

Abstract
Hollongdione is the first recorded example of the occurrence of a dammarane hexanor-triterpene in nature possessing antiviral and cytotoxic activity. Its simple one-stage transformation into compounds with terminal alkyne and vinyl chloride fragments via the interaction with phosphorus halides is reported. The copper(I)-catalyzed Mannich reaction of 3-oxo-22,23,24,25,26,27-hexanor-dammar-20(21)-in 3 led to a series of aminomethylated products, while 17-carboxylic acid was obtained by ozone oxidation of 3-oxo-22,23,24,25,26,27-hexanor-dammar-20-chloro-20(21)-en 4; the following direct amidation of the latter has been developed. The structures of all new molecules were established by spectroscopic studies that included 2D NMR correlation methods; the molecular structures of compounds 2–5 were determined by X-ray analysis.
Keywords: dammarane triterpenoids, hollongdione, alkyne, vinyl chloride, Mannich base, hollongdionoic acid
1. Introduction
The unique and complex structures of natural products make them important sources for exploring novel areas of the chemical space [1,2]. The introduction of highly effective reaction centers in such molecules allows expanding the library of semi-synthetic compounds, with the aim of their future application as units in biochemical science.
Pentacyclic triterpenoids comprise a diverse group that is widely present in plants and has stable skeleton and reactive sites suitable for structural transformations. Triterpenoids usually possess a secondary hydroxyl group or a carbon atom at position C3, and a primary hydroxyl group or carboxyl group at position C28 (the general structures of triterpenoids with the key sites for modification are presented in Figure 1). The most frequent modifications of the listed fragments include an acylation reaction resulting in various esters [3,4,5] and glycosides [6,7], since the addition of hydrosoluble moieties such as sugar substituents at the C3 and/or C28 positions can enhance both the water solubility and the pharmacological activity [8,9].
Figure 1.

The general structures of lupane, oleanane, and dammarane type triterpenoids. The cycles in the structures of triterpenoids are marked in blue.
On the other hand, there is an oxygen function bonded to the ring E or D, a number of transformations of which have also been proposed in the literature. The most unique examples are Baeyer–Villiger oxidation of 20-oxo-30-nortaraxasteryl acetate that lead to ε-lactone [10]; 30-oxobetulinic acid conversion into a set of substituted dienes by the Wittig reaction with the use of triphenylphosphonium salts [11]; and an epimerization at C19 and a condensation reaction of several platanic carboxamides with the synthesis of novel E-ring δ−lactams [12]. Another attractive method of carbonyl group modification is a reaction of dehydration, which is a route to terminal acetylenes, which are key precursors in drug discovery [2,13]. One widely used method of accessing alkynes is an interaction of methyl ketones with phosphorous-based reagents. This approach was found to be a short-stage, simple experimental procedure with no by-product formation in the synthesis of terminal lupane-type alkynes [14,15] and was more efficient compared to other methods, such as conventional Pd-catalyzed Sonogashira coupling [16,17], the Grignard reaction [18] and propargylation of the NH-group [19].
In this work, we decided to use dammarane-type triterpenoid hollongdione (22,23,24,25,26,27-hexanordammar-3,20-dione) 1 with a C17 acetyl group as an initial scaffold [20]. Hollongdione (the structure of which is presented in Figure 2) can be obtained by one-reactor synthesis from dipterocarpol, the main metabolite of Dipterocarpus alatus oleoresin or isolated from Dipterocarpus pilosus, Gardenia aubryi, Chisocheton penduliflorus, and leaves of Euphorbia leucocephala, which exhibits potent anti-influenza A virus activity [21,22,23]. Moreover, it is a crucial intermediate in the synthesis of the potent immunosuppressive agent 17α-23-(E)-dammara-20,23-diene-3β,25-diol and moderately active against small-cell lung cancer cells (NCI-H187) [24,25]. Arylidene derivatives of hollongdione, described before, induce antiproliferative activity against melanoma and breast cancer through pro-apoptotic and antiangiogenic mechanisms [26]. It is recognized as a hybrid molecule that contains cycle A, similar to triterpene, and a C17-acetyl group with a β-configuration attached to the D cycle, similar to the pregnane steroids.
Figure 2.
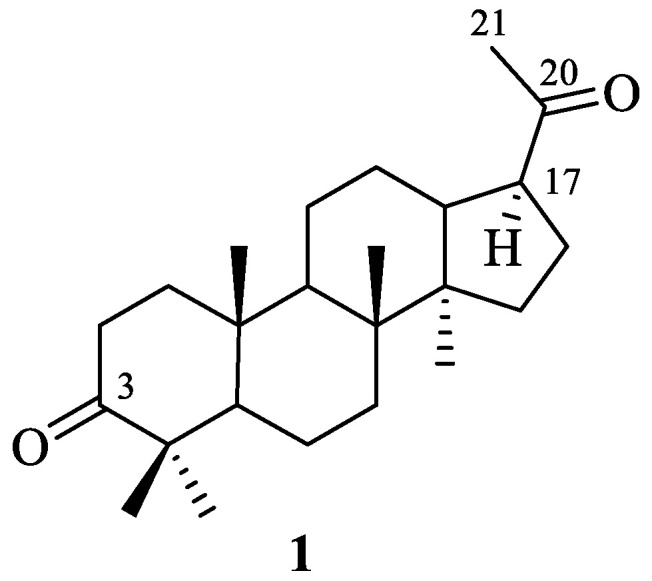
The structure of hollongdione 1.
The choice of the initial structure can be justified by the fact that there are no attempts of acetylene fragment formation as well as its further modification in the family of dammarane triterpenoids or pregnane steroids, especially among the closest analogues like panaxadiol and protopanaxadiol. Just in a few cases, changes were made based on the protopanaxadiol ketones; different kinds of functionalities including hydroxy, fluoride, aldehyde, carboxy, ester, and enol ether were introduced for diversity of ring D [27,28].
Taking into account the above, we report herein a one-stage method of hollongdione modification into compounds with terminal alkyne and vinyl chloride fragments via the interaction with phosphorus halides. The obtained unsaturated hollongdiones were transformed to aminomethylated products and carboxamide.
2. Results
Procedure of compound 2
A solution of 3β-hydroxy-dipterocarpol (0.440 g; 1.0 mmol) in AcOH (30 mL) was refluxed for 2 h; the reaction mixture was cooled to 0 °C, and ozone was passed through it until the starting compound disappeared (TLC control). The mixture was then poured into H2O (40 mL), removed by filtration, and washed with H2O. The crude product was crystallized from benzene.
3β-Acetoxy-22,23,24,25,26,27-hexanor-dammar-20-one (2)
White solid, yield: 96%, mp: 126–127 °C, [α]20D = −60 (c 0.05, CHCl3). 13C NMR (CDCl3, δ ppm): 15.59 (C18); 15.88 (C30); 16.31 (C19); 16.50 (C29); 18.15 (C6); 21.26 (C11); 21.31 (C2′); 23.71 (C2); 25.59 (C12); 25.98 (C16); 27.98 (C28); 30.06 (C21); 31.57 (C15); 35.49 (C7); 37.14 (C10); 37.92 (C4); 38.81 (C1); 40.53 (C8); 45.14 (C13); 50.07 (C14); 50.65 (C9); 54.27 (C17); 55.98 (C5); 80.87 (C3); 171.00 (C1′); 212.32 (C20). 1H NMR (CDCl3, δ ppm, J Hz): 0.83 (dd, 1H, 3J5-6ax = 11.7, 3J5-6eq = 2.6, H-5); 0.85 (s, 3H, H-29); 0.86 (s, 3H, H-28); 0.87 (s, 3H, H-30); 0.88 (s, 3H, H-19); 0.98 (s, 3H, H-18); 1.05 (td, 1H, 2J = 12.2, 3J1ax-2ax = 12.2, 3J1ax-2ax = 5.6, Hax-1); 1.16 (m, 1H, Hα-15); 1.20 (m, 1H, Hax-12); 1.26 (m, 1H, Hax-11); 1.29 (m, 1H, Heq-7); 1.32 (dd, 1H, 3J9-11ax = 11.8, 3J9-11eq = 3.3, H-9); 1.47 (m, 1H, Hax-6); 1.52 (m, 1H, Heq-11); 1.53 (m, 1H, Heq-6); 1.57 (m, 1H, Hax-7); 1.63 (m, 1H, Hax-2); 1.64 (m, 1H, Heq-12); 1.64 (m, 1H, Heq-2); 1.66 (m, 1H, Hβ-15); 1.70 (m, 1H, Heq-1); 1.71 (m, 1H, Hβ-16); 1.92 (m, 1H, H-13); 1.93 (m, 1H, Hα-16); 2.04 (s, 3H, H-2′); 2.13 (s, 3H, H-21); 2.59 (td, 1H, 3J17-13 = 10.9, 3J17-16α = 10.9, 3J17-16β = 6.2, H-17); 4.48 (dd, 1H, 3J3-2ax = 10.7, 3J3-2eq = 5.6, H-3).
General procedure of compounds 3–8
(a) Phosphoryl oxychloride (1 mL) was added to a solution of compound 1 or 2 (0.359 g or 0.403 g; 1.0 mmol) in 10 mL of anhydrous pyridine. The mixture was heated for 8 h under reflux, poured onto water, and extracted with chloroform (3 × 100 mL). The extract was washed with water (3 × 100 mL), dried over anhydrous calcium chloride, and evaporated under reduced pressure (water-jet pump). The residue was subjected to column chromatography on silica gel using eluent-petroleum ether-ethyl acetate (70:1→40:1). Compounds 6, 7 were isolated in a mixture of products, with an overall yield of 87%.
3-Oxo-22,23,24,25,26,27-hexanor-dammar-20(21)-in (3)
White solid, yield: 73%, mp: 161–162 °C, [α]20D = +12 (c 0.05, CHCl3). 13C NMR (CDCl3, δ ppm): 15.33 (C30); 15.34 (C18); 16.04 (C19); 19.62 (C6); 21.03 (C29); 21.77 (C11); 24.77 (C12); 26.75 (C28); 29.93 (C16); 31.09 (C15); 31.55 (C17); 34.08 (C2); 34.71 (C7); 36.92 (C10); 39.95 (C1); 40.25 (C8); 47.39 (C4); 49.24 (C13); 49.26 (C14); 50.11 (C9); 55.34 (C5); 67.92 (C21); 89.04 (C20); 217.87 (C3). 1H NMR (CDCl3, δ ppm, J Hz): 0.81 (s, 3H, H-30); 0.95 (s, 3H, H-19); 1.01 (s, 3H, H-18); 1.04 (s, 3H, H-29); 1.08 (s, 3H, H-28); 1.15 (m, 1H, Hα-15); 1.18 (qd, 1H, 2J = 12.4, 3J12ax-13 = 12.4, 3J12ax-11ax = 12.4, 3J12ax-11eq = 4.6, Hax-12); 1.32 (m, 1H, Heq-7); 1.35 (m, 1H, Hax-11); 1.37 (dd, 1H, 3J5-6ax = 11.7, 3J5-6eq = 2.0, H-5); 1.39 (dd, 1H, 3J9-11ax = 12.5, 3J9-11eq = 2.6, H-9); 1.46 (ddd, 1H, 2J = 13.2, 3J1ax-2ax = 9.5, 3J1ax-2eq = 7.8, Hax-1); 1.48 (m, 1H, Heq-6); 1.57 (m, 1H, Hax-6); 1.58 (m, 1H, Heq-11); 1.59 (m, 1H, Hax-7); 1.69 (m, 1H, Hβ-16); 1.71 (m, 1H, Hβ-15); 1.74 (ddd, 1H, 3J13-12ax = 12.4, 3J13-17 = 12.2, 3J13-12eq = 4.1, H-13); 1.87 (m, 1H, Heq-12); 1.95 (ddd, 1H, 2J = 13.2, 3J1eq-2ax = 7.6, 3J1eq-2eq = 4.7, Heq-1); 2.05 (d, 1H, 4J21-17 = 2.4, H-21); 2.11 (m, 1H, Hα-16); 2.34 (dddd, 1H, 3J17-13 = 12.2, 3J17-16α = 9.0, 3J17-16β = 6.4, 4J17-21 = 2.4, H-17); 2.43 (ddd, 1H, 2J = 15.6, 3J2eq-1ax = 7.8, 3J2eq-1eq = 4.7, Heq-2); 2.51 (ddd, 1H, 2J = 15.6, 3J2ax-1ax = 9.5, 3J2ax-1eq = 7.6, Hax-2).
3-Oxo-22,23,24,25,26,27-hexanor-dammar-20-chloro-20(21)-ene (4)
White solid, yield: 7%, mp: 148–149 °C, [α]20D = −30 (c 0.05, CHCl3). 13C NMR (CDCl3, δ ppm): 15.30 (C18); 15.96 (C30); 16.08 (C19); 19.66 (C6); 21.04 (C29); 21.80 (C11); 24.46 (C12); 26.75 (C28); 27.48 (C16); 31.28 (C15); 34.11 (C2); 34.79 (C7); 36.95 (C10); 39.99 (C1); 40.44 (C8); 45.14 (C13); 47.43 (C4); 49.40 (C14); 49.79 (C17); 50.28 (C9); 55.41 (C5); 111.59 (C21); 146.83 (C20); 218.00 (C3). 1H NMR (CDCl3, δ ppm, J Hz): 0.88 (s, 3H, H-30); 0.96 (s, 3H, H-19); 1.03 (s, 3H, H-18); 1.05 (s, 3H, H-29); 1.09 (s, 3H, H-28); 1.13 (m, 1H, Hax-12); 1.15 (m, 1H, Hα-15); 1.31 (m, 1H, Hax-11); 1.33 (m, 1H, Heq-7); 1.38 (dd, 1H, 3J5-6ax = 11.7, 3J5-6eq = 2.0, H-5); 1.40 (dd, 1H, 3J9-11ax = 12.6, 3J9-11eq = 2.6, H-9); 1.46 (ddd, 1H, 2J = 13.2, 3J1ax-2ax = 9.5, 3J1ax-2eq = 7.8, Hax-1); 1.47 (m, 1H, Heq-6); 1.55 (m, 1H, Heq-11); 1.56 (m, 1H, Hax-6); 1.60 (m, 1H, Hax-7); 1.63 (m, 1H, Heq-12); 1.67 (m, 1H, Hβ-15); 1.74 (m, 1H, Hβ-16); 1.88 (ddd, 1H, 3J13-12ax = 12.4, 3J13-17 = 10.6, 3J13-12eq = 3.7, H-13); 1.89 (m, 1H, Hα-16); 1.94 (ddd, 1H, 2J = 13.2, 3J1eq-2ax = 7.6, 3J1eq-2eq = 4.7, Heq-1); 2.43 (ddd, 1H, 2J = 15.6, 3J2eq-1ax = 7.8, 3J2eq-1eq = 4.7, Heq-2); 2.49 (td, 1H, 3J17-13 = 10.6, 3J17-16α = 10.6, 3J17-16β = 6.2, H-17); 2.51 (ddd, 1H, 2J = 15.6, 3J2ax-1ax = 9.5, 3J2ax-1eq = 7.6, Hax-2); 5.10 (d, 1H, 2J = 1.2, HA-21); 5.11 (d, 1H, 2J = 1.2, HB-21).
3β-Acetoxy-22,23,24,25,26,27-hexanor-dammar-20-chloro-20(21)-ene (6)
White solid, yield: 80%, mp: 138–139 °C, [α]20D = −110 (c 0.05, CHCl3). 13C NMR (CDCl3, δ ppm): 15.57 (C18); 16.02 (C30); 16.30 (C19); 16.50 (C29); 18.16 (C6); 21.28 (C11); 21.30 (C2′); 23.70 (C2); 24.42 (C12); 27.51 (C16); 27.98 (C28); 31.29 (C15); 35.38 (C7); 37.16 (C10); 37.90 (C4); 38.80 (C1); 40.54 (C8); 45.02 (C13); 49.41 (C14); 49.84 (C17); 50.83 (C9); 55.99 (C5); 80.86 (C3); 111.46 (C21); 146.93 (C20); 170.95 (C1′). 1H NMR (CDCl3, δ ppm, J Hz): 0.84 (dd, 1H, 3J5-6ax = 11.5, 3J5-6eq = 1.8, H-5); 0.85 (s, 3H, H-29); 0.86 (s, 3H, H-28); 0.86 (s, 3H, H-30); 0.88 (s, 3H, H-19); 0.99 (s, 3H, H-18); 1.05 (ddd, 1H, 2J = 11.8, 3J1ax-2ax = 11.6, 3J1ax-2eq = 4.7, Hax-1); 1.09 (qd, 1H, 2J = 12.4, 3J12ax-13 = 12.4, 3J12ax-11ax = 12.4, 3J12ax-11eq = 4.6, Hax-12); 1.12 (m, 1H, Hα-15); 1.23 (qd, 1H, 2J = 12.7, 3J11ax-9 = 12.6, 3J11ax-12ax = 12.6, 3J12ax-11eq = 4.6, Hax-11); 1.29 (m, 1H, Heq-7); 1.32 (dd, 1H, 3J9-11ax = 12.6, 3J9-11eq = 2.6, H-9); 1.46 (m, 1H, Hax-6); 1.53 (m, 1H, Heq-6); 1.54 (m, 1H, Heq-11); 1.58 (m, 1H, Hax-7); 1.60 (m, 1H, Heq-12); 1.61 (m, 1H, Hax-2); 1.64 (m, 1H, Heq-2); 1.65 (m, 1H, Hβ-15); 1.71 (m, 1H, Heq-1); 1.74 (m, 1H, Hβ-16); 1.84 (ddd, 1H, 3J13-12ax = 12.4, 3J13-17 = 10.6, 3J13-12eq = 3.7, H-13); 1.88 (m, 1H, Hα-16); 2.04 (s, 3H, H-2′); 2.47 (td, 1H, 3J17-13 = 10.6, 3J17-16α = 10.6, 3J17-16β = 6.2, H-17); 4.48 (dd, 1H, 3J3-2ax = 10.7, 3J3-2eq = 5.8, H-3); 5.08 (d, 1H, 2J = 1.2, HA-21); 5.10 (d, 1H, 2J = 1.2, HB-21).
3β-Acetoxy-22,23,24,25,26,27-hexanor-dammar-20(21)-in (7)
White solid, yield: 7%, mp: 149–150 °C, [α]20D = −61 (c 0.05, CHCl3). 13C NMR (CDCl3, δ ppm): 15.41 (C30); 15.65 (C18); 16.29 (C19); 16.51 (C29); 18.15 (C6); 21.27 (C11); 21.31 (C2′); 23.72 (C2); 24.75 (C12); 28.00 (C28); 29.98 (C16); 31.13 (C15); 31.61 (C17); 35.32 (C7); 37.16 (C10); 37.92 (C4); 38.82 (C1); 40.39 (C8); 49.17 (C13); 49.32 (C14); 50.71 (C9); 55.97 (C5); 67.80 (C21); 80.86 (C3); 89.24 (C20); 170.98 (C1′). 1H NMR (CDCl3, δ ppm, J Hz): 0.79 (s, 3H, H-30); 0.83 (dd, 1H, 3J5-6ax = 11.5, 3J5-6eq = 1.8, H-5); 0.85 (s, 3H, H-29); 0.85 (s, 3H, H-28); 0.88 (s, 3H, H-19); 0.97 (s, 3H, H-18); 1.05 (ddd, 1H, 2J = 11.8, 3J1ax-2ax = 11.6, 3J1ax-2eq = 4.7, Hax-1); 1.11 (m, 1H, Heq-15); 1.16 (m, 1H, Hax-12); 1.27 (m, 1H, Heq-7); 1.29 (m, 1H, Hax-11); 1.31 (dd, 1H, 3J9-11ax = 12.6, 3J9-11eq = 2.6, H-9); 1.46 (m, 1H, Hax-6); 1.53 (m, 1H, Heq-6); 1.56 (m, 1H, Hax-7); 1.58 (m, 1H, Heq-11); 1.61 (m, 1H, Hax-2); 1.64 (m, 1H, Heq-2); 1.68 (m, 1H, Heq-16); 1.70 (m, 1H, Hax-15); 1.71 (m, 1H, Heq-1); 1.73 (m, 1H, H-13); 1.85 (m, 1H, Heq-12); 2.03 (d, 1H, 4J21-17 = 2.4, H-21); 2.04 (s, 3H, H-2′); 2.10 (m, 1H, Hax-16); 2.32 (tdd, 1H, 3J17-13 = 10.6, 3J17-16ax = 10.6, 3J17-16eq = 6.2, 4J17-21 = 2.4, H-17); 4.48 (dd, 1H, 3J3-2ax = 10.7, 3J3-2eq = 5.8, H-3).
22,23,24,25,26,27-Hexanor-dammar-3,20-dichloro-20(21), 2(3)-diene (8)
White solid, yield: 8%, mp: 106–107 °C, [α]20D = −28 (c 0.05, CHCl3). 13C NMR (CDCl3, δ ppm): 15.14 (C18); 15.98 (C30); 16.35 (C19); 20.01 (C29); 20.33 (C6); 21.71 (C11); 24.48 (C12); 27.54 (C16); 29.06 (C28); 31.30 (C15); 34.77 (C7); 36.42 (C10); 40.22 (C4); 40.35 (C8); 42.87 (C1); 45.17 (C13); 49.38 (C14); 49.44 (C9); 49.80 (C17); 54.37 (C5); 111.51 (C21); 122.07 (C2); 141.55 (C3); 146.94 (C20). 1H NMR (CDCl3, δ ppm, J Hz): 0.87 (s, 3H, H-30); 0.92 (s, 3H, H-19); 1.02 (s, 3H, H-18); 1.04 (s, 3H, H-29); 1.09 (qd, 1H, 2J = 12.4, 3J12ax-13 = 12.4, 3J12ax-11ax = 12.4, 3J12ax-11eq = 4.6, Hax-12); 1.14 (s, 3H, H-28); 1.15 (m, 1H, Hα-15); 1.26 (dd, 1H, 3J5-6ax = 11.0, 3J5-6eq = 1.8, H-5); 1.29 (qd, 1H, 2J = 12.7, 3J11ax-9 = 12.6, 3J11ax-12ax = 12.6, 3J12ax-11eq = 4.6, Hax-11); 1.33 (m, 1H, Heq-7); 1.36 (dd, 1H, 3J9-11ax = 12.6, 3J9-11eq = 2.6, H-9); 1.50 (m, 1H, Heq-11); 1.55 (m, 1H, Hax-6); 1.58 (m, 1H, Hax-7); 1.58 (m, 1H, Heq-6); 1.62 (m, 1H, Heq-12); 1.65 (m, 1H, Hβ-15); 1.71 (dd, 1H, 2J = 16.7, 3J1α-2 = 2.2, Hα-1); 1.75 (m, 1H, Hβ-16); 1.87 (ddd, 1H, 3J13-12ax = 12.6, 3J13-17 = 10.9, 3J13-12eq = 3.7, H-13); 1.89 (m, 1H, Hα-16); 2.09 (dd, 1H, 2J = 16.7, 3J1β-2 = 6.8, Hβ-1); 2.49 (ddd, 1H, 3J17-13 = 10.9, 3J17-16α = 10.1, 3J17-16β = 6.3, H-17); 5.10 (d, 1H, 2J = 1.2, HA-21); 5.11 (d, 1H, 2J = 1.2, HB-21); 5.63 (dd, 1H, 3J2-1β = 6.8, 3J2-1α = 2.2, H-2).
(b) Phosphoryl pentachloride (1.04 g; 5.0 mmol) was added to a solution of compound 1 or 2 (0.359 g or 0.403 g; 1.0 mmol) in 10 mL of anhydrous chloroform. The mixture was heated for 8 h under reflux, poured onto water, and extracted with chloroform (3 × 100 mL). The extract was washed with water (3 × 100 mL), dried over anhydrous calcium chloride, and evaporated under reduced pressure (water-jet pump). The residue was subjected to column chromatography on silica gel using eluent-petroleum ether-ethyl acetate (70:1→40:1). Compounds 6, 7 were isolated in a mixture of products, with an overall yield of 77%.
3-Oxo-22,23,24,25,26,27-hexanor-dammar-20(21)-in (3)
White solid, yield: 14%, mp: 161–162 °C, [α]20D = +12 (c 0.05, CHCl3).
3-Oxo-22,23,24,25,26,27-hexanor-dammar-20-chloro-20(21)-ene (4)
White solid, yield: 71%, mp: 148–149 °C, [α]20D = −30 (c 0.05, CHCl3).
3-Chloro-22,23,24,25,26,27-hexanor-dammar-20-chloro-2(3),20(21)-diene (5)
White solid, yield: 8%, mp: 99–100 °C, [α]20D = +18 (c 0.05, CHCl3). 1H NMR (CDCl3, δ ppm, J Hz): 0.87 (s, 3H, H-30); 0.92 (s, 3H, H-19); 1.02 (s, 3H, H-18); 1.04 (s, 3H, H-29); 1.09 (qd, 1H, 2J = 12.4, 3J12ax-13 = 12.4, 3J12ax-11ax = 12.4, 3J12ax-11eq = 4.6, Hax-12); 1.14 (s, 3H, H-28); 1.15 (m, 1H, Heq-15); 1.26 (dd, 1H, 3J5-6ax = 11.0, 3J5-6eq = 1.8, H-5); 1.29 (qd, 1H, 2J = 12.7, 3J11ax-9 = 12.6, 3J11ax-12ax = 12.6, 3J12ax-11eq = 4.6, Hax-11); 1.33 (m, 1H, Heq-7); 1.36 (dd, 1H, 3J9-11ax = 12.6, 3J9-11eq = 2.6, H-9); 1.50 (m, 1H, Heq-11); 1.55 (m, 1H, Hax-6); 1.58 (m, 1H, Hax-7); 1.58 (m, 1H, Heq-6); 1.62 (m, 1H, Heq-12); 1.65 (m, 1H, Hax-15); 1.71 (dd, 1H, 2J = 16.7, 3J1α-2 = 2.2, Hα-1); 1.75 (m, 1H, Heq-16); 1.87 (ddd, 1H, 3J13-12ax = 12.6, 3J13-17 = 10.9, 3J13-12eq = 3.7, H-13); 1.89 (m, 1H, Hax-16); 2.09 (dd, 1H, 2J = 16.7, 3J1β-2 = 6.8, Hβ-1); 2.49 (ddd, 1H, 3J17-13 = 10.9, 3J17-16ax = 10.1, 3J17-16eq = 6.3, H-17); 5.10 (d, 1H, 2J = 1.2, HA-21); 5.11 (d, 1H, 2J = 1.2, HB-21); 5.63 (dd, 1H, 3J2-1β = 6.8, 3J2-1α = 2.2, H-2). 13C NMR (CDCl3, δ ppm): 15.14 (C18); 15.98 (C30); 16.35 (C19); 20.01 (C29); 20.33 (C6); 21.71 (C11); 24.48 (C12); 27.54 (C16); 29.06 (C28); 31.30 (C15); 34.77 (C7); 36.42 (C10); 40.22 (C4); 40.35 (C8); 42.87 (C1); 45.17 (C13); 49.38 (C14); 49.44 (C9); 49.80 (C17); 54.37 (C5); 111.51 (C21); 122.07 (C2); 141.55 (C3); 146.94 (C20).
(c) A mixture of 2 (0.403 g; 1.0 mmol), PCl5 (0.210 g; 1.0 mmol), and a catalytic amount of DMAP in dry pyridine (5 mL) was refluxed for 2 h. After completion of the reaction, the mixture was poured onto water. The precipitate that formed was collected by filtration and washed with water. The residue was purified by SiO2 column chromatography (eluent petroleum ether-ethyl acetate (70:1→40:1)).
3β-Acetoxy-22,23,24,25,26,27-hexanor-dammar-20(21)-in (7)
White solid, yield: 89%, mp: 149–150 °C, [α]20D = −61 (c 0.05, CHCl3).
General procedure of compounds 9a–c
To a solution of compound 3 (0.204 g; 0.6 mmol) in 12 mL of dry dioxane was added paraformaldehyde (0.180 g, 6 mmol), 0.72 mmol of corresponding amine (diethylamine, N-methylpiperazine, or morpholine), NaOAc (0.250 g; 3 mmol), and CuI (6 mg; 0.03 mmol). The reaction mixture was stirred under argon for 10 h at 60 °C. After the reaction was completed by TLC, the mixture was diluted with water and extracted with CHCl3 (3 × 20 mL), and the combined organic layer was washed with water, dried over CaCl2, and evaporated under reduced pressure. The residue was purified by SiO2 column chromatography (eluent petroleum ether-ethyl acetate (40:1→1:1), chloroform).
17-[3-(Diethylamino)-prop-1-yn-1-yl]-22,23,24,25,26,27-hexanor-dammar-3-one (9a)
White solid, yield: 78%, mp: 98–99 °C, [α]20D = −15 (c 0.05, CHCl3). 13C NMR (CDCl3, δ ppm): 12.21 (C3”); 15.39 (C18); 15.39 (C30); 16.08 (C19); 19.64 (C6); 21.03 (C29); 21.85 (C11); 24.92 (C12); 26.77 (C28); 30.19 (C16); 31.06 (C15); 31.90 (C17); 34.10 (C2); 34.70 (C7); 36.94 (C10); 39.98 (C1); 40.26 (C8); 41.07 (C1′); 47.14 (C2″); 47.41 (C4); 49.19 (C14); 49.42 (C13); 50.17 (C9); 55.35 (C5); 73.42 (C21); 89.88 (C20); 217.94 (C3).1H NMR (CDCl3, δ ppm, J Hz): 0.80 (s, 3H, H-30); 0.95 (s, 3H, H-19); 1.01 (s, 3H, H-18); 1.04 (s, 3H, H-29); 1.08 (s, 3H, H-28); 1.11 (t, 6H, 3J = 7.1, H-3″); 1.12 (m, 1H, Hα-15); 1.16 (m, 1H, Hax-12); 1.32 (m, 1H, Hax-11); 1.32 (m, 1H, Heq-7); 1.37 (dd, 1H, 3J5-6ax = 11.7, 3J5-6eq = 2.6, H-5); 1.39 (dd, 1H, 3J9-11ax = 12.6, 3J9-11eq = 2.6, H-9); 1.46 (ddd, 1H, 2J = 13.2, 3J1ax-2ax = 9.6, 3J1ax-2eq = 7.8, Hax-1); 1.48 (m, 1H, Heq-6); 1.56 (m, 1H, Hax-6); 1.57 (m, 1H, Heq-11); 1.59 (m, 1H, Hax-7); 1.63 (m, 1H, Hβ-16); 1.68 (m, 1H, Hβ-15); 1.69 (ddd, 1H, 3J13-12ax = 11.9, 3J13-17 = 11.4, 3J13-12eq = 3.7, H-13); 1.83 (m, 1H, Heq-12); 1.94 (ddd, 1H, 2J = 13.2, 3J1eq-2ax = 7.6, 3J1eq-2eq = 4.7, Heq-1); 2.07 (m, 1H, Hα-16); 2.35 (ddd, 1H, 3J17-13 = 11.4, 3J17-16α = 10.5, 3J17-16β = 6.4, H-17); 2.43 (ddd, 1H, 2J = 15.6, 3J2eq-1ax = 7.8, 3J2eq-1eq = 4.7, Heq-2); 2.51 (ddd, 1H, 2J = 15.6, 3J2ax-1ax = 9.6, 3J2ax-1eq = 7.6, Hax-2); 2.60 (q, 4H, 3J = 7.1, H-2″); 3.45 (s, 2H, H-1′). 15N NMR (CDCl3, δ ppm): 46.35 (N1″).
17-[3-(Pyrrolidin-1-yl)-prop-1-yn-1-yl]-22,23,24,25,26,27-hexanor-dammar-3-one (9b)
White solid, yield: 87%, mp: 100–102 °C, [α]20D = +67 (c 0.05, CHCl3). 13C NMR (CDCl3, δ ppm): 15.36 (C30); 15.39 (C18); 16.07 (C19); 19.63 (C6); 21.03 (C29); 21.79 (C11); 24.09 (C3″(4″)); 24.94 (C12); 26.75 (C28); 30.02 (C16); 31.09 (C15); 31.86 (C17); 34.08 (C2); 34.71 (C7); 36.94 (C10); 39.97 (C1); 40.29 (C8); 43.45 (C1′); 47.41 (C4); 49.27 (C13); 49.29 (C14); 50.12 (C9); 52.14 (C2″(5″)); 55.36 (C5); 73.53 (C21); 89.50 (C20); 217.83 (C3). 1H NMR (CDCl3, δ ppm, J Hz): 0.81 (s, 3H, H-30); 0.95 (s, 3H, H-19); 1.01 (s, 3H, H-18); 1.04 (s, 3H, H-29); 1.08 (s, 3H, H-28); 1.15 (m, 1H, Hα-15); 1.19 (m, 1H, Hax-12); 1.32 (m, 1H, Hax-11); 1.32 (m, 1H, Heq-7); 1.38 (dd, 1H, 3J5-6ax = 11.8, 3J5-6eq = 2.7, H-5); 1.39 (dd, 1H, 3J9-11ax = 12.7, 3J9-11eq = 2.7, H-9); 1.47 (m, 1H, Hax-1); 1.48 (m, 1H, Heq-6); 1.56 (m, 1H, Hax-6); 1.57 (m, 1H, Heq-11); 1.59 (m, 1H, Hax-7); 1.65 (m, 1H, Hβ-16); 1.71 (m, 1H, Hβ-15); 1.74 (ddd, 1H, 3J13-12ax = 11.9, 3J13-17 = 11.4, 3J13-12eq = 3.7, H-13); 1.82 (m, 1H, Heq-12); 1.94 (ddd, 1H, 2J = 13.2, 3J1eq-2ax = 7.6, 3J1eq-2eq = 4.7, Heq-1); 1.96 (m, 4H, H-3″(4″)); 2.08 (m, 1H, Hα-16); 2.36 (ddd, 1H, 3J17-13 = 11.4, 3J17-16α = 10.5, 3J17-16β = 6.4, H-17); 2.43 (ddd, 1H, 2J = 15.6, 3J2eq-1ax = 7.8, 3J2eq-1eq = 4.7, Heq-2); 2.51 (ddd, 1H, 2J = 15.6, 3J2ax-1ax = 9.6, 3J2ax-1eq = 7.6, Hax-2); 2.97 (m, 4H, H-2″(5″)); 3.63 (br.s, 2H, H-1′).
17-[(3-Morpholino-prop-1-yn-1-yl]-22,23,24,25,26,27-hexanor-dammar-3-one (9C)
White solid, yield: 82%, mp: 105–106 °C, [α]20D = −32 (c 0.05, CHCl3). 13C NMR (CDCl3, δ ppm): 15.38 (C30); 15.40 (C18); 16.07 (C19); 19.64 (C6); 21.04 (C29); 21.80 (C11); 24.94 (C12); 26.77 (C28); 30.12 (C16); 31.08 (C15); 31.91 (C17); 34.10 (C2); 34.71 (C7); 36.95 (C10); 39.98 (C1); 40.29 (C8); 47.41 (C4); 47.74 (C1′); 49.23 (C13); 49.24 (C14); 50.14 (C9); 52.05 (C2″(4″)); 55.37 (C5); 66.48 (C3″(5″)); 74.83 (C21); 89.30 (C20); 217.87 (C3). 1H NMR (CDCl3, δ ppm, J Hz): 0.81 (s, 3H, H-30); 0.95 (s, 3H, H-19); 1.00 (s, 3H, H-18); 1.04 (s, 3H, H-29); 1.08 (s, 3H, H-28); 1.13 (m, 1H, Hα-15); 1.17 (m, 1H, Hax-12); 1.29 (m, 1H, Hax-11); 1.32 (m, 1H, Heq-7); 1.37 (dd, 1H, 3J5-6ax = 11.7, 3J5-6eq = 2.7, H-5); 1.39 (dd, 1H, 3J9-11ax = 12.7, 3J9-11eq = 2.7, H-9); 1.46 (m, 1H, Hax-1); 1.48 (m, 1H, Heq-6); 1.55 (m, 1H, Hax-6); 1.57 (m, 1H, Heq-11); 1.59 (m, 1H, Hax-7); 1.66 (m, 1H, Hβ-16); 1.70 (m, 1H, Hβ-15); 1.71 (ddd, 1H, 3J13-12ax = 11.9, 3J13-17 = 11.4, 3J13-12eq = 3.7, H-13); 1.83 (m, 1H, Heq-12); 1.94 (ddd, 1H, 2J = 13.2, 3J1eq-2ax = 7.6, 3J1eq-2eq = 4.7, Heq-1); 2.08 (m, 1H, Hα-16); 2.34 (ddd, 1H, 3J17-13 = 11.4, 3J17-16α = 10.5, 3J17-16β = 6.4, H-17); 2.43 (ddd, 1H, 2J = 15.6, 3J2eq-1ax = 7.8, 3J2eq-1eq = 4.7, Heq-2); 2.51 (ddd, 1H, 2J = 15.6, 3J2ax-1ax = 9.6, 3J2ax-1eq = 7.6, Hax-2); 2.65 (m, 4H, H-2″(6″)); 3.36 (br.s, 2H, H-1′); 3.81 (m, 4H, H-3″(5″)).
General procedure of compounds 10 and 11
Through a solution of compound 4 or 6 (0.377 g or 0.421 g; 1 mmol) in 50 mL of (CH3)2CO:H2O (20:1) solution, 2 eq. of ozone at room temperature was passed until the starting substance disappeared (TLC-control). The residue was subjected to column chromatography on SiO2 using eluent-petroleum ether-ethyl acetate (10:1→1:1), chloroform.
3-Oxo-22,23,24,25,26,27-hexanor-dammar-17-carboxylic acid (10)
White solid, yield: 94%, mp: 135-137 °C, [α]20D = −48 (c 0.05, CHCl3). 13C NMR (CDCl3, δ ppm): 15.29 (C18); 15.55 (C30); 16.08 (C19); 19.64 (C6); 21.03 (C29); 21.71 (C11); 25.34 (C12); 26.26 (C16); 26.76 (C28); 31.53 (C15); 34.08 (C2); 34.87 (C7); 36.92 (C10); 39.97 (C1); 40.39 (C8); 45.56 (C17); 46.59 (C13); 47.42 (C4); 50.05 (C9); 50.06 (C14); 55.36 (C5); 181.26 (C20); 217.92 (C3). 1H NMR (CDCl3, δ ppm, J Hz): 0.87 (s, 3H, H-30); 0.95 (s, 3H, H-19); 1.03 (s, 3H, H-18); 1.04 (s, 3H, H-29); 1.09 (s, 3H, H-28); 1.20 (ddd, 1H, 2J = 12.1, 3J15α-16β = 8.6, 3J15α-16α = 2.4, Hα-15); 1.25 (qd, 1H, 2J = 12.6, 3J12ax-13 = 12.6, 3J12ax-11ax = 12.6, 3J12ax-11eq = 4.6, Hax-12); 1.32 (qd, 1H, 2J = 12.2, 3J11ax-9 = 12.2, 3J11ax-12ax = 12.2, 3J11ax-12eq = 3.7, Hax-11); 1.35 (m, 1H, Heq-7); 1.38 (dd, 1H, 3J5-6ax = 11.7, 3J5-6eq = 2.8, H-5); 1.41 (dd, 1H, 3J9-11ax = 12.5, 3J9-11eq = 2.6, H-9); 1.46 (ddd, 1H, 2J = 13.2, 3J1ax-2ax = 9.6, 3J1ax-2eq = 7.8, Hax-1); 1.48 (m, 1H, Heq-6); 1.56 (m, 1H, Heq-11); 1.57 (m, 1H, Hax-6); 1.61 (m, 1H, Hax-7); 1.72 (ddd, 1H, 2J = 12.1, 3J15β-16β = 10.8, 3J15β-16α = 8.8, Hβ-15); 1.81 (m, 1H, Heq-12); 1.92 (m, 1H, Hβ-16); 1.94 (ddd, 1H, 2J = 13.2, 3J1eq-2ax = 7.6, 3J1eq-2eq = 4.7, Heq-1); 1.98 (ddd, 1H, 3J13-12ax = 12.6, 3J13-17 = 11.5, 3J13-12eq = 4.1, H-13); 2.00 (m, 1H, Hα-16); 2.43 (ddd, 1H, 2J = 15.6, 3J2eq-1ax = 7.8, 3J2eq-1eq = 4.7, Heq-2); 2.47 (ddd, 1H, 3J17-13 = 11.5, 3J17-16α = 10.5, 3J17-16β = 6.5, H-17); 2.51 (ddd, 1H, 2J = 15.6, 3J2ax-1ax = 9.6, 3J2ax-1eq = 7.6, Hax-2).
3β-Acetoxy-22,23,24,25,26,27-hexanor-dammar-17-carboxylic acid (11)
White solid, yield: 91%, mp: 147–148 °C, [α]20D = +5 (c 0.05, CHCl3). 13C NMR (CDCl3, δ ppm): 15.56 (C18); 15.61 (C30); 16.30 (C19); 16.49 (C29); 18.14 (C6); 21.19 (C11); 21.29 (C2′); 23.69 (C2); 25.28 (C12); 26.25 (C16); 27.97 (C28); 31.54 (C15); 31.92 (C2); 35.46 (C7); 37.13 (C10); 37.90 (C4); 38.80 (C1); 40.50 (C8); 45.61 (C13); 46.51 (C14); 50.07 (C17); 50.63 (C9); 55.99 (C5); 80.85 (C3); 170.99 (C1′). 1H NMR (CDCl3, δ ppm, J Hz): 0.81 (dd, 1H, 3J5-6ax = 11.5, 3J5-6eq = 1.8, H-5); 0.84 (s, 3H, H-29); 0.87 (s, 3H, H-28); 0.90 (s, 3H, H-30); 0.92 (s, 3H, H-19); 0.99 (s, 3H, H-18); 1.02 (ddd, 1H, 2J = 11.8, 3J1ax-2ax = 11.6, 3J1ax-2eq = 4.7, Hax-1); 1.09 (qd, 1H, 2J = 12.4, 3J12ax-13 = 12.4, 3J12ax-11ax = 12.4, 3J12ax-11eq = 4.6, Hax-12); 1.12 (m, 1H, Hα-15); 1.23 (qd, 1H, 2J = 12.7, 3J11ax-9 = 12.6, 3J11ax-12ax = 12.6, 3J12ax-11eq = 4.6, Hax-11); 1.29 (m, 1H, Heq-7); 1.32 (dd, 1H, 3J9-11ax = 12.6, 3J9-11eq = 2.6, H-9); 1.46 (m, 1H, Hax-6); 1.53 (m, 1H, Heq-6); 1.54 (m, 1H, Heq-11); 1.58 (m, 1H, Hax-7); 1.60 (m, 1H, Heq-12); 1.61 (m, 1H, Hax-2); 1.64 (m, 1H, Heq-2); 1.65 (m, 1H, Hβ-15); 1.71 (m, 1H, Heq-1); 1.74 (m, 1H, Hβ-16); 1.84 (ddd, 1H, 3J13-12ax = 12.4, 3J13-17 = 10.6, 3J13-12eq = 3.7, H-13); 1.88 (m, 1H, Hα-16); 2.04 (s, 3H, H-2′); 2.48 (td, 1H, 3J17-13 = 10.6, 3J17-16α = 10.6, 3J17-16β = 6.2, H-17); 4.50 (dd, 1H, 3J3-2ax = 10.7, 3J3-2eq = 5.8, H-3).
Procedure of compound 12
Through a solution of the acid 11 (0.406 g; 1 mmol) in 50 mL of anhydrous CH2Cl2, 2 eq. of ozone at –40 °C was passed until the starting substance disappeared (TLC-control). The resulting acid chloride was dissolved in anhydrous CH2Cl2 (30 mL) and treated with pyrrolidine (0.150 g; 1.5 mmol) and three drops of Et3N. The mixture was stirred at room temperature for 3 h, washed with 5% HCl solution (2 × 100 mL) and H2O (100 mL), dried over CaCl2, and the solvent was removed under reduced pressure. The product was purified by column chromatography with SiO2 using CHCl3 and a mixture of CHCl3–EtOH (100:1) as eluents.
3β-Acetoxy-22,23,24,25,26,27-hexanor-dammar-17-N-pyrrolidine amide (12)
White solid, yield: 89%, mp: 114–115 °C, [α]20D = +23 (c 0.05, CHCl3). 13C NMR (CDCl3, δ ppm): 15.69 (C18); 16.01 (C30); 16.34 (C19); 16.50 (C29); 18.15 (C6); 21.26 (C11); 21.34 (C2′); 23.70 (C2); 24.36 (C4″); 25.54 (C12); 26.11 (C3″); 26.60 (C16); 27.97 (C28); 31.66 (C15); 35.53 (C7); 37.12 (C10); 37.89 (C4); 38.80 (C1); 40.53 (C8); 45.06 (C17); 45.81 (C5″); 45.89 (C13); 46.48 (C2″); 49.63 (C14); 50.77 (C9); 55.99 (C5); 80.94 (C3); 171.08 (C1′); 175.10 (C20). 15N NMR (CDCl3, δ ppm): 127.29 (N1″). 1H NMR (CDCl3, δ ppm, J Hz): 0.83 (dd, 1H, 3J5-6ax = 11.7, 3J5-6eq = 2.6, H-5); 0.84 (s, 3H, H-29); 0.85 (s, 3H, H-28); 0.86 (s, 3H, H-19); 0.87 (s, 3H, H-30); 1.01 (s, 3H, H-18); 1.04 (td, 1H, 2J = 12.2, 3J1ax-2ax = 12.2, 3J1ax-2ax = 5.6, Hax-1); 1.15 (m, 1H, Hα-15); 1.17 (m, 1H, Hax-12); 1.27 (m, 1H, Hax-11); 1.30 (m, 1H, Heq-7); 1.57 (m, 1H, Hax-7); 1.31 (dd, 1H, 3J9-11ax = 11.8, 3J9-11eq = 3.3, H-9); 1.47 (m, 1H, Hax-6); 1.49 (m, 1H, Heq-11); 1.53 (m, 1H, Heq-6); 1.57 (m, 1H, Hax-7); 1.62 (m, 1H, Hax-2); 1.64 (m, 1H, Heq-12); 1.64 (m, 1H, Heq-2); 1.71 (m, 1H, Heq-1); 1.74 (m, 1H, Hβ-15); 1.77 (m, 1H, Hβ-16); 1.84 (m, 2H, H-4″); 1.92 (m, 1H, Hα-16); 1.92 (m, 2H, H-3″); 3.44 (s, 3H, H-2″); 2.16 (m, 1H, H-13); 2.52 (td, 1H, 3J17-13 = 10.9, 3J17-16α = 10.9, 3J17-16β = 6.2, H-17); 3.44 (s, 3H, H-2″); 3.47 (m, 1H, H-5″); 4.47 (dd, 1H, 3J3-2ax = 10.7, 3J3-2eq = 5.6, H-3).
3. Discussion
We used hollongdiones 1 and 2 (for X-ray crystal structure, see Figure 3, for NMR data see Supplementary Material Figures S1–S8) in the reactions with POCl3 and PCl5 (Scheme 1). As a result, a series of derivatives 3–8 was obtained and identified.
Figure 3.

The molecular structure of compound 2.
Scheme 1.

Reagents and conditions: a. POCl3/Py, ∆, 8 h; b. PCl5/CHCl3, ∆, 8 h; c. PCl5/Py, DMAP (cat.), ∆, 2 h.
Alkyne 3 was synthesized by the interaction of hollongdione 1 with POCl3 in pyridine, with the yield of 73% after purification by column chromatography, while the vinyl chloride 4 was also isolated as the minor (7%) product. The alkyne synthesis can be explained by dehydrohalogenation of a gem-dihalogen intermediate formation during thermal activation under reaction conditions; vinyl chloride’s mechanism includes the cleavage of one HCl molecule step. It is worth noting that the composition of the products was different when lupane triterpenoids were used as the initial scaffolds: the presence of a molecule with a double C=C bond and tetrahydrofuran ring and a compound with a trichloroacetyl group besides the main alkyne derivative were confirmed, which differs from previous results [15]. Moreover, the abeo-lupene fragment was obtained under the action of POCl3 in pyridine [29,30].
At the same time, the reaction of 1 with PCl5 in CHCl3 led to the vinyl chloride 4 as the main product (71%); alkyne 3 was obtained among the by-products (14%), and ring A was transformed into a 3-chloro-2(3)-ene derivative 5 (8%), which could be explained as a result of the action of the Vilsmeyer–Haack reagent on the C3-oxo-group. The application of phosphorus oxychloride along with DMF to friedelin as the substrate with chlorofriedel-2 and 3-chlorofriedel-3-enes as the products was demonstrated [31].
The protection of the C3 position by the acetoxy group (compound 2) made it possible to avoid the formation of A-ring by-products. In the case of reaction with POCl3, compounds 6 and 7 were isolated only as the mixture of products, with the overall yield of 87%, and 8 became a new by-product (5%). The overall yield of compounds 6 and 7 reached 77% when the halogenating reagent was PCl5 (Conditions b at Scheme 1). Previously, the optimization of the synthetic strategy for the lup-20(30)-yne preparation by adding a catalytic amount of DMAP was proposed [30]. An attempt to streamline the synthesis in this way was also successful, yielding only compound 7 with 89% after purification by column chromatography.
The structure of all compounds was determined by 1H and 13C NMR spectroscopy data. For compound 3, the presence of the oxo-group at the C3 position was confirmed by HMBC cross-peaks of gem-dimethyl groups C28 (δH 1.08 ppm) and C29 (δH 1.04 ppm) along with δC 217.87 ppm. Also, HMBC cross-peaks of methylene protons at C1 and C2 were observed for the C3-oxo-group. For the ethynyl substituent at C17 with δC 89.04 ppm of the quaternary carbon atom and δC 67.33 ppm and δH 2.05 ppm of the methine group, a number of interactions were observed, confirming the position of substitution. Thus, HMBC proton cross-peaks of H-13, H-17, and Hβ-16, with quaternary position and cross-peak H-17/C21 were confirmed. In addition, the acetylene proton signal has a doublet with the value 4J = 2.4 Hz, with a methine proton H-17, which is confirmed in the spectrum COSY-DQF and its response splitting (Figure 4), (Supplementary Material Figures S9–S16).
Figure 4.

The key NMR assignments and significant {1H, 13C} HMBC, {1H, 1H} COSY, and NOESY correlations of compounds 3–7.
For the vinyl chloride group in compounds 4–7 with characteristic values δC ~147 ppm of the quaternary carbon atom C20 and δC ~112 ppm for the terminal methylene group, HMBC interactions with cycle D protons were observed, which confirms the position of substitution C17. In particular, HMBC cross-peaks of proton Hα-16, Hβ-16, and H-13 with quaternary carbon atom C20 and HMBC cross-peaks of terminal protons with carbon signal C17 at δC ~50 ppm were observed. Additionally, in the NOESY spectrum, cross-peaks were observed between one of the terminal protons with Heq-12, H-13 and H-17. The presence of acetate in the C3 position of cycle A for compound 4 is confirmed by the HMBC cross peak of the doublet-doublet signal H-3 (δH 4.48 ppm) with a carboxyl group signal at δC 170.95 ppm (Figure 4).
For compound 5, the presence of C2 = C3 double bond in cycle A was confirmed on the basis of HMBC cross-peaks gem-dimethyl C28 and C29 groups (δH 1.14 and 1.04 ppm, respectively) with a quaternary Cl-bearing position at δC 141.55 ppm, and H-2 protons (δH 5.63 ppm) and α,β-protons of the methylene group C1 (δH 1.71, 2.09 ppm). A double bond methine proton H-2 (δH 5.63 ppm) was represented by doublet-doublet splitting with 3J = 6.8 and 2.2 Hz on neighboring methylene protons at C1. The position of the carbon signal C1 at δC 42.87 ppm was localized on the basis of HMBC cross-peaks with methine protons H-5 (δH 1.26 ppm) and H-9 (δH 1.36 ppm) and methyl group protons at C19 (δH 0.92 ppm). The α-orientation of protons at C1 (δH 1.71 ppm) was confirmed by NOESY cross-peaks with H-5 and H-9; for the protons Hβ-1, NOE interactions with H3-19 and Heq-11 (δH 1.50 ppm) protons were also observed (Figure 5), (Supplementary Material Figures S17–S40).
Figure 5.

The key NMR assignments and significant {1H, 13C} HMBC, {1H, 1H} COSY, and NOESY correlations of compounds 5, 8. The oxygen atom is highlighted in red color in the structure of compound 8.
The formation of a 1,2-dichloro-1-hydroxyethyl group at the C17 of compound 8 is confirmed by the characteristic 13C NMR values: δC 97.32 ppm for the quaternary position C20 and δC 54.62 ppm for the methylene group C21. For OH, Cl-bearing quaternary carbon, three-bond HMBC correlations with protons Hα-16, Hβ-16 (δH 2.00 and 1.76 ppm, respectively), and H-13 (δH 1.95 ppm) are observed. The HMBC spectrum also contains cross peaks of diastereotopic methylene protons at C21 (δH 4.01 and 4.05 ppm) with carbons C20 and C17 (δC 51.18 ppm), and the protons themselves in the 1H NMR spectrum are presented as two doublets with a geminal constant of 12.2 Hz.
The molecular structures of 3-oxo-22,23,24,25,26,27-hexanor-dammar-20(21)-in 3, 3-oxo-22,23,24,25,26,27-hexanor-dammar-20-chloro-20(21)-en 4, and 22,23,24,25,26,27-hexanor-dammar-3,20-dichloro-20(21), 2(3)-dien 5 were determined by X-ray analysis (Figure 6). All the compounds have typical four-cycle moiety as in hollongdione [20], including three six- and one five-membered trans-fused cycles. All the six-membered cycles in 3, 4, and 5 adopt chair conformation, except the chlorine substituted one in 5 with double bond C2 = C3 equaling 1.307(4) Å, which has a distorted half-chair conformation. The five-membered cycles show twist conformation in all the compounds. The length of the C≡C bond in 3 slightly differs, being the same within the limit of experimental accuracy (Table 1) and very close to the average literature value of 1.174(11) [32]. The alkynyl substituent on C17 in 3 is not ideally linear, so the angle C17C20C21 is not the 180° that is usual for such moieties. The bond lengths and orientation of the vinyl chloride group on C17 in 4 and 5, describing by torsion angle C16C17C20Cl1(Cl2), are the same in both structures (Table 1). There are not any specific intermolecular interactions in the crystals under consideration. The crystal structure of compounds is stabilized by van der Waals interactions. The main XRD data and experimental details for compounds 2–5 are presented in Table 2.
Figure 6.

Molecular structures of compounds 3, 4, and 5.
Table 1.
Selected geometric parameters of compounds 3–5.
| 3 | 4 | 5 | |
|---|---|---|---|
| C20-C21 | 1.157 (13)/1.136 (16) * | 1.312 (4) | 1.315 (5) |
| C17C20C21 | 175.3 (11)/175.2 (15) | − | − |
| C20-Cl1(Cl2) | 1.750 (3) | 1.748 (4) | |
| C20-C17 | 1.493 (4) | 1.496 (4) | |
| C16C17C20Cl1 (Cl2) | −55.9 (3) | −53.1 (3) |
* geometrical parameters for two independent molecules in 3.
Table 2.
XRD data and experimental details for compounds 2–5.
| 2 | 3 | 4 | 5 | |
|---|---|---|---|---|
| Empirical formula | C26H42O3 | C24H35O | C24H37ClO | C24H36Cl2 |
| Formula weight | 402.59 | 339.52 | 376.98 | 395.43 |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Monoclinic |
| Space group | P21 | P21 | P1 | C2 |
| Unit cell dimensions a, b, c (Å) | 25.332(2), 7.6508(5), 25.543(2) | 7.5834(6), 20.548(2), 12.9331(9) | 6.3333(3), 7.4257(5), 11.8452(8) | 13.880(1), 6.3288(4), 24.409(2) |
| α, β, γ (°) | 90, 107.232(3), 90 | 90, 90.020(4), 90 | 89.026(2), 84.321(2), 69.960(2) | 90, 97.004(4), 90 |
| Volume (Å3) | 4728.1(6) | 2015.3(3) | 520.69(6) | 2128.1(3) |
| Z | 8 | 4 | 1 | 4 |
| Density (calcd.) (Mg.mm−3) | 1.131 | 1.119 | 1.202 | 1.234 |
| F(000) | 1776 | 748 | 206 | 856 |
| Absorption coefficient (mm−1) | 0.07 | 0.07 | 0.19 | 0.311 |
| Crystal size (mm3) | 1.00 × 0.08 × 0.05 | 1.00 × 0.45 × 0.01 | 1.00 × 0.72 × 0.20 | 1.00 × 0.30 × 0.02 |
| Tmin, Tmax | 0.832, 0.928 | 0.770, 0.928 | 0.764, 0.813 | 0.762, 0.801 |
| Θ range for data collection | 0.8–26.5 | 1.0–26.1 | 2.9–33.5 | 0.8–27.1 |
| Reflections collected | 101,280 | 19,801 | 14,660 | 35,210 |
| Independent reflections [Rint] | 19,379 [0.088] | 7303 [0.095] | 6349 [0.024] | 4471 [0.049] |
| Observed [I > 2 σ (I)] reflections | 11,585 | 4090 | 5142 | 4097 |
| Completeness to θ = 25° (%) |
99.9 | 99.7 | 99.9 | 98.8 |
| Data/restraints/parameters | 193,79/1/1073 | 7303/2/462 | 6349/3/240 | 4471/1/240 |
| Goodness-of-fit on F2 | 1.01 | 0.99 | 1.02 | 1.107 |
| Final R1, [I > 2σ(I)] | 0.061 | 0.064 | 0.046 | 0.041 |
| Final wR2 (all data) | 0.155 | 0.148 | 0.138 | 0.109 |
| Largest diff. peak/hole e.Å−3 | 0.20, −0.21 | 0.18, −0.17 | 0.36, −0.27 | 0.27, −0.201 |
| Absolute structure parameter | −0.1(5) | 0.5(10) | 0.039(17) | 0.029(17) |
| CCDC | 2,315,012 | 2,315,013 | 2,315,014 | 2,350,691 |
It is known that alkynes are simple and valuable precursors that act as electrophiles due to the presence of π-bonds and can be successfully utilized for the construction of various classes of carbocycles, heterocycles, complex molecular architectures, and natural products [33]. For example, the terminal carbon–carbon triple bond is an activating functional group that is required to render the substrate active in the Mannich reaction. In the last decade, new propargylamines were synthesized by the interaction of 19- and 28-alkynyltriterpenoids with N-methylpiperazine [34]. Propargylated oleanolic and glycyrrhetic acids were involved in aminomethylation by the Mannich reaction [35]. Moreover, this type of modification has been studied on the basis of steroid alkynes, and the interest keeps growing [36].
Taking into account the above, we involved alkyne 3 in the Mannich reaction with diethylamine, pyrrolidine, or morpholine, giving the conjugates 9a–c in the yields of 78–87% (Scheme 2).
Scheme 2.

Reagents and conditions: a. diethylamine, pyrrolidine, or morpholine, paraformaldehyde, NaOAc, CuI, 1,4-dioxane, 10 h, 60 °C.
The formation of aminomethylated products 9a–c is confirmed by NMR spectra due to the presence of methylene group signals in the range δC 41.07 ÷ 47.74 ppm (δH 3.36 ÷ 3.62 ppm), for which HMBC cross-peaks with carbon signals of the acetylene group (δC 73.42÷74.89 and δC 89.28 ÷ 89.88 ppm) are observed. In addition, cross-peaks of protons of the α-position of amines (δH 2.60 ÷ 2.69 ppm) with the carbon signal of the newly formed methylene group C1′ are observed in the HMBC spectrum (Supplementary Material Figures S41–S63).
In the next stage of our research, we decided to involve the hollongdione vinyl chloride for transformation to carboxylic acid. It is known that androstane methylketones are transformed to carboxylic acids by a haloform reaction [37,38]. On the other hand, dichlorovinyl insecticides by ozone oxidation in water-containing solvents afford carboxy-derivatives [39].
Vinyl chloride is a well-known monomer for the synthesis of copolymers [40], but no instances of its oxidation to acid have been found in the case of triterpenoid or steroid scaffolds. We propose here a new approach for the synthesis of acids from C17-methylketones. For this, vinyl chlorides 4 and 6 were oxidized with ozone in acetone-water medium to give acids 10 and 11. So, these acids could be called “hollongdionoic acid”-like, androstane-17-carboxylic acids were given the trivial name “etianic acids” [41]. One-pot synthesis of amide 12 in the yield of 89% was realized by the oxidation of vinyl chloride 6 in CH2Cl2 followed by the addition of pyrrolidine (Scheme 3).
Scheme 3.

Reagents and conditions: a. O3, (CH3)2C(O):H2O (20:1), rt; b. i O3, CH2Cl2, −40 °C, ii pyrrolidine, CH2Cl2, rt, 3 h.
The formation of a carboxyl group at the C17 position of compound 10 was established based on the presence of a signal at δC 181.26 ppm in the 13C NMR spectrum. The HMBC spectrum showed cross-peaks of the H-13, Hβ-16, and H-17 (δH 1.98, 1.92, and 2.47 ppm, respectively) protons of cycle D with a quaternary carbon signal at δC 181.26 ppm, (Supplementary Material Figures S64–S79).
4. Materials and Methods
NMR spectra were recorded on a Bruker “Avance-III” 500 MHz spectrometer (Bruker, Billerica, MA, USA), (500, 125, and 50 MHz for 1H, 13C, and 15N, respectively, δ, ppm, J, Hz) in CDCl3 with tetramethylsilane as the internal standard. Mass spectra were obtained on a liquid chromatograph–mass spectrometer LCMS-2010 EV (Shimadzu, Kyoto, Japan). Melting points were detected on the micro table “Rapido PHMK05” (Nagema, Dresden, Germany). Optical rotations were measured on the polarimeter “Perkin-E lmer 241 MC” (Perkin Elmer, Waltham, MA, USA) in a tube length of 1 dm. Thin-layer chromatography analyses were performed on Sorbfil plates (Sorbpolimer, Krasnodar, Russian Federation) using the solvent system chloroform-ethyl acetate, 40:1. Substances were detected by 10% H2SO4 with subsequent heating to 100–120 °C for 2–3 min. The X-ray diffraction experiments for compounds 2–5 were carried out at 296(2) K on a Bruker KAPPA APEX II diffractometer (graphite-monochromated Mo Kα radiation). Reflection intensities were corrected for absorption by the SADABS-2016 program [42]. The structure of compounds was solved by direct methods using the SHELXT-2014 program [43] and refined by the anisotropic (isotropic for all H atoms) full-matrix least-squares method against F2 of all reflections by SHELXL-2018 [44]. The positions of the hydrogen atoms were calculated geometrically and refined in the riding model. The asymmetric unit of 2 contained four molecules, the unit of 3—two molecules, and structure of this compound was refined as a twin. Crystallographic data for 2–5 have been deposited at the Cambridge Crystallographic Data Centre as supplementary publication Nos. CCDC 2315012, 2315013, 2315014, and 2350691. A copy of the data can be obtained, free of charge, on application to the CCDC, 12 Union Road, Cambridge CB21EZ, UK (fax: +44 122 3336033 or e-mail: deposit@ccdc.cam.ac.uk; internet: www.ccdc.cam.ac.uk, accessed on 14 October 2023). Compound 1 was obtained according to the method described previously [20].
5. Conclusions
Hollongdione is the naturally occurring “triterpenoid-steroid” hybrid. Its design and synthesis is deemed to be very interesting from the perspective of potent activities. Its reactivity with an emphasis on dehydrohalogenation reactions such as phosphorus-chlorine derivatives (POCl3, PCl5) was studied for the first time. As a result, compounds with C17 alkynyl and vinyl chloride substituents were synthesized, the structures of which were confirmed by NMR spectra and X-ray analysis. The choice of a dehydrohalogenating agent made it possible to regulate the structure, composition, and the yield of the products. Hollongdione C17-alkyne was successfully transformed into aminomethylated products by the Mannich reaction. An opportunity for a one-stage pathway from hollongdione vinyl chloride to the carboxylic acid with the following amide preparation is also demonstrated.
Acknowledgments
The authors would like to acknowledge the Center for the Collective Use “Chemistry” of the Ufa Institute of Chemistry of the UFRC RAS, the RCCU “Agidel” of the UFRC RAS, and the “Chemical service center for collective use” of the SB RAS for carrying out spectral and analytical measurements.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms25158356/s1.
Author Contributions
Conceptualization, O.K.; methodology, O.K. and I.S.; validation, I.S. and Z.G.; formal analysis, A.L., T.R. and D.P.; investigation, A.L. and T.R.; writing—original draft preparation, O.K., I.S. and Z.G.; writing—review and editing, O.K. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available within the article or its Supplementary Materials.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding Statement
This work was supported by the Federal Program No. 1021062311392-9-1.4.1 and No. 123011300044-5.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Chávez-Hernández A.L., Sánchez-Cruz N., Medina-Franco J.L. Fragment library of natural products and compound databases for drug discovery. Biomolecules. 2020;10:1518. doi: 10.3390/biom10111518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siddiq A., Dembitsky V. Acetylenic anticancer agents. Anti-Cancer Agents Med. Chem. 2008;8:132–170. doi: 10.2174/187152008783497073. [DOI] [PubMed] [Google Scholar]
- 3.Pęcak P., Świtalska M., Chrobak E., Boryczka G., Bębenek E. Betulin Acid Ester Derivatives Inhibit Cancer Cell Growth by Inducing Apoptosis through Caspase Cascade Activation: A Comprehensive In Vitro and In Silico Study. Int. J. Mol. Sci. 2022;24:196. doi: 10.3390/ijms24010196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heller L., Schwarz S., Obernauer A., Csuk R. Allobetulin derived seco-oleananedicarboxylates act as inhibitors of acetylcholinesterase. Bioorg. Med. Chem. Lett. 2015;25:2654. doi: 10.1016/j.bmcl.2015.04.086. [DOI] [PubMed] [Google Scholar]
- 5.Pereira V.V., Pereira N.R., Pereira R.C.G., Duarte L.P., Takahashi J.A., Silva R.R. Synthesis and antimicrobial activity of ursolic acid ester derivatives. Chem. Biodivers. 2022;19:e202100566. doi: 10.1002/cbdv.202100566. [DOI] [PubMed] [Google Scholar]
- 6.Tsepaeva O.V., Salikhova T.I., Ishkaeva R.A., Kundina A.V., Abdullin T.I., Laikov A.V., Tikhomirova M.V., Idrisova L.R., Nemtarev A.V., Mironov V.F. Bifunctionalized Betulinic Acid Conjugates with C-3-Monodesmoside and C-28-Triphenylphosphonium Moieties with Increased Cancer Cell Targetability. J. Nat. Prod. 2023;86:1939–1949. doi: 10.1021/acs.jnatprod.3c00304. [DOI] [PubMed] [Google Scholar]
- 7.Nazaryan S., Bruguière A., Hovhannisyan N., Miyamoto T., Dias A.M.M., Bellaye P.S., Collin B., Briand L., Mitaine-Offer A.C. Oleanolic Acid Glycosides from Scabiosa caucasica and Scabiosa ochroleuca: Structural Analysis and Cytotoxicity. Molecules. 2023;28:4329. doi: 10.3390/molecules28114329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gauthier C., Legault J., Piochon-Gauthier M., Pichette A. Advances in the synthesis and pharmacological activity of lupane-type triterpenoid saponins. Phytochem. Rev. 2011;10:521–544. doi: 10.1007/s11101-010-9176-y. [DOI] [Google Scholar]
- 9.Augustin J.M., Kuzina V., Andersen S.B., Bak S. Molecular activities, biosynthesis and evolution of triterpenoid saponins. Phytochemistry. 2011;72:435–457. doi: 10.1016/j.phytochem.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 10.Akhmetova V.R., Shakurova E.R., Khalilova A.Z., Khalilov L.M., Dzhemilev U.M. Synthesis and transformations of 20-oxo-30-nortaraxasteryl acetate derivatives. Russ. J. Org. Chem. 2007;43:363–369. doi: 10.1134/S1070428007030050. [DOI] [Google Scholar]
- 11.Pokorný J., Olejníková D., Frydrych I., Lišková B., Gurská S., Benická S., Šarek J., Kotulová J., Hajdúch M., Džubák P., et al. Substituted dienes prepared from betulinic acid–synthesis, cytotoxicity, mechanism of action, and pharmacological parameters. Eur. J. Med. Chem. 2021;224:113706. doi: 10.1016/j.ejmech.2021.113706. [DOI] [PubMed] [Google Scholar]
- 12.Heise N.V., Kahnt M., Wagner C., Al-Harrasi A., Csuk R. An unprecedented epimerization and annelation reaction of platanic acid amides. J. Mol. Struct. 2020;1220:128718. doi: 10.1016/j.molstruc.2020.128718. [DOI] [Google Scholar]
- 13.Habrant D., Rauhala V., Koskinen A.M.P. Conversion of carbonyl compounds to alkynes: General overview and recent developments. Chem. Soc. Rev. 2010;39:2007–2017. doi: 10.1039/b915418c. [DOI] [PubMed] [Google Scholar]
- 14.Kazakova O.B., Medvedeva N.I., Tolstikov G.A., Kukovinets O.S., Yamansarov E.Y., Spirikhin L.V., Gubaidullin A.T. Synthesis of terminal acetylenes using POCl3 in pyridine as applied to natural triterpenoids. Mendeleev Commun. 2010;20:234–236. doi: 10.1016/j.mencom.2010.06.018. [DOI] [Google Scholar]
- 15.Kazakova O.B., Yamansarov E.Y., Spirikhin L.V., Yunusov M.S., Baikova I.P., Kukovinets O.S., Musin R.Z. Effective synthesis and transformations of alkyne betulin derivatives. Russ. J. Org. Chem. 2011;47:456460. doi: 10.1134/S1070428011030249. [DOI] [Google Scholar]
- 16.Shakhmaev R.N., Sunagatullina A.S., Abdullina E.A., Zorin V.V. Pd-catalyzed synthesis of 2-alkynyl derivatives of 19β, 28-epoxy-18α-olean-1-en-3-one. Russ. J. Org. Chem. 2017;53:1705–1709. doi: 10.1134/S1070428017110173. [DOI] [Google Scholar]
- 17.Kotovshchikov Y.N., Latyshev G.V., Lukashev N.V., Beletskaya I.P. Alkynylation of steroids via Pd-free Sonogashira coupling. Org. Biomol. Chem. 2015;13:5542–5555. doi: 10.1039/C5OB00559K. [DOI] [PubMed] [Google Scholar]
- 18.Ngoc T.D., Dehaen W., Meervelt L.V., Balzarini J. Synthesis of Heterocyclic Triterpene Derivatives with Biological Activities via Click Reaction. Curr. Org. Chem. 2019;23:2969–2974. doi: 10.2174/1385272823666191212110411. [DOI] [Google Scholar]
- 19.Khusnutdinova E.F., Petrova A.V., Kukovinets O.S., Kazakova O.B. Synthesis and Cytotoxicity of 28-N-Propargylaminoalkylated 2,3-Indolotriterpenic acids. Nat. Prod. Commun. 2018;13:1934578X1801300. doi: 10.1177/1934578X1801300603. [DOI] [Google Scholar]
- 20.Smirnova I.E., Kazakova O.B., Huong D.T.T., Minnibaeva E.M., Lobov A.N., Suponitsky K.Y. One-pot synthesis of hollongdione from dipterocarpol. Nat. Prod. Commun. 2014;9:1934578X1400901005. doi: 10.1177/1934578X1400901005. [DOI] [PubMed] [Google Scholar]
- 21.Grougnet R., Magiatis P., Mitaku S., Skaltsounis A.L., Cabalion P., Tillequin F., Michel S. Dammarane Triterpenes from Gardenia aubryi Vieill. Helv. Chim. Acta. 2011;94:656–661. doi: 10.1002/hlca.201000286. [DOI] [Google Scholar]
- 22.Phongmaykin J., Kumamoto T., Ishikawa T., Suttisri R., Saifah E. A new sesquiterpene and other terpenoid constituents of Chisocheton penduliflorus. Arch. Pharm. Res. 2008;31:21–27. doi: 10.1007/s12272-008-1115-8. [DOI] [PubMed] [Google Scholar]
- 23.Chen H.T., Chuang C.W., Cheng J.C., Yeh Y.J., Chang T.H., Shi Y.T., Chao C.H. Terpenoids with anti-influenza activity from the leaves of Euphorbia leucocephala. Nat. Prod. Res. 2023;37:936–943. doi: 10.1080/14786419.2022.2098739. [DOI] [PubMed] [Google Scholar]
- 24.Révész L., Hiestand P., La Vecchia L., Naef R., Naegeli H.U., Oberer L., Roth H.J. Isolation and synthesis of a novel immunosuppressive 17α-substituted dammarane from the flour of the Palmyrah palm (Borassus flabellifer) Bioorg. Med. Chem. Lett. 1999;9:1521–1526. doi: 10.1016/S0960-894X(99)00220-6. [DOI] [PubMed] [Google Scholar]
- 25.Giaginis C., Theocharis S. Current evidence on the anticancer potential of Chios mastic gum. Nutr. Cancer. 2011;63:1174–1184. doi: 10.1080/01635581.2011.607546. [DOI] [PubMed] [Google Scholar]
- 26.Smirnova I., Drăghici G., Kazakova O., Vlaia L., Avram S., Mioc A., Mioc M., Macaşoi I., Dehelean C., Voicu A., et al. Hollongdione arylidene derivatives induce antiproliferative activity against melanoma and breast cancer through pro-apoptotic and antiangiogenic mechanisms. Bioorg. Chem. 2022;119:105535. doi: 10.1016/j.bioorg.2021.105535. [DOI] [PubMed] [Google Scholar]
- 27.Shao L.D., Bao Y., Shen Y., Su J., Leng Y., Zhao Q.S. Synthesis of selective 11β-HSD1 inhibitors based on dammarane scaffold. Eur. J. Med. Chem. 2017;135:324–338. doi: 10.1016/j.ejmech.2017.04.059. [DOI] [PubMed] [Google Scholar]
- 28.Shao L.D., Xu J., Li X.N., Zhang Z.J., Shi X., Ren J., He J., Zhao Y., Leng Y., Xia C.F., et al. Synthesis of hupehenols A, B, and E from protopanaxadiol. RSC Adv. 2016;6:35792–35803. doi: 10.1039/C6RA04236H. [DOI] [Google Scholar]
- 29.Flekhter O.B., Medvedeva N.I., Kukovinets O.S., Spirikhin L.V., Galkin E.G., Galin F.Z., Golovanov D.G., Pavlova N.I., Savinova O.V., Boreko E.I., et al. Synthesis and antiviral activity of lupane triterpenoids with modified cycle E. Russ. J. Bioorg. Chem. 2007;33:584–588. doi: 10.1134/S1068162007060088. [DOI] [PubMed] [Google Scholar]
- 30.Khusnutdinova E.F., Bremond P., Petrova A.V., Kukovinets O.S., Kazakova O.B. Synthesis of lupane mono-and bis-C19-(1, 2, 3-triazolyl)-triterpenoids by “Click” reaction. Lett. Org. Chem. 2017;14:743–747. doi: 10.2174/1570178614666170918120624. [DOI] [Google Scholar]
- 31.Das J., Sarkar A., Ghosh P. Friedelane triterpenoids: Transformations toward A-ring modifications including 2-homo derivatives. New J. Chem. 2018;42:6673–6688. doi: 10.1039/C8NJ00009C. [DOI] [Google Scholar]
- 32.Allen F.H., Kennard O., Watson D.G., Brammer L., Orpen A.G., Taylor R.J. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 1987;2:S1–S19. doi: 10.1039/p298700000s1. [DOI] [Google Scholar]
- 33.Shaw R., Elagamy A., Althagafi I., Pratap R. Synthesis of alkynes from non-alkyne sources. Org. Biomol. Chem. 2020;18:3797–3817. doi: 10.1039/D0OB00325E. [DOI] [PubMed] [Google Scholar]
- 34.Khusnutdinova E.F., Apryshko G.N., Petrova A.V., Kukovinets O.S., Kazakova O.B. The synthesis and selective cytotoxicity of new Mannich bases, derivatives of 19-and 28-alkynyltriterpenoids. Russ. J. Bioorg. Chem. 2018;44:123–127. doi: 10.1134/S1068162018010090. [DOI] [Google Scholar]
- 35.Petrova A., Tretyakova E., Khusnutdinova E., Kazakova O., Slita A., Zarubaev V., Ma X., Jin H., Xu H., Xiao S. Antiviral opportunities of Mannich bases derived from triterpenic N-propargylated indoles. Chem. Biol. Drug Des. 2024;103:e14370. doi: 10.1111/cbdd.14370. [DOI] [PubMed] [Google Scholar]
- 36.Hirai S., Harvey R.G., Jensen E.V. The Mannich reaction: Improved conditions and application to 20-ketosteroids. Tetrahedron Lett. 1963;4:1123–1126. doi: 10.1016/S0040-4039(01)90787-7. [DOI] [Google Scholar]
- 37.Zhu N., Ling Y., Lei X., Handratta V., Brodie A.M.H. Novel P45017α inhibitors: 17-(2′-oxazolyl)-and 17-(2′-thiazolyl)-androstene derivatives. Steroids. 2003;68:603–611. doi: 10.1016/S0039-128X(03)00082-5. [DOI] [PubMed] [Google Scholar]
- 38.Kovács D., Wölfling J., Szabó N., Szécsi M., Kovács I., Zupkó I., Frank É. An efficient approach to novel 17-5′-(1′,2′,4′)-oxadiazolyl androstenes via the cyclodehydration of cytotoxic O-steroidacylamidoximes, and an evaluation of their inhibitory action on 17α-hydroxylase/C17,20-lyase. Eur. J. Med. Chem. 2013;70:649–660. doi: 10.1016/j.ejmech.2013.10.038. [DOI] [PubMed] [Google Scholar]
- 39.Nagendrappa G., Griesbaum K. Degradation of cyclodiene insecticide-like vinylic chloro compounds by ozone. J. Agric. Food Chem. 1978;26:581–583. doi: 10.1021/jf60217a022. [DOI] [Google Scholar]
- 40.Kazakova O.B., Medvedeva N.I., Yamansarov E.Y., Spirikhin L.V., Khusnutdinova E.F., Kukovinets O.S., Tolstikov G.A. Synthesis of Vinyl Chloride Derivatives on the Basis of Betulin. Chem. Sustain. Dev. 2011;19:335–338. [Google Scholar]
- 41.Monder C., Bradlow H.L. Carboxylic acid metabolites of steroids. J. Steroid Biochem. 1977;8:897–908. doi: 10.1016/0022-4731(77)90101-7. [DOI] [PubMed] [Google Scholar]
- 42.Krause L., Herbst-Irmer R., Sheldrick G.M., Stalke D.J. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. Appl. Cryst. 2015;48:3–10. doi: 10.1107/S1600576714022985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheldrick G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A. 2015;71:3–8. doi: 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheldrick G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C. 2015;71:3–8. doi: 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data available within the article or its Supplementary Materials.