

Abstract
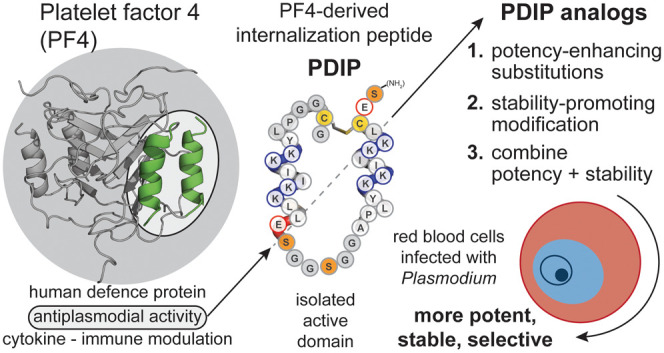
The control of malaria, a disease caused by Plasmodium parasites that kills over half a million people every year, is threatened by the continual emergence and spread of drug resistance. Therefore, new molecules with different mechanisms of action are needed in the antimalarial drug development pipeline. Peptides developed from host defense molecules are gaining traction as anti-infectives due to theood of inducing drug resistance. Human platelet factor 4 (PF4) has intrinsic activity against P. falciparum, and a macrocyclic helix–loop–helix peptide derived from its active domain recapitulates this activity. In this study, we used a stepwise approach to optimize first-generation PF4-derived internalization peptides (PDIPs) by producing analogues with substitutions to charged and hydrophobic amino acid residues or with modifications to terminal residues including backbone cyclization. We evaluated the in vitro activity of PDIP analogues against P. falciparum compared to their overall helical structure, resistance to breakdown by serum proteases, selective binding to negatively charged membranes, and hemolytic activity. Next, we combined antiplasmodial potency-enhancing substitutions that retained favorable membrane and cell-selective properties onto the most stable scaffold to produce a backbone cyclic PDIP analogue with four-fold improved activity against P. falciparum compared to first-generation peptides. These studies demonstrate the ability to modify PDIP to select for and combine desirable properties and further validate the suitability of this unique peptide scaffold for developing a new molecule class that is distinct from existing antimalarial drugs.
Keywords: malaria, Plasmodium, host defense peptide, targeted cell-penetration, rational design, drug development
Malaria is a serious global health burden, killing over 500,000 people every year. Efforts to control the disease saw a decline in the number of deaths between 2000 and 2019, but recent data suggest that the downward trend has stalled, with 608,000 deaths reported in 2022.1 Factors contributing to this change include the emergence and spread of drug resistance in Plasmodium falciparum parasites, insecticide resistance in mosquito vectors that spread the disease, environmental and economic factors, and disruptions to health services due to the COVID-19 pandemic.2 Artemisinin combination therapies (ACT) have been instrumental in controlling the disease burden since their adoption as the first-line treatment in the early 2000s,2,3 but this control is under threat with ACT treatment failure reported in South-East Asia and the Western Pacific (incidence summarized by region in the World Malaria report 2023).1 Artemisinin-resistant P. falciparum has also been reported in Africa,1,4,5 which is especially concerning as this region accounts for 94% of malaria cases globally.1
Field surveillance measures that identify resistance to artemisinin derivatives and partner drugs to inform the adoption of different drug combinations are critical in the ongoing battle against drug resistance.6−8 Rotational use of drug components in combination therapies is a promising strategy for managing resistance,3,9 and antimalarial drug partners with different mechanisms of action and resistance profiles may serve to prolong the efficacy of individual drug components and circumvent combination therapy failure.3,9 Even drugs with modest antimalarial efficacy (e.g., antibacterial drugs)10,11 can be considered if they are refractory to resistance development by P. falciparum.3 Drug discovery efforts have delivered several new small molecule drugs through to validation in clinical trials or in vivo studies (reviewed in ref (2)). However, Plasmodium parasites can rapidly acquire resistance to small-molecule drugs. For example, a single amino acid mutation (G358S) in the target P. falciparum ATP4 sodium pump produced clinically relevant resistance to cipargamin, a promising new antimalarial drug candidate.12,13 To address this rapid rate of acquired resistance, expansion of the discovery efforts to include other classes of bioactive molecules is urgently needed.
Host defense peptides (HDPs), often referred to as antimicrobial peptides (AMPs), are produced by a wide range of organisms.14,15 HDPs are promising candidates for developing targeted therapies due to their several advantageous properties. They exhibit membrane selectivity that allows distinction between the negatively charged outer membrane leaflets of bacteria,16 cancer cells,17,18 and red blood cells (RBCs) infected with P. falciparum,19,20 compared to the neutral surface of healthy mammalian cells. Rather than the discrete single-molecule targets typified by most current drugs, the inherently larger size and interacting surfaces of peptides provide improved target cell selectivity21 and reduce the likelihood of inducing drug resistance.22,23 In addition to these properties, some HDPs can cross cell membranes to access intracellular targets.24−26
The host defense protein platelet factor 4 (PF4) has intrinsic activity against blood stage P. falciparum(27) that can be attributed to short (14 amino acid) AMP-like regions at the C-terminus of each of four subunits.28,29 Inspired by the paired head-to-tail presentation of these regions in PF4, we engineered a macrocyclic helix–loop–helix peptide (cPF4PD), joined at one end by a flexible linker, and a disulfide bond between introduced Cys residues proximal to the N- and C-termini at the other.30 cPF4PD kills P. falciparumin vitro with similar low micromolar potency to PF4. The mechanism of action involves selective binding to and lysis of membranes that are rich in negatively charged phospholipid headgroups.30 This selectivity drives entry into infected RBCs, which have increased surface presentation of negatively charged phosphatidylserine (PS) relative to uninfected cells,19,20 and disruption of the parasite digestive vacuole membrane (Figure 1A), which contains negatively charged phosphatidylinositol 3-phosphate.31 Notably, cPF4PD does not enter or damage uninfected RBCs at concentrations that inhibit P. falciparum growth30 (Figure 1B).
Figure 1.

First-generation PF4-derived internalization peptide (PDIP) analogue (cPF4PD, 1) targets and selectively enters RBCs infected with P. falciparum. (A) Normal uninfected RBCs (uRBC) have an asymmetric distribution of lipids, maintaining negatively charged phosphatidylserine (PS) headgroups on the inner leaflet of the cell plasma membrane, whereas PS is also present on the outer leaflet of infected RBCs (iRBC). Percentage RBC phospholipid data from ref (32). PC, phosphatidylcholine; SM, sphingomyelin; PE, phosphatidylethanolamine. (B) PDIP analogue (1) labeled with AlexaFluor-488 (A488) has rapid (<1 h) selective entry into iRBC and accumulates inside the parasite. Scale bars are 5 μm.
In this study, we aimed to improve the antiplasmodial potency of the first-generation cPF4PD peptide while maintaining or improving beneficial features including a stable structure that resists proteolysis, and selectivity for infected compared to uninfected RBCs. To achieve these aims, we designed and chemically synthesized two sets of peptide analogues, herein called PF4-derived internalization peptides (PDIPs), to separately identify antiplasmodial potency-enhancing substitutions and scaffold modifications that favor peptide stability. In line with the need for low-cost production of new malaria medicines,2 we maintained proteinogenic amino acids in the sequence to allow future transfer to biosynthetic production. We assessed each PDIP analogue according to antiplasmodial potency—in vitro activity against P. falciparum; activity-hemolysis index—antiplasmodial activity relative to lysis of uninfected RBCs; membrane selectivity—binding affinity for negatively charged compared to neutral phospholipid membranes; structure—helicity in aqueous solution; and stability—percentage of recovered intact peptide following incubation with human serum. We combined the best potency-enhancing substitutions with the most stable and selective scaffold to produce a backbone cyclic PDIP analogue with four-fold enhanced in vitro activity against P. falciparum and a wider window between antiplasmodial activity and hemolysis of uninfected RBCs compared to first-generation analogues. These improvements are an important step toward developing peptide-based drugs to expand the repertoire of drug classes in the antimalarial drug development pipeline.
Results
Two sets of PDIP analogues were designed, with substitutions to charged and hydrophobic residues (set 1) and modification of the C-terminus, including backbone cyclization (set 2). The best potency-enhancing substitutions identified from set 1 were then incorporated onto the most stable scaffold from set 2 to produce analogues with combined optimal modifications. Peptides were produced using Fmoc solid phase synthesis, employing intramolecular native chemical ligation33,34 for backbone cyclic peptides. Amino acid sequences for the base scaffolds (1–3), together with cartoon representations of the structure of the parent and modified peptides, are shown in Figure 2. Sequences and mass data for the base scaffolds and analogues (4–27) are shown in Table S1, with purity demonstrated in Figures S1 and S2. The % helicity (structure), stability in the presence of serum proteases, in vitro activity against P. falciparum, hemolysis, and activity-hemolysis index between antiplasmodial activity and RBC lysis are summarized in Table 1.
Figure 2.

Cartoon Representation of the Structures of PDIP Parent Peptides and Analogues.
Table 1. In Vitro Antiplasmodial Activity and Characteristics of PDIP Analogues.
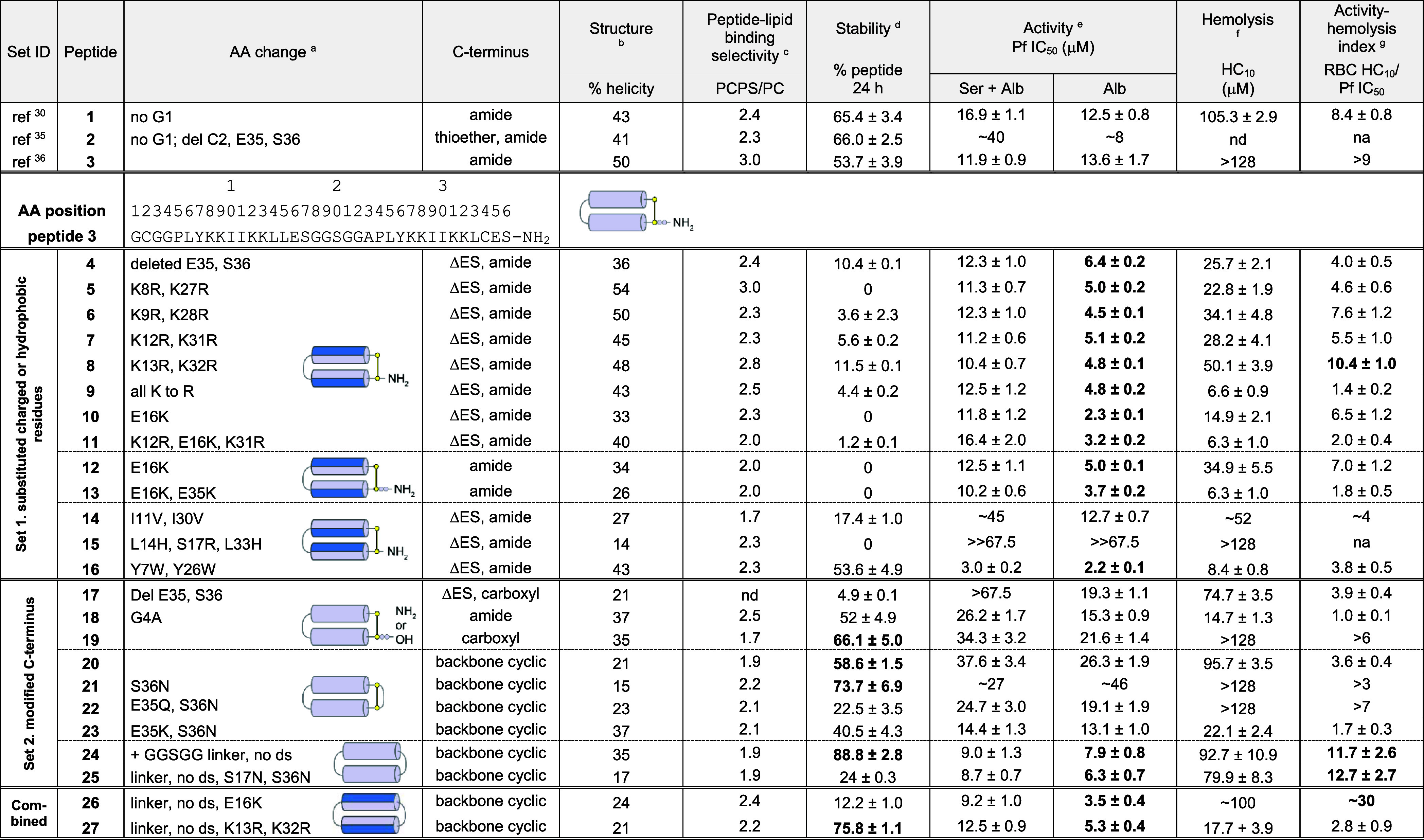
Sequence change relative to peptide 3, with the full amino acid sequence shown in Table S1; ds, disulfide bond.
% helicity was determined from the lowest MRE between 218 and 222 nm for 50 μM peptide in 100 mM NaF, 10 mM KH2PO4, pH 7.5 (Figures 4A, 5A, 6A).
Relative peptide binding PCPS/PC was determined using P/L at the end of the association phase of 16 μM peptide bound to POPC/POPS (4:1) compared to POPC bilayers (Figure 4C, 5C, 6C).
% peptide remaining was determined from area under the curve of the [M + 4]4+ peak (intact mass) using TOF-MS. Peptide analogues (50 μM) were incubated in 25% (v/v) serum in PBS at 37 °C for 24 h and compared to 0 h controls (100% peptide).
Pf IC50, peptide concentration to inhibit 50% growth of P. falciparum 3D7 compared to untreated controls, as determined from dose–response curves (Figure S5). Growth inhibition was determined from DAPI staining of parasite nuclei after 72 h incubation at 37 °C, 5% CO2, 5% O2. Culture medium was supplemented with either 5% serum, 2.5 mg/mL Albumax II (Ser + Alb), or 5 mg/mL Albumax II (Alb).
Hemolysis HC10, peptide concentration that lyses 10% of uninfected RBCs, determined from dose–response curves for released hemoglobin (Figure S6). Less than 10% hemolysis was confirmed within the active concentration range for 3, 10, 12, 24, and 26 up to 72 h incubation.
Activity-hemolysis index comparing 50% inhibition of in vitroP. falciparum (5 mg/mL Albumax II supplement) to 10% hemolysis of uninfected RBCs. Values highlighted in bold indicate improvements over the respective scaffold peptides (1–3).
Peptide Set 1: Substitution of Charged and Hydrophobic Residues
Design
The peptide scaffold used as the base for set 1 analogues was ctPF4PD (2), a variant of cPF4PD (1) with a truncated C-terminus (Glu, Ser deleted; ΔES), C-terminal amide, and a thioether linkage replacing the disulfide bond. We had previously demonstrated similar properties between 1 and 2(35) and proposed that the shorter scaffold would be more desirable for drug development. In other work, we added a Gly to the N-terminus to produce disulfide macrocyclic peptide 3,36 with improved ability to use the Cys residue for covalent bridge formation if required (e.g., acetone staple).37 Therefore, we maintained N-terminal Gly in the newly designed C-terminally truncated, disulfide macrocyclic analogues. To select residues for substitution in set 1, we examined the natural sequence diversity within the C-terminal region of PF4 subunits by performing a BLASTP search (https://blast.ncbi.nlm.nih.gov)38 of a nonredundant protein database (Figure S3). The representative sequence variants shown in Figure 3A were transposed onto the PDIP scaffold (Figure 3B,C) to produce analogues 4–16. We were interested in determining whether replacing Lys with Arg (5–9) or increasing the overall charge (10) would promote stronger interactions with negatively charged membranes and thereby increase potency against malaria parasites. Substitutions to inward facing hydrophobic residues (see Figure 3B) were included to determine the effect on membrane interaction for peptides with the shorter side chain of Val (14) in place of Ile in the parent sequence or the bulkier side chain of Trp (16) replacing Tyr in the base sequence. Previous studies have demonstrated that replacing Tyr with Trp and/or Lys with Arg can improve the membrane-binding affinity of AMPs without increasing nonselective binding or toxicity.39,40 Dual substitutions in 11 and 15 were included due to their natural occurrence in PF4 from nonhuman species. Peptides 12 and 13 were included to determine the effect of the charge reversing Glu-to-Lys substitution on the full-length peptide 3 scaffold.
Figure 3.

Sequence Diversity of the C-terminal 14 Amino Acids of PF4 Informed the Design of PDIP Analogues. (A) C-terminal 14 amino acids of PF4 subunits (PDB: 1F9Q) arrange into paired α-helices. Representative sequence of PF4 C-terminal amino acids showing naturally occurring substitutions (full alignment from BLASTP search of nonredundant protein sequences is shown in Figure S3). (B) Helical wheel diagram showing the predicted position of residues in the two helices of PDIP analogue 3; heptad position is shown (a–g), and the diagram was generated using DrawCoil 1.0 (https://grigoryanlab.org/drawcoil). (C) A cartoon of PDIP (3) shows the location of amino acid substitutions.
Structure and Stability
We previously demonstrated that cPF4PD (1) maintains a helical structure in aqueous solution, which likely explains its increased resistance to breakdown by serum proteases and improved activity against P. falciparum parasites compared to a linear 14 amino acid peptide from PF4’s C-terminus.28,30 To determine whether these desirable characteristics were maintained for the peptides in the present study, we examined their structure using circular dichroism (CD) spectroscopy (Figure 4A) and calculated the % helicity (shown in Table 1) from the minimal mean residual ellipticity (MRE) (218–222 nm) of the peptides in aqueous solution (100 mM NaF, 10 mM KH2PO4 pH 7.5). The CD spectra observed for parent peptides 1–3 confirmed an overall helical structure with spectral minima at 208 and 222 nm. Lys-to-Arg-substituted peptides 5–9 had similar % helicity to 3, but the new truncated scaffold peptide (4) and Glu-to-Lys-substituted peptides 10–13 were less helical. Ile-to-Val substitution in 14 decreased the % helicity to a similar extent, whereas Tyr-to-Trp substitution in 16 had less effect on helix formation. The CD spectrum for 15 was consistent with a random coil structure, suggesting that the Leu-to-His substitutions on the inward-facing region of the helices completely disrupted helix formation in the tested aqueous solution (see Figure 4A and Table 1). We also examined the CD spectrum for 15 under more acidic conditions (pH 5) as some AMPs with a His residue undergo pH-dependent changes in structure that confer differences in membrane-permeabilizing properties41 as well as in a less polar solvent mixture (50% trifluoroethanol, TFE) to determine whether helix formation occurs in more hydrophobic environments, akin to membrane interactions (Figure S4A). pH-dependent changes were not observed for 15 or parent peptide 2, included as the scaffold control, and 15 adopted a similar helical structure to 2 in 50% TFE, suggesting that the substituted residues did not hinder the propensity to form α-helices in more hydrophobic environments. Overall, set 1 analogues with Lys-to-Arg substitution (5–9) had similar helicity in aqueous solution to parent peptides 1–3, whereas Glu-to-Lys substitution (10–13) adversely affected helicity. Substitution of hydrophobic residues (Ile-to-Val, 14; Leu-to-His, 15) had the strongest adverse effect on helicity in an aqueous solution.
Figure 4.

Characteristics of PDIP Base Peptides and Set 1 Analogues. (A) CD spectra were collected for 50 μM peptides in aqueous solution (100 mM NaF, 10 mM KH2PO4 pH 7.5). Peptides with spectral minima at 208 and 222 nm have an overall helical structure. The MRE at 222 nm was used to calculate percentage helicity (Table 1). (B) Resistance to breakdown by serum proteases was determined from the amount of peptide remaining after 24 h of incubation with 25% (v/v) human serum. Peptides were quantified relative to time zero samples from area under the curve of intact mass m/z peaks using TOF-MS. (C) Peptide–lipid binding was compared using surface plasmon resonance (SPR) sensorgrams collected for 16 μM peptides binding to POPC/POPS (4:1) and POPC lipid bilayers. Response units (RU) were converted to P/L using (RUpeptide/mwpeptide)/(RUlipid/mwlipid). P/L at the end of the association phase (170 s) was used for comparing the peptide–lipid binding affinity for POPC/POPS (4:1) compared to that of POPC membranes in (D) (Table 1). Dashed lines or shaded regions provide a comparison to peptide 3 in each of the plots. Structure cartoons show the location of substitutions (in blue) relative to the parent scaffold.
To investigate proteolytic stability, we used time-of-flight mass spectrometry (TOF-MS) to determine the amount of peptide remaining after incubation with a protease-rich solution of 25% human serum (v/v) in phosphate buffered saline (PBS) for 24 h at 37 °C, relative to 0 h controls (100% for each peptide). The 24 h time point was chosen to compare all analogues in this study as it is close to the half-life for parent peptide 1 in these conditions.30 Most of the peptides in set 1 had lower serum stability compared to that of scaffold peptides 1–3, suggesting that removal of C-terminal Glu and Ser residues, without the thioether linkage present in 2, adversely affects the stability (Figure 4B, Table 1). Peptides with Glu-to-Lys substitutions (10–13) were the least stable. Peptide 16 (Tyr-to-Trp substitution) was the most stable set 1 analogue, having similar stability to the parent scaffolds.
Interaction with Lipid Bilayers
The ability of cPF4PD (1) to preferentially bind to and lyse membranes that are rich in negatively charged phospholipids contributes to its selective entry into infected RBCs and subsequent lysis of parasite digestive vacuole membranes.30 This selectivity was therefore a beneficial characteristic that we wished to maintain in the analogues. In the current study, we use SPR to compare the binding interactions between each peptide and two different lipid bilayers. One was composed of neutral palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) to mimic the properties of the outer leaflet of healthy uninfected RBC. The other contained a mixture of POPC with 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (POPS) in a 4:1 ratio (POPC/POPS 4:1) to mimic the properties of P. falciparum-infected RBCs, in which negatively charged PS headgroups are exposed on the outer leaflet of the host cell membrane (Figure 1A).19,20 This method reports on relative binding but not whether peptides penetrate or lyse the different lipid bilayers. However, we previously demonstrated excellent correlation between relative binding to lipid bilayers and lysis of vesicles comprising similar lipid mixtures for peptides 1 and 2.30,35
Figure 4C shows the rapid association and dissociation of peptides 1–3, with a higher P/L (ratio of peptide binding to lipid) obtained for POPC/POPS (4:1) compared to that obtained for POPC lipid bilayers, indicating the selective binding of these peptides to negatively charged compared to neutral membranes. Set 1 peptides showed similar rapid association and dissociation characteristics, except 14 (Ile-to-Val substitution), which had a slower association with POPC/POPS (4:1) bilayers. P/L values at the end of the association phase for 16 μM peptides were used to provide a comparison between relative binding to bilayers (see Figure 4D, and relative binding of POPC/POPS (4:1):POPC reported in Table 1). These values demonstrate the maintained selectivity between POPC/POPS (4:1) and POPC bilayers for all set 1 peptides. Most of the analogues with charge substitutions (5–13), together with 16 (Tyr-to-Trp substitution), had higher binding affinity (higher P/L values) for POPC/POPS (4:1) bilayers than scaffold peptides 1–3. However, the affinity for POPC bilayers was also increased for these peptides, suggesting that the increase was not completely selective.
Antiplasmodial Activity and Activity-Hemolysis Index
Despite the lower serum stability observed for set 1 analogues, we remained interested in examining whether the substitutions affect in vitro growth of P. falciparum. Therefore, we performed activity experiments in the presence (culture medium supplemented with 5% serum, 2.5 mg/mL Albumax II; Ser + Alb) or absence (5 mg/mL Albumax II; Alb) of human serum. This assay was performed in a 384-well plate format, and growth inhibition was determined after 72 h based on the detection of DAPI-stained parasite nuclei using automated analysis.42,43 As anticipated, dose–response curves for set 1 peptides showed enhanced inhibition of parasite growth when serum was excluded from the experiment, whereas parent peptides 1 and 3 had similar curves in both conditions (Figure S5). Peptide concentrations required to inhibit 50% of P. falciparum growth (IC50) were determined from the dose–response curves and are summarized in Table 1 for both assay conditions. To allow distinction between antiplasmodial activity and nonselective lysis of RBCs, the hemolytic activity of the analogues was determined by measuring the release of hemoglobin following incubation of peptides with uninfected RBCs. Peptide concentrations required to lyse 10% of RBCs (HC10) were determined from dose–response curves (Figure S6) and are summarized in Table 1.
An activity-hemolysis index (P. falciparum IC50 relative to RBC HC10) was calculated for the analogues, as summarized in Table 1, and used to assess their relative selectivity for parasite killing over RBC lysis. Analogues with higher activity-hemolysis index values were considered the most desirable.
From the parasite activity assays, truncated scaffold peptide 4 and charge variant peptides 5–13 showed between two- and six-fold enhancement in potency against P. falciparum in the serum-free experiments, consistent with their higher affinity for binding to negatively charged POPC/POPS (4:1) bilayers (see Figure S7). However, hemolysis was also increased for most of these peptides, and the activity-hemolysis index between P. falciparum IC50 and RBC HC10 observed for parent peptides 1 and 3 (8.4 – >9) was only maintained for charge-substituted peptide 8 (see Table 1). Interestingly, this peptide has similar Lys-to-Arg substitutions compared to 5–7, suggesting that positioning of the substituted Arg closest to Glu16 (Figure 3B) is best suited for improving membrane penetration without increasing nonselective disruption.
Substitution of inward-facing Ile or Leu residues adversely affected parasite growth inhibition. Ile-to-Val substitution (14) resulted in a two-fold decrease compared to parent scaffold peptide 2, and Leu-to-His (polar at neutral pH, positive charge at acidic pH e.g. inside the P. falciparum digestive vacuole) substitution resulted in no parasite killing detected for 15 up to 67 μM. For both peptides, the lower activity was consistent with decreased helicity in aqueous solution and increased susceptibility to breakdown by serum proteases. However, the loss of P. falciparum growth inhibition is not explained by diminished membrane interaction (see Figure 4D) or selective uptake into infected versus uninfected RBCs (see Figure S4B) as 15 performed similarly to parent peptides 1–3 with respect to these parameters. These observations, together with the ability of 15 to adopt a helical structure in a less polar solution with 50% TFE (see Figure S4A), suggest that the helical regions of 15 become structured when encountering membranes of infected RBCs, thereby facilitating cell penetration. The loss of activity against the intracellular target(s) remains unexplained. Substitution of Tyr with Trp (16) produced the most stable truncated analogue, with the most potent activity against P. falciparum. However, like the charge-inverted (Glu-to-Lys) peptide 10, the activity-hemolysis index between antiplasmodial activity and hemolysis of RBCs was decreased compared to parent scaffold peptides 1 and 3 (Table 1).
Peptide Set 2: Modification of Peptide C-Terminus
Design
Encouraged by the ability to improve potency against P. falciparum by substituting amino acid residues, we next sought to improve peptide stability with a focus on maintaining compatibility for the biosynthetic production of lead peptides. Peptides 17 and 19 were included to determine whether the C-terminal amide could be replaced by a carboxylic acid (akin to biosynthetic products), and 18 served as a control peptide for replacing Gly4 in the peptide 3 scaffold with Ala, the conserved amino acid at this position in PF4 (Figure S3). Cyclization is known to improve peptide stability,44,45 and peptide ligation can be achieved in biosynthetic pathways using asparaginyl endopeptidase (AEP) enzymes that leave a minimal Asn footprint in the resultant cyclic peptide.46,47 Peptides 20–23 were included to examine the effect of backbone cyclization of the peptide 3 scaffold (20) (compared to macrocyclization exclusively via the disulfide bridge), introduction of Asn into the sequence (21), and replacement of negatively charged Glu with Gln (22) or Lys (23). Peptides 24 and 25 contain a flexible linker (GGSGG) in place of the disulfide bond, with Ser-to-Asn substitution additionally explored in 25.
Structure and Stability
Disulfide-bridged macrocycles with modified C-terminal residues (17–19) and backbone cyclic peptides with a retained disulfide bond (20–23) had an overall helical structure in aqueous solution, albeit with reduced % helicity compared to scaffold peptides 1–3. However, the CD spectra of backbone cyclic peptides with a flexible linker between the helices but no disulfide bond (24–25) suggested a deviation from a well-defined helical structure (Figure 5A).
Figure 5.

Characteristics of Set 2 Analogues. (A) CD spectra were collected for 50 μM peptides in aqueous solution (100 mM NaF, 10 mM KH2PO4 pH 7.5) as above. (B) Resistance to breakdown by serum proteases was determined from the amount of peptide remaining after 24 h incubation with 25% (v/v) human serum as above. (C) Peptide–lipid binding was compared using SPR sensorgrams collected for 16 μM peptides binding to POPC/POPS (4:1) and POPC lipid bilayers as above. P/L at the end of the association phase (170 s) was used for comparing peptide–lipid binding affinity for POPC/POPS (4:1) compared to POPC membranes in (D). Peptide 3 is included, with dashed lines or shaded regions providing a comparison in each of the plots.
Proteolytic stability was maintained or improved compared to scaffold peptides (1–3) for all set 2 peptides, except for truncated peptide 17, which has a C-terminal carboxylic acid, and 22 and 25, which have both Gln and Asn substitutions or contain two Asn residues (Figure 5B). The stability of backbone cyclic analogues with a single Asn substitution (21 and 23) suggests that this inclusion is well tolerated, which is a promising outcome for future cyclization of peptides using AEP ligases during biosynthetic production.
Interaction with Lipid Bilayers
Replacement of the C-terminal amide with a carboxylic acid in 19 or through backbone cyclization in 20 lowered the binding affinity for both tested lipid bilayers, but this was partly overcome by including Asn in the peptide sequence for backbone cyclic peptides 22 and 23. Replacement of the disulfide bond with a flexible GGSGG linker in backbone cyclic peptides 24 and 25 increased the overall membrane binding affinity (Figure 5C) and retained selectivity for negatively charged POPC/POPS (4:1) compared to neutral POPC bilayers (Figure 5D). One explanation for the increased affinity of 24 and 25 is that some of the inward-facing hydrophobic residues become more surface exposed due to their more conformationally flexible and less-defined structure compared to that of the parent peptides (Figure 5A).
Antiplasmodial Activity and Activity-Hemolysis Index
Consistent with their superior serum stability profiles, set 2 peptides (except for truncated 17) had more similar IC50 values for P. falciparum growth inhibition in the presence or absence of serum in culture medium compared to set 1 analogues (Figure S5, Table 1). However, modification of the C-terminus (including carboxylic acid or backbone cyclization) of the peptide 3 scaffold (peptides 18–23) decreased the antiplasmodial potency. It is possible that increased structural rigidity combined with insufficient spacing between the joined N- and C-terminal residues of backbone cyclic peptides 20–23 caused an unfavorable presentation of residues at positions 35 and 36. Inclusion of a longer flexible linker between the terminal residues appeared to restore full peptide function, and consistent with the observed increase in membrane-binding affinity (Figure 5C), 24 and 25 had increased potency (lower IC50) for inhibiting P. falciparum growth. These analogues maintained selectivity for negatively charged membranes (Figure 5D) and had an activity-hemolysis index at least as high as that of the parent scaffold peptide 3 (Table 1). Together, these results show that backbone cyclized PDIP analogues with extended linkers between the two helices have superior ability to resist breakdown by serum proteases, maintain selectivity for negatively charged lipids, and have enhanced ability to kill P. falciparum parasites in vitro.
Combined Potency-Enhancing Substitutions and Stability-Enhancing Modifications
Design
The most promising potency-enhancing substitutions (E16K; and K13R, K32R) identified from set 1 analogues were incorporated onto the most stable and selective peptide scaffold (24) identified from set 2 analogues to produce 26 and 27, respectively.
Structure and Stability
Both 26 and 27 had a helical structure in aqueous solution (Figure 6A), but with a lower % helicity than the corresponding charge-substituted disulfide macrocyclic analogues (10–13 with E16K; 8 with K13R, K32R) or backbone cyclic scaffold peptide 24 (see Table 1). The observed changes in % helicity did not appear to affect stability as resistance to breakdown by serum proteases was improved for charge-substituted backbone cyclic analogues compared to their disulfide macrocyclic counterparts. After 24 h of incubation in 25% (v/v) serum in PBS, 12% of 26 remained compared to 0% for 10–13, and 76% of 27 remained compared to 12% of 8 (Figures 5B and 6B).
Figure 6.

Characteristics of PDIP Analogues with Combined Modifications. (A) CD spectra were collected for 50 μM peptides in aqueous solution (100 mM NaF, 10 mM KH2PO4 pH 7.5) as above. (B) Resistance to breakdown by serum proteases was determined from the amount of peptide remaining after 24 h incubation with 25% (v/v) human serum as above. (C) Peptide–lipid binding was compared using SPR sensorgrams collected for 16 μM peptides binding to POPC/POPS (4:1) and POPC lipid bilayers as above. P/L at the end of the association phase (170 s) was used for comparing peptide–lipid binding affinity for POPC/POPS (4:1) compared to POPC membranes in (D). Peptide 3 is included, with dashed lines or shaded regions providing comparison in each of the plots.
Interaction with Lipid Bilayers
Peptides 26 and 27 had similar high peptide-to-lipid binding ratios (P/L) as scaffold peptide 24 and maintained selectivity for negatively charged POPC/POPS (4:1) compared to neutral POPC lipid bilayers (Figure 6C,D and Table 1).
Antiplasmodial Activity and Activity-Hemolysis Index
Introducing the E16K substitution onto a stable backbone cyclic scaffold (26) resulted in similar increased in vitro activity against P. falciparum (IC50 3.5 μM), as observed for disulfide macrocyclic counterparts, but with greatly improved activity-hemolysis index between antiplasmodial activity and RBC lysis, and HC10 > 100 μM for 26 (Table 1). The potency enhancement observed for the K13R, K32R substitution (disulfide macrocyclic peptide 8) was also maintained on the stable backbone cyclic scaffold (IC50 5.3 μM for 27 compared to 4.8 μM for 8, no serum condition). However, this combination produced a more hemolytic peptide with an RBC HC10 of 18 μM for 27 (Table 1).
Discussion
New antimalarial candidates need to overcome many challenges as they progress along the drug development pipeline. Ideally, they kill Plasmodium parasites at multiple life cycle stages, are inexpensive and easy to administer, are effective against most circulating drug-resistant strains, and have a good resistance profile.2 We propose that peptide-based drugs have the potential to fulfill these requirements. In addition, the different mechanism of action of PF4-derived peptides compared to that of current antimalarial drugs makes them useful candidates for development as drug partners in new combination therapies.
Peptide scaffolds can be modified to enhance the structural stability and resistance to breakdown. We reasoned that improving PDIP resistance to proteolytic breakdown would enhance antiplasmodial potency by having the active peptide present for a longer treatment time. Backbone cyclization of PDIP analogues with flexible linkers between the two bioactive helical sequences produced a more stable scaffold (24), although we observed a deviation away from the overall helical structure of parent peptides 1–3. Three of the four peptides with this less-defined structure (24–26) had improved antiplasmodial potency and favorably low hemolytic activity compared to the parent peptides, but future scaffold adaptations that promote peptide helical structure in aqueous solution (e.g., reintroduction of a disulfide bond between the paired helices) might be required.
Peptides can be readily modified by substituting amino acids in the primary sequence. However, careful consideration of the locations of target residues is required. Here, we demonstrate that changes to inward-facing hydrophobic residues of the PDIP scaffold likely induced structural changes that decreased potency against P. falciparum (14 and 15). Conversely, changes to outward-facing charged residues (5–13) did not noticeably alter the helical structure and enhanced antiplasmodial potency, but many of these also increased the hemolysis of uninfected RBCs. Two promising substitutions were identified, E16K (10, 12) and K13R, K32R (8), that enhanced antiplasmodial potency and maintained a good activity-hemolysis index. However, both substitutions produced less proteolytically stable peptides compared with scaffold peptides 1–3.
Our approach of combining these desirable potency-enhancing substitutions onto the most stable cyclic peptide scaffold (24) was successful in producing an analogue (26, E16K substitution) that satisfied the requirements of enhanced antiplasmodial potency and low hemolytic activity compared to first generation peptides (1–3). Backbone cyclic analogue 26 had improved resistance to serum proteases compared to charge-inverted disulfide macrocyclic counterparts (10–13), but further improvement in stability is required for the progression of 26 as a drug candidate. There are several examples of helical peptides where substitution of Lys or Arg residues proximal to the C-terminus with their d-amino acid counterparts improves resistance to proteolytic breakdown while maintaining overall helical structure and bioactivity.48−50 Incorporation of d-Lys in positions 16 or 36 of the PDIP scaffold may provide similar benefits, but this approach would deviate from our current efforts to maintain an all-l-amino-acid scaffold to allow transfer to a biosynthetic production pathway.
Toward the goal of future biosynthetic production, we explored whether deviation from the C-terminal amide of first-generation PDIP analogues would affect the overall properties and bioactivity. Disulfide macrocyclic analogues with a C-terminal carboxylic acid (17, 19) and backbone cyclic analogues with a maintained disulfide bond (20 and 21) had lower antiplasmodial activity, but this was partially recovered by altering the charge at C-terminal position 35 from negative (Glu) to neutral polar (Gln, 22) or positive (Lys, 23). Notably, 21–23 also contained an Asn residue, confirming that this residue is well tolerated in the backbone cyclic PDIP scaffold. This is an important requirement for incorporating AEP enzymatic cyclization into biosynthetic production methods.46,47
The cost of new antimalarial drugs is a major hurdle to overcome as the regions most affected by the disease have the poorest economies and out-of-pocket treatment costs are prohibitive for low-income earners (recently reviewed51,52). Efforts to reduce the cost of antimalarial drugs include improvements in synthesis efficiency (e.g., artemisinin biosynthesis53,54) and promotion of local manufacture to bypass expenses incurred by import and distribution.55 Likewise, the cost of producing and distributing peptides is a limitation that must be addressed when developing peptide-based therapeutics. Toward this requirement, we and others are optimizing biosynthetic methods for producing therapeutic peptides.56−58 Importantly, disulfide-rich cyclic peptides can be produced with high yields via yeast57 or plant59,60 expression systems. Production and recovery of biosynthetic products is less expensive, requires simpler infrastructure, and is more environmentally friendly than chemical synthesis,57 making it amenable for local production as a means of further reducing costs.
Challenges that we have not addressed in this study include the determination of activity against drug-resistant P. falciparum, activity against other life cycle stages, and evaluation of resistance risk. Here, we examined the in vitro activity against blood-stage parasites of the drug-sensitive P. falciparum 3D7 strain. Studies on drug-resistant strains will be required, but we anticipate similar activity against drug-sensitive and -resistant strains due to the differences in the mechanism of action between PDIP and small-molecule antimalarial drugs2,30 and because the parental PF4 protein, which acts on parasites via the same membrane-active mechanism,28 is equally active on drug sensitive and resistant strains.27 In other work (unpublished), we are examining the effect of PDIP and PDIP-drug conjugates36 on the development of sexual-stage gametocytes and maturation into macrogametes.61 Measuring resistance risk is a challenging undertaking, requiring culture of P. falciparum under drug pressure for long periods of time, especially if the parasites are refractory to developing resistance to the target drug.2 On the basis of previous resistance studies with AMP-treated bacteria62 and our observations with melanoma cells (peptide versus dabrafenib acquired resistance),63 we anticipate a slower acquisition of resistance to peptides, and with a greater fitness cost to Plasmodium parasites compared to small-molecule drugs.
Conclusions
Here we describe the rational optimization of the selective, membrane-active PDIP scaffold. We identified an antiplasmodial potency-enhancing substitution (E16K) and incorporated this into a stable and selective scaffold (24) to produce a backbone cyclic PDIP analogue (26) with low micromolar activity against in vitroP. falciparum parasites (see Figure 7). The outcomes of this study represent a practical advance in developing the PDIP scaffold as an antimalarial molecule and provide a framework for monitoring and selecting desirable properties during peptide-based drug development. PDIP analogues are unique in their derivation from a human defense protein, and their selective entry into infected RBCs and membrane-active mechanism of action (lysis of the parasite digestive vacuole) is distinct from that of current antimalarial drugs. These features make the tunable PDIP scaffold a valuable candidate for expanding the range of bioactive molecules available for the development of new antimalarial drugs and combination therapies.
Figure 7.
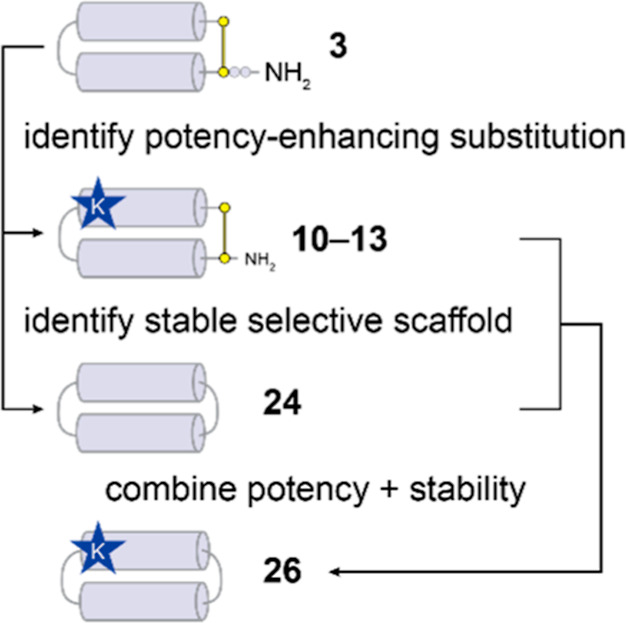
Flow diagram showing the rational improvement of selective antiplasmodial activity and stability for PDIPs.
Methods
Peptide Synthesis and Purification
All peptides were synthesized using automated Fmoc solid phase chemistry (Symphony, Protein Technologies Inc.). Rink amide resin was used to produce peptides with an amidated C-terminus (3–16 and 18), and 2-chlorotrityl chloride resin was used for peptides with a C-terminal carboxylic acid (17 and 19). Linear precursors for backbone cyclic peptides (20–27) were synthesized on Fmoc-NHNH-chlorotrityl resin prepared as previously described.64,65 Peptides were deprotected and cleaved from the resin using TFA:H2O:TIPS (95:2.5:2.5, v/v/v) and collected by precipitating with ice-cold ether. Crude peptides were purified by RP-HPLC using a Shimadzu system and Phenomenex Gemini C18 column with a gradient of solvent B (90% CH3CN, 0.05% v/v TFA) against solvent A (0.05% TFA v/v).
Disulfide Macrocyclic Peptides
Purified and lyophilized peptides were dissolved at 0.5 mg/mL in 45% CH3CN and 0.05% TFA (v/v), flushed with nitrogen gas, and oxidized with 20 mol equiv of iodine, as reported before.30
Backbone Cyclic Peptides
Linear precursors were synthesized with an N-terminal Cys and C-terminal hydrazide to facilitate cyclization via intramolecular native chemical ligation, as previously described.33,34 To convert the C-terminal hydrazide to a thioester, purified and lyophilized peptides were dissolved at 3 mM in 6 M GnHCl (pH < 3), with 4-mercaptophenylacetic acid (200 mM) and 9 mM acetyl acetone (3 equiv), and stirred at room temperature for 4 h. Head-to-tail cyclization was achieved by diluting the reaction to achieve a solution of 0.5 mM peptide in 6 M GnHCl, 100 mM NaH2PO4, and 50 mM TCEP, with pH adjusted to pH 7, and stirring overnight at room temperature. Disulfide bonds were formed for peptides 20–23 by iodine oxidation as described above. Cys residues in precursors for peptides 24–27 were desulfurized to Ala by reacting the peptides with 0.25 M TCEP, 3 M GnHCl, 50 mM NaH2PO4, 10 mM reduced glutathione, and 50 mM VA-044 (Fujifilm Wako Chemicals), with pH adjusted to 6.5, purged with argon, and incubated overnight at 65 °C.66 Oxidized or backbone cyclized peptides were purified by RP-HPLC as above. Purity and correct mass of the peptides were simultaneously determined using a Shimadzu LCMS-2020 instrument with a Phenomenex 5 μm C18/300 Å/150 × 2 mm LC column. HPLC traces are shown in Figure S1, with LC peak integration and MS characterization provided for analogues with apparent shoulder peaks or impurities (≤5%) in Figure S2. Observed masses derived from +4 m/z ions are compared to calculated masses, as shown in Table S1.
Overall Structure Determination
CD spectra between 185 and 260 nm were collected for 50 μM peptides in aqueous solution (100 mM NaF, 10 mM KH2PO4, pH 7.5) using a Jasco J810 spectropolarimeter, as reported before.30 CD spectra were also collected in 50% aqueous solution and 50% trifluoroethanol (v/v) for peptides 2 and 15. MRE was calculated using the formula
The % helicity was determined from the minimal MRE between 218 and 222 nm.67,68
Resistance to Breakdown by Serum Proteases
Peptide stability was determined by incubating 5 μM peptide in 25% (v/v) human serum (Sigma-Aldrich) in PBS for 24 h at 37 °C. The amount of peptide remaining after 24 h incubation was calculated relative to 0 h controls by precipitating serum proteins with 2% (v/v) TFA in CH3CN as before30 and running the soluble fraction (with recovered peptide) on a Triple TOF 5600 mass spectrometer (AB SCIEX). The [M + 4]4+m/z peak corresponding to full-length peptide mass was identified using SCIEX Analyst software, and the area under the curve was determined using SCIEX MultiQuant software. The average of three 0 h controls was used to determine 100% recovered peptide, and % recovery was determined for three technical repeats of each peptide.
Membrane Binding and Selectivity
Small unilamellar vesicles were prepared by resuspending dried films of synthetic POPC or a 4:1 mixture of POPC and POPS (Avanti Polar Lipids via Sigma-Aldrich) in 10 mM HEPES, 150 mM NaCl (pH 7.4) (SPR running buffer), followed by freeze–thaw cycles and extrusion through a 50 nm membrane.30 Peptide–lipid interactions were examined by using SPR with an L1 chip (Cytiva) on a Biacore T200 instrument. Lipid vesicles were deposited until a steady-state plateau was reached, when we assume that lipid bilayers have formed.69 Serial dilutions of peptides from 4 to 32 μM in SPR running buffer were injected over deposited lipid bilayers for 180 s (association). Dissociation was followed for 600 s, while SPR running buffer continued to flow over the chip surface. Response units were converted to P/L using the following equation
P/L at the end of the association phase (170 s) was used to prepare dose–response curves, which were fitted using One-Site specific binding (GraphPad Prism v10). P/L for 16 μM peptides was used to compare between binding to POPC/POPS (4:1) and POPC.
In Vitro Activity Against P. falciparum
P. falciparum 3D7 parasites were cultured in human RBCs (from screened volunteers via Red Cross Lifeblood). Culture medium comprised RPMI1640 (Sigma) supplemented with 2.5 mg/mL Albumax II (Thermo Fisher Scientific), 25 mM HEPES, 0.37 mM hypoxanthine, and 5% human serum (Sigma). Growth inhibition assays were performed in the serum-containing media above or media supplemented with 5 mg/mL Albumax II and no serum, as previously described.42 In brief, synchronized parasite cultures (sorbitol enrichment of early ring stage) were plated at 2% parasitemia and 0.3% hematocrit in 384-well CellCarrier Ultra poly-d-lysine imaging plates (Revvity), treated with serially diluted peptides in PBS (with final 0.4% v/v DMSO) starting from 67.5 μM, and incubated for 72 h at 37 °C, 5% CO2, and 5% O2. Inhibition of P. falciparum growth was determined for treated samples compared to vehicle (0.4% DMSO, 0% inhibition) and an in-plate positive control (5 μM puromycin, 100% inhibition) by staining nuclei with 2-(4-amidinophenyl)-1H-indole-6-carboxamidine (DAPI). Stained nuclei were detected using a Phenix confocal imaging system (Revvity) with images analyzed as previously described using Acapella or Harmony spot detection software.42,43 Dose–response curves were fitted with GraphPad Prism (v10) using [inhibitor] versus response with variable slope and constraining the top to 100% (See Figure S5). IC50 values derived from dose–response curves are shown in Table 1. Data represent the mean and standard error from two biological replicates.
Peptide Uptake Studies
Peptides 3, 14, and 15 were labeled in the presence of TCEP, using dichloroacetone to form an acetone bridge between Cys residues and oxime ligation using 2 mol equiv AlexaFluor-488 (A488) hydroxylamine (Thermo Fisher Scientific), as previously described.30 Labeled peptides were purified by RP-HPLC as described above.
Cultures of uninfected and trophozoite-infected human RBCs (approximately 28–36 h post-invasion; 3–5% parasitemia) were incubated with A488-labeled peptides (0.625–40 μM) in culture medium supplemented with 5 mg/mL Albumax II and no serum (as above) for 1 h (our unpublished live-cell uptake studies indicate that this is sufficient time for significant peptide uptake). Treated cells were washed twice with serum-free culture medium to remove any residual unincorporated peptide and then fixed with 1% (w/v) formaldehyde [CytofixTM (BD Biosciences) diluted 1/4 with PBS] for at least 24 h at 4 °C. For the peptide uptake studies, fixed cells were washed with PBS containing 1% (w/v) bovine serum albumin and then stained with 5 mg/mL Hoechst 33342 (Life Technologies) for 5 min at 4 °C. Fluorescence intensity signals were measured by flow cytometry using an LSR Fortessa cell analyzer (BD Biosciences); at least 100,000 events were collected per sample (350 nm laser, 450/50 filter for Hoechst; 488 nm laser, 530/30 filter for A488). Percentages of infected cells (Hoechst-stained nuclei) and mean fluorescence intensity of cells (with the A488-labeled peptide) were identified and enumerated using FlowJo software (BD Biosciences). For the microscopy image shown in Figure 1, P. falciparum (trophozoite-stage) cultured human RBCs, either untreated or treated with PDIP-A488 (20 μM for 1 h), were washed and fixed as described for the peptide uptake studies. The fixed cells were then washed and diluted into PBS, smeared onto glass slides coated with polyethylenimine (0.1% v/v), air-dried (5 min), and then washed again in PBS before mounting under coverslips (#1 from Menzel-Glaser) in SlowFade Gold Antifade Mountant with DAPI (Thermo Fisher Scientific). Slides were examined at room temperature using an Axio Observer inverted fluorescence microscope using 630× magnification and coupled to an Axiocam 503 monochrome camera (Zeiss). Images were acquired and processed using a ZEN microscope and imaging software (Zeiss).
Hemolysis of Uninfected RBCs
Blood was collected from a healthy human volunteer directly into PBS. RBCs were pelleted by centrifuging at 1000 g for 1 min; the supernatant was removed, and the cells were washed three times with PBS. RBCs were plated at 0.5% hematocrit in PBS into 96-well plates, and an equal volume of peptides serially diluted in PBS were added to the wells. PBS (0% lysis) and 0.1% (v/v) Triton-X 100 (100% lysis) controls were included. Plates were incubated for 1 h at 37 °C, 5% CO2; then intact cells were pelleted by centrifuging at 500 g for 5 min. The supernatant was transferred to another plate, and release of hemoglobin from lysed cells was detected by measuring the absorbance at 405 nm. Dose–response curves were fitted with GraphPad Prism (v10) using [inhibitor] vs response with variable slope (Figure S6). Minimum hemolytic concentrations (HC10, 10% of RBCs lysed) were interpolated from dose–response curves and are shown in Table 1. Data represent mean and standard error from two biological replicates. Hemolysis assays were also performed for peptides 3, 10, 12, 24, and 26 under similar conditions to in vitroP. falciparum activity experiments (serum-free culture medium supplemented with 5 mg/mL Albumax II), confirming <10% lysis with up to 72 h incubation.
Human Ethics Approvals
Experiments involving human RBCs and serum were performed in accordance with the following ethics approvals: Griffith University Human Research Ethics Exemption Approval 03/08/11019 (P. falciparum culture and activity assays); Australian National University Human Research Ethics Committee, approval number 2018/398 (P. falciparum culture and peptide uptake studies); and University of Queensland Human Research Ethics Approval number 2022/HE000300 (RBC hemolysis assays).
Acknowledgments
This work was supported by funding from the Australian National Health and Medical Research Council (1183927 to B. J. M., N. L., and L. R. M.), US Department of Defense grant (PR210354 to D. J. C. and N. L.), and the Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science (CE200100012). D.J.C. was supported by NHMRC grant (2009564). S.D. was supported by a Griffith University Postdoctoral Fellowship (GUPF_23_24). The authors would like to thank Yen-Hua Huang, Bhavesh Khatri, and Lai Yue Chan for peptide synthesis; and Megan Drew, Huma Sohail, and Kiran Javed for malaria parasite culture and support in performing preliminary activity assays that guided analogue design.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.4c00276.
Peptide amino acid sequence and masses of PDIP analogues; HPLC of the analogues; integration of LC trace and MS spectra for analogues with shoulder peaks; sequence homology to the PF4 AMP domain from a nonredundant protein database search; additional structural and cell-penetrating characterization performed for inactive peptide 15 compared to parent peptides 2 and 3; dose–response curves for in vitro activity against P. falciparum; dose–response curves for RBC hemolysis; and the relationship between peptide interaction with negatively charged lipid bilayers and in vitro antiplasmodial activity (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organisation . World Malaria Report 2023; Licence: CC BY-NC-SA 3.0 IGO: Geneva, 2023.
- Siqueira-Neto J. L.; Wicht K. J.; Chibale K.; Burrows J. N.; Fidock D. A.; Winzeler E. A. Antimalarial drug discovery: progress and approaches. Nat. Rev. Drug Discovery 2023, 22 (10), 807–826. 10.1038/s41573-023-00772-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowe C. V. Malaria chemoprevention and drug resistance: a review of the literature and policy implications. Malar. J. 2022, 21 (1), 104. 10.1186/s12936-022-04115-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balikagala B.; Fukuda N.; Ikeda M.; Katuro O. T.; Tachibana S.-I.; Yamauchi M.; Opio W.; Emoto S.; Anywar D. A.; Kimura E.; Palacpac N. M. Q.; Odongo-Aginya E. I.; Ogwang M.; Horii T.; Mita T. Evidence of artemisinin-resistant malaria in Africa. N. Engl. J. Med. 2021, 385 (13), 1163–1171. 10.1056/NEJMoa2101746. [DOI] [PubMed] [Google Scholar]
- Jeang B.; Zhong D.; Lee M. C.; Atieli H.; Yewhalaw D.; Yan G. Molecular surveillance of Kelch 13 polymorphisms in Plasmodium falciparum isolates from Kenya and Ethiopia. Malar. J. 2024, 23 (1), 36. 10.1186/s12936-023-04812-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M. D.; Rosenthal P. J. Antimalarial drug resistance in Africa: the calm before the storm?. Lancet Infect. Dis. 2019, 19 (10), e338–e351. 10.1016/s1473-3099(19)30261-0. [DOI] [PubMed] [Google Scholar]
- Schäfer T. M.; Pessanha de Carvalho L.; Inoue J.; Kreidenweiss A.; Held J. The problem of antimalarial resistance and its implications for drug discovery. Expert Opin. Drug Discovery 2024, 19 (2), 209–224. 10.1080/17460441.2023.2284820. [DOI] [PubMed] [Google Scholar]
- Ippolito M. M.; Moser K. A.; Kabuya J. B.; Cunningham C.; Juliano J. J. Antimalarial drug resistance and implications for the WHO global technical strategy. Curr. Epidemiol. Rep. 2021, 8 (2), 46–62. 10.1007/s40471-021-00266-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni M. F. Breaking the cycle of malaria treatment failure. Front. Epidemiol. 2022, 2, 1041896. 10.3389/fepid.2022.1041896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chico R. M.; Pittrof R.; Greenwood B.; Chandramohan D. Azithromycin-chloroquine and the intermittent preventive treatment of malaria in pregnancy. Malar. J. 2008, 7 (1), 255. 10.1186/1475-2875-7-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard T.; Boxberger M.; Madamet M.; Pradines B. Has doxycycline, in combination with anti-malarial drugs, a role to play in intermittent preventive treatment of Plasmodium falciparum malaria infection in pregnant women in Africa?. Malar. J. 2018, 17 (1), 469. 10.1186/s12936-018-2621-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu D.; Pei J. V.; Rosling J. E. O.; Thathy V.; Li D.; Xue Y.; Tanner J. D.; Penington J. S.; Aw Y. T. V.; Aw J. Y. H.; Xu G.; Tripathi A. K.; Gnadig N. F.; Yeo T.; Fairhurst K. J.; Stokes B. H.; Murithi J. M.; Kümpornsin K.; Hasemer H.; Dennis A. S. M.; Ridgway M. C.; Schmitt E. K.; Straimer J.; Papenfuss A. T.; Lee M. C. S.; Corry B.; Sinnis P.; Fidock D. A.; van Dooren G. G.; Kirk K.; Lehane A. M. A G358S mutation in the Plasmodium falciparum Na+ pump PfATP4 confers clinically-relevant resistance to cipargamin. Nat. Commun. 2022, 13 (1), 5746. 10.1038/s41467-022-33403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt E. K.; Ndayisaba G.; Yeka A.; Asante K. P.; Grobusch M. P.; Karita E.; Mugerwa H.; Asiimwe S.; Oduro A.; Fofana B.; Doumbia S.; Su G.; Csermak Renner K.; Venishetty V. K.; Sayyed S.; Straimer J.; Demin I.; Barsainya S.; Boulton C.; Gandhi P. Efficacy of cipargamin (KAE609) in a randomized, phase II dose-escalation study in adults in Sub-Saharan Africa with uncomplicated Plasmodium falciparum malaria. Clin. Infect. Dis. 2022, 74 (10), 1831–1839. 10.1093/cid/ciab716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasloff M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415 (6870), 389–395. 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- Hancock R. E. W.; Diamond G. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol. 2000, 8 (9), 402–410. 10.1016/S0966-842X(00)01823-0. [DOI] [PubMed] [Google Scholar]
- Hancock R. E. W.; Sahl H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24 (12), 1551–1557. 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- Fadok V. A.; Voelker D. R.; Campbell P. A.; Cohen J. J.; Bratton D. L.; Henson P. M. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 1992, 148 (7), 2207–2216. 10.4049/jimmunol.148.7.2207. [DOI] [PubMed] [Google Scholar]
- Sharma B.; Kanwar S. S. Phosphatidylserine: A cancer cell targeting biomarker. Semin. Cancer Biol. 2018, 52, 17–25. 10.1016/j.semcancer.2017.08.012. [DOI] [PubMed] [Google Scholar]
- Engelbrecht D.; Coetzer T. L. Plasmodium falciparum exhibits markers of regulated cell death at high population density in vitro. Parasitol. Int. 2016, 65 (6), 715–727. 10.1016/j.parint.2016.07.007. [DOI] [PubMed] [Google Scholar]
- Fraser M.; Jing W.; Bröer S.; Kurth F.; Sander L.-E.; Matuschewski K.; Maier A. G. Breakdown in membrane asymmetry regulation leads to monocyte recognition of P. falciparum-infected red blood cells. PLoS Pathog. 2021, 17 (2), e1009259 10.1371/journal.ppat.1009259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorzi A.; Deyle K.; Heinis C. Cyclic peptide therapeutics: past, present and future. Curr. Opin. Chem. Biol. 2017, 38, 24–29. 10.1016/j.cbpa.2017.02.006. [DOI] [PubMed] [Google Scholar]
- Peschel A.; Sahl H.-G. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 2006, 4 (7), 529–536. 10.1038/nrmicro1441. [DOI] [PubMed] [Google Scholar]
- Xuan J.; Feng W.; Wang J.; Wang R.; Zhang B.; Bo L.; Chen Z.-S.; Yang H.; Sun L. Antimicrobial peptides for combating drug-resistant bacterial infections. Drug Resist. Updat. 2023, 68, 100954. 10.1016/j.drup.2023.100954. [DOI] [PubMed] [Google Scholar]
- Le C. F.; Fang C. M.; Sekaran S. D. Intracellular targeting mechanisms by antimicrobial peptides. Antimicrob. Agents Chemother. 2017, 61 (4), e02340 10.1128/aac.02340-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogden K. A. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria?. Nat. Rev. Microbiol. 2005, 3 (3), 238–250. 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- Amiss A. S.; Henriques S. T.; Lawrence N. Antimicrobial peptides provide wider coverage for targeting drug-resistant bacterial pathogens. Pept. Sci. 2022, 114 (2), e24246 10.1002/pep2.24246. [DOI] [Google Scholar]
- McMorran B. J.; Wieczorski L.; Drysdale K. E.; Chan J. A.; Huang H. M.; Smith C.; Mitiku C.; Beeson J. G.; Burgio G.; Foote S. J. Platelet factor 4 and Duffy antigen required for platelet killing of Plasmodium falciparum. Science 2012, 338 (6112), 1348–1351. 10.1126/science.1228892. [DOI] [PubMed] [Google Scholar]
- Love M. S.; Millholland M. G.; Mishra S.; Kulkarni S.; Freeman K. B.; Pan W.; Kavash R. W.; Costanzo M. J.; Jo H.; Daly T. M.; Williams D. R.; Kowalska M. A.; Bergman L. W.; Poncz M.; DeGrado W. F.; Sinnis P.; Scott R. W.; Greenbaum D. C. Platelet factor 4 activity against P. falciparum and its translation to nonpeptidic mimics as antimalarials. Cell Host Microbe 2012, 12 (6), 815–823. 10.1016/j.chom.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeaman M. R.; Yount N. Y.; Waring A. J.; Gank K. D.; Kupferwasser D.; Wiese R.; Bayer A. S.; Welch W. H. Modular determinants of antimicrobial activity in platelet factor-4 family kinocidins. Biochim. Biophys. Acta 2007, 1768 (3), 609–619. 10.1016/j.bbamem.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence N.; Dennis A. S. M.; Lehane A. M.; Ehmann A.; Harvey P. J.; Benfield A. H.; Cheneval O.; Henriques S. T.; Craik D. J.; McMorran B. J. Defense peptides engineered from human platelet factor 4 kill Plasmodium by selective membrane disruption. Cell Chem. Biol. 2018, 25 (9), 1140–1150.e5. 10.1016/j.chembiol.2018.06.009. [DOI] [PubMed] [Google Scholar]
- Tawk L.; Chicanne G.; Dubremetz J. F.; Richard V.; Payrastre B.; Vial H. J.; Roy C.; Wengelnik K. Phosphatidylinositol 3-phosphate, an essential lipid in Plasmodium, localizes to the food vacuole membrane and the apicoplast. Eukaryot. Cell 2010, 9 (10), 1519–1530. 10.1128/EC.00124-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke R. J.; Hossain K. R.; Cao K. Physiological roles of transverse lipid asymmetry of animal membranes. BBA. Biomembranes 2020, 1862 (10), 183382. 10.1016/j.bbamem.2020.183382. [DOI] [PubMed] [Google Scholar]
- Dawson P. E.; Muir T. W.; Clark-Lewis I.; Kent S. B. H. Synthesis of proteins by native chemical ligation. Science 1994, 266 (5186), 776–779. 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- Cistrone P. A.; Bird M. J.; Flood D. T.; Silvestri A. P.; Hintzen J. C. J.; Thompson D. A.; Dawson P. E. Native chemical ligation of peptides and proteins. Curr. Protoc. Chem. Biol. 2019, 11 (1), e61 10.1002/cpch.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence N.; Philippe G. J. B.; Harvey P. J.; Condon N. D.; Benfield A. H.; Cheneval O.; Craik D. J.; Troeira Henriques S. Cyclic peptide scaffold with ability to stabilize and deliver a helical cell-impermeable cargo across membranes of cultured cancer cells. RSC Chem. Biol. 2020, 1 (5), 405–420. 10.1039/D0CB00099J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palombi I. R.; Lawrence N.; White A. M.; Gare C. L.; Craik D. J.; McMorran B. J.; Malins L. R. Development of antiplasmodial peptide-drug conjugates using a human protein-derived cell-penetrating peptide with selectivity for infected cells. Bioconjugate Chem. 2023, 34 (6), 1105–1113. 10.1021/acs.bioconjchem.3c00147. [DOI] [PubMed] [Google Scholar]
- Assem N.; Ferreira D. J.; Wolan D. W.; Dawson P. E. Acetone-linked peptides: a convergent approach for peptide macrocyclization and labeling. Angew. Chem., Int. Ed. Engl. 2015, 54 (30), 8665–8668. 10.1002/anie.201502607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M.; Zaretskaya I.; Raytselis Y.; Merezhuk Y.; McGinnis S.; Madden T. L. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008, 36 (Web Server), W5–W9. 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torcato I. M.; Huang Y. H.; Franquelim H. G.; Gaspar D. D.; Craik D. J.; Castanho M. A.; Henriques S. T. The antimicrobial activity of Sub3 is dependent on membrane binding and cell-penetrating ability. Chembiochem 2013, 14 (15), 2013–2022. 10.1002/cbic.201300274. [DOI] [PubMed] [Google Scholar]
- Henriques S. T.; Lawrence N.; Chaousis S.; Ravipati A. S.; Cheneval O.; Benfield A. H.; Elliott A. G.; Kavanagh A. M.; Cooper M. A.; Chan L. Y.; Huang Y. H.; Craik D. J. Redesigned spider peptide with improved antimicrobial and anticancer properties. ACS Chem. Biol. 2017, 12 (9), 2324–2334. 10.1021/acschembio.7b00459. [DOI] [PubMed] [Google Scholar]
- Wiedman G.; Kim S. Y.; Zapata-Mercado E.; Wimley W. C.; Hristova K. pH-triggered, macromolecule-sized poration of lipid bilayers by synthetically evolved peptides. J. Am. Chem. Soc. 2017, 139 (2), 937–945. 10.1021/jacs.6b11447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S.; Avery V. M. Development and optimization of a novel 384-well anti-malarial imaging assay validated for high-throughput screening. Am. J. Trop. Med. Hyg. 2012, 86 (1), 84–92. 10.4269/ajtmh.2012.11-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiguemde W. A.; Shelat A. A.; Bouck D.; Duffy S.; Crowther G. J.; Davis P. H.; Smithson D. C.; Connelly M.; Clark J.; Zhu F.; Jiménez-Díaz M. B.; Martinez M. S.; Wilson E. B.; Tripathi A. K.; Gut J.; Sharlow E. R.; Bathurst I.; Mazouni F. E.; Fowble J. W.; Forquer I.; McGinley P. L.; Castro S.; Angulo-Barturen I.; Ferrer S.; Rosenthal P. J.; Derisi J. L.; Sullivan D. J.; Lazo J. S.; Roos D. S.; Riscoe M. K.; Phillips M. A.; Rathod P. K.; Van Voorhis W. C.; Avery V. M.; Guy R. K. Chemical genetics of Plasmodium falciparum. Nature 2010, 465 (7296), 311–315. 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro J.; Grundy L.; Deiteren A.; Harrington A. M.; O’Donnell T.; Maddern J.; Moore J.; Garcia-Caraballo S.; Rychkov G. Y.; Yu R.; Kaas Q.; Craik D. J.; Adams D. J.; Brierley S. M. Cyclic analogues of α-conotoxin Vc1.1 inhibit colonic nociceptors and provide analgesia in a mouse model of chronic abdominal pain. Br. J. Pharmacol. 2018, 175 (12), 2384–2398. 10.1111/bph.14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernen F.; Harvey P. J.; Dias S. A.; Veiga A. S.; Huang Y.-H.; Craik D. J.; Lawrence N.; Troeira Henriques S. Characterization of tachyplesin peptides and their cyclized analogues to improve antimicrobial and anticancer properties. Int. J. Mol. Sci. 2019, 20 (17), 4184. 10.3390/ijms20174184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saska I.; Gillon A. D.; Hatsugai N.; Dietzgen R. G.; Hara-Nishimura I.; Anderson M. A.; Craik D. J. An asparaginyl endopeptidase mediates in vivo protein backbone cyclization. J. Biol. Chem. 2007, 282 (40), 29721–29728. 10.1074/jbc.M705185200. [DOI] [PubMed] [Google Scholar]
- Jackson M. A.; Gilding E. K.; Shafee T.; Harris K. S.; Kaas Q.; Poon S.; Yap K.; Jia H.; Guarino R.; Chan L. Y.; Durek T.; Anderson M. A.; Craik D. J. Molecular basis for the production of cyclic peptides by plant asparaginyl endopeptidases. Nat. Commun. 2018, 9 (1), 2411. 10.1038/s41467-018-04669-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia F.; Wang J.; Peng J.; Zhao P.; Kong Z.; Wang K.; Yan W.; Wang R. D-amino acid substitution enhances the stability of antimicrobial peptide polybia-CP. Acta Biochim. Biophys. Sin. 2017, 49 (10), 916–925. 10.1093/abbs/gmx091. [DOI] [PubMed] [Google Scholar]
- Böttger R.; Hoffmann R.; Knappe D. Differential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serum. PLoS One 2017, 12 (6), e0178943 10.1371/journal.pone.0178943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Xu H.; Xia J.; Ma J.; Xu J.; Li Y.; Feng J. D- and unnatural amino acid substituted antimicrobial peptides with improved proteolytic resistance and their proteolytic degradation characteristics. Front. Microbiol. 2020, 11, 563030. 10.3389/fmicb.2020.563030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayogu E. E.; Mosanya A. U.; Onuh J. C.; Adibe M. O.; Ubaka C. M.; Ukwe C. V. Direct medical cost of treatment of uncomplicated malaria after the adoption of artemisinin-based combination therapy in Nigeria. J. Appl. Pharm. Sci. 2021, 11 (9), 029–034. 10.7324/JAPS.2021.110904. [DOI] [Google Scholar]
- Ismail N. E.; Jimam N. S.; Goh K. W.; Tan C. S.; Ming L. C. Economic burdens of uncomplicated malaria in primary health care (PHC) facilities of Plateau State, Nigeria: patients’ perspectives. Int. J. Environ. Res. Public Health 2023, 20 (2), 1093. 10.3390/ijerph20021093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wani K. I.; Choudhary S.; Zehra A.; Naeem M.; Weathers P.; Aftab T. Enhancing artemisinin content in and delivery from Artemisia annua: a review of alternative, classical, and transgenic approaches. Planta 2021, 254 (2), 29. 10.1007/s00425-021-03676-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayani W. K.; Kiani B. H.; Dilshad E.; Mirza B. Biotechnological approaches for artemisinin production in Artemisia. World J. Microbiol. Biotechnol. 2018, 34 (4), 54. 10.1007/s11274-018-2432-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medicines for Malaria Venture . African Drug Manufacturing. 2024. https://www.mmv.org/our-work/access-to-medicines/african-drug-manufacturing (accessed May 31, 2024).
- Wang J.; Chen L.; Qin S.; Xie M.; Luo S.-Z.; Li W. Advances in biosynthesis of peptide drugs: Technology and industrialization. Biotechnol. J. 2024, 19 (1), 2300256. 10.1002/biot.202300256. [DOI] [PubMed] [Google Scholar]
- Yap K.; Du J.; Looi F. Y.; Tang S. R.; de Veer S. J.; Bony A. R.; Rehm F. B. H.; Xie J.; Chan L. Y.; Wang C. K.; Adams D. J.; Lua L. H. L.; Durek T.; Craik D. J. An environmentally sustainable biomimetic production of cyclic disulfide-rich peptides. Green Chem. 2020, 22 (15), 5002–5016. 10.1039/D0GC01366H. [DOI] [Google Scholar]
- Hoelscher M. P.; Forner J.; Calderone S.; Krämer C.; Taylor Z.; Loiacono F. V.; Agrawal S.; Karcher D.; Moratti F.; Kroop X.; Bock R. Expression strategies for the efficient synthesis of antimicrobial peptides in plastids. Nat. Commun. 2022, 13 (1), 5856. 10.1038/s41467-022-33516-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M. A.; Xie J.; Nguyen L. T. T.; Wang X.; Yap K.; Harvey P. J.; Gilding E. K.; Craik D. J. Plant-based production of an orally active cyclotide for the treatment of multiple sclerosis. Transgenic Res. 2023, 32 (1–2), 121–133. 10.1007/s11248-023-00341-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M. A.; Yap K.; Poth A. G.; Gilding E. K.; Swedberg J. E.; Poon S.; Qu H.; Durek T.; Harris K.; Anderson M. A.; Craik D. J. Rapid and scalable plant-based production of a potent plasmin inhibitor peptide. Front. Plant Sci. 2019, 10, 602 10.3389/fpls.2019.00602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro B. A.; McMorran B. J. Antimalarial drug strategies to target Plasmodium x2gametocytes. Parasitologia 2022, 2 (2), 101–124. 10.3390/parasitologia2020011. [DOI] [Google Scholar]
- Andersson D. I.; Hughes D.; Kubicek-Sutherland J. Z. Mechanisms and consequences of bacterial resistance to antimicrobial peptides. Drug Resist. Updat. 2016, 26, 43–57. 10.1016/j.drup.2016.04.002. [DOI] [PubMed] [Google Scholar]
- Benfield A. H.; Vernen F.; Young R. S. E.; Nadal-Bufí F.; Hammerlindl H.; Craik D. J.; Schaider H.; Lawrence N.; Blanksby S. J.; Henriques S. T. Cyclic tachyplesin I kills proliferative, non-proliferative and drug-resistant melanoma cells without inducing resistance. Pharmacol. Res. 2024, 207, 107298 10.1016/j.phrs.2024.107298. [DOI] [PubMed] [Google Scholar]
- Huang Y.-C.; Chen C.-C.; Li S.-J.; Gao S.; Shi J.; Li Y.-M. Facile synthesis of C-terminal peptide hydrazide and thioester of NY-ESO-1 (A39-A68) from an Fmoc-hydrazine 2-chlorotrityl chloride resin. Tetrahedron 2014, 70 (18), 2951–2955. 10.1016/j.tet.2014.03.022. [DOI] [Google Scholar]
- Bird M. J.; Dawson P. E. A shelf stable Fmoc hydrazine resin for the synthesis of peptide hydrazides. Pept. Sci. 2022, 114 (5), e24268 10.1002/pep2.24268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cergol K. M.; Thompson R. E.; Malins L. R.; Turner P.; Payne R. J. One-pot peptide ligation-desulfurization at glutamate. Org. Lett. 2014, 16 (1), 290–293. 10.1021/ol403288n. [DOI] [PubMed] [Google Scholar]
- Greenfield N. J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1 (6), 2876–2890. 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd N. E.; Hoang H. N.; Abbenante G.; Fairlie D. P. Single turn peptide alpha helices with exceptional stability in water. J. Am. Chem. Soc. 2005, 127 (9), 2974–2983. 10.1021/ja0456003. [DOI] [PubMed] [Google Scholar]
- Figueira T. N.; Freire J. M.; Cunha-Santos C.; Heras M.; Gonçalves J.; Moscona A.; Porotto M.; Salomé Veiga A.; Castanho M. A. R. B. Quantitative analysis of molecular partition towards lipid membranes using surface plasmon resonance. Sci. Rep. 2017, 7 (1), 45647. 10.1038/srep45647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.