


Abstract
Chimeric oligonucleotides (chimeras), consisting of RNA and DNA bases folded by complementarity into a double hairpin conformation, have been shown to alter or repair single bases in plant and animal genomes. An uninterrupted stretch of DNA bases within the chimera is known to be active in the sequence alteration while RNA residues aid in complex stability. In this study, the two strands were separated in the hope of defining the role each plays in conversion. Using a series of single-stranded oligonucleotides, comprised of all RNA or DNA residues and various mixtures, several new structures have emerged as viable molecules in nucleotide conversion. When extracts from mammalian and plant cells and a genetic readout assay in bacteria are used, single-stranded oligonucleotides, containing a defined number of thioate backbone modifications, were found to be more active than the original chimera structure in the process of gene repair. Single-stranded oligonucleotides containing fully modified backbones were found to have low repair activity and in fact induce mutation. Molecules containing various lengths of modified RNA bases (2′-O-methyl) were also found to possess low activity. Taken together, these results confirm the directionality of nucleotide conversion by the DNA strand of the chimera and further present a novel, modified single-stranded DNA molecule that directs conversion in plant and animal cell-free extracts.
INTRODUCTION
A chimeric RNA/DNA oligonucleotide (chimera) was developed to correct point or frameshift mutations in episomal or chromosomal copies of genes. These molecules were first used in experiments aimed at elucidating the role of RNA in homologous recombination (1–3). While undergoing the DNA pairing phase of the process, RNA was found to stabilize the central reaction intermediate. Due to the increase in stability, the chimera enabled nucleotide exchange in one of the pairing partners (4). This ‘gene repair’ reaction was demonstrated in an episomal target by Kmiec and colleagues (5) wherein a mutated alkaline phosphatase gene was corrected to generate a functional protein. This repair event was detected at both the genetic and phenotypic levels. The same group demonstrated correction of a chromosomal target (β globin gene) by correcting the βs mutation in lymphoblastoid cells (6).
Since these original papers appeared, a series of publications have confirmed and extended the technique and demonstrated applicability to various systems. A similar alkaline phosphatase gene was targeted in HUH7 cells (7) followed by mutation in the Factor IX gene in primary hepatocytes by the same group (8). Lai and colleagues used the chimera to correct a mutation in the carbonic anhydrase gene in kidney cells (9). Recent experiments have shown that chimera can effectively repair a mutation in the tyrosinase gene which upon correction alters the appearance of epithelial cells in culture (10) and hair color in an animal model (11).
The chimera has been used successfully to alter genes in plants. Beetham et al. (12) created a mutation in the ALS gene and was able to select converted tobacco cells in the presence of a herbicide. Zhu et al. (13) used the chimera to correct a mutation in a fusion gene using a GFP readout system. Importantly, these workers demonstrated genetic inheritance through Mendelian segregation. The data now confirm that chimeric oligonucleotides can direct a repair or mutagenic event and that these DNA sequence changes are genetically transmissible.
While the success rate of this technique is rising and application to humans is potentially at hand, the molecular and biochemical mechanism by which alteration occurs is only beginning to be understood. We developed a cell-free extract system to elucidate the reactions that enable correction (14). A mutation in a gene, which confers antibiotic resistance in Escherichia coli, was created and posed as the target for chimera-directed repair, catalyzed by a cell-free extract. Due to its high in vivo activity for repair events, the hepatocytoma cell line, HUH7, was used as the source of the extract. Correction is measured by counting and sequencing the recovered plasmid from bacterial colonies growing in the presence of the appropriate antibiotic. This system provides a way to assess DNA changes and phenotypic responses in a controlled environment. In addition, the growth of the colonies confirms genetic inheritance and stability of the repair event. Using this strategy, we demonstrated the repair of both point and frameshift mutations and that the reaction depends heavily on the presence of the hMSH2 gene product (14). Hence, it is likely that at least part of the mismatch repair pathway is involved in chimera-directed repair events, although more recent data suggest that a gene conversion pathway may also be involved (M.C.Rice, M.Bruner, K.Czymmek and E.B.Kmiec, in preparation).
Recently, we began a systematic dissection of the original chimera structure in the hope of defining its functional components (15). Several significant observations were made. First, the all-DNA strand of the chimera directs the initial correction once bound to its complementary target strand. Second, the RNA-containing strand engages the second strand of the target and stabilizes the complex joint. Third, if the RNA-containing strand attempts to direct the conversion, then a series of mutagenic reactions resulting in non-specific base alteration are derived. Finally, the chimera is an active participant in DNA pairing reactions, conjoining double strand targets into a complement-stabilized displacement loop (D-loop) (16,17). But the most significant observation for our purposes here was that the DNA strand initializes DNA repair.
Based on the above data, we pursued the possibility that the DNA strand of the chimera can act solely and independently in directing gene repair events. As a corollary series of experiments, we investigated the capacity of RNA strands and single-stranded molecules, containing varying lengths of RNA, to direct repair. Correction of both point and frameshift mutations were tested as well as the impact on reaction mixtures lacking functional MSH2. Here, we report that modified single strands of DNA can enable specific repair of mutations at a higher level than their RNA counterparts.
MATERIALS AND METHODS
Plasmids, oligonucleotides and cells
The plasmids containing the mutant kanamycin gene (pKsm4021) and the mutant tetracycline gene (pTsΔ208) and the chimeric oligonucleotides used to correct these genes have been described previously (14,15,18). Chimeric oligonucleotides and single-stranded oligonucleotides (including those with the indicated modifications) were synthesized using available phosphoramidites on controlled pore glass supports. After deprotection and detachment from the solid support, each oligonucleotide was gel-purified according to Gamper et al. (15) and the concentrations determined spectrophotometrically (33 or 40 µg/ml per A260 unit of single-stranded or hairpin oligomer). HUH7 cells were obtained from Dr Clifford Steer (University of Minnesota) and grown in DMEM, 10% FBS, 2 mM glutamine, 0.5% penicillin/streptomycin. The E.coli strain, DH10B, was obtained from Life Technologies (Gaithersburg, MD); DH10B cells contain a mutation in the RECA gene (recA).
Cell-free extracts
Extracts from HUH7, MEF+/+ and MEF–/– cells were prepared as described by Cole-Strauss et al. (14). Mouse embryonic fibroblasts (MEFs), either as wild-type (+/+) or deficient in MSH2 (–/–) or MSH3 (–/–) were a kind gift from Dr Neils de Wind (University of Leiden, Netherlands). The final aliquots (100 µg protein) were frozen at –80°C. Musa and canola cells were obtained from Dr Greg May (Noble Foundation) and extracts prepared as described by Rice et al. (18). Briefly, cell suspensions were ground with a mortar and pestle in liquid nitrogen. The plant tissue was extracted in 20 mM HEPES pH 7.5, 5 mM KCl, 1.5 mM MgCl2, 10 mM DTT, 10% (v/v) glycerol and 1% (w/v) PVP. This slurry was homogenized and plant cell debris removed by centrifugation. Extracts were dispensed in 100 µg aliquots, frozen in a dry ice–ethanol bath and stored at –80°C.
Reaction mixtures
Reaction mixtures (50 µl) contained 1 µg of plasmid (pKsm4021 or pTsΔ208) and indicated amounts of oligonucleotide in 20 mM Tris–HCl pH 7.4, 15 mM MgCl2, 0.4 mM DTT and 1.0 mM ATP. The extract (10–30 µg as indicated) was added to initiate the reaction which was incubated at 37°C for 45 min. The reaction was stopped by placing the tubes on ice and the plasmid extracted by phase partition with phenol:chloroform. Isolated plasmid was recovered by ethanol precipitation on dry ice for 2 h and centrifugation at 4°C for 30 min.
Electroporation, plating and selection
Five microliters of resuspended reaction mixtures (total volume 50 µl) were used to transform 20 µl aliquots of electro-competent DH10B bacteria using a Cell-Porator apparatus (Life Technologies). The mixtures were allowed to recover in 1 ml SOC at 37°C for 1 h at which time 50 µg/ml kanamycin or 12 µg/ml tetracycline was added for an additional 3 h. Prior to plating, the bacteria were pelleted and resuspended in 200 µl of SOC. 100 µl aliquots were plated onto kan or tet agar plates and 100 µl of a 10–4 dilution of the cultures were concurrently plated on agar plates containing 100 µg/ml of ampicillin. Plating was performed in triplicate using sterile Pyrex beads. Colony counts were determined by an Accu-count 1000 plate reader (Biologics). Each plate contained 200–500 ampicillin resistant colonies or 0–500 tetracycline or kanamycin resistant colonies. Resistant colonies were selected for plasmid extraction and DNA sequencing using an ABI Prism kit on an ABI 310 capillary sequencer (PE Biosystems).
RESULTS
Experimental design
A cell-free extract was developed recently to study the mechanism of chimera-directed gene repair (14). This strategy centers around the use of extracts from various sources to correct a mutation in a plasmid using a chimeric oligonucleotide. A mutation is placed inside the coding region of a gene conferring antibiotic resistance in bacteria, here kanamycin or tetracycline. The appearance of resistance is measured by genetic readout in E.coli grown in the presence of the specified antibiotic. The importance of this system is that both phenotypic alteration and genetic inheritance can be measured. Figure 1 illustrates the experimental approach. Plasmid pKsm4021 contains a mutation (T→G) at residue 4021 rendering it unable to confer antibiotic resistance in E.coli. This point mutation is targeted for repair by oligonucleotides designed to restore kanamycin resistance. To avoid concerns of plasmid contamination skewing the colony counts, the directed correction is from G→C rather than G→T (wild-type). After isolation, the plasmid is electroporated into the DH10B strain of E.coli, which contains inactive RecA protein. The number of kanamycin colonies is counted and normalized by ascertaining the number of ampicillin colonies, a process that controls for the influence of electroporation. The number of colonies generated from three to five independent reactions was averaged and is presented for each experiment. A fold increase number is recorded to aid in comparison.
Figure 1.

Genetic readout system for correction of a point mutation in plasmid pKsm4021. A mutant kanamycin gene harbored in plasmid pKsm4021 is the target for correction by oligonucleotides. The mutant G is converted to a C by the action of the oligo. Corrected plasmids confer resistance to kanamycin in E.coli strain DH10B after electroporation leading to the genetic readout and colony counts.
The original chimera design consists of two hybridized regions of a single-stranded oligonucleotide folded into a double hairpin configuration. The double-stranded targeting region is made up of a 5 bp DNA/DNA segment bracketed by 10 bp RNA/DNA segments. The central base pair is mismatched to the corresponding base pair in the target gene. When a molecule of this design is used to correct the kans mutation, gene repair is observed (15; I in Fig. 2A). Recently, Gamper et al. (15) demonstrated that the all-DNA strand of the chimera directs the gene correction event. Chimera II (Fig. 2B) differs partly from chimera I in that only the DNA strand of the double hairpin is mismatched to the target sequence. When this chimera was used to correct the kans mutation, it was twice as active. In the same study, repair function could be further increased by making the targeting region of the chimera a continuous RNA/DNA hybrid.
Figure 2.

Flow diagram for the generation of modified single-stranded oligonucleotides. The upper targeting strands of chimeric oligonucleotides I and II are separated into pathways resulting in the generation of single-stranded oligonucleotides that contain (A) 2′-O-methyl RNA nucleotides or (B) phosphorothioate linkages. Fold changes in repair activity for correction of kans in the HUH7 cell-free extract are presented in parentheses. Each single-stranded oligonucleotide is 25 bases in length and contains a G residue mismatched to the complementary sequence of the kans gene. The numbers 3, 6, 8, 10, 12 and 12.5 indicate how many phosphorothioate linkages (S) or 2′-O-methyl RNA nucleotides (R) are at each end of the molecule. Hence oligo 12S/25G contains an all phosphorothioate backbone, displayed as a dotted line. Smooth lines indicate DNA residues, wavy lines indicate 2′-O-methyl RNA residues and the carat indicates the mismatched base site (G).
Modified single-stranded oligonucleotides repair point mutations in HUH7 cell-free extracts
Our earlier results prompted us to test a single-stranded DNA molecule (IV), a single-stranded RNA molecule (VIII), and a single-stranded molecule (III) duplicating the top strand of chimera I. All molecules have a total length of 25 nt. The molecules containing RNA were almost devoid of gene repair activity while the all DNA construct, IV, was approximately one-fifth as active as chimera I. These results show that a single-stranded DNA molecule could direct gene repair, and suggest to us that its reduction in activity may be due to nuclease sensitivity. To address this issue, we synthesized a series of oligonucleotides with varying numbers of phosphorothioate linkages on the 3′ and 5′ ends. Molecules bearing 3, 6, 8, 10 and 12 phosphorothioate linkages at each end of a backbone with a total of 24 linkages (25 bases) were tested in the kans system. The results, presented in Table 1 and Figure 2B, illustrate an enhancement of correction activity directed by some of these modified structures. The most efficient molecules contained 3 or 6 thioate linkages at each end; the activities were approximately equal. A reduction in activity was observed as the number of modified linkages in the molecule was further increased. Interestingly, a single-strand molecule containing 24 thioate linkages was minimally active (see below) suggesting that this backbone modification when used throughout the molecule supports only a low level of targeted gene repair.
Table 1. Gene repair activity is directed by single-stranded oligonucleotides.

Plasmid pKsm4021 (1 µg), the indicated oligonucleotide (1.5 µg chimeric oligonucleotide or 0.55 µg single-stranded oligonucleotide; molar ratio of oligo to plasmid of 360:1) and either 10 or 20 µg of HUH7 cell-free extract were incubated for 45 min at 37°C. Isolated plasmid DNA was electroporated into E.coli strain DH10B and the number of kanr colonies counted. The data represent the number of kanamycin-resistant colonies per 106 ampicillin-resistant colonies generated from the same reaction and is the average of three experiments (standard deviation usually less than +/– 15%). Fold increase is defined relative to 418 kanr colonies (second reaction) and in all reactions was calculated using the 20 µg sample.
The efficiency of gene repair directed by thioate-modified, single-stranded molecules, in a length dependent fashion, led us to examine the length of the RNA modification used in the original chimera as it relates to correction. Construct III represents the ‘RNA-containing’ strand of chimera I and, as shown in Table 1 and Figure 2A, it promotes inefficient gene repair. But, as shown in the same figure, reducing the RNA residues on each end from 10 to three elevates the frequency of repair. At equal levels of modification, however, 25mers with 2′-O-methyl ribonucleotides were less effective gene repair agents than the same oligomers with phosphorothioate linkages. These results reinforce earlier observations (15) suggesting that the strand containing RNA is not as effective in promoting gene repair as the DNA strand.
Repair of the kanamycin mutation requires a G→C exchange. To confirm that the specific correction was obtained, colonies selected at random from multiple experiments were processed and the isolated plasmid DNA was sequenced. As seen in Figure 3, colonies generated through the action of the single-stranded molecules 3S/25G, 6S/25G and 8S/25G, respectively, contained plasmid molecules harboring the targeted base correction. While a few colonies appeared on plates derived from reaction mixtures containing 25mers with 10 or 12 thioate linkages on both ends, the sequences of the plasmid molecules from these colonies contained non-specific base changes. In these examples, the second base of the codon was changed (see Fig. 3). These results indicate that modified single strands can direct gene repair, but that efficiency and specificity are reduced when the 25mers contain 10 or more phosphorothioate linkages at each end. It is, however, important to note that only a small number of colonies are available for analyses and these colonies arise only if particular, heavily modified, oligonucleotides are used. These circumstances make it difficult to render a true measure of non-specific mutagenesis frequency.
Figure 3.

DNA sequences of representative kanr colonies. Confirmation of sequence alteration directed by the indicated molecule is presented along with a table outlining codon distribution. Note that 10S/25G and 12S/25G elicit both mixed and unfaithful gene repair. The number of clones sequenced is listed in parentheses next to the designation for the single-stranded oligonucleotide. A plus (+) symbol indicates the codon identified while a figure after the (+) symbol indicates the number of colonies with a particular sequence. TAC/TAG indicates a mixed peak. Representative DNA sequences are presented below the table with yellow highlighting altered residues.
Modified single-stranded oligonucleotides are not dependent on MSH2 for optimal gene repair activity
The reaction mixtures described thus far included a cell-free extract made from HUH7 cells that contains a full complement of DNA repair proteins. MEFs deficient in MSH2 (–/–) or MSH3 (–/–) due to partial deletions in both copies of either gene, are available and were a kind gift from Dr Neils de Wind. Such cells, while murine in origin, are more genetically defined relative to the human cell line, LoVo, which while having a partial deletion in hMSH2 (19) also exhibits proteolysis of useable MSH3 and MSH6. Since the MSH2 protein acts in the initialization of the mismatch repair process, the question of whether the modified single strands could direct the repair of a point mutation in the absence of MSH2 or MSH3 was therefore addressed using MEFs. The reaction mixtures were identical to those described in Table 1 except that the source of extract was varied. As seen in Table 2, directed repair is not reduced when either of two modified single-stranded molecules are used in the reaction. When a cell line containing a deletion in XPA, a central gene in the process of nucleotide excision repair, or HEC-1-A, a cell line deficient in hMSH6, was used as the source of extract, no significant difference in repair activity was observed (data not shown) thus supporting the results obtained when MEF cell-free extracts are used. Taken together, these results suggest that MSH2, MSH3 or perhaps the mismatch repair pathway itself, do not participate in a majority of the gene repair events directed by the modified single-stranded molecules presented herein.
Table 2. Modified single-stranded oligomers are not dependent on MSH2 or MSH3 for optimal gene repair activity.

Chimeric oligonucleotide (1.5 µg) or modified single-stranded oligonucleotide (0.55 µg) was incubated with 1 µg of plasmid pKsm4021 and 20 µg of the indicated extracts. MEF represents mouse embryonic fibroblasts with either MSH2 (2–/–) or MSH3 (3–/–) deleted. MEF+/+ indicates wild-type mouse embryonic fibroblasts. The other reaction components were then added and processed through the bacterial readout system. The data represent the number of kanamycin-resistant colonies per 106 ampicillin-resistant colonies.
Modified single-stranded oligonucleotides repair frameshift mutations in HUH7 cell-free extracts
A point mutation has been the target of repair for single-stranded molecules thus far, but previously, chimeric oligonucleotides were shown to direct repair of a single base deletion (14). By using plasmid pTsΔ208, the capacity of the modified single-stranded molecules that showed activity in correcting a point mutation can be tested for repair of a frameshift. To correct the mutation, a chimeric oligonucleotide (Tet I), which is designed to insert a T residue at position 208, was used previously (14). A modified single-stranded oligonucleotide (Tet IX) was therefore designed to direct the insertion of a T residue at this same site. Figure 4A illustrates the plasmid and target bases designated for change in the experiments. When all reaction components are present (extract, plasmid, oligomer), tetracycline-resistant colonies appear. The colony count increases with the amount of oligonucleotide used up to a point beyond which the count falls off (Table 3). No colonies are observed in the absence of either extract or oligonucleotide, nor when a modified single-stranded molecule bearing perfect complementarity is used. Figure 4B represents the sequence surrounding the target site and shows that a T residue is inserted at the correct site. We have isolated plasmids from 15 colonies obtained in three independent experiments and each analyzed sequence revealed the same precise nucleotide insertion. These data suggest that the single-stranded molecules used initially for point mutation correction can also repair nucleotide deletions.
Figure 4.
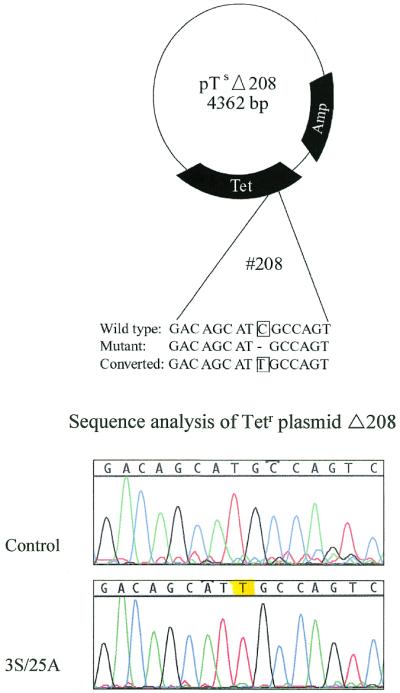
Target plasmid and sequence correction of a frameshift mutation by chimeric and single-stranded oligonucleotides. (A) Plasmid pTsΔ208 contains a single base deletion mutation at position 208 rendering it unable to confer tet resistance. The target sequence presented below indicates the insertion of a T directed by the oligonucleotides to re-establish the resistant phenotype. (B) DNA sequence confirming base insertion directed by Tet 3S/25G; the yellow highlight indicates the position of frameshift repair.
Table 3. Frameshift mutation repair is directed by single-stranded oligonucleotides.

Each reaction mixture contained the indicated amounts of plasmid and oligonucleotide. The extract used for these experiments came from HUH7 cells. The data represent the number of tetracycline resistant colonies per 106 ampicillin resistant colonies generated from the same reaction and is the average of three independent experiments. Tet I is a chimeric oligonucleotide and Tet IX is a modified single-stranded oligonucleotide that are designed to insert a T residue at position 208 of pTsΔ208. These oligonucleotides are equivalent to structures I and IX in Figure 2.
Modified single-stranded oligonucleotides direct gene repair in plant cell-free extracts
Recent reports by Beetham et al. (12) and Zhu et al. (13) have demonstrated the utility of chimeric oligonucleotides in introducing nucleotide alterations in plant cells. To explore the mechanism of correction in these cells, we developed a cell-free extract system from a variety of plant tissues (18). Based on the results reported therein, we tested the modified single-stranded constructs in plant cell extracts. Two sources were used: a dicot (canola) and a monocot (Musa). Each vector tested was found to direct gene repair of the kanamycin mutation (Table 4); however, the level of correction was elevated 2–3-fold relative to the frequency observed with the chimeric oligonucleotide. These results are similar to those observed in the mammalian system wherein a significant improvement in gene repair occurred when modified single-stranded molecules were used.
Table 4. Plant cell-free extracts support gene repair by single-stranded oligonucleotides.

Canola or Musa cell-free extracts were tested for gene repair activity on the kanamycin-sensitive gene as previously described (18). Chimeric oligonucleotide II (1.5 µg) and modified single-stranded oligonucleotides IX and X (0.55 µg) were used to correct pKsm4021. Total number of kanr colonies are present per 107 ampicillin-resistant colonies and represent an average of four independent experiments.
DISCUSSION
This manuscript extends the observations of Cole-Strauss et al. (14) and Gamper et al. (15), namely that the DNA strand of the chimeric RNA/DNA oligonucleotide directs the majority of gene repair in plasmid targets. But, here we report the successful use of modified single-stranded DNA molecules as substitutes for chimera in directing gene repair in vitro. A cell-free extract system was used to determine if the dominant functional unit of the chimera, active in gene repair, is the complete strand of DNA. The system takes advantage of the DNA repair activity present in mammalian and plant cell-free extracts and uses a genetic readout in bacteria to assess correction. Such a readout has several advantages: first, it enables a quantitative evaluation of gene repair in many different cell-types; and second, it demonstrates phenotypic change (under selection) and genetic inheritance. We envisage using this approach to screen cell lines, cell types or tissues in order to determine the level of endogenous activity responsive to chimera-directed repair.
Since the chimera basically consists of two complementary strands of nucleic acid folded into a hairpin structure, we were able to synthesize each ‘strand’ independently and evaluate its activity. Here, we demonstrate the all-DNA strand is more efficacious in gene repair events. In contrast, the strand containing RNA (originally 20 2′-O-methyl RNA residues and five DNA residues) has appreciably reduced corrective function. Modifying the terminal nucleotides of the all-DNA strand enhances activity up to 4-fold in some mammalian and plant cell-extracts by rendering the modified oligonucleotide more resistant to digestion by exonucleases. This is expected since all extracts contain some nuclease activity. As discovered by Gamper et al. (15), introduction of RNA into both strands of a chimera reduces repair capacity, suggesting that the strand containing RNA in the original (I) or second-generation chimera (II) does not support repair. Yet, a chimera containing an uninterrupted RNA sequence in one strand is 2–3-fold better in directing repair than a chimera in which the same strand contains a mixed DNA–RNA sequence (15). This paradox could be explained by presuming that RNA functions in a double-stranded conformation by simply stabilizing the joint molecule at the target site (H.B.Gamper, Y.-M.Hou and E.B.Kmiec., submitted).
A similar phenomenon is seen when phosphorothioate modified backbones are introduced into the single-stranded DNA oligos. As the interface between the flanking phosphorothioate linkages get closer, repair frequency is reduced. These data suggest that the ‘region of repair’ directed by the chimeric hairpin or single-stranded oligonucleotide requires unmodified DNA and is larger than the targeted single nucleotide.
The structure created when the oligonucleotide binds at the target site is likely to distort the helix more than when a base is misincorporated by DNA polymerase. Hence, the proteins attracted to this perturbation might include members of the mismatch repair pathway (20–24). If true, then the ‘region of repair’ may impact the path used to reach the mismatched base pair and two models have been proposed for this transaction. The first involves the MutS complex binding to a heteroduplex which initiates downstream repair events coupled to an ATP-hydrolysis-dependent translocation away from the site (25). The second centers around the binding of ATP altering the conformation of MutS complex (26) which in turn translocates in an ATP-hydrolysis-independent fashion, transducing a signal to the repair machinery. Our data align in general with the former ‘translocation’ model as opposed to the latter ‘sliding clamp’ model. To this end experimental results in human cell lines (LoVo) using the chimeric oligonucleotide implicate hMSH2 (14). But, our data presented in this work with the modified single-stranded vectors suggest that other pathways may be involved in targeted gene repair.
The spacing ‘requirement’ may also help explain why an increase in the number of modified linkage groups on each end of the single-stranded molecule reduces the efficiency of repair. DNA containing thioate backbones forms an altered B-form structure when hybridized with a complementary strand of unmodified DNA (27). A similar situation could help explain the low activity of single-stranded molecules containing high percentages of 2′-O-methyl RNA residues. A molecule having three RNA bases at each end is approximately equal in activity to the chimera I structure in directing repair, but advancing the number of RNA residues to six reduces activity significantly. Kamath-Loeb et al. (28) demonstrated inefficient repair of RNA–DNA hybrids. These authors suggested the RNA–DNA hybrid structure, known to be distinct from a DNA–DNA helix (29), may negatively impact scanning recognition and/or excision processes. These observations may also explain the lack of activity mediated by the all-RNA construct described herein. From a comparison of molecules VII and XI, it is apparent that gene repair is more subject to inhibition by RNA residues than by phosphorothioate linkages. Thus, even though both of these oligonucleotides contain an equal number of modifications to impart nuclease resistance, XI (with 16 phosphorothioate linkages) has good gene repair activity while VII (with 16 2′-O-methyl RNA residues) is inactive. Hence, the original chimeric double hairpin oligonucleotide enabled correction directed, in large part, by the strand containing a large region of contiguous DNA residues.
The intermediate joint molecule formed by modified single-stranded oligonucleotides with a double-stranded DNA target could be a D-loop, the formation of which would be mediated by a recombinase. In vitro, both prokaryotic and eukaryotic recombinases catalyze D-loop formation between oligodeoxyribonucleotides and supercoiled DNA (30–32). Little is known of how this reaction is affected by the presence of modified phosphodiester linkages in the oligonucleotide. A study conducted in our laboratory demonstrated that RecA protein is able to mediate joint molecule formation as long as the number of capping 2′-O-methyl RNA or phosphorothioate linkages is kept to a small number (data not shown). The lack of pairing capacity observed with fully modified oligonucleotides would clearly restrict the subsequent repairing phase of the reaction. This phenomenon offers a coincident explanation as to why such molecules exhibit lower repair activity. It follows that most antisense oligonucleotides would not be good substrates for recombinase mediated DNA-targeting reactions since their backbones are fully modified with nuclease resistant linkages.
It is well known that unmodified oligodeoxynucleotides are rapidly degraded in cell culture. This susceptibility to degradation is probably a major reason why earlier attempts to use synthetic oligonucleotides as gene targeting agents yielded very low levels of gene repair (33–35). Electrophoretic analysis of nucleic acid recovered from the cell-free extract reactions conducted here confirm that the unmodified single-stranded 25mer did not survive incubation whereas >90% of the terminally modified oligos did survive (as judged by photo-image analyses of agarose gels). Given the presence of nuclease activity in the extracts, it is possible that single-strand annealing (36) could provide an alternative route for joint molecule formation, especially for highly modified oligonucleotides. Our results with such oligos suggest that this pathway, if operative, is relatively inefficient and that repair of the heteroduplex occurs with low fidelity.
Targeted mutagenesis of episomal and genomic targets in cultured cells has been achieved using single-stranded triplex forming oligonucleotides (TFOs). The oligonucleotides are usually coupled to a psoralen moiety, such that triple strand formation is accompanied by cross-linkage of the DNA target in the presence of ultraviolet irradiation (37,38). Error-prone repair of the cross-link as well as processing of the triple strand itself (39,40) leads to introduction of site-specific mutations. However, with this strategy, recognition of double-stranded DNA sequences is severely restricted since triple strand formation requires an uninterrupted homopurine run in one strand of the DNA target. Unlike the approach we have described, these TFOs do not form a mismatch with the target. As a consequence the exact location and identity of the targeted mutation cannot be controlled.
An elegant modification of the TFO strategy was recently introduced by Glazer and colleagues (41) in which bi-functional oligonucleotides (42) were used. One section of the molecule took advantage of the stable binding between the TFO and the purine-rich target sequence (stability of binding function), while the other segment, tethered to the first, contained a DNA sequence specific for the target site (repair function). Through the process of nucleotide excision repair, target sequence changes up to 0.04% were seen in episomal targets. In a subsequent study using similar vectors, Culver et al. (43) reported correction of chromosomal point mutations with a frequency of 19% or greater. In these investigations the presence of both segments were required for optimal activity suggesting that untethered single-stranded oligonucleotides were inefficient in directing nucleotide alterations. But in this report, we find that untethered single-stranded oligonucleotides direct correction in cell-free extracts as long as the molecules contain a particular number of modified linkages at each end. Furthermore, the absence of XPA in the extract, and hence significantly reduced nucleotide excision repair activity, does not appear to hamper nucleotide alteration. Since Chan et al. (41) conducted their experiments in cells and our system is a cell-free extract, a direct comparison between these two types of molecular vectors awaits episomal targeting experiments, currently underway in our laboratory.
In summary, we have designed a novel class of single-stranded oligonucleotides with backbone modifications at the termini and demonstrate gene repair/conversion activity in mammalian and plant cell-free extracts. We confirm that the all-DNA strand of the chimera is the active component in the process of gene repair, the main focus of this report. In some cases, the relative frequency of repair is elevated ~3–4-fold when compared to frequencies directed by chimeric oligonucleotides. While this level still represents only a 0.1–0.2% conversion rate, we may be moving closer to direct applications of gene repair in vivo. One clear application centers around functional genomics and the possibility of making single base nucleotide changes in targeted genes.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to members of the Kmiec laboratory for comments on the manuscript and to Dr Joseph Jiricny for advice on MSH2 cell lines. We also thank Dr. Niels de Wind for supplying wild-type MEFs and MEFs lacking MSH2 and for helpful discussions. We greatly appreciate the technical and graphic skills of Eric M. Roberts. This work was supported by NIH grants HL 58563-01A1 and DK 56134-01A1.
REFERENCES
- 1.Kotani H. and Kmiec,E.B. (1994) Mol. Cell. Biol., 14, 6097–6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kotani H., Germann,M.W., Andrus,A., Vinayak,R., Mulla,B. and Kmiec,E.B. (1996) Mol. Gen. Genet., 250, 626–634. [DOI] [PubMed] [Google Scholar]
- 3.Havre P.A. and Kmiec,E.B. (1998) Mol. Gen. Genet., 258, 580–586. [DOI] [PubMed] [Google Scholar]
- 4.Kmiec E.B. (1999) Gene Ther., 6, 1–3. [DOI] [PubMed] [Google Scholar]
- 5.Yoon K., Cole-Strauss,A. and Kmiec,E.B. (1996) Proc. Natl Acad. Sci. USA, 93, 2071–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cole-Strauss A., Yoon,K., Xiang,Y., Byrne,B.C., Rice,M.C., Gryn,J., Holloman,W.K. and Kmiec,E.B. (1996) Science, 273, 1386–1389. [DOI] [PubMed] [Google Scholar]
- 7.Kren B.T., Cole-Strauss,A., Kmiec,E.B. and Steer,C.J. (1997) Hepatology, 25, 1462–1468. [DOI] [PubMed] [Google Scholar]
- 8.Kren B.T., Bandyopadhyay,P. and Steer,C.J. (1998) Nature Med., 4, 285–290. [DOI] [PubMed] [Google Scholar]
- 9.Lai L.W., O’Connor,H.M. and Lien,Y.H. (1998) In Conference Proceedings: 1stAnnual Meeting of the American Society of Gene Therapy. American Society of Gene Therapy, Seattle, WA, p. 183a.
- 10.Alexeev V. and Yoon,K. (1998) Nat. Biotechnol., 16, 1343–1346. [DOI] [PubMed] [Google Scholar]
- 11.Alexeev V., Igoucheva,O., Domashenko,A., Cotsavelis,G. and Yoon,K. (2000) Nat. Biotechnol., 18, 43–50. [DOI] [PubMed] [Google Scholar]
- 12.Beetham P.R., Kipp,P.B., Sawyky,X.L., Antzen,C. and May,G.D. (1999) Proc. Natl Acad. Sci. USA, 96, 8774–8778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu T., Peterson,D.J., Tagliani,L., St Clair,G., Boszczynski,C. and Bowen,B. (1999) Proc. Natl Acad. Sci. USA, 96, 8774–8778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cole-Strauss A., Gamper,H., Holloman,W.K., Munoz,M., Cheng,N. and Kmiec,E.B. (1999) Nucleic Acids Res., 27, 1323–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gamper H., Cole-Strauss,A., Metz,R., Parekh,H., Kumar,R. and Kmiec,E.B. (2000) Biochemistry, 39, 5808–5816. [DOI] [PubMed] [Google Scholar]
- 16.Kutyavin I.V., Rhinehart,R.L., Lukhtanov,E.A., Gorn,V.V., Meyer,R.B. and Gamper,H.B. (1996) Biochemistry, 35, 11170–11176. [DOI] [PubMed] [Google Scholar]
- 17.Jayasena V.K. and Johnston,B.H. (1993) J. Mol. Biol., 230, 1015–1024. [DOI] [PubMed] [Google Scholar]
- 18.Rice M.C., May,G.D., Kipp,P.B., Parekh,H. and Kmiec,E.B. (2000) Plant Physiol., 123, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palambo F., Iaccarino,I., Nakajima,E., Iksejima,M., Slimada,T. and Jiricny,J. (1996) Curr. Biol., 6, 1181–1184. [DOI] [PubMed] [Google Scholar]
- 20.Drummund J.T., Li,G.M., Langley,M.J. and Modrich,P. (1995) Science, 268, 1909–1912. [DOI] [PubMed] [Google Scholar]
- 21.Li G.M. and Modrich,P. (1995) Proc. Natl Acad. Sci. USA, 85, 8860–8864. [Google Scholar]
- 22.Umar A., Buermeyer,A.B., Simm,J.A., Thomas,D.C., Clark,A.B., Lishey,R.M. and Kunkel,T.A. (1996) Cell, 87, 65–73. [DOI] [PubMed] [Google Scholar]
- 23.Gu L., Hong,Y., McCulloch,S., Watanabe,H. and Li,G.M. (1998) Nucleic Acids Res., 26, 1173–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langley M.J., Pierce,A.J. and Modrich,P. (1997) J. Biol. Chem., 272, 1173–1178. [Google Scholar]
- 25.Bjornson K.P., Allen,D.J. and Modrich,P. (2000) Biochemistry, 39, 3176–3183. [DOI] [PubMed] [Google Scholar]
- 26.Gradia S., Subramanian,D., Wilson,T., Acharya,S., Makhov,A., Griffith,J. and Fishel,R. (1999) Mol. Cell., 3, 255–261. [DOI] [PubMed] [Google Scholar]
- 27.Kanaori K., Tamura,Y., Wada,T., Nishi,M., Kanehara,H., Morii,T., Tajima,K. and Makino,K. (1999) Biochemistry, 38, 16058–16066. [DOI] [PubMed] [Google Scholar]
- 28.Kamath-Loeb A.S., Hizi,A., Tabone,J., Solomon,M.S. and Loeb,L. (1997) Eur. J. Biochem., 250, 492–501. [DOI] [PubMed] [Google Scholar]
- 29.Salazar M., Federoff,O.Y., Miller,J.M., Ribeiro,N.S. and Reid,B.R. (1993) Biochemistry, 32, 4207–4215. [DOI] [PubMed] [Google Scholar]
- 30.Shibata T., DasGupta,C., Cunningham,R.P. and Radding,C.M. (1979) Proc. Natl Acad. Sci. USA, 76, 1638–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsieh P., Camerini-Otero,C.S. and Camerini-Otero,R.D. (1992) Proc. Natl Acad. Sci. USA, 89, 6492–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tracy R.B., Baumohl,J.K. and Kowalczykowski,S.C. (1997) Genes Dev., 11, 3423–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moerschell R.P., Tsunasawa,S. and Sherman,F. (1988) Proc. Natl Acad. Sci. USA, 85, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campbell C.R., Keown,W., Lowe,L., Kirschling,D. and Kucherlapati,R. (1989) New Biol., 1, 223–227. [PubMed] [Google Scholar]
- 35.Yamamato T., Moerschell,R.P., Wakem,L.P., Komar-Panicucci,S. and Sherman,F. (1992) Genetics, 131, 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Segal D.J. and Carroll,D. (1995) Proc. Natl Acad. Sci. USA, 92, 806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Majumdar A., Khorlin,A., Dyatkina,N., Lin,F.L.M., Powell,J., Liu,J., Fei,Z., Khripine,Y., Watanabe,K.A., George,J., Glazer,P.M. and Seidman,M.M. (1998) Nature Genet., 20, 212–214. [DOI] [PubMed] [Google Scholar]
- 38.Barre F.X., Ait-Si-Ali,S., Giovannangeli,C., Luis,R., Robin,Ph., Pritchard,L.L., Helene,C. and Harel-Bellan,A. (2000) Proc. Natl Acad. Sci. USA, 97, 3084–8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang G., Seidman,M.M. and Glazer,P.M. (1996) Science, 271, 812–805. [Google Scholar]
- 40.Vasquez K.M., Wang,G., Havre,P.A. and Glazer,P.M. (1999) Nucleic Acids Res., 27, 1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan P.P., Lin,M., Faruqui,A.F., Powell,J., Seidman,M.M. and Glazer,P.M. (1999) J. Biol. Chem., 274, 11541–11548. [DOI] [PubMed] [Google Scholar]
- 42.Gamper H.B., Hou,Y.M., Stamm,M.R., Podyminogin,M.A. and Meyer,R.B. (1998) J. Am. Chem. Soc., 120, 2182–2183. [Google Scholar]
- 43.Culver K.W., Hsieh,W.-T., Huyen,Y., Chen,V., Liu,J., Khripine,Y. and Khorlin,A. (1999) Nat. Biotechnol., 17, 989–993. [DOI] [PubMed] [Google Scholar]