

Abstract
Mapping of cis-diamminedichloroplatinum(II) (cis-DDP, cisplatin) DNA adducts over >3000 nucleotides was carried out using a replication blockage assay. The sites of inhibition of modified T4 DNA polymerase, also referred to as stop sites, were analyzed to determine the effects of local sequence context on the distribution of intrastrand cisplatin cross-links. In a 3120 base fragment from replicative form M13mp18 DNA containing 24.6% guanine, 25.5% thymine, 26.9% adenine and 23.0% cytosine, 166 individual stop sites were observed at a bound platinum/nucleotide ratio of 1–2 per thousand. The majority of stop sites (90%) occurred at Gn>2 sequences and the remainder were located at sites containing an AG dinucleotide. For all of the GG sites present in the mapped sequences, including those with Gn>2, 89% blocked replication, whereas for the AG sites only 17% blocked replication. These blockage sites were independent of flanking nucleotides in a sequence of N1G*G*N2 where N1, N2 = A, C, G, T and G*G* indicates a 1,2-intrastrand platinum cross-link. The absence of long-range sequence dependence was confirmed by monitoring the reaction of cisplatin with a plasmid containing an 800 bp insert of the human telomere repeat sequence (TTAGGG)n. Platination reactions monitored at several formal platinum/nucleotide ratios or as a function of time reveal that the telomere insert was not preferentially damaged by cisplatin. Both replication blockage and telomere-insert plasmid platination experiments indicate that cisplatin 1,2-intrastrand adducts do not form preferentially at G-rich sequences in vitro.
INTRODUCTION
cis-Diamminedichloroplatinum(II) (cis-DDP, cisplatin) is used in the treatment of several types of cancer including those of the genitourinary tract and certain carcinomas of the lung, head and neck. Considerable effort has been expended to characterize cisplatin–DNA cross-links and to understand how the drug functions as an antitumor agent (1–3). The principal adducts formed between cisplatin and double-stranded DNA are 1,2-intrastrand cross-links of adjacent deoxyguanosines with platinum binding to the N7 positions of the bases. The remaining lesions include 1,2-intrastrand cross-links of AG pairs and longer-range intra- and interstrand cross-links involving guanine bases. Adduct profiles in cisplatin-treated cancer patients demonstrate that tumor response correlates to levels of 1,2-intrastrand d(GpG) cross-links (4–6).
Studies of other DNA-modifying drugs, including aflatoxin (7) and nitrogen mustards (8), which also react with the N7 position of guanine, reveal that reactivity depends upon local sequence context. The number and types of cisplatin cross-links with duplex DNA are known, but the influence of sequence context on cisplatin binding has not been extensively investigated. Reactions of platinum compounds with single-stranded or hairpin oligonucleotides demonstrate that adduct formation is influenced by DNA conformation, the nature of the platinum species, and sequence (9). In addition, analysis of cellular and purified genomic DNA by using a Taq DNA polymerase or exonuclease blockage assay (10–15) reveals that cisplatin binds predominantly to G-rich sequences (16,17). None of these investigations has clarified whether platination of such G-rich regions is due to intrinsic targeting of cisplatin to these sites or simply to the statistically greater number of potential binding sites in such G-rich regions. Selective binding of cisplatin to functionally G-rich regions of the genome, such as promoter sequences or telomeres, could play an important role in the genotoxic mechanism of the drug (18,19).
In the present study we have employed double-stranded replication mapping to analyze the long-range sequence dependence of cisplatin binding to DNA. Preferential blockage occurs at Gn>2 sites, and a sufficient number of nucleotides were examined to analyze the sequence context of cisplatin binding. In addition, platination of a plasmid containing an 800 bp telomeric (TTAGGG)n repeat was examined. In agreement with data from the replication blockage experiment, the platinum content of the 800 bp insert was proportionally higher than in the remainder of the plasmid, but only as predicted by the greater number of binding sites within the insert. Together these experiments suggest that cisplatin does not specifically target G-rich regions in vitro.
MATERIALS AND METHODS
Preparation and platination of plasmid DNA
Oligodeoxyribonucleotide primers were purchased from New England Biolabs (Universal primers, reverse primers, 17mers and 24mers) or synthesized by solid phase phosphoramidite chemistry (primers for the SV40-containing genomes). Replicative form M13 DNA containing 200–400 bp inserts from the HAP4 gene of Saccharomyces cerevisiae was isolated by the cleared lysate method. Plasmid DNA from pUC19-based plasmids containing inserts from the SV40 virus was prepared by similar methods. DNA was allowed to react with cisplatin at a formal drug-to-nucleotide (D/N)f ratio (rf) of 0.00125 for 16 h at 37°C in 1 mM sodium phosphate, 3 mM NaCl, pH 7.4. Following ethanol precipitation and washing to remove the excess platinum, the DNA was quantitated by UV-visible absorption spectroscopy and the platinum content was determined by atomic absorption (AA) spectroscopy to get the bound drug-to-nucleotide ratio, (D/N)b or rb.
Replication mapping on platinated templates
Preliminary experiments using single-stranded M13mp18 DNA were carried out to determine the effect of platinum levels on the replication process. M13mp18 viral DNA was modified with cisplatin to an rb of 0.002–0.02. Dideoxynucleotide triphosphates (Bethesda Research Laboratories) were dissolved in deionized water and, after the pH was adjusted to 7.0, stored at 20°C. The DNA was primed with a 17mer Universal primer (New England Biolabs) by heating to 85°C and cooling slowly to room temperature. Initially, [α-35S]dATP (5 µCi, 600 µCi/µmol, Amersham) and unlabeled dGTP, dCTP and TTP (0.2 µM) were added to the primed template in 25 mM Tris–Cl (pH 7.6), 6.5 mM dithiothreitol (DTT), 12 mM MgCl2, and 30 mM NaCl and incubated with modified T7 polymerase (1–5 U Sequenase, US Biochemical) for 10 min at room temperature. An excess of all four dNTPs was added for a total dNTP concentration of 40 µM and the incubation was continued for 15 min. The reaction was stopped by the addition of a solution of 90% formamide, 5 mM EDTA, 1% bromophenol blue and xylene cyanol. The samples were denatured by heating to 85°C and analyzed by electrophoresis on 8% acrylamide/7 M urea gels. Sequencing reactions were carried out according to the Sequenase protocol (US Biochemical) and resolved in parallel by gel electrophoresis. The gels were dried and autoradiographed at room temperature for 2–5 days.
In subsequent replication mapping experiments an rb of 0.001–0.002 was used to allow identification of the stop sites over the greatest number of nucleotides. Double-stranded plasmid DNA was denatured with 2 N NaOH and a 2–10-fold excess of primer was added just prior to neutralization with ammonium acetate and ethanol precipitation. Replication was initiated by addition of Sequenase enzyme to the primed template in the presence of [α-35S]dATP (5 µCi, 600 µCi/mmol, Amersham) and unlabeled dGTP, dCTP and TTP (0.2 µM) in 25 mM Tris–Cl (pH 7.6), 6.5 mM DTT, 12 mM MgCl2 and 30 mM NaCl and incubated for 10 min at room temperature. An excess of all four dNTPs was then added for a total dNTP concentration of 70 µM and the incubation was continued for 15 min at 37°C. The replication reaction was stopped, the DNA was denatured and the products were resolved by electrophoresis as described above. Double-stranded templates were used for the sequencing reactions, primed in an identical manner to the platinated samples, and sequencing reactions were carried out according to the Sequenase protocol. The sequence of the various plasmid DNAs was read and verified for internal consistency by comparing the gels. An error rate of one incorrect base per 250 nucleotides for the sequenced DNA was estimated by determining the number of errors in the sequence of M13mp18 compared to the published sequence (20). A comprehensive list of the mapped sequences and the identified stop sites can be found in the Supplementary Material available at NAR Online.
Preparation of telomere-containing plasmid
pSP73.Sty11, a plasmid containing an 800 bp insert of (TTAGGG)n, was obtained as a gift from T. deLange (Rockefeller University, New York, NY). The plasmid was introduced by electroporation into XL1Blue Escherichia coli cells. A transformed colony was grown in 5 ml of LB media inoculated with 100 µg/ml ampicillin at 37°C for 8 h. The starter culture was evenly divided between 6 × 500 ml cultures that were inoculated with 150 µg/ml of ampicillin. Cultures were shaken at 200 r.p.m. and incubated at 37°C overnight. After centrifugation at ∼6000 g for 15 min at 4°C, the supernatant was decanted away from the bacterial pellet. Cells were lysed and the plasmid DNA was isolated by using a 2500 Mega Kit (Qiagen). Plasmid isolation was performed by using the procedures, buffers and columns supplied with the kit. The yield was 14.9 mg of plasmid DNA.
Platination reactions with telomere-containing plasmid
In time-course experiments, pSP73.Sty11 plasmid (3.4 mM) was combined with cisplatin (71 µM) in 0.60 mM sodium phosphate (pH 6.8) and 1.9 mM NaCl (100 µl reaction volumes). At 0.5, 1, 2, 4 and 8 h, 20 µl aliquots were removed, quenched with 5 M NaCl (1 µl), and frozen until analysis. Thawed aliquots were spin dialyzed through G-25 Sephadex Quickspin columns (Boehringer-Mannheim). Effluents were mixed with 50 U of EcoRI (Gibco BRL) in 1× REACT 3 buffer (supplied with the enzyme) at 37°C for 1 h. Reactions were stopped by enzymatic inactivation by incubation at 65°C for 30 min. Reaction mixtures were diluted with 4 µl of 6× native PAGE loading buffer and loaded onto a 1% agarose dye gel (0.5 µg/ml ethidium bromide). The gels were run at constant voltage (100 V) in 1× TAE buffer. After 1 h, gels were visualized under UV light and the 800 bp insert and pSP73 bands were excised from the gel. Gel slices were transferred to Eppendorf tubes, and the DNA was isolated via spin dialysis using a QIAEX II Gel Extraction Kit (Qiagen). DNA isolation was performed by using the procedures, buffers and columns supplied with the kit. The isolated DNA was analyzed by UV-visible and atomic absorption (AA) spectroscopy to determine the rb value of each fragment.
In a related set of experiments, pSP73.Sty11 plasmid (3.6 mM) was combined with cisplatin at several rf values (0, 0.007, 0.009, 0.011, 0.015) in 0.55 mM sodium phosphate (pH 6.8) and 1.65 mM NaCl (20 µl reaction volumes). The reaction mixtures were incubated at 37°C for 18 h. According to the methods described above, reaction mixtures were treated with EcoRI, resolved by electrophoresis, isolated by spin dialysis and analyzed by UV-visible and AA spectroscopy.
RESULTS
Replication blockage assay
Preliminary experiments were used to determine the level of platination that allowed replication to proceed over a substantial distance, while enabling blockage sites to be observed. Figure 1 shows replication mapping on templates modified with increasing levels of cisplatin. The lowest level of modification, rb = 0.002 (lane 1), gave clear blockage sites when the replication products were resolved by gel electrophoresis and stop sites could be read throughout the sequence. At higher levels of adduct formation, rb = 0.0075 (lane 2), replication inhibition was increased to such an extent that it was not possible to determine stop sites over all of the readable sequence. At higher rb values, intensity of the stop sites decreased (lane 3), owing to the high probability that the enzyme encounters platinum adducts at sites close to the primer. Therefore, for purposes of replication mapping over a large number of nucleotides, rb values of 0.001–0.002 were employed. This level of platination gave approximately one to two platinum adducts per thousand nucleotides, well below saturation of the available GG sites in a random DNA sequence.
Figure 1.
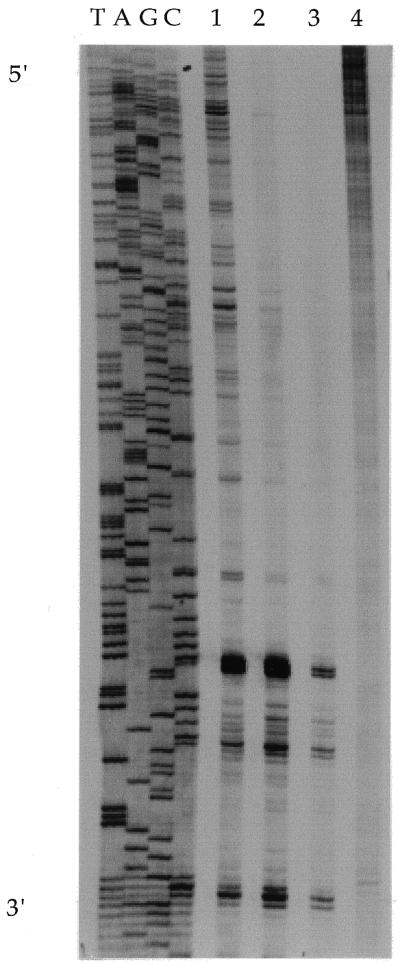
Replication mapping of cisplatin on M13 DNA at different rb values. The sequence of the template strand is read from 5′ (top) to 3′ (bottom) as indicated. The direction of DNA synthesis is from 5′ (bottom) to 3′ (top) on the daughter strand. Lane 1, rb = 0.002; lane 2, rb = 0.0075; lane 3, rb = 0.0186; lane 4, control (rb = 0).
Platinated DNA was primed, replicated and resolved by gel electrophoresis in parallel with replicated control plasmid and sequencing reactions on the control template (Fig. 2). In all, 3120 bases were resolved: 771 (24.6%) G, 793 (25.5%) T, 839 (26.9%) A and 717 (23.0%) C. The experiment identified 166 individual stop sites containing at least two adjacent deoxyguanosines. Sequences with Gn>2 were counted as one stop site although there are several possible adducts that can form and more than one band was observed on the gel. There were 195 distinct GG sites that blocked replication, including all sites for which Gn>2; for example, the sequence TG1G2G3G4 has three GG sites, G1G2, G2G3 and G3G4. The lanes corresponding to the platinated template were compared with the corresponding control lanes and bands appearing in the cisplatin-modified lane but not in the control lane were tabulated according to their sequence context (Table 1). There were a small number of stop sites that did not contain deoxyguanosines and did not appear likely to be platinum binding sites. These sites were also weak stop sites in the control lane and were always in close proximity to a strong stop site that contained multiple deoxyguanosines. Apparently, local changes in secondary structure due to the presence of nearby platinum adducts intensified natural stop sites. Only one stop site that was unique to the experimental lane appeared at a site that did not contain a deoxyguanosine.
Figure 2.
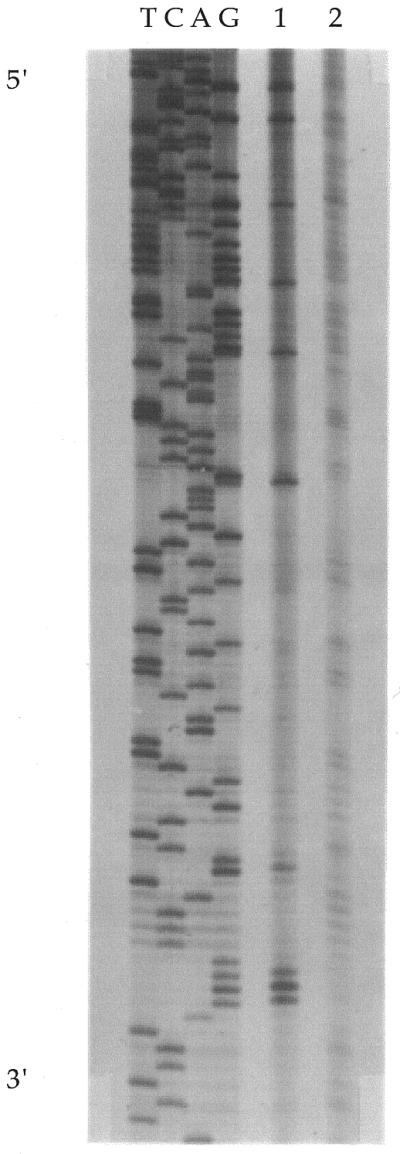
Representative replication mapping of cisplatin on double-stranded DNA. The sequence of the template strand is read from 5′ (top) to 3′ (bottom) as indicated. The direction of DNA synthesis is from 5′ (bottom) to 3′ (top) on the daughter strand. Lane 1, DNA containing cisplatin at rb = 0.0012; lane 2, control (rb = 0).
Table 1. Tabulation of all possible bifunctional platinum binding sites and their frequency of occurrence as stop sites.
| Sequence | Sites | Stop sites | Stop sites/sites |
|---|---|---|---|
| Gn>2 | 218 | 195 | 0.894 |
| AGa | 234 | 95 | 0.406 |
| AGb | 161 | 27 | 0.168 |
| AGG | 73 | 69 | 0.945 |
| GNGc | 183 | 0 | 0.000 |
aAll AG sites in the sequence were counted and all stop sites that contained an AG pair within a four base window were counted.
bOnly AG sites where there was no adjacent GG sequence were counted.
cExcluding GGG. See Table 2.
The stop sites were classified into groups according to the presence of potential platinum binding sites. The sites considered included GG, AG and GNG sites, where N = A, C or T. Because all of the observed stop sites could be classified into one of these three categories, low frequency (<4%) interstrand cross-links were not taken into account (21,22). Table 1 summarizes the frequency of different sites in the sequence and in the stop sites. The majority (∼90%) of stop sites occurred at GG pairs, however, ∼10% of stop sites were due to AG pairs (Table 2). Stop sites that involved GG pairs were evaluated according to whether they included more than two deoxyguanosine residues in tandem. Stop sites in which more than two deoxyguanosines were present accounted for ∼19% of all observed stop sites. The remaining sites were a result of a single GG pair.
Table 2. Frequency of replication blockage at probable platinum binding sites.
| Sequence | Sites | Stop sites | Stop sites/all stop sites |
|---|---|---|---|
| AG | 163a | 16 | 0.096 |
| GG | 136 | 118 | 0.711 |
| GGG | 23 | 21 | 0.126 |
| GGGG | 9 | 9 | 0.054 |
| GGGGG | 1 | 1 | 0.006 |
| GGGGGG | 1 | 1 | 0.006 |
Only unique occurrences of each sequence are included.
aThe discrepancy between this number and the number of AG sites in Table 1 is due to the fact that two AG sites were present at the 3′ edge of a region of the sequence.
The number of stops observed at each possible flanking sequence containing a central GG platinum binding site is given in Table 3. Table 4 shows analogous data for sites containing a central AG pair. Tabulation of these stop sites reveals that the number of stop sites at each possible flanking sequence for N1GGN2 is nearly equal to the frequency of that site in the total sequence (Table 3). Although there does appear to be a slightly greater number of stop sites for sequences containing adjacent purines (Table 5), there is not a substantial difference between the frequency of such sites in the whole sequence and their frequency as stop sites.
Table 3. Frequencies of each flanking sequence containing a GG pair in the total mapped sequence and the number of observed stop sites (not unique occurrences).
| Sequence | Sites | Stop sites | Stop sites/sites |
|---|---|---|---|
| AGGA | 21 | 21 | 1.000 |
| AGGT | 16 | 14 | 0.875 |
| AGGC | 25 | 23 | 0.920 |
| AGGG | 11 | 11 | 1.000 |
| TGGA | 24 | 21 | 0.875 |
| TGGT | 10 | 8 | 0.800 |
| TGGC | 9 | 6 | 0.667 |
| TGGG | 10 | 10 | 1.000 |
| CGGA | 10 | 10 | 1.000 |
| CGGT | 10 | 7 | 0.700 |
| CGGC | 11 | 8 | 0.727 |
| CGGG | 13 | 11 | 0.846 |
| GGGA | 10 | 8 | 0.800 |
| GGGT | 9 | 9 | 1.000 |
| GGGC | 15 | 15 | 1.000 |
| GGGG | 14 | 14 | 1.000 |
Table 4. Frequencies of each flanking sequence containing an AG pair in the total mapped sequence and the number of observed stop sites (not unique occurrences).
| Sequence | Sites | Stop sites | Stop sites/sites |
|---|---|---|---|
| AAGG | 7 | 7 | 1.000 |
| AAGA | 14 | 3 | 0.214 |
| AAGT | 11 | 1 | 0.091 |
| AAGC | 24 | 3 | 0.125 |
| TAGG | 12 | 10 | 0.833 |
| TAGA | 13 | 1 | 0.077 |
| TAGT | 11 | 2 | 0.182 |
| TAGC | 14 | 0 | 0.000 |
| CAGG | 31 | 30 | 0.968 |
| CAGA | 13 | 2 | 0.154 |
| CAGT | 10 | 0 | 0.000 |
| CAGC | 16 | 1 | 0.062 |
| GAGG | 23 | 20 | 0.870 |
| GAGA | 6 | 1 | 0.167 |
| GAGT | 13 | 2 | 0.154 |
| GAGC | 15 | 2 | 0.133 |
| AAGa | 2 | 0 | 0.000 |
aTwo AAG sites were present at the 3′ edge of a mapped sequence.
Table 5. Nearest neighbor analysis.
| Sequence | Sites | Sites/total GG sites | Stop sites | Stop sites/sites |
|---|---|---|---|---|
| AGG | 73 | 0.335 | 69 | 0.945 |
| TGG | 53 | 0.243 | 45 | 0.849 |
| CGG | 44 | 0.202 | 36 | 0.818 |
| GGG | 48 | 0.220 | 46 | 0.958 |
| GGA | 65 | 0.298 | 60 | 0.923 |
| GGT | 45 | 0.206 | 38 | 0.844 |
| GGC | 60 | 0.275 | 52 | 0.867 |
This table lists the frequency of all trinucleotides N1GG and N2GG in the total mapped sequence compared with the frequency of the same sequences in the stop sites (not unique occurrences).
Platination of telomere sequences
The plasmid pSP73.Sty11 comprises an 800 bp insert of the human telomere repeat (TTAGGG)n embedded in a commercially available 2464 bp vector, pSP73 (Promega). Plasmid pSP73.Sty11 was allowed to react with cisplatin, and was then subjected to digestion with EcoRI to excise the 800 bp insert from the vector. The 800 and 2464 bp fragments were separated by agarose dye gel electrophoresis. The bands were isolated and the DNA recovered using spin dialysis methods. The platinum content of each fragment was evaluated by UV-visible and AA spectroscopy as a function of time and rf.
The results of these experiments, shown in Figure 3, reveal that the 800 bp telomere insert acquires more platinum per nucleotide than the remainder of the plasmid. The double-stranded 6 bp telomere repeat TTAG1G2G3 contains two potential GG sites, G1G2 and G2G3, for every 12 nt, or 16.7% of all dinucleotide neighbor pairs in telomere repeats. A purely random DNA fragment would be expected to have 0.25 × 0.25 or 6.25% of adjacent bases as GG sites. Telomere repeats therefore contain 2.67-fold more GG target sites than random DNA. The telomere:pSP73 rb ratio found in the time-course (2.54 ± 0.04) and variable rf (2.75 ± 0.01) experiments with pSP73.Sty11 are in good agreement with the statistically calculated value of 2.67. At shorter incubation times or lower rf values, this ratio is smaller. These findings are consistent with the results of the replication blockage experiments, indicating that cisplatin does not specifically target G-rich DNA sequences in vitro.
Figure 3.

Reaction of pSP73.Sty11 plasmid with cisplatin. (A) The rb value of the 800 bp insert (squares) and the pSP73 fragment (circles) as a function of time. After 8 h, the 800:pSP73 rb ratio was 2.54 ± 0.04. (B) The rb value of the 800 bp insert (squares) and the pSP73 fragment (circles) as a function of rf. At rf = 0.015 the 800:pSP73 rb ratio was 2.75 ± 0.01. Both experiments with pSP73.Sty11 give ratios very close to the statistically predicted value of 2.67.
DISCUSSION
Replication mapping of cisplatin over a large number of nucleotides in random sequence DNA does not indicate any long-range sequence dependence of platinum adduct formation. Despite the low levels of platinum on the DNA, 89% of all possible GG sites also blocked replication in the mapping experiment. This number reflects the presence of platinum adducts at essentially all possible binding sites without notable selectivity for any particular sequence. This phenomenon is not a consequence of saturation of all possible platinum binding sites because the adduct levels, rb = 0.001–0.002, are well below the saturation (D/N) value of 0.06 for all GG sites in this sequence and in random sequence DNA. The mapped sequence carries only between three and six cisplatin adducts per strand under the experimental conditions. There are 34 sites in which Gn>3 and 11 where Gn>4 (Table 2). If cisplatin significantly preferred to bind at polydG sites, these positions should sequester all available platinum. The data reveal, however, that replication blockage also occurs at G2 sites spaced throughout the sequence, clearly indicating that cisplatin does not target polydG sequences.
The extent to which platinum binds to a given dinucleotide cannot be fully assessed because of ambiguity about the nature of the adduct present at each site. In the case of AGG or Gn>2 stop sites, there are at least three different adducts that can form when platinum binds, namely, A*G*G, AG*G* and A*GG*, where asterisks indicate the platinum-modified bases. The position of the last incorporated base suggests that in many sites more than one type of adduct may be present because blockage occurs at many or all of the potentially modified bases and also at the position 3′ to the first possible modified base (17). The likely explanation for this observation is that, within the population of platinated templates, each type of adduct is present. Certain sequences, such as those with more than two deoxyguanosines in a row, appear to give several stops within the likely platinum target site. Replication mapping on site-specifically platinated genomes containing the cis-Pt-d(GpG), cis-Pt-d(ApG), and cis-Pt-d(GpNpG) platinum adducts demonstrates that even unique adducts do not necessarily result in a single stop site (17). The multiplicity of stop sites is presumably a function of the response of the polymerase to the platinum adduct, suggesting that it is not possible to determine the amount of platinum bound at a given dinucleotide site by using replication mapping. Therefore, the observation that polydG sequences (Gn>4, Table 2) always cause blockage of replication on a platinated template is most likely due to the presence of multiple binding sites rather than a preference for flanking deoxyguanosine base pairs around a GG dinucleotide. This conclusion is supported by the presence of multiple bands at these stop sites as well as by experiments with (TTAGGG)n-containing plasmids.
Platination of the pSP73.Sty11 plasmid was used to model the overall genome, because most of the DNA, 75% of the plasmid, has random sequences, and the remainder has a repeating G-rich sequence. In eukaryotic DNA the percentage of telomeric sequence is much smaller (∼0.01%) than in the plasmid model (23,24). Nevertheless, even with this higher percentage of telomere sequence, platination of pSP73.Sty11 showed only a statistically predictable number of platinum adducts on the telomere insert, ∼2.6-fold more lesions relative to the random DNA comprising the bulk of the plasmid (Fig. 3). This result is in agreement with the replication blockage assay and suggests that enhanced damage to specific regions of the genome is unlikely in vivo as a result of only sequence context.
The absence of long-range sequence dependence for the binding of cisplatin to DNA suggests that minor changes in the secondary structure of the DNA do not strongly influence drug binding. Electrostatic considerations may be of substantially greater importance in the interaction of small electrophiles, such as aquated cisplatin, with DNA. The N7 position of guanine has a greater Lewis basicity than any other site on the DNA double helix and is therefore expected to provide the best donor capability for late transition metal ions (25). The preferential binding of cisplatin to these bases is predominantly a result of electrostatic attraction of the platinum toward the most nucleophilic sites (26,27).
It has been suggested that targeting of platinum to telomeric or G-rich regulatory regions may play an important role in the mechanism of cisplatin (18,19). SV40 viruses having deletions in the G-rich origin region of their genome are resistant to cisplatin (28). The present replication blockage experiments included the origin of replication of the SV40 virus and revealed no preferential platination of these G-rich sequences, in agreement with previous studies (29). Platination of the telomere sequence in pSP73.Sty11 is only 2.6-fold greater than random DNA. Therefore, any enhanced platination of these sequences in vivo would have to arise from increased access of cisplatin because of cellular packaging or processing of DNA. Such targeting was found for promoters of actively-transcribed genes (30). Although binding of cisplatin to these sensitive regions of the genome can clearly be capable of disrupting cellular activity, there is no intrinsic sequence preference for binding to such sites beyond the availability of multiple GG units.
Binding to an essential part of the genome could clearly be deleterious for cell survival. It is interesting to consider the probability that such targeting would occur in a patient treated with cisplatin. The human genome contains 3 × 109 base pairs of DNA of which ∼10–20% are functionally important as coding, control or regulatory sequences. The probability that a platinum adduct will be present at a given site in the human genome in vivo can be estimated from the D/N ratio in treated patients. Adduct levels of 0.05–0.2 fmol Pt/µg DNA, corresponding to an rb of 2–7 × 10–8 occurred in 40 patients whose tumors were responsive to treatment (5). Higher levels of platinum, 0.5–9 fmol/µg DNA, rb = 2–30 × 10–7, were found in a smaller set of patients when samples were taken more rapidly following administration of the drug (31). In the latter study, rapid removal of platinum from the DNA was observed in the first 24 h after administration to a level that was consistent with that observed in the former survey. The probability that platinum will bind once to a given gene is then rb × N, where N is the number of nucleotides. If an entire gene contains 1 × 104 nucleotides that are essential for expression, the probability that this gene will contain one platinum atom is between 0.0002 (rb = 2 × 10–8) and 0.03 (rb = 3 × 10–6) (32). For the purposes of this discussion, it was assumed that the average gene contains 1 × 104 nucleotides, all of which are necessary for expression. This value is likely to be an overestimate for most genes. A study that assessed the effect of platinum adducts on the expression of chloramphenicol acetyltransferase activity in transfected cells discovered that the level of platinum required to inactivate the transfected gene depended on the repair proficiency of the host cells (33). On average an rb = 1–20 × 10–4 was required to produce a hit in this system. These adduct levels are significantly higher than those observed in patients, suggesting that gene targeting may not be significant in the clinical efficacy of cisplatin.
SUPPLEMENTARY MATERIAL
Supplementary Material consisting of detailed mapping and stop-site sequences and an agarose dye gel of resolved 800 bp insert and pSP73 fragments is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Prof. T. de Lange for providing a gift of pSP73.Sty11 plasmid, Dr S. S. Marla for transforming pSP73.Sty11 into XL1Blue E.coli cells, Dr E. R. Jamieson and Prof. J. M. Essigmann for helpful discussions, and Johnson Matthey and Englehard for a gift of cisplatin. Supported by grants CA34992 (to S.J.L.) and CA86061 and T32ES07020 (to J.M.E.) from the National Cancer Institute. S.M.C. is a National Institutes of Health postdoctoral fellow.
REFERENCES
- 1. Gelasco,A. and Lippard,S.J. (1999) In Clarke,M. and Sadler,P.J. (eds), Topics in Biological Inorganic Chemistry. Springer-Verlag, Heidelberg, Germany, Vol. 1, pp. 1–43.
- 2.Jamieson E.R. and Lippard,S.J. (1999) Chem. Rev., 99, 2467–2498. [DOI] [PubMed] [Google Scholar]
- 3.Zamble D.B. and Lippard,S.J. (1999) In Lippert,B. (ed.), Cisplatin-Chemistry and Biochemistry of a Leading Anticancer Drug. Verlag Helvetica Chimica Acta, Zurich, Switzerland, pp. 73–110.
- 4.Reed E., Ozols,R.F., Tarone,R., Yuspa,S.H. and Poirier,M.C. (1987) Proc. Natl Acad. Sci. USA, 84, 5024–5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reed E., Yuspa,S.H., Zwelling,L.A., Ozols,R.F. and Poirer,M.C. (1986) J. Clin. Invest., 77, 545–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poirier M.C., Reed,E., Zwelling,L.A., Ozols,R.F., Litterst,C.L. and Yuspa,S.H. (1985) Environ. Health Perspect., 62, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benasutti M., Ejadi,S., Whitlow,M.D. and Loechler,E.L. (1988) Biochemistry, 27, 472–481. [DOI] [PubMed] [Google Scholar]
- 8.Mattes W.B., Hartley,J.A. and Kohn,K.W. (1986) Nucleic Acids Res., 14, 2971–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Villanueva J.M., Jia,X., Yohannes,P.G., Doetsch,P.W. and Marzilli,L.G. (1999) Inorg. Chem., 38, 6069–6080. [DOI] [PubMed] [Google Scholar]
- 10.Temple M.D., McFadyen,W.D., Holmes,R.J., Denny,W.A. and Murray,V. (2000) Biochemistry, 39, 5593–5599. [DOI] [PubMed] [Google Scholar]
- 11.Murray V., Motyka,H., England,P.R., Wickham,G., Lee,H.H., Denny,W.A. and McFadyen,W.D. (1992) Biochemistry, 31, 11812–11817. [DOI] [PubMed] [Google Scholar]
- 12.Murray V., Motyka,H., England,P.R., Wickham,G., Lee,H.H., Denny,W.A. and McFadyen,W.D. (1992) J. Biol. Chem., 267, 18805–18809. [PubMed] [Google Scholar]
- 13.Murray V., Whittaker,J., Temple,M.D. and McFadyen,W.D. (1997) Biochim. Biophys. Acta, 1354, 261–271. [DOI] [PubMed] [Google Scholar]
- 14.Ponti M., Forrow,S.M., Souhami,R.L., D’Incalci,M. and Hartley,J.A. (1991) Nucleic Acids Res., 19, 2929–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whittaker J., McFadyen,W.D., Wickham,G., Wakelin,L.P.G. and Murray,V. (1998) Nucleic Acids Res., 26, 3933–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tullius T.D. and Lippard,S.J. (1981) J. Am. Chem. Soc., 103, 4620–4622. [Google Scholar]
- 17.Pinto A.L. and Lippard,S.J. (1985) Proc. Natl Acad. Sci. USA, 82, 4616–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gralla J.D., Sasse-Dwight,S. and Poljak,L.G. (1987) Cancer Res., 47, 5092–5096. [PubMed] [Google Scholar]
- 19.Ishibashi T. and Lippard,S.J. (1998) Proc. Natl Acad. Sci. USA, 95, 4219–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yanisch-Perron C., Vieira,J. and Messing,J. (1985) Gene, 33, 103–119. [DOI] [PubMed] [Google Scholar]
- 21.Fichtinger-Schepman A.M.J., van der Veer,J.L., den Hartog,J.H.J., Lohman,P.H.M. and Reedijk,J. (1985) Biochemistry, 24, 707–713. [DOI] [PubMed] [Google Scholar]
- 22. Lepre,C.A. and Lippard,S.J. (1990) In Eckstein,F. and Lilly,D.M.J. (eds), Nucleic Acids and Molecular Biology. Springer-Verlag, Berlin, Germany, Vol. 4, pp. 9–38.
- 23.Colgin L.M. and Reddel,R.R. (1999) Curr. Opin. Gen. Dev., 9, 97–103. [DOI] [PubMed] [Google Scholar]
- 24.Blackburn E.H. (1991) Nature, 350, 569–573. [DOI] [PubMed] [Google Scholar]
- 25. Saenger,W. (1984) In Cantor,C.R. (ed.), Springer Advanced Texts in Chemistry. Springer-Verlag, New York, NY, pp. 201–219.
- 26.Elmroth S.K.C. and Lippard,S.J. (1994) J. Am. Chem. Soc., 116, 3633–3634. [Google Scholar]
- 27.Elmroth S.K.C. and Lippard,S.J. (1995) Inorg. Chem., 34, 5234–5243. [Google Scholar]
- 28.Buchanan R.L. and Gralla,J.D. (1990) Biochemistry, 29, 3436–3442. [DOI] [PubMed] [Google Scholar]
- 29.Heiger-Bernays W.J., Essigmann,J.M. and Lippard,S.J. (1990) Biochemistry, 29, 8461–8466. [DOI] [PubMed] [Google Scholar]
- 30.Haghighi A., Lebedeva,S. and Gjerset,R.A. (1999) Biochemistry, 38, 12432–12438. [DOI] [PubMed] [Google Scholar]
- 31.Fichtinger-Schepman A.M.J., van Oosterom,A.T., Lohman,P.H.M. and Berends,F. (1987) Cancer Res., 47, 3000–3004. [PubMed] [Google Scholar]
- 32.Lewin B. (1990) Genes IV. John Wiley & Sons, New York, NY.
- 33.Sheibani N., Jennerwein,M.M. and Eastman,A. (1989) Biochemistry, 28, 3120–3124. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.