


Abstract
Tlr1 is a member of a family of ∼20–30 DNA elements that undergo developmentally regulated excision during formation of the macronucleus in the ciliated protozoan Tetrahymena. Analysis of sequence internal to the right boundary of Tlr1 revealed the presence of a 2 kb open reading frame (ORF) encoding a deduced protein with similarity to retrotransposon integrases. The ORFs of five unique clones were sequenced. The ORFs have 98% sequence conservation and align without frameshifts, although one has an additional trinucleotide at codon 561. Nucleotide changes among the five clones are highly non-random with respect to the position in the codon and 93% of the nucleotide changes among the five clones encode identical or similar amino acids, suggesting that the ORF has evolved under selective pressure to preserve a functional protein. Nineteen T/C transitions in T/CAA and T/CAG codons suggest selection has occurred in the context of the Tetrahymena genome, where TAA and TAG encode Gln. Similarities between the ORF and those encoding retrotransposon integrases suggest that the Tlr family of elements may encode a polynucleotide transferase. Possible roles for the protein in transposition of the elements within the micronuclear genome and/or their developmentally regulated excision from the macronucleus are discussed.
INTRODUCTION
DNA is remarkably mobile. Two major categories of DNA rearrangements are those resulting from the invasion and proliferation of independent transposable elements and those which occur as part of a developmental program. The relationship between the two kinds of events is not yet understood.
Transposons are invasive elements that integrate into host genomes. Class I elements, or retrotransposons, transpose via an RNA intermediate. Class II elements transpose via a DNA intermediate. Both types of elements encode polynucleotidyltransferases, which catalyze transposition, and an integrase or a transposase in the class I and class II elements, respectively (1). The active sites of these proteins are structurally similar in that they contain regularly spaced acidic amino acids, called DDE motifs, which chelate metal ions essential for enzymatic activity.
In a variety of phylogenetically diverse organisms an integral part of cellular development involves genomic reorganization associated with developmentally regulated DNA deletion. One example is the rearrangement of immunoglobulin genes and T cell receptors of vertebrate immune systems. Recent evidence has shown that the catalytic activity of RAG1, a recombinase that mediates the rearrangement of these genes, is dependent on three acidic residues, two of which may be involved in binding metal ions (2,3). This raises the possibility that there may be mechanistic similarities between the developmentally regulated DNA rearrangement and transposition.
Ciliated protozoa are particularly good model systems for the analysis of DNA rearrangement because thousands of programmed DNA deletions occur during development of the somatic genome. Ciliates are single cell eukaryotes that contain two structurally and functionally distinct nuclei that share the same genetic origin. The transcriptionally active somatic macronucleus sustains the cell during vegetative growth while the germline micronucleus remains transcriptionally silent. When ciliates reproduce sexually, the existing macronucleus is degraded and a new macronucleus develops from a mitotic copy of the fertilization micronucleus. Macronuclear development is associated with removal of thousands of internal eliminated sequences (IESs) (4,5).
The developmentally regulated deletion of IESs has been studied extensively in two classes of ciliates. In Oxytricha and Euplotes (formerly hypotrichs, these ciliates are currently placed in the class Spirotrichea; 6) ∼93% of the DNA is eliminated during the transition from the micronuclear to the macronuclear genome (5,7). Specific elements are removed precisely and with 100% efficiency.
A substantial fraction of the eliminated DNA in the spirotrichs belongs to families of thousands of repetitive transposon-like elements. Examples of these include the Tec elements (transposon-like, Euplotes crassus) in Euplotes and the TBEs (telomere-bearing elements) of Oxytricha. Tec elements are 5.3 kb in length and have ∼700 bp of terminal inverted repeat sequence (8). TBE elements are 4.1 kb in length and have 78 bp terminal inverted repeats which include 17 bp of telomere-like G4T4 repeats (9–11). The Tec and TBE IESs are thought to be class II transposable elements. They contain terminal inverted repeats; there are short direct repeats that resemble target site duplications at the ends of the elements (5,10,12); and the presence of TBE1 is allele-specific, which is consistent with a recent transposition event (13). Furthermore, both types of elements have open reading frames encoding putative transposases that are under selective pressure to maintain a functional protein (11,14,15).
In addition to the transposon-like IESs, the micronuclear genomes of the spirotrichs contain thousands of short, 10–539 bp, non-coding IESs that are AT-rich. These IESs are unique or of low copy number. One structural feature the short IESs of Euplotes share with Tec elements is the presence of a terminal TA direct repeat (5). Deletion of the two types of elements is similar in that both are excised as circular molecules with heteroduplex junctions (12) and their removal is coordinated with DNA replication during polytenization of the DNA in the macronuclear anlagen (16). Thus it seems likely that the two types of elements are deleted by similar mechanisms.
The process of developmentally regulated DNA deletion differs between spirotrichs and the evolutionarily distant Oligohymenophora class of ciliates. The most extensively studied member of the Oligohymenophora is Tetrahymena thermophila, wherein approximately 6000 elements, constituting 15% of the genome, are deleted during macronuclear development. As in the spirotrichs, IES deletion in Tetrahymena is 100% efficient. However, in Tetrahymena excision is imprecise. Several elements have alternative excision boundaries ranging over a few hundred base pairs (17–20) and most show sequence microheterogeneity at the rearrangement junctions (20–22). Another striking difference is in the structure of the excision products. In Tetrahymena the short IESs are more likely to be excised as linear molecules, in contrast to the circular heteroduplex excision products found in Euplotes crassus (12,23).
Most of the IESs that have been analyzed to date in Tetrahymena are relatively short, ranging from 0.6 to 2.9 kb, and consist of presumably non-coding AT-rich sequence (24). The elements have no common structural features. Some have short terminal direct repeats, but these are not required for excision of the element (25). The largest deletion element characterized to date is Tlr1, for Tetrahymena long repeat 1, which deletes >25 kb of sequence. Tlr1 belongs to a family of ∼20–30 micronuclear-specific elements (19). The most striking structural characteristic of Tlr1 is an 825 bp terminal inverted repeat near the element termini. In this respect, Tlr1 structurally resembles the transposon-like elements found in the spirotrichs.
In order to determine whether the Tlr elements contain an open reading frame encoding a transposase, genomic clones of several family members were isolated and sequenced. This study describes the identification and sequence analysis of a 2 kb open reading frame within the Tlr family of elements. This open reading frame is highly conserved and encodes a putative protein with similarity to the integrases encoded by retroviruses and retrotransposons.
MATERIALS AND METHODS
Accession numbers
The Tlr Int clone sequences have been deposited in the GenBank database. Accession numbers are: Tlr1 Int, AF232246; Tlr Int A, AF232242; Tlr Int B, AF232243; Tlr Int C, AF232244; Tlr Int D, AF232245; Tlr Int E, AF232247.
Strains
Tetrahymena thermophila strain CU428 Mpr/Mpr (6-methylpurine-sensitive, VI) was obtained from Peter Bruns (Cornell University, Ithaca, NY). Strain CU399 was used as a source for the pMBR micronuclear plasmid library.
Micronuclear and macronuclear DNA isolation
Strain CU428 DNA was isolated according to the method described by Gorovsky et al. (26). Following separation by differential centrifugation, the nuclei were treated with 500 µg proteinase K in 20 mM Tris–HCl pH 7.5, 40 mM EDTA and 1% SDS at 37°C for 1 h. DNA was extracted with phenol:chloroform (1:1) and ethanol precipitated.
Plasmid DNA isolation
Plasmid DNA was isolated from bacterial cell transformants using either the Wizard Plus Maxiprep DNA purification protocol (Promega) or the Qiagen plasmid purification protocol (Qiagen).
Oligonucleotides and primers
Oligonucleotides for circularization of HindIII–EcoRI fragments of micronuclear DNA were 5′-AGCTTGAGCTCTCGAGTCGACGATCG-3′ and 5′-AATTCGATCGTCGACTCGAGAGCTCA-3′. The circularized DNA was amplified by inverse PCR with primers 1 (5′-TCTATTCATCACTTTCTTA-3′) and 2 (5′-TTAATTTTATGTAAGTGAAGCTT-3′). Primer 3, (5′-TCGATTTAAAATTATCTTTCTCTG-3′), derived from the sequence of the inverse PCR product, was used against primer 2 to amplify Tlr family sequences from micronuclear DNA. Tlr1 sequences were amplified with the Tlr1 specific primer 4 (5′-AATGTGAATTTCGATTCGAT-3′) and internal primer 5 (5′-GATGTCTACAATTTTATAGTTTCTC-3′).
Southern hybridization
Purified micronuclear DNA isolated from strain CU428 was digested with the appropriate restriction endonucleases, fractionated through 0.8–1% agarose gels and transferred via capillary action onto NEF 976 Genescreen Plus transfer membrane (NEN Research Products). DNA hybridization probes were radioactively labeled by the random primer protocol (Boehringer Mannheim). The nylon membranes were washed in 2× SSC (1× SSC is 0.15 M NaCl, 0.015 M sodium citrate) at room temperature, 2× SSC, 1% SDS at 65°C and 0.1× SSC at 58°C.
Genomic library screening
The pMBR plasmid library was constructed from strain CU399 micronuclear DNA partially digested with MboI and cloned into the plasmid vector pUC19, as described previously (27). Escherichia coli SURE (Promega) cells were transformed by electroporation with 100 ng of the library. Cells were spread onto Luria–Bertani broth (LB) plates containing 50 µg/ml carbenicillin. Colonies were screened by colony hybridization according to the Southern hybridization protocol.
DNA sequencing
DNA was sequenced on an Applied Biosystems sequencer using the ABI Prism™ dye terminator cycle sequencing ready reaction kit with AmpliTaq DNA polymerase at the University of Wisconsin, Milwaukee, automated sequencing center.
Sequence alignment
Sequences were aligned with Clustal 1.7 found under the multiple sequence alignment program using the Baylor College of Medicine search launcher (www.hgsc.bcm.tmc.edu/searchlauncher/ ) sequence utilities.
RESULTS
Cloning of the Tlr family of deleted elements
The Tlr1 element, with long inverted repeats near the termini, is structurally similar to class II transposable elements, which undergo ‘cut and paste’ transposition, often mediated by transposases. The family of sequences is micronucleus-limited. The elements are unusual in that the copy number differs for different regions of the inverted repeat. Southern blot analysis showed that the innermost half of the Tlr1 inverted repeat hybridized to a family of 20–30 elements. This defines the Tlr family. The outermost region of the Tlr1 inverted repeat has sequence homology to a subfamily of sequences with a copy number of only 6–7 (19).
In order to determine whether the Tlr elements contained open reading frames that might encode a transposase, sequences internal to the inverted repeat at the right end of Tlr1 were cloned by inverse PCR (Fig. 1). A HindIII site is located 45 bp outside the right boundary of Tlr1. Genomic restriction mapping placed an EcoRI site ∼2.5 kb inside the element. Micronuclear DNA was digested with HindIII and EcoRI and circularized by ligation of the DNA in the presence of two complementary oligonucleotides which hybridize to each other to form a linker with HindIII and EcoRI sticky ends (Materials and Methods). DNA sequences internal to the Tlr1 inverted repeat were amplified by PCR with primers 1 and 2. The resulting inverse PCR product was partially sequenced and primer 3 was synthesized from this sequence. Micronuclear DNA was PCR amplified using primers 2 and 3. Since both primers are within sequences that are conserved in the Tlr family, this reaction contained a mixture of PCR products corresponding to various members of the family. A 1.2 kb cloned fragment contained the expected sequences with homology to the Tlr1 inverted repeat and 1079 bp of additional sequence internal to the repeat.
Figure 1.

Restriction maps of the Tlr1 regions of micronuclear and macronuclear DNA. The cross-hatched line represents macronucleus-destined sequence. The thick solid line represents the micronuclear-limited sequence. The bold arrows represent the 825 bp Tlr1 inverted repeat. The symbol * marks the location of 19mer tandem repeats. The angled arrow depicts the location of the 2 kb open reading frame identified in this study. Arrowheads represent PCR primers. Restriction sites: B, BglII; C, ClaI; E, EcoRI; H, HindIII; S, Sau3A; X, XbaI. The HindIII restriction site indicated by the dashed line is not present in Tlr1, but is present in other family members. The boxes represent fragments used in hybridization for isolation of Tlr Int clones.
The innermost 369 bp of the 1.2 kb PCR product contained an open reading frame with similarity to Caenorhabditis elegans retrotransposon CER1 integrase. This open reading frame contains two Asp residues that are spaced proportionally to the Asp residues in the D39–58D35E signature motif that is present in all integrases. In order to obtain the complete open reading frame, genomic clones were isolated from a plasmid library of micronuclear DNA (Materials and Methods). The library was screened with the 264 bp XbaI–HindIII and 971 bp XbaI–XbaI fragments internal to the Tlr1 inverted repeat (Fig. 1). Four clones with large inserts were selected for sequencing. All of these clones contained a 2 kb open reading frame encoding deduced proteins with homology to retroelement integrases. In Figure 2 the open reading frames of clones Tlr Int A, Tlr Int B, Tlr Int C and Tlr Int D are aligned. The sizes of the micronuclear DNA fragments demonstrate that they were independent clones and slight differences in the nucleotide sequence between clones indicated that they were four different members of the Tlr family of elements.
Figure 2.

Alignment of Tlr clones according to shared sequence. The vertical line in clone Tlr1 Int indicates the location of the right Tlr1 boundary and the arrow the inverted repeat. Mic, micronuclear-limited sequence; mac, macronucleus-destined sequence; angled arrow, the 2 kb open reading frame; triangles, the primers used to PCR amplify clone Tlr1 Int from micronuclear DNA.
Tlr Int A–D are members of a family of elements that is repeated in the micronuclear genome. In order to obtain the integrase-like ORF associated with Tlr1, the corresponding fragment of micronuclear DNA was PCR amplified with Tlr1 specific primer 4, located in unique macronucleus-destined DNA to the right of Tlr1, and primer 5, in conserved DNA 3′ to the open reading frame of the genomic clones (Fig. 2). The sequence at the right end of the 4.4 kb PCR product matched the known sequence of Tlr1 and the clone was designated Tlr1 Int. The integrase-like open reading frame begins 1583 nt from the right Tlr1 boundary and 710 nt internal to the inverted repeat (Fig. 1).
Similarity of the conceptual protein to retroelement integrases
The Tlr1 Int clone and Tlr Int A–C each contained an open reading frame of 1998 nt encoding a conceptual protein of 666 amino acids. The open reading frame of Tlr Int D encoded one additional amino acid, due to insertion of a trinucleotide encoding Glu at residue 561. To find similarity to previously identified proteins, the DNA sequence of the 2 kb open reading frame was submitted to NCBI BLASTX (Basic Local Alignment Search Tool) (28). The closest matches to the Tlr family open reading frame were sequences found in transposons or putative transposons. An alignment of the deduced proteins from a variety of organisms is shown in Figure 3. The best characterized of these was the TYB protein of Saccharomyces cerevisiae, which encodes a retrotransposon integrase. All integrases and many transposases contain three acidic amino acids with characteristic spacing (D39–58D35E) which comprise the catalytic core of the proteins. Each of these residues is independently essential for catalytic activity involved in the reactions of transposition (29–35). The Tetrahymena open reading frame encodes these acidic residues, along with characteristic blocks of conserved amino acid residues surrounding each of the signature amino acids.
Figure 3.

Alignment of the first 227 amino acid residues in the Tlr Int consensus sequence with proteins or putative proteins that have similarity according to NCBI BLAST. Amino acids that are identical in at least five sequences are shaded black. Amino acids that are similar in at least five sequences are shaded gray. The active site DDE residues are marked with an asterisk. The integrase HHCC domain residues are indicated by circles. E values and accession numbers of the sequences are: C.elegans cosmid Y57G11C, 1e–22, Z99281; X.maculatus HASI putative protein, 3e–22, U43331; Trichopulsia ni TED retrotransposon, 7e–11, B36329; Drosophila melanogaster mdg3 retrotransposon, 1e–7, X95908; Schizosaccharomyces pombe Tf1 retrotransposon 1e–7, M38526; Zea mays Opie-2 retrotransposon, 2e–7, AF105716; S.cerevisiae Ty1 retrotransposon, 4e–5, Z47746.
Integrases and transposases can be distinguished on the basis of the conserved amino acids surrounding the DDE signature (36; Fig. 4). The first Asp of the signature is generally followed by an acidic residue in transposases and more often by a hydrophobic amino acid in integrases. The second Asp is followed by an Asn in both classes of enzymes. Most notably, the integrase Glu is in a consensus sequence ERMNR/KTI/LK. In the transposases the consensus sequence in the region of the Glu is SPDLNPIEHL/I. The alignment in Figure 4 suggests that the deduced protein encoded by the Tlr family of elements in Tetrahymena is more closely related to the integrases than transposases. Thus this sequence is referred to as the Int open reading frame.
Figure 4.

An alignment of the residues surrounding the DDE amino acids in integrases and transposases with the corresponding Tlr1 Int amino acids. The amino acids common to all types of proteins are red. Blue letters correspond to the retroviral integrase consensus, green letters correspond to the retrotransposon integrase consensus and purple letters correspond to the class II element transposase consensus. Black letters show no consensus. Similar residues are included in the consensus and similarity for this analysis is defined as a score of 5 on the SG Matrix scoring system (38).
In addition to the DDE signature motif, integrases have an N-terminal zinc finger domain (HHCC) (Fig. 3), which is not found in transposases. This domain is required for the formation of a stable complex between viral cDNA and the integrase during cDNA 3′-end processing and strand transfer (30,37). Interestingly, although the DDE signature of the Tlr Int open reading frame is more similar to integrases than transposases, it does not have this zinc finger domain. The uncharacterized elements in C.elegans and Xiphophorus maculatus that are most similar to the Tlr Int open reading frame also lack this HHCC motif (Fig. 3).
Conservation of the open reading frame
The open reading frames encoding the integrase-like proteins of Tlr1 and Tlr Int A–D were analyzed to determine the degree of conservation of the putative gene products. There were 196 nucleotide changes among the 9993 nt analyzed, for 98% conservation of sequence. According to χ2 analysis, the position of the nucleotide changes within the open reading frames was highly non-random (P << 0.001), with most of them occurring in the third position of the codon where there is the greatest likelihood of amino acid conservation (Table 1).
Table 1. Nucleotide and amino acid substitutions among the Int open reading frames.
| Amino acid | Nucleotide position | Total nucleotides | ||
|---|---|---|---|---|
| First | Second | Third | ||
| Identical | 20 | 0 | 120 | 140 (72%) |
| Similar | 13ab | 16 | 13ab | 42 (21%) |
| Dissimilar | 4c | 9cd | 1d | 14 (7%) |
| Total | 37 | 25 | 134 | 196 |
a–dFour codons with two nucleotides different from the consensus sequence.
To assess the degree of amino acid conservation, amino acid substitutions were categorized as identical, similar or dissimilar. Similarity was defined according to the Structure–Genetic (SG) Matrix scoring system (38), where amino acids are assigned a number from 0 to 6 based on the structural identity and likelihood of interchanges. A score of 6 indicates identity. For the analysis in Table 1, similarity was defined as an SG score of 4 or 5. Of the 196 nucleotide changes among the five open reading frames, 140 encoded identical amino acids, 42 encoded similar amino acids and 14 encoded dissimilar amino acids (Table 1). Therefore, 93% of the nucleotide changes altered codons to encode either identical or similar amino acids and the amino acid sequence of the putative proteins encoded by the five elements is 99% conserved.
Conservation of the amino acid sequence despite nucleotide changes suggested that the open reading frame has evolved under selective pressure to conserve a functional protein. Statistical analysis of the synonymous versus non-synonymous nucleotide changes was done in order to determine the significance of the amino acid conservation (39). According to this method, nucleotide substitutions that encode similar or identical amino acids are synonymous (ps) whereas nucleotide substitutions that encode dissimilar amino acids are non-synonymous (pn). If there is selective pressure to conserve a protein coding region, ps is expected to be greater than pn. In a one-tailed t-test the ps value was significantly greater than pn for each pair of open reading frames at P << 0.001.
One unusual feature of the Tetrahymena genetic code is that the canonical stop codons TAA and TAG encode Gln (40,41). Thus T→C transitions in the first nucleotide of T/CAA or T/CAG Gln codons are silent. Such transitions accounted for 19 of the 20 nucleotide changes in the first nucleotide of a codon that resulted in identical amino acids (Table 1), suggesting that the open reading frame has evolved in the context of the Tetrahymena genome.
The N-terminus of the Tetrahymena integrase-like protein had the highest degree of similarity to integrases of other organisms. In order to determine whether selection among the Tlr family members is uniform over the length of the putative protein, the open reading frame was arbitrarily divided into thirds, with the DDE motifs of the active site located in the N-terminal third of the protein. Interestingly, χ2 analysis indicated that although the nucleotide substitutions occur randomly across the gene, dissimilar amino acid changes were preferentially located in the C-terminal third of the open reading frame (P < 0.01). Thus, although the C-terminal region of the protein is under strong selection, it is not as stringent as the selection in the N-terminal two-thirds of the protein.
Copy number and genome specificity of the Tlr family of sequences
Whereas the innermost portion of the Tlr1 inverted repeat is repeated 20- to 30-fold in the micronuclear genome, the outermost sequences, as indicated by hybridization of the tandemly repeated 19mers, belongs to a smaller family of only six to seven elements (19). Thus it was of interest to determine the copy number of sequences internal to the inverted repeat and of the open reading frame encoding the putative integrase. Micronuclear and macronuclear DNA were digested with various restriction enzymes and hybridized in a Southern analysis with the 714 bp HindIII–XbaI fragment containing sequences between the Tlr1 inverted repeat and the integrase open reading frame (Fig. 5B). In the lanes containing micronuclear DNA digested with EcoRI, EcoRV and HindIII ∼20–30 fragments hybridized, suggesting that sequences homologous to this fragment have a copy number similar to that of the inner part of the Tlr1 inverted repeat. In micronuclear DNA digested with XbaI the majority of the hybridization was to a 1 kb fragment. This was consistent with the sequence data suggesting that sequences within the element are highly conserved among family members. There was no hybridization to macronuclear DNA, thus the family of elements is efficiently eliminated during macronuclear development. The presence of macronuclear DNA was confirmed by ethidium bromide staining of the gel prior to transfer (data not shown).
Figure 5.
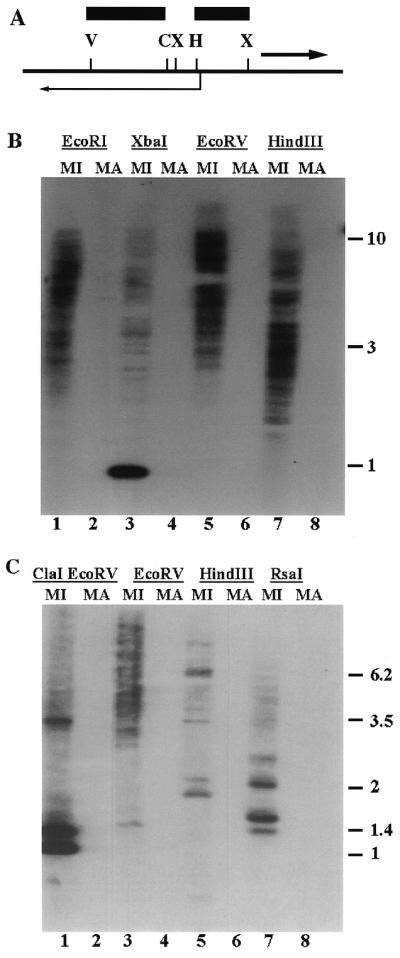
(A) Partial restriction map of restriction sites in the Tlr Int clones. Angled arrow, Int open reading frame; heavy arrow, Tlr1 inverted repeat; bars, the HindIII–XbaI and EcoRV–ClaI fragments used to probe the blots in (B) and (C), respectively. Restriction sites C, ClaI; H, HindIII; V, EcoRV; X, XbaI. (B) Southern blot of micronuclear (MI) and macronuclear (MA) DNA digested with EcoRI, XbaI, EcoRV and HindIII. (C) Southern blot of micronuclear (MI) and macronuclear (MA) DNA digested with ClaI + EcoRV, EcoRV, HindIII and RsaI.
A similar copy number was found for sequences in the open reading frame. Sequence analysis of the five clones at hand revealed a conserved EcoRV site within the open reading frame and no additional EcoRV sites in the conserved sequence 5′ to the open reading frame. Therefore, Southern analysis of DNA digested with EcoRV and probed with the ClaI–EcoRV fragment located entirely within the open reading frame provided an estimate of the number of elements that contain the open reading frame (Fig. 5C). The observed pattern of 20–30 bands was similar in number to that found for sequences 5′ to the open reading frame and for the innermost part of the Tlr1 inverted repeat. As in the previous Southern blot, the probe hybridized only to micronuclear DNA, suggesting that the 2 kb open reading frame is located within a family of micronuclear-limited elements.
The similarity in copy number and the micronucleus-limited character suggested that the integrase-like open reading frame may be generally associated with sequences in the Tlr1 inverted repeat. The limited data available for individual clones supports this model. Of the clones that cover the integrase-like open reading frame, only Tlr Int A extends far enough 5′ of the gene to contain DNA with homology to the Tlr1 inverted repeat. Another clone, Tlr Int E, which contained only the 5′-end of the integrase-like open reading frame, extended far enough to include sequences with homology to the Tlr1 inverted repeat. Sequence data from these three clones suggests that the integrase-like open reading frame is generally associated with the Tlr family, among which the innermost ∼275 bp of the Tlr1 inverted repeat are conserved.
The Southern blots also support the sequence data indicating a high degree of conservation among the integrase-like genes. Three major bands were seen in DNA digested with ClaI and EcoRI (Fig. 5C). One of these had the mobility expected for the 937 bp ClaI–EcoRV fragment that is present in all of the sequenced clones. The two other bands at 1.4 and 3.5 kb indicate that there are three major variations in the ClaI + EcoRV restriction pattern among the various family members.
DISCUSSION
The Tlr family of micronucleus-limited elements in Tetrahymena encodes a putative protein with similarity to retrotransposon integrases. The open reading frames from five of the 20–30 family members have been cloned and sequenced. Nucleotide changes among the five family members are highly non-random, suggesting that the elements are under selective pressure to maintain a functional protein in most or all of the elements.
The role of the integrase-like protein in Tetrahymena biology is unknown. One possibility is that the Tlr family of IESs encodes the machinery for its own developmentally regulated excision and the integrase-like enzyme is a polynucleotidyltransferase that is part of that machinery. A functional relationship between transposable elements and developmentally regulated DNA excision was first proposed to account for conservative selection of the open reading frames in the Tec and TBE elements of the spirotrichs (5). According to this model, IESs are degenerate transposons. Developmentally regulated DNA deletion evolved as a mechanism to remove them from the transcriptionally active macronucleus, with the transposase serving as the excisase. In Euplotes and Oxytricha, where many of the IESs occur within coding sequences, efficient IES excision might be expected to exert a powerful selective pressure to conserve the active transposase (42). This model is somewhat less compelling in Tetrahymena, where no protein coding sequences are known to be interrupted by IESs. However, it is possible that there are other selective advantages to removing IESs from the macronucleus genome. For example, they might play a role in micronuclear functions such as chromatin condensation and/or transcriptional silencing that are dispensable in the macronucleus (43).
Although conservative selection for functional protein might be expected if the integrase-like open reading frame of the Tlr family encodes excisase, it is not clear what selective mechanism would maintain functional genes in multiple copies of the element, since such an enzyme would presumably act in trans. Constructs containing the inverted repeats of Tlr1, but lacking the integrase-like gene, undergo efficient and accurate rearrangement in vivo (44). Thus developmentally regulated DNA excision of the Tlr family does not require an active integrase-like gene in cis.
Multiple genes encoding the integrase-like protein might be required to synthesize sufficient amounts of the protein to catalyze excision of the family of elements within a specific and brief developmental time period. However, transcripts of the Tec transposase gene of Euplotes were detected only by extremely sensitive methods involving Southern hybridization of RT–PCR products and the transcript levels are thought to be insufficient to account for the en masse excision of ∼106 Tec elements/polytene nucleus in a 2–4 h time period (45,46). Similarly, transcripts of the integrase-like genes in Tetrahymena have not been detected by standard northern blot analysis (J.A.Gershan, unpublished data).
A second possible function of the integrase-like gene is that the Tlr family of sequences are mobile elements and the integrase-like enzyme is responsible for their transposition in the micronuclear genome. Although the enzyme encoded by the Tlr family of elements has a DDE motif similar to that of retroelements, the Tlr elements differ from retrotransposons in several respects. At >25 kb (J.D.Wuitschick and J.A.Gershan, unpublished data), they are larger than most active retroelements and the terminal repeats are inverted rather than direct repeats. Analysis of sequences surrounding the Int open reading frame has not revealed a discernible gag–pol gene structure characteristic of retroelements and the putative integrase gene lacks the N-terminal HHCC zinc finger domain that is required for retroelement cDNA processing (30,37). (A complete analysis of the structure of these large elements is in progress and will be published elsewhere.) The long inverted repeats of Tlr1 are a structural characteristic common to many class II elements. Perhaps the Tlr elements are class II elements in which the motifs surrounding the active site DDE residues of the transposases are more similar to those in the integrases than to those of the majority of transposases. This would be unusual, but not unprecedented (36). The bacterial IS30 element is an example of a class II element with integrase-like active site motifs. The putative enzyme encoded by the Tlr family is like the transposase of the bacterial IS30 elements in that both lack the N-terminal HHCC integrase domain (47).
If the Tlr family of sequences are transposons, then it is necessary to account for the fact that these elements are found exclusively in the 15% of the micronuclear genome that undergoes developmentally regulated elimination. Excision of the Tlr elements during macronuclear development could be a secondary consequence of transposon targeting into micronuclear IESs. Two observations support this hypothesis. First, Tlr1 is removed from the macronuclear genome at the same stage of macronuclear development as the smaller, AT-rich IESs (17; Capowski and K.M.Karrer, unpublished data). Second, flanking sequences have been shown to play a role in the delineation of the rearrangement boundaries of Tlr1 (44). This is not an expected feature of transposon excision, but is consistent with the hypothesis that Tlr1 resides within an IES because, in Tetrahymena, cis-acting sequences in the flanking DNA regulate the deletion of IESs (21,22,48).
Unique features of chromatin structure might serve to target transposition of Tlr elements to IESs. There is a precedent for transposon targeting to regions of distinct chromatin structure in yeast, where Ty5 elements are selectively inserted into regions of silent chromatin by association of the integration complexes with localized host factors (49–52). Differences between the chromatin structure of eliminated sequences and macronucleus-destined sequences have been detected in Euplotes by analysis of chromatin digested with micrococcal nuclease (53). In Tetrahymena the chromatin of IES-containing regions of the genome is distinct from that of the bulk of the genome during macronuclear development. The abundant stage-specific proteins Pdd1p and Pdd2p, both of which are required for developmentally programmed DNA deletion in Tetrahymena (54,55), are preferentially associated with heterochromatic regions containing IESs (56,57).
If the integrase-like protein encoded by the Tlr family of elements is a transposase and is not functioning as the excisase, then the need to maintain sufficient enzyme for IES excision cannot be invoked to explain the apparent selection on these genes. The transposases of class II elements can be subject to selective pressure for functions other than the transposase activity, such as repression of transposition (58).
Five of the estimated 20–30 integrase-like open reading frames, a significant fraction of the total Tlr family, were analyzed in this study. Despite numerous nucleotide changes amongst the genes, all of the family members examined maintained the open reading frame and a high degree of protein similarity. Whatever the biological role of the integrase-like protein, the data indicate that there is strong selective pressure to maintain an active gene in most or all of the Tlr family members.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Inta Kalve and Kevin Miller for technical assistance. This work was supported by grant MCB-9974885 from the National Science Foundation. Jill Gershan was supported in part by a Marquette University John P. Raynor fellowship and an Arthur J. Schmitt Foundation fellowship.
DDBJ/EMBL/GenBank accession nos AF232242–AF232247
REFERENCES
- 1.Polard P. and Chandler,M. (1995) Mol. Microbiol., 15, 13–23. [DOI] [PubMed] [Google Scholar]
- 2.Landree M.A., Wibbenmeyer,J.A. and Roth,D.B. (1999) Genes Dev., 13, 3059–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim D.R., Dai,Y., Mundy,C.L., Yang,W. and Oettinger,M.A. (1999) Genes Dev., 13, 3070–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karrer K.M., (1999) In Asai,D.J. and Forney,J.D. (eds), Tetrahymena thermophila. Academic Press, San Diego, CA, pp. 127–186.
- 5.Klobutcher L.A. and Herrick,G. (1997) Prog. Nucleic Acid Res. Mol. Biol., 56, 1–62. [DOI] [PubMed] [Google Scholar]
- 6.Lynn D.H. and Small,E.B. (1997) Rev. Soc. Mex. Hist. Nat., 47. [Google Scholar]
- 7.Swanton M.T., Greslin,A.F. and Prescott,D.M. (1980) Chromosoma, 77, 203–215. [DOI] [PubMed] [Google Scholar]
- 8.Jahn C.L., Krikau,M.F. and Shyman,S. (1989) Cell, 59, 1009–1018. [DOI] [PubMed] [Google Scholar]
- 9.Herrick G., Cartinhour,S., Dawson,D., Ang,D., Sheets,R., Lee,A. and Williams,K. (1985) Cell, 43, 759–768. [DOI] [PubMed] [Google Scholar]
- 10.Hunter D.J., Williams,K., Cartinhour,S. and Herrick,G. (1989) Genes Dev., 3, 2101–2112. [DOI] [PubMed] [Google Scholar]
- 11.Doak T.G., Witherspoon,D.J., Doerder,F.P., Williams,K. and Herrick,G. (1997) Genetica, 101, 75–86. [DOI] [PubMed] [Google Scholar]
- 12.Jaraczewski J.W. and Jahn,C.L. (1993) Genes Dev., 7, 95–105. [DOI] [PubMed] [Google Scholar]
- 13.Seegmiller A., Williams,K.R., Hammersmith,R.L., Doak,T.G., Witherspoon,D., Messick,T., Storjohann,L.L. and Herrick,G. (1996) Mol. Biol. Evol., 13, 1351–1362. [DOI] [PubMed] [Google Scholar]
- 14.Jahn C.L., Doktor,S.Z., Frels,J.S., Jaraczewski,J.W. and Krikau,M.F. (1993) Gene, 133, 71–78. [DOI] [PubMed] [Google Scholar]
- 15.Doak T.G., Doerder,F.P., Jahn,C.L. and Herrick,G.H. (1994) Proc. Natl Acad. Sci. USA, 91, 942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frels J.S. and Jahn,C.L. (1995) Mol. Cell. Biol., 15, 6488–6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Austerberry C.F., Allis,C.D. and Yao,M.-C. (1984) Proc. Natl Acad. Sci. USA, 81, 7383–7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Austerberry C.F. and Yao,M.-C. (1988) Mol. Cell. Biol., 8, 3947–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wells J.M., Ellingson,J.L.E., Catt,D.M., Berger,P.J. and Karrer,K.M. (1994) Mol. Cell. Biol., 14, 5939–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patil N.S., Hempen,P.M., Udani,R.A. and Karrer,K.M. (1997) J. Eukaryot. Microbiol., 44, 518–522. [DOI] [PubMed] [Google Scholar]
- 21.Austerberry C.F., Snyder,R.O. and Yao,M.-C. (1989) Nucleic Acids Res., 17, 7263–7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J. and Pearlman,R.E. (1996) Nucleic Acids Res., 24, 1943–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao M.-C. and Yao,C.-H. (1994) Nucleic Acids Res., 22, 5702–5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wuitschick J.D. and Karrer,K.M. (1999) J. Eukaryot. Microbiol., 46, 239–247. [DOI] [PubMed] [Google Scholar]
- 25.Godiska R. and Yao,M.-C. (1990) Cell, 61, 1237–1246. [DOI] [PubMed] [Google Scholar]
- 26.Gorovsky M.A., Yao,M.-C., Keevert,J.B. and Pleger,G.L. (1975) Methods Cell Biol., 9, 311–327. [DOI] [PubMed] [Google Scholar]
- 27.Rogers M.B. and Karrer,K.M. (1989) Dev. Biol., 131, 261–268. [DOI] [PubMed] [Google Scholar]
- 28.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drelich M., Wilhelm,R. and Mous,J. (1992) Virology, 188, 459–468. [DOI] [PubMed] [Google Scholar]
- 30.Engelman A. and Craigie,R. (1992) J. Virol., 66, 6361–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kulkosky J., Jones,K.S., Katz,R.A., Mack,J.P.G. and Skalka,A.M. (1992) Mol. Cell. Biol., 12, 2331–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leavitt A.D., Shive,L. and Varmus,H.E. (1993) J. Biol. Chem., 268, 2113–2119. [PubMed] [Google Scholar]
- 33.Jenkins T.M., Esposito,D., Engelman,A. and Craigie,R. (1997) EMBO J., 16, 6849–6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiskerchen M. and Muesing,M.A. (1995) J. Virol., 69, 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Gent D.C., Oude Groeneger,A.A.M. and Plasterk,R.H.A. (1992) Proc. Natl Acad. Sci. USA, 89, 9598–9602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Capy P., Vitalis,R., Langin,T., Higuet,D. and Bazin,C. (1996) J. Mol. Evol., 42, 359–368. [DOI] [PubMed] [Google Scholar]
- 37.Ellison V. and Brown,P.O. (1994) Proc. Natl Acad. Sci. USA, 91, 7316–7320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng D.F., Johnson,M.S. and Doolittle,R.F. (1985) J. Mol. Evol., 21, 112–125. [DOI] [PubMed] [Google Scholar]
- 39.Nei M. and Gojobori,T. (1986) Mol. Biol. Evol., 3, 418–426. [DOI] [PubMed] [Google Scholar]
- 40.Horowitz S. and Gorovsky,M.A. (1985) Proc. Natl Acad. Sci. USA, 82, 2452–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanyu N., Kuchino,Y. and Susumu,N. (1986) EMBO J., 5, 1307–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Witherspoon D.J., Doak,T.G., Williams,K.R., Seegmiller,A., Seger,J. and Herrick,G. (1997) Mol. Biol. Evol., 14, 696–706. [DOI] [PubMed] [Google Scholar]
- 43.Yao M.-C. (1996) Trends Genet., 12, 26–30. [DOI] [PubMed] [Google Scholar]
- 44.Patil N. and Karrer,K.M. (2000) Nucleic Acids Res., 28, 1465–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klobutcher L.A., Turner,L.R. and LaPlante,J. (1993) Genes Dev., 7, 84–94. [DOI] [PubMed] [Google Scholar]
- 46.Jaraczewski J.W., Frels,J.S. and Jahn,C.L. (1994) Nucleic Acids Res., 22, 4535–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dalrymple B., Caspers,P. and Arber,W. (1984) EMBO J., 3, 2145–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chalker D.L., La Terza,A., Wilson,A., Kroenke,C.D. and Yao,M.C. (1999) Mol. Cell. Biol., 19, 5631–5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zou S., Ke,N., Kim,J.M. and Voytas,D. (1996) Genes Dev., 10, 634–645. [DOI] [PubMed] [Google Scholar]
- 50.Zou S. and Voytas,D.F. (1997) Proc. Natl Acad. Sci. USA, 94, 7412–7416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gai X. and Voytas,D.F. (1998) Mol. Cell, 1, 1051–1055. [DOI] [PubMed] [Google Scholar]
- 52.Zhu Y., Zou,S., Wright,D.A. and Voytas,D.F. (1999) Genes Dev., 13, 2738–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jahn C.L. (1999) Mol. Biol. Cell, 10, 4217–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Coyne R.S., Nidiforov,M., Smothers,J.F., Allis,C.D. and Yao,M.-C. (1999) Mol. Cell, 4, 865–872. [DOI] [PubMed] [Google Scholar]
- 55.Nikiforov M.A., Smothers,J.F., Gorovsky,M.A. and Allis,C.D. (1999) Genes Dev., 13, 2852–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Madireddi M.T., Coyne,R.S., Smothers,J.F., Mickey,K.M., Yao,M.-C. and Allis,C.D. (1996) Cell, 87, 75–84. [DOI] [PubMed] [Google Scholar]
- 57.Smothers J.F., Mizzen,C.A., Tubbert,M.M., Cook,R.G. and Allis,C.D. (1997) Development, 124, 4537–4545. [DOI] [PubMed] [Google Scholar]
- 58.Misra S. and Rio,D.C. (1990) Cell, 62, 269–284. [DOI] [PubMed] [Google Scholar]