


Abstract
Error-free lesion bypass and error-prone lesion bypass are important cellular responses to DNA damage during replication, both of which require a DNA polymerase (Pol). To identify lesion bypass DNA polymerases, we have purified human Polκ encoded by the DINB1 gene and examined its response to damaged DNA templates. Here, we show that human Polκ is a novel lesion bypass polymerase in vitro. Purified human Polκ efficiently bypassed a template 8-oxoguanine, incorporating mainly A and less frequently C opposite the lesion. Human Polκ most frequently incorporated A opposite a template abasic site. Efficient further extension required T as the next template base, and was mediated mainly by a one-nucleotide deletion mechanism. Human Polκ was able to bypass an acetylaminofluorene-modified G in DNA, incorporating either C or T, and less efficiently A opposite the lesion. Furthermore, human Polκ effectively bypassed a template (–)-trans-anti-benzo[a]pyrene-N2-dG lesion in an error-free manner by incorporating a C opposite the bulky adduct. In contrast, human Polκ was unable to bypass a template TT dimer or a TT (6-4) photoproduct, two of the major UV lesions. These results suggest that Polκ plays an important role in both error-free and error-prone lesion bypass in humans.
INTRODUCTION
Lesion bypass is an important cellular response to unrepaired DNA damage during replication. Two modes of lesion bypass are known, error-free and error-prone bypass. The former mechanism does not introduce mutations opposite the lesion, whereas the latter mechanism is frequently accompanied by mutations. Therefore, error-prone lesion bypass constitutes a major mechanism of DNA damage-induced mutagenesis in cells. In Escherichia coli, damage-induced mutagenesis is mainly mediated by the UmuCD pathway under the control of the SOS response (1,2). DNA polymerase (Pol) V composed of the UmuCD′2 protein complex is responsible for the translesion DNA synthesis step (3,4). This mechanism largely results in targeted mutations opposite the DNA lesion. Under genotoxic stress conditions, the SOS system additionally turns on the dinB gene that codes for DNA polymerase IV (5). This polymerase is involved in untargeted mutagenesis in E.coli, i.e. mutations that arise in undamaged regions of DNA (6).
In eukaryotes, a damage-induced mutagenesis pathway has been discovered, originally in the yeast Saccharomyces cerevisiae (7,8) and later in humans (9–13). In this mutagenesis pathway, DNA Polζ (the REV3–REV7 protein complex) and the REV1 dCMP transferase are involved in the translesion DNA synthesis step (11,14,15). More recently, it has been demonstrated that Polη is also involved in translesion synthesis opposite several DNA lesions, notably TT dimer (16–18). However, genetic analysis in yeast has indicated that Polη and the Rev3 mutagenesis pathway are two independent mechanisms of lesion bypass in response to UV radiation (19). During bypass of UV lesions, the Polη mechanism is error-free, whereas the REV3 pathway is error-prone (16,17,19,20). This feature further distinguishes these two lesion bypass mechanisms. In response to some other DNA lesions, Polη can be error-prone as well (18,21,22). In yeast, Polη is encoded by the RAD30 gene (19,23). It is now clear that the E.coli UmuC, the E.coli DinB, the yeast Rev1 and the yeast Rad30 constitute a group of related proteins called the UmuC superfamily. Human homologs of DinB (24,25), Rev1 (11,12) and Rad30 (26–28) have recently been isolated. A direct role in lesion bypass has been demonstrated in vitro for UmuC (3,4), Rev1 (11,14) and Rad30 (16–18,27). Most recently, the human DINB1 protein was shown to be a DNA polymerase, variously designated as Polκ (29) and Polθ (30). It is noted that the eighth human DNA polymerase that is different from DINB1 has been previously reported and named Polθ (31). To avoid confusion, we will refer to the human DINB1 protein as Polκ in this report.
Lesion bypass is a ubiquitous biological process and directly affects human health. It affects the cytotoxicity and mutageneity of DNA damaging agents, including many chemical carcinogens and radiations. Mutations are building blocks for evolution, hereditary diseases and cancer. A recent example for the latter two biological effects is demonstrated by the human hereditary disease xeroderma pigmentosum variant (XPV). This disease is caused by mutations that inactivate the XPV gene coding for the lesion bypass protein Polη (26,27). Loss of error-free lesion bypass of pyrimidine dimers by Polη results in elevated mutagenesis following UV radiation (32), sensitivity to the sunlight and a predisposition to skin cancer (33). Hence, understanding lesion bypass is a key to the understanding of carcinogenesis.
In addition to the REV3 pathway and Polη, it is not known whether there is another lesion bypass mechanism in humans. Most recently, in vitro studies of apurinic/apyrimidinic (AP) site bypass by human Polκ have yielded contradictory conclusions (29,30). To gain an insight into the function of Polκ in human biology, we have studied this DNA polymerase in response to six different types of DNA lesions. In this report, we demonstrate that human Polκ is a novel lesion bypass polymerase in vitro. Furthermore, we show that purified human Polκ is capable of both error-free and error-prone lesion bypass, depending on the specific lesion.
MATERIALS AND METHODS
Materials
A mouse monoclonal antibody against the His6 tag was purchased from Qiagen (Valencia, CA). Alkaline phosphatase conjugated anti-mouse IgG was from Sigma (St Louis, MO). Pfu DNA polymerase was from Stratagene (La Jolla, CA). The E.coli Fpg protein was obtained from Trevigen (Gaithersburg, MA). The yeast rad30 deletion mutant strain BY4741rad30Δ (MATa his3 leu2 met15 ura3 rad30Δ) was purchased from Research Genetics (Huntsville, AL). N-acetoxy-N-2-acetylaminofluorene (AAAF) was obtained from the Midwest Research Institute (Kansas City, MO). Human testis cDNA was purchased from Clontech Laboratories (Palo Alto, CA).
Damaged DNA templates
A 30mer DNA template containing a site-specific 8-oxoguanine was synthesized via automated DNA phosphoramidite methods by Operon (Alameda, CA). Its sequence is 5′-GGATGGACTGCAGGATCCGGAGGCCGCGCG-3′, where the position of the 8-oxoguanine is underlined. The 36mer templates containing a site-specific tetrahydrofuran (AP site analog) were also synthesized by Operon. Their sequences are 5′-GAAGGGATCCTTAAGACTXTAACCGGTCTTCGCGCG-3′, 5′-GAAGGGATCCTTAAGACAXTAACCGGTCTTCGCGCG-3′, 5′-GAAGGGATCCTTAAGACGXTAACCGGTCTTCGCGCG-3′ and 5′-GAAGGGATCCTTAAGACCXTAACCGGTCTTCGCGCG-3′, where X designates the AP site. A 49mer DNA template containing a site-specific cis-syn TT dimer or a TT (6-4) photoproduct was prepared as previously described (34). Its sequence is 5′-AGCTACCATGCCTGCACGAATTAAGCAATTCGTAATCATGGTCATAGCT-3′, where the modified TT is underlined. A 33mer DNA template, 5′-CTCGATCGCTAACGCTACCATCCGAATTCGCCC-3′, containing a site-specific (–)-trans-anti-benzo[a]pyrene [(–)-trans-anti-BPDE] adduct at the N2 position of the underlined G was prepared as previously described (35–37).
To prepare AAF-adducted DNA template, 2 nmol of the oligonucleotide 5′-CCTTCTTCTTCATACAAGCTTACTTCTTCC-3′ was incubated with 200 nmol of N- AAAF at 37°C in the dark for 3 h in 100 µl of TE buffer (10 mM Tris–HCl pH 7.5, 1 mM EDTA) containing 20% ethanol. AAAF predominantly modified the unique G in the DNA. Free AAAF was removed by extracting the reaction mixture five times with water-saturated ether. Then, damaged and undamaged oligonucleotides were separated by electrophoresis on a 20% denaturing polyacrylamide gel. Acetylaminofluorene (AAF)-modified DNA migrated slower on the gel and was sliced out of the gel. The gel slices were soaked in 150 µl water at room temperature for 4 h. Finally, AAF-damaged DNA was recovered using GenElute DNA spin column (Sigma).
Human DINB1 gene and its overexpression
The vector pECUh6 was used to express human DINB1 gene in yeast cells. To construct pECUh6, the yeast CUP1 promoter sequence (269 bp) was amplified by polymerase chain reaction (PCR) from yeast genomic DNA using Pfu DNA polymerase and two primers: 5′-CGGAATTCTGTCAAGAGATTCTTTTGCTGGC-3′ and 5′-CGGAATTCTCGAGTTTATGTGATGATTGATTGATTGATTGTAC-3′. The resulting PCR product was cloned into the EcoRI-digested pEGUh6 (38), replacing its GAL1/10 promoter sequence and yielding pECUh6. The yeast expression vector pECUh6 contains the 2 µm origin for multicopy plasmid replication, the URA3 gene for plasmid selection, the CUP1 promoter for inducible gene expression and six histidine codons for N-terminal protein tagging.
The human DINB1 cDNA was obtained by PCR amplification from human testis cDNAs using Pfu DNA polymerase and two primers: 5′-CGGGATCCATGGATAGCACAAAGGAGAAG-3′ and 5′-CCCAAGCTTCACCTTCTTATCCCATTCCTAATTTC-3′. The resulting 2.9 kb PCR product was then cloned into the BamHI and HindIII sites of the vector pECUh6, yielding pECUh6–hDINB1. The entire sequence of the cloned human DINB1 cDNA was confirmed by DNA sequencing.
Purification of human DNA Polκ
Yeast rad30 deletion mutant cells harboring pECUh6–hDINB1 were grown at 30°C for 2 days in minimum medium containing 2% dextrose. After 10-fold dilution in 16 l of YPD medium (2% bacto-peptone, 1% yeast extract, 2% dextrose), cells were grown for 6 h at 30°C. Expression of the human DINB1 gene was induced by adding CuSO4 to 0.3 mM and growth for another 3 h. Collected cells (∼80 g) were homogenized by zirconium beads in a Bead-beater in an extraction buffer containing 50 mM Tris–HCl pH 7.5, 1 M KCl, 10% sucrose, 20% glycerol, 5 mM β-mercaptoethanol and protease inhibitors (39). The clarified extract (∼130 ml) was loaded onto a HiTrap chelating column charged with NiSO4 (2 × 5 ml; Amersham Pharmacia Biotech, Piscataway, NJ), followed by washing the column sequentially with 100 ml of Ni buffer A (20 mM KH2PO4 pH 7.4, 1 M NaCl, 10% glycerol, 5 mM β-mercaptoethanol and protease inhibitors) containing 10 mM imidazole and 100 ml of Ni buffer A containing 35 mM imidazole. Bound proteins were eluted with a linear gradient of 35–108 mM imidazole. The His-tagged human DINB1 protein was identified by western blots using a mouse monoclonal antibody specific to the His6 tag. The pooled sample was concentrated by PEG 10 000 and desalted through 5 × 5 ml Sephadex G-25 columns in FPLC buffer A (50 mM Tris–HCl pH 7.5, 1 mM EDTA, 10% glycerol and 5 mM β-mercaptoethanol) containing 100 mM KCl. The resulting sample (∼40 ml) was loaded onto an FPLC Mono S HR5/5 column (Amersham Pharmacia Biotech) and eluted with a 30 ml linear gradient of 100–400 mM KCl in FPLC buffer A. Human DINB1 protein was eluted at ∼250 mM KCl. The Mono S fractions were concentrated by PEG 10 000 and then loaded onto an FPLC Superdex 200 gel filtration column that had been equilibrated in FPLC buffer A containing 300 mM KCl. Human DINB1 protein was eluted at ∼100 kDa position.
DNA lesion bypass assays
Lesion bypass assays were performed in standard DNA polymerase reactions using various damaged DNA templates as indicated in the text. The standard DNA polymerase reaction (10 µl) contained 25 mM potassium phosphate pH 7.0, 5 mM MgCl2, 5 mM dithiothreitol, 100 µg/ml bovine serum albumin, 10% glycerol, 50 µM dNTPs (dATP, dCTP, dTTP and dGTP, individually or together as indicated), 50 fmol of a DNA substrate containing a 32P-labeled primer and purified human DNA Polκ. After incubation at 30°C for 10 min, reactions were terminated with 7 µl of a stop solution (20 mM EDTA, 95% formamide, 0.05% bromophenol blue and 0.05% xylene cyanol). The reaction products were resolved on a 20% polyacrylamide gel containing 8 M urea and visualized by autoradiography. Primer extension was quantitated by scanning densitometry of the autoradiogram using the SigmaGel software (Sigma) for analysis.
RESULTS
Purification of human DNA Polκ
The human DINB1 gene codes for a DNA polymerase (24,25,29,30), designated Polκ (29,40). To investigate its potential role in DNA lesion bypass, we purified human Polκ. To facilitate its purification and detection, we tagged human Polκ with six histidine residues at its N-terminus. The tagged human Polκ was expressed in yeast cells of the rad30 deletion mutant strain to avoid potential contamination by the yeast Polη. Human Polκ was then purified to near homogeneity (Fig. 1A). The identity of the tagged human Polκ was confirmed by western blot analysis using a mouse monoclonal antibody specific to the His6 tag (Fig. 1B). A faint band below human Polκ was detected (Fig. 1A). It most likely is a C-terminal degradation product of human Polκ, because this protein band was detected by the His6-specific antibody (Fig. 1B). The purified human Polκ migrated at 81 kDa on a 10% SDS–polyacrylamide gel (Fig. 1A), faster than its calculated molecular weight of 99 kDa. Human Polκ in freshly prepared cell extracts also migrated at a similar position on the 10% SDS–polyacrylamide gel (data not shown). Furthermore, the native molecular weight of the purified human Polκ was determined to be ∼100 kDa by gel filtration chromatography on an FPLC Superdex 200 column. Hence, we conclude that our purified human Polκ is a full-length protein.
Figure 1.
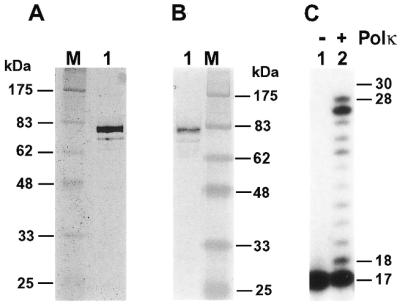
Analyses of purified human Polκ. (A) Purified human Polκ (200 ng) was analyzed by electrophoresis on a 10% SDS–polyacrylamide gel and visualized by silver staining. Protein size markers (lane M) are indicated on the left. (B) Purified human Polκ (40 ng) was analyzed by a western blot using a mouse monoclonal antibody against the His6 tag. Protein size markers (lane M) are indicated on the right. (C) DNA polymerase assays were performed without (lane 1) or with (lane 2) purified human Polκ (0.2 ng, 2 fmol), using the 30mer DNA template, 5′-CCTTCTTCATTGGAACATACTTCTTCTTCC-3′, annealed with the 5′-32P-labeled primer, 5′-GGAAGAAGAAGTATGTT-3′. DNA size markers in nucleotides are indicated on the right.
Using a 30mer DNA template and an annealed 17mer primer containing a 32P label at its 5′-end, we performed a DNA polymerase assay with the purified human Polκ. As shown in Figure 1C, the purified human Polκ possesses a DNA polymerase activity. Primer extension by human Polκ resulted in various sizes of DNA products that differed by one nucleotide in length (Fig. 1C, lane 2), indicating that it is a distributive polymerase. We consistently observed that human Polκ-catalyzed DNA synthesis stops one or two nucleotides before the end of the template (Fig. 1C). This property of human Polκ was also observed by Johnson et al. (30).
Translesion synthesis of template 8-oxoguanine by human Polκ
8-Oxoguanine is a major form of oxidative damage in DNA. To examine whether human Polκ can bypass this DNA lesion, we synthesized a 30mer DNA template containing a site-specific 8-oxoguanine (Fig. 2). This DNA was then labeled with 32P at its 5′-end, annealed to its 30mer complementary strand and incubated with the E.coli 8-oxoguanine-DNA glycosylase (Fpg protein). Complete cleavage at the lesion site was observed (data not shown), thus confirming the identity and the position of the 8-oxoguanine residue in the synthesized DNA.
Figure 2.
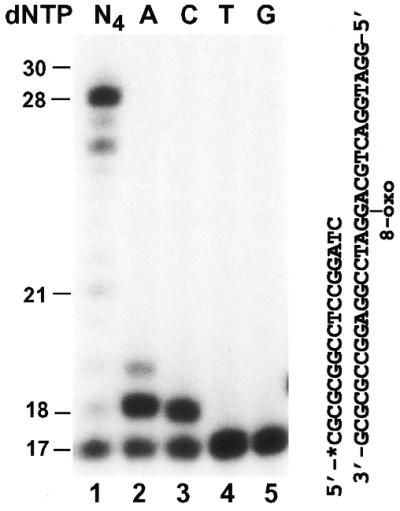
Bypass of a template 8-oxoguanine (8-oxoG) by human Polκ. Standard DNA polymerase assays were performed with purified human Polκ (0.4 ng, 4 fmol) using the indicated DNA substrate containing a template 8-oxoG and a 32P-labeled 17mer primer. Polymerase reactions were carried out in the presence of a single deoxyribonucleoside triphosphate dATP (lane 2), dCTP (lane 3), dTTP (lane 4) or dGTP (lane 5), or all four dNTPs (lane 1). DNA size markers in nucleotides are indicated on the left.
To perform translesion synthesis assays, a 17mer primer was labeled with 32P at its 5′-end and subsequently annealed to the template, right before the 8-oxoguanine residue (Fig. 2). As shown in Figure 2 (lane 1), human Polκ efficiently bypassed the template 8-oxoguanine at a low polymerase concentration (4 fmol). The presence of 8-oxoguanine did not appear to significantly inhibit the activity of human Polκ (Fig. 2, lane 1). This is in contrast to the significant blocking effect of a template 8-oxoguanine on the activity of purified human Polι (our unpublished data). It is known that 8-oxoguanine is a miscoding DNA lesion, capable of base pairing with either a C or an A residue (41). To identify the base incorporated opposite 8-oxoguanine by human Polκ, we performed DNA synthesis assays with only one deoxyribonucleoside triphosphate: dATP, dCTP, dGTP or dTTP individually. Human Polκ extended 63% of the primers using dATP (Fig. 2, lane 2) and 53% of the primers using dCTP (Fig. 2, lane 3) opposite the template 8-oxoguanine. Primer extension was not detected with dTTP and dGTP (Fig. 2, lanes 4 and 5). These results suggest that human Polκ predominantly incorporates an A residue opposite a template 8-oxoguanine. Hence, we conclude that when encountering 8-oxoguanine in DNA, human Polκ will cause mutagenic translesion synthesis.
Mutagenic bypass opposite a template AP site by human Polκ
AP sites are significant DNA lesions, which can arise in the genome spontaneously or can be induced by many environmental agents. Since an AP site can derive from A, T, C or G, AP site bypass by a DNA polymerase will be error-prone. Thus, AP sites are potent mutagenic lesions in DNA. Using purified human Polκ, we examined the response of this polymerase to an AP site in DNA. A 17mer primer was labeled at its 5′-end with 32P and annealed to a 36mer DNA template right before the template AP site. To additionally examine potential sequence context effect on AP site bypass, four templates were used that differed by one nucleotide 5′ to the AP site (Fig. 3A). As shown in Figure 3B (lane 4), human Polκ effectively bypassed the AP site in template AP–T that contained a T 5′ to the AP site. When the template nucleotide 5′ to the AP site was replaced with a C, A or G, the AP site was less efficiently bypassed by human Polκ and one nucleotide insertion opposite the AP site was accumulated as the major product even at a high Polκ concentration (20 ng, 200 fmol) (Fig. 3B, compare lanes 2, 3 and 5 with lane 4). Using the template AP–T containing a ‘running start’ primer annealed 3 nt before the template AP site, lesion bypass was also observed (Fig. 3C, lane 2). Human Polκ activity was slightly inhibited right before and opposite the AP site in template AP–T during DNA synthesis, indicated by a small fraction of aborted synthesis as 17- and 18-nucleotide fragments (Fig. 3C, compare lane 1 with lane 2). These results show that human Polκ is capable of translesion synthesis opposite a template AP site and that the bypass efficiency is greatly stimulated by a template T 5′ to the AP site.
Figure 3.

Bypass of a template AP site by human Polκ. (A) DNA templates used for AP site bypass assays. The 17mer primer was labeled with 32P at its 5′-end as indicated by an asterisk. The AP site is located at the X position. Different template bases 5′ to the AP site are underlined. The AflII restriction cleavage site on the 32P-labeled strand is shown, as well as the AflII recognition sequence. (B) Standard polymerase assays were performed with 20 ng (200 fmol) of purified human Polκ using an undamaged DNA template (lane 1) or the various AP site-containing templates as indicated (lanes 2–5). (C) Polymerase assays were performed using undamaged template 18T (lane 1) or the AP site-containing template AP–T (lane 2) containing the 32P-labeled primer, 5′-CGCGCGAAGACCGG-3′. This ‘running start’ primer was annealed 3 nt before the AP site (arrowhead) on template AP–T. Polκ used was 10 ng (100 fmol) and 20 ng (200 fmol) for lanes 1 and 2, respectively. DNA size markers in nucleotides are indicated on the left.
To identify the nucleotide incorporated opposite the template AP site, we performed lesion bypass assays with only one deoxyribonucleoside triphosphate. As shown in Figure 4A–D (lane 2), A was most frequently inserted opposite the template AP site, which occurred at a low Polκ concentration (20 fmol) with the AP–T template, but required a high Polκ concentration (200 fmol) with templates AP–G, AP–A and AP–C. The efficiency of C, T or G incorporation opposite the AP site was strongly influenced by the template base 5′ to the AP site (Fig. 4A–D, lanes 3–5). With the template AP–A, significant C, T and G incorporations by human Polκ were observed opposite the AP site, at levels only slightly lower than that of A incorporation (Fig. 4C, lanes 2–5).
Figure 4.

Nucleotide incorporation opposite the template AP site. Lesion bypass assays were performed with various AP site-containing templates (50 fmol) in a standard DNA polymerase reaction buffer containing all four dNTPs (lane 1) or dATP (lane 2), dCTP (lane 3), dTTP (lane 4) and dGTP (lane 5) individually. Templates contained a 32P-labeled 17mer primer annealed right before the template AP site. DNA sequences of the various templates are shown in Figure 3A. (A) Template AP–T, 2 ng (20 fmol) Polκ; (B) template AP–G, 20 ng (200 fmol) Polκ; (C) template AP–A, 20 ng (200 fmol) Polκ; (D) template AP–C, 20 ng (200 fmol) Polκ. DNA size markers in nucleotides are indicated on the left. Quantitation of extended primers is shown below the gels.
We consistently observed that DNA synthesis products are 1–2 nt shorter using template AP–T than those using the undamaged control template (template 18T) (Figs 3B, compare lane 1 with lane 4, 3C and 5A, compare lane 1 with lane 3). We suspected that these bypass products might contain deletions. To examine this possibility, we performed AP site bypass assays followed by digestion with the restriction endonuclease AflII. After AflII digestion, a 32P-labeled 22-nt band was expected with a normal primer extension (Fig. 3A). Indeed, using the undamaged template (template 18T), DNA synthesis products of human Polκ (Fig. 5A, lane 1) were cleaved by AflII to a 22-nt fragment (Fig. 5A, lane 2). Using the AP site template AP–T, lesion bypass products of human Polκ (Fig. 5A, lane 3) were cleaved by AflII to a major 21-nt fragment and a minor 20-nt fragment (Fig. 5A, lane 4), indicating 1- and 2-nt deletions, respectively. Using AP site templates AP–A, AP–G and AP–C containing an A, G or C, respectively, 5′ to the AP site (Fig. 3A), we performed similar AflII cleavage experiments following lesion bypass by human Polκ. As shown in Figure 5B (lanes 2, 4 and 6), the major cleavage product was the 22-nt DNA fragment. A 21-nt DNA fragment (due to –1 deletion) was additionally observed as a minor AflII cleavage product with templates AP–A and AP–G (Fig. 5B, lanes 2 and 4). A 20-nt DNA fragment (due to –2 deletion) was observed as a minor AflII cleavage product with the template AP–C (Fig. 5B, lane 6). Consistent with these results, the major lesion bypass products with templates AP–A, AP–G and AP–C ranged from 33–35 nt, whereas those with the template AP–T ranged from 32–34 nt (Fig. 5A and B, compare lane 3 with lanes 1, 3 and 5, respectively).
Figure 5.
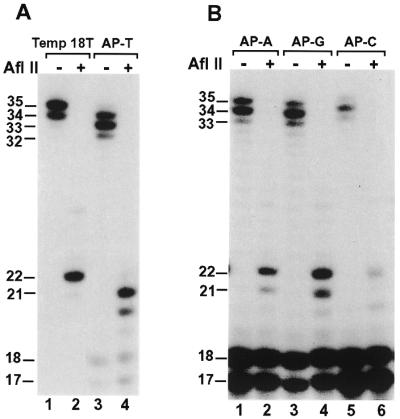
Analysis of AP site bypass products by AflII restriction digestion. (A) Standard DNA polymerase assays were performed with the undamaged DNA template (Temp 18T) or the AP site-containing template (AT–T) using 20 ng (200 fmol) of human Polκ. After the polymerase reaction, 5 µl of the reaction products were mixed with 2 µl of H2O, 1 µl of the 10× AfIII buffer (500 mM Tris–HCl pH 8.0, 100 mM MgCl2) and 2 µl of AfIII (20 U). AfIII digestions were at 37°C for 4 h. The digested products were separated by electrophoresis on a 20% denaturing polyacrylamide gel and visualized by autoradiography. Samples without (AflII, –) or with (AflII, +) AflII treatment are indicated. DNA size markers in nucleotides are indicated on the left. (B) Standard DNA polymerase assays were performed with the AP site-containing templates as indicated using 20 ng (200 fmol) of human Polκ. After the polymerase reaction, 5 µl of the reaction products were digested with AflII as in (A). DNA sequences of the various templates are shown in Figure 3A.
Taken together, these results show that human Polκ most frequently incorporates A opposite a template AP site. Efficient further extension requires T as the next template base and is mediated mainly by a one-nucleotide deletion mechanism. With an A, C or G template base 5′ to the AP site, lesion bypass by human Polκ is inefficient and is mediated mainly by a mechanism of A insertion followed by extension without deletion.
Error-prone lesion bypass opposite AAF-adducted guanine by human Polκ
In contrast to AP sites, AAF adducts are bulky lesions in DNA. AAF-adducted guanines are the major AAF lesions in DNA, which block many DNA polymerases (18,42). To examine whether human Polκ is able to bypass the bulky AAF-guanine lesion, we performed lesion bypass assays using a DNA template containing a site-specific AAF adduct. A 32P-labeled 13mer primer was annealed 4 nt before the AAF-adducted guanine (Fig. 6A). As shown in Figure 6A, human Polκ was able to bypass the AAF adduct at a high polymerase concentration (200 fmol). However, a significant amount of DNA synthesis stopped after incorporating one nucleotide opposite the AAF-guanine (18-nt product) (Fig. 6A, lane 2). DNA synthesis stopping at this position was not observed with the undamaged control template (Fig. 6A, lane 1). Thus, the rate-limiting step of AAF bypass by human Polκ is at the extension step after nucleotide incorporation opposite the lesion. Furthermore, the pattern of primer extension products is very similar at the end of both undamaged and AAF-damaged templates (Fig. 6A, compare lane 1 with lane 2). This result suggests that deletion is not a major mechanism for AAF bypass by human Polκ at this sequence context.
Figure 6.

Bypass of AAF-guanine by human Polκ. (A) DNA polymerase assays were performed with purified human Polκ using undamaged template (lane 1) or the AAF-damaged template (lane 2), both of which shared identical nucleotide sequence. The 32P-labeled ‘running start’ primer was annealed 4 nt before the AAF-G as indicated. Polκ used was 10 ng (100 fmol) and 20 ng (200 fmol) for lanes 1 and 2, respectively. The 18mer DNA band extended opposite the template AAF-G is indicated by an arrowhead. (B) DNA polymerase assays were performed with purified human Polκ (10 ng, 100 fmol) using the indicated DNA substrate containing a template AAF-G and a 32P-labeled 17mer primer. Polymerase reactions were carried out in the presence of a single deoxyribonucleoside triphosphate dATP (lane 3), dCTP (lane 4), dTTP (lane 5) or dGTP (lane 6) or all four dNTPs (lane 2). Lane 1, control reaction without Polκ. DNA size markers in nucleotides are indicated on the left.
To identify the nucleotide incorporated opposite the AAF-guanine, we annealed a 32P-labeled 17mer primer right before the lesion and performed lesion bypass assays in the presence of only one deoxyribonucleoside triphosphate. As shown in Figure 6B (lane 2), effective bypass of the AAF-guanine was also observed when human Polκ was reduced to 100 fmol in the reaction. Furthermore, human Polκ incorporated a C or a T opposite the AAF-guanine with similar efficiencies, extending 64% and 65% primers, respectively (Fig. 6B, lanes 4 and 5). Less frequently, human Polκ also incorporated an A opposite the AAF-guanine, with 40% primer extension (Fig. 6B, lane 3). Least frequently, human Polκ incorporated a G opposite the AAF-guanine, with 11% primer extension (Fig. 6B, lane 6). These results show that human Polκ is capable of translesion synthesis opposite an AAF-adducted guanine in an error-prone manner.
Error-free lesion bypass opposite a (–)-trans-anti-benzo[a]pyrene-N2-dG lesion by human Polκ
Benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide (BPDE) is a metabolite of benzo[a]pyrene, representing an ultimate carcinogen of the parent compound. The racemic anti-BPDE reacts with DNA mainly at the N2 position of guanine, forming stereoisomeric bulky adducts (+)-trans-anti-BPDE-N2-dG, (+)-cis-anti-BPDE-N2-dG, (–)-trans-anti-BPDE-N2-dG and (–)-cis-anti-BPDE-N2-dG (43,44). Using purified human Polκ, we examined the response of this polymerase to a template (–)-trans-anti-BPDE-N2-dG bulky lesion. A 19mer primer was labeled with 32P at its 5′ end and annealed right before the template (–)-trans-anti-BPDE-N2-dG lesion (Fig. 7). As shown in Figure 7 (lane 1), human Polκ efficiently bypassed the (–)-trans-anti-BPDE-N2-dG lesion at a low polymerase concentration (20 fmol). To identify the nucleotide incorporated opposite the lesion, we performed lesion bypass assays with only one deoxyribonucleoside triphosphate. As shown in Figure 7 (lanes 2–5), human Polκ incorporated a C opposite the template (–)-trans-anti-BPDE-N2-dG. These results demonstrate that human Polκ is capable of error-free lesion bypass opposite a template (–)-trans-anti-BPDE-N2-dG.
Figure 7.
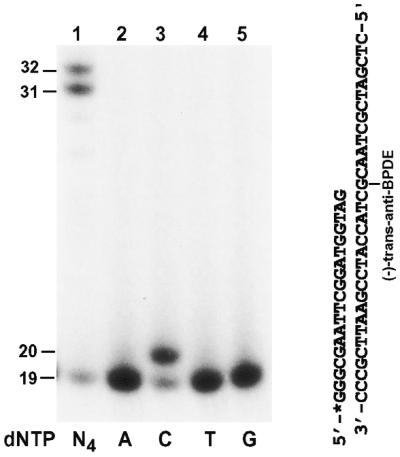
Bypass of a template (–)-trans-anti-BPDE-N2-dG lesion by human Polκ. A 19mer primer was labeled with 32P at its 5′-end and annealed right before a template (–)-trans-anti-BPDE-N2-dG as shown on the right. Polymerase reactions were performed with 2 ng (20 fmol) of human Polκ in the presence of a single deoxyribonucleoside triphosphate dATP (lane 2), dCTP (lane 3), dTTP (lane 4) or dGTP (lane 5) or all four dNTPs (lane 1). DNA size markers in nucleotides are indicated on the left.
Human Polκ is unable to bypass a TT dimer or a TT (6-4) photoproduct
Cyclobutane pyrimidine dimers (CPD) and (6-4) photoproducts are the major DNA lesions following UV radiation. Recently, it was discovered that DNA Polη is capable of error-free lesion bypass opposite a TT dimer (a CPD lesion), but is unable to bypass a TT (6-4) photoproduct (16,17,27). Like Polκ, Polη is also a member of the UmuC superfamily of proteins. Thus, it is of particular interest to examine how human Polκ would respond to a TT dimer and a TT (6-4) photoproduct in DNA. To do this, we labeled a 15mer primer at its 5′-end with 32P and annealed it to a 49mer DNA template containing a site-specific TT dimer or a TT (6-4) photoproduct (Fig. 8). Lesion bypass assays were then carried out with purified human Polκ. As shown in Figure 8 (lanes 3 and 4), human Polκ was unable to insert a nucleotide opposite the 3′ T of either the TT dimer or the TT (6-4) photoproduct, even at a high polymerase concentration (200 fmol). In contrast, purified human Polη efficiently bypassed the template TT dimer (Fig. 8, lane 2), as expected. These results show that human Polκ is unable to bypass a TT dimer or a TT (6-4) photoproduct.
Figure 8.

Response of human Polκ to a TT dimer and a TT (6-4) photoproduct in template DNA. Standard DNA polymerase assays were performed using the indicated DNA substrates containing a template TT dimer or a TT (6-4) photoproduct and a 32P-labeled 15mer primer. The modified template TT sequence is indicated. Polymerase reactions were carried out with 0.8 ng (10 fmol) of purified human Polη (lane 2) or 20 ng (200 fmol) of purified human Polκ (lanes 3 and 4). Lane 1, control reaction without DNA polymerase. DNA size markers in nucleotides are indicated on the left.
DISCUSSION
We have examined the ability of the purified human Polκ in translesion DNA synthesis opposite a template 8-oxoguanine, AP site, AAF-adducted guanine, (–)-trans-anti-BPDE-N2-dG, TT dimer and TT (6-4) photoproduct. Human Polκ effectively bypassed the first four DNA lesions in vitro. Furthermore, purified human Polκ bypassed 8-oxoguanine, AP site, AAF-adducted guanine in an error-prone manner, but bypassed (–)-trans-anti-BPDE-N2-dG in an error-free manner. Hence, we conclude that human Polκ is a novel lesion bypass DNA polymerase.
Human Polκ can incorporate either a C or an A opposite 8-oxoguanine, consistent with the miscoding property of this lesion (41). However, unlike yeast Polη that predominantly incorporates a C opposite a template 8-oxoguanine (18,45), human Polκ incorporates an A more frequently. Thus, the miscoding property of 8-oxoguanine is modulated by the interaction between the lesion and the specific DNA polymerase.
In a study by Johnson et al. (30), it was concluded that human Polκ is unable to bypass an AP site. However, in another study by Ohashi et al. (29), it was concluded just the opposite. It should be noted that while Johnson et al. (30) used a fusion human Polκ containing at its N-terminus a bulky (26 kDa) glutathione S-transferase (GST) protein, Ohashi et al. (29) used a truncated human Polκ missing 310 amino acid residues at the C-terminus. Our results using a full-length human Polκ are consistent with those of Ohashi et al. (29). The different results of Johnson et al. (30) with respect to AP site bypass might be due to the fused GST protein and/or lower concentrations of Polκ used. Human Polκ was used at 0.5 nM by Johnson et al. (30), whereas 2 nM and 4 nM were used by us (Fig. 4A) and Ohashi et al. (29), respectively.
The efficiency and specificity of nucleotide incorporations by human Polκ opposite an AP site are strongly influenced by the template base 5′ to the lesion. Nucleotide incorporation occurs most efficiently when the template base 5′ to the AP site is a T. Nevertheless, A is most frequently incorporated opposite the AP site by human Polκ, which is most dramatic when the template base 5′ to the lesion is either a T or a C. Further extension by human Polκ is greatly stimulated by a template T 5′ to the AP site. This stimulation is largely due to a one-nucleotide deletion mechanism. We propose that following incorporation of A opposite the AP site, further extension by human Polκ is efficiently achieved through misaligning the primer terminal A with the next template T, leading to –1 deletion. Supporting this model, in the absence of the 5′ template T, primer extension by human Polκ is largely stopped after incorporating one nucleotide opposite the AP site. Moreover, this model is also consistent with the observation that the E.coli DinB is prone to cause –1 deletion mutations (6). A similar model was also independently proposed by Ohashi et al. (29).
In E.coli, translesion synthesis of an AP site results in preferential incorporation of an A opposite the lesion, leading to the ‘A rule’ hypothesis (46). In mammals, such a strong bias opposite an AP site was not obvious. Opposite an AP site, incorporations of A, C and T at similar frequencies were reported in simian COS cells using a plasmid mutagenesis system (47–50). In another study in COS cells, preferential A incorporation was observed (51). In a study using human lymphoblastoid cells, preferential G incorporation was noticed (52).
In the yeast S.cerevisiae, the Polζ–REV1 bypass of AP site results in predominantly C insertion opposite the lesion (53), while yeast Polη incorporates G and less frequently A opposite the AP site (18). However, this yeast does not contain the DINB1 gene. Humans contain Polζ encoded by the REV3 (9,10) and REV7 (13) genes, REV1 (11,12) and Polη (26,27). Thus, in addition to Polκ, AP sites may also be bypassed by the Polζ–REV1 pathway and the Polη mechanism in human cells. Indeed, human REV1 efficiently incorporates a C opposite a template AP site in vitro (11) and purified human Polη is capable of incorporating an A and less efficiently a G opposite the template AP site (21,22). Possible involvement of multiple mechanisms and the sequence context effect during AP site bypass in mammals may provide an explanation to reconcile different results of AP site bypass by different studies.
The bulky AAF adduct blocks many DNA polymerases (18,42). However, human Polκ is able to effectively bypass an AAF-adducted guanine in vitro at a high polymerase concentration (100 fmol) and incorporates C, T, less frequently A, and occasionally G, at least in the sequence context tested. Similar results were also obtained by Ohashi et al. (29) with a C-terminal truncated form of human Polκ. Using a shuttle vector containing a site-specific AAF-guanine, Shibutani et al. (54) observed that A, T and G were misincorporated opposite the lesion in simian kidney cells, resulting in eight G→T, six G→A and two G→C mutations, respectively, from 156 clones analyzed. Our in vitro results are in general agreement with this in vivo study. Thus, Polκ may contribute to AAF mutagenesis in mammals. Previously, we have shown that yeast Polη specifically incorporates a C opposite an AAF-adducted guanine (18). More recently, error-free bypass of AAF-adducted guanine has been extended to human Polη (our unpublished data) (22). AAF bypass by Polη may account for the fact that the majority of recovered shuttle vector from COS cells did not contain mutations opposite the lesion (54).
Benzo[a]pyrene is a potent environmental carcinogen. The racemic anti-BPDE compounds are cellular metabolites of benzo[a]pyrene, which readily attack DNA at the N2 position of guanine (43,44). One of the four stereoisomeric guanine adducts of racemic anti-BPDE is the (–)-trans-anti-BPDE-N2-dG (43,44). This lesion is mutagenic in COS cells (55). In this study, we observed that human Polκ is capable of efficient error-free bypass opposite a template (–)-trans-anti-BPDE-N2-dG lesion at a low polymerase concentration. This result suggests that Polκ may play an important role in suppressing benzo[a]pyrene-induced mutagenesis in humans.
In contrast, human Polκ is unable to bypass a TT dimer and a TT (6-4) photoproduct, which are among the major lesions induced by UV radiation. On the other hand, human Polη efficiently catalyzes error-free translesion synthesis opposite the template TT dimer (17,27). Without functional Polη (encoded by the XPV gene in humans), XPV disease is manifested in humans (26,27), which is characterized by sun sensitivity and a predisposition to skin cancer (33). The dramatic phenotype of XPV patient suggests that error-free bypass of TT dimers and perhaps additional CPD lesions is mainly contributed by Polη and could not be substituted by other DNA polymerases in vivo. The inability of human Polκ to bypass a TT dimer supports this conclusion.
The biological consequence of an unrepaired DNA lesion is determined by whether it is bypassed or not, bypassed in an error-free or error-prone mode and the specificity of mutations induced during lesion bypass. Since the same lesion can be responded to very differently by different DNA polymerases, it seems that the biological effect of a lesion can be significantly affected by the unique interactions between the lesion and a specific DNA polymerase. Based on this notion, it appears that the structure of a lesion alone would not be sufficient to precisely predict the biological outcome of the lesion in DNA. With the identification of Polκ as a lesion bypass polymerase in vitro, it is very likely that multiple lesion bypass mechanisms exist in humans in response to DNA damage during replication.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by a THRI grant from the Tobacco and Health Research Institute of the University of Kentucky (Z.W.), a New Investigator Award in Toxicology from Burroughs Wellcome Fund (Z.W.), an NIH grant CA40463 (J.-S.T.) and an NIH grant CA20851 (N.E.G.).
REFERENCES
- 1.Walker G.C. (1995) Trends Biochem. Sci., 20, 416–420. [DOI] [PubMed] [Google Scholar]
- 2.Goodman M.F. and Tippin,B. (2000) Curr. Opin. Genet. Dev., 10, 162–168. [DOI] [PubMed] [Google Scholar]
- 3.Tang M., Shen,X., Frank,E.G., O’Donnell,M., Woodgate,R. and Goodman,M.F. (1999) Proc. Natl Acad. Sci. USA, 96, 8919–8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reuven N.B., Arad,G., Maor-Shoshani,A. and Livneh,Z. (1999) J. Biol. Chem., 274, 31763–31766. [DOI] [PubMed] [Google Scholar]
- 5.Wagner J., Gruz,P., Kim,S.R., Yamada,M., Matsui,K., Fuchs,R.P. and Nohmi,T. (1999) Mol. Cell, 4, 281–286. [DOI] [PubMed] [Google Scholar]
- 6.Kim S.R., Maenhaut-Michel,G., Yamada,M., Yamamoto,Y., Matsui,K., Sofuni,T., Nohmi,T. and Ohmori,H. (1997) Proc. Natl Acad. Sci. USA, 94, 13792–13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lemontt J.F. (1971) Genetics, 68, 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lemontt J.F. (1977) Mutat. Res., 43, 179–204. [DOI] [PubMed] [Google Scholar]
- 9.Gibbs P.E., McGregor,W.G., Maher,V.M., Nisson,P. and Lawrence,C.W. (1998) Proc. Natl Acad. Sci. USA, 95, 6876–6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin W., Wu,X. and Wang,Z. (1999) Mutat. Res., 433, 89–98. [DOI] [PubMed] [Google Scholar]
- 11.Lin W., Xin,H., Zhang,Y., Wu,X., Yuan,F. and Wang,Z. (1999) Nucleic Acids Res., 27, 4468–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gibbs P.E., Wang,X.D., Li,Z., McManus,T.P., McGregor,W.G., Lawrence,C.W. and Maher,V.M. (2000) Proc. Natl Acad. Sci. USA, 97, 4186–4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murakumo Y., Roth,T., Ishii,H., Rasio,D., Numata,S., Croce,C.M. and Fishel,R. (2000) J. Biol. Chem., 275, 4391–4397. [DOI] [PubMed] [Google Scholar]
- 14.Nelson J.R., Lawrence,C.W. and Hinkle,D.C. (1996) Nature, 382, 729–731. [DOI] [PubMed] [Google Scholar]
- 15.Nelson J.R., Lawrence,C.W. and Hinkle,D.C. (1996) Science, 272, 1646–1649. [DOI] [PubMed] [Google Scholar]
- 16.Johnson R.E., Prakash,S. and Prakash,L. (1999) Science, 283, 1001–1004. [DOI] [PubMed] [Google Scholar]
- 17.Masutani C., Araki,M., Yamada,A., Kusumoto,R., Nogimori,T., Maekawa,T., Iwai,S. and Hanaoka,F. (1999) EMBO J., 18, 3491–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan F., Zhang,Y., Rajpal,D.K., Wu,X., Guo,D., Wang,M., Taylor,J.-S. and Wang,Z. (2000) J. Biol. Chem., 275, 8233–8239. [DOI] [PubMed] [Google Scholar]
- 19.McDonald J.P., Levine,A.S. and Woodgate,R. (1997) Genetics, 147, 1557–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrison A., Christensen,R.B., Alley,J., Beck,A.K., Bernstine,E.G., Lemontt,J.F. and Lawrence,C.W. (1989) J. Bacteriol., 171, 5659–5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y., Yuan,F., Wu,X., Rechkoblit,O., Taylor,J.-S., Geacintov,N.E. and Wang,Z. Nucleic Acids Res. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masutani C., Kusumoto,R., Iwai,S. and Hanaoka,F. (2000) EMBO J., 19, 3100–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roush A.A., Suarez,M., Friedberg,E.C., Radman,M. and Siede,W. (1998) Mol. Gen. Genet., 257, 686–692. [DOI] [PubMed] [Google Scholar]
- 24.Gerlach V.L., Aravind,L., Gotway,G., Schultz,R.A., Koonin,E.V. and Friedberg,E.C. (1999) Proc. Natl Acad. Sci. USA, 96, 11922–11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogi T., Kato,T., Jr., Kato,T. and Ohmori,H. (1999) Genes Cells, 4, 607–618. [DOI] [PubMed] [Google Scholar]
- 26.Johnson R.E., Kondratick,C.M., Prakash,S. and Prakash,L. (1999) Science, 285, 263–265. [DOI] [PubMed] [Google Scholar]
- 27.Masutani C., Kusumoto,R., Yamada,A., Dohmae,N., Yokoi,M., Yuasa,M., Araki,M., Iwai,S., Takio,K. and Hanaoka,F. (1999) Nature, 399, 700–704. [DOI] [PubMed] [Google Scholar]
- 28.McDonald J.P., Rapic-Otrin,V., Epstein,J.A., Broughton,B.C., Wang,X., Lehmann,A.R., Wolgemuth,D.J. and Woodgate,R. (1999) Genomics, 60, 20–30. [DOI] [PubMed] [Google Scholar]
- 29.Ohashi E., Ogi,T., Kusumoto,R., Iwai,S., Masutani,C., Hanaoka,F. and Ohmori,H. (2000) Genes Dev., 14, 1589–1594. [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson R.E., Prakash,S. and Prakash,L. (2000) Proc. Natl Acad. Sci. USA, 97, 3838–3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharief F.S., Vojta,P.J., Ropp,P.A. and Copeland,W.C. (1999) Genomics, 59, 90–96. [DOI] [PubMed] [Google Scholar]
- 32.McGregor W.G., Wei,D., Maher,V.M. and McCormick,J.J. (1999) Mol. Cell. Biol., 19, 147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cleaver J.E. and Kraemer,K.H. (1989) In Scriver,C.R., Beaudet,A.L., Sly,W.S. and Valle,D. (eds), The Metabolic Basis of Inherited Disease, 6th edn. McGraw-Hill Book Co., New York, NY, pp. 2949–2971.
- 34.Smith C.A. and Taylor,J.S. (1993) J. Biol. Chem., 268, 11143–11151. [PubMed] [Google Scholar]
- 35.Cosman M., Ibanez,V., Geacintov,N.E. and Harvey,R.G. (1990) Carcinogenesis, 11, 1667–1672. [DOI] [PubMed] [Google Scholar]
- 36.Geacintov N.E., Cosman,M., Mao,B., Alfano,A., Ibanez,V. and Harvey,R.G. (1991) Carcinogenesis, 12, 2099–2108. [DOI] [PubMed] [Google Scholar]
- 37.Rechkoblit O., Amin,S. and Geacintov,N.E. (1999) Biochemistry, 38, 11834–11843. [DOI] [PubMed] [Google Scholar]
- 38.Wu X., Braithwaite,E. and Wang,Z. (1999) Biochemistry, 38, 2628–2635. [DOI] [PubMed] [Google Scholar]
- 39.Xin H., Lin,W., Sumanasekera,W., Zhang,Y., Wu,X. and Wang,Z. (2000) Nucleic Acids Res., 28, 2847–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Friedberg E.C., Feaver,W.J. and Gerlach,V.L. (2000) Proc. Natl Acad. Sci. USA, 97, 5681–5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shibutani S., Takeshita,M. and Grollman,A.P. (1991) Nature, 349, 431–434. [DOI] [PubMed] [Google Scholar]
- 42.Belguise-Valladier P. and Fuchs,R.P. (1995) J. Mol. Biol., 249, 903–913. [DOI] [PubMed] [Google Scholar]
- 43.Cheng S.C., Hilton,B.D., Roman,J.M. and Dipple,A. (1989) Chem. Res. Toxicol., 2, 334–340. [DOI] [PubMed] [Google Scholar]
- 44.Peltonen K. and Dipple,A. (1995) J. Occup. Environ. Med., 37, 52–58. [DOI] [PubMed] [Google Scholar]
- 45.Haracska L., Yu,S.L., Johnson,R.E., Prakash,L. and Prakash,S. (2000) Nat. Genet., 25, 458–461. [DOI] [PubMed] [Google Scholar]
- 46.Strauss B.S. (1991) Bioessays, 13, 79–84. [DOI] [PubMed] [Google Scholar]
- 47.Gentil A., Renault,G., Madzak,C., Margot,A., Cabral-Neto,J.B., Vasseur,J.J., Rayner,B., Imbach,J.L. and Sarasin,A. (1990) Biochem. Biophys. Res. Commun., 173, 704–710. [DOI] [PubMed] [Google Scholar]
- 48.Gentil A., Cabral-Neto,J.B., Mariage-Samson,R., Margot,A., Imbach,J.L., Rayner,B. and Sarasin,A. (1992) J. Mol. Biol., 227, 981–984. [DOI] [PubMed] [Google Scholar]
- 49.Neto J.B., Gentil,A., Cabral,R.E. and Sarasin,A. (1992) J. Biol. Chem., 267, 19718–19723. [PubMed] [Google Scholar]
- 50.Cabral Neto J.B., Cabral,R.E., Margot,A., Le Page,F., Sarasin,A. and Gentil,A. (1994) J. Mol. Biol., 240, 416–420. [DOI] [PubMed] [Google Scholar]
- 51.Takeshita M. and Eisenberg,W. (1994) Nucleic Acids Res., 22, 1897–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klinedinst D.K. and Drinkwater,N.R. (1992) Mol. Carcinog., 6, 32–42. [DOI] [PubMed] [Google Scholar]
- 53.Gibbs P.E. and Lawrence,C.W. (1995) J. Mol. Biol., 251, 229–236. [DOI] [PubMed] [Google Scholar]
- 54.Shibutani S., Suzuki,N. and Grollman,A.P. (1998) Biochemistry, 37, 12034–12041. [DOI] [PubMed] [Google Scholar]
- 55.Moriya M., Spiegel,S., Fernandes,A., Amin,S., Liu,T., Geacintov,N. and Grollman,A.P. (1996) Biochemistry, 35, 16646–16651. [DOI] [PubMed] [Google Scholar]