

Significance
Plants and microorganisms release metabolites that mediate rhizosphere host–microbe interactions and modulate plant adaptation to environmental stresses. However, the mechanisms underlying rhizosphere metabolite-microbiome dynamics and their functional and biological significance are largely unknown. Our study reveals that certain classes of rhizosphere metabolites exhibit a response to abiotic stressors and are linked to changes in the rhizosphere microbial community and plant phenotypes. We propose that a group of underinvestigated rhizosphere compounds can act as keystone metabolites, impacting the composition of the rhizosphere microbiome and potentially modulating plant metabolism in response to nutrient availability. These findings demonstrate the tremendous potential of harnessing plant–metabolite–microbe interactions to optimize rhizosphere microbiome function, promote plant and ecosystem health, and offer broad avenues for soil microbiome research.
Keywords: rhizosphere, microbiome, metabolome, abiotic stress, switchgrass
Abstract
Plants release a wealth of metabolites into the rhizosphere that can shape the composition and activity of microbial communities in response to environmental stress. The connection between rhizodeposition and rhizosphere microbiome succession has been suggested, particularly under environmental stress conditions, yet definitive evidence is scarce. In this study, we investigated the relationship between rhizosphere chemistry, microbiome dynamics, and abiotic stress in the bioenergy crop switchgrass grown in a marginal soil under nutrient-limited, moisture-limited, and nitrogen (N)-replete, phosphorus (P)-replete, and NP-replete conditions. We combined 16S rRNA amplicon sequencing and LC-MS/MS-based metabolomics to link rhizosphere microbial communities and metabolites. We identified significant changes in rhizosphere metabolite profiles in response to abiotic stress and linked them to changes in microbial communities using network analysis. N-limitation amplified the abundance of aromatic acids, pentoses, and their derivatives in the rhizosphere, and their enhanced availability was linked to the abundance of bacterial lineages from Acidobacteria, Verrucomicrobia, Planctomycetes, and Alphaproteobacteria. Conversely, N-amended conditions increased the availability of N-rich rhizosphere compounds, which coincided with proliferation of Actinobacteria. Treatments with contrasting N availability differed greatly in the abundance of potential keystone metabolites; serotonin and ectoine were particularly abundant in N-replete soils, while chlorogenic, cinnamic, and glucuronic acids were enriched in N-limited soils. Serotonin, the keystone metabolite we identified with the largest number of links to microbial taxa, significantly affected root architecture and growth of rhizosphere microorganisms, highlighting its potential to shape microbial community and mediate rhizosphere plant–microbe interactions.
It is well established that plants modify the chemistry and microbiology of the rhizosphere—the soil adjacent to their roots. Compared to the surrounding bulk soil, rhizosphere microbial communities have increased biomass (1, 2), and often lower diversity, and are frequently dominated by microbial taxa from specific lineages (3, 4). In addition, distinct traits are enhanced in rhizosphere microbial communities, including motility (5), cell-to-cell communication or sensing (3), and nutrient uptake (6). While this indicates strong selection for specific rhizosphere competence traits, the mechanisms of rhizosphere microbial community assembly remain ill-defined. Laboratory incubations, hydroponic systems, and greenhouse studies suggest that the chemical signatures of plant exudates and mucilage—collectively termed “rhizodeposits”—are key drivers of rhizosphere microbial community structure and function. For example, as plants develop, their exudation rates and exudate chemistry change in a consistent manner (6–8) as do their rhizosphere microbial communities (2, 9). Furthermore, experiments in hydroponic systems and with exudate additions have shown changes in gene expression of rhizosphere communities (10) and taxa recruitment/repulsion in response to specific exudate compounds (6, 11–13). These findings provide evidence for the direct impact of root-derived metabolites on rhizosphere community structure. However, the relationship between the full spectrum of the rhizosphere exometabolome and changes in microbial community structure in natural soils, particularly under environmental stress conditions, remains largely uninvestigated.
The rhizosphere exometabolome is a diverse chemical milieu of primary and secondary metabolites released by the plant host and rhizosphere microorganisms into the soil surrounding plant roots (6, 14). The exometabolome interacts with the soil environment (i.e., mineralogy, pH, water) and provides a dynamic “playground” for the cross-talk between plants and microorganisms living in soil (15–17). Plant-derived metabolites reflect plant responses to a changing environment and enable plants to modulate their metabolic interactions with microorganisms (14, 18–20), thus potentially enabling them to recruit a beneficial microbiome. At the same time, microorganisms attracted by plant-derived molecules produce metabolites that can alter the plant host’s phenotype and enhance its capacity to withstand environmental stresses (18, 20).
Switchgrass (Panicum virgatum) is a broadly distributed tallgrass native to prairies in the Eastern and Midwestern, USA (21). Drought-tolerant (22) and capable of growing in nutrient-poor marginal soils (23), the bioenergy crop switchgrass is a deep-rooted perennial with significant potential to promote long-term soil carbon (C) sequestration (24–26). Switchgrass seedlings are susceptible to a number of biotic and abiotic stresses during the establishment phase (27), a period when mutualistic plant-microbial relationships that enhance nutrient availability, reduce moisture stress, or protect against pathogens (28–30) could be critical to plant resilience and future yields. Switchgrass has a core microbiome of bacterial taxa that are consistently found in its rhizosphere across diverse soil and sampling environments (31, 32). However, it is not known how the switchgrass microbiome is recruited during establishment, what role metabolites play in this process, and how abiotic stress affects these recruitment mechanisms.
In this study, we linked dynamics of switchgrass rhizosphere metabolites and microbiomes in response to abiotic stress by growing a single switchgrass genotype in a nutrient-poor marginal soil for 18 wk under five treatments: a control, soils amended with phosphorus (+P), nitrogen (+N), both nitrogen and phosphorus (+NP), and water-limited (−W) (Fig. 1A). We identified potential “keystone metabolites”—compounds that may have functional links to specific microbial lineages or abiotic stressors that significantly alter the structure of rhizosphere microbiomes. We found that N availability was a significant determinant of both metabolite and microbial community composition, and resulted in the coenrichment of select microbial lineages and metabolites. Keystone metabolites included compounds such as organic and aromatic acids previously linked to changes in microbiome community structure (6, 12), but also N-rich compounds such as serotonin and acetylcholine that have not been investigated in the rhizosphere but are known to be strong signaling molecules in other settings (33, 34). Further, we used simplified lab experiments to show that one of these keystone metabolites, serotonin, significantly impacts both plant phenotype and the growth of specific rhizosphere microorganisms. This study identifies keystone metabolites with unexplored potential to mediate rhizosphere communities and influence plant phenotypes under nutrient stress. In addition, our study demonstrates an approach for identifying the relationships between rhizosphere metabolites, microbial communities, plant phenotypes, and abiotic stressors in complex living soils.
Fig. 1.

Greenhouse experiment investigating the effect of nutrient or moisture stress on switchgrass biomass, rhizosphere chemistry, and microbial communities. Plants were grown in one-meter-deep mesocosms containing a marginal sandy loam soil, with recreated “A”, “B,” and “C” horizons. (A) Schematic of experimental design illustrating five treatments: “Control” with nutrient-poor marginal soil, “+P,” “+N,” and “+NP” mesocosms with phosphorus and/or nitrogen amendments in the top soil horizon, and “−W” mesocosms which received 50% less water relative to the other treatments. Box-whisker plots (median and 25 to 75% quartiles) of (B) switchgrass root biomass (g dry mass), (C) soil water potential (−kPa), (D) microbial biomass (pmol PLFA/g dry soil) from bulk soil, and (E) microbial α-diversity (Fisher’s α) in rhizosphere soil are shown by treatment for the “A” horizon. Letters represent significantly different post hoc pairwise comparisons via Tukey’s test (P < 0.05, n = 6).
Materials and Methods
Experimental Design, Sample Collection, and Processing.
Soil from remnant Dust Bowl fields in Anadarko, Oklahoma, USA consisting of a nutrient-deplete Pond Creek fine sandy loam (<0.5% total carbon, <1 ppm total nitrogen, <6 ppm total phosphorus) classified as a superactive, thermic Pachic Arguistoll, was collected and used to construct one-meter-deep soil mesocosms (n = 30) in 19.7 cm diameter polycarbonate tubes. Each mesocosm contained layered soils from the “A”, “B”, and “C” horizons and one of five treatments: control (no nutrient amendment), +N (nitrogen added), +P (phosphorus added), +NP (nitrogen and phosphorus added), or −W (water-limited) (Fig. 1A). Slow-release coated urea and rock phosphate were added to the “A” horizon of the appropriate treatments (24). The mesocosms were watered from above with either 50% or 100% of the mean monthly rainfall at the field site in Oklahoma in the summer months. A clonal ramet of Alamo switchgrass genotype was planted in each mesocosm and grown at the University of California, Berkeley, Oxford Tract greenhouse. After 18 wk, the mesocosms were destructively harvested and the soil was processed by horizon. We collected roots and associated rhizosphere soil (<2 mm from root) for DNA extractions and analyzed microbial communities in the “A” horizon where most root biomass and soil nutrients were found (24). Rhizosphere soil from all three horizons was used for metabolite extractions. See Sher et al. (24) and SI Appendix for full sample processing details and assessments.
DNA Extraction, Sequencing and Analysis.
Rhizosphere soil in Lifeguard solution was centrifuged to pellet, and DNA was extracted from single 0.5 g aliquots using a modified RNA/DNA phenol chloroform coextraction protocol via bead-beating (35, 36). The microbial community composition was characterized using the V4 region of the 16S ribosomal RNA gene with the 515F and 806R primer set (37, 38), and samples were sequenced on the Illumina MiSeq platform with 2 × 250 bp format.
All initial bioinformatics processing and production of amplicon sequence variants (ASVs) by DADA2 (39) were conducted within Qiime2 (40), with taxonomy assigned via the SILVA database (release 132) (41). Subsequent processing, visualization, and statistical tests of sequence data were performed in R version 3.6.0 (R Core Team, 2020), primarily within the phyloseq package (42). Chloroplast, mitochondrial, and bacterial or archaeal sequences that lacked designation at the phylum level were discarded. Singletons and doubletons were removed for all analyses other than α-diversity, resulting in 7,093 ASVs accounting for 3,792,087 reads (104,776 to 194,526 per sample). Differentially abundant ASVs between each of the individual treatments and the control samples were determined with the DESeq2 package (43), and differentially abundant ASVs whose responses to a treatment were driven by only one sample were removed from subsequent analyses. Analyses of β-diversity were performed via permutational analysis of variance (PERMANOVA) on rarefied sets of 100,000 reads per sample using Unifrac distance matrices.
Metabolite Extraction and Analysis.
Soil metabolites were extracted separately from the “A”, “B”, and “C” soil horizons using a method described in Swenson et al. (44). Rhizosphere soil samples were shaken in ice-cold liquid chromatography-mass spectrometry (LC-MS)-grade water, centrifuged, filtered, lyophilized, and resuspended in 100% methanol with internal standards for analysis using normal-phase LC-MS with a HILIC-Z column and an Agilent 1290 LC stack. MS and MS/MS data were collected using a Q Exactive Orbitrap MS (Thermo Scientific) (see SI Appendix for additional details and Dataset S4).
Metabolomics data were analyzed using Metabolite Atlas software to obtain extracted ion chromatograms and peak heights for each metabolite (45). Metabolite identifications were verified with authentic chemical standards based on matching m/z better than 5 ppm for positive mode, 15 ppm for negative mode, retention time difference ≤0.5 min, and/or MS/MS fragment matching score of >0.6 as calculated by the Stein and Scott “composite” algorithm with modifications (46) (Dataset S4). As defined by the Metabolomics Standards Initiative (47), any two of these orthogonal measures support a level 1 identification for the identified metabolites (provided that the third measure did not invalidate the identification). All identified metabolites were detected in at least four out of six replicates from at least one treatment.
Relative abundances of each metabolite were assessed by the peak heights of this metabolite across samples. Significant differences in switchgrass rhizosphere metabolite profiles in response to treatments and between the three soil horizons were determined with PERMANOVA. The magnitude of change (Delta metabolite abundance) for all significantly changed metabolites (P < 0.05) was calculated by scaling metabolite peak heights from 0 to 1, where “1” is the highest peak height of each metabolite across all samples, and then subtracting the scaled metabolite abundances observed in control soil from the treatments. A positive Delta indicates an increase in metabolite abundance in a specific treatment.
Rhizosphere Metabolite—Microbiome Associations.
To find connections between metabolites and microbial ASVs, we built a correlation network using their relative abundances in the “A” horizon. Only ASVs present in at least 15 of the 25 samples were considered, and correlations were calculated using Spearman coefficients. We used a Random Matrix Theory approach to set a correlation coefficient cutoff of 0.710, which separated noise from nonrandom correlations (48). We only included metabolite-ASV pairs with a correlation coefficient above the threshold and discarded links within metabolite species or 16S ASVs. Positive and negative correlations were represented by positive and negative links in the network. We identified putative keystone metabolites and ASVs based on network topology using a fast greedy algorithm to calculate within-module connectivity (zi) and among-module connectivity (pi) for each node (49). Nodes with zi > 2.5 were designated as module hubs, while nodes with pi > 0.62 were designated as connectors among different modules. Nodes with both zi > 2.5 and pi > 0.62 were designated as network hubs (50). We used the igraph package (51) for correlation calculations, network construction, and topology analysis, and Cytoscape (52) for visualization. Hierarchical clustering analysis was performed using the vegan package (53) to identify associations between differentially abundant ASVs and metabolites with significant Spearman correlations (r > ± 0.7, P < 0.05).
Plant Phenotype Response to Serotonin.
Surface-sterilized Alamo switchgrass seeds were grown on 1/5 strength Murashige and Skoog (MS) basal salt mixture M524, supplemented with either 0.1 mM serotonin (n = 9) or 0 mM serotonin as a control (n = 9). After 25 d, root and shoot biomass were measured and root length and root number were quantified using the SmartRoot plugin in ImageJ (version 2.0.0) (54). Significant differences between the treatments were determined using an ANOVA test (P < 0.05).
Microbial Response to Serotonin.
We investigated the effect of serotonin on microbial growth using six bacterial isolates from Oklahoma marginal soils planted with switchgrass. To ensure that the isolates were closely related to those observed in the switchgrass rhizosphere, we mapped their 16S rRNA gene sequences to the 16S amplicon sequences (ASVs) from our study. Based on the V4 region homology with the corresponding ASVs in our correlation network, we assigned the isolates to either a positively or negatively correlated serotonin response pattern (BLASTN, Evalue < 1 × 10−10, ≥97% of gene sequence homology). These isolates, which represent common rhizosphere genera, were grown in 1/10 R2A medium with 0, 0.1, or 0.5 mM serotonin. Optical density (OD600) measurements were taken and compared to a control without serotonin. We analyzed the OD600 responses using a Kruskal–Wallis test after subtracting the OD600 of uninoculated blanks from the inoculated samples.
Further details on the experimental design, sampling, rhizosphere soil DNA and metabolite extraction and analysis, sequencing, statistical methods, and serotonin tests can be found in SI Appendix.
Results
Plant and Soil Responses to Nutrient and Water Treatments.
Switchgrass root biomass in the “A” horizon varied significantly by treatment (P < 0.001, Fig. 1B) and was highest in the +NP treatment (4.80 ± 1.04 g; mean ± SD) and lowest in the −W treatment (2.45 ± 0.24 g). The bulk soil water potential in the “A” horizon at harvest also varied significantly by treatment (P < 0.001, Fig. 1C); the control soils were the wettest (−527 ± 352 kPa), treatments with higher root biomass (+N, +NP) had generally drier soils, and the −W treatment had the driest soils (−12,100 ± 5,700 kPa). Bulk soil microbial biomass in the “A” horizon, measured by phospholipid fatty acid analysis (PLFA) (24), did not vary significantly by treatment (Fig. 1D), with an average 9.81 ± 1.25 pmol PLFA/g dry soil observed across all treatments.
Microbial Diversity in Rhizosphere Soil.
To evaluate how nutrient availability and moisture stress affected microbial diversity in the rhizosphere of switchgrass, we analyzed 7,841 ASVs from the uppermost soil horizon. Microbial α-diversity varied significantly by treatment according to a suite of metrics including Fisher’s α (P = 0.01, Fig. 1E). Phylogenetic diversity also varied significantly by treatment (P = 0.01), but mean pairwise distance between ASVs did not. In general, α-diversity metrics were significantly higher in controls and in the +P treatment relative to soils where N was added, while reduced watering did not have a significant effect relative to controls (SI Appendix, Fig. S1).
At the phylum level, rhizosphere communities were dominated by Actinobacteria and Proteobacteria; together, these phyla made up 46.4 ± 8.2% and 26.5 ± 2.3% of the sequences observed in each sample, respectively (Dataset S1). Acidobacteria and Verrucomicrobia were the only other phyla that comprised >4% of the community in each sample, on average (SI Appendix, Fig S2A). The phyla Actinobacteria and Acidobacteria were the only dominant phyla (>4% relative abundance) that varied significantly between treatments (P < 0.05). Communities from the +N and +NP treatments were significantly different (P < 0.05) from those in the control and +P treatment, and communities from the −W treatment were significantly different from those in the control and +NP treatment (SI Appendix, Fig. S2B and Dataset S2).
Impact of Nutrient and Moisture Limitation on Rhizosphere ASVs.
To assess the effects of abiotic stressors on specific ASVs and determine which ASVs were driving community-level differences between treatments, we used DESeq2 to identify lineages that were differentially abundant in the +N, +P, +NP, and −W treatments relative to the control (Fig. 2A). N addition (+N, +NP) caused the strongest shifts in community composition (Fig. 2 A and B). Specifically, the +NP treatment affected the abundance of 184 ASVs, and the +N treatment impacted 139 ASVs (Fig. 2C). The −W treatment had the least effect. While there was some overlap in the effect of treatments on ASVs (Fig. 2C), many ASVs were solely affected by the +NP and +N treatments. Notably, 83 ASVs were uniquely affected in both the +N and +NP treatment soils, evidence of a strong N addition effect. Relatively few ASVs were solely affected by the +P and −W treatments (11 and 1, respectively). DESeq responsive ASVs and their taxonomy are listed in Dataset S3.
Fig. 2.

Influence of nutrient and water limitation on switchgrass rhizosphere microbial community structure assessed by DESeq2 analysis. (A) Number of positively (+) and negatively (−) responsive ASVs in nutrient-amended and water-limited treatments (+N, +NP, +P, −W) as compared to control soils, arranged by phyla (bubble size reflects the number of responsive ASVs). Empty cells indicate no responsive ASVs from that phylum. (B) The top-50 ASVs that increased (+Log2 fold-change) versus decreased in prevalence (−Log2 fold-change) in response to the +N treatment. ASVs are presented at the highest available taxonomic resolution and are colored by class for Proteobacteria and by phylum for all other phyla. (C) Number of unique and shared ASVs that changed in prevalence in response to each treatment relative to controls.
We defined ASVs that were more likely to be found (or not found) in a given treatment as “positive” (or “negative”) responders. The taxonomic identity of ASVs that responded to the +N or +NP treatments depended on whether they were positive- (increased in abundance) or negative-responders (decreased in abundance). Positive-responders to N-application (+N or +NP) were dominated (>50%) by ASVs from the phyla Actinobacteria (e.g., Kribbella, Streptomyces, Marmoricola, Conexibacter) (Fig. 2B). ASVs with negative-responses to N were more taxonomically diverse as a group, with negative-responders coming from over 17 classes, with the majority belonging to the Acidobacteria, Alphaproteobacteria, Deltaproteobacteria, Gammaproteobacteria, Planctomycetes, and Verrucomicrobia (Fig. 2B).
Far fewer ASVs responded to the +P and −W treatments (SI Appendix, Fig. S3 A and B) and there were few taxonomic patterns. Of 16 responders to the +P treatment, only five ASVs increased in abundance in response to P amendment, all from different phyla. A majority of the ASVs (n = 5) that decreased in response to P addition belonged to Actinobacteria. The −W treatment had the fewest ASVs change in abundance relative to the control soil, but six ASVs responded positively to water limitation, with the response driven by five ASVs from Actinobacteria.
Treatment-Induced Changes in Rhizosphere Metabolite Chemistry.
LC-MS-based metabolomics was used to identify rhizosphere metabolites under different nutrient additions and water limitation. 99 unique metabolites were identified across five treatments and three soil horizons (Dataset S4). Analysis of rhizosphere metabolites revealed compositional changes in all treatments, with the +N and +NP treatments exhibiting the greatest differences compared to the nutrient-poor control soil (Fig. 3). Across all soil horizons seventeen metabolites were significantly more abundant (P < 0.05) in the rhizosphere of the marginal control soils when compared to nitrogen (+N and/or +NP) supplemented treatments (Fig. 3 A and B and SI Appendix, Fig. S4). The majority of these 17 metabolites were organic acids (n = 12) and about half contained an aromatic ring (n = 9); the remaining metabolites included pentoses and pentose alcohols (n = 3), vitamin B, and a lactone. Conversely, addition of N (+N and/or +NP) significantly increased the abundance of 35 N-containing rhizosphere metabolites and one sugar for all three soil horizons. This included amino acids, nucleosides, and quaternary amines, as well as N-containing azoles such as allantoin and N-containing indoles such as serotonin (Fig. 3 C and D and SI Appendix, Fig. S4). Moisture stress also had a significant effect on an array of rhizosphere metabolites; 17 metabolites increased in abundance in response to the −W treatment, including osmolytes, such as amino acids, quaternary amines, sugars (Fig. 3 E and F and SI Appendix, Fig. S4). P amendment had a much smaller effect on rhizosphere metabolite chemistry than the other treatments; in the +P treatment, only seven metabolites significantly changed in abundance relative to the control (SI Appendix, Fig. S4).
Fig. 3.
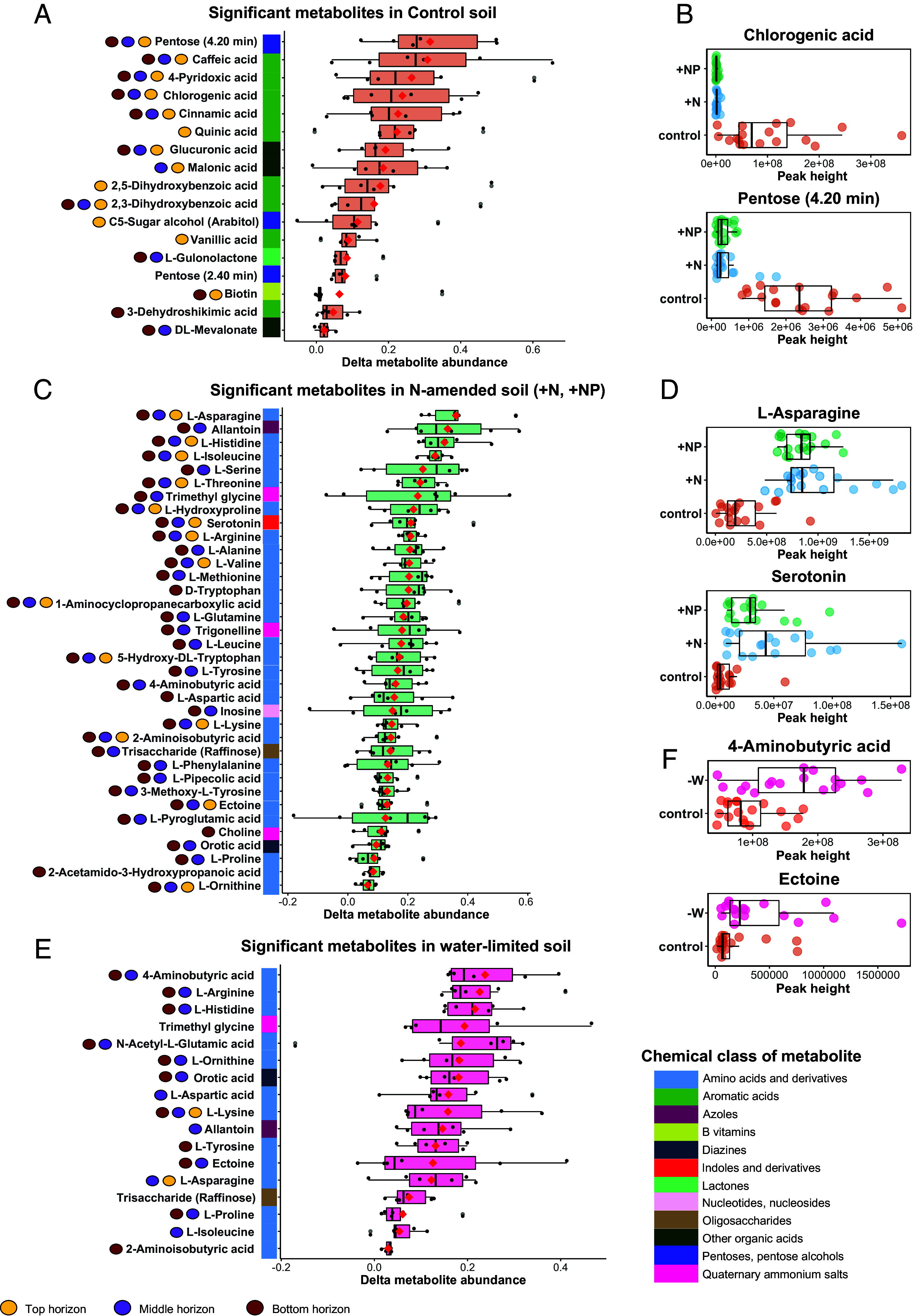
Significant changes in switchgrass rhizosphere metabolite profiles in response to five nutrient and water stress treatments (n replicates = 6), assessed by PERMANOVA (P < 0.05; Dataset S4 for details). (A) Metabolites significantly enriched (P < 0.05) in a nutrient-depleted marginal soil (control) compared to treatments where N was added (+N; +NP). Y-axis circles next to each metabolite represent the soil horizons where the metabolite had a significantly different abundance. Unresolvable metabolites are indicated by parentheses. (B) Abundance of an example metabolite enriched in nutrient-depleted soil across all three horizons. (C) Metabolites that increased (P < 0.05) in abundance in response to N addition (+N, +NP) compared to the control soil. (D) Abundance of an example metabolite enriched in N-replete soil. (E) Metabolites that increased in abundance (P < 0.05) in response to water limitation (−W) compared to the control soil. (F) Abundance of an example metabolite enriched in the water-limited treatment. The red diamond inside each box denotes the mean and the horizontal line denotes the median. Points reflect a single metabolite per sample, the outer boxes indicate the first, second, and third data quartiles, and whiskers indicate the range of the points excluding outliers.
We also analyzed metabolite changes in response to the treatments for each soil horizon. The top-soil horizon responded the most to nutrient limitation, with 13 metabolites increased in abundance when N was limited out of the 17 metabolites that changed across all horizons (Fig. 3A). Nitrogen addition resulted in the most significant changes in metabolite abundances of any treatment in the middle and bottom horizons (Fig. 3C), where nearly any metabolite with a significant response to N addition was found to increase in abundance, and very few were observed to decrease. Notably, the bottom and middle soil horizons revealed more profound metabolite responses to water limitation than the top horizon, where only two out of 17 metabolites increased (Fig. 3E).
Associations Between Metabolites, Microbial ASVs, and Abiotic Stresses.
To identify relationships between microbes and metabolites, we used Spearman’s rank correlations and hierarchical clustering of differentially abundant rhizosphere ASVs defined by the DESeq2 analysis and metabolites observed in the “A” soil horizon. This analysis groups treatment-responsive rhizosphere metabolites and ASVs by their degree of correlation, to identify clusters with similar behavior. Hierarchical clustering of the most responsive ASVs and differentially abundant metabolites revealed two large microbial-metabolite clusters. Cluster #1 (Fig. 4) contains microbial ASVs (n = 8) and rhizosphere metabolites (n = 17) that increased in abundance in the N-amended treatments (+N, +NP), including ASVs from the Actinobacteria, Alphaproteobacteria, and Gammaproteobacteria as well as metabolites with N-rich compounds (amino acids, azoles, quaternary amines). Cluster #2 (Fig. 4) includes metabolites (n = 8) and microbial ASVs (n = 29) with higher relative abundance in the unamended control soils. ASVs in this cluster were distinct from the ASVs identified in Cluster #1 and include diverse microbial classes from the Alphaproteobacteria, Deltaproteobacteria, Verrucomicrobia, Acidobacteria, as well as ASVs from Planctomycetes, Nitrospirae, Armatimonadetes, Gemmatimonadetes, Bacteroidetes, and Actinobacteria. The majority (75%) of rhizosphere metabolites that covaried with the ASVs from Cluster #2 were organic acids, particularly aromatic acids (chlorogenic, cinnamic, caffeic, 4-pyridoxic, 2,3-dihydroxybenzoic acid).
Fig. 4.

Heatmap representing the top covarying microbial taxa and metabolites in the rhizosphere of switchgrass grown with five soil nutrient and water treatments. Top associations between metabolites (columns) and ASVs (row) include i) DESeq2-determined differentially abundant ASVs (n = 37) with more than three significant positive or negative correlations (Spearman’s rank correlation, r > 0.7, P < 0.05) with metabolites; and ii) metabolites (n = 25) with more than one significant positive or negative correlation (Spearman’s rank correlation, r > 0.7, P < 0.05) with ASVs. Hierarchical clustering shows two clusters of metabolite-ASV correlations. Cluster #1 (blue lines) represents metabolites and ASVs that were more abundant in the rhizosphere when nitrogen was added (+N, +NP treatments) and Cluster #2 (brown lines) includes metabolites and ASVs that were more abundant in nitrogen-poor marginal soil (controls). Purple colors in the heatmap represent positive Spearman correlations, white represents no correlation, and green colors represent negative correlations between metabolites and ASVs.
Metabolite-Microbial Rhizosphere Community Network.
In our bipartite co-occurrence network of rhizosphere ASVs and metabolites (Fig. 5A), 117 ASVs connect to 31 metabolites via 368 links, including 153 positive and 215 negative links, with an average of five links per node (Dataset S5). We identified five module hubs and one network hub as putative “keystone metabolites” (Fig. 5B and Dataset S5). Three metabolites reflect modules dominated by negative correlations, including serotonin, acetylcholine, and ectoine (modules 2, 3, and 4, respectively), with serotonin exhibiting negative links (83%) with a wide range of bacteria, and positive links primarily with Actinobacteria ASVs. Module 1, the largest, was driven by positive interactions with chlorogenic acid, glucuronic acid, and cinnamic acid, and included 78% positive links with bacterial ASVs from a diverse range of lineages and negative links primarily with ASVs from Actinobacteria. There was no module hub observed for Module 5, which was dominated by metabolite nodes instead of ASVs.
Fig. 5.

Co-occurrence network of switchgrass rhizosphere metabolites and microbial ASVs exposed to five soil treatments in a greenhouse study. (A) An association network between 908 16S ASVs and 99 rhizosphere metabolites. Nodes with circle symbols represent 16S ASVs, and nodes with square symbols represent metabolites. Links between nodes are based on Spearman correlations (r > 0.710) of their relative abundances, red for positive correlation and blue for negative correlation. The network separates into five major modules, or highly connected groups of nodes, shown as the five numbered circles. Red filled squares highlight rhizosphere metabolites that act as network and module hubs, which are the nodes with dense connections to other nodes within the entire network (network hub) or a module (module hub). The six microbial ASV nodes at the center serve as connectors of different modules, or the nodes linking different modules. (B) Subnetworks of rhizosphere metabolites that formed module hubs and their neighboring microbial nodes. (C) Subnetworks of microbial nodes that serve as connectors, and their linked rhizosphere metabolite. Microbial ASVs are colored by class for Proteobacteria and by phylum for all other phyla.
The six connector ASVs behaved similarly to one another (Fig. 5C), forming most of their positive links (4/5) with organic acids—including the three aforementioned module hubs and 2,3-dihydroxybenzoic acid in Module 4—and forming most of their negative links (10/12) with nonorganic acid metabolites. Approximately half of the ASVs in the network were also identified as differentially abundant by DESeq (Dataset S3), and nearly all of these were responsive to the +N or +NP treatment but not the −W or +P treatments.
Serotonin Impacts on Plant Biomass and Rhizosphere Isolates.
Serotonin was identified as a key hub metabolite in the reconstructed network (Fig. 5); it formed the largest number of ASV links and had the largest number of significant ASV correlations (Figs. 4 and 5). Given serotonin’s known role in gut bacterial–host interactions (34) and phenotypic effects on Arabidopsis (55) we conducted a follow-up study to examine its effect in the switchgrass rhizosphere. Switchgrass seedlings grown with 0.1 mM serotonin had increased root and shoot biomass (SI Appendix, Fig. S5), promoted the number of secondary roots (Fig. 6A), and increased secondary root length (Fig. 6B) (P < 0.05, n = 9).
Fig. 6.

Serotonin effects on switchgrass plant phenotype and growth of rhizosphere microorganisms. (A and B) 25-d-old switchgrass seedlings (n = 9) grown with exogenous application of 0.1 mM of serotonin (+SER) or controls (−SER). Serotonin effects on secondary root number (A) and total root length (B). Significant differences between added-serotonin and controls were assessed by ANOVA, asterisks reflect P < 0.05. (C) Optical density (OD600) of rhizosphere bacteria cultures after 130 h of growth in 1/10 R2A medium with 0, 0.1, or 0.5 mM of serotonin. Values have been scaled to the highest OD for each isolate across the row. The highest OD of the isolate is 100% (dark purple) and the lowest OD is 0% (dark green), meaning that isolate growth has been completely inhibited. Orange cells indicate isolates related to ASVs with significant negative correlations with serotonin (−SER) and brown cells indicate isolates matched to ASVs with positive correlations (+SER). Positive and negative correlations between specific ASV (shown in parentheses) and serotonin shown inside of each cell. Asterisks indicate significantly different OD600 between the 0.1 and 0.5 mM serotonin treatments (n = 4) and a control treatment without serotonin (0 mM, n = 4) at P < 0.05 by means of the Kruskal–Wallis test.
We also tested serotonin’s effects on six microbial isolates purified from the switchgrass rhizosphere in marginal soils from Oklahoma (Fig. 6C), and are closely related to the ASVs identified in the switchgrass rhizosphere in this study (≥97% of 16S gene sequence homology). Three of the isolates are closely related to ASVs exhibiting negative serotonin correlations (“−SER”) and three to ASVs with positive serotonin correlations (“+SER”) (Fig. 6C). Increased serotonin concentrations (0.5 mM) in the isolate growth medium suppressed growth of the -SER isolates and did not affect growth of the +SER isolates based on the OD600 readings of this treatment compared to the control (Fig. 6C and SI Appendix, Fig. S6).
Discussion
Exometabolites Reflect Rhizosphere Abiotic Stress Conditions.
Plant exudates and microbial metabolites present in the rhizosphere play significant roles in shaping plant–microbe and microbe–microbe relationships under a range of environmental conditions, including abiotic stresses (56–59). These rhizosphere metabolites primarily consist of plant exudates, microbial products, and background C compounds present in the bulk soil. While a number of studies have analyzed metabolites present in rhizosphere soil of maize, Arabidopsis, wheat, rice, and wild oat (7, 60–63), few studies have identified changes of rhizosphere metabolites in soil in response to abiotic stressors and nutrient limitations (64, 65). Here, we demonstrate that metabolites recovered from the rhizosphere soil of switchgrass changed in response to nutrient (N and P) and water-limited conditions. We identified three major patterns in switchgrass rhizosphere metabolite shifts: i) enhanced abundances of aromatic acids when switchgrass was grown in N-limited soil (Fig. 3A); ii) enhanced abundances of N-containing compounds when N was added (Fig. 3C); and iii) enhanced abundances of osmolytes in water-limited conditions (Fig. 3E).
One of the most significant patterns observed in this study was the enhanced release of aromatic acids when switchgrass was limited by N (Fig. 3A). Numerous studies have established the crucial roles of aromatic acids in plant-soil-microbial ecology (6, 20, 66–68). In our recent work, we demonstrated that aromatic acids impact the microbiome assembly of wild oat through the metabolic synchronization of microbial substrate utilization traits and root exudation (6). We propose that the increased release of this class of metabolites under N limitation may be linked to the plant’s efforts to reshape its microbiome assembly to favor the selection of beneficial microorganisms that enhance plant nutrient availability. Further research is warranted to establish direct evidence of switchgrass’s release of these compounds as a means of recruiting beneficial microorganisms. However, other studies have demonstrated that plants exude compounds that facilitate Fe (56, 58), P (56, 59), and N (57) acquisition.
We observed greater abundances of amino acids, nucleosides, and other N-containing molecules (Fig. 3C), in response to N-amendment. The resulting alleviation of N stress led to enhanced root biomass (Fig. 1B) and C availability (24). Consequently, the effect on nutrient status influenced the composition of rhizosphere microbial communities, potentially attracting microorganisms that thrive in environments with higher nutrient availability, particularly where N-rich compounds are present.
We observed a heightened production of ectoine, choline, betaine, raffinose, and a variety of amino acids under water-limited conditions (Fig. 3E). These known osmolytes—compounds produced by plants and microorganisms to alleviate osmotic stress (69–72), are generally more abundant in soils with limited water availability (69, 73). Notably, some of the same osmolytes were produced in the rhizosphere in response to both water limitation and enhanced N availability (Fig. 3C). This convergence of metabolites responsive to reduced watering and increased N availability is reasonable, as the increased root biomass in the treatments with added N and NP resulted in soil that was drier relative to the control conditions (Fig. 1 B and C) (24). Thus, the observed response of osmolytes in our system is consistent with an increase in osmolyte abundances in response to moisture stress experienced by plant and/or microbial cells in the soil matrix.
Abiotic Stress Structures Rhizosphere Microbiomes.
The community composition observed in the switchgrass rhizosphere was in agreement with the literature regarding the taxa found in the rhizosphere of various other grasses and soil bacteria associated with switchgrass (2, 31, 74). We observed that Actinobacteria was the dominant switchgrass rhizosphere phylum and Proteobacteria (particularly class Alphaproteobacteria), Acidobacteria, and Verrucomicrobia were the next-most dominant phyla (SI Appendix, Fig S2A). While Verrucomicrobia are not generally considered to be “rhizosphere” taxa, Hestrin et al. reviewed switchgrass microbiome literature and noted that Verrucomicrobia were consistently associated with switchgrass roots and rhizosphere soil (75).
We observed that abiotic stresses and nutrient limitation caused changes in the metabolic profiles of the switchgrass rhizosphere. We suggest that changes in the switchgrass rhizosphere metabolome in response to nutrient and water availability, mediate the assembly of the rhizosphere microbiome. The shifts in rhizosphere bacterial community structure that we observed in response to changes in abiotic stress and in tandem with associated changes in metabolite profiles demonstrate close linkages between these factors and generally support this hypothesis.
Alleviating N-limitation likely resulted in the proliferation of bacteria that are adapted to environments with plentiful soil resources at the expense of more diverse taxa better adapted to nutrient-limited environments (76). In terms of taxonomic shifts, we observed the same trend consistently seen by Ramirez et al. whereby bacterial communities under N-addition have greater abundances of Actinobacteria (77). In contrast, members of Verrucomicrobia and Acidobacteria decreased in abundance under N-addition. These slow-growing, lineages have been linked to nutrient deficiencies in general, and N-limitation, in particular (77). Our findings corroborate previous field studies, in which comparable community changes were observed in the same bacterial groups (77–79).
We did not observe a strong microbial community response to P addition or watering reduction in our study. Notably, a number of rhizosphere metabolites that responded to +P treatment were also the lowest compared to other nutrient treatments. P availability was likely not as limiting in our system as N. The fewest number of ASVs (6) responded to our −W treatment, with the majority of these also from Actinobacteria, a phyla with many drought-tolerant lineages (80). We note that no ASVs decreased in prevalence in response to the −W treatment. This may reflect the frequently dry conditions at the site where the soils were originally collected, and hence the long-term adaptation of the indigenous microbial communities.
Metabolite Chemistry Associated With Changes in Abundance of Specific ASVs.
Taxon-specific responses to individual rhizosphere metabolites could be an important driver of rhizosphere bacterial community assembly. While we note that rhizosphere metabolites are not direct measures of plant exudation, we hypothesize that nonrandom covariations in the abundances of microorganisms and rhizosphere metabolites across the broad range of abiotic stresses in our treatments could indicate potential functional links between identified metabolites and microbial lineages. Our results support this hypothesis, with rhizosphere metabolites shifting with microbial community composition in a similar manner to that observed in the literature for soil microbial communities exposed to changing exudate chemistry.
It has been previously established that plants can exude organic acids in nutrient-limited conditions, at least in part to directly liberate C and nutrients into the soil matrix (16). However, it has also been shown that organic acids, and in particular aromatic acids, are exuded by plants as they develop, and that greater abundances of such acids correspond to large-scale shifts in soil microbial community composition (11, 12, 67). Several potential mechanisms of how aromatic acids may modulate rhizomicrobiomes have been proposed, including shifts in soil pH, antimicrobial effects, or preferential utilization of these metabolites as a nutrient source by specific microbial taxa geared to decompose them (6, 12, 67).
In our association networks, we found that half of the six module hubs were organic acids, with chlorogenic acid, an aromatic compound, possessing the most links to microbial ASVs of the three (Fig. 5). In addition, we observed that aromatic compounds such as chlorogenic acid, caffeic acid, and four-pyridoxic acid (among others) were most abundant in our control, N-limited marginal soils. These soils also possessed the most diverse microbial communities (Fig. 1E). Reduced concentration of these aromatic acids in our N-amended treatments also corresponded to significantly less diverse rhizosphere bacterial communities, with consistent reductions in similar ASVs (Fig. 2 B and C). The ability to metabolize organic acids, in particular, has been linked to the proliferation of taxa in the rhizosphere of a variety of plant hosts (6, 12, 81), which is notable given that organic acids are among the dominant classes of exudate compounds and many plants are known to exude them (along with other rhizodeposits) from their roots during active growth and development (6, 8, 82). Thus, aromatic acids such as chlorogenic acid are likely strong drivers of switchgrass rhizomicrobiome structure.
Two of the three remaining rhizosphere module hub metabolites, serotonin and acetylcholine, have not been extensively studied in the context of soil. In soil, serotonin can result from the degradation of tryptophan (83), itself a precursor for many essential plant metabolites including plant hormone auxin (33). Interestingly, serotonin was the largest module hub that we observed in our network. In plants, serotonin plays important roles in growth, development, and response to environmental stresses (33). However, the mechanism of action of this signaling metabolite and its role in the rhizosphere, particularly in plant–microbe interactions, are unknown. We have demonstrated that serotonin not only exhibits a correlation with a significant number of ASVs, but also influences the growth of rhizosphere microorganisms and the phenotype of the plant. We found that growing switchgrass in microcosms with applications of serotonin increased plant aboveground biomass and significantly enhanced root growth (Fig. 6 A and B and SI Appendix, Fig. S5). We observed that serotonin has the capacity to selectively inhibit rhizosphere microorganisms, thereby granting the plant the ability to sculpt its community in response to its surrounding environment. Root phenotypic modifications and substantial shifts in rhizosphere ASV abundance in correlation with serotonin levels and its impact on microbial growth suggest serotonin’s role as a keystone metabolite in mediating plant–microbe interactions in the rhizosphere. Recent studies demonstrated that gut microorganisms coevolved to induce serotonin production by the host and can sense this host-derived serotonin to increase their colonization and fitness in the intestine (34). However, it is also known that many phenylamides, such as serotonin have antibiotic properties (84) which is consistent with our observed suppression of microbial growth and negative correlations between selected microbes and serotonin. In contrast, microbes that have not been inhibited by serotonin and correspond to the ASVs positively correlating with this molecule, could possibly cometabolize serotonin as a nutrient source or have developed a mechanism to detoxify this molecule.
The final module hub metabolite we observed is ectoine, one of the most abundant osmolytes in nature and commonly produced by prokaryotes (85). As such, it may be indicative of moisture stress experienced by microbial communities in the rhizosphere. Notably, ectoine abundance was positively associated with Actinobacteria, a lineage often thought to be drought-tolerant (86), and associated with ectoine in arid environments (87). We note that the mechanisms behind links in a correlation network are difficult to assess, in the context of the broad range of abiotic conditions experienced by the plant and soil in our study, strong correlations between metabolites and microbial ASVs could well imply that they respond similarly (positive links) or differentially (negative links) to these conditions. The organic acids that were identified as module hubs in our association network had mostly positive associations with a diverse array of ASVs, which is supported by the treatment responses we observed for rhizosphere metabolites and microbiome community composition. In contrast, serotonin had negative associations with a diverse array of ASVs, but its few positive associations were almost entirely with Actinobacteria lineages, which is supported by the taxonomic responses to N-rich rhizosphere metabolites that we observed. Thus, rhizosphere microbial assembly mediated by metabolites could be important drivers of these covariations—especially when the potential relevance of the chemistry of these compounds to plant-microbial metabolism is consistent with the literature and can be demonstrated (as we did for serotonin) in controlled experiments.
Conclusion
Metabolic changes belowground play a vital role in plant stress resilience and microbial adaptations to environmental change. However, the relationships between rhizosphere metabolite chemistry and the dynamics of microorganisms in soil have been largely overlooked. Our results show that rhizosphere metabolites are sensitive indicators of abiotic conditions in the soil environment that can be linked to the shifts of specific bacterial lineages in response to such changes. We show that aromatic acids were enriched in the rhizosphere of N-limited switchgrass and identified microbial lineages associated with this N-limiting condition that were enriched in the presence of these organic acids. In contrast, N-rich metabolites were plentiful in the rhizosphere of N-replete switchgrass, as were fast-growing microbial lineages capable of responding to increased nutrient availability. We contend that the metabolites identified as module hubs in our association network—chlorogenic acid, cinnamic acid, glucuronic acid, serotonin, ectoine, and acetylcholine—merit further study as “keystone metabolites” by structuring soil microbial communities in response to abiotic stress. In conclusion, the rhizosphere metabolite response to nutrient and moisture availability and associated changes in microbiota suggest a putative mechanism of metabolite-driven microbial community assembly under abiotic stress and highlight potential keystone metabolites in the rhizosphere of switchgrass.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Acknowledgments
Funding for this project was provided by the United States Department of Energy Office of Biological and Environmental Research (DOE OBER), Genomic Science Program Sustainable Bioenergy award DE-SC0014079 to the University of California–Berkeley, with subawards to Lawrence Berkeley National Laboratory (LBNL), the University of Oklahoma, the Noble Research Institute, and Lawrence Livermore National Laboratory (LLNL, SCW1555). Research performed at LLNL was conducted under the hospices of the US department of energy under contract DE-AC52-07NA27344. For the metabolite analysis, K.Z., T.N., B.P.B., J.J., and J.S. were supported by m-CAFEs Microbial Community Analysis and Functional Evaluation in Soils, an SFA led by LBNL supported by the U.S. DOE OBER under contract number DE-AC02-05CH11231. We thank David Orme for allowing us to collect soil from his ranch in Anadarko Oklahoma, and Hugh Aljoe, Kelly Craven, and Shawn Norton of the Noble Research Institute for facilitating site access and soil collection. We thank Christina Wistrom and the Oxford Tract Greenhouse at the University of California-Berkeley for the infrastructure required to carry out the greenhouse experiment. We thank Katerina Estera-Molina, Anne Kakouridis, Sarah Baker, Steve Blazewicz, Evan Starr, Ka Ki Law, Heejung Cho, Alexa Nicholas, Eoin Brodie, Peter Nico, Caleb Herman, Ashley Campbell, and Amrita Bhattacharyya for their help with the destructive harvest of mesocosms. Thanks also to Madeline Moore, David Sanchez, Cynthia-Jeanette Mancilla, and Ilexis Jacoby for their help with processing of soil and nucleic acid samples and Suzanne Kosina for the review of the metabolite identifications.
Author contributions
N.R.B., K.Z., M.S., J.P.-R., and M.K.F. designed research; N.R.B., K.Z., M.Y., D.H., J.A.C.-N., J.S., J.S.J., L.W., C.F., A.C., Y.F., and J.P.-R. performed research; N.R.B., K.Z., M.Y., D.H., M.S., J.Z., and T.R.N. contributed new reagents/analytic tools; N.R.B., K.Z., M.Y., J.A.C.-N., J.S., and B.P.B. analyzed data; and N.R.B., K.Z., M.Y., J.A.C.-N., J.S., J.Z., J.P.-R., T.R.N., and M.K.F. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
Reviewers: G.A.B., Iowa State University; D.C., The University of Manchester; and A.S., Austrian Institute of Technology.
Data, Materials, and Software Availability
The DNA sequences of the 16S rRNA gene amplicons were deposited in the National Center for Biotechnology Information (accession no. PRJNA781222) (88). The raw metabolomics data were deposited to the Global Natural Products Social Molecular Networking (https://gnps.ucsd.edu) data repository (MSV000088543) (89).
Supporting Information
References
- 1.Haichar F. el Z., et al. , Plant host habitat and root exudates shape soil bacterial community structure. ISME J. 2, 1221–1230 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Shi S., et al. , Successional trajectories of rhizosphere bacterial communities over consecutive seasons. mBio 6, e00746 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bulgarelli D., et al. , Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 17, 392–403 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Philippot L., et al. , Loss in microbial diversity affects nitrogen cycling in soil. ISME J. 7, 1609–1619 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van de Broek A., Lambrecht M., Vanderleyden J., Bacterial chemotactic motility is important for the initiation of wheat root colonization by Azospirillum brasilense. Microbiology 144, 2599–2606 (1998). [DOI] [PubMed] [Google Scholar]
- 6.Zhalnina K., et al. , Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat. Microbiol. 3, 470–480 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Iannucci A., Fragasso M., Platani C., Papa R., Plant growth and phenolic compounds in the rhizosphere soil of wild oat (Avena fatua L.). Front. Plant Sci. 4, 509 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pett-Ridge J., et al. , “Rhizosphere carbon turnover from cradle to grave: The role of microbe-plant interactions” in Rhizosphere Biology: Interactions Between Microbes and Plants, Rhizosphere Biology, Gupta V. V. S. R., Sharma A. K., Eds. (Springer Singapore, 2021), pp. 51–73. [Google Scholar]
- 9.Nuccio E. E., et al. , Niche differentiation is spatially and temporally regulated in the rhizosphere. ISME J. 14, 999–1014 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaparro J. M., Badri D. V., Vivanco J. M., Rhizosphere microbiome assemblage is affected by plant development. ISME J. 8, 790–803 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Badri D. V., Chaparro J. M., Zhang R., Shen Q., Vivanco J. M., Application of natural blends of phytochemicals derived from the root exudates of arabidopsis to the soil reveal that phenolic-related compounds predominantly modulate the soil microbiome. J. Biol. Chem. 288, 4502–4512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi S., et al. , Effects of selected root exudate components on soil bacterial communities: Root exudate components and soil microbial communities. FEMS Microbiol. Ecol. 77, 600–610 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Compant S., Clément C., Sessitsch A., Plant growth-promoting bacteria in the rhizo- and endosphere of plants: Their role, colonization, mechanisms involved and prospects for utilization. Soil Biol. Biochem. 42, 669–678 (2010). [Google Scholar]
- 14.van Dam N. M., Bouwmeester H. J., Metabolomics in the rhizosphere: Tapping into belowground chemical communication. Trends Plant Sci. 21, 256–265 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Baran R., et al. , Exometabolite niche partitioning among sympatric soil bacteria. Nat. Commun. 6, 8289 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keiluweit M., et al. , Mineral protection of soil carbon counteracted by root exudates. Nat. Clim. Change 5, 588–595 (2015). [Google Scholar]
- 17.Swenson T. L., Karaoz U., Swenson J. M., Bowen B. P., Northen T. R., Linking soil biology and chemistry in biological soil crust using isolate exometabolomics. Nat. Commun. 9, 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Vries F. T., Griffiths R. I., Knight C. G., Nicolitch O., Williams A., Harnessing rhizosphere microbiomes for drought-resilient crop production. Science 368, 270–274 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Jung S. C., Martinez-Medina A., Lopez-Raez J. A., Pozo M. J., Mycorrhiza-induced resistance and priming of plant defenses. J. Chem. Ecol. 38, 651–664 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Rizaludin M. S., Stopnisek N., Raaijmakers J. M., Garbeva P., The chemistry of stress: Understanding the “cry for help” of plant roots. Metabolites 11, 357 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McLaughlin S. B., Adams Kszos L., Development of switchgrass (Panicum virgatum) as a bioenergy feedstock in the United States. Biomass Bioenergy 28, 515–535 (2005). [Google Scholar]
- 22.Sanderson M. A., et al. , Switchgrass cultivars and germplasm for biomass feedstock production in Texas. Bioresour. Technol. 67, 209–219 (1999). [Google Scholar]
- 23.Thomason W. E., et al. , Switchgrass response to harvest frequency and time and rate of applied nitrogen. J. Plant Nutr. 27, 1199–1226 (2007). [Google Scholar]
- 24.Sher Y., et al. , Microbial extracellular polysaccharide production and aggregate stability controlled by switchgrass (Panicum virgatum) root biomass and soil water potential. Soil Biol. Biochem. 143, 107742 (2020). [Google Scholar]
- 25.Slessarev E. W., et al. , Quantifying the effects of switchgrass (Panicum virgatum) on deep organic C stocks using natural abundance 14C in three marginal soils. GCB Bioenergy 12, 834–847 (2020). [Google Scholar]
- 26.Whitaker J., et al. , Consensus, uncertainties and challenges for perennial bioenergy crops and land use. GCB Bioenergy 10, 150–164 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beckman J. J., Moser L. E., Kubik K., Waller S. S., Big bluestem and switchgrass establishment as influenced by seed priming. Agron. J. 85, 199–202 (1993). [Google Scholar]
- 28.Ghimire S. R., Craven K. D., Enhancement of switchgrass (Panicum virgatum L.) biomass production under drought conditions by the ectomycorrhizal fungus Sebacina vermifera. Appl. Environ. Microbiol. 77, 7063–7067 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mendes R., Garbeva P., Raaijmakers J. M., The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 37, 634–663 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Roley S. S., et al. , Associative nitrogen fixation (ANF) in switchgrass (Panicum virgatum) across a nitrogen input gradient. PLOS ONE 13, e0197320 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mao Y., Li X., Smyth E. M., Yannarell A. C., Mackie R. I., Enrichment of specific bacterial and eukaryotic microbes in the rhizosphere of switchgrass (Panicum virgatum L.) through root exudates. Environ. Microbiol. Rep. 6, 293–306 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Rodrigues R. R., Rodgers N. C., Wu X., Williams M. A., COREMIC: A web-tool to search for a niche associated CORE MICrobiome. PeerJ 6, e4395 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erland L. A. E., Turi C. E., Saxena P. K., Serotonin: An ancient molecule and an important regulator of plant processes. Biotechnol. Adv. 34, 1347–1361 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Fung T. C., et al. , Intestinal serotonin and fluoxetine exposure modulate bacterial colonization in the gut. Nat. Microbiol. 4, 2064–2073 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeAngelis K. M., et al. , Selective progressive response of soil microbial community to wild oat roots. ISME J. 3, 168–178 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Griffiths R. I., Whiteley A. S., O’Donnell A. G., Bailey M. J., Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA-and rRNA-based microbial community composition. Appl. Environ. Microbiol. 66, 5488–5491 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caporaso J. G., et al. , Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108, 4516–4522 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parada A. E., Needham D. M., Fuhrman J. A., Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Callahan B. J., et al. , DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bolyen E., et al. , Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quast C., et al. , The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucl. Acids Res. 41, D590–D596 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McMurdie P. J., Holmes S., phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swenson T. L., Jenkins S., Bowen B. P., Northen T. R., Untargeted soil metabolomics methods for analysis of extractable organic matter. Soil Biol. Biochem. 80, 189–198 (2015). [Google Scholar]
- 45.Yao Y., et al. , Analysis of metabolomics datasets with high-performance computing and metabolite atlases. Metabolites 5, 431–442 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stein S. E., Scott D. R., Optimization and testing of mass spectral library search algorithms for compound identification. J. Am. Soc. Mass Spectrom. 5, 859–866 (1994). [DOI] [PubMed] [Google Scholar]
- 47.Sumner L. W., et al. , Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics Off. J. Metabolomic Soc. 3, 211–221 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deng Y., et al. , Molecular ecological network analyses. BMC Bioinformatics 13, 113–20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guimerà R., Nunes Amaral L. A., Functional cartography of complex metabolic networks. Nature 433, 895–900 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olesen J. M., Bascompte J., Dupont Y. L., Jordano P., The modularity of pollination networks. Proc. Natl. Acad. Sci. U.S.A. 104, 19891–19896 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Csardi G., Nepusz T., The igraph software package for complex network research. J. Complex Syst. 1695 (2005). [Google Scholar]
- 52.Shannon P., et al. , Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oksanen J., et al. , Vegan: Community Ecology Package. (2020), https://CRAN.R-project.org/package=vegan.
- 54.Lobet G., Pagès L., Draye X., A novel image-analysis toolbox enabling quantitative analysis of root system architecture. Plant Physiol. 157, 29–39 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pelagio-Flores R., Ortíz-Castro R., Méndez-Bravo A., Macías-Rodríguez L., López-Bucio J., Serotonin, a tryptophan-derived signal conserved in plants and animals, regulates root system architecture probably acting as a natural auxin inhibitor in Arabidopsis thaliana. Plant Cell Physiol. 52, 490–508 (2011). [DOI] [PubMed] [Google Scholar]
- 56.Carvalhais L. C., et al. , Root exudation of sugars, amino acids, and organic acids by maize as affected by nitrogen, phosphorus, potassium, and iron deficiency. J. Plant Nutr. Soil Sci. 174, 3–11 (2011). [Google Scholar]
- 57.Dong W., Song Y., The significance of flavonoids in the process of biological nitrogen fixation. Int. J. Mol. Sci. 21, 5926 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rosenkranz T., Oburger E., Baune M., Weber G., Puschenreiter M., Root exudation of coumarins from soil-grown Arabidopsis thaliana in response to iron deficiency. Rhizosphere 17, 100296 (2021). [Google Scholar]
- 59.Wang Y., Lambers H., Root-released organic anions in response to low phosphorus availability: Recent progress, challenges and future perspectives. Plant Soil 447, 135–156 (2020). [Google Scholar]
- 60.Hu L., et al. , Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat. Commun. 9, 2738 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kudjordjie E. N., Sapkota R., Steffensen S. K., Fomsgaard I. S., Nicolaisen M., Maize synthesized benzoxazinoids affect the host associated microbiome. Microbiome 7, 17–59 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Y., Xu L., Letuma P., Lin W., Metabolite profiling of rhizosphere soil of different allelopathic potential rice accessions. BMC Plant Biol. 20, 265–21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pétriacq P., et al. , Metabolite profiling of non-sterile rhizosphere soil. Plant J. 92, 147–162 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Caddell D. F., et al. , Drought shifts sorghum root metabolite and microbiome profiles and enriches for pipecolic acid. Phytobiomes J. 7, 449–463 (2023). [Google Scholar]
- 65.Mavrodi D. V., et al. , Accumulation of the antibiotic phenazine-1-carboxylic acid in the rhizosphere of dryland cereals. Appl. Environ. Microbiol. 78, 804–812 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li Z.-H., Wang Q., Ruan X., Pan C.-D., Jiang D.-A., Phenolics and plant allelopathy. Mol. Basel Switz. 15, 8933–8952 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lebeis S. L., et al. , Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science 349, 860–864 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Bhattacharya A., Sood P., Citovsky V., The roles of plant phenolics in defence and communication during Agrobacterium and Rhizobium infection. Mol. Plant Pathol. 11, 705–719 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bouskill N. J., et al. , Belowground response to drought in a tropical forest soil. II. Change in microbial function impacts carbon composition. Front. Microbiol. 7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lebre P. H., De Maayer P., Cowan D. A., Xerotolerant bacteria: Surviving through a dry spell. Nat. Rev. Microbiol. 15, 285–296 (2017). [DOI] [PubMed] [Google Scholar]
- 71.Ngumbi E., Kloepper J. W., Bacterial-mediated drought tolerance: Current and future prospects. Appl. Soil Ecol. 105, 109–125 (2016). [Google Scholar]
- 72.Schimel J., Balser T. C., Wallenstein M., Microbial stress-response physiology and its implications for ecosystem function. Ecology 88, 1386–1394 (2007). [DOI] [PubMed] [Google Scholar]
- 73.Malik A. A., et al. , Drought and plant litter chemistry alter microbial gene expression and metabolite production. ISME J. 14, 2236–2247 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Singer E., Bonnette J., Kenaley S. C., Woyke T., Juenger T. E., Plant compartment and genetic variation drive microbiome composition in switchgrass roots. Environ. Microbiol. Rep. 11, 185–195 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hestrin R., Lee M. R., Whitaker B. K., Pett-Ridge J., The switchgrass microbiome: A review of structure, function, and taxonomic distribution. Phytobiomes J. 5, 14–28 (2021). [Google Scholar]
- 76.Fierer N., et al. , Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 6, 1007–1017 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ramirez K. S., Craine J. M., Fierer N., Consistent effects of nitrogen amendments on soil microbial communities and processes across biomes. Glob. Change Biol. 18, 1918–1927 (2012). [Google Scholar]
- 78.Campbell B. J., Polson S. W., Hanson T. E., Mack M. C., Schuur E. A. G., The effect of nutrient deposition on bacterial communities in Arctic tundra soil. Environ. Microbiol. 12, 1842–1854 (2010). [DOI] [PubMed] [Google Scholar]
- 79.Nemergut D. R., et al. , The effects of chronic nitrogen fertilization on alpine tundra soil microbial communities: Implications for carbon and nitrogen cycling. Environ. Microbiol. 10, 3093–3105 (2008). [DOI] [PubMed] [Google Scholar]
- 80.Simmons T., et al. , Drought drives spatial variation in the millet root microbiome. Front. Plant Sci. 11, 599 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lugtenberg B. J., Dekkers L., Bloemberg G. V., Molecular determinants of rhizosphere colonization by Pseudomonas. Annu. Rev. Phytopathol. 39, 461–490 (2001). [DOI] [PubMed] [Google Scholar]
- 82.Jones D. L., Organic acids in the rhizosphere – A critical review. Plant Soil 205, 25–44 (1998). [Google Scholar]
- 83.Martens D. A., Frankenberger W. T., Metabolism of tryptophan in soil. Soil Biol. Biochem. 25, 1679–1687 (1993). [Google Scholar]
- 84.Morimoto N., et al. , Induced phenylamide accumulation in response to pathogen infection and hormone treatment in rice (Oryza sativa). Biosci. Biotechnol. Biochem. 82, 407–416 (2018). [DOI] [PubMed] [Google Scholar]
- 85.da Costa M. S., Santos H., Galinski E. A., An overview of the role and diversity of compatible solutes in Bacteria and Archaea. Adv. Biochem. Eng. Biotechnol. 61, 117–153 (1998). [DOI] [PubMed] [Google Scholar]
- 86.Naylor D., DeGraaf S., Purdom E., Coleman-Derr D., Drought and host selection influence bacterial community dynamics in the grass root microbiome. ISME J. 11, 2691–2704 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mohammadipanah F., Wink J., Actinobacteria from arid and desert habitats: Diversity and biological activity. Front. Microbiol. 6, 1541 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Baker N., et al. , Switchgrass rhizosphere 16S communities in response to nitrogen, phosphorus, and water stress. NCBI BioProject. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA781222. Deposited 17 November 2021.
- 89.Baker N., et al. , GNPS - Exometabolomics of Switchgrass rhizosphere. Global Natural Products Social Molecular Networking. https://gnps.ucsd.edu/ProteoSAFe/result.jsp?task=feacd4da6d8c470fb7712e6da02b9dd2&view=advanced_view. Deposited 7 December 2021.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Data Availability Statement
The DNA sequences of the 16S rRNA gene amplicons were deposited in the National Center for Biotechnology Information (accession no. PRJNA781222) (88). The raw metabolomics data were deposited to the Global Natural Products Social Molecular Networking (https://gnps.ucsd.edu) data repository (MSV000088543) (89).