

Abstract
Background
Decoding human genomic sequences requires comprehensive analysis of DNA sequence functionality. Through computational and experimental approaches, researchers have studied the genotype-phenotype relationship and generate important datasets that help unravel complicated genetic blueprints. Thus, the recently developed artificial intelligence methods can be used to interpret the functions of those DNA sequences.
Methods
This study explores the use of deep learning, particularly pre-trained genomic models like DNA_bert_6 and human_gpt2-v1, in interpreting and representing human genome sequences. Initially, we meticulously constructed multiple datasets linking genotypes and phenotypes to fine-tune those models for precise DNA sequence classification. Additionally, we evaluate the influence of sequence length on classification results and analyze the impact of feature extraction in the hidden layers of our model using the HERV dataset. To enhance our understanding of phenotype-specific patterns recognized by the model, we perform enrichment, pathogenicity and conservation analyzes of specific motifs in the human endogenous retrovirus (HERV) sequence with high average local representation weight (ALRW) scores.
Results
We have constructed multiple genotype-phenotype datasets displaying commendable classification performance in comparison with random genomic sequences, particularly in the HERV dataset, which achieved binary and multi-classification accuracies and F1 values exceeding 0.935 and 0.888, respectively. Notably, the fine-tuning of the HERV dataset not only improved our ability to identify and distinguish diverse information types within DNA sequences but also successfully identified specific motifs associated with neurological disorders and cancers in regions with high ALRW scores. Subsequent analysis of these motifs shed light on the adaptive responses of species to environmental pressures and their co-evolution with pathogens.
Conclusions
These findings highlight the potential of pre-trained genomic models in learning DNA sequence representations, particularly when utilizing the HERV dataset, and provide valuable insights for future research endeavors. This study represents an innovative strategy that combines pre-trained genomic model representations with classical methods for analyzing the functionality of genome sequences, thereby promoting cross-fertilization between genomics and artificial intelligence.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12967-024-05567-z.
Keywords: Genomic sequences, Genotype-phenotype, Fine-tuning, HERV, Motif
Background
The Human Genome Project marked the beginning of an era characterized by the assembly to resolve high-quality genome sequences, in which the complete genetic code of DNA is gradually being deciphered [1]. The improvement of high-quality reference genome sequences and their gene annotation information, enhancing molecular diagnostics capabilities and enabling advances in disease prevention and personalized treatment strategies [2]. However, the current functional studies of genome sequences focus primarily on ∼ 3% protein-coding regions, leaving the vast majority of regulatory functions largely unexplored [3]. Therefore, we need to employ more strategies to explore even the unknown functional regulatory elements in the human genome, such as the recently prominent HERV sequences, which are closely associated with gene regulation, immune modulation, carcinogenesis and the pathophysiology of complex diseases such as neurodegenerative disorders [4].
In the field of artificial intelligence, natural language processing (NLP) has made rapid progress, particularly owing to advancements in transfer learning and Transformer architectures, which has led to innovative methods for processing and analyzing large-scale complex datasets [5, 6]. The development of models like Bidirectional Encoder Representations from Transformers (BERT) and Generative Pre-trained Transformer (GPT), leveraging the Transformer architecture, has significantly boosted NLP by capturing context and data dependencies through self-attention mechanisms [7]. These advances have not only revolutionized traditional text processing tasks, but also provided new methods for fields such as bioinformatics and healthcare. In bioinformatics, the adoption of Transformer models has been particularly transformative in sequence analysis, gene expression, proteomics, and drug discovery, with notable advancements made in the latter two domains due to their ability to model long-range associations [8]. Moreover, the integration of NLP techniques with bioinformatics enhances data analysis and interpretation, opens new avenues for personalized and precision medicine, and facilitates scientific discovery.
Owing to their length and complexity, genomic DNA sequences present unique challenges for machine learning. However, their structural similarity to human language (long strings consisting of basic units such as bases or words) provides opportunities for modeling and interpreting DNA sequences using NLP methods [9, 10]. Scientists are increasingly leveraging pre-trained genomic models, leading to significant successes with Transformer-based frameworks [11, 12] and other language framework models [10]. For sequence classification evaluation, researchers have constructed benchmark datasets for DNA classification and used modified Convolutional Neural Network (CNN) models as baselines. Ultimately, they reported that fine-tuning the pre-trained genomic model DNABERT achieved better performance in classification tasks than the DNABERT model with randomly initialized weights and the CNN model [13, 14]. Nevertheless, there is a notable shortage of comprehensive datasets for evaluating the performance of large genomic models, especially for analyses involving complex genotypes [13, 15].
This study has constructed several medically significant genotype-phenotype datasets by utilizing human reference and pan-genome data [16], as well as information from the 1000 Genomes Project. We performed DNA sequence balanced binary classification and imbalanced multi-classification performance evaluation by optimizing pre-trained models such as DNA_bert_6 and human_gpt2-v1. Focusing on the HERV dataset, we extensively investigated the representability of the models on these data and identified phenotype-specific motifs within the HERV sequences. Further analysis revealed that genes associated with these motif sequences play critical roles in various biological processes, including neural development and synaptic functions, oncogenesis, cellular adhesion, and spatial localization (Fig. 1; Supplementary Fig. 1). The genotype-phenotype datasets we constructed can be used as benchmarks for DNA sequence modeling and performance evaluation of pre-trained genomic models, providing novel insights into the functionality of genomic sequences when exploring the HERV dataset based on fine-tuned models.
Fig. 1.

A schematic diagram of the article’s overall structure. (A) Generation of the multi-phenotypic classification datasets. (B) Demonstration of the model fine-tuning process using the HERV dataset. (C) The primary sequence features learned by our model. (D) Identification of motifs from high ALRW scoring sequences and their related classical analysis strategy
Methods
Pan-genomic phenotype classification datasets construction
Screening of potential phenotype-related regions
Based on existing literature and databases, we compiled potential functional phenotype datasets. According to the specific conditions of different phenotype datasets, we adopted various filtering steps, merged adjacent intervals within 10 bp using Bedtools Merge [17], and ultimately unified them to non-overlapping genomic regions on hg38, designated as phenotype-related regions. Simultaneously, we used Bedtools Shuffle to generate non-phenotype regions with similar length distributions. We then conducted a final confirmation to ensure that the identified regions did not overlap.
Phenotype dataset construction
Functional phenotypic sequence extraction within dataset-specific regions
We started using Bedtools Getfasta to extract the regional sequences from the hg38 for potential regions in each dataset. Using Bedtools Intersect, we isolated human pangenomic and 1000 Genome Project structural variation data [16, 17]. After splitting the continuous sequence with two or more consecutive Ns, retain those longer than 150 bp and further remove sequences with a similarity above 95% using Mmseqs Easy-linclust [18].
Data partitioning for model training
For the sequences extracted from each specific region of the dataset, we conducted sequence feature statistics using Seqkit Stat [19]. Subsequently, we employed Seqkit Sample (-s100) to extract approximately 20% of the dataset as an independent test set, while using the remaining approximately 80% as the training and validation set (further dividing ∼ 20% as the validation set). Subsequently, in order to conduct further research and validate our conclusions, we divided the datasets into training, validation, and test sets in accordance with the same ratio as required. In addition, for efficient management, we store all the data in a single file and provide each dataset with a numerical label (Supplementary Table 1).
Display of phenotype-related regions and datasets
We calculated and plotted the cumulative autosomal sequence length within all phenotype-related regions and datasets. Furthermore, we performed statistical analysis to compare the overall distribution and chromosome distribution between sequences in phenotypic and non-phenotypic regions. The gene enrichment rate in specific phenotypic regions was calculated as:
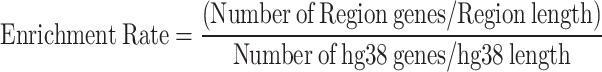 |
Fine-tuning of pre-trained models for phenotypic datasets
Model selection and Hyperparameters
Overview of the core models
The main models, DNA_bert_6 and human_gpt2-v1, are utilized to fine-tune diverse genotype-phenotype datasets, evaluating their ability to classify and recognize functional phenotypic data and assign records precisely to the corresponding labels. DNA_bert_6 processes DNA sequences using 6-mer (stride 1) tokens, with pre-training on the BERT architecture, which consists of 12 hidden layers, 12 attention heads, a 768-dimensional hidden layer, and a maximum input token limit of 512. The outputs of DNA_bert_6 include attention weights, pooling layers, and all hidden layers. In contrast, Human_gpt2-v1 handles DNA sequences via Byte Pair Encoding (BPE) tokens and is pre-trained with the gpt2 framework on the human telomere-to-telomere genome (T2T-CHM13), which consists of 12 hidden layers, 12 attention heads, a 768-dimensional hidden layer, and a maximum input token limit of 1,024. The outputs of the model, similar to those of DNA_bert_6, also include attention weights, and all hidden layers.
Sequence classification model structure
The pre-trained model can be regarded as a data compressor that identifies patterns and latent knowledge within datasets. Therefore, it is possible to incorporate additional network architectures after their hidden layers to achieve sequence classification tasks. In this study, the main approach involved using the AutoModelForSequenceClassification class provided by Huggingface to load those models for fine-tuning of multiple phenotype datasets. The AutoModelForSequenceClassification class can utilize BertForSequenceClassification and GPT2ForSequenceClassification to apply the pre-trained large BERT and GPT2 models to text classification tasks. The BertForSequenceClassification class adds a dropout layer after the output pooling layer of the BERT model and then passes it through a linear classification layer. On the other hand, the GPT2ForSequenceClassification class directly uses a linear classification layer on top of the last hidden layer.
Hyperparameters for training
The fine-tuning hyperparameters for DNA_bert_6 and human_gpt2-v1 on multiple phenotype datasets are specified as follows: 6-mer (stride 1), batch size of 10, accumulation steps of 4, a learning rate of 2E-5, 50 epochs, and a warmup ratio of 0.1.
Binary classification of functional phenotypic sequences
The fine-tuning process for binary classification was performed on balanced multi-functional phenotype datasets, ultimately achieving good robustness and high accuracy (Fig. 1B). After the sequence data related to the phenotype were processed into 6-mer, the first 300 and last 212 6-mer strings in the sequence were extracted. Phenotypic sequences consisting of 512 tokens were then used as the input for the DNA_bert_6 model using padding and truncation strategies. Meanwhile, the phenotype-related sequence data were processed via BPE, and the first 1,024 tokens of the sequence were selected using padding and truncation strategies as the inputs for the human_gpt2-v1 model. Subsequently, independent fine-tuning trials (RUN0, RUN1, RUN2) process these data under specified models and parameters, with evaluations across training, validation, and test sets following an approximate 6:2:2 ratio. In each round of independent fine-tuning, we selected the optimal model based on the minimum validation loss and calculated various evaluation metrics. After the three rounds of fine-tuning, we averaged these metrics to assess the model’s performance. Additionally, a voting mechanism was employed, with two identical predicted labels being retained. Otherwise, the predicted label from RUN0 was used, resulting in the final RUN_Vote. The entire training process, facilitated by Huggingface and PyTorch, is streamlined by the Trainer function’s embedded optimization strategies for efficient fine-tuning of large models.
Multiclass classification of functional phenotypic sequences
The multi-classification of phenotypes was carried out via a strategy similar to binary classification. To attenuate the effects of class imbalance on classification efficiency, the ratio of each data category within the training set labels was calculated and integrated into the CrossEntropyLoss function as a customized Trainer function. Subsequently, the independent fine-tuning of pre-trained genomic models (RUN0, RUN1, RUN2) was achieved using Huggingface and PyTorch. To validate the efficacy of our training model strategy and the reasonableness of the related conclusions, we utilized the DNA_bert_6 model for cross-validation (Cross-Valid) and random data splitting strategies (Split1, Split2, Split3_1, Split3_2, Split3_3) on the dataset with the best classification performance. The cross-validation modeling strategy involved splitting the original dataset into training, validation, and test sets. The model was then trained using 5-fold cross-validation, with performance assessed based on the cross-validation and initial validation sets. To select the best model, it needed to achieve the highest F1 score on the cross-validation validation set and the lowest loss on the initial validation set. If the current model performed well on both selection criteria, the state dictionary of the best model was updated. Subsequently, the optimal model was then evaluated on the test set to validate its generalization capability. Additionally, the random data splitting strategy involves multiple random splits of the dataset, followed by modeling using the original method, to further confirm the reliability of the conclusions.
Model evaluation metrics
All multiple phenotypic datasets are evaluated using the following metrics while recording the loss and runtime of the model on the test set:
Accuracy is the quotient of the number of correctly predicted samples by the total sample count.
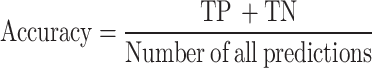 |
Recall is the ratio of correctly predicted true positives to all actual positives.
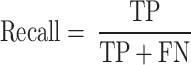 |
Precision measures the ratio of true positives in all positive predictions.
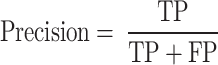 |
The F1 score, the harmonic mean of precision and recall, usually used to measure the accuracy of classification models, especially in cases of imbalanced class distributions.
 |
The ROC AUC (Auroc) is the area under the ROC curve and is used to measure model performance across various classification thresholds. Auroc can be calculated in two ways: Auroc_macro, which averages the AUC scores for each class equally, while Auroc_weighted, which assigns weights on the basis of the number of true instances per class to account for class imbalance.
Pr_auc represents the area under the Precision-Recall Curve. It focuses on the predictive power of a small number of classes (positive classes) without being affected by a large number of negative samples, and is suitable for the case of class imbalance.
The MCC (Matthews correlation coefficient) quantifies the performance of binary classification models, considering all varieties of positives and negatives.
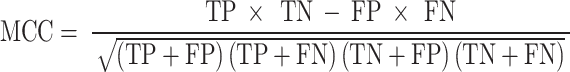 |
The impact of different lengths of HERV datasets on phenotypic classification
To evaluate the classification performance of the models at different sequence lengths, we further evaluate the changes in the prediction results of the DNA_bert_6 and human_gpt2-v1 models within the HERV dataset for three independent fine-tunings (RUN0, RUN1, RUN2 and RUN_Vote). To mitigate the significant differences in sequence length distribution, a logarithmic transformation of the sequence lengths was applied, supplemented by uniformly distributed random noise from − 0.1 to 0.1 for length correction. We then sort the corrected sequence lengths and select 20 equally spaced values as the interval divisions. For the last intervals with fewer than 10 data points, we merge them into a single interval to maintain reliability and visualization. Next, we present the results of fine-tuning the DNA_bert_6 and human_gpt2-v1 models on the HERV dataset, highlighting the models’ maximum token input threshold and the proportional distribution of data points across intervals. Similarly, a statistical analysis of various classification metrics across different chromosomes was conducted.
Model feature learning effect evaluation in the HERV dataset
Representativeness of classification labels
The DNA sequences in the test set were processed into 6-mer nucleotide fragments. Their frequencies were calculated via CountVectorizer to convert the sequences into numerical feature vectors. The sparse matrices of these feature vectors in a high-dimensional feature space were subjected to dimensionality reduction via TruncatedSVD (n_components = 10) and SparsePCA (n_components = 10) for further data visualization.
Feature extraction by the fine-tuned model
The DNA_bert_6 and human_gpt2-v1 models fine-tuned with the HERV dataset (RUN0, Cross-Valid, Split1, Split2, Split3_3) were used to extract features in batches from sequences in their test sets. We utilized the final layer of the hidden layer features (batch_size, sequence_length, hidden_size) to calculate the mean feature values across all positions within a sequence, obtaining the representation vector (hidden_size = 768) of the entire sequence. A test set feature matrix (sequence_number, 768) was compiled by combining the extracted features from all test set sequences. Subsequently, PCA and t-SNE were applied to reduce the dimensions and visualize this feature matrix.
Visualization of HERV subtype information
The complete HERV dataset was retrieved from the database, and data from HervD_Atlas [20] were matched with the HERV test set based on hg38 coordinates. The HervD_Atlas fragment with the longest overlap with the test set sequence was selected as the subtype for that sequence. The largest overlapping segment of HervD_Atlas was designated as the subtype for the corresponding sequence. These overlapping sequences were used as reference points in the HERV test set, along with the associated subtype information to interpret t-SNE results.
Analysis of phenotype-specific high ALRW in the HERV dataset
Extraction of attention matrices from the fine-tuned model using the test set
Using the DNA_bert_6 model fine-tuned on the HERV dataset (RUN0, Cross-Valid, Split1, Split2, Split3_1, Split3_2, Split3_3), the final layer attention matrix scores (batch_size, num_heads, sequence_length, sequence_length) for the test set data were extracted. To calculate and enhance the representation of the entire 6-mers token scores, we first iterated through the attention matrices to calculate the attention scores at each position. Next, these scores were aggregated with those of the subsequent five positions, resulting in the cumulative score for the entire 6-mer tokens. Given that each position could be included in multiple different 6-mer combinations during cumulative score calculation, it was necessary to record the count frequency at each position to calculate the mean score, which was then normalized across all 6-mer tokens using L2 normalization. Following this approach, we calculated the overall average attention score (single attention) for the 12 attention heads, as well as the individual attention scores (multiple attention) for each of the 12 attention heads, more accurately capturing the relative significance of each 6-mer token in the DNA sequence. During the calculation of single and multiple attention for test set data in batches, if the computed length is shorter than 512 tokens, a padded array filled with zeros can be used to avoid errors.
Statistical analyses of attention matrices by phenotype-specific labels
To preliminarily characterize the phenotypic sequence features learned by different models after fine-tuning (RUN0, Cross-Valid, Split1, Split2, Split3_1, Split3_2, Split3_3), we conducted statistical analyses of the single attention matrices specific to the phenotypic sequences [“Non-HERV_Coding”, “HERV_Coding”, “Non-HERV_Non-Coding”, “HERV_Non-Coding”], focusing on the minimum, median, mean and third quartile values to characterize the overall distribution and variability of the matrices. Furthermore, the multiple attention score matrices learned by the RUN0 model were extracted, and these statistical values for the 12 headers were calculated to represent their learning phenotype-specific sequence characteristics.
Visualization attention scores and sequence analysis by phenotype-specific labels
The single and multiple attention score matrices, derived from the above methods and integrated with their respective test set labels, were used to investigate the distribution patterns of single and multi-head attentions across various token lengths for different phenotypes (RUN0, Split1, Split2, Split3_3). Notably, there was minimal overlap among these test sets, highlighting their distinctiveness and enabling a more nuanced analysis of attention patterns across diverse phenotypic labels. For single attention, the average attention score was calculated for each phenotype and referred to as the phenotype-specific average local representation weighted (ALRW) score, which is the average of certain specific phenotype-related sequences and multi-head attention layers. In the case of multiple attention, the average attention score was calculated for each head and subsequently displayed. To further characterize the sequence feature variations across different token intervals and explain the DNA sequence reasons for phenotypic attentions distribution differences, specific token regions [(0, 50), (50, 150), (150, 250), (250, 350), (350, 450), (450, 512)] were analyzed within the BERT model. The DNA feature statuses in the corresponding token regions for different phenotype sequences [“Non-HERV_Coding”, “HERV_Coding”, “Non-HERV_Non-Coding”, “HERV_Non-Coding”] in the test set were calculated as follows:
GC Content: The proportion of guanine (G) and cytosine (C) in the DNA sequence, typically denoted by:
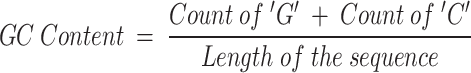 |
6-mer(6mer) frequency: refers to the number of unique 6-mer sequences in DNA, calculated as:
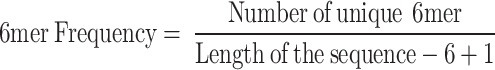 |
Shannon Entropy: A measure of DNA sequence complexity, computed using the probabilities of each base’s occurrence and applying the concept of information entropy: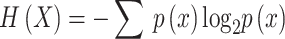 , where
, where 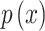 is the probability of the occurrence of base x.
is the probability of the occurrence of base x.
CpG Island Score: The observed-to-expected frequency ratio of CpG dinucleotides, which is calculated on the basis of the occurrence probabilities of C and G within the DNA sequence:
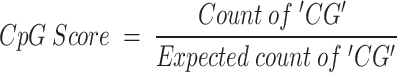 |
where the expected count of ‘CG’ is the product of the frequency of ‘C’, the frequency of ‘G’, and the sequence length.
Line graphs depicting the variation in DNA sequence features in the different token regions [(0, 50), (50, 150), (150, 250), (250, 350), (350, 450), (450, 512)] were plotted.
Analysis of phenotype-specific HERV subtypes in the dataset
In the HERV test set, data that overlapped with the HervD_Atlas database were selected to calculate the percentage of various HERV classes (ERV1, ERV2, ERV3, ERVL-MaLR, Gypsy, LTR) within coding and non-coding regions.
The data were further analyzed to determine the representation of different HERV functional groups. The relative enrichment rates were calculated as the ratio of each group’s percentage in non-coding regions to coding regions. This ratio allows us to understand the tendency of each group for being in non-coding versus coding regions, thereby providing insight into the functional dynamics of HERV elements within the genome.
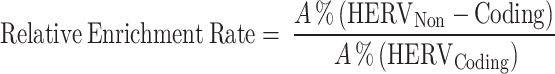 |
Here, A denotes an individual element within the HERV functional group.
Motif analysis using high ALRW scores in the HERV dataset
Enrichment of motifs in continuously high ALRW score sequences
Extraction of continuously high ALRW score sequences
In order to extract the continuous high ALRW score regions in specific phenotypic DNA sequences, each sequence within every phenotypic subset [“Non-HERV_Coding”, “HERV_Coding”,“Non-HERV_Non-Coding”,“HERV_Non-Coding”] is processed. Regions in each sequence with scores exceeding the average value and the minimum value by 10 times were identified as potential areas of interest, represented as Boolean arrays with high attention parts set to true. Subsequently, consecutive true regions with lengths exceeding 5 bp are identified from the Boolean array as continuous high attention score regions. These segments were aligned to the corresponding DNA sequences, which were extended 4 bp upstream and downstream to capture maximum potential information. Furthermore, the above procedure was also applied to the non-overlapping HERV dataset test sets (RUN0-Related, Split1-Related, Split2-Related, Split3-Related).
Motif analysis of high ALRW scoring sequences
DNA sequences from the HERV dataset associated with the identified high ALRW score sequences across different phenotypes [“Non-HERV_Coding”, “HERV_Coding”, “Non-HERV_Non-Coding”, “HERV_Non-Coding”] underwent motifs analysis using Fimo software in conjunction with non-redundant transcription factor binding sites sourced from the JASPAR database in both the MEME and TRANSFAC formats [21, 22]. Motif sequences identified in the above four phenotypes were combined and filtered to retain those with q.value ≤ 0.05 for further analysis. Firstly, Venn diagrams are used to represent the number of intersections of identified motifs among different phenotypes. Secondly, the specificity enrichment of motif sequences in the HERV group [“HERV_Coding”, “HERV_Non-Coding”] relative to the Non-HERV group [“Non-HERV_Coding”, “Non-HERV_Non-Coding”] is statistically analyzed. The frequency of motif sequences occurring in the HERV and Non-HERV groups is normalized by dividing by the cumulative number of motif sequences in each group. Then, the hypergeometric distribution test is used to perform enrichment analysis on specific motifs, with the calculation method being phyper(k-1, m, total_motifs-m, total_case, lower.tail = FALSE), where k is the normalized frequency of each specific motif in the HERV group, m is the normalized frequency of each specific motif in the HERV and Non-HERV groups, total_motifs is the total number of motifs in the HERV and Non-HERV groups, and total_case is the total number of motifs in the HERV group. Next, the Benjamini-Hochberg method was used to correct the p-values. The frequency of enriched motifs in HERV_Coding and HERV_Non-Coding facilitated the categorization of motifs into three categories [“HERV_NonCoding”, “HERV_Coding”, “HERV_Both”]. The selected motif sequences are shown for enrichment results (P_hyper_adjust ≤ 0.05; HERV_Rate ≥ 0.5; HERV_Num > 10, where P_hyper_adjust is the adjusted p-value, HERV_Rate is the proportion of the specific motif’s normalized frequency in the HERV group, and HERV_Num is the normalized frequency of the specific motif in the HERV group). This analysis method was applied to the high ALRW scoring sequences identified in the four non-overlapping HERV dataset test sets as described above, which served as independent replicates to further increase the reliability of our findings.
Motif analysis of the full-length HERV dataset sequences
To validate the reliability of motif sequences identified by high ALRW scores, we performed a similar statistical analysis and evaluation on the full-length HERV test sets as previously described. Furthermore, we conducted a comparative analysis of specifically enriched motifs by high ALRW scores and those enriched in full-length sequences within the four non-overlapping HERV dataset test sets.
Functional enrichment and pathogenicity of phenotype-specific motifs
We compiled the HERV sequence motifs identified in each of the four experiments and matched the selected motifs to the hg38 genome, combining adjacent intervals within a 10 bp range to define HERV sequence-specific motif regions. Initially, we characterized the basic characteristics of the four HERV-specific motifs. Then, using regulatory element annotation information from hg38, we quantified the functional element ratios within these specific motif regions. Subsequently, we performed functional enrichment and related disease analyses on protein-coding genes via Metascape [23], which associated with HERV-specific motifs extracted from high ALRW sequences in four independent experiments. Although many common HERV-special motifs were identified across the four independent test sets, we selected RUN0-related motifs for comparison of the detailed characteristics and pathogenicity between the high ALRW score and full-sequence analysis method due to their genomic non-overlapping. Pathogenicity scores for variants in these regions were predicted using PrimateAI and AlphaMissense [24, 25], and subsequently checked for intersection with HervD_Atlas.
Species conservation of HERV sequence-specifically enriched motif sequences
Polymorphism in specific motif regions of the human pangenome
The genomic diversity within HERV sequence-specific enriched motif regions of the human pangenome was assessed via Odgi Depth [26]. Gene annotations that overlapped with these regions were categorized by chromosome and gene category using Bedtools Intersect. We identified genes that exhibited both high divergence and high conservation in the human population.
Conservation of specific motifs in primates
Genome-wide alignment data from 27 primate species, obtained from UCSC with hg38 as the reference, were extracted using MafSpeciesSubset. The primate evolutionary tree was reconstructed with PhyloFit, facilitating the evaluation of primate conservation scores using PhyloP and PhastCons. The conservation scores within motif-enriched regions were determined using BigWigAverageOverBed, complemented by gene annotations from Bedtools Intersect to present a comprehensive conservation landscape and annotate specific genes [27, 28]. Moreover, to further explore the potential functions of motif sequences located in non-gene regions, we annotated HERV-related motifs by integrating regulatory elements, including those located 1 kb upstream and downstream of genes, promoters, enhancers, and open chromatin regions. Additionally, we compiled the basic information, population diversity, and primate conservation of specific motifs as needed.
Results
Phenotypic regions collection and dataset construction
Functional phenotypic regions of the human genome
We consolidated five biomedically significant phenotypes from existing knowledge datasets: potential disease susceptible regions (Diseases_GWAS), regulatory element regions (Regulatory), endogenous viral regions (HERV), immunogenetic regions (Immunogenetic), and highly specifically expressed gene regions (Highly_Specifically _Gene). By organizing the genome coordinates linked to these phenotypes, we collected essential information about the specific sequences associated with each phenotype (Supplementary Table 2). For instance, the intervals within the HERV dataset cover nearly 302.9 Mb of the genome, with the longest interval length exceeding 153 kb. There are a total of 382,317 intervals within the genome, covering approximately 98.47% of the HERV intervals from the HervD_Atlas dataset, and spanning a total of 29.68 Mb [20]. Using the genomic coordinates of these five phenotypes, we illustrated the cumulative length distribution of the phenotype sequence regions across different chromosomes (Fig. 2A). The cumulative distributions of Diseases_GWAS and Regulatory Regions are relatively consistent across different chromosome lengths, whereas HERV, Immunogenetic and Highly_Specifically_Gene show distinct chromosomal enrichments. Notably, the X chromosome has extremely long intervals, whereas the Y chromosome has very few intervals in most phenotypic datasets, especially in the HERV dataset which accounts for ∼ 6.4% (Supplementary Tables 3, 4). This phenomenon may be closely related to the greater concentration of regulatory elements and genes on the X chromosome. To further emphasize the genes as critical functional units, the study also revealed gene enrichment ratios within the phenotypic regions (Fig. 2B), highlighting an enrichment in the number of genes compared with the genome-wide average, particularly for protein-coding genes.
Fig. 2.

Illustrates the distribution and genetic characteristics of the selected phenotypic regions. (A) The cumulative chromosomal distribution patterns of five regions with phenotypic data. (B) The enrichment rates of all genes (red) and coding genes (blue) within the five phenotypic regions compared with the entire genome. A black color (y = 1) or below indicates the absence of enrichment
Construction of a multi-phenotype classification dataset for humans
We constructed multiple functional phenotypic classification datasets by linking specific interval features of genomic phenotypic sequences with randomly selected non-phenotypic genomic regions as controls. We also inspected the length distributions of functional and non-functional random regions for different phenotypes (Supplementary Fig. 2A). The results revealed similar length distributions between the two regions in all datasets. The HERV and Regulatory phenotype datasets, which maintain the original interval lengths, allowed us to analyze the chromosomal distribution of the corresponding functional and non-functional random regions, thereby confirming the uniformity of the constructed datasets across all chromosomes (Supplementary Fig. 2B).
Subsequently, we extracted corresponding sequences from the selected specific functional and non-functional random regions, using databases like hg38, the human pangenome, and the 1000 Genomes Project. After removing redundancy, we performed sequence statistics on the constructed functional phenotype datasets. Furthermore, we classified these datasets into binary and multi-classification categories considering biological factors (Table 1; Supplementary Tables 4, 5). The HERV dataset, for example, includes both balanced binary classification (HERV: Non-HERV = 417,416: 362,907) and imbalanced multi-classification (HERV_Coding: Non-HERV_Coding: HERV_Non-Coding: Non-HERV_Non-Coding = 16,556: 18,994: 400,860: 343,913). This dataset features approximately 22.64 Mb of coding sequences and roughly 295.69 Mb of non-coding sequences within the HERV functional regions, enabling the development of a multi-classification task through binary classification. Furthermore, the Dataset_Regulatory is the most diverse, requiring a six-class classification task to distinguish TF_binding_site, Enhancer, CTCF_binding_site, Promoter, Open_chromatin_region, and Non-Regulatory element sequences within the genome.
Table 1.
Display of the functional phenotypic dataset sequence
| Dataset | Classification | Numbers | Length | |||
|---|---|---|---|---|---|---|
| (Multiclass/Binary) | Sum | Average | Max | |||
|
Dataset_HERV (HERV) |
HERV_Coding/HERV | 16,556 | 22,643,342 | 1,368 | 125,188 | |
| HERV_Non-Coding/HERV | 400,860 | 295,693,006 | 738 | 112,801 | ||
| Non-HERV_Coding/ Non-HERV | 18,994 | 23,490,411 | 1,237 | 83,426 | ||
| Non-HERV_Non-Coding/ Non-HERV | 343,913 | 251,939,874 | 733 | 93,893 | ||
|
Dataset_Immuno (Immunogenetic) |
Immuno_KIR/Immuno | 1,723 | 3,096,500 | 1,797 | 39,269 | |
| Immuno_Others/Immuno | 16,719 | 83,307,498 | 4,983 | 71,249 | ||
| Non-Immuno | 18,326 | 78,935,043 | 4,307 | 93,519 | ||
|
Dataset_Regulatory (Regulatory) |
TF_binding_site/Regulatory | 22,860 | 13,391,832 | 586 | 93,893 | |
| Enhancer/Regulatory | 320,389 | 335,938,843 | 1,049 | 93,893 | ||
| CTCF_binding_site/Regulatory | 94,738 | 52,273,466 | 552 | 93,893 | ||
| Promoter/Regulatory | 52,413 | 79,591,023 | 1,519 | 99,371 | ||
| Open_chromatin_region/Regulatory | 130,006 | 54,838,694 | 422 | 93,893 | ||
| Non-Regulatory | 494,389 | 435,738,664 | 881 | 96,745 | ||
|
Dataset_Diseases_GWAS (GWAS_loci/PrimateAI-3D scores) |
Diseases-GWAS_Coding/Diseases_GWAS | 175,920 | 566,540,577 | 3,220 | 99,371 | |
| Diseases-GWAS_Non-Coding/Diseases_GWAS | 51,427 | 132,677,910 | 2,580 | 92,731 | ||
| Non_Diseases-GWAS_Coding/Non_Diseases_GWAS | 148,235 | 550,157,564 | 3,711 | 93,893 | ||
| Non_Diseases-GWAS_Non-Coding/Non_Diseases_GWAS | 31,298 | 104,453,319 | 3,337 | 93,893 | ||
|
Dataset_Highly_Specifically_Gene (Defensins/Olfactory Receptor) |
Defensins/Highly_Specifically_Gene | 415 | 418,877 | 1,009 | 9,191 | |
| Olfactory_Receptor/ Highly_Specifically_Gene | 5,400 | 5,305,444 | 983 | 40,645 | ||
| Others | 5,610 | 6,653,032 | 1,186 | 94,155 | ||
Binary and multi-classification of functional phenotype datasets
Binary classification performance of multiple phenotype datasets
To evaluate the performance of fine-tuning pre-trained genomic models on multiple phenotype datasets, we conducted three independent fine-tuning iterations using the DNA_bert_6 and human_gpt2-v1 models for each phenotype dataset, including both the training and validation sets. The model performance was then evaluated on the test set with various metrics for all datasets (Table 2), using the random genomic region dataset (Dataset_Random) serving as a foundational baseline. The constructed multiple phenotype datasets demonstrated differential classification performance, with Dataset_HERV, Dataset_ Diseases-GWAS, and Dataset_Immuno models performed well for all metrics, especially Dataset_HERV with accuracy and F1 values were above 0.935. In comparison, the DNA_bert_6 and human_gpt2-v1 models showed negligible performance variance, although with a notable increase in fine-tuning time for human_gpt2-v1. In summary, the models’ performance across various phenotypic datasets highlights that the phenotypic labels assigned to specific DNA sequences can partially reflect the inherent information within the data, implying distinctive DNA sequence patterns associated with the HERV, Diseases-GWAS, and Immuno phenotypes. Moreover, the Dataset_Random outcomes imply that current those models struggle to distinguish genomic regions in a non-selective manner. This discrepancy is likely due to the incongruence between the information contained in the sequence data and the assigned labels.
Table 2.
Binary classification performance of functional phenotypic datasets using fine-turned models
| Model | Accuracy | F1 | Loss | Precision | Recall | Auroc_macro | Pr_auc | MCC | Time(h) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Dataset_HERV | A | 0.9354 ± 0.0025 | 0.9386 ± 0.0025 | 0.1748 ± 0.0026 | 0.9544 ± 0.0027 | 0.9234 ± 0.0051 | 0.9851 ± 0.0007 | 0.9883 ± 0.0006 | 0.8710 ± 0.0048 | 13.2393 ± 0.3802 |
| B | 0.9363 ± 0.0010 | 0.9395 ± 0.0010 | 0.1554 ± 0.0012 | 0.9566 ± 0.0035 | 0.9230 ± 0.0040 | 0.9859 ± 0.0003 | 0.9888 ± 0.0003 | 0.8731 ± 0.0019 | 17.7381 ± 1.2064 | |
| Dataset_Immuno | A | 0.8122 ± 0.0002 | 0.8051 ± 0.0037 | 0.3211 ± 0.0013 | 0.8414 ± 0.0132 | 0.7729 ± 0.0182 | 0.9238 ± 0.0007 | 0.9298 ± 0.0007 | 0.6274 ± 0.0017 | 0.7026 ± 0.0165 |
| B | 0.8129 ± 0.0008 | 0.8128 ± 0.0039 | 0.3198 ± 0.0021 | 0.8182 ± 0.0167 | 0.8095 ± 0.0244 | 0.9238 ± 0.0013 | 0.9300 ± 0.0011 | 0.6274 ± 0.0018 | 1.9810 ± 0.0044 | |
| Dataset_Regulatory | A | 0.7438 ± 0.0003 | 0.7764 ± 0.0028 | 0.5016 ± 0.0013 | 0.7540 ± 0.0060 | 0.8005 ± 0.0127 | 0.8270 ± 0.0005 | 0.8602 ± 0.0004 | 0.4787 ± 0.0009 | 18.4481 ± 0.0137 |
| B | 0.7614 ± 0.0012 | 0.7911 ± 0.0020 | 0.4744 ± 0.0015 | 0.7704 ± 0.0061 | 0.8134 ± 0.0104 | 0.8462 ± 0.0014 | 0.8763 ± 0.0012 | 0.5147 ± 0.0027 | 27.2533 ± 1.6940 | |
| Dataset_Diseases_GWAS | A | 0.8380 ± 0.0007 | 0.8537 ± 0.0021 | 0.3023 ± 0.0023 | 0.8642 ± 0.0095 | 0.8442 ± 0.0130 | 0.9368 ± 0.0008 | 0.9541 ± 0.0006 | 0.6731 ± 0.0022 | 7.2913 ± 0.0123 |
| B | 0.8443 ± 0.0010 | 0.8609 ± 0.0014 | 0.2976 ± 0.0019 | 0.8622 ± 0.0048 | 0.8597 ± 0.0070 | 0.9393 ± 0.0008 | 0.9555 ± 0.0006 | 0.6843 ± 0.0018 | 21.3534 ± 0.0286 | |
| Dataset_Highly_Specifically_Gene | A | 0.6346 ± 0.0044 | 0.6081 ± 0.0180 | 0.6075 ± 0.0029 | 0.6688 ± 0.0081 | 0.5604 ± 0.0352 | 0.7076 ± 0.0034 | 0.7372 ± 0.0043 | 0.2758 ± 0.0054 | 0.2051 ± 0.0003 |
| B | 0.6237 ± 0.0056 | 0.5861 ± 0.0147 | 0.6349 ± 0.0040 | 0.6668 ± 0.0189 | 0.5264 ± 0.0332 | 0.6894 ± 0.0011 | 0.7261 ± 0.0011 | 0.2572 ± 0.0144 | 0.1858 ± 0.0098 | |
| Dataset_Random | A | 0.5205 ± 0.0031 | 0.6503 ± 0.0118 | 0.6953 ± 0.0008 | 0.5299 ± 0.0006 | 0.8436 ± 0.0384 | 0.4983 ± 0.0021 | 0.5296 ± 0.0006 | -0.0003 ± 0.0026 | 0.7690 ± 0.0038 |
| B | 0.5134 ± 0.0059 | 0.5895 ± 0.0242 | 0.7010 ± 0.0014 | 0.5327 ± 0.0019 | 0.6656 ± 0.0606 | 0.4997 ± 0.0012 | 0.5290 ± 0.0014 | 0.0085 ± 0.0067 | 2.4468 ± 0.0038 |
Note The A and B represent the DNA_bert_6 model and the human_gpt2-v1 model, respectively
Multi-classification performance of phenotypic datasets
We adopted a training and evaluation strategy similar to that employed in assessing multi-classification performance across various phenotype datasets. Given the significant class imbalance in certain multiclass phenotype datasets (Table 1), we optimized the cross-entropy loss function in the DNA_bert_6 and human_gpt2-v1 models during fine-tuning by adjusting it according to the label proportions in the training set. Moreover, we performed three independent fine-tuning rounds for each dataset, including both the training and validation sets, and subsequently evaluated the classification outcomes via the test set (Table 3). Notably, when comparing the model’s performance in multi-classification fine-tuning versus binary classification within the same phenotype dataset, we observed a decrease in metrics, which may be attributed to the increased difficulty of the multi-classification task. Consistently, the model’s performance exceeded that of the genomic random region dataset (Dataset_Random) across Dataset_HERV, Dataset_Immuno, and Dataset_Disease-GWAS, with Dataset_HERV achieving accuracy and F1 scores above 0.888. Furthermore, we used cross-validation and random data splitting strategies for sequence classification in Dataset_HERV, achieving similar results after fine-tuning DNA_bert_6 (Supplementary Table 6). In specific functional phenotypic tasks, a significant decrease in performance between multi-classification and binary classification tasks was observed in Dataset_Regulatory and Dataset_Diseases-GWAS, suggesting that too many class labels and inconsistencies between data and label information can detrimentally affect the final sequence classification performance.
Table 3.
Multi-classification performance of functional phenotypic datasets using fine-turned models
| Model | Accuracy | F1 | Loss | Precision | Recall | Auroc_weighted | Pr_auc | MCC | Time(h) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Dataset_HERV | A | 0.8880 ± 0.0125 | 0.8895 ± 0.0077 | 0.3352 ± 0.0177 | 0.8989 ± 0.0045 | 0.8880 ± 0.0125 | 0.90625 ± 0.0047 | 0.8455 ± 0.0063 | 0.7967 ± 0.0197 | 14.6616 ± 0.4380 |
| B | 0.8954 ± 0.0024 | 0.8956 ± 0.0017 | 0.3061 ± 0.0087 | 0.8985 ± 0.0012 | 0.8954 ± 0.0024 | 0.9122 ± 0.0010 | 0.8515 ± 0.0014 | 0.8078 ± 0.0040 | 18.8373 ± 1.7596 | |
| Dataset_Immuno | A | 0.8096 ± 0.0029 | 0.8091 ± 0.0033 | 0.3432 ± 0.0025 | 0.8164 ± 0.0033 | 0.8096 ± 0.0029 | 0.8197 ± 0.0020 | 0.7487 ± 0.0017 | 0.6549 ± 0.0023 | 0.6727 ± 0.0035 |
| B | 0.8057 ± 0.0022 | 0.8049 ± 0.0029 | 0.3396 ± 0.0040 | 0.8180 ± 0.0063 | 0.8057 ± 0.0022 | 0.8180 ± 0.0012 | 0.7471 ± 0.0017 | 0.6534 ± 0.0056 | 1.9903 ± 0.0047 | |
| Dataset_Regulatory | A | 0.5937 ± 0.0033 | 0.6055 ± 0.0029 | 0.9905 ± 0.0073 | 0.6491 ± 0.0028 | 0.5937 ± 0.0033 | 0.7366 ± 0.0017 | 0.5107 ± 0.0024 | 0.4634 ± 0.0038 | 22.4265 ± 1.1836 |
| B | 0.6263 ± 0.012 | 0.6355 ± 0.0099 | 0.9275 ± 0.0309 | 0.6662 ± 0.0008 | 0.6263 ± 0.0128 | 0.7536 ± 0.0038 | 0.5324 ± 0.0047 | 0.4966 ± 0.0098 | 31.2417 ± 2.6290 | |
| Dataset_Diseases_GWAS | A | 0.7563 ± 0.0062 | 0.7579 ± 0.0044 | 0.5693 ± 0.0049 | 0.7698 ± 0.0029 | 0.7563 ± 0.0062 | 0.8193 ± 0.0005 | 0.6599 ± 0.0018 | 0.6355 ± 0.0049 | 7.7064 ± 0.2058 |
| B | 0.7511 ± 0.0015 | 0.7585 ± 0.0012 | 0.5747 ± 0.0036 | 0.7761 ± 0.0006 | 0.7511 ± 0.0015 | 0.8293 ± 0.0003 | 0.6683 ± 0.0006 | 0.6376 ± 0.0011 | 22.5575 ± 0.6066 | |
| Dataset_Highly_Specifically_Gene | A | 0.6002 ± 0.0048 | 0.6014 ± 0.0034 | 0.8146 ± 0.0092 | 0.6029 ± 0.0031 | 0.6002 ± 0.0048 | 0.6312 ± 0.0029 | 0.5498 ± 0.0024 | 0.2551 ± 0.0059 | 0.2179 ± 0.0122 |
| B | 0.5652 ± 0.0091 | 0.5673 ± 0.0129 | 0.8564 ± 0.0164 | 0.5725 ± 0.0166 | 0.5652 ± 0.0091 | 0.6003 ± 0.0152 | 0.5270 ± 0.0107 | 0.1947 ± 0.0274 | 0.1933 ± 0.0159 | |
| Dataset_Random | A | 0.2864 ± 0.012 | 0.2793 ± 0.0025 | 1.3934 ± 0.0011 | 0.2881 ± 0.0053 | 0.2864 ± 0.0126 | 0.4943 ± 0.0038 | 0.2935 ± 0.0014 | -0.0109 ± 0.0074 | 0.7665 ± 0.0030 |
| B | 0.3321 ± 0.0095 | 0.2984 ± 0.0071 | 1.3955 ± 0.0029 | 0.2992 ± 0.0018 | 0.3321 ± 0.0095 | 0.5026 ± 0.0009 | 0.2969 ± 0.0004 | 0.0052 ± 0.0018 | 2.4553 ± 0.0020 |
Note The A and B represent the DNA_bert_6 model and the human_gpt2-v1 model, respectively
Binary and multi-classification performance across HERV sequence lengths
When fine-tuning the DNA_bert_6 and human_gpt2-v1 models with new datasets, it is necessary to adjust the length of the input DNA sequences within the maximum allowed tokens for each respective model. Considering the model’s outstanding performance on the HERV dataset and the preservation of original functional phenotype interval lengths, we delved into how sequence length influences model effectiveness within this specific dataset. The test set was partitioned into multiple subsets based on sequence length, and the changes in HERV sequence classification scoring metrics within different length ranges were evaluated in three independent fine-tuning experiments (Fig. 3; Supplementary Fig. 3). The fine-tuning experiments on the HERV dataset, which utilizing the DNA_bert_6 (Fig. 3A; Supplementary Fig. 3A) and the human_gpt2-v1 (Fig. 3B; Supplementary Fig. 3B) models, revealed that as the sequence length surpasses the maximum token limit of the model, the model’s classification metrics gradually decrease. The trend observed in the multiclass evaluation metrics is consistent with the change in sequence length frequency, likely due to the varying number of test samples for each class in the multiclass task, thereby leading to a significant metric variability, unlike the more stable trend seen in binary classification metrics. Furthermore, the human_gpt2-v1 model slightly outperformed the DNA_bert_6 in handling longer sequences due to an increased input length within a permissible range. Nevertheless, considering that the majority of test sequences adhere to the token limit of the DNA_bert_6 model, the differences in the final evaluation metrics are not statistically significant, and overall performance remained high. Moreover, a voting strategy across classification trials may improve model consistency (RUN_Vote), in split of a decrease in the MCC metric was observed in RUN1. Additionally, we evaluated the model’s performance across different chromosomes and found that while some chromosomes presented slightly lower metrics, the overall classification effect remained unaffected (Supplementary Fig. 4). Therefore, it is crucial to thoroughly assess both the data distribution characteristics and the complexity of the classification task in a comprehensive manner. Besides, conducting multiple independent experiments to assess the model’s overall classification efficacy is crucial in strengthening its robustness.
Fig. 3.

Multi-classification effects of sequences across different length ranges in the HERV dataset. (A) DNA_bert_6 model fine-tuning results; (B) human_gpt2-v1 model fine-tuning results. The top grey line is the percentage of sequences in different length ranges, the red line is the maximum number of tokens allowed by the model, and the RUN_Vote is the result of three independent runs of voting (RUN0, RUN1, RUN2)
Model feature learning performance in the HERV dataset
After fine-tuning on the HERV dataset, the advanced pre-trained models DNA_bert_6 and human_gpt2-v1 demonstrated effective sequence classification performance. To gain a deeper understanding of the representation learning ability of fine-tuned large models, we visualized the latent patterns learned through unsupervised learning from the dataset, as well as the representations learned by the models respectively (Fig. 4). The DNA sequences in the test set were parsed into 6-mers, which yielded high-dimensional sparse matrices that were subsequently dimensionally reduced and visualized using SparsePCA and TruncatedSVD (Fig. 4A, B). The visualization revealed that specific sequence patterns matching the labels were present in the dataset, HERV and Non-HERV sequences in non-coding regions exhibited clear differences, whereas HERV and Non-HERV sequences in coding regions showed smaller differences and clustered together with the points in non-coding regions. To evaluate the models’ ability to recognize sequence features, we utilized the last hidden layer of the fine-tuned DNA_bert_6 model for the HERV multi-classification dataset. We employed PCA and t-SNE methods to visualize the hidden layer representations, and the results showed that our fine-tuned DNA_bert_6 model successfully learned the patterns specific to HERV sequences and could distinguish HERV sequences within coding regions from HERV Non-coding regions more distinctly. PCA clearly captured differences between multiple classification labels, whereas the t-SNE method revealed differences between classification labels and also highlighted subgroups within specific label types (Fig. 4C, D; Supplementary Fig. 5). We marked the ∼ 3.92% overlapping HERV sequences in the test set with the HervD_Atlas database as reference points, and further visualization of the t-SNE results indicated that the model learned the specific patterns of the ERV1, ERV2, and ERV3 subtypes in the HERV sequences through unsupervised learning, and could differentiate them. However, it is crucial to note that it remains unclear whether the model classified subgroups based on the length differences of these three types of HERV sequences [29] (Supplementary Fig. 6). Furthermore, the last layer feature visualization of the fine-tuned human_gpt2-v1 model also demonstrated that this pre-trained model learned the latent patterns from the HERV dataset (Supplementary Fig. 7).
Fig. 4.

The RUN0 model representation learning of potential feature patterns in the HERV dataset. Visualization of (A) SparsePCA and (B) TruncatedSVD downscaled components of the original DNA sequences. Visualization of (C) PCA and (D) t-SNE downscaled components of the last hidden layer in the fine-tuned model
Phenotype-specific ALRW scores in the HERV dataset
In our investigation of the attention scores assigned to different regions within DNA sequences by the DNA_bert_6 model after fine-tuning on the HERV dataset, we extracted the attention score matrices from the final layer. Initially, we calculated the single and multiple attention score matrices for phenotype-specific sequences and computed basic statistics, including the mean and other values. The results revealed that the scores for HERV_Non-Coding sequences were lower than those for other phenotype-specific sequences (Supplementary Tables 7, 8). We then calculated the phenotype-specific ALRW score, which represents the overall average attention score of related sequences, taking into account the mean values from all heads. The ALRW scores of different phenotype-specific labels in the HERV dataset exhibited variation across the 512 tokens of the input sequences (Fig. 5A; Supplementary Fig. 8). All the phenotypic label-specific sequences showed higher ALRW scores at the beginning and end of the tokens. Compared with those of the other sequences, the ALRW scores of the non-coding HERV sequences were highest at the beginning and end, but lower in almost all other regions. In contrast, the overall distribution of ALRW scores for HERV sequences closely matched that of Non-HERV sequences within the coding regions. Notably, the ALRW scores for the 12 multi-head attention blocks on the non-coding HERV sequences within the test dataset demonstrated a distinct distribution (Supplementary Fig. 9). Furthermore, the distinct distributions of ALRW scores within the 12 multi-head attention blocks indicated that different attention heads captured various feature representations from the sequences (Supplementary Fig. 10).
Fig. 5.

In-depth analysis of ALRW scores for specific phenotypic sequences in the RUN0 model. (A) The general distribution of ALRW scores across specific phenotypic sequences; (B) the characterization of sequences within different tokens positional regions; (C) Relative enrichment rates of specific group sequences within different HERV subtypes (ERV1, ERV2, ERV3, ERVL-ma) in overlapping sequences of the HervD Atlas database
To further investigate the reasons for the differences in ALRW scores among the various phenotype-specific labels, we truncated the HERV phenotypic sequence in the corresponding regions based on token positions. Then, we calculated the GC content, unique 6-mer frequency, sequence information entropy, and potential CpG island scores of these sequences. It can be observed that the non-coding HERV sequences had higher sequence information entropy and unique 6-mer frequency, as well as lower CpG island scores, whereas both the non-coding and coding region HERV sequences presented higher GC content (Fig. 5B; Supplementary Fig. 11). Moreover, the test set of HERV sequences that could be classified by the HervD_Atlas database revealed that compared with the coding region sequences, the non-coding region sequences had a greater proportion of ERV3-type HERVs and a lower proportion of ERV2-type HERVs (Supplementary Fig. 12). The phylogenetic tree of ERV sequences indicated that ERV3-type sequences were older than ERV1 and ERV2 [29], implying that their prevalence in non-coding regions might result from evolutionary silencing, which is consistent with the finding that the ALRW score distribution is significantly different from that of other phenotype-specific sequences. To further explore this phenomenon, we calculated the relative enrichment rates of non-coding versus coding region HERV sequences in the HervD_Atlas dataset (Fig. 5C). The results indicated a marked enrichment of ERV3-type HERVL sequences in non-coding regions, characterized by the loss of viral coding capabilities. These sequences are highly conserved and ubiquitously distributed among mammals [30, 31]. Conversely, ERV2-type HERVK sequences, preserving the original provirus structure, were significantly enriched in coding regions. It was also shown that HERVK (HML-2) is not fixed in the gorilla genome but is present in the Neanderthal and Denisovan genomes [30], highlighting its recent positive selection and integration into the human genome.
Based on the characteristics observed in these sequences, we can roughly summarize the potential reasons for the overall low ALRW of the non-coding region HERV sequences as follows: (1) Enhanced complexity and diversity: The high sequence information entropy and unique 6-mer frequency of non-coding HERV sequences indicate their complexity and diversity, which makes it challenging for the model to capture useful features, resulting in lower scores; (2) Evolutionary silencing and balance: Non-coding HERV sequences may have been subjected to more extensive silencing events during evolution, leading to differences in their activity, expression, or functionality compared with other sequences, such as low CpG island scores, loss of HERVL viral coding sequences, etc.; (3) A bias towards learning prominent sequence features: The non-coding HERV region contains more solitary LRT sequences, and the model tends to focus on assimilating these prominent features, thereby reducing the ability to capture complex features in other HERV sequences.
Motif analysis of HERV dataset with high ALRW scores
Enrichment and pathogenicity analysis of motifs in high ALRW score regions
The above analysis demonstrated that pre-trained genomic models can capture distinctive internal features of HERV sequences, suggesting their potential associations with functional elements such as regulatory motifs. Further exploration using four independent test sets revealed that sequences with high ALRW scores are effective in identifying motifs that exhibit both specific and shared features across various phenotype-specific labels. Our analysis generally found that these non-overlapping genomic sequences identified a limited number of HERV-specific motifs in both coding and non-coding regions, while unique motifs were also discernible in these regions (Supplementary Fig. 13). To assess motif enrichment specificity in HERV sequences, a hypergeometric test was employed to statistically analyze motifs in both coding and non-coding regions across all test sets. The significant enrichment of 42 overlapping HERV sequence motifs identified in the four experiments indicates a similarity in regulatory elements within different genomic regions containing HERV sequences (Fig. 6A; Supplementary Table 9). Additionally, using high ALRW scores, we identified these overlapping motifs in phenotype-specific sequences, most of which could also be identified via the full-length test set sequences, where analyzing motifs with the complete sequences exhibited similar trends (Supplementary Figs. 14, 15 A; Supplementary Table 10). Furthermore, high ALRW scores in phenotype-specific sequences identified certain phenotype-specific sequence patterns learned by the model (Supplementary Figs. 14, 15). The results revealed a significant enrichment of HERV-specific motifs when identifying motifs through high ALRW scores compared to using full-length sequences. This indicates that high ALRW scores can effectively capture and filter unique patterns in HERV sequences.
Fig. 6.

Identification analysis of motifs within phenotype-specific high ALRW scores sequences. (A) The overlap of motifs was determined for four non-overlapping HERV dataset test sets (RUN0-Related, Split1-Related, Split2-Related, Split3-Related); (B) HERV phenotype sequence-specific motif enrichment analysis demonstrated, where the x-axis of the analysis represents the overall rate of HERV type motifs; (C) The multiple motifs-enriched AKR1E2 gene screened by two strategies in RUN0-related test set. ominoidae-specific HERV sequences chr10_ERV1_01040 are highlighted in orange (Hominoidae: Human, Chimp, Bonobo, Gorilla, Gibbon)
Using the motifs identified in the above four experiments and their corresponding non-redundant genomic regions, we further explored the potential regulatory functions of these HERV-specific enriched motifs. Based on the HERV sequence encoding types, we found that the 42 overlapping specific-enriched HERV motifs identified across the four experiments are associated with DNA-binding and gene regulation. These motifs include factors such as C2H2 zinc finger factors, tryptophan cluster factors, and AP2/EREBP, which are enriched in both coding and non-coding regions [32–35]. Conversely, motifs such as nuclear receptors with C4 zinc fingers were predominantly found in coding regions, whereas transcription-related motifs, such as Fork head/winged helix factors and TATA-binding proteins, were abundant in non-coding regions [36–40] (Fig. 6B; Supplementary Fig. 16; Supplementary Table 9). In summary, specific enriched motifs present in HERV sequences are implicated in gene expression regulation, with some unique motifs involved in recognizing nuclear receptors within coding regions. Additionally, motifs identified from the full-length sequences also demonstrated enrichment, with those like Rbpjl, vertebrates, Rel homology region (RHR) factors playing roles in immune regulation, development, and inflammation regulation processes in organisms [41–43] (Supplementary Fig. 17; Supplementary Table 10). Furthermore, combining gene annotation information, we analyzed the genomic regions of HERV-specific enriched motifs and found that the major genes overlapping within these intervals were located primarily in the protein-coding and lncRNA regions (Supplementary Table 11). We also observed specific enrichment of motifs in genes related to neurodevelopment and synaptic function, cellular processes and cancer, cell adhesion and intercellular communication, and signal transduction. These regions involve functional sequence elements such as enhancers, CTCF binding sites, and open chromatin regions (Supplementary Fig. 18). Functional enrichment analysis of all protein-coding genes and disease relevance analysis revealed that genes associated with the specific enriched motifs were involved in processes such as neural activity, cellular morphology and transport, metabolic regulation, and GPCR signaling. Moreover, these genes were closely associated with spinal health, neural development and intelligence, and exhibited specific enrichment in DRG neuronal cells (Supplementary Fig. 19).
To further investigate the induction of disease by specific enriched motifs, we integrated information from the HervD_Atlas database with the pathogenicity scores predicted by AlphaMissense [25] and PrimateAI [24]. By utilizing the HERV-specific non-redundant motif sequence intervals identified by high ALRW score sequences, a cumulative length of 47,759 bp (∼ 15.31%) mainly from the ERV1 family overlapped with sequences in the HervD_Atlas, which are involved in diseases such as cancer and the nervous system. Compared with the motifs identified based on the full-length sequences, the motifs identified on the basis of high ALRW scores lean toward high-frequency HERV sequences, resulting in the loss of low-frequency sequence information but also the identification of new motifs. Employing both strategies, we successfully identified 8 shared motifs within the AKR1E2 gene. Within this gene, there is a Hominoidae-specific HERV sequence chr10_ERV1_01040 exhibited low expressed in 8 cancerous tissues but demonstrated high pathogenicity scores at specific sites (Supplementary Tables 12–14). The motif sequences AT3G46070, SP8, and KLF17 within this interval play important roles in processes such as cell proliferation and metabolism, brain development [44], and cancer invasion [45] (Fig. 6C; Supplementary Fig. 20). Furthermore, motifs like Eor-1, identified between genes in the chr6_ERV2_01565 sequence, which is significantly upregulated in liver cancer (Supplementary Table 12). Regarding HERV-specific motifs identified through high ALRW scores, it is worth mentioning that the Hominoidae-specific HERV sequence chr9_ERV2_01861 contains the CTFC motifs identified by high ALRW scores within the PRUNE2 gene. This particular HERV sequence is expressed at low levels in 4 cancerous tissues and encompasses a CRE regulatory element (Supplementary Fig. 21A; Supplementary Tables 12, 14). Lastly, specific motifs identified through full-length sequence analysis, motifs like ZNF135 within PPIL1, which are associated with protein folding and activation processes, closely correlation with a wide range of biological functions and diseases [46, 47] (Supplementary Fig. 21B; Supplementary Tables 13, 14).
Species conservation of HERV-specific enriched motif sequences
Using fine-tuned genomic models to obtain the ALRW of DNA sequences, our study pinpointed motifs uniquely enriched within human HERV. These HERV-specific motif sequences are involved in regulating essential biological activities and are closely related to nervous system diseases and tumors. To further evaluate the characteristics of HERV-specific motifs, we investigated their diversity among human populations and their conservation across primate species (Fig. 7; Supplementary Fig. 22; Supplementary Table 15). By integrating published human pan-genomic data, we analyzed the sequence polymorphism (alignment depth) of HERV-specific motifs within their corresponding intervals and visualized them based on overlapping gene types. This analysis indicated that lncRNA and protein-coding genes exhibit higher polymorphism (Fig. 7A). We also identified highly conserved motif sequences in the CCDC200 gene within the human population, although significant differentiation was observed in primates. Notably, the CCDC family proteins are believed to be involved in various physiological and pathological processes, including gametogenesis, embryonic development, hematopoiesis, angiogenesis, ciliogenesis, and cancer [48, 49] (Fig. 7B; Supplementary Fig. 23). Additionally, regions with highly differentiated motifs in the human population or genes containing multiple repetitive segments within the genome are associated with immune function and inflammatory responses, signal transduction, cell communication, the nervous system, and metabolic processes. These findings suggest their role in the transcriptional regulation of divergent traits within primates and their co-evolution with environmental pathogens [30, 50, 51] (Fig. 7). Moreover, motifs that are conserved among primates but polymorphic in humans may be involved in the development of human organs and immune system adaptability. By comparing brain organ samples from humans and great apes, it was found that the epithelial-mesenchymal transition regulator ZEB2 promotes the transformation of the neural epithelium, ultimately leading to the expansion of the human brain [52]. The membrane receptor ROR1, which is crucial for embryonic development and is overexpressed in multiple cancers, has been shown to be a safe and practical target for CAR-T cell immunotherapy in clinical trials involving non-human primates [53, 54]. Additionally, our annotation of HERV-related motifs within non-gene regions revealed that ∼ 47.6% were enhancers, over 10% were genes upstream /downstream and CTCF binding sites, and over 7% were open chromatin regions, highlighting their potential biological significance (Supplementary Fig. 24). Further exploration revealed that only motifs within 1 kb of genes presented predictive conservation scores, indicating higher species-specific variation in non-gene sequences, possibly due to early viral retrotransposon fixation (Supplementary Table 16). Most motifs enriched in gene upstream/downstream regions were not conserved in primates but showed high diversity in human populations, with some showing potential population-level conservation (requiring broader validation). These motifs need further investigation to understand their roles in regulatory mechanisms.
Fig. 7.

Conservation of species within phenotype-specific RUN0-related enriched motif regions. (A) Pan-genomic representation of population polymorphisms within phenotype-specific enriched motif regions; (B) The conservation of these motifs is investigated in primates
Discussion
In the post-genomic era, the functional analysis of DNA sequences has paramount importance. We constructed multiple genotype-phenotype (individual and molecular phenotypes) dataset by integrating accumulated data and knowledge. After fine-tuning the genomic model with DNA_bert_6 and human_gpt2-v1, we achieved balanced binary and imbalanced multiclass phenotypic classifications with exceptional efficacy. Notably, the fine-tuning of the HERV dataset not only demonstrated an enhanced capability in identifying and delineating various informational types within DNA sequences, but also pinpointed specific motifs associated with neurological disorders and cancers regions with high ALRW scores. Further analysis of these conserved motifs shed light on the adaptive responses of species to their environment and co-evolution with pathogens. These sequence-specific motifs could revolutionize nucleic acid vaccine development and targeted therapeutics for genomic diseases, such as liver cancer intervention vaccines and drug development based on the expanded sequence of the Eor-1 motif within HERVK.
The main challenges of DNA sequence analysis include the complex sequence features and model input length limitations. DNA sequences usually encapsulate vast amounts of information and complex structures, such as repetitive sequences, etc., making high-quality sequence resolution of these regions challenging and making it difficult to extract useful knowledge via traditional methods. Moreover, variability in loss function results due to different sequence complexities may necessitate sequence-type-specific model pre-training. In addition, the population biases and sequence frequency may generate learning bias in the model and affect downstream tasks. Currently, the commonly used pre-trained BERT and GPT models have a maximum input token limitation, possibly resulting in loss of spatial information of the genome and important regulatory elements, such as the long-distance enhancers. While DNA controls complex life activities, research has focuses predominantly on just 3% of protein-coding sequences in the genome. The deployment of pre-trained genomic models can improve the understanding of the DNA genetic blueprint, which is vital for deciphering gene control of biological traits and disease.
Future research will focus on developing incremental pre-training models for the human pangenome, incorporating model knowledge distillation, and integrating knowledge graphs, etc. We will also explore innovative frameworks capable of handling extended input tokens, such as HyenaDNA and LongNet, and further evaluate their genomic representation capabilities [10, 55]. Ultimately, it is expected that our pangenome incremental pre-trained model can surpass the capabilities of the hg38 reference, potentially transforming the computational biology landscape. This advancement in genome-level representation learning will deepen our understanding of life and potentially impact related fields, including NLP technologies. Furthermore, the findings and data from this research will contribute to personalized therapy strategies, including vaccine and drug development targeting HERVK sequences [56]. Additionally, using pre-trained genomic models to explore the high proportion of HERV sequences on chromosomes X and Y, this will prompt us to consider their genetic patterns, chromosomal evolution, and embryonic development.
Conclusion
This study employs a pre-trained genome model to identify and interpret genetic signals associated with specific phenotypes. The findings can be categorized into three main aspects:1) Genotype-Phenotype Dataset: We utilized various genotype-phenotype datasets to assess the effectiveness of fine-tuned large models in balanced binary classification and imbalanced multi-class sequence challenges. Using the HERV dataset, we evaluated the variation in model classification metrics across different sequence length distributions, exploring the correlation between data distribution patterns and model performance. These datasets play a critical role in assessing the efficacy of those models and they highlight the importance of model selection based on data distribution, requirements, and costs. 2) Representation Learning in Pre-trained Genomic Models: The fine-tuned HERV dataset reveals that hidden layer features enable the model to recognize phenotypic information in sequences and reduce noise. To investigate how the model isolates phenotypic label-specific signals, we calculated the ALRW for phenotypic labels using average attention matrices. The distribution of ALRW scores aligns with the fundamental characteristics of coding and non-coding areas in HERV sequences and their evolutionary subtypes, validating the utility of pre-trained models in the deep analysis of biological sequences. 3) Novel attempts to integrating Pre-trained Genomic Models with Classical Omics Analysis: By selecting sequences with high ALRW scores specific to phenotypes for motif enrichment analysis, we identified HERV-specific motifs that are implicated in neurological diseases, tumors, and other biological processes. Therefore, they have potential applications in vaccine and targeted drug discovery. Furthermore, the polymorphism of these motifs in human populations and their conservation in primates provide insights into primate adaptation to environmental pressures and integration of pathogens into the host genome. This insight aids in selecting motifs for further research and development in vaccines and drugs, using non-human primates, exemplified by HERVK sequences.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We acknowledge the support provided by the project of Shanghai’s Double First-Class University Construction, the Development of High-Level Local Universities: Intelligent Medicine Emerging Interdisciplinary Cultivation Project, and Medical Science Data Center of Fudan University.
Abbreviations
- HERV
Human Endogenous Retrovirus
- ALRW
Average Local Representation Weight
- NLP
Natural Language Processing
- CNN
Convolutional Neural Network
- BERT
Bidirectional Encoder Representations from Transformers
- GPT
Generative Pre-trained Transformer
- BPE
Byte Pair Encoding
- GWAS
Genome-Wide Association Studies
Author contributions
Conceptualization: DD, FZ, LL; Formal analysis: DD; Funding Acquisition: LL; Investigation: DD; Methodology: DD; Project administration: FZ, LL; Resources: LL; Supervision: FZ, LL; Visualization: DD; Writing – original draft: DD; Writing – review & editing: DD, FZ, LL.
Funding
This work was supported by the Peak Disciplines (Type IV) of Institutions of Higher Learning in Shanghai.
Data availability
We utilized two NVIDIA A100 GPUs and a large-memory multi-core CPU system for all analyses. The code for this project can be accessed at https://github.com/GeorgeBGM/Genome_Fine-Tuning, and the relevant key result files are accessible for download via https://zenodo.org/records/10342199.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Generative AI in scientific writing
AI-assisted technologies have been employed to increase readability and improve language proficiency. Nevertheless, the final results were exclusively produced by the authors, who meticulously edited the language to conform to domain terminology. Consequently, we are wholly responsible and accountable for the content of this study.
Competing interests
Not applicable.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Fan Zhong, Email: zonefan@163.com.
Lei Liu, Email: liulei_sibs@163.com.
References
- 1.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–51. 10.1126/science.1058040 [DOI] [PubMed] [Google Scholar]
- 2.Manolio TA, Chisholm RL, Ozenberger B, Roden DM, Williams MS, Wilson R, Bick D, Bottinger EP, Brilliant MH, Eng C, et al. Implementing genomic medicine in the clinic: the future is here. Genet Med. 2013;15:258–67. 10.1038/gim.2012.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hatje K, Muhlhausen S, Simm D, Kollmar M. The protein-coding Human Genome: Annotating High-hanging fruits. BioEssays. 2019;41:e1900066. 10.1002/bies.201900066 [DOI] [PubMed] [Google Scholar]
- 4.Jakobsson J, Vincendeau M. SnapShot: human endogenous retroviruses. Cell. 2022;185:400–400. e401. 10.1016/j.cell.2021.12.028 [DOI] [PubMed] [Google Scholar]
- 5.Malte A, Ratadiya P. Evolution of transfer learning in natural language processing. pp. arXiv:1910.07370; 2019:arXiv:1910.07370.
- 6.Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez AN, Kaiser L, Polosukhin I. Attention Is All You Need. pp. arXiv:1706.03762; 2017:arXiv:1706.03762.
- 7.Onat Topal M, Bas A, van Heerden I. Exploring Transformers in Natural Language Generation: GPT, BERT, and XLNet. pp. arXiv:2102.08036; 2021:arXiv:2102.08036.
- 8.Zhang S, Fan R, Liu Y, Chen S, Liu Q, Zeng W. Applications of transformer-based language models in bioinformatics: a survey. Bioinform Adv. 2023;3:vbad001. 10.1093/bioadv/vbad001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang B, Xie Q, Pei J, Chen Z, Tiwari P, Li Z, fu J. Pre-trained Language models in Biomedical Domain: a systematic survey. pp arXiv:2110 05006. 2021. arXiv:2110.05006.
- 10.Nguyen E, Poli M, Faizi M, Thomas A, Birch-Sykes C, Wornow M, Patel A, Rabideau C, Massaroli S, Bengio Y et al. HyenaDNA: long-range genomic sequence modeling at single Nucleotide Resolution. pp. arXiv:2306.15794; 2023:arXiv:2306.15794.
- 11.Ji Y, Zhou Z, Liu H, Davuluri RV. DNABERT: pre-trained bidirectional encoder representations from transformers model for DNA-language in genome. Bioinformatics. 2021;37:2112–20. 10.1093/bioinformatics/btab083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang D, Zhang W, Zhao Y, Zhang J, He B, Qin C, Yao J. DNAGPT: a generalized pre-trained Tool for versatile DNA sequence analysis tasks. pp. arXiv:2307.05628; 2023:arXiv:2307.05628.
- 13.Gresova K, Martinek V, Cechak D, Simecek P, Alexiou P. Genomic benchmarks: a collection of datasets for genomic sequence classification. BMC Genom Data. 2023;24:25. 10.1186/s12863-023-01123-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinek V, Cechak D, Gresova K, Alexiou P, Simecek P. Fine-tuning transformers for genomic tasks. bioRxiv. 2022;2022(2002):2007–479412. [Google Scholar]
- 15.Dalla-Torre H, Gonzalez L, Mendoza-Revilla J, Carranza NL, Grzywaczewski AH, Oteri F, Dallago C, Trop E, Almeida BPd, Sirelkhatim H et al. The Nucleotide Transformer: building and evaluating Robust Foundation models for Human Genomics. bioRxiv 2023:2023.2001.2011.523679.
- 16.Liao WW, Asri M, Ebler J, Doerr D, Haukness M, Hickey G, Lu S, Lucas JK, Monlong J, Abel HJ, et al. A draft human pangenome reference. Nature. 2023;617:312–24. 10.1038/s41586-023-05896-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siren J, Monlong J, Chang X, Novak AM, Eizenga JM, Markello C, Sibbesen JA, Hickey G, Chang PC, Carroll A, et al. Pangenomics enables genotyping of known structural variants in 5202 diverse genomes. Science. 2021;374:abg8871. 10.1126/science.abg8871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hauser M, Steinegger M, Soding J. MMseqs software suite for fast and deep clustering and searching of large protein sequence sets. Bioinformatics. 2016;32:1323–30. 10.1093/bioinformatics/btw006 [DOI] [PubMed] [Google Scholar]
- 19.Shen W, Le S, Li Y, Hu F. SeqKit: a cross-platform and Ultrafast Toolkit for FASTA/Q file manipulation. PLoS ONE. 2016;11:e0163962. 10.1371/journal.pone.0163962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li C, Qian Q, Yan C, Lu M, Li L, Li P, Fan Z, Lei W, Shang K, Wang P et al. HervD Atlas: a curated knowledgebase of associations between human endogenous retroviruses and diseases. Nucleic Acids Res 2023. [DOI] [PMC free article] [PubMed]
- 21.Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27:1017–8. 10.1093/bioinformatics/btr064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Lemma RB, Turchi L, Blanc-Mathieu R, Lucas J, Boddie P, Khan A, Manosalva Perez N, et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022;50:D165–73. 10.1093/nar/gkab1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10:1523. 10.1038/s41467-019-09234-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao H, Hamp T, Ede J, Schraiber JG, McRae J, Singer-Berk M, Yang Y, Dietrich ASD, Fiziev PP, Kuderna LFK, et al. The landscape of tolerated genetic variation in humans and primates. Science. 2023;380:eabn8153. 10.1126/science.abn8197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng J, Novati G, Pan J, Bycroft C, Zemgulyte A, Applebaum T, Pritzel A, Wong LH, Zielinski M, Sargeant T, et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science. 2023;381:eadg7492. 10.1126/science.adg7492 [DOI] [PubMed] [Google Scholar]
- 26.Guarracino A, Heumos S, Nahnsen S, Prins P, Garrison E. ODGI: understanding pangenome graphs. Bioinformatics. 2022;38:3319–26. 10.1093/bioinformatics/btac308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fan K, Moore JE, Zhang XO, Weng Z. Genetic and epigenetic features of promoters with ubiquitous chromatin accessibility support ubiquitous transcription of cell-essential genes. Nucleic Acids Res. 2021;49:5705–25. 10.1093/nar/gkab345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiao Y, Ren C, Huang S, Yuan J, Liu X, Fan J, Lin J, Wu S, Chen Q, Bo X, et al. High-resolution annotation of the mouse preimplantation embryo transcriptome using long-read sequencing. Nat Commun. 2020;11:2653. 10.1038/s41467-020-16444-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jern P, Sperber GO, Blomberg J. Use of endogenous retroviral sequences (ERVs) and structural markers for retroviral phylogenetic inference and taxonomy. Retrovirology. 2005;2:50. 10.1186/1742-4690-2-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson WE. Origins and evolutionary consequences of ancient endogenous retroviruses. Nat Rev Microbiol. 2019;17:355–70. 10.1038/s41579-019-0189-2 [DOI] [PubMed] [Google Scholar]
- 31.Benit L, Lallemand JB, Casella JF, Philippe H, Heidmann T. ERV-L elements: a family of endogenous retrovirus-like elements active throughout the evolution of mammals. J Virol. 1999;73:3301–8. 10.1128/JVI.73.4.3301-3308.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Najafabadi HS, Mnaimneh S, Schmitges FW, Garton M, Lam KN, Yang A, Albu M, Weirauch MT, Radovani E, Kim PM, et al. C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat Biotechnol. 2015;33:555–62. 10.1038/nbt.3128 [DOI] [PubMed] [Google Scholar]
- 33.Kanei-Ishii C, Sarai A, Sawazaki T, Nakagoshi H, He DN, Ogata K, Nishimura Y, Ishii S. The tryptophan cluster: a hypothetical structure of the DNA-binding domain of the myb protooncogene product. J Biol Chem. 1990;265:19990–5. 10.1016/S0021-9258(17)45472-X [DOI] [PubMed] [Google Scholar]
- 34.Feng K, Hou XL, Xing GM, Liu JX, Duan AQ, Xu ZS, Li MY, Zhuang J, Xiong AS. Advances in AP2/ERF super-family transcription factors in plant. Crit Rev Biotechnol. 2020;40:750–76. 10.1080/07388551.2020.1768509 [DOI] [PubMed] [Google Scholar]
- 35.Li P, Chai Z, Lin P, Huang C, Huang G, Xu L, Deng Z, Zhang M, Zhang Y, Zhao X. Genome-wide identification and expression analysis of AP2/ERF transcription factors in sugarcane (Saccharum spontaneum L). BMC Genomics. 2020;21:685. 10.1186/s12864-020-07076-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fan X, Shi H, Adelman K, Lis JT. Probing TBP interactions in transcription initiation and reinitiation with RNA aptamers that act in distinct modes. Proc Natl Acad Sci U S A. 2004;101:6934–9. 10.1073/pnas.0401523101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akhtar W, Veenstra GJ. TBP-related factors: a paradigm of diversity in transcription initiation. Cell Biosci. 2011;1:23. 10.1186/2045-3701-1-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maity SN, de Crombrugghe B. Biochemical analysis of the B subunit of the heteromeric CCAAT-binding factor. A DNA-binding domain and a subunit interaction domain are specified by two separate segments. J Biol Chem. 1992;267:8286–92. 10.1016/S0021-9258(18)42440-4 [DOI] [PubMed] [Google Scholar]
- 39.Kim JE, Nam H, Park J, Choi GJ, Lee YW, Son H. Characterization of the CCAAT-binding transcription factor complex in the plant pathogenic fungus fusarium graminearum. Sci Rep. 2020;10:4898. 10.1038/s41598-020-61885-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herman L, Todeschini AL, Veitia RA. Forkhead Transcription Factors in Health and Disease. Trends Genet. 2021;37:460–75. 10.1016/j.tig.2020.11.003 [DOI] [PubMed] [Google Scholar]
- 41.Leger MM, Ros-Rocher N, Najle SR, Ruiz-Trillo I. Rel/NF-kappaB Transcription Factors Emerged at the Onset of Opisthokonts. Genome Biol Evol 2022, 14. [DOI] [PMC free article] [PubMed]
- 42.Duan X, Lv M, Liu A, Pang Y, Li Q, Su P, Gou M. Identification and evolution of transcription factors RHR gene family (NFAT and RBPJ) involving lamprey (Lethenteron reissneri) innate immunity. Mol Immunol. 2021;138:38–47. 10.1016/j.molimm.2021.07.017 [DOI] [PubMed] [Google Scholar]
- 43.Yuan Z, VanderWielen BD, Giaimo BD, Pan L, Collins CE, Turkiewicz A, Hein K, Oswald F, Borggrefe T, Kovall RA. Structural and functional studies of the RBPJ-SHARP Complex reveal a conserved corepressor binding site. Cell Rep. 2019;26:845–e854846. 10.1016/j.celrep.2018.12.097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gaborieau E, Hurtado-Chong A, Fernandez M, Azim K, Raineteau O. A dual role for the transcription factor Sp8 in postnatal neurogenesis. Sci Rep. 2018;8:14560. 10.1038/s41598-018-32134-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu FY, Deng YL, Li Y, Zeng D, Zhou ZZ, Tian DA, Liu M. Down-regulated KLF17 expression is associated with tumor invasion and poor prognosis in hepatocellular carcinoma. Med Oncol. 2013;30:425. 10.1007/s12032-012-0425-3 [DOI] [PubMed] [Google Scholar]
- 46.Chai G, Webb A, Li C, Antaki D, Lee S, Breuss MW, Lang N, Stanley V, Anzenberg P, Yang X, et al. Mutations in spliceosomal genes PPIL1 and PRP17 cause neurodegenerative Pontocerebellar Hypoplasia with Microcephaly. Neuron. 2021;109:241–e256249. 10.1016/j.neuron.2020.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee A, Park HJ, Jo SH, Jung H, Kim HS, Lee HJ, Kim YS, Jung C, Cho HS. The spliceophilin CYP18-2 is mainly involved in the splicing of retained introns under heat stress in Arabidopsis. J Integr Plant Biol. 2023;65:1113–33. 10.1111/jipb.13450 [DOI] [PubMed] [Google Scholar]
- 48.Liu Z, Yan W, Liu S, Liu Z, Xu P, Fang W. Regulatory network and targeted interventions for CCDC family in tumor pathogenesis. Cancer Lett. 2023;565:216225. 10.1016/j.canlet.2023.216225 [DOI] [PubMed] [Google Scholar]
- 49.Priyanka PP, Yenugu S. Coiled-Coil Domain-Containing (CCDC) proteins: functional roles in General and Male Reproductive Physiology. Reprod Sci. 2021;28:2725–34. 10.1007/s43032-021-00595-2 [DOI] [PubMed] [Google Scholar]
- 50.Shao Y, Zhou L, Li F, Zhao L, Zhang BL, Shao F, Chen JW, Chen CY, Bi X, Zhuang XL, et al. Phylogenomic analyses provide insights into primate evolution. Science. 2023;380:913–24. 10.1126/science.abn6919 [DOI] [PubMed] [Google Scholar]
- 51.Ning Z, Tan X, Yuan Y, Huang K, Pan Y, Tian L, Lu Y, Wang X, Qi R, Lu D, et al. Expression profiles of east-west highly differentiated genes in Uyghur genomes. Natl Sci Rev. 2023;10:nwad077. 10.1093/nsr/nwad077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benito-Kwiecinski S, Giandomenico SL, Sutcliffe M, Riis ES, Freire-Pritchett P, Kelava I, Wunderlich S, Martin U, Wray GA, McDole K, Lancaster MA. An early cell shape transition drives evolutionary expansion of the human forebrain. Cell 2021, 184:2084–2102 e2019. [DOI] [PMC free article] [PubMed]
- 53.Osorio-Rodriguez DA, Camacho BA, Ramirez-Segura C. Anti-ROR1 CAR-T cells: Architecture and performance. Front Med (Lausanne). 2023;10:1121020. 10.3389/fmed.2023.1121020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berger C, Sommermeyer D, Hudecek M, Berger M, Balakrishnan A, Paszkiewicz PJ, Kosasih PL, Rader C, Riddell SR. Safety of targeting ROR1 in primates with chimeric antigen receptor-modified T cells. Cancer Immunol Res. 2015;3:206–16. 10.1158/2326-6066.CIR-14-0163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ding J, Ma S, Dong L, Zhang X, Huang S, Wang W, Zheng N, Wei F. LongNet: Scaling Transformers to 1,000,000,000 Tokens. pp. arXiv:2307.02486; 2023:arXiv:2307.02486.
- 56.Wang J, Lu X, Zhang W, Liu GH. Endogenous retroviruses in development and health. Trends Microbiol 2023. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We utilized two NVIDIA A100 GPUs and a large-memory multi-core CPU system for all analyses. The code for this project can be accessed at https://github.com/GeorgeBGM/Genome_Fine-Tuning, and the relevant key result files are accessible for download via https://zenodo.org/records/10342199.