (51 to 82) | ||||||
| 68 | 64 (45 to 80) | ||||||
| Creatine kinase increase | Deterioration | 8 studies (389/1605) | 18 (13 to 24) | 78 | 61 (29 to 86) | Very lowa,c,d | Creatine kinase increase can result from muscle injury or damage. Many patients will be missed. |
| 86 | 38 (14 to 70) | ||||||
| 87 | 35 (12 to 68) | ||||||
GRADE Working Group grades of evidence High certainty: we believe strongly that the true effect lies close to the estimated effect. Moderate certainty: we have moderate confidence in the estimated effect; the true effect is likely to be close to the estimate of the effect, but possibly is substantially different. Low certainty: we have limited confidence in the effect estimate; the true effect could differ significantly from the estimated effect. Very low certainty: there is very little confidence in the effect estimate; it's highly likely that the actual effect is substantially different from the estimated effect.
Abbreviations: CI: confidence interval, IQR: interquartile range, Q1: first quartile, Q3: third quartile.
1We used the first quartile, median and third quartile specificity of all specificities of the included studies to estimate the corresponding sensitivity estimates from the HSROC model. 2Combined target condition of death due to any cause or deterioration, or both, from mild or moderate to severe or critical COVID‐19 cases.
We started at a high certainty of the evidence.
aWe downgraded the evidence by one level for risk of bias when at least half of the studies had high risk of bias on one or more domains. bWe downgraded the evidence by one level for indirectness when at least half of the studies in the meta‐analyses had high concerns regarding applicability on at least one domain. cWe downgraded the evidence by one level for imprecision when fewer people with the target condition were included than would have been needed to achieve the sensitivity estimates listed with a width of the confidence interval of at most 10% points. dWe downgraded the evidence by one level for inconsistency when study estimates differed more than 20% points from each other. Publication bias was not considered to be a problem.
Summary of findings 3. Comparisons of routine laboratory tests for COVID‐19 with sensitivity and specificity higher than 50%.
| Comparisons (*reference test) | Number of studies (number of cases/number of non‐cases)a | Median fixed specificityb (IQR) | Comparisons (*reference test) | RDOR | Pr > |z| | Lower limit 95% CI | Upper limit 95% CI |
| Indirect comparisons: mortality | |||||||
| CRP increase, D‐dimer increase, and LDH increase | |||||||
| CRP increase | 14 studies (1977/9834) | 56% (36% to 74%) | CRP/LDH* | 1.62 | 0.1473 | 0.8439 | 3.0984 |
| D‐dimer increase | 14 studies (5565/28,556) | DD/LDH* | 1.26 | 0.2565 | 0.8470 | 1.8627 | |
| LDH increase | 10 studies (2786/14,208) | ||||||
| CRP increase, D‐dimer increase, and lymphocyte count decrease | |||||||
| CRP increase | 14 studies (1977/9834) | 57% (40% to 70%) | CRP/LYMP* | 3.17 | 0.1967 | 0.5495 | 18.2844 |
| D‐dimer increase | 14 studies (5565/28,556) | DD/LYMP* | 1.08 | 0.6321 | 0.7834 | 1.4943 | |
| Lymphocyte count decrease | 13 studies (3241/21,207) | ||||||
| CRP increase, LDH increase, and lymphocyte count decrease | |||||||
| CRP increase | 14 studies (1977/9834) | 61% (28% to 77%) | CRP/LYMP* | 1.55 | 0.501 | 0.4343 | 5.5019 |
| LDH increase | 14 studies (5565/28,556) | LDH/LYMP* | 1.07 | 0.8528 | 0.5033 | 2.2932 | |
| Lymphocyte count decrease | 13 studies (3241/21,207) | ||||||
| CRP increase, NLR increase, and d‐dimer increase | |||||||
| CRP increase | 14 studies (1977/9834) | 58% (44% to 73%) | CRP/DD* | 2.83 | 0.12 | 0.7622 | 10.506 |
| NLR increase | 11 studies (1369/6092) | NLR/DD* | 2.05 | 0.0022 | 1.2971 | 3.2449 | |
| D‐dimer increase | 14 studies (5565/28,556) | ||||||
| CRP increase, NLR increase, and lymphocyte count decrease | |||||||
| CRP increase | 14 studies (1977/9834) | 61% (42% to 76%) | CRP/LYMP* | 2.19 | 0.3684 | 0.3956 | 12.1581 |
| NLR increase | 11 studies (1369/6092) | NLR/LYMP* | 2.63 | 0.0003 | 1.5504 | 4.4558 | |
| Lymphocyte count decrease | 13 studies (3241/21,207) | ||||||
| D‐dimer increase, LDH increase, and lymphocyte count decrease | |||||||
| D‐dimer increase | 14 studies (5565/28,556) | 56% (40% to 70%) | DD/LDH* | 1.42 | 0.0212 | 1.0535 | 1.9008 |
| Lymphocyte count decrease | 13 studies (3241/21,207) | LYMP/LDH* | 1.24 | 0.2578 | 0.8529 | 1.8095 | |
| LDH increase | 14 studies (5565/28,556) | ||||||
| D‐dimer increase, lymphocyte count decrease, and NLR increase | |||||||
| D‐dimer increase | 14 studies (5565/28,556) | 60% (49% to 70%) | DD/LYMP* | 1.49 | <.0001 | 1.2346 | 1.7999 |
| NLR increase | 11 studies (1369/6092) | NLR/LYMP* | 2.41 | <.0001 | 1.7926 | 3.2499 | |
| Lymphocyte count decrease | 13 studies (3241/21,207) | ||||||
| Direct comparisons: mortality | |||||||
| D‐dimer increase and CRP increase* | 9 studies (1460/6949) | 48% (27% to 62%) | DD/CRP* | 1.31 | 0.0255 | 1.0334 | 1.6531 |
| CRP increase and LDH increase* | 7 studies (1156/5934) | 45% (14% to 79%) | CRP/LDH* | 1.01 | 0.9763 | 0.7039 | 1.436 |
| CRP increase and lymphocyte count decrease* | 7 studies (1305/6962) | 57% (27% to 74%) | CRP/LYMP* | 1.23 | 0.0635 | 0.9885 | 1.5286 |
| NLR increase and CRP increase* | 5 studies (1037/5035) | 57% (38% to 75%) | NLR/CRP* | 2.64 | <.0001 | 2.0888 | 3.3274 |
| D‐dimer increase and LDH increase* | 7 studies (2680/13,822) | 54% (31% to 65%) | DD/LDH* | 1.05 | 0.5529 | 0.9022 | 1.2118 |
| D‐dimer increase and lymphocyte count decrease* | 7 studies (2712/14,170) | 51% (42% to 66%) | DD/LYMP* | 1.28 | 0.0003 | 1.1214 | 1.4608 |
| LDH increase and lymphocyte count decrease* | 6 studies (2443/13,369) | 60% (43% to 73%) | LDH/LYMP* | 1.30 | 0.0002 | 1.1323 | 1.491 |
| NLR increase and lymphocyte count decrease* | 5 studies (970/3796) | 58% (53% to 63%) | NLR/LYMP* | 1.84 | < 0.0001 | 1.386 | 2.4412 |
| Indirect comparisons: deterioration | |||||||
| CRP increase, D‐dimer increase, and lymphocyte count decrease | |||||||
| CRP increase | 18 studies (1221/4819) | 68% (60% to 75%) | CRP/DD* | 1.76 | 0.0013 | 1.2478 | 2.4956 |
| Lymphocyte count decrease | 14 studies (1101/8698) | LYMP/DD* | 1.54 | 0.0198 | 1.0715 | 2.2233 | |
| D‐dimer increase | 10 studies (651/1970) | ||||||
| CRP increase, LDH increase, and lymphocyte count decrease | |||||||
| CRP increase | 18 studies (1221/4819) | 69% (60% to 78%) | CRP/LDH* | 1.33 | 0.1472 | 0.9053 | 1.9413 |
| Lymphocyte count decrease | 14 studies (1101/8698) | LYMP/LDH* | 1.48 | 0.1164 | 0.9077 | 2.3988 | |
| LDH increase | 12 studies (780/2838) | ||||||
| CRP increase, D‐dimer increase, and LDH increase | |||||||
| CRP increase | 18 studies (1221/4819) | 68% (55% to 77%) | CRP/LDH* | 1.97 | 0.1126 | 0.8521 | 4.5661 |
| D‐dimer increase | 10 studies (651/1970) | DD/LDH* | 1.01 | 0.9694 | 0.5955 | 1.7143 | |
| LDH increase | 12 studies (780/2838) | ||||||
| CRP increase, D‐dimer increase, and NLR increase | |||||||
| CRP increase | 18 studies (1221/4819) | 70% (61% to 79%) | CRP/DD* | 1.77 | 0.0166 | 1.11 | 2.8341 |
| NLR increase | 9 studies (673/2034) | NLR/DD* | 1.79 | 0.041 | 1.0241 | 3.1113 | |
| D‐dimer increase | 10 studies (651/1970) | ||||||
| CRP increase, NLR increase, and lymphocyte count decrease | |||||||
| CRP increase | 18 studies (1221/4819) | 70% (61% to 82%) | CRP/LYMP* | 1.13 | 0.3719 | 0.8652 | 1.4724 |
| NLR increase | 9 studies (673/2034) | NLR/LYMP* | 1.32 | 0.1246 | 0.9253 | 1.8938 | |
| Lymphocyte count decrease | 14 studies (1101/8698) | ||||||
| D‐dimer increase, NLR increase, and lymphocyte count decrease | |||||||
| NLR increase | 9 studies (673/2034) | 68% (61% to 75%) | NLR/DD* | 2.77 | 0.0004 | 1.5832 | 4.8431 |
| Lymphocyte count decrease | 14 studies (1101/8698) | LYMP/DD* | 1.70 | 0.0101 | 1.1355 | 2.5578 | |
| D‐dimer increase | 10 studies (651/1970) | ||||||
| D‐dimer increase, LDH increase, and lymphocyte count decrease | |||||||
| LDH‐increase | 12 studies (780/2838) | 66% (57% to 76%) | LDH/DD* | 1.21 | 0.4457 | 0.7404 | 1.9795 |
| Lymphocyte count decrease | 14 studies (1101/8698) | LYMP/DD* | 2.02 | 0.0003 | 1.3870 | 2.9402 | |
| D‐dimer increase | 10 studies (651/1970) | ||||||
| Direct comparisons: deterioration | |||||||
| CRP increase, and D‐dimer* | 7 studies (552/1623) | 67% (46% to 73%) | CRP/DD* | 1.64 | 0.0021 | 1.1973 | 2.2463 |
| CRP increase, and LDH increase* | 7 studies (372/1345) | 72% (65% to 77%) | CRP/LDH* | 1.13 | 0.5352 | 0.7717 | 1.6466 |
| CRP increase, and lymphocyte count decrease* | 9 studies (640/2420) | 69% (62% to 82%) | CRP/LYMP* | 1.04 | 0.7934 | 0.7886 | 1.3644 |
| LDH increase and D‐dimer increase* | 5 studies (338/1076) | 62% (49% to 70%) | LDH/DD* | 1.21 | 0.3795 | 0.7937 | 1.8331 |
| Lymphocyte count decrease and D‐dimer increase* | 6 studies (379/1237) | 64% (55% to 73%) | LYMP/DD* | 2.10 | 0.0001 | 1.4411 | 3.0679 |
| Lymphocyte count decrease and LDH increase* | 7 studies (393/1443) | 66% (49% to 69%) | LYMP/LDH* | 2.22 | < 0.0001 | 1.5154 | 3.2625 |
For the target conditions, mortality and deterioration to severe/critical disease, we found 5 tests with median specificity and corresponding summary sensitivity, including their 95% CI, above 50%. The test accuracies of these index tests were compared with a direct, head‐to‐head comparison (i.e. assessed 2 of the biomarkers in the same participants), and an indirect comparison, in which all studies reporting on these tests were included. The different comparisons are displayed in this table. The relative diagnostic odds ratio (RDOR) was calculated as a summary of the relative accuracy of the 2 biomarkers at hand. Statistical significant differences in test accuracy were assessed at P < 0.05.
Test performances are compared to the reference test, which is indicated with * and is the test with the lowest test performance.
aCases are defined as participants who reached the outcome, mortality or deterioration to severe or critical disease. bMedian specificity of all studies included in the comparison.
Abbreviations: CI: confidence interval, CRP: C‐reactive protein, DD: D‐dimer, IQR: interquartile range, LDH: lactate dehydrogenase, LYMP: lymphocyte count, NLR: neutrophil‐lymphocyte ratio, RDOR: relative diagnostic odds ratio
Background
On 30 December 2019, a report about a cluster of people with pneumonia of unknown origin in Wuhan, China, was publicly described in ProMED (promedmail.org/promed-posts). In January 2020, it became clear that this was caused by a new coronavirus, and was also spreading to other countries. In March 2020, the World Health Organization (WHO) declared that the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS‐CoV‐2) and the resulting novel coronavirus disease‐2019 (COVID‐19) was a worldwide pandemic. Prior to 23 October 2023, there had been 771,407,825 confirmed cases, including 6,972,152 deaths, reported to the WHO (WHO 2023a). This pandemic presented important challenges, such as assessing whether existing biochemical and imaging tests could identify people who need critical care. Although WHO declared that the pandemic was subsiding on 5 May 2023 (WHO 2023b), our review question remains relevant, given the persistence of the virus necessitating continued vigilance. Despite different variants of concern and vaccination, COVID‐19 disease can still lead to hospitalisation and mortality. Our review can aid physicians to triage patients with COVID‐19 disease, even in a post‐pandemic era.
Prognostic accuracy studies evaluate the ability of medical tests to predict disease, or to identify people who are likely to experience an adverse medical event amongst those who have a disease. These studies typically present results in terms of sensitivity and specificity, or the area under the curve. The current review focuses on the prognostic accuracy of individual biomarkers to predict mortality and deterioration to severe or critical COVID‐19. This is fundamentally different from (1) prediction model reviews, which focus on the predictive performance of models, and critically appraise model development studies (including discrimination and calibration) and external validation studies of prediction models (Wynants 2020); and (2) prognostic factor studies, which investigate the association between a test, biomarker, or personal characteristic and a future outcome, either by itself, or over and above other known predictors. Prognostic factor studies typically present the results as a measure of association, rather than the accuracy at a given test threshold for test positivity.
This Cochrane review concentrates on the accuracy of routine, blood‐based laboratory tests to predict death and deterioration to severe or critical COVID‐19 disease in people with SARS‐CoV‐2 infection. In clinical care, routine laboratory markers, such as white blood cell count, measures of anticoagulation, C‐reactive protein (CRP), and procalcitonin, are used to assess a person's health status. These laboratory markers are also used in people with COVID‐19, and may be useful for hospital triage, to assess whether a person with COVID‐19 should receive outpatient treatment or more intensive treatment, which usually requires hospital admission. Other meta‐analyses on prognostic accuracy of biomarkers have been performed. Most of them included only one biomarker, such as d‐dimer (Nugroho 2021; Simadibrata 2020), platelet (Pranata 2021), procalcitonin (Vazzana 2022), and troponin (Wibowo 2021). Others also included more than one biomarker (Zare 2020). However, our review is unique in its comprehensiveness. It includes data on 53 different biomarkers from a total of 71,170 participants. Many studies have reported different cut‐off values for the same biomarker; however, by using HSROC analysis, we can account for within and between‐study variations.
The protocol of this Cochrane review was published in 2021 (Verbakel 2021). This review follows a generic protocol that covers the full series of Cochrane Diagnostic Test Accuracy (DTA) Reviews for the diagnosis of COVID‐19 (Deeks 2020a). Therefore, we used some text in the Background and Methods sections that was originally published in that protocol, and some text overlaps with some of our other reviews (Deeks 2020b; Dinnes 2020; Islam 2021; Stegeman 2020; Struyf 2021).
Target condition being diagnosed
COVID‐19 is the disease caused by infection with SARS‐CoV‐2. SARS‐CoV‐2 infection can (1) be asymptomatic (no symptoms), (2) cause mild or moderate signs and symptoms, such as fever, cough, aches, lethargy, breathlessness, and fast breathing, (3) cause severe signs and symptoms, which include severe respiratory distress and low oxygen saturation, indicative of severe pneumonia, (4) cause critical signs and symptoms, which require respiratory support due to Acute Respiratory Distress Syndrome (ARDS), or (5) can lead to organ dysfunction (indicative of sepsis). People with severe or critical COVID‐19 require distinctive management of their signs and symptoms; it is, therefore, important to identify them.
At present, polymerase chain reaction (PCR) testing is more prevalent compared to when the pandemic started, and diagnosing SARS‐CoV‐2 infection is no longer challenging. Clinicians now look at the added value of routine laboratory tests to decide to admit people with a suspected or confirmed SARS‐CoV‐2 infection to the hospital, or to adapt a watchful‐waiting approach. Therefore, we aim, on the one hand, to focus on the distinction between mild or moderate COVID‐19, and severe or critical COVID‐19; and, on the other hand, on mortality from COVID‐19.
Index test(s)
Routinely available blood‐based biomarkers for infection and inflammation may be considered in the investigation of people with confirmed SARS‐CoV‐2 infection. Evaluation of commonly available tests may be helpful to predict death or deterioration of a person with mild or moderate COVID‐19 to severe or critical COVID‐19.
We collated evidence on all routine blood, plasma, and serum biomarker tests reported in the identified studies.
Clinical pathway
The standard workup for people suspected of having COVID‐19 consists of assessing signs and symptoms, and performing a PCR test. However, as people with COVID‐19 present a variety of symptoms of varying severity and, as they may deteriorate quickly, it is important to be able to predict who will deteriorate and who may not. Therefore, it is common practice to perform routine laboratory tests whenever people are assessed at the hospital (either outpatient or inpatient).
Routine laboratory tests may be used to predict deterioration from mild or moderate disease to severe or critical outcomes in people with a suspected or confirmed SARS‐CoV‐2 infection. In ambulatory care, the decision to refer a person with a SARS‐CoV‐2 infection implies the potential breach of quarantine measures. Routine laboratory tests might help to inform the decision to treat the person at home, to reduce the workload of already burdened hospitals and intensive care units (ICU), requiring highly sensitive tests able to rule out severe disease or deterioration. More favourable laboratory test results could support ambulatory care management of people with COVID‐19, providing clinicians with information on which signs and symptoms might trigger a further diagnostic workup. For people who are hospitalised, routine laboratory tests may inform the decision to refer them to the ICU, or confirm that they are stable enough to remain in the general ward.
Prior test(s)
Prior testing consists of assessment of signs and symptoms.
Role of index test(s)
The role of the index tests as add‐ons or triage tests is considered in this Cochrane review.
Alternative test(s)
In emergency departments, chest X‐rays, ultrasounds, and computed tomography (CT) are additionally widely used to assess the severity of a person’s condition, especially in the case of pneumonia. Which imaging test is available may depend on the type of hospital and available resources. A tertiary care hospital in a high‐income country may have a mobile CT scanner available, while smaller hospitals may only have an X‐ray and ultrasound machine. These imaging tests have the advantage that they can enable a visual assessment of the condition of the lungs.
Rationale
It is essential to understand the prognostic accuracy of tests to inform clinicians on how to use them optimally in different settings and to help to develop effective management pathways. New evidence about routine laboratory testing is becoming available quickly.
The use of prediction models may be an alternative, rather than performing alternative tests. However, not all laboratories measure all biomarkers and the tests required to estimate a specific model. Furthermore, it would be very useful if there was a biomarker that could serve as a ‘red flag’: if this biomarker is positive, it means that the person needs extra care to prevent deterioration.
While we wanted to examine the accuracy of routine laboratory markers to predict person‐related outcomes in this review, the DTA framework still applies, with the addition of some adaptations to the risk of bias assessment to account for the prognostic nature of our research objectives.
Objectives
To assess the accuracy of routine blood‐based laboratory tests to predict mortality and deterioration to severe or critical (from mild or moderate) COVID‐19 in people with SARS‐CoV‐2 infection.
Secondary objectives
Where data were available, we investigated whether prognostic accuracy varied according to a specific measurement or test, reference standard, timing of outcome verification, sample type, study design, and setting, including prevalence of the target condition (either by stratified analysis or meta‐regression).
Methods
Criteria for considering studies for this review
Types of studies
We kept the eligibility criteria broad to include all groups of people and all variations of a test. If the participant population was unclear, i.e. when the study did not provide details regarding the inclusion criteria of the participants, such as age, the study was included.
We included studies of all designs that produced estimates of prognostic accuracy, or provided data from which estimates could be computed: prospective and retrospective cohort studies, case‐control designs (using participants from a single original cohort), and consecutive series of participants assessing the prognostic accuracy of routine laboratory tests.
We included only single‐gate designs, in which a single group of participants who may develop the target condition or event is recruited. We included studies that based their results on individual participants and studies that based their results on laboratory samples. We carefully considered the limitations of different study designs using quality assessment and analysis. Both studies that reported on a single biomarker or a range of biomarkers were considered eligible for our review question, allowing indirect or direct head‐to‐head comparisons, respectively.
Participants
We included studies recruiting people who presented to outpatient services, or were admitted to general hospital wards with confirmed SARS‐CoV‐2 infection, and studies that were based on serum banks of samples from people with confirmed SARS‐CoV‐2.
Studies required a minimum of 10 samples or 10 participants for inclusion.
Index tests
Evidence on all reported routine blood‐based laboratory tests was collected. Tests were performed during the first assessment of the person as part of the initial routine diagnostic workup (e.g. during admission to hospital). The tests were classified into the following groups:
Markers of inflammation;
Complete blood count;
Liver function tests;
Biochemistry;
Coagulation markers;
Kidney function tests;
Cardiac markers;
Other tests.
We interpreted the term 'routine' broadly, considering that some markers can be more routine in some settings or countries than in others. Most routine laboratory tests provide test results as continuous measurements. That means that an explicit threshold is needed to provide positive and negative results. A positive test was defined as an increased or decreased value compared to the normal range, which was defined by the included study, at the moment of first assessment. This was indicated in this review with an increase and decrease, e.g. CRP increase.
Target conditions
To be eligible, studies had to identify a current SARS‐CoV‐2 infection. SARS‐CoV‐2 infection had to be confirmed by either PCR testing or (negative) PCR testing in combination with other findings (clinical or radiological). Studies that only included patients with suspected, non‐laboratory confirmed, SARS‐CoV‐2 infection were excluded. Studies that reported confirmed SARS‐CoV‐2 infection, but did not explicitly report the method of testing were included.
First target condition
Mortality: death due to any cause (30‐day mortality, unless otherwise specified), in participants with different disease severity at baseline.
Second target condition
Deterioration: deterioration from mild or moderate to severe or critical COVID‐19 disease (14‐day assessment, unless otherwise specified). If patients already presented with severe or critical disease on admission, they were not included for this target condition.
Third target condition
Combined target condition of death due to any cause or deterioration, or both, from mild or moderate to severe or critical COVID‐19 disease.
Reference standards
We expected the definitions for mild, moderate, severe, and critical COVID‐19 to vary between publications, and to be poorly reported. Therefore, we included any reference standard used to define severity that was provided by the authors, and documented the definitions.
Unless otherwise provided by the original paper, the study would need to make a distinction between mild (to moderate) and severe (to critical) cases as defined by the WHO Clinical Management of COVID‐19 interim guidance report (WHO 2020).
Mild disease: people with symptoms who meet the case definition for COVID‐19, without evidence of viral pneumonia or hypoxia;
-
Moderate disease (pneumonia):
adolescent or adult with clinical signs of pneumonia (fever, cough, dyspnoea, fast breathing), but with SpO₂ ≥ 90% on room air, and no signs of severe pneumonia;
child with clinical signs of non‐severe pneumonia (cough or difficulty breathing plus fast breathing or chest indrawing, or both), and no signs of severe pneumonia. Fast breathing is defined as (breaths/minute): < 2 months old: ≥ 60; 2 to 11 months old: ≥ 50; 1 to 5 years old: ≥ 40;
-
Severe disease (severe pneumonia):
adolescent or adult with clinical signs of pneumonia (fever, cough, dyspnoea, fast breathing) plus one of the following: respiratory rate > 30 breaths/minute; severe respiratory distress; or SpO₂ < 90% on room air;
-
child with clinical signs of pneumonia (cough or difficulty in breathing) plus at least one of the following:
central cyanosis or SpO₂ < 90%; severe respiratory distress (e.g. fast breathing, grunting, very severe chest indrawing); general danger sign: inability to breastfeed or drink, lethargy or unconsciousness, or convulsions;
fast breathing is defined as (breaths/minute): < 2 months old: ≥ 60; 2 to 11 months old: ≥ 50; 1 to 5 years old: ≥ 40;
Critical disease: acute respiratory distress syndrome (ARDS), sepsis, or septic shock. We will also categorise the following outcomes as critical disease: ICU admission, need for ventilation, and need for intubation.
While these diagnoses can be made on clinical grounds, chest imaging (radiograph, CT scan, ultrasound) may assist in making the diagnosis, and identify or exclude pulmonary complications.
We assessed and extracted, if available, the prediction horizon of the biomarkers identified in the included studies, aiming to allow reasonable assessment and comparison of the laboratory markers measured at baseline (e.g. admission to hospital or first assessment).
We assessed the quality of these definitions according to the criteria listed in the Quality Assessment of Prognostic Accuracy Studies (QUAPAS) Table. The QUAPAS tool is an adaptation of the Quality Assessment of Diagnostic Accuracy Studies‐2 (QUADAS‐2) tool and used for prognostic accuracy studies (Lee 2022). We provided a qualitative overview of the reference standards used and reported in the included studies.
Search methods for identification of studies
Electronic searches
We searched the Cochrane COVID‐19 Study Register (covid-19.cochrane.org/).
The Cochrane COVID‐19 Study Register is a specialised register built within the Cochrane Register of Studies (CRS) and is maintained by Cochrane Information Specialists. The register contains study reports from several sources, including:
Monthly searches of the Cochrane Central Register of Controlled Trials (CENTRAL);
Daily searches of MEDLINE via PubMed;
Weekly searches of Embase.com;
Weekly searches of medRxiv;
Weekly searches of the WHO International Clinical Trials Registry Platform (ICTRP);
Daily searches of ClinicalTrials.gov.
Complete data sources and search methods for the register are available at: community.cochrane.org/about-covid-19-study-register.
We performed the search using the CRS web interface (crsweb.cochrane.org), using a strategy that combines a search for diagnostic studies, a search for prognostic characteristics, and a search for severely ill patients. See Appendix 1 for search terms.
Searching other resources
No resources other than the ones described above were searched.
Data collection and analysis
Selection of studies
We conducted a pilot screening of 802 titles and abstracts, performed independently by two reviewers. Subsequently, we compared these results to the pilot screening of the same title and abstracts by a single review author, supported by an established machine‐learning‐based priority screening module in EPPI‐Reviewer, to assess the equivalence of both methods. If the machine‐learning method was deemed suitable and equivalent, this method was used for further title and abstract screening. As part of this method, the records were ordered for manual screening: records more relevant to the research question were presented at the beginning of the screening process and likely irrelevant records were presented towards the end. In the next step, two review authors independently screened the full text of each possibly relevant article. From the final list of included studies; we performed both forward and backward citation tracking, using Microsoft Academic through EPPI‐Reviewer (EPPI‐Reviewer 2020).
For articles only available in languages other than English, we used Google Translate, or review authors who could read and understand that language performed translations. We solved disagreements by discussion. If discussion could not solve the dispute, we consulted a third review author.
Data extraction and management
Two review authors independently extracted data from each included study. We assigned multiple studies with the same first author to one extractor, so that they could detect preprints from already peer‐reviewed, published articles.
We extracted data on the country and region, the setting, the time period of the study, funding, and information needed for the Characteristics of included studies tables. Studies defined a positive test as an increased or decreased value, or both, compared to the normal range. Where possible, we adapted the 2 x 2 tables so that all studies included in the analyses reported on the same definition of test positivity. However, if studies reported both an increase and a decrease relative to the normal range of test results as a positive test result, we included both separately. We resolved disagreements by discussion between the two review authors, and the results were entered into Review Manager (RevMan) (RevMan 2024).
Assessment of methodological quality
Two review authors independently assessed the risk of bias and applicability concerns using the Quality Assessment of Prognostic Accuracy Studies (QUAPAS) tool. This tool incorporates elements of the QUADAS‐2 tool, supplemented by elements of the Quality in Prognostic Studies (QUIPS) tool and Prediction model Risk Of Bias Assessment Tool (PROBAST) and adds a fifth domain of 'analysis' to the quality appraisal (Hayden 2013; Lee 2022; Whiting 2011; Wolff 2019). We resolved disagreements by discussion.
The focus of our review was on prognostic accuracy and not on the predictive performance of models, so we did not critically appraise model development studies (including discrimination and calibration), and external validation studies of prediction models. Therefore, we decided that using QUIPS (prognostic factors) or PROBAST (models) as such, was unsuitable.
The other four domains of the QUAPAS tool are identical to the QUADAS‐2 tool: participant selection, index test, reference standard, and flow and timing (Lee 2022, Whiting 2011). Each domain was assessed for risk of bias, and the domains were also assessed for concerns of applicability. Signalling questions were included to help judge bias.
Statistical analysis and data synthesis
Although this review focuses on prognostic accuracy to predict patient‐related outcomes, the same approach as for DTA reviews applies, given the nature of the data identified in the primary studies (2 x 2 tables for each test in each study).
Most routine laboratory tests provide test results as continuous measurements. That means that an explicit threshold is needed to provide positive and negative results, to estimate diagnostic characteristics, such as sensitivity and specificity. Some tests indicate mild (or moderate) versus severe (or critical) disease if the value is decreased relative to the normal range. For other tests, mild versus severe disease is indicated when the value is increased. For another group of tests, both an increased and a decreased value may indicate the presence of mild or severe disease. For each test in each study, we reported the threshold used in our analyses, and whether an increased or decreased value was regarded as a positive test result.
From each study, we included one threshold for each test. If a study reported multiple thresholds for the same biomarker, the threshold closest to a value of clinical relevance was chosen through discussion by the study team. We presented the sensitivity and specificity results in forest plots, and provided positive and negative predictive values for each study. We reported the median and interquartile range (IQR) of pre‐test probability of the target condition in 2 x 2 tables.
We considered that a meta‐analysis was appropriate when four or more studies reported on a particular test. As studies might report different thresholds for the same test, we used the Hierarchical Summary Receiver Operator Curve (HSROC) model for meta‐analyses, to estimate summary curves, recommended by the Cochrane Handbook for Systematic Reviews of Diagnostic Test Accuracy (Macaskill 2010). Since summary sensitivities and specificities are only clinically interpretable when the studies included in a meta‐analysis use a common cut‐off value, we estimated sensitivity at points on the Summary Receiver Operator Curves (SROC) that corresponded to the median specificity observed in the studies included in the meta‐analysis. We reported the estimates for the first and third quartile specificity in the summary of findings tables. We used SAS 9.4, using PROC NLMIXED, for the meta‐analyses (SAS 2015).
To identify the most discriminative test in the situation, we compared the prognostic accuracy of biomarkers with a minimum estimated sensitivity of 50% (median and IQR), at a minimum specificity of 50% (median and IQR). We performed these analyses on all studies that evaluated one of these tests (indirect comparison). We also performed analyses that were restricted to studies that made head‐to‐head comparisons (i.e. assessed two of the biomarkers in the same participant) when at least four studies were included that enabled these direct comparisons. We made test comparisons by adding a covariate for test type to the HSROC model to assess the effect of test type on the accuracy, cut‐off, or shape parameters of the model. Whenever the estimated SROC curves had the same shape, we calculated the relative diagnostic odds ratio (RDOR) as a summary of the relative accuracy of the two biomarkers at hand. To assess the statistical significance of differences in test accuracy, we used likelihood ratio tests for comparisons of models with and without covariate terms. If fewer than 10 primary studies were available for the head‐to‐head comparison, we assumed that the shape parameter of the model was equal for the biomarkers under evaluation.
Investigations of heterogeneity
If adequate data were available, we investigated the following sources of heterogeneity: measurement technique or test type, reference standard, timing of outcome verification, sample type, study design, and setting, including prevalence of the target condition. This could be done by either using stratification (where we believe it is inappropriate to combine studies), or with meta‐regression models.
Summary of findings and assessment of the certainty of the evidence
We developed a list of key findings in the 'summary of findings' tables, and determined the certainty in the summary estimates for each test and findings using the GRADE approach (Schünemann 2020a; Schünemann 2020b).
Starting at high certainty, we downgraded by one level when at least half of the studies were at high risk of bias for one or more domains. We downgraded for indirectness when we had high concerns about applicability for at least one domain in at least half of the studies. We downgraded for imprecision when fewer people with the target condition were included than would have been needed to achieve the sensitivity estimates listed, and the confidence interval was wider than 10 percentage points. Lastly, we downgraded for inconsistency when study estimates differed more than 20 percentage points from each other.
Updating
Although it has been declared that the current pandemic has subsided, we do not exclude the possibility that updating our review could become highly relevant, especially in the context of emerging variants, the development of new diagnostic technologies, or the occurrence of any unforeseen challenges in the management of infectious diseases. We will consider updating depending on the relevance of our research question and the number of new and important studies found in each search update. We will consider updating this review with each search, if resources allow.
Sensitivity analyses
When sufficient data were available, we aimed to undertake sensitivity analyses considering the impact of unpublished studies. We aimed to perform sensitivity analyses to investigate the impact of prospective versus retrospective data collection.
Assessment of reporting bias
We aimed to publish lists of studies that we knew existed but for which we have not managed to locate reports, and requested information to include in review updates.
Results
Results of the search
The overall search was performed on 25 August 2022, and is described in Appendix 1. A total of 22,818 records were identified from the Cochrane COVID‐19 Study Register. A total of 22,705 records were included for title and abstract screening, of which we excluded 20,823. We performed full‐text screening on 1882 articles. Of these articles, we excluded three duplicates, 40 trial register records, 236 conference proceedings, 11 reports without original data (for example, reviews, and meta‐analyses), five studies with an ineligible study design, and two studies with fewer than 10 participants. Furthermore, we excluded 667 studies that only included participants admitted to tertiary care or ICU, 289 studies that did not have dichotomisation of test results, 265 studies that did not measure prognostic accuracy, 159 studies that measured the wrong outcome, 82 studies that did not assess a routine laboratory test, and 48 studies that included a specific group of participants (such as participants with a transplant organ). We included a total of 64 studies in this review (Figure 1).
1.
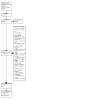
Study flow diagram
Included studies
Most studies (n = 57) were retrospective cohort studies, and seven studies were prospective cohort studies. Of all studies, 24 were published in 2020, 28 studies in 2021, and 12 studies in 2022. The time period in which participants were included was 2020 for 57 studies, 2021 for two studies, and was not reported for five studies. All studies were conducted in a hospital setting.
A total of 71,170 participants were included in this review. Different target conditions were assessed. Mortality was assessed in 36 studies, deterioration in 38 studies, and a combined outcome, including both mortality and deterioration, was assessed in six studies. Some studies assessed mortality and deterioration separately in the same participant population. Deterioration to severe disease was defined in different ways, such as ICU admission, intubation/invasive ventilation, or progression to severe disease following national clinical guidelines. The total sample size of studies assessing mortality was 63,691 participants, of which 8169 participants died. Out of a total of 32,723 participants, 4031 participants were classified as participants with deterioration to severe disease. A combined outcome was assessed in 2229 participants, of which 313 participants experienced the combined outcome.
Adult participants were included in 31 studies, two studies reported on participants more than 60 years old, two studies included a mix of children and adults, and one study included paediatric participants only. In the remaining 28 studies, inclusion criteria for age were not reported. However, the age of included participants was described with a range of the mean/median age from 28 to 71 years (Characteristics of included studies). Of the included studies, 15 studies were performed in China, seven in Spain, six in Italy, five each in Turkey and the USA, four each in France and the United Kingdom, two each in Indonesia, and Pakistan, and one each in Argentina, Bangladesh, Belgium, Greece, India, Iraq, Israel, Japan, Malaysia, Peru, Poland, Saudi Arabia, South Korea, and the United Arab Emirates.
In 42 studies, SARS‐CoV‐2 infection was confirmed by reverse transcriptase polymerase chain reaction (RT‐PCR) testing; in 12 studies, the definition of SARS‐CoV‐2 cases also included clinical or radiological symptoms and signs; and in 10 studies the method to confirm the diagnosis of SARS‐CoV‐2 was not clearly reported. No studies reported the variant of concern during the study period; one study mentioned that the alpha variant was the dominant variant during the study period. Only one study reported on vaccine status, but did not specify the vaccine type.
A total of 53 different laboratory tests were included; for some tests, both increases and decreases relative to the normal range were included in the review. For mortality and deterioration, 47 different laboratory tests were included and, for the combined outcome, 21 laboratory tests were assessed. The number of included biomarkers per study, if increase and decrease were taken into account separately, ranged from one to 33. For 18 studies, one biomarker was included and, in 18 other studies, ten or more biomarkers were included. In 50 studies, information about the method used to analyse the index test was poorly or not reported. All index tests were tested on individual participants. In 55 studies, index tests were assessed on admission and, in the other studies, this was unclear or not reported.
The time horizon for mortality ranged from 15 to 90 days, and was not reported in 27 studies. The time horizon for deterioration ranged from 7 to 30 days, and was not reported in 35 studies. The time horizon for the combined outcome was 28 days in one study, and was not reported in five studies.
Excluded studies, ongoing studies, and studies awaiting classification
We excluded a total of 22,641 studies. The complete list is available upon request. There are no ongoing studies or studies awaiting classification.
Methodological quality of included studies
Of the 64 included studies, there were no studies with a low risk of bias in all domains. One study had an unclear risk of bias in all domains. All other studies had a high risk of bias in at least one domain (Figure 2). A summary graph is presented in Figure 3.
2.

Figure 2: Risk of bias and applicability concerns
3.

Summary graph presenting the distribution of risk of bias and applicability concerns across all studies
The risk of bias for the domain participants was high in 15 studies. This was mainly due to inappropriate selection criteria, such as the inclusion of participants with a probable SARS‐CoV‐2 infection based on clinical presentation. For 31 studies, we judged the risk of bias to be unclear. In these studies, it was unclear whether a consecutive or random sample was used (n = 26 studies), or the selection criteria were unclear (n = 17 studies), or both. In one study, the study design was unclear. The risk of bias for the index test was high in 30 studies, and unclear in 34 studies. In these studies, the method to perform the index test was often not reported; therefore, it was not clear if it was valid and reliable (n = 48 studies), or the same in all participants (n = 32 studies). For 60 studies, it was unclear if the results were interpreted without knowledge of the outcome. The threshold was not prespecified in 29 studies, leading to a high risk of bias for the domain index test in those studies. We judged the risk of bias for the outcome to be low in 21 studies, unclear in 26 studies, and high in two studies. In 15 studies, both mortality and deterioration to severe disease were reported. The risk of bias in this domain could therefore be different in the same study depending on the outcome. This is illustrated in Figure 2 as low/unclear. Most studies had a valid and reliable measurement of the outcome (n = 62 studies), and in the same manner for all participants (n = 63 studies). It was unclear if the outcome was measured without knowledge of the index test results in 27 studies, and it was not the case in two studies. The risk of bias for flow and timing was high in most studies (n = 56 studies), because treatment was often not avoided after the index test was performed (n = 52 studies). In other studies, it was unclear whether participants received treatment or not. In 37 studies, not all participants received the index test. The time horizon, between index test and outcome, was often not reported (n = 52 studies). In 40 studies, the risk of bias for analysis was unclear, and it was high in 18 studies. It was unclear if all enroled participants were included in the analysis (n = 39 studies), and if appropriate methods were used for missing data (n = 33 studies), for censoring (n = 40 studies), or for competing events (n = 34 studies). In 15 studies with missing data, it was reported that imputation was not used to correct for the missing data.
One study had low concern regarding applicability on all domains, and 62 studies had high concern on at least one domain (Figure 2). Most studies (n = 29 studies) had high concern regarding applicability of the index test, because the threshold used was not prespecified. Most studies (n = 52 studies) had unclear concern for flow and timing, due to an unclear time horizon. We judged low concern for the outcome in 60 studies, and low concern for participants in 36 studies. In studies with inappropriate participant selection, the concern about applicability was high.
An overview of all signalling questions used in the QUAPAS tool and how they were answered in this review is shown in Table 4. The answer per study is provided for every study under the study characteristics section except for the domain analysis, which can be found in Table 5. Depending on the assessed index test or outcome, risk or bias could change. This is illustrated in Figure 2.
1. QUAPAS checklist.
| Participants | |
| S1.1 Was a consecutive or random sample of participants enroled? | YES: if convenience sampling, non‐randomised stratified sampling, self‐selection of participants, or cluster sampling related to a variable of interest was avoided. NO: if the above was not avoided. UNCLEAR: if the selection procedure was not clear or not reported. |
| S1.2 Was a case‐control design avoided? | YES: if the study enroled a single group of participants likely to undergo the index test in practice. Case‐control designs can overestimate accuracy and should be avoided. NO: if the study did not avoid a case‐control design. If, in this case, it was possible to extract data on cases only, we estimated it was unlikely to result in a high risk of bias. UNCLEAR: if the selection procedure was not clear or not reported. |
| S1.3 Did the study avoid inappropriate study criteria? | YES: if the inclusion and exclusion criteria are formulated in such a way that the study participants will reflect the population in which the index test will be used. NO: if a high proportion of eligible patients was excluded without providing a reason; if patients without confirmed SARS‐CoV‐2 or other viral infections were included. UNCLEAR: if the exclusion criteria were not reported, if it was unclear if SARS‐CoV‐2 infection was confirmed with RT‐PCR test. |
| Could the selection of participants have introduced bias? | HIGH: if one or more signalling questions were answered with NO. LOW: if all signalling questions were answered with YES. UNCLEAR: all other instances. |
| Are there concerns that the participants do not match the review question? | HIGH: if prognostic accuracy of laboratory tests was assessed in an already highly selected group of participants, for example, participants with severe or critical disease. LOW: if the study included participants who presented to outpatient services, or were admitted to general hospital wards with confirmed SARS‐CoV‐2 infection. UNCLEAR: if the description of participants was not clear or not reported. |
| Index test | |
| S2.1 Was the method used to perform the index test valid and reliable? | YES: if the study used an adequately validated assay or other measurement method that is reliable, reproducible and fit for intended use, e.g. blood‐based markers are measured using a validated ELISA kit. NO: if the measurement method of the index test was not valid or reliable. UNCLEAR: if the method and setting for performing the index test was not clear or not reported. |
| S2.2 Was the method for performing the index test the same for all participants? | YES: if the study explicitly reported that the same technique (or assay kit) for the index tests was used in all study participants. NO: if different techniques were used for the same index test in different study participants. UNCLEAR: if the method and setting for performing the index test was not clear or not reported. |
| S2.3 Was the index test interpreted without knowledge of the outcome? | YES: if the study explicitly reported blinding for the outcome information at the time of interpreting the index test results. Only applicable for retrospectively or previously collected data, as blinding naturally occurs in prospective study designs. NO: if the study explicitly reported that the index test results were interpreted with knowledge of the outcome. UNCLEAR: if blinding was unclearly reported. |
| S2.4 If a threshold was used, was it prespecified? | YES: if data‐driven threshold selection was avoided, if the threshold was prespecified in the methodology. NO: if the threshold was selected post hoc, e.g. via the Youden index, or to match desired sensitivity or specificity. UNCLEAR: if threshold selection was not clearly reported. |
| Could the conduct or interpretation of the index test have introduced bias? | HIGH: if one or more signalling questions were answered with NO. LOW: if all signalling questions were answered with YES. UNCLEAR: all other instances. |
| Are there concerns that the index test or its conduct, interpretation, or threshold differ from the review question? | HIGH: if the threshold was unclear or not defined beforehand. LOW: if the study included routine blood‐based laboratory tests performed during the first encounter with the participant as part of the initial routine diagnostic workup (e.g. during admission to hospital, or during first assessment) UNCLEAR: if the method to measure the index tests was unclear or not reported. |
| Outcome | |
| S3.1 Was the method used to measure the outcome valid and reliable? | YES: if the outcome was measured using an adequately validated method to avoid misclassification of participants. If the outcome was mortality, it can be assumed that the measurement is valid and reliable. NO: if the method to measure the outcome was not valid. UNCLEAR: if the method to measure the outcome was not reported; if the outcome was not defined. |
| S3.2 Was the method used for measuring the outcome the same for all participants? | YES: if all participants received the same method of measuring the outcome regardless of other factors such as the index test results. NO: if the method to measure the outcome was not the same for all participants. UNCLEAR: if the method to measure the index tests was unclear or not reported. |
| S3.3 Was the outcome measured without knowledge of the index test results? | YES: if the outcome assessment was done blind to the index test results. This was not applicable if measurement of the outcome does not involve subjective interpretation. In some cases, knowledge of the index test results would not influence the measurement/interpretation of the outcome, e.g. all‐cause mortality. NO: if the outcome assessment was not done blind to the index test results, e.g. the index test was known when the participant was admitted to the ICU. UNCLEAR: not reported |
| Could measurement of the outcome have introduced bias? | HIGH: if one or more signalling questions were answered with NO. LOW: if all signalling questions were answered with YES. UNCLEAR: all other instances. |
| Are there concerns that the outcome does not match the review question? | HIGH: if death due to any cause or deterioration from mild or moderate to severe or critical COVID‐19 cases was not assessed as an outcome, for example, if deterioration in severe or critical participants was assessed. LOW: if death due to any cause or deterioration from mild or moderate to severe or critical COVID‐19 cases was assessed as an outcome. UNCLEAR: if the description of the target event was not clear or not reported. |
| Flow and timing | |
| S4.1 Did all participants receive the index test? | YES: if there were no enroled participants with incomplete or missing index test information. NO: if not all eligible participants received the index test. UNCLEAR: if the flow and timing of participants was not clear or not reported. |
| S4.2 Was treatment avoided after the index test was performed? | YES: if no participants received any treatment between the index test measurement and occurrence of the outcome and the time horizon. This will be very rare, since the participants included in the studies are admitted to the hospital and will most likely always receive some kind of treatment. NO: if any participant received treatment between measurement of the index test and occurrence of the outcome. UNCLEAR: if treatment was not reported. |
| S4.3 Was the time horizon sufficient to capture the outcome? | YES: if the time horizon was sufficient for occurrence of the outcome; a follow‐up period that is too long for capturing acute disease incidence may skew the proportion of those experiencing the outcome. NO: if the time horizon was not sufficient to capture the outcome; less than one month for mortality, and less than 14 days for deterioration. On the other hand, when the time horizon was too long, this could also lead to risk of bias; for example, mortality assessed 2 or 3 months after index test measurement, and deterioration more than 14 days after deterioration. UNCLEAR: if the time horizon was not reported. |
| S4.4 Was information on the outcome available for all participants? | YES: if no enroled participants were lost to follow‐up, or if there were no significant differences between those who were lost to follow‐up and those who completed the study. NO: if outcome data were unavailable, for example, due to participants being transferred to another hospital. UNCLEAR: if the availability of the outcome was not reported. |
| Could the study flow have introduced bias? | HIGH: if one or more signalling questions were answered with NO. LOW: if all signalling questions were answered with YES. UNCLEAR: all other instances. |
| Are there concerns that the time horizon does not match the review question? | HIGH: if the time horizon to assess the target event was too long or short. LOW: if the time horizon to assess mortality was one month or less, and the time horizon to assess deterioration was less than 14 days. UNCLEAR: if the time horizon was unclear or not reported |
| Analysis | |
| S5.1 Were all enroled participants included in the analysis? | YES: if all eligible participants were included in the analysis. NO: if any participants were excluded from the analysis, for reasons besides participant selection and missing index test/outcome data, e.g. exclusion of those with intermediate or out‐of‐range index test results. Excluding eligible participants can introduce bias. UNCLEAR: if this was not reported. |
| S5.2 If data were missing, were appropriate methods used? | YES: if multiple imputations were used to handle missing index test results or outcome data or if reasons for or consequences of missing data were identified. Not applicable in the absence of missing data. NO: if complete analysis is used, in which missing data were omitted. UNCLEAR: if information about missing data was not reported. |
| S5.3 Were appropriate methods used to account for censoring? | YES: if the time dependent ROC curve analysis (or equivalent method) was used to account for censoring. Selection of an appropriate method may depend on characteristics of the study data. NO: if censoring was ignored or simple Kaplan‐Meier analysis was used. UNCLEAR: if information about censoring was not reported. |
| S5.4 In the case of competing events, were appropriate methods used to account for them? | YES: if the Cox Proportional Hazard (cause‐specific) model, cumulative incidence function or other competing risk models were used. Not applicable when the risk of competing events is rare or non‐existent. NO: if competing events were ignored or not handled appropriately. UNCLEAR: if the method used for competing events is not reported. |
| Could the analysis have introduced bias? | HIGH: if one or more signalling questions were answered with NO. LOW: if all signalling questions were answered with YES. UNCLEAR: all other instances. |
Abbreviations: ELISA: enzyme‐linked immunosorbent assay, ICU: intensive care unit, ROC: receiver operating characteristics, RT‐PCR: reverse transcriptase polymerase chain reaction, SARS‐CoV‐2: severe acute respiratory syndrome coronavirus 2.
QUAPAS: explanation
The Quality Assessment of Prognostic Accuracy Studies (QUAPAS) tool was initiated to address the need for a risk of bias and applicability assessment tool in systematic reviews of prognostic accuracy studies. While tools such as QUADAS‐2 exist to assess methodological quality in diagnostic accuracy reviews, as well as QUIPS (prognostic factors) and PROBAST (models) for prognostic reviews, no comparable tool exists for reviews of prognostic accuracy studies. Based on this need and availability of reliable existing resources, the QUAPAS tool was created by mapping the relevant items from QUIPS and PROBAST to the existing domain‐based framework and logic of QUADAS‐2. This way, the format of QUADAS‐2 could be used, with which many reviewers of diagnostic accuracy studies are familiar: domains, signalling questions, and a judgement on risk of bias and applicability concerns of a given primary study.
With QUAPAS, the aim was to focus on the key distinguishing factor between diagnostic and prognostic test accuracy research: the longitudinal study design and time‐dependent occurrence of the outcome, inherent to the research question. The domains of QUADAS‐2 were modified to account for factors unique to prognostic research, while keeping intact signalling questions where the potential risk of bias assessment does not differ significantly between diagnostic or prognostic questions (Participants and Index Test domains). Changes were applied to the Reference Standard (now called Outcome) and Flow and Timing domains to better account for bias introduced from longitudinal research questions. Inspired by QUIPS, PROBAST, and our understanding of prognostic research, a fifth domain was added, called Analysis, as there are complexities introduced from time‐dependent analysis. In this fifth domain, two signalling questions from PROBAST were added, and one from QUIPS, as neglecting methods for handling censoring, competing events, and missing data raise concerns about bias in prognostic accuracy studies. Other domains, such as Confounding (from QUIPS), were excluded entirely from QUAPAS, as causal questions are not relevant for test accuracy studies. Despite the changes to the QUADAS‐2 domain names and signalling questions, QUAPAS is intended to be used in the same manner. Reviewers assess the risk of bias for each of the five domains, with signalling questions to aid the grading as high, low, or unclear. Concerns for applicability are graded on the same scale for four of the five domains.
2. Methodological quality domain 5: Analysis.
| Study | Were all enroled participants included in the analysis? | If data were missing, were appropriate methods used? | Were appropriate methods used to account for censoring? | In the case of competing events, were appropriate methods used to account for them? | Could the analysis have introduced bias? |
| Al‐Samkari 2020 | No | No | No | Unclear | High |
| An 2021 | No | No | Unclear | Unclear | High |
| Arnold 2021 | Yes | Unclear | Unclear | Unclear | Unclear |
| Arshad 2020 | Yes | No | Unclear | Yes | High |
| Asaduzzaman 2022 | Unclear | Unclear | Unclear | Unclear | Unclear |
| Asghar 2020 | Unclear | Unclear | Unclear | Unclear | Unclear |
| Aydin 2022 | Unclear | Unclear | Unclear | Unclear | Unclear |
| Ayus 2021 | Yes | Yes | Yes | Yes | Low |
| Azad Allarakia 2022 | Unclear | Unclear | Unclear | Unclear | Unclear |
| Bahl 2020 | Yes | Yes | Unclear | Unclear | Unclear |
| Barrett 2021 | Unclear | Unclear | Unclear | Unclear | Unclear |
| Bassoli 2020 | Yes | Yes | Unclear | Unclear | Unclear |
| BG 2021 | Unclear | Yes | Yes | Yes | Unclear |
| Cazzaniga 2021 | No | Yes | Yes | Unclear | High |
| Chen 2020a | Yes | Yes | Yes | Yes | Low |
| Chen 2020b | Unclear | Unclear | Yes | Yes | Unclear |
| Chen 2021 | Yes | Yes | Yes | Yes | Low |
| Chen 2020c | Unclear | Unclear | Unclear | Unclear | Unclear |
| Chen 2020d | Unclear | No | Yes | Yes | High |
| Chocron 2021 | Yes | Unclear | Yes | Yes | Unclear |
| Detsika 2021 | Unclear | Unclear | Unclear | Unclear | Unclear |
| Dong 2020 | Unclear | No | Yes | Yes | High |
| Duan 2020 | Unclear | Unclear | Unclear | Unclear | Unclear |
| Elhadad 2020 | Unclear | Unclear | Yes | Unclear | Unclear |
| Fernandez‐Botran 2021 | Unclear | No | Unclear | Unclear | High |
| Gao 2020 | Unclear | Unclear | Yes | Unclear | Unclear |
| García de Guadiana‐Romualdo 2021a | Yes | Unclear | Yes | Yes | Unclear |
| García de Guadiana‐Romualdo 2021b | Unclear | Unclear | Unclear | Yes | Unclear |
| García de Guadiana‐Romualdo 2022 | Unclear | No | Unclear | Unclear | High |
| Goudot 2020 | Unclear | Unclear | Unclear | Unclear | Unclear |
| Guan 2020 | Unclear | Unclear | Unclear | Yes | Unclear |
| Haymana 2021 | Yes | Unclear | Unclear | Unclear | Unclear |
| Ibraheem 2021 | Unclear | Unclear | Unclear | Unclear | Unclear |
| López‐Escobar 2021 | No | Yes | Unclear | Yes | High |
| Marwah 2021 | Unclear | No | Unclear | Unclear | High |
| Masotti 2021 | Unclear | Unclear | Unclear | Unclear | Unclear |
| Mateos González 2021 | Unclear | Unclear | Yes | Unclear | Unclear |
| Meng 2021 | Yes | Yes | Unclear | Yes | Unclear |
| Muñoz‐Rodríguez 2021 | Unclear | Unclear | No | Yes | High |
| Öktem 2020 | Unclear | No | Unclear | Yes | High |
| Olivieri 2022 | Yes | Yes | Yes | Yes | Low |
| Ortega‐Rojas 2022 | Unclear | No | No | Yes | High |
| Ouldali 2021 | Unclear | Unclear | Unclear | Unclear | Unclear |
| Pan 2021 | Unclear | Unclear | Yes | Yes | Unclear |
| Papageorgiou 2022 | Yes | Yes | Unclear | Yes | Unclear |
| Para 2022 | Yes/Noa | Yes/Nob | Unclear | Yes | Unclear/Highc |
| Prasetya 2021 | Yes | No | Yes | No | High |
| Qu 2021 | Yes | No | Yes | Yes | High |
| Rasyid 2020 | Unclear | Unclear | Unclear | Unclear | Unclear |
| Şan 2021 | Unclear | Unclear | Unclear | Unclear | Unclear |
| Şener 2022 | Unclear | No | Unclear | Unclear | High |
| Sepulchre 2022 | Yes | Yes | Unclear | Unclear | Unclear |
| Simon 2022 | Unclear | Unclear | Unclear | Yes/Uncleard | Unclear |
| Sisó‐Almirall 2020 | Unclear | Unclear | Unclear | Unclear | Unclear |
| Soh 2020 | Unclear | Unclear | Yes | Yes | Unclear |
| Statsenko 2021 | Unclear | Yes | Yes | Yes | Unclear |
| Suardi 2020 | Unclear | Unclear | Unclear | Yes | Unclear |
| Suzuki 2022 | Yes | No | Unclear | Yes | High |
| Ullah 2020 | Unclear | Unclear | Unclear | Unclear | Unclear |
| Xia 2020 | Unclear | Unclear | Unclear | Unclear | Unclear |
| Zafar 2021 | Yes | Yes | Yes | Yes | Low |
| Zarębska‐Michaluk 2021 | Unclear | Unclear | Unclear | Yes | Unclear |
| Zhang 2020 | Yes | Yes | Unclear | Unclear | Unclear |
| Zhou 2020 | Yes | Yes | Yes | Yes | Low |
This table contains the answers to the signalling questions and risk of bias assessment of the fifth domain, Analysis of the QUAPAS tool.
aAll enroled participants were included in the analysis of ferritin increase, haemoglobin decrease, leucocytes count decrease, and platelet count decrease, but not LDH increase. bAppropriate methods were used for missing data in the analysis of ferritin increase, haemoglobin decrease, leucocytes count decrease, and platelet count decrease, but not LDH increase. cRisk of bias for analysis was unclear for ferritin increase, haemoglobin decrease, leucocytes count decrease, and platelet count decrease, and high LDH increase. dAppropriate methods were used for competing events for mortality; for deterioration this was unclear.
Findings
The findings of tests that were assessed in four or more studies are described below. A total of 47 laboratory tests were included for meta‐analyses, 16 for mortality, 26 for deterioration, and five for the combined outcome. The following laboratory tests were assessed for the different target conditions: alanine aminotransferase (ALT) increase, activated partial thromboplastin time (APTT) increase, aspartate aminotransferase (AST) increase, bilirubin increase, blood urea nitrogen increase, creatine kinase (CK) increase, creatinine increase, CRP increase, d‐dimer increase, eosinophile count decrease, ferritin increase, fibrinogen increase, haemoglobin decrease, lactate dehydrogenase (LDH) increase, lymphocyte absolute count decrease and increase, neutrophils absolute count increase, neutrophil‐to‐lymphocyte ratio (NLR) increase, platelet count decrease and increase, potassium decrease, procalcitonin increase, prothrombin time (PT) increase, sodium decrease, troponin I increase, troponin increase, and white blood cell count increase and decrease.
An overview of tests and cut‐off values is provided in Figure 4 and Figure 5. The summary findings, including the number of studies, median prevalence, fixed specificity and corresponding summary sensitivity can be found in the Table 1 (featuring, amongst others, hyperinflammatory markers) and Table 2 (including, amongst others, hypercoagulability markers).
4.

List of tests, cut‐off values, positive and negative predictive values, and prevalence per study (part A)
5.

List of tests, cut‐off values, positive and negative predictive values, and prevalence per study (part B)
Markers of inflammation
CRP increase
CRP increase was assessed for the target conditions: mortality, deterioration, and combined outcome.
For mortality (Figure 6), the cut‐off values to define CRP increase varied between 1 and 112 mg/L (14 studies). Sensitivity ranged from 36% to 100%, and specificity from 2% to 92%. At a fixed specificity of 27% (Q1), 59% (median), and 76% (Q3), meta‐analysis found a sensitivity of 90% (95% CI 86% to 92%), 76% (95% CI 73% to 79%), and 63% (95% CI 57% to 69%), respectively.
6.

Summary ROC plot to predict mortality: CRP increase and ferritin increase
CRP increase was assessed to predict deterioration (Figure 7). The cut‐off values in these studies varied between 6 and 101.15 mg/L (18 studies). Sensitivity ranged from 43% to 100%, and specificity from 34% to 94%. At a fixed specificity of 62% (Q1), 72% (median), and 81% (Q3), meta‐analysis found a sensitivity of 85% (95% CI 75% to 91%), 78% (95% CI 67% to 86%), and 67% (95% CI 53% to 78%), respectively.
7.

Summary ROC plot to predict deterioration: CRP increase and ferritin increase
CRP increase to predict a combined outcome (4 studies) had cut‐off values varying between 7.6 and 70 mg/L. Sensitivity ranged from 57% to 91%, and specificity from 41% to 69%. At a fixed specificity of 54% (Q1), 60% (median), and 64% (Q3), meta‐analysis found a sensitivity of 78% (95% CI 55% to 91%), 70% (95% CI 49% to 85%), and 64% (95% CI 39% to 83%), respectively.
Procalcitonin increase
Procalcitonin increase was assessed for the target conditions: mortality and deterioration.
For mortality (9 studies), the cut‐off values varied between 0.12 and 0.5 ng/mL. Sensitivity ranged between 17% and 85%, specificity between 59% and 98%. At a fixed specificity of 82% (Q1), 84% (median), and 93% (Q3), meta‐analysis found a sensitivity of 54% (95% CI 51% to 57%), 52% (95% CI 49% to 55%), and 38% (95% CI 31% to 46%), respectively.
For deterioration (7 studies), the cut‐off values to define procalcitonin increase ranged from 0.04 to 0. 5 ng/mL. Specificity ranged from 65% to 98%. At a fixed specificity of 86% (Q1), 93% (median), and 96% (Q3), meta‐analysis found a sensitivity of 40% (95% CI 25% to 58%), 25% (95% CI 17% to 35%), and 16% (95% CI 8% to 29%), respectively.
Ferritin increase
Ferritin increase was assessed for the target conditions: mortality and deterioration.
For mortality (Figure 6), the cut‐off values varied between 275 and 1658 ng/mL (9 studies). Sensitivity ranged from 20% to 98% and specificity from 22% to 87%. At a fixed specificity of 35% (Q1), 64% (median), and 79% (Q3), meta‐analysis found a sensitivity of 88% (95% CI 65% to 97%), 64% (95% CI 40% to 83%), and 42% (95% CI 17% to 73%), respectively.
For deterioration (Figure 7), the cut‐off values ranged from 275 to 1288.5 ng/mL (5 studies). Sensitivity ranged from 35% to 71%, and specificity from 67% to 90%. At a fixed specificity of 74% (Q1), 75% (median), and 77% (Q3), meta‐analysis found a sensitivity of 58% (95% CI 38% to 76%), 57% (95% CI 37% to 74%), and 53% (95% CI 35% to 71%), respectively.
Complete blood count
White blood cell count increase
White blood cell count increase was assessed for the target conditions: mortality, and deterioration.
For mortality (10 studies), the cut‐off values to define white blood cell count increase varied between 9.31 and 11 x 109 cells/L. Sensitivity in the included studies ranged from 16% to 74%, and specificity ranged from 50% to 94%. The median specificity was 82%, with the interquartile range from 61% to 89%. The summary estimate of sensitivity corresponding with a specificity of 61%, was 38% (95% CI 35% to 40%). The summary estimate of sensitivity corresponding with the median specificity of 82%, was 37% (95% CI 34% to 40%) and the summary estimate of sensitivity corresponding with a specificity of 89%, was 36% (95% CI 33% to 39%).
For deterioration (13 studies), the cut‐off values varied between 5.27 and 11 x 109 cells/L. At a fixed specificity of 75% (Q1), 89% (median), and 94% (Q3), meta‐analysis found a sensitivity of 40% (95% CI 35% to 44%), 32% (95% CI 28% to 36%), and 27% (95% CI 22% to 32%), respectively.
White blood cell count decrease
White blood cell count decrease was assessed for the target conditions: mortality, and deterioration.
For mortality (5 studies), four studies used a cut‐off value of 4 x 109 cells/L, and one study used 5.025 x 109 cells/L as the cut‐off value. Sensitivity ranged from 0% to 40%, and specificity from 39% to 97%. At a fixed specificity of 68% (Q1), 83% (median), and 87% (Q3), the summary estimates of sensitivity were 9% (95% CI 6% to 14%), 9% (95% CI 6% to 14%), and 9% (95% CI 6% to 14%), respectively.
For deterioration (6 studies), the cut‐off values ranged from 3.5 to 4 x 109 cells/L. Sensitivity ranged from 0% to 43%; two studies reported a sensitivity of 0%. Specificity ranged from 37% to 91%. Meta‐analysis yielded a sensitivity of 15% (95% CI 6% to 33%), 12% (95% CI 5% to 26%) and 8% (95% CI /) at a fixed specificity of 83% (Q1), 85% (median) and 88% (Q3), respectively.
Neutrophil count increase
For deterioration (5 studies), cut‐off values to define neutrophil count increase ranged between 4.7 and 10 x 109 cells/L. Sensitivity ranged from 17% to 62%, and specificity from 70% to 93%. The median specificity in these studies was 89% with an IQR from 88% to 93%. The summary estimates of sensitivity corresponding to these numbers were: 62% (95% CI 46% to 75%) at a specificity of 88%, 49% (95% CI 37% to 62%) at a specificity of 89%, and 41% (95% CI 28% to 55%) at a specificity of 93%.
Lymphocyte count increase
For deterioration (4 studies), cut‐off values ranged from 3.2 to 4.8 x 109 cells/L. Sensitivity in all four studies was 0%. The studies did not include any true positive cases at the reported cut‐off value. Specificity ranged from 98% to 100%. The summary estimate of sensitivity was 7% (95% CI 0% to 83%) at a specificity of 99% (identical for Q1, median and Q3).
Lymphocyte count decrease
Lymphocyte count decrease was assessed for the target conditions: mortality, deterioration, and combined outcome.
For mortality (Figure 8), the cut‐off values to define lymphocyte count decrease ranged from 0.7 to 1.5 x 109 cells/L (13 studies). Sensitivity ranged from 41% to 98%. Specificity ranged from 18% to 89%. At a fixed specificity of 48% (Q1), 61% (median), and 70% (Q3), meta‐analyses produced a sensitivity of 79% (95% CI 68% to 87%), 67% (95% CI 56% to 77%), and 56% (95% CI 43% to 68%), respectively.
8.

Summary ROC plot to predict mortality: lymphocyte count decrease and NLR increase
For deterioration (Figure 9), the cut‐off values ranged from 0.8 to 1.5 x 109 cells/L (14 studies). Sensitivity ranged from 40% to 88%. Specificity ranged from 32% to 91%. At a fixed specificity of 61% (Q1), 67% (median), and 74% (Q3), meta‐analysis yielded a sensitivity of 74% (95% CI 64% to 81%), 69% (95% CI 60% to 76%), and 62% (95% CI 52% to 71%), respectively.
9.

Summary ROC plot to predict deterioration: lymphocyte count decrease and NLR increase
For the combined outcome (4 studies), the cut‐off values ranged between 0.9 and 1.5 x 109 cells/L. Sensitivity ranged from 59% to 93%, and specificity from 18% to 69%. At a fixed specificity of 20% (Q1), 29% (median), and 46% (Q3), meta‐analysis yielded a sensitivity of 87% (95% CI 66% to 96%), 83% (95% CI 67% to 92%), and 76% (95% CI 58% to 88%), respectively.
NLR increase
NLR increase was assessed for both mortality and deterioration.
For mortality (Figure 8), the cut‐off values used in the different studies ranged from 3.5 to 11 (11 studies). Sensitivity ranged from 22% to 92%, and specificity from 38% to 92%. At a fixed specificity of 54% (Q1), 63% (median), and 78% (Q3), meta‐analysis yielded a sensitivity of 73% (95% CI 69% to 77%), 69% (95% CI 66% to 72%), and 59% (95% CI 52% to 66%), respectively.
For deterioration (Figure 9), the cut‐off values ranged from 3.5 to 11 (9 studies). Sensitivity ranged from 21% to 91%, and specificity from 42% to 89%. At a fixed specificity of 64% (Q1), 71% (median), and 82% (Q3), meta‐analysis yielded a sensitivity of 82% (95% CI 65% to 91%), 75% (95% CI 59% to 87%), and 59% (95% CI 38% to 77%), respectively.
Platelet count increase
For deterioration (6 studies), the cut‐off values ranged between 100 and 450 x 109 cells/L. One study reported a cut‐off value of 100 x 109 cells/L, and the cut‐off values of the other five studies ranged from 335 to 450 x109 cells/L. Sensitivity ranged from 1% to 29%; four studies reported a sensitivity below 10%. Specificity ranged from 88% to 98%. At a fixed specificity of 93% (Q1), 96% (median), and 98% (Q3), meta‐analysis yielded a sensitivity of 15% (95% CI 12% to 20%), 10% (95% CI 7% to 15%), and 6% (95% CI 5% to 8%), respectively.
Platelet count decrease
Platelet count decrease was assessed for the target conditions: mortality, deterioration, and the combined outcome.
For mortality (12 studies), the cut‐off values for platelet count decrease ranged from 100 to 209 x 109 cells/L. Sensitivity ranged from 7% to 60%, and specificity from 60% to 97%. At a fixed specificity of 79% (Q1), 88% (median), and 90% (Q3), meta‐analysis yielded a sensitivity of 32% (95% CI 26% to 39%), 21% (95% CI 17% to 26%), and 19% (95% CI 14% to 26%), respectively.
For deterioration (12 studies), the cut‐off value ranged from 125 to 174 x 109 cells/L. Sensitivity ranged from 17% to 100%. One study, with a cut‐off value 174 x 109 cells/L, reported a sensitivity of 100%; all other studies had a sensitivity below 40%. Specificity ranged from 56% to 97%. At a fixed specificity of 77% (Q1), 83% (median), and 90% (Q3), meta‐analysis yielded a sensitivity of 47% (95% CI 27% to 68%), 37% (95% CI 22% to 54%), and 22% (95% CI 12% to 38%), respectively.
For the combined outcome (4 studies), the cut‐off values ranged from 100 to 200 x 109 cells/L. Sensitivity ranged from 12% to 63%, and specificity from 57% to 96%. At a fixed specificity of 63% (Q1), 73% (median), and 86% (Q3), meta‐analysis yielded a sensitivity of 52% (95% CI 28% to 75%), 41% (95% CI 21% to 64%), and 24% (95% CI 1% to 88%), respectively.
Haemoglobin decrease
Haemoglobin decrease was assessed for the target conditions: mortality, and deterioration.
For mortality (5 studies), three studies used a different cut‐off value for female and male participants. The cut‐off value for female patients ranged from 11.5 to 12.1 g/dL, and from 12.5 to 13.8 g/dL for male participants. The other two studies reported a cut‐off value of 12.6 and 13.0 g/dL, respectively. Sensitivity ranged from 34% to 63%. Specificity ranged from 30% to 83%. At a fixed specificity of 41% (Q1), 60% (median), and 75% (Q3), meta‐analysis yielded a sensitivity of 52% (95% CI 43% to 61%), 45% (95% CI 40% to 51%), and 39% (95% CI 34% to 45%), respectively.
For deterioration (5 studies), one study reported a different cut‐off value for female and male participants, 12.1 g/dL and 13.8 g/dL, respectively. The cut‐off value of the other studies ranged from 10 to 12.8 g/dL. Sensitivity ranged from 3% to 66%, and specificity from 71% to 95%. At a fixed specificity of 77% (Q1), 77% (median), and 88% (Q3), meta‐analysis yielded a sensitivity of 47% (95% CI 5% to 94%), 47% (95% CI 5% to 94%), and 0% (95% CI 0% to 99%), respectively.
Eosinophil count decrease
For mortality (4 studies), one study did not report the cut‐off value for eosinophil count decrease. The cut‐off values of the other studies ranged from 0.01 to 0.15 x 109 cells/L. Sensitivity ranged from 37% to 96%, and specificity from 5% to 37%. At a fixed specificity of 13% (Q1), 25% (median), and 36% (Q3), meta‐analysis yielded a sensitivity of 91% (95% CI 14% to 100%), 75% (95% CI 5% to 99%), and 57% (95% CI 1% to 99%), respectively.
Liver function tests
ALT increase
ALT increase was assessed for the target conditions: mortality and deterioration.
For mortality (Figure 10), the cut‐off values ranged from 22 to 60 U/L (7 studies). One study used a different cut‐off value for female and male participants, 33 and 41 U/L, respectively. Sensitivity ranged from 13% to 85%, and specificity from 42% to 83%. At a fixed specificity of 57% (Q1), 65% (median), and 73% (Q3), meta‐analysis found a sensitivity of 60% (95% CI 37% to 79%), 45% (95% CI 29% to 61%), and 30% (95% CI 19% to 44%), respectively.
10.

Summary ROC plot to predict mortality: ALT increase and AST increase
For deterioration (Figure 11), the cut‐off values ranged from 22 to 78 U/L (11 studies). Sensitivity ranged from 13% to 90%. One study reported a sensitivity of 90%; the sensitivity of the other studies ranged from 13% to 46%. Specificity ranged from 54% to 90%. At a fixed specificity of 75% (Q1), 81% (median), and 88% (Q3), meta‐analysis found a sensitivity of 56% (95% CI 36% to 74%), 47% (95% CI 33% to 62%), and 34% (95% CI 26% to 43%), respectively.
11.

Summary ROC plot to predict deterioration: ALT increase and AST increase
AST increase
AST increase was assessed for the target conditions: mortality and deterioration.
For mortality (Figure 10), the cut‐off values varied between 40 and 60 U/L (6 studies). One study reported a separate cut‐off value for female and male participants, 32 and 40 U/L respectively. Sensitivity ranged from 27% to 69%, and specificity from 33% to 83%. At a fixed specificity of 51% (Q1), 64% (median), and 80% (Q3), meta‐analysis found a sensitivity of 69% (95% CI 41% to 87%), 58% (95% CI 36% to 77%), and 40% (95% CI 18% to 66%), respectively.
For deterioration (Figure 11), the cut‐off value varied between 34 and 40 U/L (11 studies). Sensitivity ranged from 23% to 76%, and specificity from 31% to 93%. At a fixed specificity of 71% (Q1), 81% (median), and 86% (Q3), meta‐analysis found a sensitivity of 60% (95% CI 47% to 71%), 48% (95% CI 36% to 60%), and 40% (95% CI 28% to 54%), respectively.
Total bilirubin increase
For deterioration (5 studies), the cut‐off values ranged between 11.9 and 21 μmol/L. Sensitivity ranged from 13% to 49%. Specificity ranged from 65% to 94%. At a fixed specificity of 74% (Q1), 90% (median), and 91% (Q3), meta‐analysis found a sensitivity of 40% (95% CI 16% to 70%), 16% (95% CI 6% to 35%), and 14% (95% CI 5% to 35%), respectively.
Biochemistry
Potassium decrease
For deterioration (Figure 12) (4 studies), one study reported a cut‐off value of 3.7 mmol/L; the other three studies reported a cut‐off value of 3.5 mmol/L. Sensitivity ranged from 16% to 85%. Specificity ranged from 55% to 95%. At a fixed specificity of 69% (Q1), 76% (median), and 83% (Q3), meta‐analysis found a sensitivity of 64% (95% CI 11% to 96%), 51% (95% CI 9% to 91%), and 35% (95% CI 5% to 84%), respectively.
12.

Summary ROC plot to predict deterioration: potassium decrease and sodium decrease
Sodium decrease
For deterioration (Figure 12). The cut‐off values ranged from 134 to 138 mmol/L (4 studies). Sensitivity ranged from 18% to 95%. Specificity ranged from 39% to 91%. At a fixed specificity of 52% (Q1), 59% (median), and 69% (Q3), meta‐analysis found a sensitivity of 75% (95% CI 14% to 98%), 67% (95% CI 13% to 97%), and 53% (95% CI 8% to 93%), respectively.
Coagulation markers
APTT increase
For deterioration (5 studies), the cut‐off values ranged between 37.05 and 40 sec. Sensitivity ranged from 3% to 80%, and specificity from 27% to 98%. At a fixed specificity of 73% (Q1), 80% (median), and 90% (Q3), meta‐analysis found a sensitivity of 41% (95% CI 28% to 56%), 34% (95% CI 23% to 48%), and 22% (95% CI 12% to 37%), respectively.
PT increase
For deterioration (4 studies), the cut‐off values to define PT increase ranged between 9.78 and 16 sec. Sensitivity ranged from 0% to 79%. One study had a sensitivity of 0%. This study did not report any severe participants above the reported cut‐off value. Specificity ranged from 40% to 99%. At a fixed specificity of 77% (Q1), 92% (median), and 95% (Q3), meta‐analysis found a sensitivity of 37% (95% CI 18% to 60%), 13% (95% CI 4% to 34%), and 8% (95% CI 2% to 28%), respectively.
D‐dimer increase
D‐dimer increase was assessed in the target conditions: mortality, deterioration, and the combined outcome.
For mortality (Figure 13), the cut‐off values ranged between 0.5 and 1.71 mg/L (14 studies). Sensitivity ranged from 46% to 95%, and specificity from 8% to 74%. At a fixed specificity of 48% (Q1), 56% (median), and 63% (Q3), meta‐analysis found a sensitivity of 77% (95% CI 71% to 83%), 70% (95% CI 64% to 76%), and 62% (95% CI 56% to 69%), respectively.
13.

Summary ROC plot to predict mortality: LDH increase and D‐dimer increase
For deterioration (Figure 14), the cut‐off values in these studies ranged between 0.14 and 1.15 mg/L (10 studies). Sensitivity ranged from 30% to 90%. Specificity ranged from 32% to 80%. At a fixed specificity of 46% (Q1), 63% (median), and 71% (Q3), meta‐analysis found a sensitivity of 80% (95% CI 69% to 88%), 65% (95% CI 56% to 74%), and 56% (95% CI 44% to 67%), respectively.
14.

Summary ROC plot to predict deterioration: D‐dimer increase and LDH increase
For a combined outcome, the cut‐off values ranged between 0.5 and 1.3 mg/L (4 studies). Sensitivity ranged from 55% to 73%, and specificity from 49% to 60%. At a fixed specificity of 52% (Q1), 54% (median), and 57% (Q3), meta‐analysis found a sensitivity of 67% (95% CI 55% to 76%), 65% (95% CI 52% to 76%), and 62% (95% CI 46% to 76%), respectively.
Fibrinogen increase
For deterioration (Figure 15), the cut‐off values ranged between 4.34 and 8.43 g/L (5 studies). Sensitivity ranged between 40% and 97%. Specificity ranged between 5% and 91%. At a fixed specificity of 22% (Q1), 49% (median), and 78% (Q3), meta‐analysis found a sensitivity of 90% (95% CI 79% to 95%), 77% (95% CI 66% to 86%), and 56% (95% CI 40% to 71%), respectively.
15.

Summary ROC plot to predict deterioration: fibrinogen increase
Kidney function tests
Serum creatinine increase
For mortality (Figure 16), the cut‐off values varied between 68.63 and 133 μmol/L (7 studies). One study used a different threshold for female and male cases, 68.63 and 91.51 μmol/L respectively. Sensitivity ranged from 17% to 68%, and specificity from 70% to 100%. At a fixed specificity of 73% (Q1), 79% (median), and 94% (Q3), meta‐analysis found a sensitivity of 43% (95% CI 40% to 45%), 43% (95% CI 40% to 45%), and 42% (95% CI 40% to 45%), respectively.
16.

Summary ROC plot to predict mortality: creatinine increase and troponin I increase
For deterioration (6 studies), cut‐off values ranged between 68 and 133 μmol/L. Sensitivity ranged between 5% and 53%. Specificity ranged between 58% and 99%. At a fixed specificity of 93% (Q1), 97% (median), and 97% (Q3), meta‐analysis found a sensitivity of 19% (95% CI 1% to 81%), 9% (95% CI 0% to 67%), and 9% (95% CI 0% to 67%), respectively.
Blood urea nitrogen increase
For deterioration (5 studies), the cut‐off values varied between 8.2 and 20 mmol/L. Sensitivity ranged between 3% and 50%. Specificity ranged between 53% and 100%. At a fixed specificity of 90% (Q1), 94% (median), and 96% (Q3), meta‐analysis found a sensitivity of 32% (95% CI 23% to 42%), 27% (95% CI 16% to 42%), and 23% (95% CI 12% to 40%), respectively.
Cardiac markers
Troponin increase
Troponin I was assessed for mortality (Figure 16). The cut‐off values ranged from 0.13 to 300 pg/mL (4 studies). One study defined troponin I increase as values above the sex‐specific 99th percentile upper reference limit. Sensitivity ranged from 12% to 98%, and specificity from 13% to 97%. At a fixed specificity of 57% (Q1), 78% (median), and 88% (Q3), meta‐analysis found a sensitivity of 84% (95% CI 65% to 93%), 64% (95% CI 47% to 78%), and 42% (95% CI 28% to 58%), respectively.
Troponin was assessed for deterioration (4 studies). The studies did not differentiate between troponin I or T. The cut‐off values ranged from 0.35 to 20 ng/mL. One study defined troponin increase as values above the sex‐specific 99th percentile upper reference limit. Sensitivity ranged from 8% to 40%. Specificity ranged from 74% to 100%. At a fixed specificity of 75% (Q1), 76% (median), and 83% (Q3), meta‐analysis found a sensitivity of 34% (95% CI 22% to 50%), 34% (95% CI 22% to 49%), and 30% (95% CI 18% to 45%), respectively.
Other tests
LDH increase
LDH can be indicative of tissue damage, or inflammation. LDH increase was assessed for the target conditions: mortality, deterioration, and the combined outcome.
For mortality (Figure 13), the cut‐off values ranged from 5 to 629.50 U/L (10 studies). One study reported a cut‐off value of 5 U/L; without this study, cut‐off values ranged from 225 to 629.50 U/L. One study reported a different cut‐off value for females and males, 225 and 241 U/L respectively. The sensitivity ranged from 55% to 95%, and specificity from 0% to 89%. The study with a cut‐off value of 5 U/L reported a specificity of 0%. At a fixed specificity of 19% (Q1), 60% (median), and 80% (Q3), meta‐analysis found a sensitivity of 91% (95% CI 80% to 96%), 82% (95% CI 66% to 91%), and 74% (95% CI 53% to 88%), respectively.
For deterioration (Figure 14), the cut‐off values ranged from 190 to 285 U/L (12 studies). Sensitivity ranged from 53% to 97%. Specificity ranged from 5% to 82%. At a fixed specificity of 51% (Q1), 66% (median), and 78% (Q3), meta‐analysis found a sensitivity of 80% (95% CI 76% to 83%), 79% (95% CI 76% to 82%), and 79% (95% CI 75% to 81%), respectively.
For a combined outcome (4 studies), the cut‐off values ranged between 250 and 400 U/L. Sensitivity ranged from 54% to 79%, and specificity from 45% to 82%. At a fixed specificity of 57% (Q1), 62% (median), and 68% (Q3), meta‐analysis found a sensitivity of 72% (95% CI 52% to 86%), 69% (95% CI 51% to 82%), and 64% (95% CI 45% to 80%), respectively.
CK increase
An increase in CK levels in the blood can indicate muscle or heart damage. For deterioration (8 studies), the cut‐off values ranged from 92.5 to 308 U/L. Sensitivity ranged from 19% to 87%. Specificity ranged from 66% to 91%. At a fixed specificity of 78% (Q1), 86% (median), and 87% (Q3), meta‐analysis found a sensitivity of 61% (95% CI 29% to 86%), 38% (95% CI 14% to 70%), and 35% (95% CI 12% to 68%), respectively.
Comparisons between tests
For the target condition mortality, we found five tests with median specificity and corresponding summary sensitivity, including their 95% CI, above 50%. These were CRP increase, d‐dimer increase, LDH increase, lymphocyte count decrease, and NLR increase. For deterioration, we found the same five tests with median specificity and corresponding summary sensitivity, including their 95% CI, above 50%. For the combined outcome, we found two tests with median specificity and corresponding summary sensitivity, including their 95% CI, above 50%. The tests were d‐dimer increase and LDH increase. The test accuracies of these index tests were compared with a direct, head‐to‐head comparison (i.e. assessed two of the biomarkers in the same participants), and an indirect comparison, in which all studies reporting on these tests were included (Table 3).
Mortality: indirect comparisons
CRP increase, LDH increase, and d‐dimer increase
We compared the test performance of CRP increase (14 studies, 1977 cases/9834 non‐cases), LDH increase (10 studies, 2786 cases/14,208 non‐cases), and d‐dimer increase (14 studies, 5565 cases/28,556 non‐cases) using all available studies in an indirect comparison. The median specificity of these three tests was 56% (IQR 36% to 74%). Within the indirect comparisons, LDH was used as a reference test, and therefore the test performance of LDH increase was compared to CRP and to d‐dimer increase, respectively. The RDOR for CRP and LDH increase was 1.62 (P = 0.1473, 95% CI 0.8439 to 3.0984). The RDOR for d‐dimer and LDH increase was 1.26 (P = 0.2565, 95% CI 0.8470 to 1.8627).
CRP increase, d‐dimer increase, and lymphocyte count decrease
We compared the test performance of CRP increase (14 studies, 1977 cases/9834 non‐cases), d‐dimer increase (14 studies, 5565 cases/28,556 non‐cases), and lymphocyte count decrease (13 studies, 3241 cases/21,207 non‐cases) using all available studies in an indirect comparison. The median specificity of these three tests was 57% (IQR 40% to 70%). Within the indirect comparisons, lymphocyte count decrease was used as a reference test, and therefore the test performance of lymphocyte count decrease was compared to CRP and to d‐dimer increase, respectively. The RDOR for CRP increase and lymphocyte count decrease was 3.17 (P = 0.1967, 95% CI 0.5495 to 18.2844). The RDOR for d‐dimer and lymphocyte count decrease increase was 1.08 (P = 0.6321, 95% CI 0.7834 to 1.4943).
CRP increase, LDH increase, and lymphocyte count decrease
The test performance of CRP increase (14 studies, 1977 cases/9834 non‐cases), LDH increase (10 studies, 2786 cases/14,208 non‐cases), and lymphocyte count decrease (13 studies, 3241 cases/21,207 non‐cases) was compared in an indirect comparison. The median specificity of these three tests was 61% (IQR 28% to 77%). Lymphocyte count decrease was used as the reference test, and therefore the test performance of lymphocyte count decrease was compared to CRP and to LDH increase, respectively. The RDOR CRP increase and lymphocyte count decrease was 1.55 (P = 0.501, 95% CI 0.4343 to 5.5019). The RDOR for LDH increase and lymphocyte count decrease was 1.07 (P = 0.8528, 95% CI 0.5033 to 2.2932).
CRP increase, d‐dimer increase, and NLR increase
17.

Summary ROC plot to predict mortality: NLR increase, CRP increase, and D‐dimer increase
We compared the test performance of CRP increase (14 studies, 1977 cases/9834 non‐cases), d‐dimer increase (14 studies, 5565 cases/28,556 non‐cases), and NLR increase (11 studies, 1369 cases/6092 non‐cases). The median specificity of these three tests was 58% (IQR 44% to 73%). D‐dimer was used as a reference test. The indirect comparison of CRP increase and d‐dimer yielded an RDOR of 2.83 (P = 0.12, 95% CI 0.7622 to 10.506). RDOR was 2.05 (P = 0.0022, 95% CI 1.2971 to 3.2449) within the indirect comparison of NLR and d‐dimer. Therefore, NLR increase was found to have a higher test accuracy to predict mortality in participants with SARS‐CoV‐2 compared to d‐dimer increase.
CRP increase, lymphocyte count decrease, and NLR increase
We compared the test performance of CRP increase (14 studies, 1977 cases/9834 non‐cases), lymphocyte count decrease (13 studies, 3241 cases/21,207 non‐cases), and NLR increase (11 studies, 1369 cases/6092 non‐cases). The median specificity of these three tests was 61% (IQR 42% to 76%). Lymphocyte count decrease was used as the reference test. The RDOR for CRP increase and lymphocyte count decrease was 2.19 (P = 0.3684, 95% CI 0.3956 to 12.1581). The RDOR for NLR increase and lymphocyte count decrease was 2.63 (P = 0.0003, 95% CI 1.5504 to 4.4558). So, NLR increase was found to have a higher test performance compared to lymphocyte count decrease to predict mortality.
D‐dimer increase, LDH increase, and lymphocyte count decrease
Test performance of d‐dimer increase (14 studies, 5565 cases/28,556 non‐cases), LDH increase (10 studies, 2786 cases/14,208 non‐cases), and lymphocyte count decrease (13 studies, 3241 cases/21,207 non‐cases) was compared. The median specificity of these three tests was 56% (IQR 40% to 70%). LDH was used as the reference test. The RDOR for d‐dimer increase and LDH increase was 1.42 (P = 0.0212, 95% CI 1.0535 to 1.9008), so d‐dimer increase had a higher test accuracy to predict mortality. The RDOR for lymphocyte count decrease and LDH increase was 1.24 (P = 0.2578, 95% CI 0.8529 to 1.8095).
D‐dimer increase, lymphocyte count decrease, and NLR increase
Test performance of d‐dimer increase (14 studies, 5565 cases/28,556 non‐cases), lymphocyte count decrease (13 studies, 3241 cases/21,207 non‐cases), and NLR increase (11 studies, 1369 cases/6092 non‐cases) was compared. The median specificity of these three tests was 60% (IQR 49% to 70%). Lymphocyte count decrease was the reference test. The RDOR for d‐dimer increase and lymphocyte count decrease was 1.49 (P < 0.0001, 95% CI 1.2346 to 1.7999). The RDOR of NLR increase and lymphocyte count decrease was 2.41 (P < 0.0001, 95% CI 1.7926 to 3.2499). Both d‐dimer increase and NLR increase had higher test accuracy to predict mortality compared to lymphocyte count decrease.
Mortality: direct comparisons
CRP increase and d‐dimer increase
18.

Summary ROC plot to predict mortality in head‐to‐head comparisons: CRP increase and D‐dimer increase
We compared the test performance of CRP increase and d‐dimer increase using a head‐to‐head comparison. Nine studies (1460 cases/6949 non‐cases) assessed the two tests for mortality. The median specificity of the two tests in these studies was 48% (IQR 27% to 62%). CRP increase was used as a reference test for the comparison of the test performance. The RDOR was 1.31 (P = 0.0255, 95% CI 1.0334 to 1.6531), meaning the test accuracy of d‐dimer increase to predict mortality was slightly higher compared to CRP increase.
CRP increase and LDH increase
Seven studies (1156 cases/5934 non‐cases) directly compared CRP and LDH increase. The median specificity in these studies was 45% (IQR 14% to 79%). With LDH as the reference test, RDOR was 1.01 (P = 0.9763, 95% CI 0.7039 to 1.436).
CRP increase and lymphocyte count decrease
Seven studies (1305 cases/6962 non‐cases) assessed CRP increase and lymphocyte count decrease. The median specificity was 57% (IQR 27% to 74%). With lymphocyte count decrease as the reference test, RDOR was 1.23 (P = 0.0635, 95% CI 0.9885 to 1.5286).
CRP increase and NLR increase
Five studies (1037 cases/5035 non‐cases) assessed both CRP increase and NLR increase. The median specificity was 57% (IQR 38% to 75%). CRP increase was the reference test, and RDOR was 2.64 (P < 0.0001, 95% CI 2.0888 to 3.3274). Therefore, the test accuracy of NLR increase to predict mortality was higher compared to the test accuracy of CRP increase.
D‐dimer increase and LDH increase
Seven studies (2680 cases/13,822 non‐cases) assessed d‐dimer and LDH increase. The median specificity was 54% (IQR 31% to 65%). The direct comparison with LDH as reference tests yielded a RDOR of 1.05 (P = 0.5529, 95% CI 0.9022 to 1.2118).
D‐dimer increase and lymphocyte count decrease
19.

Summary ROC plot to predict mortality in head‐to‐head comparisons: lymphocyte count decrease and D‐dimer increase
Seven studies (2712 cases/14,170 non‐cases) assessed both d‐dimer increase and lymphocyte count decrease. The median specificity was 51% (IQR 42% to 66%). Lymphocyte count decrease was the reference test. The direct comparison yielded a RDOR of 1.28 (P = 0.0003, 95% CI 1.1214 to 1.4608). Therefore, test accuracy of d‐dimer was higher compared to the lymphocyte count decrease.
LDH increase and lymphocyte count decrease
Six studies (2443 cases/13,369 non‐cases) assessed LDH increase and lymphocyte count decrease. The median specificity was 60% (IQR 43% to 73%). Lymphocyte count decrease was the reference test. The direct comparison yielded a RDOR of 1.30 (P = 0.0002, 95% CI 1.1323 to 1.491), and therefore, the test accuracy of LDH increase was higher compared to lymphocyte count decrease.
Lymphocyte count decrease and NLR increase
Five studies (970 cases/3796 non‐cases) assessed lymphocyte count and NLR increase. The median specificity was 58% (IQR 53% to 63%). With lymphocyte count decrease as the reference test, RDOR was 1.84 (P < 0.0001, 95% CI 1.386 to 2.4412). Meaning that the test accuracy of NLR increase was higher than the test accuracy of lymphocyte count decrease.
D‐dimer increase and NLR increase, and LDH increase and NLR increase were not compared in a direct comparison, because less than four studies were available for analyses.
Deterioration: indirect comparisons
CRP increase, d‐dimer increase, and lymphocyte count decrease
We compared the test performance of CRP increase (18 studies, 1221 cases/4819 non‐cases), d‐dimer increase (10 studies, 651 cases/1970 non‐cases), and lymphocyte count decrease (14 studies, 1101 cases/8698 non‐cases) using all available studies in an indirect comparison. The median specificity of the tests in these studies was 68% (IQR 60% to 75%). D‐dimer was the reference test. The RDOR for CRP and d‐dimer increase was 1.76 (P = 0.0013, 95% CI 1.2478 to 2.4956). The RDOR for lymphocyte count decrease and d‐dimer increase was 1.54 (P = 0.0198, 95% CI 1.0715 to 2.2233). Therefore, both CRP increase and lymphocyte count decrease had a higher test accuracy to predict deterioration to severe disease compared to d‐dimer increase.
CRP increase, LDH increase, and lymphocyte count decrease
Test performance of CRP increase (18 studies, 1221 cases/4819 non‐cases), LDH increase (12 studies, 780 cases/2838 non‐cases), and lymphocyte count decrease (14 studies, 1101 cases/8698 non‐cases) was compared. The median specificity was 69% (IQR 60% to 78%). LDH was the reference test. The RDOR for CRP and LDH increase was 1.33 (P = 0.1472, 95% CI 0.9053 to 1.9413). The RDOR for lymphocyte count decrease and LDH increase was 1.48 (P = 0.1164, 95% CI 0.9077 to 2.3988).
CRP increase, d‐dimer increase, and LDH‐increase
Test performance of CRP increase (18 studies, 1221 cases/4819 non‐cases), d‐dimer increase (10 studies, 651 cases/1970 non‐cases), and LDH increase (12 studies, 780 cases/2838 non‐cases) was compared in an indirect comparison. The median specificity was 68% (IQR 55% to 77%). LDH was the reference test. The RDOR for CRP and LDH increase was 1.97 (P = 0.1126, 95% CI 0.8521 to 4.5661). The RDOR for d‐dimer and LDH increase was 1.01 (P = 0.9694, 95% CI 0.5955 to 1.7143).
CRP increase, d‐dimer increase, and NLR increase
20.

Summary ROC plot to predict deterioration: CRP increase, D‐dimer increase and NLR increase
Test performance of CRP increase (18 studies, 1221 cases/4819 non‐cases), d‐dimer increase (10 studies, 651 cases/1970 non‐cases), and NLR increase (9 studies, 673 cases/2034 non‐cases) was compared. The median specificity was 70% (IQR 61% to 79%). D‐dimer was the reference test. The RDOR for CRP and d‐dimer increase was 1.77 (P = 0.0166, 95% CI 1.1100 to 2.8341). The RDOR for NLR and d‐dimer increase was 1.79 (P = 0.041, 95% CI 1.0241 to 3.1113). Both CRP increase and NLR increase had a higher test accuracy compared to d‐dimer increase to predict deterioration in COVID‐19 disease.
CRP increase, NLR increase, and lymphocyte count decrease
Test performance of CRP increase (18 studies, 1221 cases/4819 non‐cases), NLR increase (9 studies, 673 cases/2034 non‐cases), and lymphocyte count decrease (14 studies, 1101 cases/8698 non‐cases) was compared. The median specificity was 70% (IQR 61% to 82%). Lymphocyte count was the reference test. The RDOR for CRP increase and lymphocyte count decrease was 1.13 (P = 0.3719, 95% CI 0.8652 to 1.4724). The RDOR for NLR increase and lymphocyte count decrease was 1.32 (P = 0.1246, 95% CI 0.9253 to 1.8938).
D‐dimer increase, NLR increase, and lymphocyte count decrease
Test performance of d‐dimer increase (10 studies, 651 cases/1970 non‐cases), NLR increase (9 studies, 673 cases/2034 non‐cases), and lymphocyte count decrease (14 studies, 1101 cases/8698 non‐cases) was compared. The median specificity was 68% (IQR 61% to 75%). D‐dimer was the reference test. The RDOR for NLR increase and d‐dimer increase was 2.77 (P = 0.0004, 95% CI 1.5832 to 4.8431). The RDOR for lymphocyte count decrease and d‐dimer increase was 1.70 (P = 0.0101, 95% CI 1.1355 to 2.5578). Therefore, both NLR increase and lymphocyte count decrease had a higher test accuracy compared to d‐dimer increase to predict deterioration in COVID‐19 disease.
D‐dimer increase, LDH‐increase, and lymphocyte count decrease
21.

Summary ROC plot to predict deterioration: D‐dimer increase, LDH increase, and lymphocyte count decrease
Test performance of d‐dimer increase (10 studies, 651 cases/1970 non‐cases), LDH increase (12 studies, 780 cases/2838 non‐cases), and lymphocyte count decrease (14 studies, 1101 cases/8698 non‐cases) was compared. The median specificity was 66% (IQR 57% to 76%). D‐dimer was the reference test. The RDOR for LDH and d‐dimer increase was 1.21 (P = 0.4457, 95% CI 0.7404 to 1.9795). The RDOR for lymphocyte count decrease and d‐dimer increase was 2.02 (P = 0.0003, 95% CI 1.3870 to 2.9402). Lymphocyte count decrease had a higher test accuracy compared to d‐dimer increase.
Deterioration direct comparisons
CRP increase and d‐dimer increase
22.

Summary ROC plot to predict deterioration in head‐to‐head comparisons: CRP increase and D‐dimer increase
Seven studies (552 cases/1623 non‐cases) directly compared CRP and d‐dimer increase for deterioration. The median specificity in these studies was 67% (IQR 46% to 73%). With d‐dimer as the reference test, RDOR was 1.64 (P = 0.0021, 95% CI 1.1973 to 2.2463). This meant that the test accuracy of CRP increase to predict mortality was slightly higher compared to d‐dimer increase.
CRP increase and LDH increase
Seven studies (372 cases/1345 non‐cases) directly compared CRP and LDH increase for deterioration. The median specificity was 72% (IQR 65% to 77%). LDH was the reference test. RDOR was 1.13 (P = 0.5352, 95% CI 0.7717 to 1.6466).
CRP increase and lymphocyte count decrease
Nine studies (640 cases/2420 non‐cases) directly compared CRP increase and lymphocyte count decrease. The median specificity was 69% (IQR 62% to 82%). Lymphocyte count was the reference test. RDOR was 1.04 (P = 0.7934, 95% CI 0.7886 to 1.3644).
D‐dimer and LDH increase
Five studies (338 cases/1076 non‐cases) directly compared d‐dimer increase and LDH increase. The median specificity was 62% (IQR 49% to 70%). D‐dimer was the reference test. RDOR was 1.21 (P = 0.3795, 95% CI 0.7937 to 1.8331).
D‐dimer increase and lymphocyte count decrease
23.

Summary ROC plot to predict deterioration in head‐to‐head comparisons: D‐dimer increase and lymphocyte count decrease
In six studies (379 cases/1237 non‐cases), both d‐dimer increase and lymphocyte count decrease were assessed. The median specificity was 64% (IQR 55% to 73%). D‐dimer was the reference test. RDOR was 2.10 (P = 0.0001, 95% CI 1.4411 to 3.0679). Therefore, lymphocyte count decrease had a higher test accuracy compared to d‐dimer increase.
LDH increase and lymphocyte count decrease
Seven studies (393 cases/1443 non‐cases) assessed both LDH increase and lymphocyte count decrease. The median specificity was 66% (IQR 49% to 69%). LDH was the reference test. RDOR was 2.22 (P < 0.0001, 95% CI 1.5154 to 3.2625), meaning lymphocyte count decrease had a higher test accuracy compared to LDH increase.
CRP increase and NLR increase, d‐dimer increase and NLR increase, LDH increase and NLR increase, lymphocyte count decrease and NLR increase were not compared in a direct comparison, because fewer than four studies were available for analyses.
For the combined outcome, not enough studies were available to perform comparisons.
Due to data scarcity, we did not investigate whether prognostic accuracy varied according to specific measurement or test, reference standard, baseline severity, timing of outcome verification, sample type, study design, and setting, including prevalence of the target condition. Moreover, we were not able to assess the difference in test accuracy using likelihood ratio tests for comparisons of models with and without covariate terms, because fewer than 10 primary studies were available for the direct comparisons. We did not have enough data available to investigate the following sources of heterogeneity: measurement technique or test type, reference standard, timing of outcome verification, sample type, study design, and setting, including prevalence of the target condition. Furthermore, there were insufficient data available to perform sensitivity analyses considering the impact of unpublished studies, and to investigate the impact of prospective versus retrospective data collection. Finally, we did not identify studies that we knew existed but for which we have not managed to locate reports.
Discussion
Summary of main results
We aimed to assess the prognostic accuracy of routine laboratory tests for predicting mortality and deterioration to severe or critical disease in patients with SARS‐CoV‐2 infection. Our analysis included data from 64 studies, evaluating 53 different laboratory tests. Within our meta‐analysis, we did not find any tests with high sensitivity or specificity, or both. So none of the tests were able to safely rule out an adverse outcome. Five tests that demonstrated sensitivity‐specificity above 50% for predicting adverse outcomes were: CRP increase, NLR increase, lymphocyte count decrease, d‐dimer increase, and LDH increase. Within the comparisons of the target condition of mortality, we found NLR increase to have a higher prognostic accuracy compared to d‐dimer increase, CRP‐increase, and lymphocyte count decrease. D‐dimer increase had a higher prognostic accuracy compared to lymphocyte count decrease, CRP increase, and LDH increase. LDH increase had a higher prognostic accuracy compared to lymphocyte count decrease. Within the comparisons of the target condition deterioration to severe disease, we found CRP increase to have a higher prognostic accuracy compared to d‐dimer increase. NLR increase had a higher prognostic accuracy compared to d‐dimer increase. Lymphocyte count decrease had a higher prognostic accuracy compared to d‐dimer increase and LDH increase. For the combined outcome, not enough studies were available for performing comparisons.
CRP increase, NLR increase, lymphocyte count decrease, D‐dimer increase, and LDH increase demonstrated good accuracy for predicting mortality and deterioration to severe or critical disease in patients with SARS‐CoV‐2 infection. Severe COVID‐19 often presents a high inflammatory response, which can lead to a cascade of events, such as tissue damage, impaired immune function, and hypercoagulability (Sriram 2021).
First, CRP is an acute‐phase protein that increases in response to systemic inflammation. In patients with severe COVID‐19 disease, there is often an important inflammatory response, which is found to be associated with critical illness, and mortality in COVID‐19 disease (Smilowitz 2021). Furthermore, NLR is calculated by dividing the absolute neutrophil count by the absolute lymphocyte count, and reflects the balance between immunity and systemic inflammation. An increase in NLR suggests an increased inflammatory state, which can be seen in severe COVID‐19 (Feng 2020). NLR is known as a prognostic biomarker in different diseases (Song 2021). Lymphocyte count measures the number of lymphocytes in the blood. They play an important role in the immune response. In patients with severe COVID‐19, a decrease, particularly in CD4+ and CD8+ T cells, can often be observed. Furthermore, it is seen to be associated with worse disease (Lai 2022). D‐Dimer is a small protein fragment, produced when a blood clot degrades. Increased d‐dimer levels can be indicative of increased blood coagulation and fibrinolysis, which can lead to thrombotic complications in COVID‐19 disease, such as pulmonary embolism and disseminated intravascular coagulation (Abou‐Ismail 2020; Kichloo 2020). LDH is a non‐specific marker of tissue damage and inflammation. It can increase because of tissue damage due to injury, disease, or infection. An increase in patients with severe COVID‐19 can reflect extensive pulmonary damage in patients with severe COVID‐19 (Nakakubo 2023). Moreover, LDH can identify lung injury and severe COVID‐19 pneumonia (Han 2020; Kojima 2023).
In summary, these five laboratory tests can provide information on the inflammatory status and thrombotic tendencies of a patient. However, because most tests present with low sensitivity and specificity, it is important to note that in many cases, sometimes 50% or more, adverse outcomes will be missed. Furthermore, the results of this review should be interpreted with caution since we found certainty of evidence to be from very‐low to low in many of the included biomarkers.
Strengths and weaknesses of the review
The prognostic accuracy of different routine laboratory tests to predict mortality or deterioration to severe or critical COVID‐19 disease in patients with SARS‐CoV‐2 was described. We performed a comprehensive review of the literature, with the use of meta‐analysis to pool data. A range of different cut‐off values for the laboratory tests was reported in the included studies. However, by using the HSROC analysis, summary sensitivity at a chosen specificity could be computed per test. The HSROC model takes into account within and between‐study variations.
The review results, however, presented some important limitations. There was a significant amount of heterogeneity in study design, patient population, and laboratory test methods. The reporting on flow and timing in the different studies was limited. In many studies, the time horizon between assessment of the index test and the target condition was not reported. This could lead to unreliable results concerning the prognostic accuracy of the tests. Furthermore, deterioration to severe disease was described in various ways, leading to heterogeneity between studies. Amongst other things, this leads to an important risk of bias and concern for applicability in almost all studies, leading to low to very low certainty of evidence.
Furthermore, following screening, we were unable to include studies focusing on outpatients. This limitation may have affected our comprehensive understanding of the prognostic accuracy of various biomarkers across different participant groups. Additionally, different biomarkers included in this review can also be increased or decreased in other infectious diseases or in non‐communicable diseases. In this review, we excluded studies that only recruited participants with a specific pre‐existing condition, such as diabetes or kidney failure. However, in the included studies, we did not assess the effect of comorbidity within our analyses. Moreover, in the specific context of our review, signalling question 4.2 of the QUAPAS tool (“Was treatment avoided after the test was performed?”) will unlikely ever score positive. Omitting the question would change the risk of bias in this domain to unclear in 18 studies; however, it would not change the risk of bias to low in any study. We believe the QUAPAS tool to be the correct tool to assess the methodological quality within our review, given the focus on prognostic accuracy.
Nowadays, most patients are vaccinated, which can influence mortality or deterioration towards severe COVID‐19 disease, or both. In this review, most patients were presumed to be unvaccinated, given the time period of participant recruitment for the individual studies prior to widespread vaccination campaigns. This could therefore be a limitation to the applicability of this review. Lastly, within our study we could not assess the effect of different variants of concern on disease progression. The study of Meletis 2023 found that laboratory tests may change over the course of the pandemic when different variants of concern emerge.
Applicability of findings to the review question
These routine laboratory tests can be used for the risk‐stratification of patients presenting with a SARS‐CoV‐2 infection. Data on different laboratory tests were included in this review. Although we tried to only include routine laboratory tests, the availability of the tests can depend on the type of hospital, department and other available resources. We included participants with a SARS‐CoV‐2 infection. Although it is well‐established that comorbidities and age can significantly impact the prognosis of patients with COVID‐19 disease, our review intentionally only focused on patients representative of those seeking help in ambulatory care settings, excluding patients with specific comorbidities, such as end‐stage renal disease and cancer.
Since the beginning of the pandemic, the SARS‐CoV‐2 virus has been known to have different variants of concern, which can influence the severity of the disease. We tried to include this in our review, but data on the variant of concern were not reported in the included studies, except for one. Furthermore, in December 2020, the first SARS‐CoV‐2 vaccine was administered. The vaccine has been proven to be effective against mortality and severe disease. In this review, almost all participants were included in 2020. Information about laboratory tests in vaccinated participants is therefore limited.
Although the WHO has declared that the pandemic has subsided (WHO 2023b), the insights gleaned from our analysis could serve as a valuable resource in preparing for potential future waves of the pandemic or other infectious disease outbreaks. By comprehensively evaluating the prognostic accuracy of various laboratory tests during the COVID‐19 crisis, we aimed to provide clinicians and researchers with evidence‐based insights that could inform more effective diagnostic and prognostic approaches for similar infectious diseases in the future. Our review not only examined the accuracy of various laboratory tests, but also critically evaluated the methodological quality of the included articles. Through the assessment of different biases, we identified crucial areas for improvement in future research endeavours. Our emphasis on standardised cut‐off values and test methodologies serves as a guideline for the design of robust studies, ensuring the reliability and comparability of results in the field.
Authors' conclusions
Implications for practice.
These five tests (CRP, NLR, lymphocyte count, d‐dimer, and LDH) are often part of an initial work‐up plan when patients with a SARS‐CoV‐2 infection present at the emergency room. Physicians can consider these laboratory tests as valuable tools for risk stratification in patients with COVID‐19 disease, together with clinical signs and symptoms. Early identification of patients with high risk of mortality or of severe or critical disease can guide clinical decision‐making and resource allocation. To safely rule out severe disease, a triage test should have a high sensitivity (> 90%). Within our review, we could not identify laboratory tests with such a high sensitivity.
In clinical practice, a more comprehensive assessment of a patient's health status is often performed by combining multiple tests (as part of a clinical prediction model) with clinical signs and symptoms, radiological findings, and the patient's characteristics, such as age and comorbidities. Prognostic models have looked into these combinations to see if they can predict adverse outcomes in patients with SARS‐CoV‐2 (Wynants 2020).
Implications for research.
The findings of the test accuracy of laboratory tests in this review could be used in prognostic models, as it can help to decide what tests should be included. Furthermore, we believe it is important for future research to focus on the use of standardised test methods, and cut‐off values. Moreover, while we were unable to include studies from Africa, we encourage research from a wider range of geographical areas, such as Africa.
History
Protocol first published: Issue 9, 2021
Acknowledgements
Members of the Cochrane COVID‐19 Diagnostic Test Accuracy Review Group include:
the project team (Deeks JJ, Dinnes J, Leeflang MMG, Spijker R, Van den Bruel A, Verbakel JY, McInnes MDF, Emperador D, Dittrich S, Cunningham J);
-
the systematic review teams for each review:
Molecular, antigen, and antibody tests (Adriano A, Arevalo‐Rodriguez I, Beese S, Buitrago DC, Ciapponi A, Domen J, Dretzke J, Ferrante di Ruffano L, Harris I, Mateos M, Price M, Taylor M, Taylor‐Phillips S)
Signs and symptoms (Struyf T, Horn S)
Routine laboratory markers (Yang B, Langendam M, Ochodo EA, Guleid F, Holtman GA, Wang J, Stegeman I)
Accuracy of routine laboratory tests (De Rop L)
Imaging tests (Salameh JP, McGrath TA, van der Pol CB, Frank RA, Prager R, Hare SS, Dennie C, Jenniskens K, Korevaar DA, Cohen JF, van de Wijgert J, Damen JAAG);
the wider team of systematic reviewers from the University of Birmingham, UK who assisted with title and abstract screening across the entire suite of reviews for the diagnosis of COVID‐19 (Agarwal R, Baldwin S, Berhane S, Herd C, Kristunas C, Quinn L, Scholefield B).
Jonathan Deeks is a United Kingdom National Institute for Health Research (NIHR) Senior Investigator Emeritus. Yemisi Takwoingi is supported by a NIHR Postdoctoral Fellowship. Jonathan Deeks and Jacqueline Dinnes are supported by the NIHR Birmingham Biomedical Research Centre at the University Hospitals Birmingham NHS Foundation Trust and the University of Birmingham. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health and Social Care.
The Cochrane Infectious Diseases Group (CIDG) supported the authors in the development of this diagnostic test accuracy review. The CIDG editorial base is funded by UK aid from the UK government for the benefit of low‐ and middle‐income countries (project number 300342‐104). The views expressed do not necessarily reflect the UK government’s official policies.
Editorial and peer‐reviewer contributions
The Cochrane Diagnostic Test Accuracy (DTA) editorial team and Central Editorial Service editorial team coordinated a dual editorial process.
The following people conducted the editorial process for this article:
Sign‐off Editors (final editorial decision): Michael D Brown, Emergency Medicine, Michigan State University College of Human Medicine, USA (clinical); and Daniëlle AWM van der Windt, School of Medicine, Keele University, UK (DTA).
Managing Editor (selected peer reviewers, provided editorial guidance to authors, edited the article): Luisa Fernandez Mauleffinch, Cochrane Central Editorial Service.
Editorial Assistant (conducted editorial policy checks, collated peer‐reviewer comments and supported editorial team): Lisa Wydrzynski, Cochrane Central Editorial Service.
Copy Editor (copy editing and production): Anne Lethaby, Cochrane Central Production Service.
Peer‐reviewers (provided comments and recommended an editorial decision): Cochrane DTA editorial team (methods and search); Olabisi Oduwole (clinical); and Pernille Just Vinholt, MD, Odense University Hospital, Denmark (clinical).
Appendices
Appendix 1. Search strategy
| Search strategy | Hits | |
| 1 | MESH DESCRIPTOR Biomarkers EXPLODE ALL AND COVID19:INREGISTER | 235 |
| 2 | MESH DESCRIPTOR Sensitivity and Specificity EXPLODE ALL AND COVID19:INREGISTER | 555 |
| 3 | MESH DESCRIPTOR Cross‐Sectional Studies EXPLODE ALL AND COVID19:INREGISTER | 988 |
| 4 | ((biomarker* or marker* or test or tests or diagn* or discrimin* or detect* or sensitivity or specificity or auc or predictive‐value or NPV or PPV or accuracy or case‐control* or cross‐sectional):AB OR (biomarker* or marker* or test or tests or diagn* or discrimin* or detect* or sensitivity or specificity or auc or predictive‐value or NPV or PPV or accuracy or case‐control* or cross‐sectional):TI) AND COVID19:INREGISTER | 87536 |
| 5 | #1 OR #2 OR #3 OR #4 | 87707 |
| 6 | MESH DESCRIPTOR Prognosis EXPLODE ALL AND COVID19:INREGISTER | 1635 |
| 7 | MESH DESCRIPTOR Clinical Decision Rules EXPLODE ALL AND COVID19:INREGISTER | 6 |
| 8 | ((prognos* OR cohort OR validat* OR predict* OR follow‐up):AB OR ( prognos* OR cohort OR validat* OR predict* OR follow‐up):TI) AND COVID19:INREGISTER | 56335 |
| 9 | #6 OR #7 OR #8 | 57286 |
| 10 | MESH DESCRIPTOR Mortality EXPLODE ALL AND COVID19:INREGISTER | 607 |
| 11 | MESH DESCRIPTOR Intensive Care Units EXPLODE ALL AND COVID19:INREGISTER | 434 |
| 12 | MESH DESCRIPTOR Continuous Positive Airway Pressure AND COVID19:INREGISTER | 21 |
| 13 | MESH DESCRIPTOR Critical Illness EXPLODE ALL AND COVID19:INREGISTER | 200 |
| 14 | MESH DESCRIPTOR Survival Analysis EXPLODE ALL AND COVID19:INREGISTER | 244 |
| 15 | MESH DESCRIPTOR Intubation, Intratracheal EXPLODE ALL AND COVID19:INREGISTER | 61 |
| 16 | ((sever* or mortal* Or critical* Or ventilation OR intub* or intensive‐care or icu or survival):AB OR (sever* or mortal* Or critical* Or ventilation OR intub* or intensive‐care or icu or survival):TI) AND COVID19:INREGISTER | 88870 |
| 17 | #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR #16 | 88963 |
| 18 | #5 AND #9 AND #17 | 21701 |
| 19 | ("Cross‐sectional"):SG AND COVID19:INREGISTER | 36780 |
| 20 | #19 OR #5 | 103681 |
| 21 | #20 AND #17 AND #9 | 22818 |
| Date | 25/08/2022 |
Data
Presented below are all the data for all of the tests entered into the review.
Tests. Data tables by test.
1. Test.

CRP increase deterioration
2. Test.

APTT increase deterioration
3. Test.

ALT increase deterioration
4. Test.

AST increase deterioration
5. Test.

Blood urea nitrogen increase deterioration
6. Test.

D‐dimer increase deterioration
7. Test.

Creatinine increase deterioration
8. Test.

Ferritin increase deterioration
9. Test.

Platelet count decrease deterioration
10. Test.

Platelet count increase deterioration
11. Test.

Fibrinogen increase deterioration
12. Test.

LDH increase deterioration
13. Test.

Haemoglobin decrease deterioration
14. Test.

White blood cell count decrease deterioration
15. Test.

White blood cell count increase deterioration
16. Test.

Lymphocyte count decrease deterioration
17. Test.

Lymphocyte count increase deterioration
18. Test.

Neutrophil count increase deterioration
19. Test.

NLR increase deterioration
20. Test.

Potassium decrease deterioration
21. Test.

Sodium decrease deterioration
22. Test.

Procalcitonin increase deterioration
23. Test.

CK increase deterioration
24. Test.

ALT increase mortality
25. Test.

AST increase mortality
26. Test.

Creatinine increase mortality
27. Test.

LDH increase mortality
28. Test.

White blood cell count decrease mortality
29. Test.

White blood cell count increase mortality
30. Test.

Lymphocyte count decrease mortality
31. Test.

NLR increase mortality
32. Test.

Platelet count decrease mortality
33. Test.

Procalcitonin increase mortality
34. Test.

Bilirubin increase deterioration
35. Test.

CRP increase mortality
36. Test.

D‐dimer increase mortality
37. Test.

Ferritin increase mortality
38. Test.

Haemoglobin decrease mortality
39. Test.

Troponin I increase mortality
40. Test.

Eosinophil count decrease mortality
41. Test.

Troponin increase deterioration
42. Test.

CRP increase combined outcome
43. Test.

D‐dimer increase combined outcome
44. Test.

LDH increase combined outcome
45. Test.

Lymphocyte count decrease combined outcome
46. Test.

Platelet count decrease combined outcome
47. Test.

Erythrocyte sedimentation rate increase deterioration
48. Test.

Prothrombin time increase deterioration
49. Test.

Haematocrit decrease deterioration
50. Test.

Haematocrit increase deterioration
51. Test.

Haemoglobin increase deterioration
52. Test.

Eosinophil count decrease deterioration
53. Test.

APTT decrease deterioration
54. Test.

Basophil count decrease deterioration
55. Test.

Basophil count increase deterioration
56. Test.

Eosinophil count increase deterioration
57. Test.

INR increase deterioration
58. Test.

Monocyte count decrease deterioration
59. Test.

Monocyte count increase deterioration
60. Test.

Neutrophil count decrease deterioration
61. Test.

Potassium increase deterioration
62. Test.

Prothrombin time decrease deterioration
63. Test.

Sodium increase deterioration
64. Test.

Albumin decrease deterioration
65. Test.

CK–MB fraction increase deterioration
66. Test.

Chlorine decrease deterioration
67. Test.

Prealbumin decrease deterioration
68. Test.

Brain natriuretic peptide increase deterioration
69. Test.

IL‐6 increase deterioration
70. Test.

Troponin I increase deterioration
71. Test.

Glucose increase deterioration
72. Test.

Blood urea nitrogen decrease deterioration
73. Test.

Fibrinogen decrease deterioration
74. Test.

NLR decrease deterioration
75. Test.

Serum amyloid protein A increase deterioration
76. Test.

Creatinine decrease deterioration
77. Test.

Troponin T increase deterioration
78. Test.

Mean platelet volume increase deterioration
79. Test.

eGFR decrease deterioration
80. Test.

CK isoenzyme increase deterioration
81. Test.

Lymphocytes % decrease deterioration
82. Test.

Myoglobin increase deterioration
83. Test.

Neutrophils % increase deterioration
84. Test.

APTT increase mortality
85. Test.

Erythrocyte sedimentation rate increase mortality
86. Test.

Fibrinogen increase mortality
87. Test.

Prothrombin time increase mortality
88. Test.

Troponin increase mortality
89. Test.

Haematocrit decrease mortality
90. Test.

Haematocrit increase mortality
91. Test.

Haemoglobin increase mortality
92. Test.

Lymphocyte count increase mortality
93. Test.

Platelet count increase mortality
94. Test.

Lymphocytes % decrease mortality
95. Test.

Neutrophils % increase mortality
96. Test.

Sodium increase mortality
97. Test.

Urea increase mortality
98. Test.

Sodium decrease mortality
99. Test.

Lactate increase mortality
TST-100. Test.

Basophil count decrease mortality
TST-101. Test.

IL‐6 increase mortality
TST-102. Test.

Serum amyloid protein A increase mortality
TST-103. Test.

Bilirubin increase mortality
TST-104. Test.

CK increase mortality
TST-105. Test.

Troponin T increase mortality
TST-106. Test.

Albumin decrease mortality
TST-107. Test.

Blood urea nitrogen increase mortality
TST-108. Test.

Brain natriuretic peptide increase mortality
TST-109. Test.

NT‐proBNP increase mortality
TST-110. Test.

INR increase mortality
TST-111. Test.

Neutrophil count increase mortality
TST-112. Test.

Glucose increase mortality
TST-113. Test.

Alkaline phosphatase increase mortality
TST-114. Test.

HbA1c increase mortality
TST-115. Test.

Potassium decrease mortality
TST-116. Test.

Neutrophil count decrease mortality
TST-117. Test.

Potassium increase mortality
TST-118. Test.

NLR decrease mortality
TST-119. Test.

Myoglobin increase mortality
TST-120. Test.

IL‐6 increase combined outcome
TST-121. Test.

ALT increase combined outcome
TST-122. Test.

AST increase combined outcome
TST-123. Test.

Bilirubin increase combined outcome
TST-124. Test.

CK increase combined outcome
TST-125. Test.

Creatinine increase combined outcome
TST-126. Test.

White blood cell count decrease combined outcome
TST-127. Test.

White blood cell count increase combined outcome
TST-128. Test.

Procalcitonin increase combined outcome
TST-129. Test.

Fibrinogen increase combined outcome
TST-130. Test.

NLR increase combined outcome
TST-131. Test.

Ferritin increase combined outcome
TST-132. Test.

Haemoglobin decrease combined outcome
TST-133. Test.

Glucose increase combined outcome
TST-134. Test.

cGFR decrease combined outcome
TST-135. Test.

Sodium decrease combined outcome
TST-136. Test.

Potassium decrease combined outcome
TST-137. Test.

CK isoenzyme increase mortality
TST-138. Test.

Albumin increase deterioration
TST-139. Test.

eGFR decrease mortality
TST-140. Test.

Eosinophils % decrease mortality
TST-141. Test.

Basophils % decrease mortality
TST-142. Test.

Basophil count increase mortality
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Al‐Samkari 2020.
| Study characteristics | |||
| Patient Sampling | All patients, aged 18 years and older, with confirmed COVID‐19 from 1 March 2020 through 5 April 2020, who had a D‐dimer test performed on initial presentation were identified using the Research Patient Data Registry at Partners Healthcare, a large multi‐institutional patient data registry | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: five Partners Healthcare institutions: Massachusetts General Hospital, Brigham and Women’s Hospital, North Shore Medical Center, Newton‐ Wellesley Hospital, and Brigham and Women’s Faulkner Hospital
Country: USA Symptoms and severity: The rate of radiographically confirmed VTE was 4.8%. The arterial thrombosis rate was 2.8%. The overall thrombotic complication rate was 9.5%. The overall bleeding rate was 4.8%. Three participants were diagnosed with DIC. Demographics: 144 critical cases and 256 non‐critical cases; critical cases: mean age 65 years (range 32‐97), non‐critical cases: mean age 60 years (range 23‐99); 35.4% females in critical cases, and 47.3% females in non‐critical cases Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: The rate of radiographically confirmed VTE was 4.8%. All but one participant received anticoagulation with standard prophylactic doses of Unfractionated Heparin (UFH) or Low Molecular Weight Heparin (LMWH) at the time of the event. One participant received a therapeutic‐dose of apixaban at the time of the event. The arterial thrombosis rate was 2.8%. All participants received anticoagulation with prophylactic doses of UFH or LMWH at the time of the event. Twelve critically ill participants placed on Continuous Veno‐Venous Hemofiltration (CVVH), eight had recurrent clotting of the CVVH circuit while receiving prophylactic‐dose anticoagulation, resulting in a change to therapeutic‐dose heparin infusion; two had continued recurrent clotting of the circuit despite therapeutic‐dose heparin infusion. Four participants receiving CVVH did not have recurrent clotting of the CVVH circuit, three were on a therapeutic‐dose heparin infusion for other indications at the time the CVVH was initiated. The overall thrombotic complication rate was 9.5%. Forty‐one participants (10%) were transitioned from prophylactic‐dose to therapeutic‐dose anticoagulation during admission to manage thrombotic complications or new‐onset atrial fibrillation, or both. Four participants with thrombotic complications also developed bleeding complications. Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: positive RT‐PCR, no further specification |
||
| Index tests | Routine laboratory tests were performed at admission (Figure 4; Figure 5). Data were obtained retrospectively by manual chart review of the electronic medical record with a data cut‐off date of 8 April 2020. Reference ranges for each test were identical or very similar at each institution, and are summarised below.
Unit of analysis: individual participants Activated partial thromboplastin time: 22.0‐36.0 sec; performed using reagents from Stago (APTT) except for one laboratory that used reagents from Instrumentation Laboratory (SynthASil). Sample not reported C‐reactive protein: < 8.0 mg/L; performed with Cobas (Roche) in all laboratories. Sample not reported D‐dimer: < 500 ng/mL; all tests reported results in FEU with a reference range of < 500 ng/mL FEU; performed using Vidas (bioMerieux) in two laboratories, Sta‐Liatest (Stago) in two laboratories, and HS 500 (Instrumentation Laboratory) in one laboratory. Sample not reported. Erythrocyte sedimentation rate: < 13 mm/h (men) and < 20 mm/h (women); performed using iSed (Alcor) except for one laboratory that used Inversa (Mechatronics). Sample not reported Ferritin: 20‐300 μg/L (men) and 10‐200 μg/L (women); performed with Cobas (Roche) in all laboratories. Sample not reported Fibrinogen: 150‐450 mg/dL; performed using reagents from Stago (Sta‐ fibrinogen) except for one laboratory that used reagents from Instrumentation Laboratory (Q.F.A. Bovine Thrombin). Sample not reported Troponin: high‐sensitivity cardiac troponin: < 14 ng/L (men) and < 9 ng/L (women); performed with Cobas (Roche) in all laboratories. Sample not reported Platelet count: 150‐450 × 109/L; obtained using Sysmex analysers in all laboratories. Sample not reported Procalcitonin: < 0.10 ng/mL; performed with Cobas (Roche) in all laboratories. Sample not reported Prothrombin time: 11.5‐14.5 sec; performed using reagents from Stago (Neoplastine Cl Plus) except for one laboratory that used reagents from Instrumentation Laboratory (HemosIL RecombiPlasTin 2G). Sample not reported |
||
| Target condition and reference standard(s) | Both mortality and deterioration were assessed. Deterioration was defined as critical illness; throughout the study it was seen as a requirement for endotracheal intubation and mechanical ventilation. This also included participants for whom intubation was clinically indicated but who chose to forego it (those with a “do not intubate” status). Survival was assessed for participants reaching a terminal end point (hospital discharge or death) by the end of the study period (n = 252). The total sample size was 400 cases, of which 144 critical and 256 non‐critical; 29 participants died. | ||
| Flow and timing | All participants received D‐Dimer testing. The missing data of other tests were not imputed. Treatment after index tests was not avoided (described above). The time horizon, defined as the timing between assessment of biomarker and outcome, was not reported. Only 252 participants reached a terminal end point (hospital discharge or death) by the end of the study period. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | H.A.‐S. has acted as a consultant for Agios, Dova, and Moderna and has received research funding from Agios, Dova, and Amgen. D.J.K. has received research funding from Protalex, Bristol‐ Myers Squibb, Rigel, Bioverativ, Agios, Syntimmune, Principia, and Alnylam and has acted as a consultant for ONO, Pfizer, 3SBios, Eisai, GlaxoSmithKline, Genzyme, Shire, Amgen, Shionogi, Rigel, Syntimmune, MedImmune, Novartis, Alexion, Bioverativ, Argenx, Zafgen, Fujifilm, Principia, Kyowa Kirin, Takeda, and Platelet Disorders Support Association. R.P.R. has acted as a consultant for Bristol‐Myers Squibb, Dova, Janssen, and Portola and has received research funding from Bristol‐Myers Squibb and Janssen. The remaining authors declared no competing financial interests. | ||
| Analysis | For mortality, only participants reaching a terminal end point (hospital discharge or death) by the end of the study period (n = 252) were included. Missing data were not imputed. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Yes | ||
| Was the method for performing the index test the same for all participants? | No | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
An 2021.
| Study characteristics | |||
| Patient Sampling | The study included 5628 patients in South Korea, who were either cured or died from COVID‐19 infection by April 30, 2020. The data, containing epidemiologic and clinical information of patients with COVID‐19, were provided by the Korea Disease Control and Prevention Agency. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: not specified
Country: South Korea Symptoms and severity: frequency of symptoms (n (%)); that were present in participants in the supportive care, O2 therapy, critical care, and mortality group, respectively: Subjective fever (3818 (88.9%), 296 (6.9%), 128 (3.4%), 32 (0.7%)); cough (2836 (86.9%), 239 (7.3%), 160 (4.9%), 30 (0.9%)); sputum (3456 (86.7%), 319 (8%), 169 (4.2%), 41 (1%)); dyspnoea (4445 (90.1%), 332 (6.7%), 128 (2.6%), 26 (0.5%)); sore throat (3989 (84.4%), 446 (9.4%), 228 (4.8%), 61 (1.3%)); rhinorrhoea (4216 (84.7%), 468 (9.4%), 228 (4.8%), 61 (1.3%)); myalgia (4005 (85.6%), 400 (8.6%), 220 (4.7%), 52 (1.1%)); fatigue (4606, (585.9%), 475 (8.9%), 224 (4.2%), 58 (1.1%)); headache (3931 (84.8%), 421 (9.1%), 228 (4.9%) 53 (1.1%)); nausea/vomiting (4598 (85.9%), 470 (8.8%), 225 (4.2%), 59 (1.1%)); diarrhoea (4354 (85.7%), 446 (8.8%) 223 (4.4%), 57 (1.1%)); altered consciousness (4772 (85.8%), 512 (9.2%), 218 (3.9%), 59 (1.1%)) Demographics: frequency of cases amongst different age groups was: 0‐9 years: 66, 10‐19 years: 205, 20‐29 years: 1109, 30‐39 years: 562, 40‐49 years: 739, 50‐59 years: 1140, 60‐69 years: 906, 70‐79 years: 545, ≥ 80 years: 324 Female: 86.9%, 8.8%, 3.5%, and 0.9% of cases of supportive care, O2 therapy, critical care and mortality, respectively. Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: 85.4% of cases received no particular therapy, 9.1% received oxygen therapy, and 5.4% received critical care (mechanical ventilation, ECMO). Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: not specified |
||
| Index tests | Routine laboratory tests (Figure 4; Figure 5) A dataset containing the epidemiologic and clinical information of participants diagnosed with COVID‐19 in South Korea was provided and anonymised by the Korea Disease Control and Prevention Agency collected for the public interest. Unit of analysis: individual participants Sample used for the index test was not reported. Timing of assessment of index test was not reported. | ||
| Target condition and reference standard(s) | Both mortality and deterioration were assessed. The worst severity during the disease course was determined by the type of treatment required: none or supportive treatment, oxygen therapy, critical care such as mechanical ventilation or ECMO, or death from COVID‐19 infection. For the target condition deterioration, the groups: none or supportive care and O2 were compared to critical care. The total sample size was 5596; 241 participants received critical care, 4780 received supportive care, and 512 received O2 therapy; 63 participants died. |
||
| Flow and timing | 32 participants were excluded because of missing data on disease severity or the presence or absence of symptoms. Haemoglobin: 1516 missing (1457 (96.1%), 43 (2.8%), 12 (0.8%), and 4 (0.3%) for supportive care, O2 therapy, critical care, and mortality, respectively); haematocrit: 1521 missing (1464 (96.3%), 42 (2.8%), 11 (0.7%), and 4 (0.3%) for supportive care, O2 therapy, critical care, and mortality, respectively); white blood cell count: 1515 missing (1456 (96.1%), 42 (2.8%), 12 (0.8%), and 5 (0.3%) for supportive care, O2 therapy, critical care, and mortality, respectively); lymphocyte count: 1540 missing (1469 (95.4%), 49 (3.2%), 17 (1.1%), 5 (0.3%) for supportive care, O2 therapy, critical care, and mortality, respectively); platelet count: 1515 missing (1456 (96.1%), 43 (2.8%), 12 (0.8%), 4 (0.3%) for supportive care, O2 therapy, critical care, and mortality, respectively). The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | The authors declared no competing interests. This work was supported by a grant from National Health Insurance Service Ilsan Hospital (NHIMC2021‐ CR017). | ||
| Analysis | The authors reported that missing values were not imputed, because imputation for missing values did not provide significant differences in the preliminary analysis. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Arnold 2021.
| Study characteristics | |||
| Patient Sampling | Patients with COVID‐19 disease (adults, 18 years and older) were included. These were both cases from the emergency department (ED) and as newly diagnosed hospitalised cases, at North Bristol NHS Trust recruited via the DISCOVER study from 30 March 2020 until 29 June 2020. Patients with the inability to consent were excluded. Patients were recruited on the basis of a positive PCR result for SARS‐CoV‐2, a clinico‐radiological diagnosis of COVID‐19 disease or a history of positive testing in the community. | ||
| Patient characteristics and setting | Setting: ED and hospitalised cases
Site: North Bristol NHS Trust, Bristol
Country: UK Symptoms and severity: not reported Demographics: the median age in all participants was 58 years (IQR 46‐73); in survivors (n = 148) 55 years (IQR 44‐71); and in non‐survivors, intensive care unit (ICU)/non‐invasive ventilation (NIV)‐participants (n = 39) 66 years (IQR 60‐76); 46% of all participants, 47% of survivors, and 44% of non‐survivors, ICU/NIV participants were female. Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: positive PCR result for SARS‐CoV‐2, using the established phenylalanine assay in use at the time, or a clinico‐radiological diagnosis of COVID‐19 disease. During the pandemic, community testing became widely available, although the results were not available to hospital staff. As such, later participants were often recruited on the basis of a history of positive testing in the community. |
||
| Index tests | Routine laboratory tests (Figure 4 and Figure 5). Analysis was performed on frozen samples in batch analysis. Laboratory staff were unaware of the clinical outcome and were therefore functionally blinded. The earliest initial sample was extracted from the blood sciences laboratory after routine testing had been performed. In admitted cases, this was the initial sample taken in the ED, and in hospitalised cases, this was the sample from the day of diagnosis. IL‐6 analysis was performed on the Fujirebio Lumipulse. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | 28‐day composite outcome of ICU admission, NIV with CPAP or BIPAP outside the ICU, or death. The total sample size was 187 cases, with 39 non‐survivors, and ICU/NIV‐participants combined, and 148 survivors. | ||
| Flow and timing | 187 participants had 28‐day outcomes at the time of analysis. | ||
| Comparative | |||
| Study design | Prospective cohort study | ||
| Funding a/o conflicts of interest | The DISCOVER study was supported by grants from the Southmead Hospital Charity and Elizabeth Blackwell Institute. DTA is funded by a National Institute for Health Research (NIHR) Doctoral Research Fellowship (DRF‐2018‐11‐ST2‐065). FH is funded via the National Institute for Health Academic Clinical Fellowship scheme. The suPARnostic ELISA kits and IL‐6/KL‐6 assays were gifts from ViroGates (Birkeroed, Denmark) and Fujirebio Europe, respectively, for unrestricted research activity. | ||
| Analysis | Missing data were not reported. Complete case analysis was performed for each individual biomarker. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Yes | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | No | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Yes | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Arshad 2020.
| Study characteristics | |||
| Patient Sampling | Patients with confirmed COVID‐19 infection, admitted to Combined Military Hospital, Peshawar Cantt, from March to June 2020, were included. Probable cases, defined as patients with findings suggestive of COVID‐19 infection on HRCT scan chest but negative PCRs for SARS‐CoV‐2 on two consecutive occasions, patients with incomplete data on laboratory parameters and patients leaving against medical advice were excluded. | ||
| Patient characteristics and setting | Setting: Department of Medicine
Site: Combined Military Hospital, Peshawar Cantt
Country: Pakistan Symptoms and severity: disease severity at admission was mild in 157 (65.97%) participants, moderate in 36 (15.13%) participants, and severe/critical in 45 (18.91%) participants. Mild cases were defined as symptomatic participants fulfilling the case definition for COVID‐19 without evidence of pneumonia or hypoxia. Moderate disease was defined as clinical signs of pneumonia (fever, cough, dyspnoea) without signs of severe pneumonia, including SpO2 ≥ 90% in room air. Severe disease was defined as clinical signs of pneumonia (fever, cough, dyspnoea) plus one of the following: respiratory rate > 30 breaths/min; severe respiratory distress; or SpO2 < 90% in room air. Lastly, critical disease was defined as acute respiratory distress syndrome. Demographics: the mean age was 41.18 years (SD 16.74); 12.61% were female. Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: SARS‐CoV‐2 was confirmed by RT‐PCR. Nasopharyngeal swabs were taken prior to admission using standard technique for determination of SARS‐CoV‐2 by PCR. |
||
| Index tests | Routine laboratory tests were performed (Figure 4 and Figure 5). As per the Institutional protocol, blood samples were collected within the first half hour of admission for tests including: quantitative CRP, serum ferritin and serum LDH. Data on these laboratory parameters as well as duration of hospital stay were collected from the medical paper records. Laboratory information management system software was used to search for missing reports. The unit of analysis was individual participants. | ||
| Target condition and reference standard(s) | The study assessed mortality in discharged versus deceased participants. The total sample size was 238 participants, 22 participants died, and 216 participants were discharged. | ||
| Flow and timing | The time horizon was not defined. During the study period, 431 participants with COVID‐19 infection were admitted. Complete results for inflammatory markers were missing for 117 participants. They were excluded from data analysis. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Missing results were excluded from data analysis. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Yes | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | Unclear | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Asaduzzaman 2022.
| Study characteristics | |||
| Patient Sampling | Patients were admitted with a diagnosis of COVID‐19 between October 2020 and January 2021 in four hospitals of Sylhet, Bangladesh (a major city in northeastern Bangladesh) during the COVID‐19 pandemic. The study included patients above the age of 18 years, who were PCR‐positive for SARS‐CoV‐2, and who were PCR‐negative but had typical clinical and radiographic findings of COVID‐19. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: four hospitals in Sylhet
Country: Bangladesh Symptoms and severity: fever was present in 52 (94.5%) non‐survivors, and 353 (91.2%) survivors; cough in 36 (65.5%) non‐survivors, and 286 (73.9%) survivors; shortness of breath in 42 (76.4%) non‐survivors, and 252 (65.1%) survivors; fatigability in 33 (60%) non‐survivors, and 213 (55%) survivors; loss of smell in 9 (16.4%) non‐survivors, and 78 (20.2%) survivors; diarrhoea in 11 (20%) non‐survivors, and 60 (15.5%) survivors; sore throat in 13 (23.6%) non‐survivors and 34 (8.8%) survivors; anorexia in 2 (3.6%) non‐survivors, and 11 (2.8%) survivors; chest pain in 0 non‐survivors, and 9 (2.3%) survivors; vomiting in 0 non‐survivors; and 4 (1%) survivors; headache in 1 (1.8%) non‐survivors, and 2 (0.5%) survivors. Demographics: the mean age of all participants was 60 years (SD 14); in non‐survivors this was 69 years (SD 13), and in survivors 59 years (SD 14); 34.16% of all participants, 38.2% of non‐survivors, and 33.6% of survivors were female. Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported SARS‐CoV‐2 cases were defined as positive PCR, or negative PCR with typical clinical and radiographic findings of COVID‐19. |
||
| Index tests | Data were extracted from the hospital record of participants. Clinical, demographic, and laboratory data from all adult participants were recorded at the time of hospital admission (Figure 4 and Figure 5). The blood samples were sent soon after hospital admission, preferably within 1 hour. Cell count was done by a fully automated analyser SYSMEX‐XT2000i (made in Japan). Unit of analysis: individual participants Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | The study assessed mortality, defined as in‐hospital death. In‐hospital death refers to those participants who died at least 24 hours after hospital admission due to COVID‐19. COVID‐19 death was defined as certified by WHO, which states “A death due to COVID‐19 is defined as a death resulting from a clinically compatible illness, in a probable or confirmed COVID‐19 case, unless there is a clear alternative cause of death that cannot be related to COVID disease.” A total sample size of 442 participants, with 55 non‐survivors, and 387 survivors was included. | ||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Yes | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | |||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | Unclear | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Asghar 2020.
| Study characteristics | |||
| Patient Sampling | This study was conducted as a retrospective, observational, multi‐centric study, and included all admitted COVID‐19‐positive patients. The time period during which inclusion took place was not reported; however, the article was received for publication on 28/07/2020. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: multi‐centric, not specified which hospitals
Country: Pakistan Symptoms and severity: most participants experienced mild‐to‐moderate symptoms and were admitted to the isolation ward (65.7%); 34.3% of participants experienced more severe disease and were admitted to the intensive care unit. Demographics: mean age was 52.69 years (SD 15.88); the study included 118 females. Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported The study did not specify which definition was used for COVID‐19‐positive participants. |
||
| Index tests | Baseline laboratory investigations of all participants were monitored both at admission and discharge (Figure 4 and Figure 5). Unit of analysis: individual participants Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Mortality was assessed in a total of 364 participants with 263 survivors, and 101 non‐survivors. | ||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | |||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | Unclear | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Aydin 2022.
| Study characteristics | |||
| Patient Sampling | All patients (adults > 18 years) testing positive for and being hospitalised with COVID‐19 between 18 March and 4 August 2020, at the Boyabat 75th Year State Hospital in Sinop, Turkey, were enroled in this study. Patients with atopic dermatitis, urticaria, allergic contact dermatitis, and known allergies were excluded. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: the Boyabat 75th Year State Hospital in Sinop
Country: Turkey Symptoms and severity: In all participants, 161 (79.7%) participants had no symptoms, 105 (52%) participants had a cough, 62 (30.7%) fatigue, 51 (25.2%) shortness of breath, 36 (17.8%) fever, 36 (17.8%) myalgia, 30 (14.9%) headache, 18 (8.9%) throat ache, 12 (5.9%) sputum, 8 (4%) vomiting, 7 (3.5%) diarrhoea, 3 (1.5%) chest tightness, and 2 (1%) smell and taste dysfunction. In stable participants, 39 (24.2%) participants had no symptoms, 78 (48.4%) cough, 47 (29.2%) fatigue, 33 (20.5%) shortness of breath, 30 (18.6%) fever, 27 (16.8%) myalgia, 28 (17.4%) headache, 13 (8.1%) throat ache, 8 (5%) sputum, 5 (3.1%) vomiting, 6 (3.7%) diarrhoea, 2 (1.2%) chest tightness, and 1 (0.6%) smell and taste dysfunction; and in participants with clinical worsening 2 (4.9%) participants had no symptoms, 27 (65.9%) cough, 15 (36.6%) fatigue, 18 (43.9%) shortness of breath, 6 (14.6%) fever, 9 (22%) myalgia, 2 (4.9%) headache, 5 (12.2%) throat ache, 4 (9.8%) sputum, 3 (7.3%) vomiting, 1 (2.4%) diarrhoea, 1 (2.4%) chest tightness, and 1 (2.4%) smell and taste dysfunction. Amongst the COVID‐19 participants, 41 (20.3%) showed clinical progression. Clinical worsening was observed on day 7 (4–10). During follow up, 20.3% (n = 41) of participants needed supplemental oxygen, 3.5% (n = 7) required intensive care, 3% (n = 6) were intubated, and 2% (n = 4) died. The median time from admission to death was 13.5 (10–28) days. Demographics: age ranged from 18 to 87 years, and mean age was 50.17 years (SD 19.68); 100 (49.5%) were female. Exposure history: not reported The median time from symptom onset to admission was 3 days (range: 0‐30) in all participants, 3 days (range: 0‐30) in stable participants, and 4 days (range: 0‐15) in clinical worsening participants. Treatment before target condition: from hospital admissions to treatment protocols, all clinical decisions adhered to the national guidelines published by the Turkish Ministry of Health. Participants with clinical worsening and hypoxaemia received favipiravir (1600 mg twice daily as a loading dose, followed by 600 mg twice daily as a maintenance dose). Given the influenza season, participants were administered oseltamivir (75 mg twice daily) and empirical antibiotics were also applied as initial therapy against the possibility of bacterial aetiology. In all participants, 81 (40.1%) received oseltamivir, 25 (12.4%) favipiravir, 201 (99.5%) plaquenil, and 196 (97%) azithromycin; in stable participants, 67 (41.6%) received oseltamivir, 4 (2.5%) favipiravir, 160 (99.4%) plaquenil, and 157 (97.5%) azithromycin; in clinical worsening participants 14 (16.4%) received oseltamivir, 21 (51.2%) received favipiravir, 41 (100%) plaquenil, and 39 (95.1%) azithromycin. Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Routine laboratory tests were assessed on the first day of hospitalisation (Figure 4 and Figure 5). Data consisting of laboratory results were obtained from the hospital’s electronic medical records system. The unit of analysis was individual participants. The sample used for absolute eosinophiles was serum; for the other laboratory tests this was not reported. | ||
| Target condition and reference standard(s) | The study assessed deterioration. The primary outcome was clinical worsening, defined by an ordinal clinical improvement scale. Scores on the scale were defined as follows: (1) discharged or ready for discharge, (2) in (or ready for) a non‐intensive care unit (ICU) hospital ward and not receiving supplemental oxygen, (3) in (or ready for) a non‐ICU hospital ward and receiving supplemental oxygen, (4) in the ICU or a non‐ICU hospital ward and receiving non‐invasive ventilation or high‐flow oxygen, (5) in the ICU, intubated and receiving mechanical ventilation, (6) in the ICU and receiving extracorporeal membrane oxygenation or mechanical ventilation, and additional organ support, and (7) death. Worsening was defined as an increase of the score by at least 1 point for participants receiving supplemental oxygen at baseline, and by at least 2 points for those not receiving supplemental oxygen at baseline. The study included 202 participants, with 161 stable participants, and 41 clinical worsening. | ||
| Flow and timing | A time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Ayus 2021.
| Study characteristics | |||
| Patient Sampling | Patients hospitalised for COVID‐19 infection at the Posadas Hospital in Buenos Aires, Argentina, between 7 March 2020 and 7 November 2020 were included. Patients were hospitalised if they were older than 60 years, had one or more comorbidities, were immunosuppressed, had uni‐ or bilateral pulmonary compromise or had oxygen desaturation (< 95%). Of these patients, only those who had in their admission biochemistry serum sodium and high‐sensitivity C‐reactive protein were included in this study. Eighty‐four patients who were admitted to the hospital for different pathologies and who got infected with SARS‐CoV‐2 were also included in the study. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: Posadas Hospital in Buenos Aires
Country: Argentina Symptoms and severity: different symptoms were reported for participants with serum sodium ≤ and > 135 mEq/L: anosmia in participants with serum sodium ≤ 135 mEq/L: 41 (11.3%), and in participants with serum sodium > 135 mEq/L: 68 (15.9%); arthralgia in participants with serum sodium ≤ 135 mEq/L: 16 (4.4%), and in participants with serum sodium > 135 mEq/L: 17 (4.0%); headaches in participants with serum sodium ≤ 135 mEq/L: 83 (23.0%), and in participants with serum sodium > 135 mEq/L: 85 (19.9%); coma in participants with serum sodium ≤ 135 mEq/L: 1 (0.3%), and in participants with serum sodium > 135 mEq/L: 3 (0.7%); confusion in participants with serum sodium ≤ 135 mEq/L: 9 (2.5%), and in participants with serum sodium > 135 mEq/L: 12 (2.8%); seizures in participants with serum sodium ≤ 135 mEq/L: 4 (1.1%), and in participants with serum sodium > 135 mEq/L: 3 (0.7%); diarrhoea in participants with serum sodium ≤ 135 mEq/L: 50 (13.9%), and in participants with serum sodium > 135 mEq/L: 47 (11.0%); dysgeusia in participants with serum sodium ≤ 135 mEq/L: 42 (11.6%), and in participants with serum sodium > 135 mEq/L: 48 (11.2%); dyspnoea in participants with serum sodium ≤ 135 mEq/L: 216 (59.8%), and in participants with serum sodium > 135 mEq/L: 240 (56.2%); abdominal pain in participants with serum sodium ≤ 135 mEq/L: 22 (6.1%), and in participants with serum sodium > 135 mEq/L: 15 (3.5%); chest pain in participants with serum sodium ≤ 135 mEq/L: 37 (10.2%), and in participants with serum sodium > 135 mEq/L: 43 (10.1%); conjunctivitis in participants with serum sodium ≤ 135 mEq/L: 1 (0.3%), and in participants with serum sodium > 135 mEq/L: 4 (0.9%); general discomfort in participants with serum sodium ≤ 135 mEq/L: 122 (33.8%), and in participants with serum sodium > 135 mEq/L: 129 (30.2%); myalgia in participants with serum sodium ≤ 135 mEq/L: 75 (20.8%), and in participants with serum sodium > 135 mEq/L: 89 (20.9%); odynophagia in participants with serum sodium ≤ 135 mEq/L: 93 (25.8%), and in participants with serum sodium > 135 mEq/L: 125 (29.3%); cough in participants with serum sodium ≤ 135 mEq/L: 240 (66.5%), and in participants with serum sodium > 135 mEq/L: 282 (66.0%); vomit in participants with serum sodium ≤ 135 mEq/L: 34 (9.4%), and in participants with serum sodium > 135 mEq/L: 30 (7.0%); fever (37°5–37°9) in participants with serum sodium ≤ 135 mEq/L: 5 (1.4%), and in participants with serum sodium > 135 mEq/L: 20 (4.7%); fever (> 38°) in participants with serum sodium ≤ 135 mEq/L: 244 (67.6%), and in participants with serum sodium > 135 mEq/L: 261 (61.1%); pneumonia (radiological‐clinical) in participants with serum sodium ≤ 135 mEq/L: 18 (5.0%), and in participants with serum sodium > 135 mEq/L: 9 (2.1%); oxygen saturation (%; median (IQR)) in participants with serum sodium ≤ 135 mEq/L: 93 (89–96), and in participants with serum sodium > 135 mEq/L: 94 (90–97). Demographics: median age in participants with serum sodium ≤ 135 mEq/L was 59.25 years (IQR 48.53–66.60), and in participants with serum sodium > 135 mEq/L was 53.96 years (IQR 41.68‐66.37); there were 122 (33.3%) females in the group participants with serum sodium ≤ 135 mEq/L, and 191 (44.1%) in the group participants with serum sodium > 135 mEq/L. Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Sodium concentration at admission was assessed (Figure 4 and Figure 5). All plasma sodium concentration measurements were performed using the ion‐selective electrode method (normal range 136–145 mmol/L). Plasma sodium levels were corrected for plasma glucose using the following formula: corrected plasma sodium (mEq/L) = measured plasma sodium (mEq/L) + 0.016 – (plasma glucose [mg/dL] – 100). Using plasma sodium data available at hospital admission, participants were classified as hyponatraemic if plasma sodium was ≤ 135 mmol/L. Data sources for the study included information extracted from the institution’s electronic health record (EHR) system (including data from the central laboratory). The unit of analysis was individual participants. | ||
| Target condition and reference standard(s) | Both mortality and deterioration were assessed. Deterioration was defined as ICU admission during hospitalisation, and for mortality in‐hospital deaths were measured. The total sample size was 799 participants; 182 participants were admitted to ICU during hospitalisation, and there were 132 in‐hospital deaths. | ||
| Flow and timing | Participants were included between 7 March 2020 and 7 November 2020, and follow‐up occurred on 7 November 2020. In‐hospital outcomes were assessed within 30 days after symptom onset. | ||
| Comparative | |||
| Study design | Prospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | All enroled participants were included in the analysis. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Was the method used to perform the index test valid and reliable? | Yes | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Yes | ||
| Was treatment avoided after the index test was performed? | Unclear | ||
| Was the time horizon sufficient to capture the outcome? | Yes | ||
| Was information on the outcome available for all participants? | Unclear | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Azad Allarakia 2022.
| Study characteristics | |||
| Patient Sampling | Patients who were diagnosed with COVID‐19 and admitted to East Jeddah General Hospital, Saudi Arabia were included from July to 30 August 2020. | ||
| Patient characteristics and setting | Setting: Hospital ward and ICU
Site: East Jeddah General Hospital
Country: Saudi Arabia Symptoms and severity: cough, fever, shortness of breath, diarrhoea and hypoxia were the most common signs reported at admission. Demographics: mean age in all participants was 47.21 years (SD 15), in ICU participants it was 55.35 years (SD 13), and in non‐ICU participants it was 45.98 years (SD 14); the study included 65 females, 6 (16.2%) ICU participants and 59 (24.3%) non‐ICU participants were female. Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Routine laboratory tests were included (Figure 4 and Figure 5). Data were extracted from medical records. Unit of analysis: individual participants Sample used for the index test was not reported. Timing of assessment of index test was not reported. | ||
| Target condition and reference standard(s) | Deterioration was assessed, participants were categorised into two groups based on admission status to the ICU or isolation ward (severe = ICU admission, mild = ward admission). Criteria used for admission to ICU were not reported. The total sample size was 280, including 27 ICU admissions and 243 participants on the hospital ward. | ||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Unclear | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Unclear | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Unclear | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Yes | ||
| Was treatment avoided after the index test was performed? | Unclear | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Bahl 2020.
| Study characteristics | |||
| Patient Sampling | Patients (> 18 years of age) who were admitted with COVID‐19 disease from March 1st through March 31st 2020 were included. Patients who were transferred within Beaumont Health were included. Patients leaving the hospital against medical advice, transfers to external hospitals, and hospital course ongoing beyond April 23, 2020 were excluded. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: Beaumont Health, an eight‐hospital acute care regional health system, ranging from a large tertiary care academic centre to intermediate‐sized and smaller community hospitals, of which one was converted to a complete COVID‐19 centre in the Metro Detroit catchment area
Country: USA Symptoms and severity: cough was present in 122 (39.0%) non‐survivors and in 562 (50.7%) survivors, shortness of breath in 188 (60.1%) non‐survivors and 599 (54.0%) survivors, fever in 115 (36.7%) non‐survivors and 493 (44.5%) survivors, gastrointestinal symptoms in 35 (11.2%) non‐survivors and 148 (13.4%) survivors, altered mental status in 45 (14.4%) non‐survivors and 46 (4.2%) survivors, chest pain in 8 (2.6%) non‐survivors and 58 (5.2%) survivors, weakness in 43 (13.7%) non‐survivors and 126 (11.4%) survivors, chills in 5 (1.6%) non‐survivors and 23 (2.1%) survivors, body aches in 11 (3.5%) non‐survivors and 71 (6.4%) survivors, syncope in 6 (1.9%) non‐survivors and 55 (5.0%) survivors, neurological symptoms in 5 (1.6%) non‐survivors and 24 (2.2%) survivors, and other symptoms were present in 26 (8.3%) non‐survivors and 78 (7.0%) survivors. Demographics: the median age was 62 years (IQR 50‐74); in non‐survivors it was 73 years (IQR 63‐81), and in survivors 59 years (IQR 48‐70); 6911 (47.3%) females were included; in non‐survivors this was 147 (45%), and in survivors 544 (48%). Exposure history: not reported Time since onset of symptoms: the median length of illness (symptom onset) for survivors was 12 days compared to 15 days for non‐survivors. Treatment before target condition: hydroxychloroquine in 290 (88.7%) non‐survivors and 783 (69.1%) survivors, corticosteroids in 211 (64.5%) non‐survivors and 406 (35.8%) survivors, NSAIDs in 12 (3.7%) non‐survivors and 91 (8.0%) survivors, vitamin C in 116 (35.5%) non‐survivors and 291 (25.7%) survivors, zinc in 111 (33.9%) non‐survivors and 261 (23.0%) survivors, ACE inhibitors in 28 (8.6%) non‐survivors and 181 (16.0%) survivors, angiotensin receptor blockers (ARBs) or sartanics in 23 (7.0%) non‐survivors and 81 (7.1%) survivors, renal replacement therapy in 50 (15.3%) non‐survivors and 32 (2.8%) survivors, non‐invasive ventilation in 53 (16.2%) non‐survivors and 53 (4.7%) survivors, high‐flow oxygen therapy in 206 (63.0%) non‐survivors and 213 (18.8%) survivors, invasive mechanical ventilation in 219 (67.0%) non‐survivors and 89 (7.9%) survivors. Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Routine laboratory tests conducted within 24 hours of admission were included (Figure 4 and Figure 5). Data were obtained from the integrated electronic health records (Epic Systems, Verona, WI). Epidemiological, demographic, radiological, therapeutic, clinical, and outcomes data were extracted. The integrity of the data were verified by two attending emergency medicine physicians. Laboratory and radiological testing was conducted at the discretion of the treating physicians. Unit of analysis: individual participants Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Mortality was assessed, and the study included a total of 1461 participants, with 327 non‐survivors, and 1134 survivors. | ||
| Flow and timing | The data were collected until April 23, 2020. The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Missing data were imputed by multiple imputation using PROC MI in SAS. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Yes | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Barrett 2021.
| Study characteristics | |||
| Patient Sampling | All patients, > 16 years, that were tested for COVID‐19 before March 29th 2020 and who were admitted to the hospital within 1 week of that test were screened. Patients were included in the study if they were admitted to the hospital before April 5th, 2020. Patients were included between March 6, 2020 and April 4, 2020. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: five hospitals in one of the most socioeconomically depressed urban counties in the US. These five hospitals included a quaternary referral centre, two community hospitals, a paediatric hospital, and a freestanding ED.
Country: USA Symptoms and severity: not reported Demographics: the mean age was 62 years (SD 16); the study included 515 (46%) females. Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: 269 (24%) participants were intubated, and 161(14%) participants required renal replacement therapy. Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Routine laboratory tests were collected (Figure 4 and Figure 5). Both automatic and manual data abstraction was used. Abstracted data were located automatically using Clinical Looking Glass (CLG), a proprietary search and abstraction program. For data not amenable to automatic search, the authors manually abstracted data. They standardised manual data abstraction by conducting training sessions for all data abstractors and providing periodic feedback on randomly selected cases. The first lab value drawn within 48 hours of arrival to the ED was recorded. If the laboratory test was not drawn within 48 hours after ED arrival, they considered the data to be missing. As with the outcome variables, a separate reviewer obtained laboratory values from a randomly selected 10% of charts. They also reported sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), positive likelihood ratio, and negative likelihood ratio for laboratory values and clinical outcomes. For these latter analyses, laboratory values were dichotomised at the upper limit of normal, based on standard laboratory cut‐offs used at all hospital sites during data collection. Unit of analysis: individual patients Serum was used to analyse the index test. | ||
| Target condition and reference standard(s) | Both mortality and deterioration were assessed in this study. Deterioration was defined as ICU admission. Death was automatically determined from the discharge code in the electronic health record; the other variables were manually abstracted. A total of 1123 participants were included; 199 (17%) were admitted to the ICU, and 305 (27%) died. |
||
| Flow and timing | Electronic health record review was completed on Sept 24th, 2020. The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Data were considered to be missing if the laboratory test was not drawn within 48 hours after ED arrival. No more information about missing data was reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Bassoli 2020.
| Study characteristics | |||
| Patient Sampling | Patients with confirmed COVID‐19 disease hospitalised at the Infectious Disease Ward (IDW) or intensive care unit (ICU) of Fatebenefratelli‐Sacco Hospital in Milan were included. Patients with a history of kidney diseases were excluded from this study. | ||
| Patient characteristics and setting | Setting: infectious disease ward and intensive care unit
Site: Fatebenefratelli‐Sacco Hospital in Milan
Country: Italy Symptoms and severity: not reported Demographics: the median age of all participants was 60.9 years (IQR 49.17‐71.29); in participants with albumin < 30 g, the median age was 68.13 years (IQR 59.43‐75.16), and in participants with albumin > 30 g, the median age was 55.59 years (IQR 44.55‐64.10); a total of 65 (31.4%) female participants were included, albumin < 30 g: 34 (32.4%), and albumin ≥ 30 g: 31(30.4%). Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: no oxygen support therapy was given to 89 (43.0) participants, albumin < 30: 24 (22.9%), and albumin ≥ 30: 65 (63.7%); nasal cannula to 41 (19.8%) participants, albumin < 30: 20 (19%), and albumin ≥ 30: 21 (20.6%); venturi‐type mask to 46 (22.2%), albumin < 30: 38 (36.2%), and albumin ≥ 30: 8 (7.8%); CPAP: to 26 (12.6) participants, albumin < 30: 18 (17.1%), and albumin ≥ 30: 8 (7.8%); invasive ventilation to 5 (2.4%), albumin < 30: 5 (4.8%), and albumin ≥ 30: 0 Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: not reported |
||
| Index tests | Data were extracted from the hospital records of participants. Clinical, demographic, and laboratory data from all adult participants were recorded at the time of hospital admission. Serum albumin measurements were available for all the participants at three time points within the first week of hospitalisation (admission, on day 3 and day 7 from admission) (Figure 4 and Figure 5). Unit of analysis: individual participants | ||
| Target condition and reference standard(s) | Deterioration was assessed; it was classified according to the Chinese Guidelines for the Diagnosis and Treatment of Novel Coronavirus (2019‐nCoV) Infection into four grades: (a) mild, with slight clinical symptoms and no evidence of pneumonia, (b) moderate, with fever, respiratory symptoms and confirmed pneumonia, (c) severe, with any of the following: respiratory distress with RR > 30 times/minutes, oxygen saturation at rest < 93% or PaO2/FiO2 < 300 mmHg, (d) critically severe, with any of the following: respiratory failure needing mechanical ventilation, shock, or a combination of other organ failure requiring intensive care. The total sample size was 207 participants, of which 30 participants had mild disease, 100 had moderate disease, 72 had severe disease, and 5 had critically severe disease. For the analyses in this review, the participants with mild and moderate disease were grouped together and participants with severe and critically severe disease were grouped together. |
||
| Flow and timing | The time horizon was not reported; 15 subjects were excluded because of kidney disease. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | All enroled participants were included in the analysis. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
BG 2021.
| Study characteristics | |||
| Patient Sampling | Patients, 18 years and older, admitted from July to August 2020 to the Chigateri General Hospital, Davangere, were included. Admitted patients with a respiratory rate exceeding 24 cycles per minute and oxygen saturation below 94% without oxygen supplementation were included in the study. Cases were defined based on the interim guidelines issued by the Ministry of Health and Family Welfare, Government of India. Patients receiving mechanical ventilation, patients on tocilizumab, pirfenidone, azathioprine, and cyclophosphamide, and those with a previous diagnosis of malignancy were excluded from the study. Patients with other febrile illnesses like dengue, malaria, leptospirosis, and rickettsial fevers were also excluded from the study. | ||
| Patient characteristics and setting | Setting: wards of Chigateri General Hospital, not specified
Site: the Chigateri General Hospital, Davangere
Country: India Symptoms and severity: fever was present in 10 non‐survivors and 15 survivors, dry cough in 9 non‐survivors and 23 survivors, productive cough in 12 non‐survivors and 43 survivors, breathlessness in 17 non‐survivors and 57 survivors (n = 75); Demographics: in all participants, the mean age was 47.1 years (SD 14.8, range 20–78), in non‐survivors 59.1 years (SD 11.5, range 40–78), and in survivors 43.0 years (SD 13.6, range 20–71); non‐survivors: 12 females; survivors: 31 females Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: the treatment protocol for COVID‐19 participants falling under Categories B and C (defined by the interim guidelines issued by the Ministry of Health and Family Welfare, Government of India) was followed, not reported how many participants were treated, or which treatment was given Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Routine laboratory tests were assessed (Figure 4 and Figure 5). A complete haemogram was ordered within one hour of admission, before the initiation of treatment. The laboratory records were collected and screened to match the criteria of the study. The cut‐off value for each parameter was considered as a suitable value around the median. Unit of analysis: individual participants Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Mortality was assessed in 100 participants, with 25 non‐survivors, and 75 survivors. | ||
| Flow and timing | Time horizon not reported | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Missing data not reported | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Yes | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | Unclear | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Cazzaniga 2021.
| Study characteristics | |||
| Patient Sampling | Patients older than 18 years, with COVID‐19 pneumonia admitted to the emergency room‐COVID‐19 area with a positive COVID‐19 nasopharyngeal swab were included. Patients transferred from other hospitals were excluded. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: not reported
Country: Italy Symptoms and severity: fever (> 37.5°C) was present in participants with absolute eosinopenia: 70 (93.3%) and participants without absolute eosinopenia: 29 (90.6%); high respiratory rate (> 20 min) was present in participants with absolute eosinopenia: 24 (32%) and participants without absolute eosinopenia: 5 (15.6%); clinically severe disease at admission (defined as: concurrent presence of: fever > 38°C; lower respiratory symptoms (cough, difficulty breathing, shortness of breath, high respiratory rate) and radiographic evidence of lung infiltrates consistent with interstitial pneumonia or Acute Respiratory Distress Syndrome) was present in participants with absolute eosinopenia: 43 (57.3%) and participants without absolute eosinopenia: 9 (29.3%) Demographics: the overall mean age was 64.3 years (SD 15.3, range 18‐96); in participants with eosinopenia the mean age was 66.3 years (SD 14.8, range 20‐96), and in participants without eosinopenia the mean age was 59.8 years (SD 15.7, range 18‐86); overall: 32 (29.9%) females, participants with eosinopenia: 20 (26.6%) females, and participants without eosinopenia: 12 (37.5%) females Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: 49.3% of participants with absolute eosinopenia and 13.3% of participants without absolute eosinopenia needed intensive respiratory treatment Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | The absolute eosinophile count was assessed at admission (Figure 4 and Figure 5). The records of admitted participants with COVID‐19 pneumonia were revised in order to compare demographic, biochemical, radiological and clinical characteristics at hospital admission and outcomes of participants with and without eosinopenia. Undetectable eosinophil count was defined as absolute eosinopenia. Unit of analysis: individual participants Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Mortality and deterioration were assessed. Four‐week survival was evaluated. Deterioration was defined as the need for intensive respiratory support: high‐flow nasal cannula oxygen support, non‐invasive ventilation or mechanical ventilation or both. The total sample size was 107 participants; 24 participants died, and 38 participants needed intensive respiratory treatment. |
||
| Flow and timing | The outcome was assessed four weeks after admission. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Yes | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Yes | ||
| Was information on the outcome available for all participants? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Chen 2020a.
| Study characteristics | |||
| Patient Sampling | Patients older than 14 years, with COVID‐19 who were admitted to the hospital in Wenzhou, China from January 11, 2020 to February 15, 2020, were included. All patients received a diagnosis of COVID‐19 according to the criteria issued by the National Health and Health. All cases were screened according to the presence of cough, fever, and radiographic presentations and were confirmed by reverse transcriptase–polymerase chain reaction on respiratory tract samples to test for a sequence of SARS‐CoV‐2. The study was conducted during the COVID‐19 outbreak. Patients with severe renal failure, type 1 diabetes, and cancer were excluded. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: hospital in Wenzhou, not specified
Country: China Symptoms and severity: amongst 175 participants, dry cough (109 (62%) participants), fever (71 (41%) participants), and diarrhoea (35 (20%) participants) were frequent symptoms. Demographics: mean age was 45 years (SD 14, range 15‐85); 87 (50%) female participants. Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: the common treatments involved interferon‐α (170 participants (97%)), lopinavir‐ritonavir (151 participants (86%)), umifenovir (140 participants (80%)), and oxygen inhalation (51 participants (29%)). Twelve participants (7%) were given glucocorticoids; participants with severe hypokalemia received 3 g (40 mEq) potassium chloride per day, with a total mean of 34 (SD 4) g (mean 453 (SD 53) mEq) potassium chloride during the whole hospital stay for each participant. Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR according to the criteria issued by the National Health and Health according to the criteria issued by the National Health and Health |
||
| Index tests | Routine laboratory tests were assessed (Figure 4 and Figure 5). A trained team of medical staff reviewed and collected the demographic, epidemiological, clinical, and laboratory data from electronic medical records. Unit of analysis: individual participants The sample used for potassium was plasma. For the other index tests, this was not reported. Potassium was assessed at admission. For the other index tests, this was not reported. | ||
| Target condition and reference standard(s) | Deterioration was assessed; the following definition was used. Mild cases presented with mild clinical manifestations and no pneumonia, moderate cases presented with respiratory symptoms and mild pneumonia, severe cases presented with respiratory distress (30 breaths/min), oxygen saturation (93% at rest), or arterial partial pressure of oxygen–fraction of inspired oxygen less than or equal to 300 mmHg, critically ill cases were those that met any of respiratory failure criteria and required mechanical ventilation, or those with shock or other organ failure that required intensive care unit care.
Mild and moderate cases were grouped together, and severe and critically ill cases were grouped together. The total sample size was 175 participants; severely and critically ill: 40 participants, and moderately and mildly ill: 135 participants. |
||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | This work was supported by the Key Scientific and Technological Innovation Projects of Wenzhou (grant ZY202004 to Dr Chen) and Natural Science Foundation of Ningbo (grant 2017A610273 to Dr Song). The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. | ||
| Analysis | All enroled participants were included in the analysis. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Unclear | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Yes | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Chen 2020b.
| Study characteristics | |||
| Patient Sampling | Consecutive patients (children and adults) from 25 hospitals in Jiangsu Province, China were enroled in this study from 23 January 2020 to 11 March 2020. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: 25 hospitals in Jiangsu Province
Country: China Symptoms and severity: conjunctival congestion was present in 1 (1.2%) participant of mild cases, and 1 (0.2%) of ordinary cases; nasal congestion was present in 21 (25.9%) participants of mild cases, and 30 (6.2%) of ordinary cases; headache was present in 4 (4.9%) participants of mild cases, 52 (10.7%) of ordinary cases, and 1 participant (3.2%) of severe or critical cases; cough was present in 29 (35.8%) of mild cases, 327 (67.3%) of ordinary cases, and 23 (74.2%) of severe or critical cases; sore throat was present in 8 (9.9%) participants of mild cases, 57 (11.7%) participants of ordinary cases, and 6 (19.4%) participants of severe or critical cases; sputum production was present in 15 (18.5%) participants of mild cases, 185 (38.1%) participants of ordinary cases, and 14 (45.2%) of severe or critical cases; fatigue was present in 6 (7.4%) participants of mild cases, 154 (31.7%) of ordinary cases, and 12 (38.7%) of severe or critical cases; haemoptysis was present in 1 (1.2%) participant of mild cases, and 6 (1.2%) of ordinary cases; shortness of breath was present in 2 (2.5%) participants of mild cases, 96 (19.8%) of ordinary cases, and 22 (71.0%) of severe or critical cases; nausea or vomiting was present in 1 (1.2%) participants of mild cases, 31 (6.4%) of ordinary cases, and 3 (9.7%) of severe or critical cases; diarrhoea was present in 5 (6.2%) participants of mild cases, 56 (11.5%) of ordinary cases, and 8 (25.8%) of severe or critical cases; myalgia or arthralgia was present in 9 (11.1%) participants of mild cases, 83 (17.1%) of ordinary cases, and 4 (12.9%) of severe or critical cases; chills were present in 2 (2.5%) participants of mild cases, 65 (13.4%) of ordinary cases, and 6 (19.4%) of severe or critical cases. Demographics: mean age in mild cases was 39.85 years (SD 18.08), in ordinary cases 46.03 years (SD 16.12), and in severe and critical cases 60.84 years (SD 13.55); 303 female participants, mild cases: 46 (53.8%) females, ordinary cases: 219 (45.1%) females, and severe and critical 11 (35.5%) females Exposure history: participants living in Wuhan: mild + ordinary: 68/576, severe and critical: 1/31; contact with wildlife: mild + ordinary: 1/576, severe and critical: 0/31;, visited Wuhan recently: mild + ordinary: 106/576, severe and critical: 4/31; and contact with Wuhan residents: mild + ordinary: 135/576, severe and critical: 4/31 Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: laboratory‐confirmed COVID‐19, not specified |
||
| Index tests | The medical records and compiled data were obtained for hospitalised participants with laboratory‐confirmed COVID‐19; data were extracted from the medical records. C‐reactive protein was assessed on admission (Figure 4). Unit of analysis: individual participants Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Deterioration was assessed as mild and ordinary versus severe and critical cases. The Guidance for Coronavirus Disease 2019: Prevention, Control, Diagnosis and Management (sixth edition), which was issued by China’s National Health Commission, was used to classify participants with mild, ordinary, severe, and critical disease according to the severity of the COVID‐19 symptoms.
(1) mild disease: mild symptoms only without radiographic features;
(2) ordinary disease present with fever, respiratory symptoms, and radiographic features;
(3) severe disease met one of the following three criteria, namely, dyspnoea, which is defined as a respiratory rate > 30 times/min, an oxygen saturation of < 93% in ambient air, or a ratio of arterial oxygen partial pressure to fractional inspired oxygen < 300 mmHg;
(4) critical disease met one of the following three criteria, namely, respiratory failure, septic shock, or multiple organ failure. The study included 598 cases, with 567 mild and ordinary cases, and 31 severe or critical cases with COVID‐19. |
||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | This study received support from the National Natural Science Foundation of China (No. 81970302, 81170180, 30400173 and 30971257) and the Priority Academic Program Development of Jiangsu Higher Education Institutions. | ||
| Analysis | Some of the data of the included participants were missing or incomplete. No other information was reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | Unclear | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Chen 2020c.
| Study characteristics | |||
| Patient Sampling | Patients with laboratory‐confirmed diagnosis of COVID‐19 were included from 575 hospitals throughout China in this study led by the China National Health Commission until March 22, 2020. The diagnosis of COVID‐19 was made on the basis of World Health Organization interim guidance. Patients with incomplete medical records or still in the hospitals were excluded. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: 575 hospitals throughout China, not specified
Country: China Symptoms and severity: fever was present in 413 (92.8%) survivors and in 100 (97.1%) non‐survivors; the median highest temperature in survivors was 38.6°C (IQR 38.0‐39.0) and in non‐survivors: 38.5°C (IQR 38.0‐39.0); rhinobyon was present in 9 (2.0%) survivors and 5 (4.9%) non‐survivors; headache in 29 (6.5%) survivors and in 7 (6.8%) non‐survivors; cough in 329 (73.9%) survivors and 87 (84.5%) non‐survivors; sore throat in 12 (2.7%) survivors and 6 (5.8%) non‐survivors; sputum in 126 (28.3%) survivors and 44 (42.7%) non‐survivors; fatigue in 155 (34.8%) survivors and 31 (30.1%) non‐survivors; haemoptysis in 5 (1.1%) survivors and 4 (3.9%) non‐survivors; dyspnoea in 170 (38.2%) survivors and 72 (69.9%) non‐survivors; vomiting in 13 (2.9%) survivors and 5 (4.9%) non‐survivors; diarrhoea in 13 (2.9%) survivors and 1 (1.0%) non‐survivor; myalgia in 56 (12.6%) survivors and 11 (10.7%) non‐survivors; and shivering in 39 (8.8%) survivors and 9 (8.7%) non‐survivors; 345 participants had mild/moderate, 155 severe, and 48 critical disease. Demographics: the mean age was 56.0 years (SD 14.5); in survivors this was 53.5 years (SD 13.9), and in non‐survivors 66.9 years (SD 12.1); there was a total of 235 (42.9%) females, survivors: 201 (45.2%) females, and non‐survivors: 34 (33.0%) females. Exposure history: not reported Time since onset of symptoms: median days from symptom onset to admission for all participants was 13 days (IQR 9‐18), for survivors 13 days (IQR 10‐18), and for non‐survivors 9 days (IQR 6‐17). Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: laboratory‐confirmed COVID‐19, not further specified |
||
| Index tests | Routine laboratory tests were assessed on admission (Figure 4 and Figure 5). All medical records were copied and sent to the data processing centre in Guangzhou, under the coordination of the National Health Commission. A team of experienced respiratory clinicians reviewed and abstracted the data. Data were entered into a computerised database and cross‐checked. The clinical data (including demographic data, clinical symptoms and signs, comorbidities, laboratory findings, treatments during hospitalisation, and clinical outcome) were extracted from electronic medical records. Unit of analysis: individual participants Plasma ferritin was assessed, and the samples of the other index tests were not reported. |
||
| Target condition and reference standard(s) | Mortality was assessed by categorising participants into discharged or deceased; 548 participants were included, with 445 survivors and 103 non‐survivors. | ||
| Flow and timing | The time horizon was not reported. The study ended on March 22, 2020; start date was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | A total of 548 cases with a clarified outcome (discharged or deceased) from the cohort were included and participants with incomplete medical records or still in the hospitals were excluded. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Yes | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | Unclear | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Chen 2020d.
| Study characteristics | |||
| Patient Sampling | Patients that were admitted for COVID‐19 disease in 575 hospitals throughout China until January 31, 2020, were included. The diagnosis of COVID‐19 was made based on World Health Organization interim guidance. The diagnosis was confirmed by a positive result of real‐time reverse transcriptase‐polymerase chain reaction assay or high‐throughput sequencing findings from nasal or pharyngeal swab specimens. Patients with incomplete medical records were excluded. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: study led by the National Health Commission of the People’s Republic of China in 575 hospitals, not specified
Country: China Symptoms and severity: fever was present in 1309 (88.0%) survivors and 42 (87.5%) non‐survivors; cough in 1015 (70.0%) survivors and 37 (77.1%) non‐survivors; headache in 196 (15.3%) survivors and 9 (20.9%) non‐survivors; sputum production in 490 (35.6%) survivors and 23 (50.0%) non‐survivors; fatigue in 565 (42.7) survivors and 19 (44.2%) non‐survivors; dyspnoea in 294 (19.1%) survivors and 37 (74.0%) non‐survivors; nausea or vomiting in 76 (5.7%) survivors and 4 (8.9%) non‐survivors; diarrhoea in 56 (4.3%) survivors and 1 (2.2%) non‐survivor; chills in 161 (12.5%) survivors and 2 (4.5%) non‐survivors; myalgia or arthralgia 226 (17.5%) survivors and 8 (18.6%) non‐survivors. Demographics: median age in survivors was 48 years (range 1‐94) and in non‐survivors 69 years (range 51‐86); there were 663 (43.4%) females in survivors, and 11 (22/0%) females in non‐survivors. Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: in survivors: antibiotics 867/1015 (85.4%), oseltamivir 510/1061 (48.1%), other antiviral 824/1107 (74.4%), anti‐fungal 41/907 (4.5%), glucocorticoid 269/973 (27.6%), non‐invasive ventilation 91/946 (9.6%), invasive mechanical ventilation 25/1135 (2.2%), ECMO 0, continuous renal replacement therapy 9/899 (1%), globulin 197/908 (21.7%), ICU 75 (10.1%); in non‐survivors: antibiotics 47/47 (100%), oseltamivir 13/36 (36.1%), other antiviral 26/36 (72.2%), anti‐fungal 8/36 (22.2%), glucocorticoid 18/32 (56.3%), non‐invasive ventilation 31/40 (77.5%), invasive mechanical ventilation 24/40 (60%), ECMO 4/37 (10.8%), continuous renal replacement therapy 8/38 (21.1%), globulin 18/37 (48.6%), ICU 24/41 (58.5%) Vaccine status: not reported Variant of concern: not reported Definition of SARS‐CoV‐2 cases: RT‐PCR, high‐throughput sequencing findings from nasal or pharyngeal swab specimens |
||
| Index tests | Routine laboratory tests were assessed on admission (Figure 4 and Figure 5). Data were extracted from patient records in multiple hospitals in China. Clinical, demographic, and laboratory data from all participants were recorded at the time of hospital admission. All medical records were copied and sent to the data‐processing centre in Guangzhou Institute of Respiratory Health, under the coordination of the National Health Commission. A team of experienced respiratory clinicians reviewed and abstracted the data, and data were entered into a computerised database and cross‐checked. Unit of analysis: individual participants Sample used for the index test was not reported. |
||
| Target condition and reference standard(s) | Mortality was assessed. The study included 1590 participants; 1540 survived and 50 died. | ||
| Flow and timing | The start date of the study was not reported; the end date was January 31, 2020. The time horizon was not reported; 417 cases were excluded because of incomplete medical records. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | |||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | |||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Chen 2021.
| Study characteristics | |||
| Patient Sampling | Patients with moderate COVID‐19 admitted to the hospital from January to March 2020 were assessed. Patients were included if they had a positive nucleic acid result for SARS‐CoV‐2 under RT‐PCR, were inpatients making their first visit because of COVID‐19; had moderate COVID‐19 defined using clinical classification criteria; and had complete CT follow‐up images and laboratory data. Patients with mild, severe, or critically severe COVID‐19 upon admission, other serious illness at the time of admission, or incomplete imaging or laboratory data were excluded. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: not specified in the article, but the affiliated hospital of the authors is the General Hospital of the Yangtze River Shipping, Wuhan Brain Hospital in Wuhan
Country: China Symptoms and severity: not reported Demographics: the mean age was 57.69 years (SD 17.62), in the moderate‐stage group mean age was 52.25 years (SD 16.8), and in the severe/critically severe group 64.25 years (SD 16.4); there were 20 females in the moderate‐stage group, and 15 females in the severe/critically severe group. Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: positive nucleic acid result for SARS‐CoV‐2 under RT‐PCR |
||
| Index tests | C‐reactive protein was assessed (Figure 4). Laboratory examinations were obtained from participants upon admission. High‐sensitivity C‐reactive protein (hs‐CRP) was measured using an automatic haematology analyser. Unit of analysis: individual participants Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | The study assessed deterioration. Participants were clinically classified according to the Diagnosis and Treatment Plan of Coronavirus Disease 2019 (trial edition 7) issued by the National Health Commission of the Peoples Republic of China. Clinical stages were divided into four groups according to the following criteria. Mild stage: the clinical symptoms are mild, and imaging shows no manifestations of pneumonia. Moderate stage: participants have symptoms such as fever and respiratory tract symptoms, and pneumonia manifestations can be seen on imaging. Severe stage: participants show signs of respiratory distress, including respiratory rate ≥ 30 breaths/min, pulse oxygen saturation (SpO2) ≤ 93% on room air in the resting state, or arterial partial pressure of oxygen (PaO2)/fraction of inspired oxygen (FiO2) ≤ 300 mmHg. At higher altitudes (above 1 km), PaO2/FiO2 values should be adjusted based on the equation PaO2/FiO2 × [atmospheric pressure (mmHg)/760], and participants with > 50% lesion progression within 24 h to 48 h as shown by pulmonary imaging should be treated as severe cases. Critically severe stage: respiratory failure occurs, and mechanical ventilation is required; shock occurs; and the condition is complicated by other organ failure that requires monitoring and treatment in the intensive care unit. During hospitalisation, participants with moderate‐stage COVID‐19 who had not undergone any change in clinical classification were included in the moderate‐stage group, while those who had passed into the severe or critically severe stage were included in the severe and critically severe group. There were 97 participants included, 53 (54.6%) in the moderate‐stage group, and 44 cases (45.3%) in the severe and critically severe group. |
||
| Flow and timing | The time horizon was not reported. Participants with incomplete imaging or laboratory data were excluded. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | Funding: Wuhan municipal health commission emergency research projects (code: EX20E19) | ||
| Analysis | All enroled participants were included in the analysis. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Yes | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Chocron 2021.
| Study characteristics | |||
| Patient Sampling | Patients admitted between February 26 and April 20, 2020 to 24 centres with a diagnosis of SARS‐CoV‐2 (RT‐PCR or typical imaging on CT when laboratory testing was inconclusive), and the value of d‐dimer available on admission were included in this study. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: 24 hospitals, not specified
Country: France Symptoms and severity: not reported Demographics: the median age was 67 years (IQR 56‐78 years); 40.2%(464/1154) female; d‐dimer ≤ 1128 ng/mL: 42.9% (261/609) female, and d‐dimer > 1128 ng/mL 37.2% (203/545) female Exposure history: not reported Time since onset of symptoms: mean in all participants 7.12 days (SD 4.76); in d‐dimer ≤ 1128 ng/mL 7.14 days (SD 4.61), and in d‐dimer > 1128 ng/mL 7.10 days (SD 4.92) Treatment before target condition: type of anticoagulation used at admission: no use of anticoagulation in all participants: 1025 (88.8%), in d‐dimer ≤ 1128 ng/mL: 539 (88.5%), and in d‐dimer > 1128 ng/mL: 486 (89.2%); NOAC in all participants: 74 (6.4%), in d‐dimer ≤ 1128 ng/mL: 40 (6.6%), and in d‐dimer > 1128 ng/mL: 34 (6.2%); VKA in all participants: 50 (4.3%), d‐dimer ≤ 1128 ng/mL: 27 (4.4%), and in d‐dimer > 1128 ng/mL: 23 (4.2%); unfractionated heparin in all participants: 5 (0.4%), in d‐dimer ≤ 1128 ng/mL: 3 (0.5%), and in d‐dimer > 1128 ng/mL: 2 (0.4%); use of oral anticoagulation (NOAC or VKA): in all participants: 124 (10.7%), in d‐dimer ≤ 1128 ng/mL: 67 (11.0%), and in d‐dimer > 1128 ng/mL: 57 (10.5%) Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: positive results from RT‐PCR of nasal and pharyngeal swabs or lower respiratory tract aspirates (confirmed case), or by typical imaging characteristics on chest computed tomography scan when laboratory testing was inconclusive (probable case) |
||
| Index tests | D‐dimer on admission was assessed (Figure 4). All data were collected by local investigators in an electronic case report form via REDCap software (ResearchElectronic Data Capture; Vanderbilt University, Nashville, TN, USA), hosted by a secured server from the French institute of Health and Medical research at the Paris Cardiovascular Research centre. Unit of analysis: individual participants Sample used for the index test was not reported. |
||
| Target condition and reference standard(s) | The study assessed in‐hospital mortality, and included 1154 participants with a d‐dimer test available. There were 121 in‐hospital deaths. | ||
| Flow and timing | During the study period, a total of 2878 consecutive participants who were hospitalised in a medical ward for SARS‐CoV‐2 infection were included. At admission, 1154/2878 (40.1%)participants had d‐dimer measurements. They were included in the study. The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | David M. Smadja's COVID team has been funded with grants from the French National Agency for Research ANR SARCODO (Fondation de France) and Mécénat Covid AP‐HP. R.C. receives consultant fees from the company Aspen, without any relation to the current manuscript. NG receives consultant and lecture fees or travel awards from the companies Aspen, Bayer, Alliance BMS‐Pfizer, LEO‐Pharma and Boehringer Ingelheim, without any relation to the current manuscript. A.C. receives research grants from RESICARD (research nurses), consultant and lecture fees from the companies Amgen, AstraZeneca, Bayer Pharma, AllianceBMS‐Pfizer, Novartis and Sanofi‐Aventis, without any relation to the current manuscript. D.M.S. receives consultant and lecture fees or travel awards from the companies Aspen, Bayer, Carmat, Alliance BMS‐Pfizer, LEO‐Pharma and Boehringer Ingelheim, without any relation to the current manuscript. The other authors declared that they had no competing interests. | ||
| Analysis | All enroled participants were included in the analysis. The study performed a sensitivity analysis to take into account the retrospective design and avoid bias caused by censored data. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | |||
| Did the study avoid inappropriate selection criteria? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | |||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Detsika 2021.
| Study characteristics | |||
| Patient Sampling | Patients from the second wave of COVID‐19 were recruited. All patients had a positive polymerase chain reaction (PCR) test for SARS‐COV‐2 from a nasopharyngeal sample. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: Evangelismos Hospital, Athens
Country: Greece Symptoms and severity: temperature (> 37.3°C) in non‐intubated patients: 44 (61.91%) and intubated participants: 28 (57.14%); cough in non‐intubated participants: 34 (47.88%) and intubated participants: 27 (55.10%); myalgia and fatigue in non‐intubated participants: 21 (29.57%) and intubated participants: 6 (12.24%); survival in non‐intubated participants: 69 (97.10%) and in intubated participants: 32 (65.30%) Demographics: mean age in non‐intubated participants was 56.31 years (SD 15.99) and in intubated participants 63.69 years (SD 10.95); female: non‐intubated: 23 (32.4%) and intubated: 12 (24.5%) Exposure history: not reported Time since onset of symptoms: mean days of illness before admission in non‐intubated participants 6.77 days (SD 4.43) and in intubated participants 6.76 days (SD 2.20) Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | NLR was assessed on admission (Figure 5). Blood samples were obtained on admission for all participants and analysed on the same day. Blood cell counts on admission were used to calculate the NLR. More specifically, the absolute neutrophil count was divided by the absolute lymphocyte count to obtain the NLR. Unit of analysis: individual participants Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Deterioration was assessed, defined as participants who did not require mechanical ventilation (non‐intubated) and those who required mechanical ventilation (intubated). The study included 120 participants, of which 71 were non‐intubated and 49 were intubated. |
||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Prospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | No imputation was performed for missing data. Frequencies of missing data were not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | |||
| Was the outcome (mortality) measured without knowledge of the index test results? | Unclear | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Unclear | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Dong 2020.
| Study characteristics | |||
| Patient Sampling | Consecutive adult patients who were admitted between January 29, 2020 and March 5, 2020 to the hospital in Wuhan, China, with laboratory‐confirmed COVID‐19 and were diagnosed as severe and critically ill according to Chinese Clinical Guidance for COVID‐19 Pneumonia Diagnosis and Treatment. Patients with primary liver disease, acute myocardial infarction, decompensate heart failure, chronic kidney disease, or those with missing demographic or laboratory data were excluded. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: hospital in Wuhan, not specified
Country: China Symptoms and severity: not reported Demographics: mean age in non‐survivors was 70.2 years (SD 10.2) and in survivors 54.2 years (SD 14.9); male/female (%) in non‐survivors: 70.4% and in survivors 46.2% Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: laboratory‐confirmed COVID‐19, not specified |
||
| Index tests | Routine laboratory tests were assessed on admission (Figure 4 and Figure 5). The baseline laboratory data were collected on admission. All the data were independently reviewed and entered into the computer database by two analysts. Unit of analysis: individual participants Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Mortality was assessed. Participants were divided into two groups according to the clinical outcomes of survival (= discharge) or death. The total sample size was 119 adults, 54 non‐survivors and 65 survivors. | ||
| Flow and timing | Participants with missing demographic or laboratory data were excluded. 26 days was chosen as the observation time point. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | Funding: Xuan Wu Hospital, Capital Medical University No. XWJL‐2019027 | ||
| Analysis | Participants who died or discharged prior to observation time were defined as censored; 26 days was chosen as the observation time point. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | |||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | |||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Yes | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Duan 2020.
| Study characteristics | |||
| Patient Sampling | Non‐severe patients with COVID‐19 were enroled in this study from 1st January to 29th February 2020. Only the non‐severe cases at admission were enroled. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: Chongqing Three Gorges Central Hospital and Chongqing Public hospital
Country: China Symptoms and severity: not reported Demographics: mean age in non‐severe cases was 44 years (SD 15) and in severe cases 58 years (SD 15); female: non‐severe cases: 158 (48%), and severe cases: 6 (30%) Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: all participants were managed according to the protocols made by National Health Commission of China. Of the 20 severe cases, eight received conventional oxygen therapy, nine received non‐invasive ventilation, two received high flow nasal cannula, and one received intubation. Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Routine laboratory tests were assessed on admission (Figure 4 and Figure 5). Data were extracted from the medical records. Three kinds of data were collected: basic information, laboratory tests, and radiographic imaging. Unit of analysis: individual participants Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Deterioration was assessed from mild to severe disease. Based on protocols made by National Health Commission of China, the severe participants met one of three criteria: (a) dyspnoea, respiratory rate > 30 breaths/min, (b) oxygen saturation < 93% in ambient air, (c) PaO2/FiO2 < 300mm Hg. The total sample size was 348 cases; 20 cases deteriorated to severe disease, and 328 cases remained non‐severe. | ||
| Flow and timing | The time horizon was not reported; participants were followed up until discharge or deterioration. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | Funding information: Chongqing Science and Technology Bureau “new crown pneumonia epidemic emergency science and technology special”, the fourth batch of projects; Chongqing Education Board “new coronavirus infection and prevention” emergency scientific research project | ||
| Analysis | Some participants had incomplete laboratory tests at admission due to the shortage of medical staff and lack of management protocols at the early stage. Other information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Unclear | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Elhadad 2020.
| Study characteristics | |||
| Patient Sampling | All adult (18 years and older) patients with laboratory‐confirmed SARS‐CoV‐2 infection referred to Laniado Hospital and subsequently admitted between 14 March 2020 and 14 May 2020 were included. Patients lacking documented clinical symptoms and laboratory findings at the time of admission with at least three consecutive laboratory tests during hospitalisation were excluded. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: Sanz Medical Center‐Laniado Hospital, Netanya
Country: Israel Symptoms and severity: fever was present in participants with invasive ventilation: 8 (33%) and non‐invasive ventilation: 16 (67%); dry cough in participants with invasive ventilation: 10 (33%) and non‐invasive ventilation: 20 (67%); expectoration in participants with invasive ventilation: 1 (33%) and non‐invasive ventilation: 2 (67%); dyspnoea in participants with invasive ventilation: 8 (44%) and non‐invasive ventilation: 10 (56%); myalgia in participants with non‐invasive ventilation: 1 (100%); confusion in participants with invasive ventilation: 2 (50%) and non‐invasive ventilation: 2 (50%); headache in participants with non‐invasive ventilation: 1 (100%); fatigue in participants with invasive ventilation: 6 (32%) and non‐invasive ventilation: 13 (68%); rhinorrhoea in participants with non‐invasive ventilation: 3 (100%); pharyngalgia in participants with non‐invasive ventilation: 4 (100%); loss of appetite in participants with non‐invasive ventilation: 2 (100%); nausea and vomiting in participants with non‐invasive ventilation: 5 (100%); diarrhoea in participants with non‐invasive ventilation: 4 (100%); abdominal pain in participants wit non‐invasive ventilation: 4 (100%); at hospital admission, 1 participant presented with respiratory distress and was immediately intubated; 57 participants presented with mild‐to‐moderate disease according to community‐acquired pneumonia guidelines (American Thoracic Society guideline). Mortality rate in invasive ventilation was 60% and 2% in non‐invasive ventilation Demographics: mean age in all participants was 70.7 years (SD 16.9), in invasive ventilation it was 78.18 years (SD 10.35), and in non‐invasive ventilation 68.14 years (SD 18); in all participants: 27 (47%) females, invasive ventilation: 3 (20%) females, and non‐invasive ventilation 27 (47%) females Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Routine laboratory tests on admission were included (Figure 4 and Figure 5). Patient electronic records were reviewed for laboratory parameters (complete blood count and biochemical panel). Routine blood tests included complete blood count and serum biochemical panel (including CRP and CK) with the frequency of test determined by the treating physician. The optimal cut‐off points for prediction were determined by Youden's index. Serum was used, and the unit of analysis was individual participants. | ||
| Target condition and reference standard(s) | Deterioration, defined as respiratory failure, was assessed. The study looked at invasive ventilation during hospitalisation. Criteria for intubation were defined as tachypnoea with respiratory rate ≥ 30 per minute, desaturation with peripheral oxygen saturation (SpO2) ≤ 90% with participant on high flow oxygen (18 litres/min). A total of 58 participants were included; 15 participants (26%) developed respiratory failure and required invasive ventilation. | ||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | Not reported | ||
| Analysis | Patients with missing laboratory findings at the time of admission with at least three consecutive laboratory tests during hospitalisation were excluded. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | |||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Yes | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Unclear | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Fernandez‐Botran 2021.
| Study characteristics | |||
| Patient Sampling | Patients with a diagnosis of SARS‐CoV‐2 and biomarker data, hospitalised in any of the eight adult hospitals in the city of Louisville, KY, were included. The study group was part of a larger study of hospitalised COVID‐19 patients and included patients hospitalised between March 5th, 2020 and July 1st, 2020. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: eight adult hospitals in the city of Louisville, KY
Country: USA Symptoms and severity: not reported Demographics: the median age was 61 years (IQR 45–73); 55% female participants Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR; no further specification |
||
| Index tests | Routine laboratory tests were included (Figure 4 and Figure 5). Data were extracted from hospital electronic medical records. Laboratory data included in this study were the first recorded results obtained within the first 24 hours of hospital admission. Biomarker results were categorised according to cut‐offs representing established normal versus abnormal levels, except for IL‐6. Based on several studies looking at IL‐6 levels and COVID‐19 disease severity, a cut‐off of 37.65 pg/mL was reported to be highly predictive of intra‐hospital mortality. Unit of analysis: individual participants Sample used for the index test was not reported. |
||
| Target condition and reference standard(s) | Both in‐hospital mortality and deterioration were assessed. Deterioration was defined as admission to an intensive care unit. The total sample size was 700 participants, 259 (37%) of the participants were admitted to the ICU, and 108 (15%) died while in the hospital. | ||
| Flow and timing | The time horizon was not reported. Not all laboratory values were available for all participants within 24 hours of admission. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | This work was funded by the Center of Excellence for Research in Infectious Diseases (CERID) of the University of Louisville. | ||
| Analysis | In adjusted logistic regression analysis, participants who were missing laboratory values were given their own factor level. No groups with missing laboratory values were found to be statistically significantly different from their normal level counterparts. No imputations were performed for missing values. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Gao 2020.
| Study characteristics | |||
| Patient Sampling | Patients with severe COVID‐19 from a continuous sample in Hubei General Hospital during the management by a national medical team. Criteria for severe conditions included respiratory rate ≥ 30/min or rest oxyhaemoglobin saturation (SpO2) ≤ 93% or oxygenation index (arterial oxygen tension/inspired oxygen fraction, PaO2/FiO2) ≤ 300mmHg. Patients lacking NT‐proBNP results, patients who had a stroke had an acute myocardial infarction, patients with malignant tumours, and pregnant patients were excluded. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: Hubei General Hospital
Country: China Symptoms and severity: not reported Demographics: mean age was 60.4 years (SD 16.1); female 30 participants Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: not reported |
||
| Index tests | NT‐proBNP was assessed (Figure 5). All data were collected using the same protocol by well‐trained researchers with a double‐blind method. Data collection of laboratory results was defined using the first‐time examination at admission (within 24 h after admission). All laboratory data were tested in the same laboratory with the same standard. The best NT‐proBNP cut‐off was that of the highest product of sensitivity and specificity for in‐hospital death prediction. Sample used was serum and the unit of analysis was individual participants. | ||
| Target condition and reference standard(s) | The study assessed in‐hospital death. The total sample size was 54 participants, of which 18 died. | ||
| Flow and timing | Patients were followed up from admission to discharge (1 to 15 days). | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | Funding: National Natural Science Foundation of China, 81570212; National Natural Science Foundation of China, 31800976; Chongqing Science and Health Joint Medical Research Project, 2018QNXM024 | ||
| Analysis | Information on missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | |||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Yes | ||
| Was treatment avoided after the index test was performed? | Unclear | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Unclear | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
García de Guadiana‐Romualdo 2021a.
| Study characteristics | |||
| Patient Sampling | Patients hospitalised between 1 March 2020 and 30 April 2020 with a diagnosis of COVID‐19 and recruited from 32 hospitals of Health National System in 9 autonomous communities of Spain, were assessed. All consecutive adult patients (≥ 14 years) discharged or dead after hospital admission, with SARS‐CoV‐2 infection, were eligible for enrolment in the study. Pregnant women, patients transferred from or to other hospitals, patients transferred from nursing homes, patients discharged from the ED for at‐home treatment, patients with intensive care unit admission criteria who were not admitted due to lack of availability, and patients in whom troponin was not measured within 24 hours from admission to ED or when it was measured using a contemporary assay, were excluded. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: 32 hospitals of National Health System in 9 autonomous communities
Country: Spain Symptoms and severity: not reported Demographics: the median age was 67 years (IQR 55‐77); 530 (41.4%) female participants Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Troponin levels were measured (Figure 5) using a cardiac troponin T assay in 494 (38.6%) participants and a cardiac troponin I assay in 786 (61.4%). A value above the 99th percentile upper reference limit of a normal reference population is the recommended threshold which defines an increased cardiac troponin indicative of myocardial injury. Data collection was performed retrospectively from electronic medical records and laboratory information systems by two researchers for each hospital. Baseline laboratory testing was defined as the first result available within 24 hours from admission to ED. A multicentre database was prepared for the register of collected data, and all participant identities were coded for blindness. Unit of analysis: individual participants Sample used for the index test was serum. | ||
| Target condition and reference standard(s) | Both deterioration and mortality were assessed. Deterioration was defined as ICU admission, and mortality was assessed 30 days after admission 1280 participants were included; 1093 (85.4%) survivors, and 187 (14.6%) non‐survivors; 249 (19.5%) participants were admitted to the ICU. | ||
| Flow and timing | According to exclusion criteria, 179 participants were excluded because troponin levels were measured using contemporary assays and 1414 because troponin levels were not available within 24 hours from admission to ED. The time horizon for mortality was 30 days; for deterioration, the time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | The follow‐up censoring date of the study was 20 May 2020; 108 participants who were still hospitalised on May 20, 2020 were excluded from analysis. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Was the method used to perform the index test valid and reliable? | Yes | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | Unclear | ||
| Was the time horizon sufficient to capture the outcome? | Yes | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
García de Guadiana‐Romualdo 2021b.
| Study characteristics | |||
| Patient Sampling | All consecutive adult patients, ≥ 14 years, discharged or dead after hospital admission to 32 Spanish hospitals from 1 March 2020 to April 30, 2020, with SARS‐CoV‐2 infection, were eligible for inclusion in the study. Exclusion criteria were: pregnant women, patients transferred from or to another hospital, patients transferred from nursing homes, patients discharged from the ED for at‐home treatment, and patients with Intensive Care Unit admission criteria who were not admitted due to lack of availability. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: 32 hospitals of National Health System in 9 autonomous communities
Country: Spain Symptoms and severity: not reported Demographics: the median age of all participants was 66 years (IQR 54–76), in survivors it was 63 years (IQR 52–74), and in non‐survivors 76 years (IQR 68–83); a total of 1174 (40.9%) females, 1041 (42.3%) in survivors, and 133 (32.3%) in non‐survivors Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification, or by a positive result of serological testing and a clinically compatible presentation |
||
| Index tests | Routine laboratory tests were assessed within the first 24 h from presentation to the ED (Figure 4; Figure 5). Data collection was performed retrospectively from electronic medical records and laboratory information systems by two researchers for each hospital. A multicenter database was prepared for registration of collected data. To achieve an adjustment of laboratory test values, a harmonisation factor was calculated for each methodology and analyser, using a category 1 external quality assurance (EQA) scheme, provided by the Spanish Society of Laboratory Medicine (SEQC‐ML), which includes results that are interchangeable and traceable to reference standards. The value measured with the most common methodology and analyser amongst participating laboratories was considered as reference to generate the factor. Unit of analysis: individual participants Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | All‐cause in‐hospital mortality was assessed. Patients were categorised as being discharged or dead after hospital admission. The total sample size was 2873 participants, 2461 survivors and 412 non‐survivors. | ||
| Flow and timing | The time horizon was not reported. The study ended April 30, 2020 and the follow‐up censoring date was 20 May 2020; 108 participants who were still hospitalised on May 20, 2020 were excluded from analysis. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Not all participants received the index test; more information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Yes | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | |||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
García de Guadiana‐Romualdo 2022.
| Study characteristics | |||
| Patient Sampling | All consecutive adult patients (≥ 14 years) discharged or dead after hospital admission to 32 Spanish hospitals, with SARS‐CoV‐2 infection, from March 1st, 2020, to April 30th, 2020. Exclusion criteria were: pregnant women, patients transferred from or to another hospital, patients transferred from nursing homes, patients discharged from the ED for at‐home treatment, patients with Intensive Care Unit admission criteria who were not admitted due to lack of availability and patients in whom D‐dimer levels were not measured on admission to ED. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: 32 hospitals of the National Health System in 9 autonomous communities
Country: Spain Symptoms and severity: not reported Demographics: median age in all participants was 65 years (IQR 54–76), in survivors 63 years (IQR 52–74), and in non‐survivors 76 years (IQR 68–83); 1103 (41.4%) were female participants, 982 (42.7%) female survivors, and 121 (33.2%) female non‐survivors Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: COVID‐19 was diagnosed by a positive result of RT‐PCR testing of a nasopharyngeal specimen or by a positive result of serological testing and a clinically compatible presentation. |
||
| Index tests | D‐dimer on admission was assessed (Figure 4). Data collection was performed retrospectively from electronic medical records and laboratory information systems by two researchers for each hospital. For measurement of d‐dimer levels, four immunoturbidimetric assays were used. For the harmonisation of results, four citrate plasma pools were prepared with a range of d‐dimer levels. Normal pooled plasma was prepared from blood samples collected from 10 individuals without known thrombotic events or prior antiplatelet or anticoagulant treatment. Blood from the cubital vein was collected into tubes containing sodium citrate as an anticoagulant, thoroughly mixed and immediately centrifugated at 2000 × g for 15 min. As pathological pooled plasma, a set of three plasma pools with increasing concentration of d‐dimer were prepared, collecting for each pool blood samples from 10 participants with recent known thromboembolic events. Pools were analysed within two hours of collection using the ACL TOP 700 analyser (Instrumentation Laboratory, US) and HemosIL D‐Dimer HS assay. Results were converted to FEU units. Plasma was immediately separated, aliquoted and frozen at −20°C to be transported to other participating laboratories using the other d‐dimer assays included in the BIOCOVID study. These laboratories were blinded to d‐dimer concentrations. The storage time of the pools was less than 1 week until analysis. After thawing, samples were analysed in duplicate within two hours. In this study, the strategy for harmonisation was based on a mathematical transformation of regression lines through the assay‐specific values of a set of plasma samples with different d‐dimer levels to a reference regression line. Because HemosIL d‐dimer HS‐500 was the assay mostly used in the BIOCOVID study, this was taken as reference, and four lines were generated for the values previously reported. The harmonisation model was based on a linear transformation of the majority method's harmonised reference line (yh = 2.197 xh − 2.843) and calculating xh by means of the line obtained in each method different from the reference one as xh = [(ym − bm)/am]. For the external validation of the harmonisation model, results were collected from two levels of quality control for d‐dimer included in the External Quality Assurance (EQA) scheme, provided by the Spanish Society of Hematology and Hemostasis (SEHH) in November 2020. | ||
| Target condition and reference standard(s) | All‐cause in‐hospital mortality was assessed in discharged or dead participants after hospital admission. Total sample size of 2663 participants with 364 non‐survivors and 2299 survivors |
||
| Flow and timing | The time horizon was not reported; the follow‐up censoring date was May 20th, 2020. Patients with no d‐dimer available on admission were excluded. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | 108 participants who were still hospitalised on May 20th, 2020, were excluded from the analysis. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Yes | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | |||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Goudot 2020.
| Study characteristics | |||
| Patient Sampling | Consecutive included adult (18 years and older) patients from March and April 2020 with confirmed SARS‐CoV‐2 infection were assessed. Inclusion criteria were patients with COVID‐19 who consulted the ED with hospitalisation criteria. | ||
| Patient characteristics and setting | Setting: ED
Site: Georges Pompidou European Hospital in Paris
Country: France Symptoms and severity: Participants in the Medicine Department presented with fever: 44 (91.7%), headache: 10 (20.8%), cough: 33 (68.8%), productive cough: 6 (12.5%), dyspnoea: 19 (39.6%), myalgia: 14 (29.2%), diarrhoea: 3 (6.2%), and pneumonia: 32 (66.7%); participants first admitted to the Medicine Department and then ICU presented with fever: 20 (95.2%), headache: 8 (38.1%), cough: 19 (90.5%), productive cough: 1 (4.8%), dyspnoea: 15 (71.4%), myalgia: 6 (28.6%), diarrhoea: 7 (33.3%), pneumonia: 19 (90.5%), and ARDS: 2 (9.5%). Demographics: median age of participants in the Medicine Department was 62.5 years (IQR 50.8–80.0), in participants: first Medicine Department then ICU 67.0 years (IQR 55.0–75.0); 17 (35.4%) in female participants in the Medicine Department, and 4 (19.0%) in female participants: first Medicine Department then ICU Exposure history: not reported Median time from illness onset to hospital admission was 4.5 days (IQR 3.0 to 7.0) in participants in the Medicine Department, and 7.0 days (IQR 4.0 to 8.0) in participants who went first to the Medicine Department, then ICU. Treatment before target condition in participants in the Medicine Department: statins: 11 (22.9%), oral antidiabetic agents: 5 (10.4%), insulin: 2 (4.2%); β‐blocker: 5 (10.4%), calcium channel blockers: 8 (16.7%), ACEi or ARBs: 13 (27.1%), ARBs: 6 (12.5%), diuretics: 4 (8.3%), central acting agent: 1 (2.1%), and in participants who went first to the Medicine Department then ICU: statins: 5 (23.8%), oral antidiabetic agents: 6 (28.6%), insulin: 4 (19.0%), β‐blocker: 3 (14.3%), calcium channel blockers: 6 (28.6%), ACEi or ARBs: 6 (28.6%), ARBs: 3 (14.3%), diuretics: 4 (19.0%) Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Routine laboratory tests were assessed on admission (Figure 4; Figure 5). All samples were collected in ethylenediaminetetraacetic acid (EDTA), sodium heparin, and 0.129 M trisodium citrate tubes (9NC BD Vacutainer©). High‐ sensitivity cardiac troponin (Hs‐cTnI, Beckman) was analysed on a DXI analyser. D‐Dimer concentrations were determined using the Vidas D‐Dimer assay (BioMérieux©) according to the manufacturer’s instructions. Data were retrieved from the medical records using a standardised data collection. Unit of analysis: individual participants Index tests were analysed on plasma. | ||
| Target condition and reference standard(s) | Deterioration was assessed; it was defined as referral to ICU of participants first admitted to the Medicine Department. The ICU referral criteria for COVID‐19 participants were: respiratory failure requiring mechanical ventilation at 6 to 8 L/min of oxygen to maintain SpO2 > 90 to 92% or signs of respiratory distress (≧ 30 breaths/min), or both; thoraco‐abdominal swaying; inspiratory depression of the suprasternal trough or other associated failure(s) or both: loss of consciousness with Glasgow Coma Scale < 12; systolic arterial pressure < 90 mmHg; signs of peripheral hypoperfusion. We included 69 participants for the analyses of this review, 48 participants in the Medicine Department, and 21 participants who were referred to ICU. The study also assessed participants who were referred to ICU immediately after emergency room admission; they were excluded from the review since they were not eligible according to the review question. | ||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Prospective cohort study | ||
| Funding a/o conflicts of interest | This work was funded by grants from the French National Research Agency and the Fondation de France ANR SARCODO. | ||
| Analysis | Information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Yes | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Guan 2020.
| Study characteristics | |||
| Patient Sampling | Hospitalised patients and outpatients with laboratory‐confirmed COVID‐19, as reported to the National Health Commission, between December 11, 2019, and January 29, 2020, from 552 hospitals in 30 provinces, autonomous regions, and municipalities in mainland China | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: 552 hospitals in 30 provinces, autonomous regions, and municipalities in mainland China
Country: China Symptoms and severity: participants presented with fever on admission: 473 (43.1%), conjunctival congestion: 9 (0.8%), nasal congestion: 53 (4.8%), headache: 150 (13.6%), cough: 745 (67.8%), sore throat: 153 (13.9%), sputum production: 370 (33.7%), fatigue: 419 (38.1%), haemoptysis: 10 (0.9%), shortness of breath: 205 (18.7%), nausea or vomiting: 55 (5.0%), diarrhoea: 42 (3.8%), myalgia or arthralgia: 164 (14.9%), chill: 126(11.5%); participants with the composite endpoint presented with fever on admission: 24 (36.4%), headache: 2 (3.0%), nasal congestion: 8 (11.9%), cough: 46 (68.7%), sore throat: 6 (9.0%), sputum production: 20 (29.9%), fatigue: 22 (32.8%), haemoptysis: 2 (3.0%), shortness of breath: 36 (53.7%), nausea or vomiting: 3 (4.5%), diarrhoea: 4 (6.0%), myalgia or arthralgia: 6 (9.0%), chill: 8 (11.9%). The severity on admission was defined based on the international guidelines for community‐acquired pneumonia; there were 926 non‐severe and 173 severe participants on admission. Demographics: the median age in all participants was 47.0 years (IQR 35.0– 58.0), in participants with the composite endpoint it was 63.0 years (IQR 53.0–71.0); female: all participants: 459 (41.9%), and composite endpoint: 22 (32.8%) Exposure to source of transmission within 14 days: in all participants: living in Wuhan: 483 (43.9%), contact with wildlife: 13 (1.9%), recently visited Wuhan: 193 (31.3%), and had contact with Wuhan residents: 442 (72.3%); in participants with composite endpoint: living in Wuhan: 39 (58.2%), contact with wildlife: 1 (2.4%), recently visited Wuhan: 10 (35.7%), and had contact with Wuhan residents: 19 (67.9%) Time since onset of symptoms: the median incubation period was 4.0 days (IQR 2.0–7.0) in all participants and 4.0 days (IQR 1.0–7.5) in participants with composite endpoint. Treatment before target condition: in all participants: intravenous antibiotics: 637 (58.0%), oseltamivir: 393 (35.8%), antifungal medications: 31 (2.8%), systemic glucocorticoids: 204 (18.6%), oxygen therapy: 454 (41.3%), mechanical ventilation: 67 (6.1%): invasive: 25 (2.3%), and non‐invasive: 56 (5.1%), use of extracorporeal membrane oxygenation: 5 (0.5%), use of continuous renal replacement therapy: 9 (0.8%), use of intravenous immunoglobin: 144 (13.1%), intensive care unit admission: 55/1099 (5.0%); in participants with the composite endpoint: intravenous antibiotics: 60 (89.6%), oseltamivir: 36 (53.7%), antifungal medications: 8 (11.9%), systemic glucocorticoids: 35 (52.2%), oxygen therapy: 59 (88.1%), mechanical ventilation: 40 (59.7%): invasive: 25 (37.3%) and non‐invasive: 29 (43.3%), use of extracorporeal membrane oxygenation: 5/67 (7.5%), use of continuous renal replacement therapy: 8/67 (11.9%), use of intravenous immunoglobin: 27/67 (40.3%), and intensive care unit admission: 55/67 (82.1%) Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: COVID‐19 was diagnosed on the basis of the WHO interim guidance. A confirmed case of COVID‐19 was defined as a positive result on high throughput sequencing or RT‐PCR assay of nasal and pharyngeal swab specimens. Only laboratory‐confirmed cases were included in the analysis. |
||
| Index tests | Routine laboratory tests performed on admission were included (Figure 4 and Figure 5). Laboratory findings were extracted from electronic medical records; a team of experienced respiratory clinicians reviewed and abstracted the data. Data were entered into a computerised database and cross‐checked. Major disagreement between two reviewers was resolved by consultation with a third reviewer. All medical records were copied and sent to the data‐processing centre in Guangzhou, under the coordination of the National Health Commission. If the core data were missing, requests for clarification were sent to the coordinators, who subsequently contacted the attending clinicians. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | The study assessed a composite endpoint consisting of admission to an intensive care unit, mechanical ventilation, or death. The total sample size was 1099 participants, with 67 with the composite endpoint, and 1032 without. | ||
| Flow and timing | The time horizon was not reported. The data cut‐off for the study was January 31, 2020. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | Supported by the National Health Commission of China, the National Natural Science Foundation, and the Department of Science and Technology of Guangdong Province | ||
| Analysis | No imputation was made for missing data. The risk of composite endpoints amongst hospitalised cases and the potential risk factors were analysed using Fine‐Gray competing‐risk models in which recovery is a competing risk. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Unclear | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Haymana 2021.
| Study characteristics | |||
| Patient Sampling | This nationwide study was conducted from 11 March through 30 May 2020 using the COVID‐19 registry of the Turkish Ministry of Health National Electronic Database. The study population included confirmed COVID‐19 adult patients with glucose measurements at the time of COVID‐19 diagnoses and without previously known diabetes. Patients with type 1, 2, or unclassified diabetes mellitus, blood glucose level above 200 mg/dL and no glucose measurement at the time of COVID‐19 diagnosis were excluded from the analysis. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: Turkish Ministry of Health National Electronic Database, hospitals not specified
Country: Turkey Symptoms and severity: not reported Demographics: the median age in all participants was 44 years (IQR 25). Participants were categorised according to their glucose range. In participants with glucose < 100 mg/dL, the median age was 35 years (IQR 19), in participants with glucose 100 to 139 mg/dL, median age was 45 years (IQR 22), and in participants with glucose 140 to 199 mg/dL, it was 55 years (IQR 20). The study included 50.9% female participants. In participants with glucose < 100 mg/dL, there were 53.1% female participants, in participants with glucose 49.7%, and in participants with glucose 140 to 199 mg/dL, 45.2%. Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: RAS blocker in participants with glucose < 100 mg/dL: 960 (16.2%), in participants with glucose 100–139 mg/dL: 1601 (26.4%), in participants with glucose 140–199 mg/dL: 330 (38.9%); statins in participants with glucose < 100 mg/dL: 236 (4.0%), in participants with glucose 100–139 mg/dL: 378 (6.2%), in participants with glucose 140–199 mg/dL: 108 (12.7%), acetylsalicylic acid in participants with glucose < 100 mg/dL: 701 (11.9%), in participants with glucose 100–139 mg/dL: 1011 (16.7), in participants with glucose 140–199 mg/dL: 209 (24.6%) Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Blood glucose was obtained from the national database. Blood glucose levels measured within 24 hours of COVID‐19 diagnosis were recorded regardless of fasting state (Figure 4). Unit of analysis: individual participants. | ||
| Target condition and reference standard(s) | Mortality and deterioration, defined as ICU admission were assessed. The study included 12,817 participants, 1043 participants were admitted to ICU, and 542 participants died. | ||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Patients with no glucose measurement at the time of COVID‐19 diagnosis were excluded from the analysis. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Ibraheem 2021.
| Study characteristics | |||
| Patient Sampling | Confirmed COVID‐19 patients who were admitted to the Al‐Furat General Hospital, Iraq from March to August 2020. At the day of admission, according to the World Health Organization (WHO) guidelines, all patients were classified as moderate‐to‐severe COVID‐19 patients. Patients with abnormal renal and hepatic tests were excluded. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: Al‐Furat General Hospital, Baghdad
Country: Iraq Symptoms and severity: all participants were symptomatic with cough, fever, tiredness and dyspnoea for a median duration of 5 days, ranging from 3‐8 days. Demographics: the median age in all participants was 40 years (range 2‐84); females: 38.2% of participants Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | NLR was assessed on the day of admission (Figure 5). Up to 2 mL of venous blood was drawn into an ethylene diamine tetra acetic acid (EDTA) tube. The blood count was estimated using the QBC Autoread Plus Hematology System (Drucker Diagnostics, USA). NLR was estimated as a neutrophil‐lymphocyte ratio. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Mortality was assessed in 123 participants, with 100 survivors, and 23 non‐survivors. | ||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Prospective cohort study | ||
| Funding a/o conflicts of interest | The study was funded by Al‐Karkh Health General Directorate (Funding Order No.: KH‐0125). | ||
| Analysis | Information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Yes | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | |||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | Unclear | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
López‐Escobar 2021.
| Study characteristics | |||
| Patient Sampling | The study included COVID‐19 patients that were hospitalised at any of the 10 hospitals of the HM Hospitales Group across different regions in Spain (including Madrid, Barcelona, and Galicia, Spain) from 1 March to 10 June 2020. Patients with missing SaO2 at admission or missing laboratory data in the first 24 hours of admission, under 18 years old, or who died at hospital admission were excluded from the analysis. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: 10 hospitals of HM Hospitales group across different regions in Spain
Country: Spain Symptoms and severity: not reported Demographics: median age was 69 years (IQR 57‐80); 39.9% female participants Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR or clinical characteristics and radiological criteria |
||
| Index tests | Routine laboratory tests were assessed at hospital admission, within 24h (Figure 4 and Figure 5). Unit of analysis: individual participants, and the sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | In‐hospital mortality was assessed. The total sample size was 1310 participants with 194 non‐survivors, and 1116 survivors (= discharged participants). | ||
| Flow and timing | The time horizon was not reported. Clinical and laboratory data measurements were available up to and including 24 June 2020. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | CRP was missing in 24 (1.8%) participants. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | No | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | |||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Marwah 2021.
| Study characteristics | |||
| Patient Sampling | The study included all admitted adult (18 years and older) patients who tested positive for COVID‐19 from a single centre National Health Trust in the UK from 3rd February 2020 to the 4th September 2020. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: secondary care, a single centre, National Health Trust
Country: UK Symptoms and severity: 46 (10%) patients were admitted to intensive care unit. Demographics: the median age in survivors was for Caucasian participants: 78 years (IQR 68‐87) and for BAME (Black (African and Afro‐Caribbean), Asian and Minority Ethnic) participants: 56 years (IQR 41.75‐73.25); and in fatalities for Caucasian participants: 81 years (IQR 73‐87.5) and for BAME participants: 79 year (IQR 67‐85); 193 female participants, 118 in survivors, and 75 in non‐survivors Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: All participants were treated using the local trust and the NHS protocols irrespective of their ethnicity. Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Routine laboratory tests were assessed on admission (Figure 4 and Figure 5). An Abbot Architect™ analyser was used to determine quantitative biochemical analysis. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Mortality was assessed. The total sample size was 445 participants, 255 participants survived, and 190 participants died. | ||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | The sample size of the index tests varied because of missing data. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Yes | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | |||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Masotti 2021.
| Study characteristics | |||
| Patient Sampling | Patients were admitted to a non‐intensive unit for respiratory failure associated with SARS‐CoV‐2‐related pneumonia during the first and second wave of the pandemic, from March to April 2020 and from October to November 2020, in San Giuseppe Hospital, Florence, Italy. | ||
| Patient characteristics and setting | Setting: non‐intensive care unit
Site: San Giuseppe Hospital, Empoli
Country: Italy Symptoms and severity: not reported Demographics: the mean age in all participants was 69.4 years (SD 13.3); 46% female participants. Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: 6.2% of participants were intubated. Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 case: not reported |
||
| Index tests | Routine laboratory tests were assessed on admission (Figure 4 and Figure 5). Quantitative determination of IL‐6 levels was performed by using an immuno‐enzymatic chemiluminescent assay (Access Immunoassay System, Beckman Coulter, USA, lowest limit of detection 0.5 pg/mL). No information reported on the analyses of the other laboratory tests. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Combined outcome was assessed, defined as in‐hospital mortality or intensive care unit admission requiring oro‐tracheal intubation or both. The study included 223 participants, 41 participants died or were admitted to ICU admission requiring oro‐tracheal intubation, or both, and 182 patients were alive and did not require ICU with oro‐tracheal intubation. | ||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Mateos González 2021.
| Study characteristics | |||
| Patient Sampling | Adult (18 years and older) patients with confirmed COVID‐19 were admitted to 147 hospitals throughout Spain. Patients included in the SEMI‐COVID‐19 Registry as of 31 May 2020, were selected for inclusion in this study if they had: (a) all epidemiological data recorded, (b) data on lymphocyte and eosinophil counts upon admission and on the secondary analysis at seven days after admission, and (c) onset of symptoms prior to admission. The SEMI‐COVID‐19 Registry was a nationwide, retrospective cohort that includes consecutive patients with a confirmed COVID‐19 infection who have been hospitalised and discharged from Spanish hospitals. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: 147 hospitals throughout Spain, from the Spanish Society of Internal Medicine’s SEMI‐COVID‐19 Registry
Country: Spain Symptoms and severity: non‐survivors presented with cough: 1093 (15.0%) participants, dyspnoea: 1100 (19.9%) participants, arthromyalgia: 309 (10.0%) participants, asthenia: 640 (15.2%) participants, anorexia: 373 (19.7%) participants, fever at home: 1266 (15.4%) participants; pneumonia was present in 1048 participants, mild ARDS in 846 participants, moderate ARDS in 687 participants, severe ARDS in 1524 participants, acute kidney failure in 1300 participants, sepsis in 564 participants; 857 participants were admitted to the ICU Demographics: the mean age in non‐survivors was 78.7 years (SD 10.5), and in survivors 63.8 years (SD 15.7); 4135 female participants included Exposure history: not reported Time from onset of symptoms at admission: mean in non‐survivors 5.7 days (SD 5.0), and in survivors: 7.2 days (SD 6.1) Treatment before target condition: non‐survivors were treated with lopinavir/ritonavir in 929 (15.0%) participants, hydroxychloroquine in 1246 (14.7%) participants, systemic corticosteroids in 532 (21.6%) participants, tocilizumab in 214 (22.8%) participants, azithromycin in 914 (15.3%) participants, inhaled corticosteroids in 117 (20.7%) participants, and low‐molecular‐weight heparin in 1324 (16.4%) participants. Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: laboratory tests, not specified |
||
| Index tests | Routine laboratory tests were assessed on admission (Figure 4 and Figure 5). Data from medical records were collected retrospectively at discharge by clinical investigators all over the country, using a standardised online data capture system. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Mortality was assessed. The total sample size was 9644 participants, including 8049 survivors, and 1595 non‐survivors. | ||
| Flow and timing | The time horizon was not reported. In the study, 533 participants did not have all demographic and epidemiological data recorded and thus were excluded, and 282 were excluded because they did not have eosinophil counts upon admission. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | This research received no external funding. The Spanish Society of Internal Medicine is the sponsor of this registry. The researchers who coordinate the study at each hospital are SEMI members and were asked to participate in this study on a voluntary basis; they did not receive any remuneration for their participation. | ||
| Analysis | All enroled participants were included in the analysis. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | |||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Meng 2021.
| Study characteristics | |||
| Patient Sampling | Patients hospitalised from January to February 2020 at the Public Health Clinical Centre of Chengdu were included. All patients were diagnosed with COVID‐19, and disease classifications were made according to the Diagnosis and Treatment Plan of New Coronavirus Pneumonia (Trial Version 6) released by the National Health Commission and National Administration of Traditional Chinese Medicine. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: Public Health Clinical Centre of Chengdu
Country: China Symptoms and severity: fever was present in the mild and moderate group in 39 (69.64%) participants, and in the severe and critically‐severe group in 21 (91.30%) participants; cough in the mild and moderate group in 34 (60.71%) participants, and in the severe and critically‐severe group in 15 (65.22%) participants; shortness of breath in the mild and moderate group in 8 (14.29%) participants, and in the severe and critically‐severe group in 7 (30.43%) participants; myalgia or fatigue in the mild and moderate group in 8 (14.29%) participants, and in the severe and critically‐severe group in 4 (17.39%) participants. Demographics: the mean age of all cases was 50.50 years; in the mild group, this was 40.14 years (SD 15.94), in the moderate group 51.00 years (SD 16.26), in the severe group 57.25 years (SD 18.26), and in the critically‐severe group 64.09 years (SD 15.04); 38 female participants included, in the mild group: 6 females, moderate group: 23 females, severe group: 5 females, and critically‐severe: 4 females Exposure history within the past 14 days: in the mild and moderate group (n = 56): in 28 (50.00%) participants and in the severe and critically‐severe group: 15 (65.22%) participants; 43 (54.43%) participants had a history of going to or living in Wuhan or coming into contact with people from Wuhan Time since onset of symptoms: not reported Treatment before target condition: oxygen inhalation: mild and moderate group: 35 (62.50%) participants, and severe and critically‐severe group: 20 (86.96%) participants; glucocorticoid: severe and critically‐severe group: 2 (8.70%); antiviral treatment: mild and moderate group: 54 (96.43%) participants, and severe and critically‐severe group: 13 (56.52%) participants; interferon atomisation: mild and moderate group: 50 (89.29%) participants, and severe and critically‐severe group: 21 (91.30%) participants; thymalfasin: mild and moderate group: 15 (26.79%) participants, and severe and critically‐severe group: 13 (56.52%) participants Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: epidemiological history, clinical signs and symptoms, imaging results, and laboratory indicators (especially SARS‐CoV‐2 nucleic acid‐positive), for the suspected participants, continuously detecting SARS‐CoV‐2 nucleic acid within 2 weeks |
||
| Index tests | Routine test were measured on admission (Figure 4 and Figure 5). Whole blood with EDTA‐K2 anticoagulant was used for complete blood count analysis, which was conducted on a Mindray BC‐6900 automatic blood cell analyser (Shenzhen Mindray Bio Medical Electronics Co., Shenzhen, China) with the corresponding reagents. Blood with lithium heparin anticoagulant was used for the peripheral lymphocyte subset count analysis, which was conducted on a Beckman Coulter Dxflex flow cytometer with Cyto‐STAT tetraCHROME CD45‐FITC/CD4‐RD1/CD8‐ECD/CD3‐PC5 and Cyto‐STAT tetraCHROME CD45‐FITC/CD56‐RD1/CD19‐ECD/ CD3‐PC5 reagents (Beckman Coulter Company, USA). All flow cytometry data were analysed by CytExpert for Dxflex version 2.0 software. Sample detection and quality control were performed according to the standard operating procedures and requirements of the clinical laboratory. Unit of analysis: individual participants | ||
| Target condition and reference standard(s) | Deterioration was assessed. Disease classifications were made according to the Diagnosis and Treatment Plan of New Coronavirus Pneumonia (Trial Version 6) released by the National Health Commission and National Administration of Traditional Chinese Medicine. According to the guidelines, participants were categorised into four subtypes: (1) mild: participants with mild symptoms without pneumonia manifestation in imaging, (2) moderate: participants with some symptoms, such as fever, respiratory tract symptoms, etc., with radiological manifestations of pneumonia, (3) severe: participants who fulfil any of the following: respiratory distress, respiratory rate ≥ 30 times/min, oxygen saturation (SpO2) ≤ 93% in resting state, partial pressure of oxygen in arterial blood/fraction of inspired oxygen (PaO2/FiO2 ratio) ≤ 300 mmHg (1 mmHg = 0.133652 kPa), greater than 50% lesion progression in pulmonary imaging within 24 to 48 h, and (4) critically severe: participants who fulfil any of the following: respiratory failure and requiring mechanical ventilation, shock, admission to the ICU with other organ failure. Participants in the mild and moderate group were compared to the severe and critically severe group. The total sample size was 79 participants, with 14 mild, 42 moderate, 12 severe and 23 critically‐severe cases. | ||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | This work was funded by grants from the National Natural Science Foundation (No. 81702002), Sichuan Science and Technology Program (2020YFS0137), and Chengdu Technology Innovation Research and Development Project (No. 2020‐YF05‐0026‐SN). | ||
| Analysis | All enroled participants were included in the analysis. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Yes | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Unclear | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Yes | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Muñoz‐Rodríguez 2021.
| Study characteristics | |||
| Patient Sampling | The study included adult patients (18 years and older), meeting one or more laboratory criteria or one or more clinical criteria of suspected COVID‐19, or both. Patients attended, from 11 February to 11 May 2020, one of the 14 public hospitals of the Health Service of Castilla‐La Mancha (SESCAM) in Spain. Patients with subsequent admissions, transfers or duplicates for the same patient, and paediatric patients (< 18 years old) were excluded. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: 14 public hospitals of the Health Service of Castilla‐La Mancha (SESCAM), Spain, covering 30,680 square miles and serving two million residents in urban, suburban, and rural areas. None of the hospitals were specifically designated for COVID‐19 and the distribution of participants in the centres was similar to under normal conditions.
Country: Spain Symptoms and severity: anosmia was present in non‐survivors: 18 (0.9%), and survivors: 635 (6.3%), diarrhoea in non‐survivors: 198 (9.8%), and survivors: 1850 (18.3%), vomiting in non‐survivors: 125 (6.2%), and survivors: 774(7.7%), fever in non‐survivors: 1250 (61.9%), and survivors: 6293(62.3%), dyspnoea in non‐survivors: 1421 (70.3%), and survivors: 5449 (53.9%), cough in non‐survivors: 980 (48.5%) and survivors: 5849 (57.9%); 596 participants were admitted to the ICU Demographics: the mean age in all participants was 66.4 years (SD 17.3), and the median age in all participants was 68 years (IQR 54‐81); 18–39 years: non‐survivors: 8/2020 (0.4%); survivors: 896/10,106 (8.9%); 40–49 years: non‐survivors: 27/2020 (1.3%); survivors: 1235/10,106 (12.2%); 50‐59 years: non‐survivors: 94/2020 (4.7%); survivors:1934/10,106 (19.1%); 60‐69 years: non‐survivors: 253/2020 (12.5%); survivors: 1917/10,106 (19%); 70‐79 years: non‐survivors: 516/2020 (25.5%); survivors: 1866/10,106 (18.5%); ≥ 80 years: non‐survivors: 1122/2020 (55.5%); survivors: 2258/10,106 (22.3%); 5667 (46.7%) female participants, in: non‐survivors: 800(39.6%), and in survivors: 4867 (48.2%) Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: invasive ventilation in non‐survivors 203 (10%), and survivors 327 (3.2%); non‐invasive ventilation in non‐survivors 316 (15.6%), and survivors: 448 (4.4%); antiretroviral treatment in non‐survivors: 597 (29.6%), and survivors: 2740 (27.1%); chloroquine in non‐survivors: 1245 (61.7%), and survivors 6665 (66%); interferon β‐1b in non‐survivors: 73 (3.6%), and survivors: 219 (2.2%); azithromycin in non‐survivors 1102 (54.6%), and survivors: 6639 (65.7%); corticosteroids in non‐survivors: 1004 (49.8%), and survivors: 3781(37.4%); tocilizumab in non‐survivors: 115 (5.7%), and survivors: 255 (2.5%) Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: laboratory and clinical criteria. The laboratory criteria included a positive detection of 2019‐nCoV nucleic acid by RT‐PCR or IgM from human serum with rapid test for SARS‐CoV‐2. The clinical criteria included acute respiratory infection with fever, cough or dyspnoea, or radiographic characteristics of pneumonia, such as multiple ground‐glass shadows, infiltrative shadows and consolidation in both lungs, or other symptoms such as odynophagia, anosmia, ageusia, vomiting, muscle aches, diarrhoea, chest pain or headaches. |
||
| Index tests | The initial laboratory tests (first test results available) of D‐dimer were assessed (Figure 4). A trained team of thirty physicians collected data from electronic medical records, using a standard data form for epidemiological surveillance, within the health information system. These physicians performed this operation after a daily updating of the admitted list in each hospital, after which, three clinical coordinators supervised and checked for errors in the data collection. Unit of analysis: individual participants Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Mortality was assessed in 12,126 participants, with 2020 non‐survivors, and 10,106 survivors. | ||
| Flow and timing | Mortality was assessed 90 days after admission. Not all participants received the index test. | ||
| Comparative | |||
| Study design | Prospective cohort study | ||
| Funding a/o conflicts of interest | This work is supported by the Spanish Ministry of Health and the General Directorate of Public Health of Castilla‐La Mancha. Leticia Serrano‐Oviedo is the recipient of a contract made by the Directorate of Public Health of Castilla‐La Mancha specifically devoted to study the COVID‐19 pandemic. | ||
| Analysis | The study did not take into account censoring. The final date of the study was May 11. The status of all participants was reviewed on that date. There was no follow‐up after this date. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Yes | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Olivieri 2022.
| Study characteristics | |||
| Patient Sampling | Consecutive geriatric patients (65 years and older) from 1st March 2020 to 24th June 2021 were hospitalised at the INRCA hospital (Ancona, Italy) due to COVID‐19. All patients aged 65 years and above with confirmed COVID‐19 who were admitted to the INRCA hospital were included in the study. The only exclusion criterion was the lack of informed consent. The study used data from the Report‐Age COVID project, an observational study conducted at the Italian National Center on Aging (IRCCS INRCA). | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: the INRCA hospital, Ancona
Country: Italy Symptoms and severity: cough in survivors: 107 (25.4%), and non‐survivors: 53 (24.2%); dyspnoea in survivors: 195 (46.3%), and non‐survivors: 156 (70.9%); diarrhoea in survivors: 44 (10.5%), and non‐survivors: 19 (8.6%); nausea in survivors: 17 (4.1%), and non‐survivors: 4 (2.0%); vomiting in survivors: 21 (5.1%), and non‐survivors: 9 (4.0%); conjunctivitis in survivors: 3 (0.7%), and non‐survivors: 2 (0.7%); ageusia in survivors: 10 (2.4%), and non‐survivors: 3 (1.3%); anosmia in survivors: 7 (1.7%), and non‐survivors: 3 (1.3%) Demographics: mean age in all cases was 86.6 years (SD 6.8); in non‐survivors: 88.5 years (SD 5.5), and in survivors: 85.6 years (SD 7.2); 375 (58.5%) female participants; in non‐survivors: 113 (51.4%), and in survivors: 262 (62.2%) Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: glucocorticoids in survivors: 301 (71.5%), and non‐survivors: 161 (73.2%); heparin in survivors: 367 (87.2%), and non‐survivors: 189 (85.9%) Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Routine laboratory tests were assessed on admission (Figure 4 and Figure 5). Clinical and epidemiological data were collected in a retrospective manner and were anonymised before release. Blood concentrations of haemoglobin, C‐reactive protein, IL‐6, d‐dimer, N‐terminal pro‐brain natriuretic peptide, serum ferritin, sodium, potassium, procalcitonin, platelet, and white blood cell count were measured by standard procedures. Unit of analysis: individual participants | ||
| Target condition and reference standard(s) | The study assessed mortality; 220 participants deceased and 421 survived. The total sample size was 641. | ||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | This work was supported by the Italian Ministry of Health – Ricerca Corrente to IRCCS INRCA and by Universita` Politecnica delle Marche (RSA grant to FO). | ||
| Analysis | Information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | |||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Ortega‐Rojas 2022.
| Study characteristics | |||
| Patient Sampling | Adults (60 years and older) hospitalised with a diagnosis of COVID‐19 in three hospitals in Lambayeque, Peru from March 18 to May 13, 2020 were assessed. All included patients were admitted to the hospital solely for treatment of COVID‐19. Older adults who did not have the variables of interest (NLR, PLR, and confounders) were excluded. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: the Hospital Almanzor Aguinaga Asenjo (third level), Hospital Luis Hey‐sen Incháustegui (second level), and the Hospital Clínica EsSalud Chepén (second level)
Country: Peru Symptoms and severity: shortness of breath in survivors: 73 participants, and in non‐survivors: 151 participants; cough in survivors: 62 participants, and in non‐survivors: 135 participants; fever in survivors: 37 participants, and non‐survivors: 93 participants; sore throat in survivors: 6 participants, and non‐survivors: 17 participants; diarrhoea in survivors: 7 participants, and non‐survivors: 13 participants; headache: in survivors: 5 participants, and in non‐survivors: 8 participants; nasal congestion: in non‐survivors: 3 participants; anosmia in non‐survivor: 1 participant; and ageusia: in non‐survivor: 1 participant; 7 participants were admitted to ICU Demographics: median age was 70 years (IQR 65‐78); in survivors: 67 years (IQR 64‐72), and in non‐survivors: 72 years (IQR 66‐80) ≤ 70 years: survivors: 57 participants, and in non‐survivors: 82 participants; 71‐80 years: survivors: 19 participants, and non‐survivors: 55 participants; ≥ 81 years: survivors: 6 participants, and non‐survivors: 43 participants; Female participants in all cases: 74 (28.2%) participants; in non‐survivors: 51 (68.9%) participants, and in survivors: 23 (31.1%) participants Exposure history: contact with a confirmed case of COVID‐19 in 8 (3.1%) participants; in non‐survivors: 5 (62.5%) participants, and in survivors: 3 (37.5%) participants Time since onset of symptoms (median days before hospital admission): in all participants: 7 days (IQR 4‐10), in survivors: 7 days (IQR 4‐10), and in non‐survivors: 7 (IQR 5‐10) Treatment before target condition: antibiotic therapy: all participants: 259 (98.9%), survivors: 81 (31.3%), non‐survivors: 178 (68.7%); types of antibiotic therapy: azithromycin + cephalosporins: all participants: 205 (79.1%), survivors: 67 (32.7%), non‐survivors: 138 (67.3%); azithromycin: all participants: 21 (8.1%), survivors: 4 (19.1%), non‐survivors: 17 (80.9%); cephalosporins: all participants: 13 (5%), survivors: 5 (38.5%), non‐survivors: 8 (61.5%); azithromycin + carbapenems: all participants: 16 (6.2%), survivors: 4 (25.0%), non‐survivors: 12 (75.0%); carbapenems: all participants: 3 (1.2%), survivors: 1 (33.3%), non‐survivors: 2 (66.7%); azithromycin + others: all participants: 1 (0.4%), non‐survivors: 1 (100%); corticosteroids: all participants: 181 (69.1%), survivor: 54 (29.8%), non‐survivors: 127 (70.2%); type of corticosteroids: methylprednisolone: all participants: 110 (60.8%), survivors: 26 (23.6%), non‐survivors: 84 (76.4%); dexamethasone: all participants: 56 (30.9%); survivors: 26 (46.4%), non‐survivors 30 (53.6%); hydrocortisone: all participants: 13 (7.2%), non‐survivors: 13 (100%); prednisone: all participants 2 (1.1%), survivors: 2 (100%); hydroxychloroquine: all participants: 192 (73.3%); survivors: 60 (31.3%), non‐survivors: 132 (68.7%); ivermectin: all participants: 71 (27.1%), survivors: 29 (40.9%), non‐survivors: 42 (59.1%); enoxaparin: all participants: 208 (79.4%), survivors: 59 (28.4%), non‐survivors: 149 (71.6%); lopinavir/ritonavir: all participants: 57 (21.8%), survivors: 17 (29.8%), non‐survivors: 40 (70.2%) Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: rapid lateral flow test or molecular testing (RT‐PCR) and suspected diagnosis based on a clinical or radiological pattern profile plus an epidemiological link (having had contact with a confirmed case during the last 14 days) despite having a non‐reactive rapid lateral flow test for COVID‐19 or a negative RT‐PCR |
||
| Index tests | Routine laboratory tests were assessed in the first 24 hours of hospital admission (Figure 4 and Figure 5). The NLR was calculated by dividing the neutrophil and lymphocyte counts. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | In‐hospital all‐cause mortality was assessed; 262 participants were included, 82 survivors, and 180 non‐survivors | ||
| Flow and timing | An administrative censoring time at 60 days was considered with hospital admission defined as zero time. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | 45 participants were excluded due to missing values (neutrophils, platelets, or lymphocytes). | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Yes | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | No | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Ouldali 2021.
| Study characteristics | |||
| Patient Sampling | Patients were recruited from 60 hospitals throughout France, using the French paediatric bacterial meningitis network. All paediatric patients with SARS‐CoV‐2 infection who were hospitalised in one of these centres from February 15 to June 1, 2020 were included. Children with positive serology alone were excluded. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: 60 hospitals throughout France
Country: France Symptoms and severity: fever was present in severe forms: 18 (82%) participants, and non‐severe form: 215 (79%) patients; cough in severe forms: 12 (52%) participants, and non‐severe form: 127 (46%) participants; rhinitis in severe forms: 6 (26%) participants, and non‐severe form: 136 (49%) participants; shortness of breath or dyspnoea in severe forms: 12 (55%) participants, and non‐severe form: 55 (20%) participants; hypoxaemia in severe forms: 8 (42%), and non‐severe form: 17 (6%) participants; abdominal pain in severe forms: 1 (6%), and non‐severe form: 25 (16%) participants; diarrhoea or vomiting in severe forms: 7 (32%) participants, and non‐severe form: 79 (29%) participants; neurologic symptoms in severe forms: 4 (18%) participants, and non‐severe form: 20 (7%) participants Demographics: median age of all cases was 16 months (IQR 51 days–134 months); female participants in all cases: 171 (43%), in severe forms: 13 (57%), and in non‐severe forms: 116 (41%) Exposure history: identified contact case: 89 (23%) participants, suspected contact case: 132 (34%) participants, and no contact case: 165 (43%) participants Time since onset of symptoms: not reported Treatment before target condition: antibiotics: 167 (43%) participants, corticosteroids: 17 (4%) participants, antiviral treatment: 7 (2%) participants, hydroxychloroquine: 2 (1%) participants, admission to PICU: 81 (20%) participants, haemodynamic support: 21 (28%) participants, non‐invasive ventilation support: 18 (23%) participants, invasive ventilation support: 17 (23%) Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: confirmed SARSCoV‐2 infection was defined by a positive RT‐PCR result for SARS‐CoV‐2 on a nasopharyngeal swab; and computed tomography scan–based cases were defined by characteristic chest CT scan lesions according to European Centre for Disease Prevention and Control and French National Health Agency case definition. |
||
| Index tests | Routine laboratory tests on admission were assessed (Figure 4 and Figure 5). For each participant, an electronic case report form was prospectively completed on a secure database. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | The study assessed deterioration to severe form. Severe form was defined by the need for either ventilatory or haemodynamic support during hospitalisation, or death. Ventilatory support was defined by use of non‐invasive ventilation, including high‐flow oxygen via nasal cannula, continuous positive airway pressure, and bilevel positive airway pressure or the use of invasive ventilation. The study included 397 participants, 23 participants had a severe form, and 283 non‐severe form. | ||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Prospective cohort study | ||
| Funding a/o conflicts of interest | Funding: Association Clinique et Thérapeutique Infantile du Val de Marne and the Clinical Research Center, Centre Hospitalier Intercommunal de Créteil, Créteil, France | ||
| Analysis | Information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Pan 2021.
| Study characteristics | |||
| Patient Sampling | All the confirmed patients who were diagnosed with COVID‐19 in Fuyang city as of April 8, 2020 were enroled in this study. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: second hospital of Fuyang city, Centers for Disease Control and Prevention in Fuyang and Anhui province
Country: China Symptoms and severity: fever in all participants: 131 (84.52%), moderate: 102 (81.6%), and severe (critical): 29 (96.7%); cough in all participants: 127 (81.94%), moderate: 101 (80.8%), severe (critical): 26 (86.7%); fatigue in all participants: 54 (34.84%), moderate: 39 (31.2%), and severe (critical): 15 (50.0%); shortness of breath in all participants: 64 (41.29%), moderate: 35 (28.0%), and severe (critical): 29 (96.7%); sputum production in all participants: 63 (40.65%), moderate: 49 (39.2%), and severe (critical): 14 (46.7%); sore throat in all participants: 25 (16.13%), moderate: 21 (16.8%), and severe (critical): 4 (13.3%); myalgia in all participants: 4 (2.58%), moderate: 3 (2.4%), and severe (critical): 1 (3.3%); chest pain in all participants: 4 (2.58%), moderate: 3 (2.4%), and severe (critical): 1 (3.3%); runny nose in all participants: 6 (3.87%), moderate: 4 (3.2%), and severe (critical): 2 (6.7%); diarrhoea in all participants: 55 (35.48%), moderate: 49 (39.2%), and severe (critical): 6 (20.0%); nausea and vomiting in all participants: 17 (10.97%), moderate: 16 (12.8%), and severe (critical): 1 (3.3%); headache/dizziness in all participants: 16 (10.32%), and moderate: 16 (12.8%); erythema in all participants: 2 (1.29%), and moderate: 2 (1.6%) Demographics: in all participants, mean age was 41.95 years (SD 15.34), in moderate participants, mean age: 39.78 (SD 15.0), and in severe participants, mean age: 50.97 (SD 13.55); the range of age was 1 year 4 months to 82 years; female: all participants: 68 (43.87%), moderate participants 58 (46.4%), and severe (critical) participants: 10 (33.3%) Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: oxygen therapy: nasal cannula in all participants: 97 (62.58%), moderate participants: 89 (71.2%), severe (critical) participants: 8 (26.67%); non‐invasive ventilation or high‐flow nasal cannula: all participants: 18 (11.61%), severe (critical) participants: 18 (60.0%); invasive ventilation: all participants : 4 (2.58%), severe (critical) participants: 4 (13.33%); antiviral therapy: all participants: 153 (98.7%), moderate participants: 123 (98.4%), severe (critical) participants: 30 (100%); antibiotic therapy: all participants : 95 (61.29%), moderate participants: 72 (57.6%), severe (critical) participants: 23 (76.67%); glucocorticoids: all participants : 42 (27.1%), moderate participants: 20 (16.0%), severe (critical) participants: 22 (73.3%); intravenous immunoglobulin therapy: all participants : 27 (17.42%), moderate participants: 8 (6.4%), severe (critical) participants: 19 (63.3%); tocilizumab: all participants: 15 (9.68%), moderate participants: 3 (2.4%), severe (critical) participants: 12 (40.0%) Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: SARS‐CoV‐2 nucleic acid detection |
||
| Index tests | Routine laboratory tests were assessed on admission (Figure 4 and Figure 5). Epidemiological characteristics, clinical manifestations, laboratory results, treatment, and radiologic data were obtained from the electronic medical record system of the hospital or by face‐to‐face interviews with participants or their relatives. Unit of analysis: individual participants. The sample used for bilirubin, serum amyloid protein A, and creatinine was serum. For the other laboratory tests, this was not reported. | ||
| Target condition and reference standard(s) | Deterioration was assessed. Deterioration was defined depending on the diagnosis and treatment program of COVID‐19 (trial seventh version). The total sample size was 155 participants, with 125 moderate participants, and 30 severe (critical) participants. | ||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | This work was supported by the National Natural Science Foundation of China (grant numbers U1803126) and Key Projects of Academic Support for Top Talents in Colleges and Universities, China (grant numbers gxbjZD2016036). | ||
| Analysis | Information about missing data was not reported. In this study, all 155 participants were cured to discharge. None of the participants died. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Papageorgiou 2022.
| Study characteristics | |||
| Patient Sampling | Adult patients (16 years and older) with a diagnosis of COVID‐19 and at least one troponin measurement admitted to six hospitals in London, UK from 16th to the 30th of March 2020 were included. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: multi‐centre study, six hospitals in London
Country: UK Symptoms and severity: cough: all: 27.4% (119 participants), negative hsTrop < 15 ng/L: 15.8% (23 participants), hsTrop > 15 ng/L: 33.3% (96 participants); fever: all: 17.1% (74 participants), negative hsTrop < 15 ng/L: 19.9% (29 participants), hsTrop > 15 ng/L: 15.6% (45 participants); both: all: 36.4% (158 participants), negative hsTrop < 15 ng/L: 52.7% (77 participants), hsTrop > 15 ng/L: 28.1% (81 participants); other: all: 191.1% (83 participants), negative hsTrop < 15 ng/L: 11.6% (17 participants), hsTrop > 15 ng/L: 22.9% (66 participants) Demographics: the median age was 66 years (IQR 56–80) in all cases, 59 years (IQR 51–64) in cases with negative hsTrop < 15 ng/L, and 72 years (IQR 58–81) in cases with hsTrop > 15 ng/L; female participants: 37.1% (161), in negative hsTrop < 15 ng/L: 41.2% (60), and in hsTrop > 15 ng/L: 35.1% (101) Exposure history: not reported Time of symptom onset before admission: all cases: median of 7 days (IQR 3–10), negative hsTrop < 15 ng/L: 7 days (IQR 2–9), and hsTrop > 15 ng/L: 4 days (IQR 1–7) Treatment before target condition: non‐invasive ventilation, mechanical ventilation, ECMO and renal replacement therapy Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: Twice RT‐PCR testing |
||
| Index tests | Troponin was measured with high‐sensitivity assay on and during admission as per Trusts’ protocols (Figure 5). Laboratory results were extracted from the electronic records and paper notes. Troponin measurements were performed based on clinical indication or based on local protocols for risk stratification of COVID‐19 participants. The term “myocardial injury” was used for participants with positive troponin levels (defined as ≥ 15 ng/mL – the 99th percentile in the normal population according to our lab). Peak troponin for each was measured and based on these values, three troponin levels were defined, utilising tertiles to define the cut‐offs, as: “negative” if < 15 ng/mL, “low‐positive” if levels ≥ 15 ng/mL and < 47 ng/mL and “high‐positive” level if ≥ 47 ng/mL. Unit of analysis: individual participants Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | The study assessed both mortality and deterioration. Deterioration was defined by ventilation status: non‐severe participants received non‐invasive and severe participants received mechanical ventilation or ECMO. The study included 434 cases, of which 140 participants died; 80 participants received non‐invasive ventilation, 99 received mechanical ventilation and 4 participants ECMO. These 179 participants were used for the assessment of deterioration in this study. |
||
| Flow and timing | The time horizon was not reported. Information about missing data was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | |||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Para 2022.
| Study characteristics | |||
| Patient Sampling | Consecutive patients with SARS‐CoV‐2 infection admitted to the Internal Medicine COVID‐19 Units of four Italian centres were included. | ||
| Patient characteristics and setting | Setting: Internal Medicine COVID‐19 Units
Site: four Italian centres: University Hospital Careggi, Florence, Department of Medicine and Surgery, University of Insubria, Varese, U. O.C. General Medicine, Versilia Hospital, Viareggio, Department of Internal Medicine, Hospital of Legnano, Legnano
Country: Italy Symptoms and severity: fever was present in participants with a favourable outcome: 122 (81.9%), and an adverse outcome: 41 (82%); dyspnoea in participants with a favourable outcome: 97 (64.7%), and an adverse outcome: 34 (68%); cough in participants with a favourable outcome: 88 (58.7%), and an adverse outcome: 20 (40%); tachypnoea in participants with a favourable outcome: 33 (22%), and an adverse outcome: 17 (34%); diarrhoea in participants with a favourable outcome: 21 (14%), and an adverse outcome: 2 (4%); myalgia or fatigue in participants with a favourable outcome: 7 (4.7%), and an adverse outcome: 2 (4%); ageusia or anosmia, or both, in participants with a favourable outcome: 7 (4.7%), and an adverse outcome: 3 (6%) Demographics: mean age in all participants: 68.75 years (SD 13.22), in participants with a favourable outcome: 67.85 years (SD 13.65), and in participants with an adverse outcome: 71.44 years (SD 11.54); 70 (35%) female participants, favourable outcome: 56 (37.3%), and adverse outcome: 14 (28%) Exposure history: not reported Time between symptoms onset and hospitalisation was on average 8.98 days (SD 4.90), favourable outcome: 9.50 days (SD 64.69), and adverse outcome: 6.97 days (SD 5.23) Treatment before target condition: continuous positive air pressure or non‐invasive ventilation, or both, was used in 36.5% of participants Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: PCR‐RNA detection of SARS‐CoV‐2 on nasopharyngeal swabs or bronchoalveolar lavage |
||
| Index tests | Routine laboratory tests were assessed on admission (Figure 4 and Figure 5). Case report forms were prepared by the coordinating centre (Florence) and were sent to all participating centres. Local investigators were asked to fill out the form and to send it back to the coordinating centre. All data were cross‐checked and centrally validated. Unit of analysis: individual participants. Ferritin was assessed in serum. For other laboratory tests, the used sample was not reported. | ||
| Target condition and reference standard(s) | Combined outcome of mortality and deterioration, in‐hospital mortality or the need for intensive care treatment or both were considered as adverse outcomes. Participants who did not experience the combined outcome were categorised as having a favourable outcome. The total sample size was 200 participants, 50 participants experienced adverse outcomes, and 150 favourable outcomes. |
||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | LDH was missing in more than 10% of participants. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Prasetya 2021.
| Study characteristics | |||
| Patient Sampling | The study included confirmed COVID‐19 patients who were hospitalised in three Siloam Hospitals located in Jakarta, Banten, and West Java provinces of Indonesia between 20 March 2020 to 30 October 2020. Exclusion criteria were age younger than 18 years, pregnant patients, patients with early admission (< 48 hours) to ICU, patients with missing clinical and haematology data, and without clinical outcome data. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: three Siloam Hospitals located in Jakarta, Banten, and West Java provinces
Country: Indonesia Symptoms and severity: fever was present in 222 (56.8%) participants, fatigue: 77 (19.7%) participants, cough: 233 (59.6%) participants, anorexia: 66 (16.9%) participants, anosmia: 35 (9%) participants, myalgia: 49 (12.5%) participants, dyspnoea: 139 (35.5%) participants, sore throat: 61 (15.6%) participants, diarrhoea: 44 (11.3%) participants, abdominal pain: 45 (11.5%) participants, nausea: 93 (23.8%) participants, vomiting: 24 (6.1%) participants, headache: 60 (15.3%) participants Demographics: median age was 43 years (IQR 32–54); female: 148 (38%) Exposure history: not reported Time since onset of symptoms to admission: median 7 days (IQR 5–7) Treatment before target condition: mechanical ventilation Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Routine laboratory tested on admission were assessed (Figure 4 and Figure 5). A trained team of physicians reviewed and collected the epidemiological, clinical, laboratory, and clinical outcome data from the electronic medical records. NLR was calculated by dividing the neutrophil count with lymphocyte count. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Deterioration was assessed based on worse clinical progression experienced in each case during hospitalisation. The end point for severe COVID‐19 was defined as admission to ICU. The criteria for ICU admission were: (1) respiratory rate > 30 counts per minute, (2) resting pulse oxygen saturation < 93%, (3) signs of severe respiratory distress, and (4) respiratory failure or signs of shock or both. The total sample size was 391 participants, 54 participants were admitted to ICU, and 337 were non‐ICU participants. |
||
| Flow and timing | The time horizon was not reported, however, participants who were admitted to ICU within 48 hours after hospital admission were excluded. Patients with missing haematology data were excluded. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | All enroled participants were included in the analysis. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Yes | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Qu 2021.
| Study characteristics | |||
| Patient Sampling | The study included patients who were admitted to the Changsha Public Health Treatment Center and tested positive for novel coronavirus nucleic acid in two respiratory specimens, according to the diagnosis and treatment guidelines of COVID‐19 in China. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: Changsha Public Health Treatment Center
Country: China Symptoms and severity: fever in mild group: 108 (46.35%), severe group: 28 (90.32%); cough in mild group: 166 (70.64%), severe group: 12 (38.71%); dyspnoea in mild group: 44 (18.72%), severe group: 15 (48.39%); diarrhoea in mild group: 35 (14.89%), severe group: 2 (6.46%); anorexia in mild group: 17 (7.23%), severe group: 6 (19.35%); headache in mild group: 23 (9.79%), severe group: 4 (12.90%) Demographics: the mean age in the mild group was 43.86 years (SD 15.71), and 57.42 years (SD 14.28) in the severe group; females in the mild group: 121 (51.49%) participants, and in the severe group: 13 (41.9%) participants Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: nucleic acid in two respiratory specimens, according to the diagnosis and treatment guidelines of COVID‐19 in China |
||
| Index tests | Routine laboratory tests were tested for the first time after admission (Figure 4 and Figure 5). Baseline clinical data were collected through the electronic medical record system. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Deterioration was assessed. Mild participants were defined as follows: mild clinical symptoms and no pneumonia manifestations on imaging, or participants presenting with symptoms, such as fever and respiratory tract symptoms, and pneumonia manifestations were seen on imaging.
Severe participants were defined as those who met any of the following criteria: respiratory rate ≥ 30 breaths/min; oxygen saturation ≤ 93% at a rest state; arterial partial pressure of oxygen (PaO2)/oxygen concentration (FiO2) ≤ 300 mm Hg; occurrence of respiratory failure requiring mechanical ventilation; the presence of shock; other organ failures that require monitoring and treatment in the Intensive Care Unit. The study included 266 participants, divided into 235 mild participants and 31 severe participants. |
||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | This study was supported by the grants of the novel coronavirus pneumonia major project of Hunan (2020SK3014), novel coronavirus pneumonia major project of Changsha Science and technology project (kq2001017) and the Planned Science and Technology Innovation Project of Hunan Province, China (2018SK50503). | ||
| Analysis | Cases without complete clinical data were excluded from this study. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Rasyid 2020.
| Study characteristics | |||
| Patient Sampling | Confirmed COVID‐19 patients between April and August 2020, and patients with confirmed diagnosis by nasopharyngeal and oropharyngeal PCR swabs and complete medical records were included. Patients with a history of cancer, kidney failure, hypertension, and diabetes mellitus were excluded. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: Siloam Hospitals Makassar, South Sulawesi
Country: Indonesia Symptoms and severity: not reported Demographics: in all participants, mean age was 47.4 years (SD 15.3, range, 20–90); in participants admitted to the ward 45.28 years (SD 15.09), admitted to ICU 56.78 years (SD 12.21), in survivors: 46.24 years (SD 15.11), and non‐survivors: 56.58 years (SD 13.83); female participants: 29.7% Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Routine laboratory tests were assessed on admission (Figure 4 and Figure 5). Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | In‐hospital mortality and deterioration were assessed. Severe participants were defined as participants who were admitted to the ICU. Participants were treated in intensive care if any of the following was present: respiratory distress with respiratory rates ≥ 30 times per min, mean oxygen saturation room air ≤ 93%, indication for mechanical ventilation, shock, and multiple organ failure. The total sample size was 295 participants; 264 participants survived, and 31 deceased; 250 participants were admitted to the COVID−19 isolation ward and 45 to the ICU. |
||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Sepulchre 2022.
| Study characteristics | |||
| Patient Sampling | The study included patients hospitalised for COVID‐19 from 1 March 2020 to 16 April 2020 in the Regional Hospital Center (RHC) Citadelle of Liège in Belgium. Ambulatory patients, patients under 15 years old, asymptomatic patients, and patients under anticoagulant therapy prior to hospitalisation were excluded. Outpatients, caregivers, screening and subcontracting analyses were not considered. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: Regional Hospital Center Citadelle of Liège
Country: Belgium Symptoms and severity: fever in all participants: 72.24%, favourable evolution: 75.36 %, and unfavourable evolution: 71.67%; myalgia: in all participants 14.14%, favourable evolution: 16.67%, and unfavourable evolution: 8.33%; cough: all participants: 62.62%, favourable evolution: 63.77%, and unfavourable evolution: 60%; dyspnoea: all participants: 75.75%, favourable evolution: 71.01%, and unfavourable evolution: 85%; intestinal disorders: all participants: 26.6%, favourable evolution: 31.16, and unfavourable evolution: 15% Demographics: median age in all participants was 62 years (IQR 50‐74); in participants with a favourable evolution: 60 years (IQR 47–74), and 66.5 years (IQR 57–76) in participants with an unfavourable evolution; female: all participants: 41.9%, participants with a favourable evolution: 47.8%, and participants with an unfavourable evolution: 28.3% Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: all participants were treated according to the national recommendations. Amongst ICU participants, a total of 28 participants received invasive ventilation (76%) and two of them were treated with ECMO. Participants receiving hydroxychloroquine therapy: all participants: 78.79%; participants with a favourable evolution: 80.44%, and participants with an unfavourable evolution: 75% Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Routine laboratory tests were assessed on admission (Figure 4 and Figure 5). Demographic and baseline characteristics were extracted from the participant’s medical record. All data were anonymised in order to protect the participant’s privacy and confidentiality. All blood samples were processed within two hours of collection. Other biochemical parameters i.e. sodium, potassium, total protein, glycaemia, C‐reactive protein, creatinine, alanine aminotransferase, aspartate aminotransferase, total bilirubin, lactate dehydrogenase, ferritin and high sensitive‐troponin T were obtained through COBAS® 8000 Modular analyser (Roche Diagnostics, Belgium) from serum or heparin plasma. The unit of analysis was individual participants. | ||
| Target condition and reference standard(s) | A combined outcome of mortality and deterioration was assessed in this study. An unfavourable evolution was defined as admission to ICU or death during hospitalisation, or both, and a favourable evolution as non‐ICU surviving participants. Intensive care unit admission was decided on the basis of Belgian Society of Intensive Care Medicine recommendations. The total sample size was 198 participants, 138 participants experienced a favourable evolution, and 60 an unfavourable evolution. | ||
| Flow and timing | The follow‐up took place until discharge or death. The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | All enroled participants were included in the analysis. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Yes | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Simon 2022.
| Study characteristics | |||
| Patient Sampling | All adult patients who were hospitalised for COVID‐19 after presenting to the ED in the first wave of the epidemic in France (between 1 March and 30 April 2020) were included. The inclusion criteria were patients with a laboratory‐confirmed diagnosis of COVID‐19 by RT‐PCR on a nasopharyngeal swab and hospitalised after admission to the ED in the participating centres. The exclusion criteria were patients who received palliative therapy or limitation of therapeutic effort upon admission to the ED, patients with a medical history or treatment that altered their blood cell counts (e.g. chemotherapy, immunosuppressive therapy, oral or inhaled long‐ and short‐term corticosteroid therapy, pre‐admission antibiotic therapy, active cancer, or haematological malignancies). | ||
| Patient characteristics and setting | Setting: ED
Site: Regional University Hospital of Strasbourg, Regional University Hospital of Reims, Colmar Hospital, Nord Franche‐Comteé Hospital, Metz‐Thionville Regional Hospital, and Haguenau Hospital
Country: France Symptoms and severity: not reported Demographics: the median age in all participants was 69 years (IQR 58–79), in moderate participants it was 70 years (IQR 58–81), in severe 66 years (IQR 57–72); participants aged above 75 years: all participants: 328 (32.1%), survivors: 247 (27.9%), and non‐survivors: 81 (58.3%) participants; female participants: all: 426 (41.2%), moderate participants: 356 (45.1%), and severe participants: 67 (27.2%) Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: the initial management guidelines included broad indications for mechanical ventilation. This impacted the final cohort as it included many intubated and deceased participants; Vaccine status: not reported Variant of concern: study was conducted in the first wave, when the alpha variant was in the majority Definition SARS‐CoV‐2 cases: RT‐PCR on a nasopharyngeal swab |
||
| Index tests | Routine laboratory tests were assessed on admission (Figure 4 and Figure 5). The participants’ electronic medical records were studied retrospectively for epidemiological, clinical, and biochemical data and then standardised in a report file. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Both in‐hospital mortality and deterioration were assessed in this study. Deterioration to severe disease was defined by admission to the ICU (participants requiring invasive mechanical ventilation), and moderate disease or not deteriorating disease was defined by admission to conventional hospitalisation units (most participants with oxygen therapy). The total sample size was 1035 participants; 139 died, and 896 participants survived, 246 participants experienced severe disease, and 789 moderate disease. | ||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | It was unclear if all enroled participants were included in the analysis. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Sisó‐Almirall 2020.
| Study characteristics | |||
| Patient Sampling | All consecutive adult (15 years and older) patients from 29 February to 4 April 2020 with COVID‐19 confirmed by polymerase chain reaction (PCR) from nasal and pharyngeal samples in 3 urban primary healthcare centres were included. Patients from nursing homes were excluded. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: three urban primary healthcare centres in Barcelona with one reference hospital
Country: Spain Symptoms and severity in participants with and without the composite endpoint (death or ICU admission): fever (> 37.5°C) in patients without the endpoint: 153 (61.0%), and participants with: 41 (77.4%); cough: without: 198 (74.4%), and with: 40 (71.4%); general malaise: without: 114 (42.9%), and with: 26 (46.4%); fatigue: without: 82 (30.8%), and with: 17 (30.4%); myalgia or arthralgia: without: 90 (33.8%), and with: 7 (12.5%); dyspnoea: without: 60 (22.6%), and with: 22 (39.3%); diarrhoea: without: 64 (24.1%), and with: 10 (17.9%); headache: without: 62 (23.3%), and with: 5 (8.9%); anosmia: without: 53 (19.9%), and with: 3 (5.4%); dyseusia: without: 45 (16.9%), and with: 3 (5.4%); sore throat: without: 33 (12.4%), and with: 5 (8.9%); blocked nose: without: 34 (12.8%), and with: 4(7.1%); nausea or vomiting: without: 30 (11.3%), and with: 8 (14.3%); sputum production: without: 21 (7.9%), and with: 8 (14.3%); chills: without: 17 (6.4%), and with: 4 (7.1%); asthenia: without: 10 (3.8%) and with: 2 (3.6%); chest pain: without: 7 2.6%), and with: 2 (3.6%); auscultatory alterations: without: 112 (65.5%), and with: 45 (80.8%); tachypnoea: without: 43(25.1%), and with: 21(40.4%); tachycardia: without: 23 (13.5%), and with: 6 (11.5%); pharyngitis: without: 22 (12.9%); oxygen saturation > 92%: without: 24 (15.6%), and with: 18 (35.3%) Symptoms and severity in participants without and with hospitalisation: fever (> 37.5°C): no hospitalisation: 73 (48.0%), hospitalisation: 121 (79.6%); cough: no hospitalisation: 114 (69.5%), hospitalisation: 124 (78.5%); general malaise: no hospitalisation: 65 (39.6%), hospitalisation: 75 (47.5%); fatigue: no hospitalisation: 41 (25.0%), hospitalisation: 58 (36.7%); myalgia or arthralgia: no hospitalisation: 53 (32.3%), hospitalisation 44 (27.8%); dyspnoea: no hospitalisation: 24 (14.6%), hospitalisation: 58 (36.7%); diarrhoea: no hospitalisation: 29 (17.7%), hospitalisation: 45 (28.5%); headache: no hospitalisation: 37 (22.6%), hospitalisation: 30 (19.0%); anosmia: no hospitalisation: 45 (27.4%), hospitalisation: 11 (7.0%); dysgeusia: no hospitalisation: 35 (21. %3), hospitalisation: 13 (8.2%); sore throat: : no hospitalisation: 25 (15.2%), hospitalisation: 13 (8.2%); blocked nose: no hospitalisation: 24 (14.6%), hospitalisation: 14 (8.9%); nausea or vomiting: no hospitalisation: 15 (9.1%), hospitalisation: 23 (14.6%); sputum production: no hospitalisation: 13 (7.9%), hospitalisation: 16 (10.1%); chills: no hospitalisation: 4 (2.4%), hospitalisation: 17 (10.8%); asthenia: no hospitalisation: 3 (1.8%), hospitalisation: 9 (5.7%); chest pain: no hospitalisation: 7 (4.3%), hospitalisation: 2 (1.3%); auscultatory alterations: no hospitalisation: 42 (56.0%), hospitalisation: 112 (75.7%); tachypnoea: no hospitalisation: 15 (20.0%), hospitalisation: 49 (33.1%); tachycardia: no hospitalisation: 7 (9.3%), hospitalisation: 22 (14.9%); pharyngitis: no hospitalisation: 9 (12.0%), hospitalisation: 13 (8.8%); oxygen saturation ≤ 92%: no hospitalisation: 8(12.1), hospitalisation: 34 (24.5%) Demographics: the mean age in all participants was 56.7 years (SD 17.8), in participants without the composite endpoint: 54.3 years (SD 17.4), participants with the composite endpoint: 68.2 years (SD 14.9), participants that were not hospitalised: 48.6 years (SD 16.0), and participants that were hospitalised: 65.1 years (SD 15.6); female sex: all participants: 161 (50%), without the composite endpoint: 145 (54.5%), with the composite endpoint: 16 (28.6%), not hospitalised: 97 (59.1%), and hospitalised: 64 (40.5%) Exposure history: Health professional: without the composite endpoint: 123 (46.2%), with the composite endpoint: 0, not hospitalised: 106 (64.6%), and hospitalised: 17 (10.8%) Other health workers: without the composite endpoint: 10 (3.8%), with the composite endpoint: 0, not hospitalised:10 (6.1%, and hospitalised: 0 Another type of exposure: without the composite endpoint: 133 (50.0%), with the composite endpoint: 56 (100%), not hospitalised: 48 (29.3%), and hospitalised: 141 (89.2%) Occupational contact with persons with confirmed or suspected COVID‐19 infection was reported by 71 (22.0%) participants, while 51 (15.8%) reported that contact occurred in the family setting. Any cohabitant: without the composite endpoint: 39 (14.7%), with the composite endpoint: 12 (21.4%), not hospitalised: 25 (15.2%); hospitalised: 26 (16.5%) Any work colleague: without the composite endpoint: 70 (26.3%), with the composite endpoint: 1 (1.8%), not hospitalised: 57 (34.8), and hospitalised: 14 (8.9%) Any contact person in other settings: without the composite endpoint: 6 (2.3%), with the composite endpoint: 5 (8.9%), not hospitalised: 2 (1.2%), and hospitalised: 9 (5.7%) Time since onset of symptoms to medical visit: all participants: 3.9 days (SD 4.6), without the composite endpoint: mean 3.9 days (SD 4.8), with the composite endpoint: mean 3.9 days (SD 3.7), not hospitalised: 3.5 days (SD 4.9), and hospitalised: 4.3 days (SD 4.3); Treatment before target condition: Hydroxychloroquine: without the composite endpoint: 125 (47.0%), with the composite endpoint: 37 (66.1%), not hospitalised: 45 (27.4%), and hospitalised: 117 (74.1%) Azithromycin: without the composite endpoint: 120 (45.1%), with the composite endpoint: 29 (51.8%), not hospitalised: 42 (25.6%), hospitalised: 107 (67.7%) Lopinavir/ritonavir: without the composite endpoint: 100 (37.6%), with the composite endpoint: 31 (55.4%), not hospitalised: 30 (18.3%), and hospitalised: 101 (63.9%) Oxygen therapy: without the composite endpoint: 48 (18.0%), with the composite endpoint:U 38 (67.9%), not hospitalised: 18 (11.0%), and hospitalised: 68 (43.0%) Intravenous antibiotics: without the composite endpoint: 48 (18.0%), with the composite endpoint: 29 (51.8%), not hospitalised: 18 (11.0%), and hospitalised: 59 (37.3%) Glucocorticoids: without the composite endpoint: 16 (6.0%), with the composite endpoint: 18 (32.1%), not hospitalised: 5 (3.0%), and hospitalised: 29 (18.4%) Tocilizumab: without the composite endpoint: 17 (6.4%), with the composite endpoint: 10 (17.9%), not hospitalised: 7 (4.3%), and hospitalised: 20 (12.7%) Cephalosporins: without the composite endpoint: 18 (6.8%), with the composite endpoint: 4 (7.1%), not hospitalised: 1 (0.6%), and hospitalised: 21 (13.3%) Low molecular weight heparin: without the composite endpoint: 12 (4.5%), with the composite endpoint: 7 (12.5%), not hospitalised: 1 (0.6%), and hospitalised: 18 (11.4%) Remdesivir: without the composite endpoint: 4 (1.5%), with the composite endpoint: 2 (3.6%), not hospitalised: 2 (1.2%), and hospitalised: 4 (2.5%) Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Routine laboratory tests were assessed (Figure 4 and Figure 5). The data were obtained from the electronic medical records. Missing data were collected by telephone interviews with participants when possible. Unit of analysis: individual participants Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | A composite endpoint with a participant that died or admitted to ICU was assessed. Furthermore, participants were divided into participants who were hospitalised and participants who were not hospitalised. A total of 322 were included, 266 without and 56 participants with the composite endpoint; 158 participants were hospitalised, 164 were not. |
||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Unclear | ||
| Was the method for measuring the outcome the same for all participants? | Unclear | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Unclear | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Soh 2020.
| Study characteristics | |||
| Patient Sampling | All laboratory‐confirmed SARS‐CoV‐2 infected patients admitted to the Hospital Tengku Ampuan Afzan from March 9 to April 15, 2020 were included. The list of the subjects was obtained from the Hospital Tengku Ampuan Afzan registry and extracted from the medical records. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: HTAA, Hospital Tengku Ampuan Afzan, Ministry of Health Malaysia, HTAA is a public hospital with budget constraints that limit the choices of laboratory and imaging testing.
Country: Malaysia Symptoms and severity: fever ≥ 37.5°C was present in: all participants: 14 (5.7%), stage 1: 0, stage 2: 6 (6.2%), stage 3: 3 (11.1%), stage 4: 2 (28.6%), stage 5: 3 (37.5%), myalgia: all participants: 14 (5.7%), stage 1: 0, stage 2: 9 (9.3%), stage 3: 4 (14.8%), stage 4: 0, stage 5: 1 (12.5%), headache: all participants: 7 (2.8%), stage 1: 0, stage 2: 4 (4.1%), stage 3: 1 (3.7%), stage 4: 1 (14.3%), stage 5: 1 (12.5%), rhinorrhoea: all participants: 34 (13.8%), stage 1: 0, stage 2: 25 (25.8%), stage 3: 7 (25.9%), stage 4: 2 (28.6%), stage 5: 0, sore throat: all participants: 33 (13.4%), stage 1: 0, stage 2: 27 (27.8%), stage 3: 4 (14.8%), stage 4: 1 (14.3%), stage 5: 1 (12.5%), dyspnoea: all participants: 14 (5.7%), stage 1: 0, stage 2: 4 (4.1%), stage 3: 0, stage 4: 4 (57.1%), stage 5: 6 (75.0%), cough: all participants: 80 (32.4%), stage 1: 0 (0%), stage 2: 55 (56.7%), stage 3: 14 (51.9%), stage 4: 5 (71.4%), stage 5: 6 (75.0%), sputum: all participants: 34 (13.8%), stage 1: 0, stage 2: 23 (23.7%), stage 3: 6 (22.2%), stage 4: 1 (14.3%), stage 5: 4 (50.0%), nausea and vomiting: all participants: 7 (2.8%), stage 1: 0, stage 2: 2 (2.1%), stage 3: 2 (7.4%), stage 4: 1 (14.3%), stage 5: 2 (25.0%), diarrhoea: all participants: 24 (9.7%), stage 1: 0, stage 2: 12 (12.4%), stage 3: 6 (22.2%), stage 4: 3 (42.9%), stage 5: 3 (37.5%), anosmia: all participants: 34/193 (17.6%), stage 1: 0, stage 2: 32/74 (43.2%), stage 3: 2/11 (18.2%), stage 4: 0, stage 5: 0 Clinically diagnosed pneumonia: all participants: 11 (4.5%), stage 1: 0, stage 2: 0, stage 3: 1 (3.7%), stage 4: 4 (57.1%), stage 5: 6 (75.0%); radiologically diagnosed pneumonia: all participants: 41 (16.6%), stage 1: 0, stage 2: 0, stage 3: 26 (96.3%), stage 4: 7 (100%), stage 5: 8 (100%); oxygen therapy: all participants: 14 (5.7%), stage 1: 0, stage 2: 0, stage 3: 0, stage 4: 7 (100%), stage 5: 7 (87.5%); ICU admission: all participants: 6 (2.4%), stage 1: 0, stage 2: 0, stage 3: 0, stage 4: 0, stage 5: 6 (75.0%); mechanical ventilation: all participants: 6 (2.4%), stage 1: 0, stage 2: 0, stage 3: 0, stage 4: 0, stage 5: 6 (75.0%) Demographics: median age in: all participants: 28 years (IQR 20‐45), stage 1: 24 years (IQR 18‐35), stage 2: 27 years (IQR 20‐35), stage 3: 41 years (IQR 31‐55), stage 4: 59 years (IQR 49‐60), stage 5: 58 years (IQR 51‐66); female sex: all participants: 75 (30.4%), stage 1: 31 (28.7%), stage 2: 37 (38.1%), stage 3: 7 (25.9%), stage 4: 0, stage 5: 0 Exposure history: last contact to illness onset (last contact was defined as the last day of a positive contact according to the history of the participants): all participants: median 3 days (IQR ‐1‐7), stage 1: /, stage 2: 3 days (IQR ‐2‐7), stage 3: 2 days (IQR 0‐5), stage 4: 3 days (IQR 3‐5), stage 5: 4 days (IQR 0‐14) Time since onset of symptoms: not reported Treatment before target condition: Osetamivir: all participants: 11 (4.5%), stage 1: 1 (0.9%), stage 2: 3 (3.1%), stage 3: 3 (11.1%), stage 4: 3 (42.9%), stage 5: 1 (12.5%) Antibiotics: all participants: 26 (10.5%), stage 1: 2 (1.9%), stage 2: 8 (8.2%), stage 3:2 (7.4%), stage 4: 6 (85.7%), stage 5: 8 (100%) Lopinavir/ritonavir: all participants: 37 (15.0%), stage 1: 0, stage 2: 7 (7.2%), stage 3: 17 (63.0%), stage 4: 6 (85.7%), stage 5: 7 (87.5%) Chloroquine or hydroxychloroquine: all participants: 68 (27.5%), stage 1: 2 (1.9%), stage 2: 34 (35.1%), stage 3: 23 (85.2%), stage 4: 4 (57.1%), stage 5: 5 (62.5%) Interferon beta 1a: all participants: 11 (4.5%), stage1: 0, stage 2: 0, stage 3: 0, stage 4: 4 (57.1%), stage 5: 7 (87.5%) Ribavirin: all participants: 4 (1.6%), stage 1: 0, stage 2: 0, stage 3: 1 (3.7%), stage 4: 1 (14.3%), stage 5: 2 (25.0%) Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Routine laboratory tests were assessed on presentation (Figure 4 and Figure 5). Data were obtained from the medical report including laboratory findings. Unit of analysis: individual participants Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | The study assessed deterioration. Clinical staging was defined as the worst clinical stage according to the fifth edition of Malaysia COVID‐19 management guidelines. Stage 1: was asymptomatic, stage 2: was symptomatic without pneumonia, stage 3: was pneumonia without hypoxia, stage 4: was pneumonia requiring oxygen supplement, and stage 5: was critically ill with multiorgan involvement. Stage 1, 2 and 3 were grouped together for non‐severe disease, and stage 4 and 5 were grouped together for severe disease. The total sample size was 247 participants, including stage 1 (n = 108), stage 2 (n = 97), stage 3 (n = 27), stage 4 (n = 7), and stage 5 (n = 8); 15 participants were categorised as severe and 232 as non‐severe. |
||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | No | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Statsenko 2021.
| Study characteristics | |||
| Patient Sampling | The study included all consecutive patients, 18 years or older, admitted to Dubai Mediclinic Parkview Hospital from 24 February to 1 July 2020 with COVID‐19 confirmed by the PCR. All patients with COVID‐19 verified by PCR were hospitalised in the Mediclinic even if they did not present any symptoms. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: Dubai Mediclinic, primary and secondary care
Country: United Arab Emirates Symptoms and severity: clinical severity was: asymptomatic or mild in all participants: 431 (76.96%), participants not admitted to ICU: 431 (88.32%), and participants admitted to ICU: 0; severe in all participants: 83 (14.82%), participants not admitted to ICU: 54 (11.07%), and participants admitted to ICU: 29 (40.28%); critical in all participants: 46 (8.21%), participants not admitted to ICU: 3 (0.61%), and participants admitted to ICU: 43 (59.72%) Demographics: median age in all participants 39.0 years (IQR 33.0–49.0), mean age in participants not admitted to ICU: 38.0 years (SD 11.97), participants admitted to ICU 51.0 years (SD 13.08); female participants: all participants: 189 (33.75%), participants not admitted to ICU: 175 (35.86%), participants admitted to ICU: 14 (19.44%) Exposure history: not reported Onset to hospitalisation (days): all: median 14.0 days (IQR 8.0–19.0), participants not admitted to ICU: mean 12.0 days (SD 7.07), participants admitted to ICU: mean 22.0 days (SD 16.5) Treatment before target condition: the treatment was administered in full accordance with ‘National Guidelines for Clinical Management and Treatment of COVID‐19’. The indications for supportive oxygen therapy were (a) the oxygen saturation level below 94%, (b) the respiratory rate (RR) above 30 breaths per minute, (c) both of them. In case of suspicion of super‐imposed bacterial pneumonia, physicians ordered empirical broad‐spectrum antibiotics. The administration of antiviral and antimalarial drugs followed the national guidelines. Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: positive SARS‐CoV‐2 RT‐PCR from nasopharyngeal swabs |
||
| Index tests | Routine laboratory tests were assessed on admission (Figure 4 and Figure 5). Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Deterioration was assessed in this study by the risk of deterioration that would require transferring to the ICU. Therefore, 72 ICU and 488 non‐ICU participants were compared in 560 cases. | ||
| Flow and timing | All the participants were discharged at the time of writing the paper. The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Missing values were reported for ALT: 10, AST: 10, D‐dimer: 86, aPTT: 73, creatinine: 6, CK: 126, CRP: 5, LDH: 95, troponin: 135, ferritin: 53, and fibrinogen: 153 participants. The missing data for the comparative analysis were treated with the complete‐case analysis method. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Suardi 2020.
| Study characteristics | |||
| Patient Sampling | All consecutive patients presenting to the ED admitted to the Infectious Diseases Unit (IDU) and Intensive Care Unit (ICU) of Santa Maria Annunziata Hospital, Florence, Italy, with a confirmed COVID‐19 diagnosis from February 25 to April 25, 2020 were enroled in this retrospective cohort study. | ||
| Patient characteristics and setting | Setting: ED, Infectious Diseases Unit and Intensive Care Unit (ICU)
Site: Santa Maria Annunziata Hospital, a medium‐size hospital with 300 beds in the southern area of Florence
Country: Italy Symptoms and severity: fever (> 37.5 C): all participants: 85 (87.6%), low‐flow oxygen support group: 51 (91.1%), non‐invasive/invasive ventilatory support group: 34 (82.9%); cough: all participants: 68 (70.1%), low‐flow oxygen support group: 40 (71.4%), non‐invasive/invasive ventilatory support group: 28 (68.3%); pharyngodynia: all participants: 8 (8.2%), low‐flow oxygen support group: 6 (10.7%), non‐invasive/invasive ventilatory support group: 2 (4.9%); dyspnoea: all participants: 43 (44.3%), low‐flow oxygen support group: 20 (35.7%), non‐invasive/invasive ventilatory support group: 23 (56.1%); haemoptysis: all participants: 1 (1%), low‐flow oxygen support group: 0, non‐invasive/invasive ventilatory support group: 1 (1.8%); headache: all participants: 2 (2.1%), low‐flow oxygen support group: 1 (1.8%), non‐invasive/invasive ventilatory support group: 1 (1.8%); coryza: all participants: 3 (3.1%), low‐flow oxygen support group: 1 (1.8%), non‐invasive/invasive ventilatory support group: 2 (4.9%); diarrhoea: all participants: 4 (4.1%), low‐flow oxygen support group: 1 (1.8%), non‐invasive/invasive ventilatory support group: 3 (5.4%); myalgia: all participants: 8 (8.2%), low‐flow oxygen support group: 4 (7.1%), non‐invasive/invasive ventilatory support group: 4 (7.1%); asthenia: all participants: 28 (28.9%), low‐flow oxygen support group: 7 (12.5%), non‐invasive/invasive ventilatory support group: 21 (51.2%); vomiting: all participants: 4 (4.1%), low‐flow oxygen support group: 2 (3.6%), non‐invasive/invasive ventilatory support group: 2 (4.9%); rash: participants: 1 (1%), low‐flow oxygen support group: 1 (1.8%), non‐invasive/invasive ventilatory support group: 0 Clinical course (on May 14, 2020): Cured and discharged: all: 86 (88.7%); low‐flow oxygen support group: 53 (94.6%); non‐invasive/invasive ventilatory support group: 33 (80.5%) Still admitted: all: 2 (2.1%); low‐flow oxygen support group: 1 (1.8%); non‐invasive/invasive ventilatory support group: 1 (1.8%) Died: all: 9 (9.3%); low‐flow oxygen support group: 2 (3.6%); non‐invasive/invasive ventilatory support group: 7 (17.1%) Demographics: the median age in all participants was 64 years (IQR 54.5–75), median age in low‐flow oxygen support group: 64.5 years (IQR 53–78), and median age in non‐invasive/invasive ventilatory support group: 63 years (IQR 57.5–72.5); female participants: all: 37 (38.1%), low‐flow oxygen support group: 25 (44.6%), and non‐invasive/invasive ventilatory support group: 12 (29.3%) Exposure history: an exposure history was reported for 11 (16.5%) participants from long‐term care facilities; three (3.1%) participants had nosocomial transmission, and three (3.1%) were healthcare workers Time since onset of symptoms: median in all participants: 6 days (IQR 3–9), in low‐flow oxygen support group: 6 days (IQR 2–8), and in non‐invasive/invasive ventilatory support group: 6 days (IQR 4–9.5) Treatment before target condition: Antiviral: all: 89 (91.8%), low‐flow oxygen support group: 49 (87.5%), non‐invasive/invasive ventilatory support group: 40 (97.6%) Hydroxychloroquine: all: 88 (90.7%), low‐flow oxygen support group: 48 (85.7%), non‐invasive/invasive ventilatory support group: 40 (97.6%) Corticosteroids: all: 44 (45.4%), low‐flow oxygen support group: 14 (25%), non‐invasive/invasive ventilatory support group: 30 (73.2%) Tocilizumab: all: 24 (24.7%), low‐flow oxygen support group: 3 (5.4%), non‐invasive/invasive ventilatory support group: 21 (51.2%) Antibiotics: all: 91 (93.8%), low‐flow oxygen support group: 50 (89.3%), non‐invasive/invasive ventilatory support group: 41 (100%) Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Routine laboratory tests were assessed upon arrival in the ED (Figure 4; Figure 5). Data were obtained from the participant's electronic medical records and were reviewed by a trained team of five physicians (three IDU and two ICU specialists) and one IDU nurse. Unit of analysis: individual participants Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Deterioration was assessed. Participants in the low‐flow oxygen support group were compared to participants in the non‐invasive/invasive ventilatory support group. Non‐invasive/invasive ventilatory support was defined as the need for CPAP or BPAP (non‐invasive ventilation) or mechanical ventilation, excluding low‐flow oxygen support, such as Venturi mask or nasal cannula use. The total sample size was 97 participants, with 56 participants in the low‐flow oxygen support group, and 41 participants in the non‐invasive/invasive ventilatory support group. |
||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | CP has received funds for speaking at symposia on behalf of Zambon, Angelini, and MSD. PB has participated on the board of and has received funds for speaking at symposia on behalf of Abbvie, MSD, Gilead, and ViiV. The authors declared no competing interests. | ||
| Analysis | Information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Suzuki 2022.
| Study characteristics | |||
| Patient Sampling | Outpatients newly diagnosed with COVID‐19 who visited Asahikawa City Hospital as new patients were enroled in this study between April 2021 and September 2021. Five patients who were admitted to other hospitals after the initial visit were excluded because the clinical course data were missing. | ||
| Patient characteristics and setting | Setting: hospital, all blood samples were collected at the outpatient setting
Site: Asahikawa City Hospital
Country: Japan Symptoms and severity: not reported Demographics: median age in participants without oxygen therapy was 44.5 years (IQR 34.3‐52.8), and in participants with oxygen therapy: 55 years (IQR 45.5‐72.25); female participants: no oxygen therapy: 43 (42.2%) participants, and oxygen therapy: 17 (42.5%) Exposure history: not reported Time since onset of symptoms to blood sampling: no oxygen therapy: median 5 days (IQR 3–7), and oxygen therapy: median 6 days (IQR 3–7.5) Treatment before target condition: not reported Vaccine status: no oxygen: 4 (3.9%) participants, and oxygen therapy: 3 (7.5%) participants; the type of vaccine was not specified Variant of concern: not reported Definition SARS‐CoV‐2 cases: all participants were diagnosed by polymerase chain reaction for severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) genes or antigen quantitation testing or both. |
||
| Index tests | Routine laboratory tests were assessed (Figure 4; Figure 5), 138 participant samples were collected at the first visit to the hospital’s outpatient unit, and four participant samples were collected at the second or third visit. All demographic, clinical, and outcome data were retrospectively extracted from the Asahikawa City Hospital record files. All blood samples were taken before participants received any treatment for COVID‐19. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Deterioration was assessed in participants with (= severe) and without (non‐severe) oxygen therapy. Participants with percutaneous oxygen saturation levels of ≤ 92% received oxygen therapy. A total of 142 participants were included, 40 received oxygen therapy, and 102 did not. |
||
| Flow and timing | Participants who required oxygen therapy started this treatment within 7 days after blood sampling. A total of 287 new outpatients were eligible for the study; five were excluded because they were admitted to other hospitals after they visited the hospital and the clinical course data were missing. Only 142 participants underwent blood testing and were included in the study. |
||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Ferritin and D‐dimer were missing for 14.7% of participants. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Yes | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Ullah 2020.
| Study characteristics | |||
| Patient Sampling | Consecutive adult inpatients (≥ 18 years old) with a confirmed diagnosis of COVID‐19 between March 1, 2020 and May 10, 2020 were included in this study at Abington Hospital‐Jefferson Health, PA, USA. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: Abington Hospital‐Jefferson Health, PA
Country: USA Symptoms and severity: not reported Demographics: the mean age in participants with neutrophil‐lymphocyte ratio (NLR) < 10 was 63.6 years, and 61.6 years in participants with NLR > 11; female participants in NLR < 10: 75 (88.20%) participants, and in NLR > 11: 10 (11.80%) participants Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: Hydroxychloroquine: NLR < 10: 114 (83.80%) participants; NLR > 11: 22 (16.20%) participants Tocilizumab: NLR < 10: 24 (82.80%) participants; NLR > 11: 5 (17.20%) participants Steroid: NLR < 10: 26 (89.70%) participants; NLR > 11: 3 (10.30%) participants Anticoagulation: NLR < 10: 26 (78.80%) participants; NLR > 11: 7 (21.20%) participants Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Neutrophil‐lymphocyte ratio (NLR) was assessed on admission (Figure 5). Clinical, demographic, laboratory, treatment, and outcome data were extracted from electronic medical records (Sunrise) using a standardised data collection form. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Mortality and deterioration were assessed in this study. Deterioration was defined as the need to upgrade care to ICU. The total sample size was 167 participants, 18 non‐survivors and 149 survivors; 53 participants were upgraded to ICU, and 114 participants who were not. | ||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Xia 2020.
| Study characteristics | |||
| Patient Sampling | The study included patients with COVID‐19 who were admitted to the respiratory ward of Wuhan Hospital of Integrative Medicine in Hubei from January 26 to February 20, 2020. Inclusion criteria were patients who met the diagnostic criteria of the "Diagnosis and Treatment Plan for Novel Coronavirus Infected Pneumonia". The exclusion criteria were as follows: including but not limited to other types of bacterial pneumonia, interstitial pneumonia, heart failure with pulmonary oedema, hypersensitivity pneumonitis, active tumours, patients taking immunosuppressive therapy, patients with negative nucleic acid test for novel coronavirus, patients without chest CT examination, patients without blood oxygen saturation monitoring, and mild and critical patients. | ||
| Patient characteristics and setting | Setting: the fourth respiratory ward and the first ward of Wuhan Hospital
Site: Wuhan Hospital of Integrated Traditional Chinese and Western Medicine, Wuhan, Hubei
Country: China Symptoms and severity: Fever: moderate cases: 26 participants, and severe cases: 25 participants; Cough: moderate cases: 22 participants, and severe cases: 21 participants; Chest tightness: moderate cases: 9 participants, and severe cases: 12 participants; Dyspnoea: moderate cases: 14 participants, and severe cases: 20 participants; Rales: moderate cases: 19 participants, and severe cases: 16 participants Demographics: age (mean +/‐ SD; range): mean age in moderate cases was: 62.25 years (SD 15.07), and 64 years (SD 14.88) in severe cases; female participants: moderate cases:17/32, and severe cases: 13/31 Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: the diagnostic criteria of the "Diagnosis and Treatment Plan for Novel Coronavirus Infected Pneumonia": the clinical manifestations are consistent with fever or respiratory symptoms, the total number of white blood cells is normal or decreased in the early stage of onset, and the lymphocyte count is decreased; chest CT shows the imaging manifestations of pneumonia; real‐time fluorescent RT‐PCR detection of respiratory or blood samples is positive for the new coronavirus nucleic acid |
||
| Index tests | Neutrophil‐lymphocyte ratio (NLR) was assessed within 24 hours of admission (Figure 5). Blood was drawn according to the standard operating procedures. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Deterioration was assessed. According to the diagnostic criteria and classification criteria of the "Pneumonia Diagnosis and Treatment Plan for Novel Coronavirus Infection" (Trial Version 5) issued by the General Office of the National Health and Health Commission, the selected participants were divided into two groups: moderate and severe cases.
Criteria for moderate cases: fever, respiratory symptoms, and manifestations of pneumonia on imaging;
Criteria for severe cases: meeting the diagnostic criteria of moderate cases and any of the following: respiratory distress, respiratory rate (RR) ≥ 30 breaths/min; resting state, refers to pulse oxygen saturation (SPO 2) ≤ 93%; arterial partial pressure of oxygen (PaO2)/inhaled oxygen concentration (FIO2) ≤ 300 mmHg. The study included 63 participants, 32 participants in the moderate group and 31 participants in the severe group. |
||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | Not reported | ||
| Analysis | Information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | Unclear | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Zafar 2021.
| Study characteristics | |||
| Patient Sampling | Patients presenting with symptoms and signs of SARS‐CoV‐2 disease, at the Conquest Hospital and Eastbourne District General Hospital in East Sussex, United Kingdom between February 10, 2020 and May 1, 2020, were assessed. This study included only those patients who had both swabs and blood lymphocyte levels tested, including those who were admitted and treated or discharged from the ED. Patients who had swabs taken but did not have blood lymphocyte levels taken were excluded. Patients who had a COVID‐19 swab test but were not admitted to the hospital were also excluded as their blood lymphocyte levels were not available. The study included swab positive and negative patients; for the review, only swab‐positive patients were included. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: Conquest Hospital and Eastbourne District General Hospital in East Sussex
Country: United Kingdom Symptoms and severity: diarrhoea in 33 (29.5%) participants Demographics: the mean age was 70.9 years (SD 17.6), in survivors it was 66.4 years (SD 18.6), and in non‐survivors 79.7 years (SD 11.1); female sex: all cases: 49 (44%), survivors: 35 (47%), non‐survivors: 14 (37%) Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: all the participants who had positive COVID‐19 swab results were managed as per NHS England guidelines. Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Absolute lymphocyte count was assessed on admission (Figure 5). All the relevant blood tests, comorbidities, hospital admissions, and other relevant information were collected from hospital records. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Mortality was assessed. The total sample size was 112 participants, 74 participants survived, and 38 participants died. | ||
| Flow and timing | Mortality during the first 30 days was assessed. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | All enroled participants were included in the analysis. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Unclear | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | |||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Yes | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Zarębska‐Michaluk 2021.
| Study characteristics | |||
| Patient Sampling | The study population consisted of patients included between 1 March and 31 December 2020 in the national database SARSTer, which is a project supported by the Polish Association of Epidemiologists and Infectiologists and covers 2784 adult individuals treated for COVID‐19 in 30 Polish centres. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: national database SARSTer, project supported by the Polish Association of Epidemiologists and Infectiologists, in 30 Polish centres
Country: Poland Symptoms and severity: disease severity at the baseline: Oxygen saturation 91–95%: participants with eGFR > 60 mL/min/1.73 m2: 596 (31.9%) participants, eGFR 30‐60 mL/min/1.73 m2: 129 (34.6%) participants, and eGFR < 30: 24 (29.3%) participants Oxygen saturation ≤ 90%: eGFR > 60 mL/min/1.73 m2: 526 (28.2%), eGFR 30‐60 mL/min/1.73 m2: 169 (45.3%); eGFR < 30 mL/min/1.73 m2: 43 (52.4%) Hospitalised, does not require oxygen supplementation and does not require medical care: eGFR > 60: 131 (7%), eGFR 30‐60: 3 (1.9%), eGFR < 30: 1 (1.2%), non‐survivors: 1 (0.4%), and survivors: 138 (6.7%) Hospitalised, requiring no oxygen supplementation, but requiring medical care: eGFR > 60: 833 (44.6%), eGFR 30‐60: 108 (29%), eGFR < 30: 21 (25.6%), non‐survivors: 36 (14.5%), and survivors: 926 (44.7%) Hospitalised, requiring normal oxygen supplementation: eGFR > 60: 835 (44.7%), eGFR 30‐60: 244 (65.4%), eGFR < 30: 54 (65.9%), non‐survivors: 174 (69.9%), and survivors: 959 (46.3%) Hospitalised, on non‐invasive ventilation with high‐flow oxygen equipment: eGFR > 60: 61 (3.3%), eGFR 30‐60: 14 (3.7%), eGFR < 30: 3 (3.7%), non‐survivors: 30 (12%), and survivors: 48 (2.3%) Hospitalised, for invasive mechanical ventilation or ECMO: eGFR > 60: 6 (0.3%), eGFR 30‐60: 0, eGFR < 30: 3 (3.7%), non‐survivors: 8 (3.2%), and survivors: 1 (0.05%) Demographics: mean age in participants with eGFR > 60: 57.1 years (SD 16.5), eGFR 30–60: 73.4 years (SD 12.5), eGFR < 30: 76.5 years (SD 12.9), in non‐survivors: 74.2 years (SD 11.9), and survivors: 58.7 years (SD 16.9); female sex: eGFR > 60: 869 (46.5%), eGFR 30–60: 177 (47.5%), eGFR < 30: 44 (53.7%), non‐survivors: 95 (38.2%), and survivors: 995 (48%) Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: Remdesivir: eGFR > 60: 454 (24.3%), eGFR 30‐60: 81 (21.7%), eGFR < 30: 5 (6.1%), non‐survivors: 61 (24.5%), and survivors: 479 (23.1%) Tocilizumab: eGFR > 60: 186 (9.9%), eGFR 30‐60: 79 (21.1%), eGFR < 30: 14 (17.1%), non‐survivors: 55 (22.1%), and survivors: 224 (10.8%) Dexamethason: eGFR > 60: 492 (26.3%), eGFR 30‐60: 137 (36.7%), eGFR < 30: 35 (42.7%), non‐survivors: 135 (54.2%), and survivors: 529 (25.5%) Convalescent plasma: eGFR > 60: 216 (11.6%), eGFR 30‐60: 44 (11.8%), eGFR < 30: 16 (19.5%), non‐survivors: 50 (20.1%), and survivors: 226 (10.9%) Low molecular weight heparin: eGFR > 60: 1306 (70%), eGFR 30‐60: 299 (80.2%), eGFR < 30: 69 (84.1%), non‐survivors: 203 (81.5%), and survivors: 1471 (71%) Antibiotics: non‐survivors: 183 (73.5%), and survivors: 1045 (50.4%) Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: all participants were diagnosed with COVID‐19 based on positive results of RT‐PCR from the nasopharyngeal swab specimen. |
||
| Index tests | Estimated glomerular filtration rate (eGFR) was assessed on admission (Figure 4). It was calculated with the MDRD Study equation and, using this measure, CKD was defined as eGFR < 60 mL/min/1.73 m2 along with a history of kidney disease from medical records. According to renal function on admission, participants were stratified into three groups: eGFR < 30 mL/min/1.73 m2, eGFR 30–60 mL/min/1.73 m2, and eGFR > 60 mL/min/1.73 m2. Patients’ data were retrieved retrospectively from hospital files and completed online. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Mortality and deterioration were assessed in this study. For mortality, the 28‐day survival was assessed. Deterioration was defined as mild and severe disease.
Mild participants = hospitalised, do not require oxygen supplementation, or hospitalised, requiring no oxygen supplementation, but requiring medical care, or hospitalised, requiring normal oxygen supplementation.
Severe patents = hospitalised, on non‐invasive ventilation with high‐flow oxygen equipment, or hospitalised, for invasive mechanical ventilation or ECMO. The total sample size was 2322 participants, 249 participants died and 2073 participants survived. The study included 2230 mild participants and 87 severe participants. |
||
| Flow and timing | Mortality was assessed after 28 days. Severity of the participants was scored at baseline and then every 7 days during the following 28 days after admission. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | This research was funded by the Medical Research Agency, grant number 2020/ABM/ COVID19/PTEILCHZ, and the Polish Association of Epidemiologists and Infectiologists. Conflicts of Interest: D.Z.‐M. reports personal fees from Gilead and Abbvie, outside the submitted work. R.F. reports grants from Abbvie, Gilead, Merck, personal fees from Gilead, Abbvie, Merck, Roche, and non‐financial support from Abbvie, Gilead, and Merck outside the submitted work. J.J. reports personal fees from Gilead, Abbvie, Bausch Health, Merck, Promed, Roche, and non‐financial support from Abbvie, Gilead, and Merck outside the submitted work. D.K. reports personal fees from Gilead and Abbvie outside the submitted work. J.K. reports personal fees from Gilead, Merck, ViiV, Janssen outside the submitted work. R.P. reports personal fees from Gilead outside the submitted work. W.M. reports grants and personal fees from Gilead, Abbvie, Abbott, Roche, Janssen outside the submitted work. P.L. reports grants and personal fees from Abbvie, Roche, UCB, Lilly, Novartis, BMS, Amgen, Janssen, Abivax, Viela‐Bio outside the submitted work. BS reports grants from Abbvie, Gilead, Janssen, personal fees from Gilead, Abbvie, Janssen outside the submitted work. MRog, B.L., MRor, A.S.‐P, A.P., A.B.‐K., B.B., M.T.‐Z., D.K., K.K., K.R., P.C., B.O.‐G., K.S. declared no competing interests. | ||
| Analysis | Amongst 2784 adult participants included in the SARSTer project, the data on kidney function were provided for 2322 individuals. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Yes | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Yes | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Zhang 2020.
| Study characteristics | |||
| Patient Sampling | Laboratory‐confirmed COVID‐19 patients admitted to The First People’s Hospital of Jiangxia District in Wuhan, China between February 1 and March 15, 2020 | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: The First People’s Hospital of Jiangxia District in Wuhan
Country: China Symptoms and severity: cough: all participants: 135 (100%), non‐ICU participants: 105 (100%), and ICU participants: 30 (100%); fever: all participants: 128 (94.8%), non‐ICU participants: 98 (93.3%), and ICU participants: 30 (100%); fatigue: all participants: 82 (60.7%), non‐ICU participants: 62 (59.0%), and ICU participants: 20 (66.7%); chest pain: all participants: 46 (34.2%), non‐ICU participants: 32 (30.4%), and ICU participants: 14 (46.7%); diarrhoea: all participants: 32 (23.7%), non‐ICU participants: 24 (22.9%), and ICU participants: 8 (26.7%); vomiting: all participants: 24 (17.8%), non‐ICU participants: 18 (17.1%), and ICU participants: 6 (20.0%); dyspnoea: all participants: 10 (7.4%), non‐ICU participants: 4 (3.8%), and ICU: 6 (20.0%) Demographics: the median age in all participants was 56 years (IQR 42‐68), in non‐ICU participants: 54 years (IQR 41‐67.5), and in ICU participants: 64 years (IQR 50‐72.3); female participants: all participants: 68 (50.4%), non‐ICU participants: 53 (50.5%), and ICU participants: 15 (50%) Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: confirmed participants were treated with antiviral drugs, including oseltamivir, arbidol, and ribavirin; respiratory support or invasive mechanical ventilation, and blood purification in case of liver/kidney injuries. They received antibiotic treatment (sulperazone, linezolid), antifungal therapy (fluconazole, caspofungin), corticosteroid therapy, respiration‐assisted ventilation, and continuous renal replacement therapy. Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: laboratory confirmed: nucleic acid tests of SARS‐CoV‐2 pathogens |
||
| Index tests | Routine laboratory tests were assessed on admission (Figure 4 and Figure 5). For all participants, blood cell analysis was detected by automated haematology analyser (SYSMEX 800i, Japan), and the biochemical indicator was analysed (Toshiba TAB2000, Japan). The biochemical markers of myocardial injury were measured (Roche Cobas 6000 Analyser, Switzerland). Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | The study assessed deterioration and mortality. Deterioration was defined by admission to ICU. Mortality was assessed in ICU participants. A total of 135 participants were included, 105 non‐ICU participants and 30 ICU participants. Of these 30 participants, 18 died and 12 survived. |
||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | Financial support from Jiangsu Province Professorship (to ZT) and Jiangsu University Jinshan Professorship (to ZT) | ||
| Analysis | Information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Yes | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Yes | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Zhou 2020.
| Study characteristics | |||
| Patient Sampling | The study included all patients, aged 18 years or older with a confirmed diagnosis of mild or moderate COVID‐19, who were admitted to Chongqing Public Health Medical Center, China, from January 24, 2020, to February 7, 2020. | ||
| Patient characteristics and setting | Setting: Public Health Medical Center
Site: Chongqing Public Health Medical Center
Country: China Symptoms and severity: symptoms at admission present in participants without progression: 90 (90.9%), and in participants with progression: 16 (94.1%) Fever: without progression: 67 (67.7%), and with progression: 11 (64.7%) Rigours: without progression: 7 (7.1%), with progression: 0 Fatigue: without progression: 28 (28.3%), and with progression: 4 (23.5%) Cough: without progression: 58 (58.6%), and with progression: 11 (64.7%) Dyspnoea: without progression: 1 (1%), and with progression: 2 (11.7%) Sputum: without progression: 36 (36.4%), and with progression: 8 (47.1%) Sore throat: without progression: 17 (17.2), and with progression: 3 (17.6) Xerostomia: without progression: 1 (1.0%), and with progression: 0 Haemoptysis: without progression: 0, and with progression: 1 (5.9%) Palpitations: without progression: 0, and with progression: 1 (5.9%) Myalgia: without progression: 9 (9.2%), and with progression: 1 (6.7%) Arthralgia: without progression: 3 (3.0%), and with progression: 1 (5.9%) Low back pain: without progression: 2 (2.0%), and with progression: 0 Abdominal pain: without progression: 3 (3.0%), and with progression: 1 (5.9%) Nausea and vomiting: without progression: 3 (3.0%), and with progression: 1 (5.9%) Diarrhoea: without progression: 9 (9.1%), and with progression: 1 (5.9%) Anorexia: without progression: 22 (22.2%), and with progression: 5 (29.4%) Headache: without progression: 12 (12.1%), and with progression: 3 (17.6%) Dizziness: without progression: 11 (11.1%), and with progression: 3 (17.6%) Hyposmia: without progression: 1 (1.0%), and with progression: 0 Moist rales: without progression: 6 (6.1%), and with progression: 3 (33.3%) Severity on admission was mild/moderate. Demographics: median age in all participants was 46 years (IQR 36–56), in participants without progression: 41 years (IQR 35–54), and in participants with progression: 59 years (IQR 50–70); female sex: all participants: 57 (49.1%); participants without progression: 48 (48.5%), and participants with progression: 9 (52.9%) Exposure history: participants with a history of stay in Wuhan: all participants: 37 (31.9%), participants without progression: 34 (34.3%), and participants with progression: 3 (17.6%) Time since onset of symptoms to hospital admission: all participants: 4 days (IQR 2–7), participants without progression: median 4 days (IQR 2–7), and participants with progression: median 5 (IQR 4–7) Treatment of COVID‐19 participants within 14 days after admission Lopinavir/tironavir: participants without progression: 74 (74.7%); participants with progression: 14 (82.4%) Ribavirin: participants without progression: 42 (42.4%); participants with progression: 4 (23.5%) Antibiotics: participants without progression: 10 (10.1%); participants with progression: 6 (35.3%) Traditional Chinese medicine: participants without progression: 26 (26.3%); participants with progression: 4 (23.5%) Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: confirmed diagnosis, not reported |
||
| Index tests | Routine laboratory tests were assessed on admission (Figure 4 and Figure 5). Laboratory test results of eligible participants were transcribed from the electronic hospital medical record system onto case record forms. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | The study assessed deterioration, defined as progression to severe COVID‐19 after admission. Participants exhibiting one or more of the following conditions were classified as having severe COVID‐19: (a) respiratory distress (≥ 30 breaths/min); (b) oxygen saturation ≤ 93% at rest; (c) arterial partial pressure of oxygen (PaO2)/fraction of inspiration O2 (FiO2) ≤ 300 mmHg (1 mmHg = 0.133 kPa); (d) respiratory failure requiring mechanical ventilation; (e) development of septic shock; and (f) critical organ failure requiring ICU care. The total sample size was 116 participants, 99 participants had no progression to severe disease, and 17 participants did progress to severe disease. |
||
| Flow and timing | Time to develop severe COVID‐19 was analysed over the duration of 14 days of hospitalisation. For some participants, the index test results for routine laboratory tests were not available. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | Chongqing Special Research Project for Novel Coronavirus Pneumonia Prevention and Control (No. cstc2020jscx‐fyzxX0005). The funding body had no role in the collection, analysis and interpretation of data, the writing of the report, and the decision to submit for publication. | ||
| Analysis | All the eligible participants were included in the analysis. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Yes | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Öktem 2020.
| Study characteristics | |||
| Patient Sampling | The study included patients with COVID‐19 findings, clinically or radiologically, and diagnosed as COVID‐19 with PCR admitted to the Kastamonu State Hospital in Turkey from March 1 to April 31, 2020. Patients with missing medical file information, active smokers, and negative PCR results were excluded from the study. | ||
| Patient characteristics and setting | Setting: policlinics and ED
Site: second‐level public hospital, Kastamonu State Hospital, Kastamonu
Country: Turkey Symptoms and severity: 21.6% (27) of participants were asymptomatic, 18.4% (23) had fever, 18.4% (23) had cough, 8.8% (11) had myalgia, 15.2% (19) had shortness of breath, 4% (5) had sore throat, 1.6% (2) had gastrointestinal complaints and 1.6% (2) had loss off smell Demographics: mean age in all cases: 50.2 years (SD 19.8), in participants that needed intensive care: 59.2 years (SD 16.9), and in participants that did not need intensive care: 49.1 years (SD 19.8); 59 (47.2%) female participants Exposure history: 54.4% of all cases had contact with a COVID‐19 diagnosed participant. Time since onset of symptoms: not reported Treatment before target condition: 14 participants needed intensive care, and 6 participants were intubated. Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: positive RT‐PCR |
||
| Index tests | Initial blood results of routine laboratory tests were assessed (Figure 4 and Figure 5). Data were obtained from participants’ files from policlinics and EDs. Lactate was analysed on venous blood gas. The unit of analysis was individual participants. | ||
| Target condition and reference standard(s) | Mortality was assessed in 125 participants, 120 participants survived, and 5 participants died. | ||
| Flow and timing | The time horizon was not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Patients with missing file data were excluded. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | |||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Şan 2021.
| Study characteristics | |||
| Patient Sampling | Adult (18 years and older) patients, who were admitted to Ankara City Hospital due to COVID‐19, were included in this study. Patients with active malignancy and pregnant women were excluded from the study. Hospitalisation, treatment, management, and discharge decisions of the patients were made according to the guidelines of the Turkish Ministry of Health. | ||
| Patient characteristics and setting | Setting: Internal Medicine and Infectious Diseases wards
Site: Ankara City Hospital, Ankara
Country: Turkey Symptoms and severity: cough was present in 396 (52.8%) participants, fever: 284 (37.9%) participants, dyspnoea: 219 (29.2%) participants, headache: 69 (9.2%) participants, nausea: 47 (6.3%) participants, myalgia: 166 (22.1%) participants, diarrhoea: 43 (5.7%) participants, back pain: 3 (0.4%) participants, anosmia: 37 (4.9%) participants, ageusia: 33 (4.4%) participants, abdominal pain: 11 (1.5%) participants, and arthralgia: 25 (3.3%) participants Demographics: median age was 49 years (IQR 28); female participants: 308 (41.1%) Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: according to the diagnosis, participants were classified into two groups. Confirmed COVID‐19: positive RT‐PCR, and probable COVID‐19: negative PCR and two or three features present: history, symptoms, or radiological findings |
||
| Index tests | Routine laboratory tests were assessed on admission (Figure 4 and Figure 5). Laboratory data were collected using a standardised case‐report form. All data were checked by 2 physicians, and a third researcher determined any differences in interpretation between the 2 primary reviewers. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Deterioration was assessed. Non‐deteriorating participants were defined as participants with slight symptoms, fever, respiratory tract symptoms, and no radiological findings or pneumonia findings on radiological examination. Deteriorating participants were participants with any of the following signs or symptoms: tachypnoea with a respiration rate > 30 beats/min, resting oxygen saturation < 92%, arterial partial oxygen pressure (PaO2)/fraction of inspired oxygen (FiO2) < 301 mmHg, radiological aggravation greater than 50% within 24–48 hours, respiratory failure and mechanical ventilation, shock, and organ failure requiring intensive care unit admission. The total sample size was 750 participants. This included 362 probable and 388 confirmed cases; 119 participants experienced severe disease and 631 non‐severe. |
||
| Flow and timing | The time horizon was not reported, the end of follow‐up time was 1st of June 2020. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the authors | ||
| Analysis | Information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Unclear | ||
| Did the study avoid inappropriate selection criteria? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Unclear | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | |||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | Unclear | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Şener 2022.
| Study characteristics | |||
| Patient Sampling | Patients, aged 18 years and older, with confirmed COVID‐19 were admitted to Başakşehir Çam and Sakura City Hospital in İstanbul between 15 and 30 November 2020. Patients with a positive RT‐PCR test who were hospitalised for follow‐up and treatment were included. Patients with haematological disease, who had a blood transfusion while hospitalised, or with incomplete data were excluded. | ||
| Patient characteristics and setting | Setting: hospital, not specified which department
Site: Başakşehir Çam and Sakura City Hospital in İstanbul
Country: Turkey Symptoms and severity: not reported Demographics: the mean age of non‐survivors was 66.79 years (SD 13.97), and 61 years (SD 17.01) in survivors; the mean age in the severe group was 60.22 years (SD 14.29) and 62.86 years (SD 16.25) in the critical group; in all participants (which also included a control group); 133/264 (50.4%) participants were female; in the severe group: 53 (52.5%) females, and critical group: 33 (37.9%) females; non‐survivors: 32.1% females, and survivors: 40.7% females Exposure history: not reported Time since onset of symptoms: not reported Treatment before target condition: not reported Vaccine status: not reported Variant of concern: not reported Definition SARS‐CoV‐2 cases: RT‐PCR, no further specification |
||
| Index tests | Routine laboratory tests were measured on admission (Figure 4 and Figure 5). Laboratory results were obtained retrospectively from electronic medical records. The laboratory tests were studied in the Laboratory of Başakşehir Çam and Sakura City Hospital. Blood samples of the participants were taken into a citrate tube for coagulation, a gel tube for biochemical parameters, and an EDTA tube for a complete blood count. Coagulation analysis was performed on an automatic coagulation analyser (Roche Cobas t 711 America), haematology analysis on (Sysmex XN‐900, Japan) device, biochemistry analysis on SF‐8200 (Roche Cobas 8000 America) brand device using original reagents. Unit of analysis: individual participants. Sample used for the index test was not reported. | ||
| Target condition and reference standard(s) | Mortality and deterioration were assessed. In this study, deteriorating participants were those admitted to the ward, and critical participants were admitted to the ICU. The study included 188 participants, 101 severe and 87 critical. Mortality was assessed in the critical participants. In this group, 59 participants survived and 28 participants died. |
||
| Flow and timing | Time horizon not reported. | ||
| Comparative | |||
| Study design | Retrospective cohort study | ||
| Funding a/o conflicts of interest | None declared by the author | ||
| Analysis | Information about missing data was not reported. | ||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate selection criteria? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index test (All outcomes) | |||
| If a threshold was used, was it pre‐specified? | No | ||
| Was the method used to perform the index test valid and reliable? | Yes | ||
| Was the method for performing the index test the same for all participants? | Yes | ||
| Were the index test results interpreted without knowledge of the outcome? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference standard | |||
| Was the method used to measure the outcome valid and reliable? | Yes | ||
| Was the method for measuring the outcome the same for all participants? | Yes | ||
| Was the outcome (deterioration or combined) measured without knowledge of the index test results? | Unclear | ||
| Was the outcome (mortality) measured without knowledge of the index test results? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and timing | |||
| Did all participants receive the index test? | No | ||
| Was treatment avoided after the index test was performed? | No | ||
| Was the time horizon sufficient to capture the outcome? | Unclear | ||
| Was information on the outcome available for all participants? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Abbreviations: ACEi: angiotensin‐converting enzyme inhibitors; ALT: alanine transaminase; aPTT: activated partial thromboplastin time; ARB: angiotensin receptor blocker; ARDS: acute respiratory distress syndrome; AST: aspartate aminotransferase; BAME: black, Asian, and ethnic minority; BIPAP: bilevel positive airway pressure; CK: creatine kinase; CPAP: continuous positive airway pressure; CRP: C‐reactive protein; CT: computer tomography; CVVH: continuous veno‐venous hemofiltration; DIC: disseminated intravascular coagulation; ECMO: extracorporeal membrane oxygenation; ED: emergency department; EDTA: ethylenediaminetetraacetic acid; EHR: electronic health record; EQA: external quality assurance; FEU: fibrinogen equivalent units; HRCT: high‐resolution computed tomography; hs‐CRP: high‐sensitivity C‐reactive protein; ICU: intensive care unit; IDW: infectious disease ward; IgM: immunoglobulin M; IL‐6: interleukin 6; IQR: interquartile range; LDH: lactate dehydrogenase; LMWH: low molecular weight heparin; NIV: non‐invasive ventilation; NLR: neutrophil to lymphocyte ratio; NOAC: non‐Vitamin‐K‐antagonist oral anticoagulants; NPV: negative predictive value; NT‐proBNP: N‐terminal pro b‐type natriuretic peptide; O2: oxygen; PaO2/Fi02: ratio of arterial oxygen partial pressure (PaO2 in mmHg) to fractional inspired oxygen (FiO2 expressed as a fraction, not a percentage); PCR: polymerase chain reaction; PLR: platelet‐to‐lymphocyte ratio; PPV: positive predictive value; RAS: radioallergosorbent; RNA: ribonucleic acid; RR: risk ratio; RT‐PCR: reverse transcriptase polymerase chain reaction; Sa02: oxygen saturation of arterial blood; SD: standard deviation; SEHH: Spanish Society of Hematology and Hemostasis; SEQC‐ML: Spanish Society of Laboratory Medicine; Sp02: oxygen saturation; UFH: unfractionated heparin; VKA: vitamin K antagonists; VTE: venous thromboembolism.
Differences between protocol and review
While we reported in the published protocol that we would perform this (Verbakel 2021), due to data scarcity, we did not investigate whether prognostic accuracy varied according to specific measurement or test, reference standard, timing of outcome verification, sample type, study design, and setting, including prevalence of the target condition. Moreover, we were unable to assess the difference in test accuracy using likelihood ratio tests for comparisons of models with and without covariate terms, because fewer than 10 primary studies were available for the direct comparisons. We did not have sufficient available data to investigate the following sources of heterogeneity: measurement technique or test type, reference standard, timing of outcome verification, sample type, study design, and setting, including prevalence of the target condition. Furthermore, there was insufficient data available to perform sensitivity analyses considering the impact of unpublished studies, and to investigate the impact of prospective versus retrospective data collection. Finally, we did not identify studies that we know exist but for which we have not managed to locate reports.
We defined a time horizon for mortality and deterioration to severe or critical disease of 30 and 14 days, respectively. This was defined to correctly assess the time horizon in the methodological quality assessment.
Contributions of authors
Liselore De Rop: study selection, data‐extraction and quality assessment, meta‐analyses, comparisons, co‐ordinated the review process; drafted all non‐automatic Tables; GRADE assessment; contributed to the first draft and subsequent revisions of the review.
David Bos: study selection, data‐extraction and quality assessment, multiple revisions of the review.
Inge Stegeman: study selection, multiple revisions of the review.
Gea Holtman: study selection, multiple revisions of the review.
Eleanor Ochodo: study selection, multiple revisions of the review.
Rene Spijker: contributed clinical, methodological/technical expertise to drafting the protocol; co‐ordinated and conducted the study retrieval and initial selection steps; contributed to multiple revisions of the review.
Jenifer Otieno: study selection, data‐extraction and quality assessment, multiple revisions of the review.
Fade Alkhlaileh: study selection, data‐extraction and quality assessment, multiple revisions of the review.
Jon J Deeks: contributed clinical, methodological/technical expertise to drafting the protocol; contributed to multiple revisions of the review and co‐ordinated all contributions to all Cochrane Rapid DTA reviews;
Jacqueline Dinnes: contributed clinical, methodological/technical expertise to drafting the protocol; did the initial screening titles and abstracts for all reviews; contributed to multiple revisions of the review.
Ann Van den Bruel: contributed clinical, methodological/technical expertise to drafting the protocol; contributed to multiple revisions of the review.
Matthew D F McInnes: contributed clinical, methodological/technical expertise to drafting the protocol; contributed to multiple revisions of the review;
Mariska Leeflang: study selection, multiple revisions of the review.
Cochrane COVID‐19 Diagnostic Test Accuracy Group: contributed clinical, methodological/technical expertise to drafting the protocol; contributed to multiple revisions of the review.
Jan Y Verbakel: study selection, contributed clinical, methodological/technical expertise to drafting the protocol; supervised the meta‐analyses; contributed to multiple revisions of the review.
Sources of support
Internal sources
-
Liverpool School of Tropical Medicine, UK, UK
No grant number available
External sources
-
Foreign, Commonwealth, and Development Office (FCDO), UK
Project number 300342‐104
-
National Institute for Health and Care Research (NIHR), UK
Jonathan Deeks is a United Kingdom National Institute for Health Research (NIHR) Senior Investigator Emeritus. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health and Social Care.
-
NIHR Birmingham Biomedical Research Centre at the University Hospitals Birmingham NHS Foundation Trust and the University of Birmingham, UK
Jonathan Deeks and Jacqueline Dinnes are supported by the NIHR Birmingham Biomedical Research Centre at the University Hospitals Birmingham NHS Foundation Trust and the University of Birmingham. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health and Social Care.
Declarations of interest
LDR has no known conflicts of interest.
DB has no known conflicts of interest.
IS has no known conflicts of interest.
GAH has no known conflicts of interest.
EO is a Cochrane editor, she was not involved in the editorial process of this research.
RS has no known conflicts of interest.
JO has no known conflicts of interest.
FA has no known conflicts of interest.
JJD has published or been quoted in opinion pieces in scientific publications, and in the mainstream and social media related to diagnostic testing.
JD is a Cochrane editor, she was not involved in the editorial process of this research.
AVdB has no known conflicts of interest.
MM has no known conflicts of interest.
MMGL is a Cochrane editor, she was not involved in the editorial process of this research.
JYV has no known conflicts of interest.
New
References
References to studies included in this review
Al‐Samkari 2020 {published data only}
- Al-Samkari H, Karp Leaf RS, Dzik WH, Carlson JCT, Fogerty AE, Waheed A, et al. COVID-19 and coagulation: bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood 2020;136(4):489-00. [DOI] [PMC free article] [PubMed] [Google Scholar]
An 2021 {published data only}
- An C, Oh HC, Chang JH, Oh SJ, Lee JM, Han CH, et al. Development and validation of a prognostic model for early triage of patients diagnosed with COVID‐19. Scientific Reports 2021;11(1):21923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Arnold 2021 {published data only}
- Arnold DT, Attwood M, Barratt S, Morley A, Elvers KT, McKernon J, et al. Predicting outcomes of COVID-19 from admission biomarkers: a prospective UK cohort study. Emergency Medicine Journal 2021;38(7):543-8. [DOI] [PubMed] [Google Scholar]
Arshad 2020 {published data only}
- Arshad AR, Khan I, Shahzad K, Arshad M, Haider SJ, Aslam MJ. Association of inflammatory markers with mortality in COVID-19 infection. Journal of the College of Physicians and Surgeons Pakistan 2020;30(10):158-63. [DOI] [PubMed] [Google Scholar]
Asaduzzaman 2022 {published data only}
- Asaduzzaman MD, Romel Bhuia M, Nazmul Alam Z, Zabed Jillul Bari M, Ferdousi T. Significance of hemogram‐derived ratios for predicting in‐hospital mortality in COVID‐19: a multicenter study. Health Science Reports 2022;5(4):e663. [DOI] [PMC free article] [PubMed] [Google Scholar]
Asghar 2020 {published data only}
- Asghar MS, Haider Kazmi SJ, Khan NA, Akram M, Hassan M, Rasheed U, et al. Poor prognostic biochemical markers predicting fatalities caused by COVID-19: a retrospective observational study from a developing country. Cureus 2020;12(8):e9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Aydin 2022 {published data only}
- Aydin Guclu O, Goktas SS, Gorek Dilektasli A, Acet Ozturk NA, Demirdogen E, Coskun F, et al. Pilot study for immunoglobulin E as a prognostic biomarker in coronavirus disease 2019. Internal Medicine Journal 2022;52(9):1495-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ayus 2021 {published data only}
- Ayus JC, Negri AL, Moritz ML, Lee KM, Caputo D, Borda ME, et al. Hyponatremia, inflammation at admission, and mortality in hospitalized COVID-19 patients: a prospective cohort study. Frontiers in Medicine 2021;8:748364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Azad Allarakia 2022 {published data only}
- Azad Allarakia BM, Gattan HS, Abdeen RH, Al-Ahmadi BM, Shater AF, Bazaid MB, et al. Predicting intensive care unit admission for COVID-19 patients from laboratory results. Disease Markers 2022;2022:4623901. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bahl 2020 {published data only}
- Bahl A, Van Baalen MN, Ortiz L, Chen NW, Todd C, Milad M, et al. Early predictors of in‐hospital mortality in patients with COVID‐19 in a large American cohort. Internal and Emergency Medicine 2020;15(8):1485-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Barrett 2021 {published data only}
- Barrett B, Pamphile S, Yang F, Naeem F, Kim J, Annam J, et al. Inflammatory markers are poorly predictive of clinical outcomes among hospitalized patients with COVID-19. American Journal of Emergency Medicine 2021;46:595-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bassoli 2020 {published data only}
- Bassoli C, Oreni L, Ballone E, Foschi A, Perotti A, Mainini A, et al. Role of serum albumin and proteinuria in patients with SARS-CoV-2 pneumonia. International Journal Of Clinical Practice 2021;75(4):e13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
BG 2021 {published data only}
- Bg S, Gosavi S, Ananda Rao A, Shastry S, Raj SC, Sharma A, et al. Neutrophil-to-lymphocyte, lymphocyte-to-monocyte, and platelet-to-lymphocyte ratios: prognostic significance in COVID-19. Cureus 2021;13(1):e12622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cazzaniga 2021 {published data only}
- Cazzaniga M, Fumagalli LAM, D'Angelo L, Cerino M, Bonfanti G, Fumagalli RM, et al. Eosinopenia is a reliable marker of severe disease and unfavourable outcome in patients with COVID-19 pneumonia. International Journal Of Clinical Practice 2021;75(7):e14047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chen 2020a {published data only}
- Chen D, Li X, Song Q, Hu C, Su F, Dai J, et al. Assessment of hypokalemia and clinical characteristics in patients with coronavirus disease 2019 in Wenzhou, China. JAMA Network Open 2020;3(6):e2011122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chen 2020b {published data only}
- Chen J, Han T, Huang M, Yang Y, Shang F, Zheng Y, et al. Clinical characteristics of asymptomatic carriers of novel coronavirus disease 2019: a multi-center study in Jiangsu Province. Virulence 2020;11(1):1557-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chen 2020c {published data only}
- Chen R, Sang L, Jiang M, Yang Z, Jia N, Fu W, et al. Longitudinal hematologic and immunologic variations associated with the progression of COVID-19 patients in China. Journal of Allergy and Clinical Immunology 2020;146(1):89-00. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chen 2020d {published data only}
- Chen R, Liang W, Jiang M, Guan W, Zhan C, Wang T, et al. Risk factors of fatal outcome in hospitalized subjects with coronavirus disease 2019 from a nationwide analysis in China. Chest 2020;158(1):97-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chen 2021 {published data only}
- Chen P, Fan N, Liang Y, Fan W. Analysis of predictive factors for moderate-stage COVID-19 outcome and maximal extent of lung injury. Iranian Journal of Radiology 2021;18(1):e106351. [Google Scholar]
Chocron 2021 {published data only}
- Chocron R, Duceau B, Gendron N, Ezzouhairi N, Khider L, Trimaille A, et al. D-dimer(s) at hospital admission for COVID-19 are associated with in-hospital mortality, independent of venous thromboembolism: insights from a French multicenter cohort study [Les D-dimères à l’admission pour COVID-19 sont associés à la mortalité hospitalière indépendamment du risque de thrombose veineuse]. Archives of Cardiovascular Disease 2021;114(5):381-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Detsika 2021 {published data only}
- Detsika MG, Grigoriou E, Psarra K, Jahaj E, Tsipilis S, Athanassiou N, et al. Combination of the CD8+:B-cell and neutrophil-to-lymphocyte ratio as a novel prediction model for intubation need and disease severity in COVID-19 patients. In Vivo 2021;35(6):3305-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Dong 2020 {published data only}
- Dong X, Sun L, Li Y. Prognostic value of lactate dehydrogenase for in-hospital mortality in severe and critically ill patients with COVID-19. International Journal of Medical Sciences 2020;17(14):2225-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Duan 2020 {published data only}
- Duan J, Wang X, Chi J, Chen H, Bai L, Hu Q, et al. Correlation between the variables collected at admission and progression to severe cases during hospitalization among patients with COVID‐19 in Chongqing. Journal of Medical Virology 2020;92(11):2616-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elhadad 2020 {published data only}
- Elhadad D, Bronstein Y, Yana M, Baris H, Levinger U, Shapiro M, et al. Characteristics and outcomes of patients infected with SARS-CoV-2 in Israel: correlation between laboratory findings on admission to emergency department and subsequent respiratory failure. Israel Medical Association Journal 2020;22(10):605-11. [PubMed] [Google Scholar]
Fernandez‐Botran 2021 {published data only}
- Fernandez-Botran R, Furmanek S, Ambadapoodi RS, Expósito González E, Cahill M, Carrico R, et al. Association and predictive value of biomarkers with severe outcomes in hospitalized patients with SARS-CoV-2 infection. Cytokine 2022;149:155755. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gao 2020 {published data only}
- Gao L, Jiang D, Wen XS, Cheng XC, Sun M, He B, et al. Prognostic value of NT-proBNP in patients with severe COVID-19. Respiratory Research 2020;21(1):83. [DOI] [PMC free article] [PubMed] [Google Scholar]
García de Guadiana‐Romualdo 2021a {published data only}
- García de Guadiana-Romualdo L, Morell-García D, Rodríguez-Fraga O, Morales-Indiano C, María Lourdes Padilla Jiménez A, Gutiérrez Revilla JI, et al. Cardiac troponin and COVID-19 severity: results from BIOCOVID study. European Journal of Clinical Investigation 2021;51(6):e13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
García de Guadiana‐Romualdo 2021b {published data only}
- García de Guadiana-Romualdo L, Morell-García D, Morales-Indiano C, Bauça JM, Alcaide Martín MJ, Esparza Del Valle C, et al. Characteristics and laboratory findings on admission to the emergency department among 2873 hospitalized patients with COVID-19: the impact of adjusted laboratory tests in multicenter studies. A multicenter study in Spain (BIOCOVID-Spain study). Scandinavian Journal of Clinical and Laboratory Investigation 2021;81(3):187-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
García de Guadiana‐Romualdo 2022 {published data only}
- García de Guadiana-Romualdo L, Morell-García D, Favaloro EJ, Vílchez JA, Bauça JM, Alcaide Martín MJ, et al. Harmonized D-dimer levels upon admission for prognosis of COVID-19 severity: results from a Spanish multicenter registry (BIOCOVID-Spain study). Journal of Thrombosis and Thrombolysis 2022;53(1):103-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Goudot 2020 {published data only}
- Goudot G, Chocron R, Augy JL, Gendron N, Khider L, Debuc B, et al. Predictive factor for COVID-19 worsening: insights for high-sensitivity troponin and D-dimer and correlation with right ventricular afterload. Frontiers in Medicine 2020;7:586307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Guan 2020 {published data only}
- Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical characteristics of 2019 novel coronavirus infection in China. New England Journal o f Medicine 2020;382(18):1708-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Haymana 2021 {published data only}
- Haymana C, Demirci I, Tasci I, Cakal E, Salman S, Ertugrul D, et al. Clinical outcomes of non-diabetic COVID-19 patients with different blood glucose levels: a nationwide Turkish study (TurCoGlycemia). Endocrine 2021;73(2):261-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ibraheem 2021 {published data only}
- Ibraheem AI, Nasir HM, Abdulamir AS, Ali CL, Kabah KK, Hussein IA, et al. Early identification of high risk COVID-19 patients using hematological indices. Journal of Health and Translational Medicine 2021;1:1-6. [Google Scholar]
López‐Escobar 2021 {published data only}
- López-Escobar A, Madurga R, Castellano JM, Velázquez S, Suárez Del Villar R, Menéndez J, et al. Risk score for predicting in-hospital mortality in COVID-19 (RIM score). Diagnostics 2021;11(4):596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Marwah 2021 {published data only}
- Marwah M, Marwah S, Blann A, Morrissey H, Ball P, Wandroo FA. Analysis of laboratory blood parameter results for patients diagnosed with COVID-19, from all ethnic group populations: a single centre study. International Journal of Laboratory Hematology 2021;43(5):1243-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Masotti 2021 {published data only}
- Masotti L, Grifoni E, Pelagalli G, Cioni E, Mattaliano C, Cioffi E, et al. Prognostic role of interleukin-6/lymphocytes ratio in SARS-CoV2 related pneumonia. International Immunopharmacology 2022;103:108435. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mateos González 2021 {published data only}
- Mateos González M, Sierra Gonzalo E, Casado Lopez I, Arnalich Fernández F, Beato Pérez JL, Monge Monge D, et al. The prognostic value of eosinophil recovery in COVID-19: a multicentre, retrospective cohort study on patients hospitalised in Spanish hospitals. Journal of Clinical Medicine 2021;10(2):305. [DOI] [PMC free article] [PubMed] [Google Scholar]
Meng 2021 {published data only}
- Meng L, Xu J, Zhang J, Zhang K, Shang P, Li Q, et al. Monitoring peripheral neutrophil and T-lymphocyte subsets could assist in differentiating the severity and disease progression of coronavirus disease 2019. Aging 2021;13(6):7723-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Muñoz‐Rodríguez 2021 {published data only}
- Muñoz-Rodríguez JR, Gómez-Romero FJ, Pérez-Ortiz JM, López-Juárez P, Santiago JL, Serrano-Oviedo L, et al. Characteristics and risk factors associated with mortality in a multicenter Spanish cohort of patients with COVID-19 pneumonia. Archivos de Bronconeumología 2021;57:34-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Öktem 2020 {published data only}
- Öktem B, Üzer F, Kukul Güven FM, Kırhan İ, Topal M. Role of methemoglobin and carboxyhemoglobin levels in predicting COVID-19 prognosis: an observational study. Medical Gas Research 2020;10(4):174-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Olivieri 2022 {published data only}
- Olivieri F, Sabbatinelli J, Bonfigli AR, Sarzani R, Giordano P, Cherubini A, et al. Routine laboratory parameters, including complete blood count, predict COVID-19 in-hospital mortality in geriatric patients. Mechanisms of Ageing and Development 2022;204:111674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ortega‐Rojas 2022 {published data only}
- Ortega-Rojas S, Salazar-Talla L, Romero-Cerdán A, Soto-Becerra P, Díaz-Vélez C, Urrunaga-Pastor D, et al. The neutrophil-to-lymphocyte ratio and the platelet-to- lymphocyte ratio as predictors of mortality in older adults hospitalized with COVID-19 in Peru. Disease Markers 2022;2022:2497202. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ouldali 2021 {published data only}
- Ouldali N, Yang DD, Madhi F, Levy M, Gaschignard J, Craiu I, et al. Factors associated with severe SARS-CoV-2 infection. Paediatrics 2021;147(3):e2020023432. [DOI] [PubMed] [Google Scholar]
Pan 2021 {published data only}
- Pan M, Wang RR, Chen X, Han J, Li Q, Miao M, et al. Laboratory predictors of severe coronavirus disease 2019 and lung function in followed-up. Clinical Respiratory Journal 2021;15(8):904-14. [DOI] [PubMed] [Google Scholar]
Papageorgiou 2022 {published data only}
- Papageorgiou N, Sohrabi C, Prieto Merino D, Tyrlis A, Atieh AE, Saberwal B, et al. High sensitivity troponin and COVID-19 outcomes. Acta Cardiologica 2022;77(1):81-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Para 2022 {published data only}
- Para O, Caruso L, Pestelli G, Tangianu F, Carrara D, Maddaluni L, et al. Ferritin as prognostic marker in COVID-19: the FerVid study. Postgraduate Medicine 2022;134(1):58-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Prasetya 2021 {published data only}
- Prasetya IB, Cucunawangsih, Lorens JO, Sungono V, El-Khobar KE, Wijaya RS. Prognostic value of inflammatory markers in patients with COVID-19 in Indonesia. Clinical Epidemiology and Global Health 2021;11:100803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Qu 2021 {published data only}
- Qu J, Zhu HH, Huang XJ, He GF, Liu JY, Huang JJ, et al. Abnormal indexes of liver and kidney injury markers predict severity in COVID-19 patients. Infection and Drug Resistance 2021;14:3029-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rasyid 2020 {published data only}
- Rasyid H, Sangkereng A, Harjianti T, Soetjipto AS. Impact of age to ferritin and neutrophil-lymphocyte ratio as biomarkers for intensive care requirement and mortality risk in COVID-19 patients in Makassar, Indonesia. Physiological Reports 2021;9(10):e14876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Şan 2021 {published data only}
- Şan İ, Gemcioğlu E, Davutoğlu M, Çatalbaş R, Karabuğa B, Kaptan E, et al. Which hematological markers have predictive value as early indicators of severe COVID-19 cases in the emergency department? Turkish Journal of Medical Sciences 2021;51(6):2810-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Şener 2022 {published data only}
- Şener G. The effectiveness of coagulation parameters in classifying patients and predicting mortality in Covid-19. Journal of Experimental and Clinical Medicine 2022;39(1):232-6. [Google Scholar]
Sepulchre 2022 {published data only}
- Sepulchre E, Pittie G, Stojkovic V, Haesbroek G, Crama Y, Schyns M, et al. Covid-19: contribution of clinical characteristics and laboratory features for early detection of patients with high risk of severe evolution. Acta Clinica Belgica 2022;77(2):261-7. [DOI] [PubMed] [Google Scholar]
Simon 2022 {published data only}
- Simon M, Le Borgne P, Lefevbre F, Chabrier S, Cipolat L, Remillon A, et al. Lymphopenia and early variation of lymphocytes to predict in-hospital mortality and severity in ED patients with SARS-CoV-2 infection. Journal of Clinical Medicine 2022;11(7):1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sisó‐Almirall 2020 {published data only}
- Sisó-Almirall A, Kostov B, Mas-Heredia M, Vilanova-Rotllan S, Sequeira-Aymar E, Sans-Corrales M, et al. Prognostic factors in Spanish COVID-19 patients: a case series from Barcelona. PLOS One 2020;15(8):e0237960. [DOI] [PMC free article] [PubMed] [Google Scholar]
Soh 2020 {published data only}
- Soh TV, Dzawani M, Noorlina N, Nik F, Norazmi A. Clinical characteristics of severe acute respiratory syndrome Coronavirus 2 (SARS-CoV2) patients in Hospital Tengku Ampuan Afzan. Medical Journal of Malaysia 2020;75(5):479-84. [PubMed] [Google Scholar]
Statsenko 2021 {published data only}
- Statsenko Y, Al Zahmi F, Habuza T, Gorkom KN, Zaki N. Prediction of COVID-19 severity using laboratory findings on admission: informative values, thresholds, ML model performance. BMJ Open 2021;11(2):e044500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Suardi 2020 {published data only}
- Suardi LR, Pallotto C, Esperti S, Tazzioli E, Baragli F, Salomoni E, et al. Risk factors for non-invasive/invasive ventilatory support in patients with COVID-19 pneumonia: a retrospective study within a multidisciplinary approach. International Journal of Infectious Diseases 2020;100:258-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Suzuki 2022 {published data only}
- Suzuki K, Ichikawa T, Suzuki S, Tanino Y, Kakinoki Y. C-reactive protein and lactate dehydrogenase as prognostic indicators in COVID-2019 outpatients. medrxiv.org 2022 (accessed prior to 19 June 2024). [DOI: ]
Ullah 2020 {published data only}
- Ullah W, Basyal B, Tariq S, Almas T, Saeed R, Roomi S, et al. Lymphocyte-to-C-reactive protein ratio: a novel predictor of adverse outcomes in COVID-19. Journal of Clinical Medicine Research 2020;12(7):415-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Xia 2020 {published data only}
- Xia X, Wen M, Zhan S, He J, Chen W. An increased neutrophil/lymphocyte ratio is an early warning signal of severe COVID-19 [中性粒细胞/淋巴细胞比值可作为重型COVID-19的预警信号]. Journal of Southern Medical University 2020;40(3):333-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zafar 2021 {published data only}
- Zafar M, Shahbaz M, Karkhanis M, Abdelbagi M, Makanjuola OA, Pun B, et al. A retrospective observational study: is absolute lymphocyte count a prognostic marker in COVID-19? Cureus 2021;13(7):e16554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zarębska‐Michaluk 2021 {published data only}
- Zarębska-Michaluk D, Jaroszewicz J, Rogalska M, Lorenc B, Rorat M, Szymanek-Pasternak A, et al. Impact of kidney failure on the severity of COVID-19. Journal of Clinical Medicine 2021;10(9):2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2020 {published data only}
- Zhang J, Ding D, Cao C, Zhang J, Huang X, Fu P, et al. Myocardial characteristics as the prognosis for COVID-19 patients. medRxiv.org 2020 (accessed prior to 19 June 2024). [DOI: ]
Zhou 2020 {published data only}
- Zhou YH, Li H, Qin YY, Yan XF, Lu YQ, Liu HL, et al. Predictive factors of progression to severe COVID-19. Open Medicine 2020;15(1):805-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional references
Abou‐Ismail 2020
- Abou-Ismail MY, Diamond A, Kapoor S, Arafah Y, Nayak L. The hypercoagulable state in COVID-19: incidence, pathophysiology, and management. Thrombosis Research 2020;194:101-15. [DOI: 10.1016/j.thromres.2020.06.029] [DOI] [PMC free article] [PubMed] [Google Scholar]
Deeks 2020a
- Deeks JJ, Dinnes J, Takwoingi Y, Davenport C, Leeflang MMG, Spijker R, et al. Diagnosis of SARS-CoV-2 infection and COVID-19: accuracy of signs and symptoms; molecular, antigen, and antibody tests; and routine laboratory markers. Cochrane Database of Systematic Reviews 2020, Issue 4. Art. No: CD013596. [DOI: 10.1002/14651858.CD013596] [DOI] [Google Scholar]
Deeks 2020b
- Deeks JJ, Dinnes J, Takwoingi Y, Davenport C, Spijker R, Taylor-Phillips S, et al. Antibody tests for identification of current and past infection with SARS-CoV-2. Cochrane Database of Systematic Reviews 2020, Issue 6. Art. No: CD013652. [DOI: 10.1002/14651858.CD013652] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dinnes 2020
- Dinnes J, Deeks JJ, Berhane S, Taylor M, Adriano A, Davenport C, et al. Rapid, point-of-care antigen and molecular-based tests for diagnosis of SARS-CoV-2 infection. Cochrane Database of Systematic Reviews 2021, Issue 3. Art. No: CD013705. [DOI: 10.1002/14651858.CD013705.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
EPPI‐Reviewer 2020 [Computer program]
- EPPI-Reviewer: advanced software for systematic reviews, maps and evidence synthesis. Thomas J, Graziosi S, Brunton J, Ghouze Z, O'Driscoll P, Bond M. London: EPPI-Centre Software. UCL Social Research Institute, 2020.
Feng 2020
- Feng X, Li S, Sun Q, Zhu J, Chen B, Xiong M, et al. Immune-inflammatory parameters in COVID-19 cases: a systematic review and meta-analysis. Frontiers in Medicine 2020;7:301. [DOI: 10.3389/fmed.2020.00301] [DOI] [PMC free article] [PubMed] [Google Scholar]
Han 2020
- Han Y, Zhang H, Mu S, Wei W, Jin C, Tong C, et al. Lactate dehydrogenase, an independent risk factor of severe COVID-19 patients: a retrospective and observational study. Aging 2020;12(12):11245-58. [DOI: 10.18632/aging.103372] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hayden 2013
- Hayden JA, Van der Windt DA, Cartwright JL, Côté P, Bombardier C. Assessing bias in studies of prognostic factors. Annals of Internal Medicine 2013;158(4):280-6. [DOI] [PubMed] [Google Scholar]
Islam 2021
- Islam N, Ebrahimzadeh S, Salameh J-P, Kazi S, Fabiano N, Treanor L, et al. Thoracic imaging tests for the diagnosis of COVID-19. Cochrane Database of Systematic Reviews 2021, Issue 3. Art. No: CD013639. [DOI: 10.1002/14651858.CD013639.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kichloo 2020
- Kichloo A, Dettloff K, Aljadah M, Albosta M, Jamal S, Singh J, et al. COVID-19 and hypercoagulability: a review. Clinical and Applied Thrombosis/Hemostasis 2020;26:1076029620962853. [DOI: 10.1177/1076029620962853] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kojima 2023
- Kojima K, Yoon H, Okishio K, Tsuyuguchi K. Increased lactate dehydrogenase reflects the progression of COVID-19 pneumonia on chest computed tomography and predicts subsequent severe disease. Scientific Reports 2023;13(1):1012. [DOI: 10.1038/s41598-023-28201-2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lai 2022
- Lai KL, Hu FC, Wen FY, Chen JJ. Lymphocyte count is a universal predictor of health outcomes in COVID-19 patients before mass vaccination: a meta-analytical study. Journal of Global Health 2022;12:05041. [DOI: 10.7189/jogh.12.05041] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lee 2022
- Lee J, Mulder F, Leeflang M, Wolff R, Whiting P, Bossuyt PM. QUAPAS: an adaptation of the QUADAS-2 tool to assess prognostic accuracy studies. Annals of Internal Medicine 2022;175(7):1010-8. [DOI: ] [DOI] [PubMed] [Google Scholar]
Macaskill 2010
- Macaskill P, Gatsonis C, Deeks JJ, Harbord RM, Takwoingi Y. Chapter 10: Analysing and presenting results. In: Deeks JJ, Bossuyt PM, Gatsonis C, editor(s). Cochrane Handbook for Systematic Reviews of Diagnostic Test Accuracy Version 1.0.0. The Cochrane Collaboration, 2010. Available from srdta.cochrane.org.
Meletis 2023
- Meletis G, Tychala A, Ntritsos G, Verrou E, Savvidou F, Dermitzakis I, et al. Variant-related differences in laboratory biomarkers among patients affected with alpha, delta and omicron: a retrospective whole viral genome sequencing and hospital-setting cohort study. Biomedicines 2023;11(4):1143. [DOI: 10.3390/biomedicines11041143] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nakakubo 2023
- Nakakubo S, Unoki Y, Kitajima K, Terada M, Gatanaga H, Ohmagari N, et al. Serum lactate dehydrogenase level one week after admission is the strongest predictor of prognosis of COVID-19: a large observational study using the COVID-19 registry Japan. Viruses 2023;15(3):671. [DOI: 10.3390/v15030671] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nugroho 2021
- Nugroho J, Wardhana A, Maghfirah I, Mulia EPB, Rachmi DA, A'yun MQ, et al. Relationship of d-dimer with severity and mortality in SARS-CoV-2 patients: a meta-analysis. International Journal of Laboratory Hematology 2021;43(1):110-5. [DOI: 10.1111/ijlh.13336] [DOI] [PubMed] [Google Scholar]
Pranata 2021
- Pranata R, Lim MA, Yonas E, Huang I, Nasution SA, Setiati S, et al. Thrombocytopenia as a prognostic marker in COVID-19 patients: diagnostic test accuracy meta-analysis. Epidemiology and Infection 2021;149:e40. [DOI: 10.1017/S0950268821000236] [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan 2024 [Computer program]
- Review Manager (RevMan). Version 7.9.2. The Cochrane Collaboration, 2024. Available at revman.cochrane.org.
SAS 2015 [Computer program]
- SAS. Version 9.4. Cary, NC, USA: SAS Institute, 2015. Available at www.sas.com.
Schünemann 2020a
- Schünemann HJ, Mustafa RA, Brozek J, Steingart KR, Leeflang M, Murad MH, et al. GRADE guidelines: 21 part 1. Study design, risk of bias and indirectness in rating the certainty across a body of evidence for test accuracy. Journal of Clinical Epidemiology 2020;122:129-41. [DOI] [PubMed] [Google Scholar]
Schünemann 2020b
- Schünemann HJ, Mustafa RA, Brozek J, Steingart KR, Leeflang M, Murad MH, et al. GRADE guidelines: 21 part 2. Test accuracy: inconsistency, imprecision, publication bias and other domains for rating the certainty of evidence for test accuracy and presenting it in evidence profiles and summary of findings tables. Journal of Clinical Epidemiology 2020;122:142-52. [DOI] [PubMed] [Google Scholar]
Simadibrata 2020
- Simadibrata DM, Lubis AM. D-dimer levels on admission and all-cause mortality risk in COVID-19 patients: a meta-analysis. Epidemiology and Infection 2020;148:e202. [DOI: 10.1017/S0950268820002022] [DOI] [PMC free article] [PubMed] [Google Scholar]
Smilowitz 2021
- Smilowitz NR, Kunichoff D, Garshick M, Shah B, Pillinger M, Hochman JS, et al. C-reactive protein and clinical outcomes in patients with COVID-19. European Heart Journal 2021;42(23):2270-9. [DOI: 10.1093/eurheartj/ehaa1103] [DOI] [PMC free article] [PubMed] [Google Scholar]
Song 2021
- Song M, Graubard BI, Rabkin CS, Engels EA. Neutrophil-to-lymphocyte ratio and mortality in the United States general population. Scientific Reports 2021;11(1):464. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sriram 2021
- Sriram K, Insel PA. Inflammation and thrombosis in COVID-19 pathophysiology: proteinase-activated and purinergic receptors as drivers and candidate therapeutic targets. Physiological Reviews 2021;101(2):545-67. [DOI: 10.1152/physrev.00035.2020] [DOI] [PMC free article] [PubMed] [Google Scholar]
Stegeman 2020
- Stegeman I, Ochodo EA, Guleid F, Holtman GA, Yang B, Davenport C, et al. Routine laboratory testing to determine if a patient has COVID-19. Cochrane Database of Systematic Reviews 2020, Issue 11. Art. No: CD013787. [DOI: 10.1002/14651858.CD013787] [DOI] [PMC free article] [PubMed] [Google Scholar]
Struyf 2021
- Struyf T, Deeks JJ, Dinnes J, Takwoingi Y, Davenport C, Leeflang MMG, et al. Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19. Cochrane Database of Systematic Reviews 2021, Issue 2. Art. No: CD013665. [DOI: 10.1002/14651858.CD013665.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Vazzana 2022
- Vazzana N, Dipaola F, Ognibene S. Procalcitonin and secondary bacterial infections in COVID-19: association with disease severity and outcomes. Acta Clinica Belgica 2022;77(2):268-72. [DOI: 10.1080/17843286.2020.1824749] [DOI] [PubMed] [Google Scholar]
Whiting 2011
- Whiting PF, Rutjes AW, Westwood ME, Mallett S, Deeks JJ, Reitsma JB, et al. QUADAS-2: a revised tool for the quality assessment of diagnostic accuracy studies. Annals of Internal Medicine 2011;155(8):529-36. [DOI] [PubMed] [Google Scholar]
WHO 2020
- World Health Organization. Clinical management of COVID-19: interim guidance, 27 May 2020. apps.who.int/iris/handle/10665/332196 (accessed prior to 5 August 2021).
WHO 2023a
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. covid19.who.int/ (accessed prior to 23 October 2023).
WHO 2023b
- World Health Organization. Coronavirus disease (COVID-19) pandemic. www.who.int/europe/emergencies/situations/covid-19 (accessed prior to 26 October 2023) (accessed prior to 19 June 2024).
Wibowo 2021
- Wibowo A, Pranata R, Akbar MR, Purnomowati A, Martha JW. Prognostic performance of troponin in COVID-19: a diagnostic meta-analysis and meta-regression. International Journal of Infectious Diseases 2021;105:312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wolff 2019
- Wolff RF, Moons KGM, Riley RD, Whiting PF, Westwood M, Collins GS, et al. PROBAST: a tool to assess the risk of bias and applicability of prediction model studies. Annals of Internal Medicine 2019;170(1):51-8. [DOI] [PubMed] [Google Scholar]
Wynants 2020
- Wynants L, Van Calster B, Collins GS, Riley RD, Heinze G, Schuit E, et al. Prediction models for diagnosis and prognosis of COVID-19: systematic review and critical appraisal. BMJ 2020;369:m1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zare 2020
- Zare ME, Wang Y, Nasir Kansestani A, Almasi A, Zhang J. Procalcitonin has good accuracy for prognosis of critical condition and mortality in COVID-19: a diagnostic test accuracy systematic review and meta-analysis. Iranian Journal of Allergy, Asthma and Immunology 2020;19(6):557-69. [DOI: 10.18502/ijaai.v19i6.4926] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Verbakel 2021
- Verbakel JY, De Rop L, Stegeman I, Holtman GA, Ochodo EA, Yang B, et al. Accuracy of routine laboratory tests to predict mortality and deterioration to severe or critical COVID‐19 in people with SARS‐CoV‐2. Cochrane Database of Systematic Reviews 2021, Issue 9. Art. No: CD015050. [DOI: 10.1002/14651858.CD015050] [DOI] [PMC free article] [PubMed] [Google Scholar]