

Abstract
Background
While the association of chronological age with DNA methylation (DNAm) in whole blood has been extensively studied, the tissue-specificity of age-related DNAm changes remains an active area of research. Studies investigating the association of age with DNAm in tissues such as brain, skin, immune cells, fat, and liver have identified tissue-specific and non-specific effects, thus, motivating additional studies of diverse human tissue and cell types.
Results
Here, we performed an epigenome-wide association study, leveraging DNAm data (Illumina EPIC array) from 961 tissue samples representing 9 tissue types (breast, lung, colon, ovary, prostate, skeletal muscle, testis, whole blood, and kidney) from the Genotype-Tissue Expression (GTEx) project. We identified age-associated CpG sites (false discovery rate < 0.05) in 8 tissues (all except skeletal muscle, n = 47). This included 162,002 unique hypermethylated and 90,626 hypomethylated CpG sites across all tissue types, with 130,137 (80%) hypermethylated CpGs and 74,703 (82%) hypomethylated CpG sites observed in a single tissue type. While the majority of age-associated CpG sites appeared tissue-specific, the patterns of enrichment among genomic features, such as chromatin states and CpG islands, were similar across most tissues, suggesting common mechanisms underlying cellular aging. Consistent with previous findings, we observed that hypermethylated CpG sites are enriched in regions with repressed polycomb signatures and CpG islands, while hypomethylated CpG sites preferentially occurred in non-CpG islands and enhancers. To gain insights into the functional effects of age-related DNAm changes, we assessed the correlation between DNAm and local gene expression changes to identify age-related expression quantitative trait methylation (age-eQTMs). We identified several age-eQTMs present in multiple tissue-types, including in the CDKN2A, HENMT1, and VCWE regions.
Conclusion
Overall, our findings will aid future efforts to develop biomarkers of aging and understand mechanisms of aging in diverse human tissue types.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13072-024-00546-6.
Background
Aging is characterized by the gradual decline of physiological function over time. The human aging process is a major risk factor for cancer, diabetes, cardiovascular diseases and neurodegenerative disorders [1]. Therefore, there is growing interest to understand the cellular and molecular mechanisms underlying aging. In recent years, studies have described several hallmarks of aging including cellular senescence [2], telomere attrition [3, 4], gene expression changes [5, 6], dysregulation of nutrient sensing [7] and epigenetic modifications [8]. Specifically, DNA methylation (DNAm) is proposed to play an important role in the aging process, with DNAm-based biomarkers commonly used as predictors of age and age-related health outcomes [9, 10].
Previous epigenome-wide association studies (EWAS) in whole blood [11–20], saliva [11], adipose [21], brain [22, 23], and breast [24] have shown that age is associated with DNAm across thousands of cytosine-guanine (CpG) dinucleotides in the human genome. Specifically, DNAm at the promoter of ELOVL2, an enzyme involved in fatty acid elongation, has been found to be significantly associated with chronological age across different populations, tissue types, and studies [11, 12, 17, 21, 25]. While, this is an example of an age-associated change that occurs in many tissue types, studies have also identified age-associated CpG sites that are differentially methylated only in specific tissue types [25–27]. Nonetheless, the patterns of enrichment of age-associated DNAm changes within specific genomic features, such as CpG islands (CGI) and chromatin states, are found to be similar across most tissue types [25, 28]. The effects of aging on gene expression have also been well-characterized, with discovery of age-altered genes involved in inflammation, metabolism, cancer, and mitochondrial activity [29, 30]. Therefore, in addition to characterizing the tissue-specific and non-specific variation in age-associated DNAm patterns, understanding the functional consequences of epigenetic aging, via regulation of gene expression, remains an area of interest. Though, studies are often limited due to the lack of both DNAm and gene expression data collected from the same tissue samples.
In this study, we assessed the association between age and genome-wide measures of DNAm for 961 tissue samples representing 9 tissue types (lung, colon, ovary, prostate, whole blood, testis, kidney, muscle and breast) from the Genotype-Tissue Expression (GTEx) project. Additionally, to gain further insights into the consequences of epigenetic aging, we investigated the correlation between age-associated DNAm and local gene expression changes to identify age-assocciated expression quantitative trait methylathion (age-eQTMs).
Materials and methods
The genotype-tissue expression project (GTEx)
The GTEx project is a publicly available biobank that has collected multiple unique tissue types (up to 54 types) from ~ 960 post-mortem donors. Medical history of donors and characteristics of tissue samples were extensively reviewed, and any sample with suggestive findings (e.g., cancer) were excluded from the GTEx normal database [31]. The GTEx v8 database consists of RNA sequencing and genotyping data from 838 donors (17,382 samples from 52 tissue types) [32]. The database additionally provides metadata collected through questionnaires (e.g., sex, age, race/ethnicity) as well as measurements of ischemic time for all samples.
We collected DNAm measurements for 961 tissue samples representing 9 tissue types (colon, lung, ovary, prostate, skeletal muscle, kidney, whole blood, breast, and testis). Tissue types were selected based on several criteria including relevance to cancer (colon, lung, prostate, kidney, breast, testis), tissues with unique aging biology (breast, testis, muscle), and common use in epidiemiological studies (whole blood). With resources to collect DNAm for ~ 1000 samples, we selected larger number of samples for tissues types with larger public health interest (lung, colon, ovary) as well as to assess the effect of sample size on the power to detect DNAm quantitative trait loci (mQTLs), as previously reported [33].
DNA methylation measurement and quality control
DNA extraction from 1000 unique GTEx tissue samples was performed using the Qiagen Gentra Puregene method at GTEx Laboratory Data, Analysis and Coordinating Center (LDACC). The extracted DNA was shipped to the University of Chicago. The 1000 samples represent 424 GTEx donors and 9 tissue types. For each tissue type, all samples were obtained from distinct donors.
For the 1000 unique DNA samples, DNAm at > 850,000 CpG sites was measured using the Infinium MethylationEPIC array (Illumina, San Diego, CA, USA) at the University of Chicago. All DNA samples were prepared and analyzed following the manufacturer’s guidelines and protocols. For quality control, we excluded 3 samples with undetectable methylation values (detection P > 0.01) in ≥ 5% of CpG sites, 6 samples with mismatched sex, and 14 samples that did not clearly cluster with their tissue type. Using the measurements of 59 high-frequency SNPs in the EPIC array, we identified one sample that did not match the donor’s existing genotype data. 15 breast tissue obtained from men were excluded. Following quality control, there were 961 remaining samples used for analysis (representing 9 tissue types and 417 GTEx donors).
For quality control of CpG sites, we followed guidelines from Pidsley et al. [34]. CpG sites measured by probes with potential non-specific binding (43,254 sites), sites overlapping genetics variants or variants that overlap single-base extension sites for Type 1 probes (7708 sites), sites mapping to the sex chromosomes (16,037 sites), and poorly performing sites based on guidance from Illumina (167 sites) were excluded. We also excluded CpGs that had detection P > 0.01 in at least one sample (44,135 sites). A total of 754,119 CpGs passed QC, and were retained for analyses. Genomic positions for all CpGs (and for all SNP and gene expression analyses described below) are mapped to human reference genome build hg19/GRCh37.
GTEx gene expression data
Gene expression data, collected via RNA-sequencing, from GTEx v8 was obtained from the GTEx portal. The expression values for each gene was estimated as reads per kilobase of transcript per million mapped reads (RPKM) using RNA-SeQC. GTEx v8 dataset provides expression levels recorded as both read counts and transcripts per million (TPM) [35].
Read counts from these genes were normalized across samples using the Trimmed Mean of M-values (TMM) normalization method in edgeR to generate TMM-normalized TPM for each gene [36]. Following TMM normalization, genes were selected based on the expression threshold of > 0.1 TPM in at least 20% of samples and ≥ 6 reads in at least 20% of the samples. We restricted to the fully processed, filtered, and normalized autosomal genes from the GTEx v8 dataset which resulted in 26,095 genes expressed in lung (n = 546), 25,379 genes expressed in colon (n = 406), 25,026 genes expressed in breast (n = 459), 24,028 genes expressed in kidney (n = 90), 20,356 genes expressed in muscle (n = 803), 24,472 genes expressed in ovary (n = 180), 25,680 genes expressed in prostate (n = 245), 33,923 genes expressed in testis (n = 361), and 20,315 genes expressed in whole blood (n = 755).
Association of age with DNAm and gene expression
Beta values for each CpG was logit transformed in M-values prior to analyses using the formula: log2[beta/(1 − beta)]. The association between chronological age and DNAm at each CpG site was assessed using a linear model implemented by the R (R/4.2.1) package limma (3.54.2) [37]. Sex, BMI, race/ethnicity, ischemic time, batch/place, and surrogate variables (SVs) were included as covariates in our model. The R (R/4.2.1) sva (3.46.0) package [38] was used to generate the SVs for each tissue type. We included the age variable in the full model matrix but omitted the age variable from the null model matrix to prevent the effects of age from being captured by SVs. The resulting SVs were used to control unknown sources of variability (e.g., technical variation and cell type composition). As a rule, we adjusted for 10 SVs for tissue types with n > 100 and 5 SVs for tissue types with n < 100.
Similarly, association between age and expression for each gene was estimated using a linear model implemented in limma, adjusting for sex, BMI, race/ethnicity, ischemic time, and 10 SVs (created using expression data).
Enrichment and pathway analyses for age-associated CpG sites
We first selected age-associated CpG sites, false discovery rate (FDR) 0.05, found in more than one tissue type, without considering directionality. We then compared the percentage of clock CpGs from Horvath [39] (353 CpGs) and AltumAge [40] (20,318 CpGs) that are age-associated to the percentage of CpGs from our EWAS (754,119 CpGs) that we identified as age-associated in multiple tissue types (n > 1). Additionally, we computed Fisher’s exact P-values.
For each tissue type, we compared the distribution of age-associated CpG sites, FDR 0.05, assigned to CpG island, shore, shelf, and open sea (based on Illumina annotations) to the distribution in the entire Infinium MethylationEPIC array (754,119 CpG sites) using chi-square tests. We assessed enrichment of age-associated CpG sites (FDR 0.05) among chromatin segmentation features. CpG sites were assigned to chromatin segmentation features using reference data from the Roadmap Epigenomics project database [41]. Background CpGs in this analysis were all CpGs assayed in the Infinium MethylationEPIC array (754,119 CpG sites). We performed this analyses for GTEx tissue types that have a closely matched reference dataset in Roadmap Epigenomics (primary tissue colonic mucosa, primary tissue lung, primary culture vHMEC mammary epithelial, and primary tissue ovary). We used the R (R/4.2.1) package ‘oddsratio’ to calculate enrichment and fisher’s exact P values.
CpGs were assigned to genes (based on Illumina annotations), and genes were assigned to pathways and biologic processes using the KEGG pathways (~ 330 pathways) [42]. We conducted gene set enrichment analysis (GSEA) using the “gsameth” function in the R package (R/4.2.1) missMethyl [43] for all tissue types using age-associated CpG sites (FDR 0.05). This function accounts for the potential bias in GSEA due to the number of CpGs per gene by computing prior probabilities and evaluates enrichment using a hypergeometric test. Enriched gene sets were defined as those passing FDR 0.05.
Identification of age-related expression quantitative trait methylation (eQTMs)
For age-associated CpG sites, we investigated the association of DNAm with expression of nearby genes (with age-associated expression). Using the tool bedtools intersect, we assigned CpG-gene pairs if a CpG site overlapped a gene region ± 10 Kb [44]. 10 Kb flanking regions of genes were assigned using Galaxy.org’s get flanks tool [45]. Gene body annotations are based on GENCODE version 26 (https://www.gencodegenes.org/human/release_26.html). Note each CpG site could be assigned to multiple genes and each gene could have multiple CpG sites assigned to it. We then assessed association between DNAm and gene expression levels using R’s lm function, adjusting for sex, BMI, EPISCORE cell-type estimates (breast, colon, lung, and prostate), ischemic time, sample group, and ethnicity. EPISCORE is a methylation-based method to estimate cell-type composition [46]. We used the “wRPC” function, with the pan-tissue DNAm atlas as input for the reference dataset. The pan-tissue DNAm atlas includes reference data for breast, colon, lung, kidney, and prostate. Except kidney, which shows a lack of age-associated signals, the other four tissue types were analyzed to detect age-eQTMs. eQTMs (P < 0.05) where the CpG site (P < 10–3, FDR < 0.05) and expression of the associated gene (P < 10–3, FDR < 0.05) is highly associated with age were considered age-eQTMs. Note we have 149 lung samples, 86 colon samples, 50 prostate samples, 30 breast samples with both DNAm and expression data.
Results
Summary of GTEx tissue samples
We generated DNAm data for 961 unique methylomes from 417 donors, spanning 9 tissue types (Table 1). The sample sizes for the tissues ranged from 38 (breast) to 223 (lung and colon). The number of tissues samples collected per donor ranged from 1 to 6. The age distribution for each tissue type ranged from 20 to 70 years (Supplementary Fig. 1), with the mean age of sample donors being 53.68 years (12.67). For tissues that are not sex-specific, approximately 70% of samples collected were from male donors and 86.3% of donors self-reported as white.
Table 1.
Characteristics of GTEx tissue samples used for DNA methylation analysis
| Lung (n=223) | Colon (n = 223) | Ovary (n = 157) | Prostate (n = 119) | Whole blood (n = 54) | Testis (n = 50) | Kidney (n = 50) | Muscle (n = 47) | Breast (n = 38) | |
|---|---|---|---|---|---|---|---|---|---|
| Age (yrs) | 56.1 (11.3) | 55.2 (11.1) | 50.5 (13.6) | 54.3 (12.7) | 50.3 (12.9) | 54.2 (12.2) | 59.7 (8.3) | 57.1 (10.5) | 50.0 (11.9) |
| BMI (kg/m2) | 27.1 (3.9) | 27.6 (3.9) | 26.7 (4.2) | 27.1 (3.8) | 27.4 (4.2) | 27.2 (3.8) | 26.4 (3.7) | 26.8 (4.4) | 25.4 (3.9) |
| Sex | |||||||||
| Male | 155 (69.5) | 160 (71.7) | 0 (0) | 119 (100) | 45 (83.3) | 50 (100) | 39 (78) | 28 (59.6) | 0 (0) |
| Female | 68 (30.5) | 63 (28.3) | 157 (100) | 0 (0) | 9 (16.7) | 0 (0) | 11 (22) | 19 (40.4) | 38 (100) |
| Race | |||||||||
| White | 196 (87.9) | 190 (85.2) | 128 (81.5) | 108 (90.8) | 49 (90.7) | 47 (94) | 44 (88) | 41 (87.2) | 32 (84.2) |
| Afr. American | 22 (9.9) | 28 (12.6) | 26 (16.6) | 9 (7.6) | 5 (9.3) | 3 (6) | 6 (12) | 6 (12.8) | 6 (15.8) |
| Others | 5 (2.2) | 5 (2.2) | 3 (1.9) | 2 (1.7) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Metrics in table are formatted as follows [mean (std) or n (%)]. Report of the donor’s race was either reported by the donor, the donor’s family/next of kin, or abstracted from medical records
Identification of age-associated differentially methylated CpG sites
We performed an EWAS examining the relationship between age and genome-wide DNAm levels, adjusting for sex, race/ethnicity, SVs, and other covariates. Our analysis identified age-associated differentially methylated CpG sites passing Bonferroni threshold and FDR 0.01 (Supplementary Table 1) and 0.05 (Table 2) in all tissues except skeletal muscle. The tissue type with the highest number of age-associated CpG sites identified was ovary (n = 157), with 134,986 CpG sites passing FDR 0.05 (P < 0.009). The signals identified in ovary account for over half of the tissue-specific hypermethylated sites (60.2%) and nearly half of the tissue-specific hypomethylated sites (39.5%). The lowest number of age-associated CpG sites was found for kidney (n = 50), with 41 CpG sites passing FDR 0.05 (P < 2.7e−6) (Supplementary Data Files 1–8; Supplementary Fig. 2). For subsequent analyses, we focused on CpG sites passing FDR 0.05. The alternative Bonferroni method is conservative and will miss many true signals that will be useful for pathway analyses. FDR provides better sensitivity to detect true associations while allowing only a small percentage of detected associations to be false positives.
Table 2.
Number of age-associated CpG sites identified in each tissue type passing false-discovery rate (FDR) 0.05
| Tissue | Age-associated CpG sites (P-value threshold) | ||
|---|---|---|---|
| Hypermethylated | Hypomethylated | Total | |
| Lung (n = 223) | 29,432 (0.003) | 21,422 (0.003) | 50,084 (0.003) |
| Colon (n = 223) | 40,029 (0.004) | 17,140 (0.004) | 57,169 (0.004) |
| Ovary (n = 157) | 96,210 (0.009) | 38,776 (0.009) | 134,986 (0.009) |
| Prostate (n = 119) | 15,624 (0.002) | 15,220 (0.002) | 30,844 (0.002) |
| Whole blood (n = 54) | 109 (3.5e-5) | 438 (3.6e-5) | 547 (3.5e−5) |
| Testis (n = 50) | 1,278 (3.4e-4) | 3,786 (3.4e-4) | 5,064 (3.4e−4) |
| Kidney (n = 50) | 41 (2.7e-6) | 0 (NA) | 41 (2.7e−6) |
| Muscle (n = 47) | 0 (NA) | 0 (NA) | 0 (NA) |
| Breast (n = 38) | 24,766 (0.003) | 13,220 (0.003) | 37,986 (0.003) |
Total counts and counts stratified by hypermethylated and hypomethylated age-associated CpG sites are shown
The age-associated CpGs identified were classified as either showing increased methylation levels with age (hypermethylated CpGs sites) or showing decreased methylation levels with age (hypomethylated CpGs). In most tissue types, we observed that hypermethylated CpG sites are more abundant than hypomethylated CpG sites (Fig. 1A). We further examined the distribution of effect size estimates, calculated as log2 fold-change, for age-associated CpG sites passing FDR 0.05 (Fig. 1B). For both hypermethylated and hypomethylated CpG sites, tissues with larger sample sizes tend to exhibit lower median effect size estimates, except for breast (n = 38), where hypermethylated CpGs have a lower median effect size estimate than estimates in kidney (n = 50), testis (n = 50), and whole blood (n = 54). Nevertheless, across all tissue types, those with smaller sample sizes (i.e., n⪅50) tend to have limited statistical power to detect weaker associations relative to tissues with larger sample sizes (n > 100) (Fig. 1B, C).
Fig. 1.

Identification of age-associated CpG sites. A Number of age-associated CpG sites identified in each tissue type passing false discovery rate (FDR) 0.05, stratified by hypermethylated and hypomethylated status. B Boxplot showing the effect size (log2 fold-change in DNA methylation per year increase in age) distribution of hypermethylated age-associated CpGs sites in each tissue type. C Boxplot showing the effect size (absolute value of log2 fold-change in DNA methylation per year increase in age) distribution of hypomethylated age-associated CpGs sites in each tissue type
Next, we investigated the tissue-specificity and non-specificity of differentially methylated CpG sites. We observed some evidence of overlap between pairs of tissues (Fig. 2A, B), with a few age-associated CpG sites shared across most tissues (n > 5), such as ELOVL2 (cg16867657 and cg21572722 hypermethylated in 8 tissue types) and ZNF549 (cg06458239 hypermethylated in 7 tissue types) (Supplementary Table 2). Due to the large differences in sample sizes across tissues, it is difficult to rigorously assess the extent of overlap between all tissue pairs. Though, for tissues with relatively large sample sizes (n > 100) and similar sample sizes (lung, colon, ovary, and prostate), we observed that 46.8% of hypermethylated CpG sites in lung (FDR 0.05) are also hypermethylated in colon (FDR 0.05), while 34% of hypermethylated CpG sites (FDR 0.05) in colon are also hypermethylated in lung (FDR 0.05). Nearly half of the hypermethylated CpG sites in prostate (FDR 0.05) are also hypermethylated in lung (49.2%), colon (49.4%) as well as in ovary (32.8%) (FDR 0.05). Comparatively, the proportion of hypomethylated CpG sites that are shared between these pairs of tissues is lower than hypermethylated CpG sites. Nonetheless, majority of the hypermethylated (~ 80.3%) and hypomethylated (~ 82.4%) CpG sites are present in a single tissue suggesting clear differences in DNAm aging across tissue types (Fig. 2C, D; Supplementary Table 3).
Fig. 2.

Pairwise overlap of age-associated CpG sites between tissues, including (A) hypermethylated and (B) hypomethylated CpG sites. Overlapping CpG sites passed false discovery rate (FDR) 0.05 in both tissue types. The intensity of the color (red shade for hypermethylated and blue shade for hypomethylated) reflects the number of CpG sites that overlap. Number of (C) hypermethylated (D) hypomethylated CpG sites passing FDR 0.05 against the number of tissues the CpG was identified in
Additionally, given that DNAm-based biomarkers are commonly used as predictors of age, we reasoned that CpGs used in pan-tissue aging clocks should be among the age-associated CpGs found in multiple tissue types (n > 1) from our analysis. Focusing on two well-known pan-tissue aging clocks, Horvath [39] (353 CpGs) and AltumAge [40] (20,318 CpGs), we tested enrichment of clock CpGs within our set of multi-tissue age-associated CpGs (60,665 CpGs). 1,764 AltumAge clock CpGs and 98 Horvath clock CpGs are amongst our set of multi-tissue age-associated CpGs. The percentage of clock CpGs from Horvath (27.7%; P < 2.2e−16) and AltumAge (8.6%; P = 8.098e−06) that are age-associated is larger than the percentage we identified in our EWAS as multi-age associated (8.04%). These results highlight that our analysis captures previously characterized DNAm age predictors.
Enrichment of CpG sites within genomic features
We examined the distribution of age-associated CpG sites within genomic features, observing distinct patterns for hypermethylated and hypomethylated CpG sites. We found that hypermethylated CpG sites were enriched in CGIs (P < 0.0001) in 5 tissue types (Fig. 3A). However, this pattern of enrichment was not observed in the sex-specific tissues, ovary, testis, and breast. For hypomethylated CpG sites, we observed enrichment in non-CGIs (P < 0.0001), specifically “open sea” for all tissues except testis, where we observed enrichment in CGIs (P < 0.0001) (Fig. 3B). Differences in sample sizes and the underlying biology of the tissue could contribute to the observed differences in patterns of enrichment.
Fig. 3.

Locational distribution of age-associated CpGs sites (FDR 0.05) in relation to CpG islands (CGI) stratified by (A) hypermethylated and (B) hypomethylated CpG sites. Colors represent location of CpGs with respect to CGIs. Background CpGs are defined as all CpGs assayed in the Infinium MethylationEPIC array included in our analyses (754,119 CpGs)
To further characterize the genomic context of age-associated CpG sites, we examined enrichment within chromatin states. We performed this analysis for breast, colon, lung, and ovary only, because for these tissue types, we were able to identify a closely matched reference tissue dataset from the Roadmap Epigenomics project database [41]. We observed distinct patterns of enrichment for hypermethylated and hypomethylated CpG sites. Consistent with prior studies, hypermethylated CpGs sites were enriched in the polycomb repressive complex epigenomic signature (enrichment in repressed polycomb present in colon and lung), while hypomethylated CpG sites were enriched in active regions, such as enhancers (enrichment in enhancers and genic enhancers present in breast, colon, lung, and ovary) and active transcription (enrichment in flanking transcription and flanking active TSS present in ovary) (Fig. 4A, B) [25]. We observed weaker enrichment among hypomethylated CpG sites compared to hypermethylated CpG sites in all tissue types. Colon and lung tissue demonstrated more similar patterns of enrichment compared to ovary and breast.
Fig. 4.

Enrichment of age-associated CpG sites (FDR < 0.05) among chromatin segmentation features stratified by (A) hypermethylated (B) hypomethylated CpG sites. Enrichment expressed as odds ratio. Background CpGs are defined as all CpGs assayed in the Infinium MethylationEPIC array included in our analyses (754,119 CpGs). Fisher’s exact P value * < 0.05, ** < 0.01, *** < 0.001. Active chromatin states: active transcription start site (TssA), flanking active TSS (TssAFlnk), transcription at gene 5′ and 3′ showing both promoter and enhancer (TxFlnk), strong transcription (Tx), weak transcription (TxWk), genic enhancers (EnhG), enhancers (Enh), zinc finger protein genes and repeats (ZNF/Rpts ZNF). Inactive chromatin states: heterochromatin (Het), bivalent/poised TSS (TssBiv), flanking bivalent TSS/Enh (BivFlnk), bivalent enhancer (EnhBiv), repressed polycomb (ReprPC), weak repressed polycomb (ReprPCWk), quiescent/low (Quies)
Pathway enrichment of age-associated CpG sites
To assess pathways related to age-related epigenetic changes we first assigned CpGs to genes based on Illumina annotations (544,631 CpGs assigned to 25,604 UCSC RefSeq annotations). We then conducted a pathway enrichment analysis of age-associated CpG sites (FDR 0.05) assigned to annotated genes from all 8 tissue types. We observed significantly enriched biological pathways (FDR < 0.05) using the KEGG database for 5 tissue types (colon, lung, prostate, ovary, and breast) (Fig. 5). Seven pathways showed evidence of enrichment in all five tissue types (FDR 0.05): arrhythmogenic right ventricular cardiomyopathy, axon guidance, focal adhesion, hippo signaling pathway, proteoglycans in cancer, Rap1 signaling pathway, and Wnt signaling pathway.
Fig. 5.
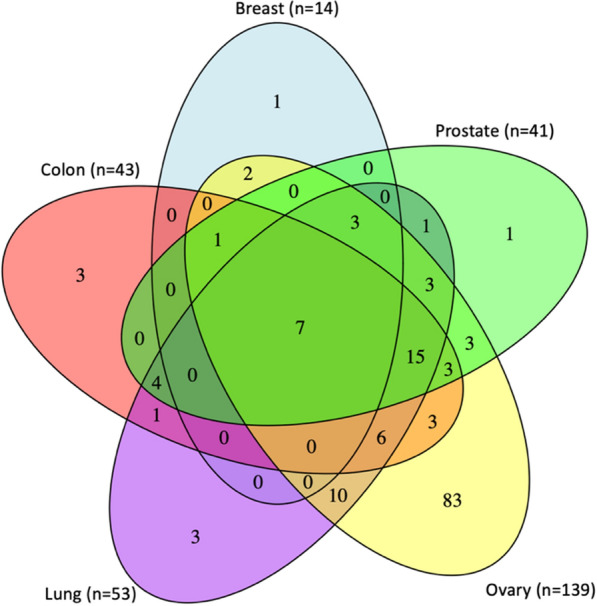
Pathway analysis of age-associated CpG sites detected in breast, prostate, ovary, lung, and colon. Venn diagram showing the overlap of enriched pathway between tissue types. ‘n’ corresponds to the number of pathways identified at FDR < 0.05
Several additional pathways, such as breast cancer, circadian entrainment, gastric cancer, dilated cardiomyopathy, and the MAPK signaling pathway, showed evidence of enrichment in at least two tissue types. While the breast cancer pathway was significantly enriched (FDR < 0.05) in all other tissue types, it was only nominally significant in breast tissue (unadjusted P < 0.05). Similarly, although gastric cancer was enriched in colon tissue (FDR < 0.05), it showed stronger enrichment in lung and ovary tissues. Nevertheless, cell signaling pathways were consistently enriched across all five tissue types, aligning with their role in cellular function throughout the human body. Many of these signaling pathways, including Wnt and MAPK signaling, are associated with cancer development [47–49]. Ovary, with the highest number of enriched KEGG pathways, showed evidence of enrichment in pathways associated with hormone production and secretion, such as estrogen, thyroid, prolactin, aldosterone, and GnRh. Colon, prostate, and lung displayed similar numbers of enriched pathways, whereas breast showed the fewest enriched pathways (Fig. 5; Supplementary Tables 4–8).
Functional characterization of age-associated CpG sites
We investigated the association between age-associated CpG sites and expression of nearby genes using linear regression. Age-eQTMs were defined as CpG sites associated with gene expression (P < 0.05), and where both the CpG site (P < 10–3, FDR < 0.05) and expression of the associated gene (P < 10–3, FDR < 0.05) were highly associated with age. We identified several examples of age-associated eQTMs that were unique to specific tissue types and those that are found in multiple tissue types (Supplementary Table 9). For example, we identified the CDKN2A region as an age-eQTM locus that is shared across 3 tissue types (colon, lung, and prostate). Four age-associated CpGs in that region were associated with CDKN2A expression (which was also associated with age) (Fig. 6). We found that CDKN2A expression is positively associated with age in all 3 tissue types (FDR < 0.05). We also observed greater variability in gene expression with increasing age (Fig. 6). Similarly, hypermethylation at the associated CpG sites (FDR < 0.05), cg1811914, cg26349275, cg08686553, and cg2422208, is observed with increasing age in all three tissue types. Examples of age-eQTMs found in a single tissue include CpGs sites annotated to ZNF518B (lung), HENMT1 (colon), ZNF154 (prostate), and HAPLN3 (breast) (Supplementary Figs. 3–6, respectively). We observed a consistent relationship for these age-eQTMs, whereby there is a negative correlation between DNAm and gene expression. Additionally, for these CpG-gene pairs, we observed that the CpG sites (hypermethylated with age) are clustered at CGIs near the gene start, consistent with the downregulation of these genes with increasing age.
Fig. 6.

Association of age with DNA methylation and expression of CDKN2A in three tissue types. Pearson’s correlation coefficient (R) and Pearson’s correlation P value reported for expression scatterplots (right). Red dot indicates hypermethylation at CpG site with increasing age. The vertical, shaded, red rectangles denote regions of age-associated CpG sites found in all three tissue types. The y-axis represents the -log10 of the P value (left)
Discussion
In this study, we collected DNAm data for 961 tissue samples, representing 9 human tissue types (lung, colon, ovary, prostate, testis, kidney, muscle, whole blood and breast) from the GTEx project. We tested the association of age with genome-wide measures of DNAm, identifying differentially methylated CpG sites (FDR 0.05) in 8 tissue types (all except skeletal muscle). We identified age-associated CpG sites that were tissue-specific (Supplementary Table 3) as well as sites shared across multiple tissue types (e.g., ELOVL2 and CDKN2A). While majority of the CpG sites identified appear tissue-specific, the patterns of enrichment within genomic features, such as CGIs and chromatin states, is largely shared across tissue types. We performed pathway enrichment analysis to identify pathways related to epigenetic aging, and our results showed clear enrichment of aging relevant pathways, such as cancer and cell-signaling. To gain insights regarding the functional consequences of age-related DNAm changes, we assessed the correlation of age-associated sites with local gene expression. We identified several regions showing correlation in multiple tissue types (> 1 tissue), including age-eQTMs in the CDKN2A region, the HENMT1 region, and the VCWE region.
For tissue types with larger sample sizes (n > 100), including lung, colon, ovary, and prostate, increased power enabled detection of more age-associated CpG sites compared to tissue types with smaller sample sizes (n ~ 50), except in the case of breast tissue (n = 38), where we observed a relatively large number of age-associated CpG sites. The abundance of associations observed for breast tissue could be explained by the underlying biology of this tissue type. It is well established that breast development and risk of breast-related diseases (e.g., breast cancer) is tightly linked to age, with breast tissue undergoing various biological changes, including regression of terminal duct lobular units, increased breast density and fat pads, hormonal fluctuations (perimenopause, menopause), breast milk composition, and cellular transformation with age [24, 50, 51]. However, as expected, for the tissue types with smaller sample sizes, we were only able to capture age-associated CpG sites with relatively large effect sizes. The differences in sample sizes limited our ability to characterize the extent of shared age-associated effects between tissue types. However, consistent with prior studies, we did identify that hypermethylation of CpG sites (cg16867657 and cg21572722) in the ELOVL2 region is shared across all 8 tissue types.
Our results show that patterns of enrichment of hypermethylated and hypomethylated age-associated CpG sites are generally consistent across tissue types. For most tissue types, we observed that, for CpGs measured in the EPIC array, hypermethylation with increasing age is more common than hypomethylation. Additionally, hypermethylated CpG sites tend to show enrichment in CGIs while hypomethylated CpG sites tend to show enrichment in non-CGIs (“open sea”). Prior studies have shown that hypermethylation in promoter regions overlapping CGIs is often observed in aging and aging-related diseases (e.g., cancer). This hypermethylation is associated with gene silencing, and is proposed to interfere with proper regulation of genes [52]. The EPIC array has extensive coverage of CGIs, genic, and enhancer regions, which likely contributes to the greater number of hypermethylated age-associated CpG sites identified in our study. Interestingly, the patterns of enrichment observed in testicular tissue show a striking difference to patterns observed in the other 7 tissues, which can be attributed to the unique aging biology and gene expression patterns of the testis [53, 54].
Next, we assessed enrichment of age-associated CpG sites in chromatin segmentation features. Consistent with multiple prior studies, we observed differing patterns of enrichment for hypermethylated sites compared to hypomethylated sites across tissues. Hypermethylated CpGs (colon and lung) showed enrichment in “repressed” polycomb and “poised” transcription states (bivalent domains including bivalent enhancers, flanking bivalent TSS/enhancers, and bivalent TSS). Hypomethylated CpGs (colon, lung, ovary, and breast) showed enrichment in enhancers and genic enhancers [13, 16, 25, 28, 55]. The polycomb group proteins (PrG) are chromatin-associated, multimeric proteins that regulate a diverse set of genes. PRC2 (one of the enzymatic forms of PrG) is a histone methyltransferase, which targets developmental genes and is involved in the silencing of bivalent genes, marked by repressive (H3K27me3 and H3K9me3) and activating (H3K4me1, H3K4me2, H3K4me3) histone modifications [56, 57]. As such, it is plausible that PRC2 contributes to age-related transcriptomic and epigenomic changes. Future studies using single-cell chromatin immunoprecipitation sequencing (ChIP-seq) and transposase-accessible chromatin with sequencing (ATAC-seq) data will provide tissue- and cell-type specific defined chromatin states, further improving our understanding of the distribution of age-associated in the epigenome.
Pathway enrichment analysis of age-associated CpG sites assigned to annotated genes showed clear enrichment of cancer-related and cell-signaling pathways in multiple tissue types. Hypermethylation at bivalent domains is also a signature found in various cancers [55, 58–60]. Aging is one of the major risk factors for cancer development [61–63]. Alterations in the epigenomic landscape with increasing age, can lead to chromatin conformations primed with oncogenic potential (e.g., aberrant silencing of tumor suppressor genes and activation of pro-oncogenes). This increased plasticity of the chromatin can additionally disrupt other processes, including DNA repair mechanisms and telomere maintenance, which taken together contribute to the development of cancer [64].
To provide functional insights into the consequences of epigenetic aging, we assessed the correlation between age-associated DNAm and local gene expression changes using a subset of samples with both gene expression and DNAm data. Our analysis identified several new multi-tissue signals, most interestingly, age-eQTMs in the CDKN2A region (colon, prostate, and lung). CDKN2A, cyclin dependent kinase inhibitor 2A, encodes the cell-cycle inhibitor p16. This gene has been extensively studied in the context of cancer and cellular senescence, with cellular damage and stress leading to activation of CDKN2A. Exponential increase in expression of CDKN2A has been observed with aging [65–67]. In our work, we find an increase in expression of CDKN2A and hypermethylation of the associated-CpG sites (located close to the gene and within the gene body) with increasing age. Although these results highlight a link between age-related DNAm and age-related gene expression changes, it does not address the mechanistic or causal relationships driving these changes.
While our study identified and characterized the effects of age on DNAm patterns, the DNAm measures used were obtained using the EPIC array, which covers a small proportion (~ 2%) of CpG sites in the human genome. Additionally, we were limited by small sample sizes (n ~ 50) for some tissues and thus, were unable to detect many associations in these tissue types and make conclusions on the extent of shared age effects between tissue types. Another limitation of this study is the skewed age distribution, with generally lower number of samples from younger donors compared to older ones. This potentially limited our ability to detect age effects on DNAm that are more prominent at younger ages. For some of our analyses, namely enrichment among chromatin segmentation features and identification of age-eQTMs, we were only able to perform them in a select number of tissues due the lack of matching reference datasets.
Conclusion
Our work highlights the importance of multi-tissue analyses to gain insights into effects of age on the epigenome. Future studies should use whole-genome data on DNAm, larger sample sizes of diverse tissue/cell types, and additional epigenetic features to validate and expand our findings to better understand the effects of aging in the human genome.
Supplementary Information
Supplementary Table 1. Number of age-associated CpG sites identified in each tissue type passing Bonferroni threshold and FDR 0.01.
Supplementary Table 2. Age-related CpGs with associations at FDR <0.05 for hypermethylated CpGs (n > 6) and hypomethylated CpGs (n > 5).
Supplementary Table 3. Age-related CpGs with associations at FDR <0.05 in a single tissue.
Supplementary Table 4. Top KEGG gene sets detected in the pathway analysis of age-associated CpGs in breast tissue.
Supplementary Table 5. Top KEGG gene sets detected in the pathway analysis of age-associated CpGs in colon tissue.
Supplementary Table 6. Top KEGG gene sets detected in the pathway analysis of age-associated CpGs in lung tissue.
Supplementary Table 7. Top KEGG gene sets detected in the pathway analysis of age-associated CpGs in ovary tissue.
Supplementary Table 8. Top KEGG gene sets detected in the pathway analysis of age-associated CpGs in prostate tissue.
Supplementary Table 9. Age-related expression quantitative trait methylation at P < 0.05 with correlation in multiple tissue types (n>1).
Acknowledgements
We thank the donors and their families for their generous gifts of biospecimens to the GTEx research project; the Genomics Platform at the Broad Institute for data generation; F. Aguet, J. Nedzel and K. Ardlie for sample delivery logistics and data release management. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author contributions
NJ performed analyses, interpreted the data, and wrote the main manuscript text. JLL contributed to data analysis, manuscript writing, and interpreting results. LIT contributed to manuscript writing, data analysis, and interpreting results. MGK and FJ generated DNA methylation data. KD and MO performed data processing and quality control. LSC advised on statistical analyses. BLP conceived the project and contributed to writing/editing and data interpretation.
Funding
This work was supported by grants U01 HG007601 (to B.L.P.), R35ES028379 (to B.L.P.), R01 GM108711 (to L.S.C) and U24 CA210993-SUB (to L.S.C) and was completed in part with computational resources provided by the Center for Research Informatics at the University of Chicago. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS.
Availability of data and materials
DNAm normalized data is available at GEO (GSE213478). All GTEx protected data are available via dbGaP (phs000424.v9); access to the DNAm raw data is provided through the AnVIL platform (https://anvil.terra.bio/#workspaces/anvil-datastorage/AnVIL_GTEx_V9_hg38). https://gtexportal.org/home/datasets. R package “oddsratio” is available through https://cran.r-project.org/web/packages/epitools. Scripts to perform analyses are located at https://github.com/niyati1211/AgingEWAS.
Declarations
Ethics approval and consent to participate
While deceased individuals do not require consent for research, GTEx consent is described in detail in (68).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–217. 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hudgins AD, Tazearslan C, Tare A, Zhu Y, Huffman D, Suh Y. Age- and tissue-specific expression of senescence biomarkers in mice. Front Genet. 2018. 10.3389/fgene.2018.00059. 10.3389/fgene.2018.00059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345(6274):458–60. 10.1038/345458a0 [DOI] [PubMed] [Google Scholar]
- 4.Heidinger BJ, Blount JD, Boner W, Griffiths K, Metcalfe NB, Monaghan P. Telomere length in early life predicts lifespan. Proc Natl Acad Sci. 2012;109(5):1743–8. 10.1073/pnas.1113306109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peters MJ, Joehanes R, Pilling LC, Schurmann C, Conneely KN, Powell J, et al. The transcriptional landscape of age in human peripheral blood. Nat Commun. 2015;6(1):8570. 10.1038/ncomms9570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Magalhães JP, Curado J, Church GM. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. 2009;25(7):875–81. 10.1093/bioinformatics/btp073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nogueiras R, Habegger KM, Chaudhary N, Finan B, Banks AS, Dietrich MO, et al. Sirtuin 1 and Sirtuin 3: physiological modulators of metabolism. Physiol Rev. 2012;92(3):1479–514. 10.1152/physrev.00022.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Talens RP, Christensen K, Putter H, Willemsen G, Christiansen L, Kremer D, et al. Epigenetic variation during the adult lifespan: cross-sectional and longitudinal data on monozygotic twin pairs. Aging Cell. 2012;11(4):694–703. 10.1111/j.1474-9726.2012.00835.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, Zhang J, et al. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010;20(3):332–40. 10.1101/gr.096826.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salameh Y, Bejaoui Y, El Hajj N. DNA methylation biomarkers in aging and age-related diseases. Front Genet. 2020. 10.3389/fgene.2020.00171. 10.3389/fgene.2020.00171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gopalan S, Carja O, Fagny M, Patin E, Myrick JW, McEwen LM, et al. Trends in DNA methylation with age replicate across diverse human populations. Genetics. 2017;206(3):1659–74. 10.1534/genetics.116.195594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johansson Å, Enroth S, Gyllensten U. Continuous aging of the human DNA methylome throughout the human lifespan. PLoS ONE. 2013;8(6): e67378. 10.1371/journal.pone.0067378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, et al. Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci USA. 2012;109(26):10522–7. 10.1073/pnas.1120658109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teschendorff AE, West J, Beck S. Age-associated epigenetic drift: implications, and a case of epigenetic thrift? Hum Mol Genet. 2013;22(R1):R7-15. 10.1093/hmg/ddt375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2):359–67. 10.1016/j.molcel.2012.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan T, Jiao Y, de Jong S, Ophoff RA, Beck S, Teschendorff AE. An integrative multi-scale analysis of the dynamic DNA methylation landscape in aging. PLOS Genet. 2015;11(2): e1004996. 10.1371/journal.pgen.1004996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zaghlool SB, Al-Shafai M, Al Muftah WA, Kumar P, Falchi M, Suhre K. Association of DNA methylation with age, gender, and smoking in an Arab population. Clin Epigenetics. 2015;7(1):6. 10.1186/s13148-014-0040-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bollati V, Schwartz J, Wright R, Litonjua A, Tarantini L, Suh H, et al. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev. 2009;130(4):234–9. 10.1016/j.mad.2008.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boks MP, Derks EM, Weisenberger DJ, Strengman E, Janson E, Sommer IE, et al. The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PLoS ONE. 2009;4(8): e6767. 10.1371/journal.pone.0006767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jansen RJ, Tong L, Argos M, Jasmine F, Rakibuz-Zaman M, Sarwar G, et al. The effect of age on DNA methylation in whole blood among Bangladeshi men and women. BMC Genomics. 2019;10(20):704. 10.1186/s12864-019-6039-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rönn T, Volkov P, Gillberg L, Kokosar M, Perfilyev A, Jacobsen AL, et al. Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum Mol Genet. 2015;24(13):3792–813. [DOI] [PubMed] [Google Scholar]
- 22.Hernandez DG, Nalls MA, Gibbs JR, Arepalli S, van der Brug M, Chong S, et al. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum Mol Genet. 2011;20(6):1164–72. 10.1093/hmg/ddq561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J, Yu L, Gaiteri C, Srivastava GP, Chibnik LB, Leurgans SE, et al. Association of DNA methylation in the brain with age in older persons is confounded by common neuropathologies. Int J Biochem Cell Biol. 2015;1(67):58–64. 10.1016/j.biocel.2015.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson KC, Koestler DC, Cheng C, Christensen BC. Age-related DNA methylation in normal breast tissue and its relationship with invasive breast tumor methylation. Epigenetics. 2014;9(2):268–75. 10.4161/epi.27015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slieker RC, Relton CL, Gaunt TR, Slagboom PE, Heijmans BT. Age-related DNA methylation changes are tissue-specific with ELOVL2 promoter methylation as exception. Epigenetics Chromatin. 2018;11(1):25. 10.1186/s13072-018-0191-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5(8): e1000602. 10.1371/journal.pgen.1000602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Day K, Waite LL, Thalacker-Mercer A, West A, Bamman MM, Brooks JD, et al. Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol. 2013;14(9):R102. 10.1186/gb-2013-14-9-r102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dozmorov MG. Polycomb repressive complex 2 epigenomic signature defines age-associated hypermethylation and gene expression changes. Epigenetics. 2015;10(6):484–95. 10.1080/15592294.2015.1040619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anisimova AS, Meerson MB, Gerashchenko MV, Kulakovskiy IV, Dmitriev SE, Gladyshev VN. Multifaceted deregulation of gene expression and protein synthesis with age. Proc Natl Acad Sci. 2020;117(27):15581–90. 10.1073/pnas.2001788117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Glass D, Viñuela A, Davies MN, Ramasamy A, Parts L, Knowles D, et al. Gene expression changes with age in skin, adipose tissue, blood and brain. Genome Biol. 2013;14(7):R75. 10.1186/gb-2013-14-7-r75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45(6):580–5. [DOI] [PMC free article] [PubMed]
- 32.The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369(6509):1318–30. [DOI] [PMC free article] [PubMed]
- 33.Oliva M, Demanelis K, Lu Y, Chernoff M, Jasmine F, Ahsan H, et al. DNA methylation QTL mapping across diverse human tissues provides molecular links between genetic variation and complex traits. Nat Genet. 2022;55(1):112–22. 10.1038/s41588-022-01248-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pidsley R, Zotenko E, Peters TJ, Lawrence MG, Risbridger GP, Molloy P, et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17(1):1–17. 10.1186/s13059-016-1066-1. 10.1186/s13059-016-1066-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Consortium Gte. Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204–13. 10.1038/nature24277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11(3):R25. 10.1186/gb-2010-11-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7): e47. 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882–3. 10.1093/bioinformatics/bts034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):3156. 10.1186/gb-2013-14-10-r115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Lima Camillo LP, Lapierre LR, Singh R. A pan-tissue DNA-methylation epigenetic clock based on deep learning. Npj Aging. 2022;8(1):1–15.35927252 10.1038/s41514-022-00085-y [DOI] [Google Scholar]
- 41.Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317–30. 10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353–61. 10.1093/nar/gkw1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phipson B, Maksimovic J, Oshlack A. missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics. 2016;32(2):286–8. 10.1093/bioinformatics/btv560 [DOI] [PubMed] [Google Scholar]
- 44.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.The Galaxy Community. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 2022;50(W1):W345–51. 10.1093/nar/gkac247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu T, Liu J, Beck S, Pan S, Capper D, Lechner M, et al. A pan-tissue DNA methylation atlas enables in silico decomposition of human tissue methylomes at cell-type resolution. Nat Methods. 2022;19(3):296–306. 10.1038/s41592-022-01412-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36(11):1461–73. 10.1038/onc.2016.304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuan J, Dong X, Yap J, Hu J. The MAPK and AMPK signalings: interplay and implication in targeted cancer therapy. J Hematol OncolJ Hematol Oncol. 2020;13(1):113. 10.1186/s13045-020-00949-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Groenewald W, Lund AH, Gay DM. The role of WNT pathway mutations in cancer development and an overview of therapeutic options. Cells. 2023;12(7):990. 10.3390/cells12070990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin J, Ye S, Ke H, Lin L, Wu X, Guo M, et al. Changes in the mammary gland during aging and its links with breast diseases. Acta Biochim Biophys Sin. 2023;55(6):1001–19. 10.3724/abbs.2023073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Denić M, Sunarić S, Genčić M, Živković J, Jovanović T, Kocić G, et al. Maternal age has more pronounced effect on breast milk retinol and β-carotene content than maternal dietary pattern. Nutrition. 2019;1(65):120–5. 10.1016/j.nut.2019.02.019 [DOI] [PubMed] [Google Scholar]
- 52.Johnson AA, Akman K, Calimport SRG, Wuttke D, Stolzing A, de Magalhães JP. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res. 2012;15(5):483–94. 10.1089/rej.2012.1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo J, Grow EJ, Mlcochova H, Maher GJ, Lindskog C, Nie X, et al. The adult human testis transcriptional cell atlas. Cell Res. 2018;28(12):1141–57. 10.1038/s41422-018-0099-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oakes CC, La Salle S, Smiraglia DJ, Robaire B, Trasler JM. A unique configuration of genome-wide DNA methylation patterns in the testis. Proc Natl Acad Sci. 2007;104(1):228–33. 10.1073/pnas.0607521104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20(4):434–9. 10.1101/gr.103101.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Piunti A, Shilatifard A. The roles of Polycomb repressive complexes in mammalian development and cancer. Nat Rev Mol Cell Biol. 2021;22(5):326–45. 10.1038/s41580-021-00341-1 [DOI] [PubMed] [Google Scholar]
- 57.Jadhav U, Manieri E, Nalapareddy K, Madha S, Chakrabarti S, Wucherpfennig K, et al. Replicational dilution of H3K27me3 in mammalian cells and the role of poised promoters. Mol Cell. 2020;78(1):141-151.e5. 10.1016/j.molcel.2020.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, et al. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39(2):157–8. 10.1038/ng1941 [DOI] [PubMed] [Google Scholar]
- 59.Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39(2):232–6. 10.1038/ng1950 [DOI] [PubMed] [Google Scholar]
- 60.Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and silencing in adult cancers. Nat Genet. 2007;39(2):237–42. 10.1038/ng1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Berben L, Floris G, Wildiers H, Hatse S. Cancer and aging: two tightly interconnected biological processes. Cancers. 2021;13(6):1400. 10.3390/cancers13061400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu M, Zhang H, Wang B, Zhang Y, Zheng X, Shao B, et al. Key signaling pathways in aging and potential interventions for healthy aging. Cells. 2021;10(3):660. 10.3390/cells10030660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tabibzadeh S. Signaling pathways and effectors of aging. Front Biosci Landmark Ed. 2021;26(1):50–96. 10.2741/4889 [DOI] [PubMed] [Google Scholar]
- 64.Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357(6348):eaal2380. 10.1126/science.aal2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baker DJ, Jin F, van Deursen JM. The yin and yang of the Cdkn2a locus in senescence and aging. Cell Cycle Georget Tex. 2008;7(18):2795–802. 10.4161/cc.7.18.6687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Florl AR, Franke KH, Niederacher D, Gerharz CD, Seifert HH, Schulz WA. DNA methylation and the mechanisms of CDKN2A inactivation in transitional cell carcinoma of the urinary bladder. Lab Invest. 2000;80(10):1513–22. 10.1038/labinvest.3780161 [DOI] [PubMed] [Google Scholar]
- 67.Mori H, Funahashi Y, Yoshino Y, Kumon H, Ozaki Y, Yamazaki K, et al. Blood CDKN2A gene expression in aging and neurodegenerative diseases. J Alzheimers Dis. 2021;82(4):1737–44. 10.3233/JAD-210483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carithers LJ, Ardlie K, Barcus M, Branton PA, Britton A, Buia SA, et al. A novel approach to high-quality postmortem tissue procurement: the GTEx project. Biopreservation Biobanking. 2015;13(5):311–9. 10.1089/bio.2015.0032 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Number of age-associated CpG sites identified in each tissue type passing Bonferroni threshold and FDR 0.01.
Supplementary Table 2. Age-related CpGs with associations at FDR <0.05 for hypermethylated CpGs (n > 6) and hypomethylated CpGs (n > 5).
Supplementary Table 3. Age-related CpGs with associations at FDR <0.05 in a single tissue.
Supplementary Table 4. Top KEGG gene sets detected in the pathway analysis of age-associated CpGs in breast tissue.
Supplementary Table 5. Top KEGG gene sets detected in the pathway analysis of age-associated CpGs in colon tissue.
Supplementary Table 6. Top KEGG gene sets detected in the pathway analysis of age-associated CpGs in lung tissue.
Supplementary Table 7. Top KEGG gene sets detected in the pathway analysis of age-associated CpGs in ovary tissue.
Supplementary Table 8. Top KEGG gene sets detected in the pathway analysis of age-associated CpGs in prostate tissue.
Supplementary Table 9. Age-related expression quantitative trait methylation at P < 0.05 with correlation in multiple tissue types (n>1).
Data Availability Statement
DNAm normalized data is available at GEO (GSE213478). All GTEx protected data are available via dbGaP (phs000424.v9); access to the DNAm raw data is provided through the AnVIL platform (https://anvil.terra.bio/#workspaces/anvil-datastorage/AnVIL_GTEx_V9_hg38). https://gtexportal.org/home/datasets. R package “oddsratio” is available through https://cran.r-project.org/web/packages/epitools. Scripts to perform analyses are located at https://github.com/niyati1211/AgingEWAS.