


Abstract
The human Ku86 gene and an isoform, KARP-1 (Ku86 autoantigen related protein-1), encode overlapping, but differentially regulated, transcripts. Ku86 is constitutively transcribed at high levels and, although it plays a seminal role in DNA double-strand break repair, its expression is not induced by DNA damage. KARP-1, in contrast, is expressed constitutively only at low levels and its expression is induced by DNA damage in a p53-dependent fashion. The regulatory elements promoting KARP-1 gene expression and p53 responsiveness, however, were unknown. Here, we report that a strong DNase I hypersensitive site (DHS) resides ∼25 kb upstream from the Ku86 promoter. This DHS is encompassed by a hypomethylated CpG island. Reporter assays demonstrated that this region corresponded to a promoter(s), which promoted transcription of peroxisomal trans-2-enoyl CoA reductase in the centromeric direction and KARP-1 in the telomeric direction. KARP-1 primer extension products were mapped to this CpG island in the correct transcriptional orientation confirming that KARP-1 transcription initiates from this site. Moreover, a p53 response element within the first intron of the KARP-1 transcriptional unit was identified using chromatin immunoprecipitation and antibodies specific to activated forms of p53. These data expand our understanding of this important DNA repair locus.
INTRODUCTION
Among the many forms of damage that can cause genomic instability, DNA double strand breaks (DSBs) appear to be the most insidious (reviewed in 1,2). DSBs can occur spontaneously during DNA replication, are formed transiently during meiosis and lymphoid V(D)J recombination and can arise in patients exposed to chemo- or radiotherapeutic agents (1). Improper repair of DSBs results in gross chromosomal rearrangements involving translocations, inversions and fusions, which invariably lead to oncogenic transformation or cell death (reviewed in 3). Mammalian cells have evolved at least two independent pathways for repairing DSBs: homologous recombination and non-homologous DNA end joining (NHEJ) (reviewed in 1,4). Homologous recombination ensures relatively error-free repair by using an undamaged sister chromatid or homologous chromosome as a template. NHEJ, on the other hand, uses limited sequence homology to rejoin ends in a manner that is often error prone. In mammalian cells, NHEJ is the preferred mechanism of DSB repair (4). The gene products involved in this pathway minimally include Ku70, Ku86, the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), XRCC4 and DNA ligase IV (reviewed in 4,5).
Ku is a heterodimeric DNA end-binding complex composed of 70 and 86 kDa subunits (Ku70 and Ku86, respectively; reviewed in 5,6) that binds in a sequence non-specific fashion to virtually all double-stranded DNA ends (7,8). The binding of Ku to broken DNA ends is required to prevent unnecessary DNA degradation (9) and juxtapose the DNA ends (10–12). One other unequivocal role for Ku is as a DNA-binding subunit of the DNA-PK complex (DNA-PK), a serine–threonine protein kinase that is composed of the Ku heterodimer and DNA-PKcs (13). Extensive genetic and molecular studies have identified the DNA-PK complex as an integral component of mammalian DNA DSB repair and the fundamental mediator of NHEJ (5). The binding of Ku to free DNA ends recruits and activates DNA-PKcs (14,15), DNA ligase IV (16,17) and XRCC4, a DNA ligase IV accessory factor (18,19), which are required for the rejoining of DNA DSBs (20–23).
Murine knockouts for each of the components of the DNA-PK complex have been generated. Mice deficient for Ku70 (24,25), Ku86 (26,27) or DNA-PKcs (28–31) are viable and exhibit the expected immune deficiencies and ionizing radiation (IR) hypersensitivity. In addition, inactivation of the Ku86 gene results in murine (26) cells with a severe growth retardation, premature senescence (32), a marked increase in chromosomal aberrations (33–35) and elevated telomeric fusions (36–38). Some of these phenotypes are also observed in human somatic cells lacking Ku86 expression (39).
In primates in general, the situation is a bit more complex as the Ku86 locus is bifunctional and, through the use of an unidentified upstream promoter, encodes a Ku86 isoform, KARP-1 (Ku86 autoantigen related protein-1) (40). The KARP-1 message is ∼0.4 kb longer than Ku86 and the KARP-1 protein contains a 9 kDa N-terminal extension not found on Ku86 (40). Overexpression of the KARP-1-specific domain conferred a dominant negative IR sensitive phenotype to human cells and a KARP-1 polyclonal antibody inhibited DNA-PK activity in vitro, suggesting a role for KARP-1 in the regulation of the DNA-PK complex and in human DNA DSB repair. Subsequently, it was shown that KARP-1 expression was strongly induced following DNA damage and that the up-regulation of KARP-1 mRNA and protein was dependent upon functional p53 and ataxia telangiectasia mutated (ATM) gene products (41).
Intriguingly, Ku86 is not induced by IR (42,43). Specifically, Ku86 appears to be a constitutively transcribed ‘housekeeping’ gene, which is expressed from a TATA-less, CpG-rich promoter (41,44). Primer extension analysis of the Ku86 gene coupled with standard promoter bashing experiments mapped the putative +1 transcription start site to a CpG-rich region (40,44). In addition, a putative transcription start site for KARP-1 was identified in the nearby, flanking upstream sequences based upon 5′-RACE reactions (40). We demonstrate here, however, that these cDNAs represented incomplete extension products. Instead, through the use of a thermostable reverse transcriptase at elevated temperatures, which resulted in the successful elimination of significant secondary structures, complete extension of the KARP-1 message was obtained resulting in the identification of ∼150 bp of additional cDNA sequence. Importantly, the 5′-most terminal 17 nt of this additional sequence mapped to a second CpG-rich region that resided ∼25 kb upstream of the Ku86 promoter. This second CpG-rich region was hypomethylated, coincident with a strong, constitutive DNase I hypersensitive site (DHS) and was ultimately shown to promote transcription for KARP-1 and the biochemically unrelated gene, TERP (trans-2-enoyl-co- enzymeA reductase, peroxisomal). Interestingly, this promoter configuration is similar to that observed for breast cancer allele-1 (BRCA-1) (45–47), DNA-PKcs (48) and ATM (49,50), all of which are divergently transcribed from bidirectional promoters, which also promote transcription of biologically unrelated genes. Lastly, we identified two consensus putative p53 responsive elements (REs) that potentially could have been responsible for inducing KARP-1 gene expression following DNA damage, and demonstrate, using chromatin immunoprecipitation and antibodies specific for activated forms of p53, that one of these REs is biologically responsive whereas the other is not.
MATERIALS AND METHODS
Cells
The human corectal carcinoma HCT116 (51,52) and HCT116+Chr3 (53) cell lines were generously provided to us by Drs Bert Vogelstein and Kyungjae Myung, respectively. Cells were cultured in McCoy’s 5A medium supplemented with 100 µg/ml penicillin and streptomycin and 2 mM l-glutamine. The cells were incubated in a humidified 37°C incubator containing 5% CO2.
DNase I hypersensitivity assays
The basic protocol has been described (54,55) and all preparatory steps were carried out at 4°C. Briefly, 1.5–2 × 108 trypsinized cells were pelleted by centrifugation at 2000 r.p.m. (Beckman Allegra 6R) for 10 min. The cells were washed twice by resuspension in 25 mM Mg/Ca-free phosphate buffered saline (PBS) and centrifugation at 1500 r.p.m. for 10 min. The cells were then resuspended in freshly made 20 mM Tris–HCl pH 7.4, 3 mM CaCl2, 2 mM MgCl2, 0.3% NP-40 and transferred to a dounce homogenizer. Following incubation on ice for 10 min and removal of an aliquot to quantitate cellular breakage, cells were dounced by approximately six hard strokes and checked by visual observation under a microscope. Following quantitative breakage, nuclei were isolated by centrifugation at 900 r.p.m. for 7 min. Nuclei were washed twice in 25 ml of resuspension buffer (RSB) (10 mM Tris–HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2) followed by centrifugation at 900 r.p.m. for 7 min. Nuclei were then resuspended in 1 ml of RSB and a 25 µl aliquot was diluted into 475 µl of 1% SDS for an OD260 measurement. The nuclear stock was diluted with RSB until the OD260 of a 1:20 dilution equaled 0.5. A ‘quenched’, i.e. control, nuclei sample was removed and added directly to cell lysis solution (CLS) (Gentra Systems, Minneapolis, MN). The purified, diluted nuclei were divided into untreated and treated aliquots and warmed to 37°C. For the untreated aliquot, an ‘endogenous’ DNase I activity sample was collected at 5 min and added to CLS. For the treated aliquot, RQDNase I (∼1 µg/5 µl; Clontech, Palo Alto, CA) was added to a final concentration of 1 µg/ml of nuclei. Aliquots of the treated nuclei were removed at 30 s, 2 min and 5 min and added to CLS and mixed well. Genomic DNA was isolated from each sample as described in the DNA Isolation Kit protocol (Gentra Systems; scaled up based on cell number) and then resuspended in 10 mM Tris–HCl pH 8.0 at 0.5 µg/µl. Genomic DNA was then analyzed by Southern blotting to detect DHSs at particular loci.
Southern blots
All blots were performed with 10 µg of genomic DNA/lane in agarose gels (0.6–2.0% agarose) following treatment with the appropriate restriction enzyme(s) as shown in each figure legend. Double digestions were performed simultaneously. Ethidium bromide-stained agarose gels were photographed and subjected to capillary transfer to nitrocellulose membranes by deaminating with 0.25 N HCl for 20 min, followed by two 20 min treatments with 1.5 M NaCl, 0.5 M NaOH and two 20 min treatments with 1.0 M Tris–HCl pH 8.0, 1.5 M NaCl. Capillary transfer was carried out in 20× SSC (3.0 M NaCl, 0.3 M Na citrate) overnight. DNA was UV crosslinked to the nitrocellulose with a Stratalinker (Stratagene, La Jolla, CA) and washed with 5× SSC for 5 min at room temperature before pre-hybridization. Hybridizations were performed according to the PerfectHyb Plus protocol (Sigma, St Louis, MO) with a 5 min prehybridization at 64°C with sheared salmon sperm DNA followed by the addition of 32P-radiolabeled probe prepared via the random hexamer Klenow synthesis Prime-It II protocol (Stratagene) and subsequently purified over a G-50 Sephadex column. Following overnight hybridizations, the blots were washed twice at room temperature with 2× SSC for 5 min, twice (once at room temperature, once at 64°C) with 1× SSC for 10–20 min and two to four times at 64°C with 0.1× SSC. The exposure of the blots to Kodak autoradiographic film at –80°C in the presence of an intensifying screen varied from 1 h to 3 days.
RNA isolation
Messenger RNA isolation from tissue culture cells was performed with Invitrogen’s FastTrack mRNA isolation kit utilizing poly(A) binding resin as described by the manufacturer.
Reverse transcriptase–PCR (RT–PCR) and 5′-RACE
RT–PCR was performed essentially as described (56). 5′-RACE reactions utilized an mRNA template for a single round of hybridization and reverse transcription with a message-specific primer using Gibco/BRL’s Thermoscript™ reverse transcriptase at 55°C to remove secondary structures. The resulting single-stranded cDNA was then extended with terminal deoxynucleotidyl-transferase and dATP to create a poly(A) tail. Reiterative PCR cycles were subsequently performed using nested message-specific primers and a poly(T) + adaptor primer (5′-GACTCGAGTCGACATCGATTTTTTTTTTTTTTTTT). Secondary PCR was performed, as needed, with an additional message-specific primer and an adaptor-specific primer (5′-GACTCGAGTCGACATCG). PCR products were gel purified using Qiagen gel elution columns and were subcloned into pCR2.1, a TA-overhang PCR cloning vector (Invitrogen, Carlsbad, CA). Ampicillin-resistant bacterial colonies were expanded in liquid culture and plasmid DNAs were isolated using miniprep columns (Qiagen, Valencia, CA) and the DNAs were subjected to restriction endonuclease digestion to assay for presence of adaptor-specific and message-specific restriction enzyme sites. Appropriate DNAs were subjected to automated DNA sequencing (Davis- Sequencing, Davis, CA) with the vector-specific primers, Rev and T3 (Operon, Alameda, CA).
Additional confirmatory 5′-RACE was performed with the FirstChoice™ RLM-RACE Kit (Ambion, Austin, TX) as recommended by the supplier with the exception that Thermoscript™ reverse transcriptase was utilized. Briefly, preparations of mRNA were treated with calf intestinal phosphatase to remove 5′-terminal phosphates from degraded mRNA, rRNA, tRNA and DNA. Subsequent tobacco alkaline phosphatase treatment removed the guanine caps from full-length mRNA and adapters were ligated onto the decapped mRNA. Reverse transcription reactions were then performed with a message-specific primer and reiterative PCR was performed with nested message-specific primers and an adaptor-specific primer. PCR products were subcloned and characterized as described above.
For the DNA damage inducibility studies, cDNA was made from mRNA harvested 2 h post mock or IR (10 Gy) treatment of HCT116 cells. The cDNA was used as template in RT–PCR reactions with primers specific to different exons within actin (to control for quantification of cDNA pools), TERP or KARP-1 messages. Serial 3-fold dilutions were prepared to allow visual quantitation of message level differences. Twenty-four PCR cycles were utilized for the actin control, 26 for TERP and 33 for KARP-1.
Chromatin immunoprecipitations
Modifications of this method (57) included harvesting in PBS solution pH 7.6 and crosslinking with 2% formaldehyde. Crosslinking was quenched by the addition of 125 mM glycine for 5 min followed by two ice-cold washes with PBS. Cells were then permeablized in lysis buffer (50 mM HEPES–KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate/protease inhibitors) and kept on ice. Chromatin was sheared by sonication until the DNA was an average length of 600–1000 bp as assessed by agarose gel electrophoresis. Cellular debris was removed by centrifugation for 5 min at 13 000 r.p.m. (Beckman centrifuge Model 5415C) and then the supernatant was centrifuged again for 15 min. A one-twentieth aliquot of the cleared extract was reserved as an input control for PCR and the remaining extract was incubated overnight with antibodies at 4°C on a rotator. A protein A sepharose slurry (50 µl) was added, incubated for 1.5 h at 4°C while rotating and then the sepharose beads were pelleted by centrifugation. The beads were washed twice at room temperature with lysis buffer, once with lysis buffer containing 500 mM NaCl, once with 10 mM Tris–HCl pH 8.0, 250 mM LiCl, 0.5% NP-40, 0.5% Na-deoxycholate, 1 mM EDTA and once with 10 mM Tris–HCl pH 8.0, 1 mM EDTA. The beads were then resuspended in 200 µl of 50 mM Tris–HCl pH 8.0, 10 mM EDTA and the precipitated protein:DNA complexes were eluted from the antibodies:beads by incubation at 65°C for 30 min. The resulting supernatants, along with input aliquots, were subjected to crosslink reversal by heating to 65°C overnight followed by treatment with 100 µg of proteinase K for 2 h at 37°C. DNA was purified by phenol:chloroform extraction followed by EtOH precipitation after the addition of 2 µg of glycogen carrier and a one-tenth volume of 3 M NaOAc. Precipitated DNA was resuspended in 100 µl of water and one-fiftieth of the immunoprecipitates or 1/10 000 of the input were used as templates in PCR. For the KARP-1 10(1)10 chromatin immunoprecipitation (ChIP)–PCR, which generated a 278 bp product, annealing was carried out at 53°C. PCR was performed for 35 cycles for Ab421 and the phospho-specific mAb and for 30 cycles for the polyclonal Ab and the acetyl-specific mAb.
Antibodies, primer sequences and Southern blot probes
Antibodies used in the chromatin immunoprecipitation analysis included a p53 polyclonal (Santa Cruz, Santa Cruz, CA) antibody, anti-acetyllysine373-p53 (Upstate Biotechnology, Lake Placid, NY), anti-phosphoserine15-p53 (Cell Signaling Technology, Beverly, MA) and epitope-specific p53 (Ab421, CalBioChem, Palo Alto, CA) antibodies and a p21/WAF1 (CalBioChem) antibody. The PCR primers used for the KARP-1 10(1)10 chromatin immunoprecipitation assays were KP-ChIP-9 (5′-AAGATGAGGAAGAGATGGGG) and KP-ChIP-10 (5′-TGAGTCAGAAGTGTGAGAGTG). DNase I hypersensitivity assay Southern blot probes were generated as follows. The probe used to detect the Ku86 promoter locus was an EagI–PstI 439 bp restriction fragment subcloned from a genomic plasmid clone of the locus. The probe used to detect the KARP-1 promoter locus was an XmaI–BglII 1064 bp restriction fragment derived from a 1812 bp ScaI GenomeWalker (Clontech) product generated with the primers K63-3 (5′-TCTTGACACCCGAACTAAAAACTTGAC) and K63-4 (5′-CTCCCTGCTCTGCCTCTCATTATTC). The primer pairs utilized for the DNA damage inducibility studies were for actin, ACTIN5′ (5′-ATCTGGCACCACACCTTCTACAATGAGCTGCG-3′) and ACTIN3′ (5′-CGTCATACTCCTGCTTGCTGATCCACATCTGC-3′); for TERP, TERP-2 (5′-CGGAAAAGCCATCGTGAAG-3′) and TERP-3 (5′-CGGATGTTGCATTGTATGGG-3′); for KARP-1, ASPEK12 (5′-CGTACAAGAAGGGAGACAAGGACCACTGAC-3′) and PHYGW01 (5′-CTTATTCCCCGACCGCACCATGTTGCCGGT-3′).
RESULTS
Characterization of the Ku86 upstream region as the potential start of KARP-1 transcription
Extensive 5′-RACE analyses were used to map the 5′-end of KARP-1 transcription to a region ∼3 kb upstream from the start of Ku86 transcription (exon 2, Fig. 1B) (40). Since no longer 5′-RACE products could be obtained initially, we investigated whether this area corresponded to the start of KARP-1 transcription even though there were no obvious signature promoter motifs. To test this hypothesis, a variety of upstream sequences were subcloned into a luciferase reporter vector. These vectors were then transfected into human colorectal carcinoma HCT116 cells (51,52) and assayed for promoter activity with and without prior X-irradiation. None of the constructs showed detectable promoter activity (data not shown). To confirm this negative data we carried out DNase I hypersensitivity assays (58). These assays, which measure chromatin accessibility, have the ability to scan for changes in chromatin structure between normal and induced states and have been extensively utilized to identify promoter regions (reviewed in 55,59). In particular, RNA polymerase II is an extraordinarily large complex that must be able to interact physically with sequences directly upstream of transcriptional start sites. Access of such a large complex to chromatin usually requires the complete disruption or shifting of nucleosomes in the immediate vicinity, generating a region that invariably becomes accessible to DNA cleaving enzymes, such as DNase I. Active transcriptional initiation regions therefore are associated with hyperaccessibility of DNase I to the chromatin in that immediate area (59). Thus, nuclei from unirradiated and X-irradiated HCT116 cells were treated with DNase I at various times post-irradiation. Genomic DNA was isolated from these cells, treated with an appropriate restriction enzyme and analyzed by Southern blotting with a probe from the Ku86 upstream region to reveal preferred sites of DNase I cutting. The region analyzed, which encompassed the Ku86 promoter and two additional upstream non-coding exons (exons 2 and 3, Fig. 1A and B) of KARP-1 revealed that the Ku86 promoter was hypersensitive to DNase I in an X-irradiation independent manner (Fig. 1C). Comparison of molecular sizes of hybridizing bands within genomic DNA digested with multiple restriction enzymes permitted the accurate placement of two closely spaced, constitutive DHSs to just upstream of the Ku86 transcriptional start site (Fig. 1C). The location of the DHSs was virtually coincident with the +1 of Ku86 transcription determined by primer extension studies (40,44, C. D. Braastad, unpublished data). Similarly, the constitutive presence of the DHSs was consistent with the constitutive, high level, non-inducible nature of Ku86 expression (42,43). Importantly, however, no DHSs were detected in the vicinity of exon 2 (Fig. 1C and data not shown). The lack of a detectable open chromatin structure, especially following X-irradiation, coupled with the negative data from the reporter constructs strongly indicated that the region directly upstream of exon 2 was not likely to be the KARP-1 promoter.
Figure 1.

A DHS(s) resides at the Ku86 promoter. (A) Skeletal diagram of the human Ku86 locus. The blue hatched rectangles represent exons. The Ku86 +1 of transcription is shown as an arrow. The green circles represent the putative p53 response elements. The orange rectangles correspond to CpG islands. (B) Exploded view of the Ku86 promoter region. The exons are numbered (yellow circles) in accord with the KARP-1 transcription unit. Thus, KARP-1 exon 4, which includes the initiator ATGs for both KARP-1 and Ku86, is the first exon of the Ku86 transcript (40). The location of the DHSs are shown by vertical arrows. The EagI to PstI restriction fragment that was used as a probe is shown as a red rectangle. Relevant restriction enzyme sites are also shown. (C) Two closely spaced DHSs map in the vicinity of the +1 for Ku86 transcription. Nuclei from control HCT116 cells (Non-Irr.) or cells which had been X-irradiated (10 Gy) were treated with DNase I for the indicated lengths of time and then processed, restricted with EcoRI and analyzed by Southern blotting as described in Materials and Methods. The endogenous band is ∼5 kb and the position of the bands resulting from DNase I cutting at ∼2.1 and ∼2.0 kb (horizontal arrows) are shown.
Identification of a DHS upstream of the Ku86 promoter by DNase I hypersensitivity walking
The experiments described above suggested that the authentic KARP-1 promoter must lie further upstream of exon 2. To identify the promoter, a series of DHS assays were carried out over the upstream 50 kb in overlapping steps of 6–10 kb (Fig. 2A and B and data not shown). A single, strong DHS was identified ∼25 kb upstream of exon 2 (Fig. 2B and C). This DHS, like those observed at the Ku86 promoter (Fig. 1), was also constitutive and not X-irradiation inducible (Fig. 2C). To confirm this result, nuclei isolated at various intervals following X-irradiation up to 1.5 h post-irradiation were also analyzed. Although KARP-1 transcription is known to be greatly induced during this time interval (41), no salient differences were detected in the DHS profile nor were any novel DHS sites identified (Fig. 2D). Thus, a constitutive, non-DNA damage inducible DHS site exists ∼25 kb upstream of the last known KARP-1 exon.
Figure 2.

A DHS site resides ∼25 kb upstream of the Ku86 promoter. (A) Skeletal diagram of the human Ku86 locus and the flanking upstream genomic DNA. The blue hatched rectangles represent exons. The Ku86 +1 of transcription is shown as an arrow. The orange rectangles correspond to CpG-rich regions. The green circles represent the putative p53 response elements. (B) Exploded view of the upstream CpG-rich region. The location of the DHSs are shown by vertical arrows. The approximate location of the restriction fragment that was used as a Southern blot probe is shown as a red rectangle. Relevant restriction enzyme sites are also shown. (C) A DHS resides in the vicinity of the CpG-rich region. Nuclei from control HCT116 cells (Non-Irr) or cells which had been X-irradiated (10 Gy) were treated with DNase I for the indicated lengths of time and then processed, restricted with either KpnI and BamHI (top two blots) or just KpnI (bottom two blots) and then analyzed by Southern blotting as described in Materials and Methods. The positions of the bands resulting from DNase I cutting are shown as horizontal arrows on the right. (D) No alterations to the DHS profile are observed post-irradiation. All conditions and symbols are as indicated in (C) except that the nuclei were first prepared at the indicated times post-irradiation and treated with DNase I for 5 min.
The upstream DHS corresponds to a hypomethylated CpG island
Sequence analysis of the region encompassing the upstream DHS revealed that it was coincident with a potential strong CpG island (Fig. 3B). The CpG dinucleotide is under-represented in the human genome and it has a tendency to cluster in regions known as ‘CpG islands’ (60,61). These islands often correspond to the promoters of genes (61) and indeed the only other CpG island within 100 kb is the one associated with the Ku86 promoter (Fig. 3A and data not shown). Importantly, however, in order for a CpG island to actively promote transcription, the cytosine residues need to be hypomethylated, since random CpG dinucleotides and inactive promoters are often hypermethylated (62,63). Thus, a series of Southern blot analyses were performed using a variety of methyl-sensitive and methyl-insensitive restriction enzyme pairs (AvaI and BsoBI, HpaII and MspI, KasI and SfoI, SmaI and XmaI; Fig. 3D–F, lanes 3 and 4, 7 and 8, 9 and 10, 13 and 14, respectively) as well as restriction enzymes whose recognition sequences were enriched in the CpG dinucleotide (e.g. EagI and SacII; Fig. 3D–F, lanes 5 and 12, respectively). These experiments showed that an ∼1 kb region (corresponding precisely to the middle of the CpG island and the location of the DHS; Fig. 3C) was completely hypomethylated, whereas upstream and downstream CpGs were semi or fully methylated (Fig. 3E and data not shown). To control for the efficiency of cutting and to ascertain if all of the restriction enzymes were functioning as expected, genomic DNA, which had been treated with the SssI CpG-specific cytosine methylase, was also digested. This DNA was refractory to cleavage by methyl-sensitive restriction enzymes and cleaved to completion by restriction enzymes that were methyl-insensitive with the exception of SfoI, which only partially cleaved the DNA (Fig. 3F). From these experiments we concluded that this CpG island was extensively hypomethylated and that this was consistent with it being a putative promoter region.
Figure 3.

The upstream CpG island is hypomethylated. (A) Identical to Figure 2. (B) Exploded view of the upstream CpG-rich region. Thin vertical lines represent CpG dinucleotides. The methylation status of the CpGs is designated as completely unmethylated (open circles), partially methylated (gray circles) and completely methylated (closed circles). The probe used for the Southern blot shown in (D) is shown as a red rectangle and the probe used for the Southern blots shown in (E) and (F) is shown as a green rectangle. (C) Exploded view of the central section of the upstream CpG-rich region. All symbols are as in (B). Methylation-sensitive restriction enzymes are shown directly beneath CpG dinucleotides contained within the restriction site. (D and E) A methylation status assay using cleavage by methylation-sensitive and -insensitive restriction enzymes. Genomic DNA was digested with BamHI and BglII, the digested DNA was purified and resuspended, and digested with the indicated third restriction enzyme. Four different isoschizomer pairs of methyl-sensitive or methyl-insensitive restriction enzymes were loaded adjacent to one another and are indicated by superscripts (1, 2, 3 or 4). (D) was probed with the distal portion of the CpG island whereas (E) was probed with the proximal portion [see (B)]. (F) A control for restriction enzyme cutting. Purified genomic DNA was methylated in vitro and then subjected to the same analysis as described for (D) and (E). Only restriction enzymes (BsoBI, MspI, SfoI and XmaI) which were methyl insensitive were able to cleave the DNA [lanes 4, 8, 10 (partially) and 14].
The CpG island promotes divergent transcription of TERP and KARP-1
When the 1.7 kb putative promoter region was subcloned into a luciferase reporter vector it exhibited low, albeit reproducible, promoter activity, although, confusingly, it appeared to be active in either orientation (data not shown). The explanation for this became clear with the publication of the sequence of the human genome which showed that this CpG island corresponded to a promoter(s) which directs transcription in the centromeric direction of TERP [an enzyme involved in fatty acid biosynthesis that is expressed primarily in the kidney and liver (64)] and transcription in the telomeric direction of an unknown gene (Fig. 4A). To confirm that this unknown gene corresponded to KARP-1, additional 5′-RACE experiments using primers to KARP-1 exons 2, 3 and 4 were performed. Importantly, unlike our earlier 5′-RACE attempts (40), these experiments utilized a thermostable reverse transcriptase at high temperature in order to penetrate the now known CpG-rich region (see Materials and Methods). Four of the resulting cDNAs contained ∼150 nt of additional novel sequence that extended exon 2 in the 5′ direction ∼130 nt. More importantly, these cDNAs contained at their 5′-ends a 14–17 nt sequence identical to a sequence within the CpG island (Fig. 4B and C). Thus, this ∼17 bp sequence defines exon 1 for KARP-1 and it is contained within, though in the opposite transcriptional orientation of, the first exon of TERP (Fig. 4A and B). There are no obvious TATA- or CCAAT-like sequences in the region immediately upstream of KARP-1 exon 1. There is, however a five out of seven match and a six out of seven match for a consensus initiator (Inr) sequence [5′-YYAN(T/A)YY-3′; (65)] from –57 to –51, TCACGAT and –13 to –7, CCACTTG, respectively (Fig. 4C). These sequences contain the C at +2 (counting the 5′ position of the consensus sequence as +1) and the invariant A residue at +3 that most functional Inr elements possess, whereas only the latter element contains the T at +5 that the most active Inr elements contain (66). A similar Inr sequence exists at –54 to –48 in the DNA-PKcs promoter (48). All of the KARP-1 exon/intron boundaries are flanked by canonical splice donor and acceptor sequences (Fig. 4C). From these experiments we concluded that the KARP-1 gene is transcribed from a CpG-rich promoter.
Figure 4.

The upstream CpG island encodes bidirectional promoter. (A) Skeletal diagram of the human Ku86 and TERP loci. The blue hatched rectangles represent exons. The Ku86, KARP-1 and TERP +1s of transcription are shown as arrows. The orange rectangles correspond to CpG-rich regions. The green circles represent the putative p53 response elements. (B) Exploded view of the bidirectional promoter region. The sequences and location of the four independent KARP-1 cDNAs, cloned with two independent 5′-RACE techniques, are shown as well as the inferred exon (uppercase letters)/intron (lowercase letters) boundary. (C) DNA sequence of the 5′-ends of the KARP-1 and Ku86 genes (bold capital letters), including the genomic sequence immediately upstream of the KARP-1 transcriptional start sites (plain capital letters) and genomic nucleotides critical for mRNA splicing (shown in red lower case letters in parentheses). Two putative Inr elements are boxed and discussed in the text. Transcription start sites obtained with 5′-RACE analyses for KARP-1 are indicated with black arrowheads and those for Ku86 with gray arrowheads. The initiator methionines for KARP-1 and Ku86 are shown in outline.
Identification of the p53 response element needed for p53-dependent KARP-1 expression
KARP-1 is induced by DNA damage in a completely p53-dependent fashion (41). Wild-type p53 binds, albeit with varying affinities (67), to consensus response elements (p53 REs) within genetic regulatory loci. p53 REs consist of tandem palindromic decamers of 5′-PuPuPuC(A/T)(A/T)GPyPyPy-3′ (68,69), which in vivo are invariably separated from each other by no more than a single nucleotide. We had previously identified a putative p53 binding site within intron 3 of the KARP-1 transcriptional locus (41) (Fig. 5A). Although p53 bound well to this site in vitro (41), the fact that the decamers are separated from each other by 9 nt made it unlikely that this site corresponded to an in vivo p53 RE. DNA sequence analysis of the genomic DNA flanking the CpG island for 25 kb in either direction identified only two additional potential p53 REs, both of which lay within the KARP-1 first intron (Fig. 5A and B). These REs consisted of a site in which the two decamers were immediately adjacent to one another and contained 9 out of 10 nt that matched the consensus sequence [9(0)9] and a site in which the decamers were separated by a single nucleotide and each decamer was a perfect match with the consensus [10(1)10] (Fig. 5B). ChIP (70) was then used to analyze the in vivo occupancy of these putative p53 REs in HCT116+Chr3 cells exposed to IR. HCT116+Chr3 cells are a daughter cell line of ‘wild-type’ HCT116 cells that have been complemented for the parental MLH1 mismatch repair deficiency (71,72) by the stable inclusion of a wild-type copy of human chromosome 3 (53). A polyclonal p53 antibody was able to co-immunoprecipitate the 10(1)10 p53 RE from HCT116+Chr3 cells that had been X-irradiated (Fig. 5C). In addition, antibodies specific to DNA-damage inducible forms of p53, including phosphoserine15 (73,74), acetyllysine373 (75–77) and mAb421 (78), which recognizes an epitope specific to the active binding form of p53, were also able to immunoprecipitate the putative 10(1)10 p53 RE (Fig. 5C). The 10(1)10 element was not immunoprecipitated from non-damaged cells, when an irrelevant antibody (to p21) was used (Fig. 5C) or when the same experiment was carried out in p53-null cells (data not shown). Lastly, the ChIP pattern for the putative 10(1)10 p53 RE at the KARP-1 locus was identical to the ChIP patterns for other well-characterized p53 REs located in the promoter regions of the p53 target genes, p21 and 14-3-3σ (C. D. Braastad, Z. Han and E. A. Hendrickson, manuscript submitted). In contrast, the distal 9(0)9 element could not be immunoprecipitated under any condition nor in any cell line (data not shown; ‘non-responsive’, Fig. 5B). From these experiments we concluded that the 10(1)10 site corresponds to a p53 RE in vivo.
Figure 5.

Identification of a putative p53 RE in the KARP-1 first intron. (A) Skeletal diagram of the human Ku86 and TERP loci. Most symbols are as indicated in Figure 2. The PCR primers utilized in the ChIPs are shown as blue triangles. (B) Nucleotide sequence of putative p53 responsive elements and immediate flanking region. Within the putative p53 REs, consensus nucleotides are represented as uppercase and non-consensus nucleotides (mismatches) are represented as lowercase. (C) p53 interacts with the 10(1)10 putative KARP-1 RE in DNA-damaged HCT116 cells. HCT116 cells were either X-irradiated (10 Gy; +IR) or mock treated (–IR). The cells were harvested 30 min later and ChIP analysis was carried out using either a polyclonal Ab to p53 or three epitope-specific p53 monoclonal antibodies: mAb421, acetyllysine373 or phosphoserine15 or a non-specific antibody to p21 as detailed in the Materials and Methods. After reversal of the protein cross links and DNA purification, a PCR analysis was carried out. Enrichment of the p53 RE in the immunoprecipitated DNA pool indicates the interaction of p53 with the RE.
KARP-1 gene expression is induced by IR, whereas TERP expression is not
The existence of an overlapping region for transcriptional initiation and a single, functional p53 RE, begged the question of whether TERP expression might also be inducible by DNA damage. To experimentally address this issue, HCT116 cells, which are wild-type for p53 expression (51,52) were either mock irradiated or exposed to 10 Gy of IR. Two hours later, the cells were harvested, RNA was prepared and first-strand cDNA synthesized. The mRNA levels of TERP, KARP-1 or β-actin, the latter of which was utilized as a negative control, were then determined by RT–PCR reactions using serial 3-fold dilutions of the cDNA preparation to ensure that the PCR responses were within the linear range. As we have described (41), IR exposure resulted in a ∼4-fold elevation of KARP-1 mRNA (Fig. 6). In contrast, no increase in TERP or β-actin expression was observed (Fig. 6). From this experiment, we concluded that KARP-1 gene expression is preferentially induced by IR, whereas TERP expression is not.
Figure 6.
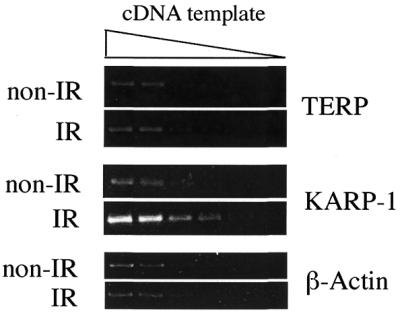
KARP-1 gene expression is induced by IR, whereas TERP expression is not. HCT116 cells were either mock-irradiated (non-IR) or exposed to 10 Gy (IR) and then 2 h later the cells were harvested, RNA was prepared and first strand cDNA was prepared. The samples were then subjected to RT–PCR with the use of pairs of gene-specific primers and 3-fold serial dilutions of the cDNA template to ensure that the signal was in the linear range.
DISCUSSION
We have identified a CpG island that directs the transcription, in opposite orientations, of two independent messenger RNAs, TERP and KARP-1. This CpG island was originally identified based upon its hypersensitivity to DNase I and was consistent with an open chromatin region that normally surrounds promoter elements. The subsequent demonstration that virtually all of the CpG dinucleotides in a ∼1 kb stretch internal to the CpG island were hypomethylated was again consistent with this region acting as a promoter. RT–PCR was then utilized to demonstrate that the first ∼17 nt of the KARP-1 mRNA transcript originated from this CpG island, proving that KARP-1 transcription is initiated from this site. Finally, chromatin immunoprecipitation experiments identified a p53 RE within the first intron of the KARP-1 locus which was bound by p53 in a completely DNA damage-inducible fashion. These experiments expand our knowledge about how the human Ku86/KARP-1 locus is regulated.
The Ku86 promoter region is constitutively DNase I hypersensitive (Fig. 1). This is consistent with the known constitutive, non-DNA damage-inducible, high level of expression of Ku86 (42,43). The transcriptional machinery that actively utilizes this site for Ku86 gene expression is almost certainly responsible for maintaining it in an open chromatin conformation (55,59). In the upstream 50 kb only a single other DHS was identified (Fig. 2). This site corresponded to the TERP/KARP-1 promoter(s). Again, this DHS was constitutive and not DNA damage inducible, which is consistent with the constitutive, albeit very low, levels of KARP-1 expression (40) and with the presumptive constitutive levels of expression of the metabolic housekeeping gene TERP (64). The only reproducible alteration observed following X-irradiation was an increased resistance to DNase I immediately after irradiation (Fig. 2D, compare lane 0′ with 15′). The mechanistic basis for this increased resistance is unknown. One possibility is that the exposure of cells to IR and general radioresistance has been correlated with the phosphorylation of the histones H2AX (79) and H3 (80), respectively. Histone phosphorylation, in turn, has long been associated with chromatin condensation, which would be consistent with the observed reduction in nuclease accessibility. Importantly, however, no other significant alterations in the DHS profile at the promoter were observed following X-irradiation (Fig. 2) even over a time interval when KARP-1 transcription is known to be induced at least 5-fold (40) (Fig. 6). Presumably the pre-existing transcriptional machinery was activated without a concomitant alteration of the surrounding chromatin. While it could be argued that the constitutive nature of the DHS at the KARP-1 promoter might be due solely to the expression of the TERP gene, it should be noted that an identical profile of hypersensitivity was recently observed for the IR-inducible genes p21 and 14-3-3σ (C. D. Braastad, Z. Han and E. A. Hendrickson, manuscript submitted), both of which have canonical, unidirectional promoters. Thus, the DHS patterns at these loci imply that p53- and IR-inducible promoters are generally occupied by an engaged transcriptional apparatus even in the absence of DNA damage. The mechanism by which this occurs is not known, but it is intriguingly similar to the ‘stalled’ or ‘paused’ RNA polymerase II complexes that are observed at the promoters of heat shock responsive genes in the absence of heat shock (reviewed in 81). Importantly, no DHS was observed at the KARP-1 p53 RE either before or after X-irradiation (Figs 1 and 2 and data not shown). Since we subsequently demonstrated that p53 is indeed binding to this site following DNA damage (Fig. 5C) these data together imply that p53 binding does not cause a detectable alteration of the chromatin structure at its cognate RE. This conclusion is consistent with the very recent demonstration that p53, in distinct contrast to most transcription factors, actually prefers to bind to nucleosomal DNA rather than naked DNA (82).
KARP-1 is transcribed from a CpG island (Fig. 5A). There is ample precedence for CpG islands serving as the cis-acting initiation core for bidirectional or divergent transcription. Indeed, some of the earliest observations linking CpG islands to promoters noted that these sites often induced the expression of divergent genes (83). Like bacterial operons, the genes involved occasionally may share a common biological function such as the histone H2A and H2B genes (84) or the avian GPAT and AIRC genes, which are both involved in purine nucleotide metabolism (85,86). Much more frequently, however, the genes share no obvious biological function and the transcripts may or may not be coordinately controlled (87,88). Within the family of DNA DSB repair proteins, this regulatory strategy/genomic configuration seems to have been utilized extensively. Thus, the DNA-PKcs and MCM4 genes are separated by 700 bp within a TATA-less CpG island (48). Likewise, the ATM and E14(NPAT) genes are separated by only 513 bp and orientated oppositely (49,50). A mere 218 bp separates the BRCA1 and NBR2 genes and they too are orientated in opposite directions (45,46). To this list, we can now add the KARP-1 and TERP genes (Fig. 5A).
The evolutionary forces that have led to this common genomic configuration are unclear. One possibility is that these important DNA repair genes (DNA-PKcs, ATM, BRCA-1 and KARP-1), which, when mutated, can often lead to cancer predisposition syndromes (reviewed in 1) have evolved to be physically connected to essential metabolic genes to limit mutational processes. Thus, while still vulnerable to small genetic alterations, any large chromosomal alteration would likely be selected against by the concomitant loss of the linked essential gene. However, it should be pointed out that BRCA-1 is by itself an essential gene (89) as are the human KARP-1/Ku86 genes (39). Correspondingly, it is more likely that these genomic configurations evolved as the result of molecular scavenging of the promoters. Alternative usage of a promoter resulting in transcription in the opposite direction might have occasionally resulted in the production of a functional message that was subsequently selected for during evolution. This hypothesis makes the prediction that the non-DNA repair half of a transcriptional pair will be evolutionarily older than its divergently transcribed DNA repair partner. This is consistent with the fact that genes like MCM4, NPAT, NBR2 and TERP are found in almost all eukaryotes whereas homologs of DNA-PKcs, BRCA-1 and KARP-1 are recently evolved genes that appear to exist only in higher eukaryotes.
KARP-1 transcription is induced by DNA damage and is dependent upon functional p53 (41). We had previously identified a putative p53 RE in the third intron of the KARP-1 transcriptional locus (41) (Fig. 1) based largely upon its proximity to the putative assignment of the KARP-1 promoter (here shown to be incorrect) and its ability to bind p53 protein in vitro. Given that the two decamers that constitute the RE were, however, separated by 9 nt—a configuration not found at any other authentic p53 RE—it seemed unlikely that this site would be used in vivo. Once the authentic KARP-1 promoter was identified, a sequence analysis revealed two additional putative p53 REs which resided within the first intron of the KARP-1 transcriptional unit and which conformed more closely to an in vivo consensus site, with the decamers being separated by 0 and 1 nt, respectively (Fig. 5B). Subsequent ChIP analysis demonstrated that the 10(1)10 site was biologically responsive (Fig. 5C). This conclusion was confirmed by the use of monoclonal antibodies directed against p53 epitopes (mAB421, phosphoserine-15, acetyllysine-373) that were specific for the activated form of p53. The existence of this p53 RE is consistent with and explains the previously demonstrated dependence on ATM and p53 for KARP-1 induction following DNA damage (41). There was no a priori reason to suspect that TERP expression would be DNA damage inducible and a lack of induction was indeed observed (Fig. 6). Thus, while these two transcriptional units physically overlap, at least a portion of their regulation does not. This latter observation implies, although certainly does not prove, that KARP-1 and TERP gene expression may be regulated from separate, closely spaced promoters, rather than regulated through a common, bidirectional promoter. Ultimately, it will be important to identify the upstream regulatory sequences in the KARP-1 promoter and determine how the transcription factors that bind to these sites interact with p53 and the RNA polymerase II machinery as well as how they establish a constitutively open chromatin structure around the promoter.
Acknowledgments
ACKNOWLEDGEMENTS
We are deeply indebted to Drs Ken Zaret, Anja-Katrin Bielinsky and Ms Jaqueline Brooks for their advice and help with the DNase I hypersensitivity, chromatin immunoprecipitation assays and 5′-RACE protocols, respectively. We thank Dr Bielinsky for her comments and criticisms of the manuscript. This work was supported in part by a grant from the NIH (AI35763).
REFERENCES
- 1.Hoeijmakers J.H. (2001) Genome maintenance mechanisms for preventing cancer. Nature, 411, 366–374. [DOI] [PubMed] [Google Scholar]
- 2.Schaer P. (2001) Spontaneous DNA damage, genome instability and cancer—when DNA replication escapes control. Cell, 104, 329–332. [DOI] [PubMed] [Google Scholar]
- 3.Norbury C.J. and Hickson,I.D. (2001) Cellular responses to DNA damage. Annu. Rev. Pharmacol. Toxicol., 41, 367–401. [DOI] [PubMed] [Google Scholar]
- 4.Barnes D.E. (2001) Non-homologous end joining as a mechanism of DNA repair. Curr. Biol., 11, 455–457. [DOI] [PubMed] [Google Scholar]
- 5.Smith G.C.M. and Jackson,S.P. (1999) The DNA-dependent protein kinase. Genes Dev., 13, 916–934. [DOI] [PubMed] [Google Scholar]
- 6.Tuteja R. and Tuteja,N. (2000) Ku autoantigen: a multifunctional DNA-binding protein. Crit. Rev. Biochem. Mol. Biol., 35, 1–33. [DOI] [PubMed] [Google Scholar]
- 7.Mimori T. and Hardin,J.A. (1986) Mechanism of interaction between Ku protein and DNA. J. Biol. Chem., 261, 10375–10379. [PubMed] [Google Scholar]
- 8.Falzon M., Fewell,J.W. and Kuff,E.L. (1993) EBP-80, a transcription factor closely resembling the human autoantigen Ku, recognizes single- to double-strand transitions in DNA. J. Biol. Chem., 268, 10546–10552. [PubMed] [Google Scholar]
- 9.Liang F. and Jasin,M. (1996) Ku80-deficient cells exhibit excess degradation of extrachromosomal DNA. J. Biol. Chem., 271, 14405–14411. [DOI] [PubMed] [Google Scholar]
- 10.Bliss T.M. and Lane,D.P. (1997) Ku selectively transfers between DNA molecules with homologous ends. J. Biol. Chem., 272, 5765–5773. [DOI] [PubMed] [Google Scholar]
- 11.Pang D., Yoo,S., Dynan,W.S., Jung,M. and Dritschilo,A. (1997) Ku proteins join DNA fragments as shown by atomic force microscopy. Cancer Res., 57, 1412–1415. [PubMed] [Google Scholar]
- 12.Walker J.R., Corpina,R.A. and Goldberg,J. (2001) Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature, 412, 607–614. [DOI] [PubMed] [Google Scholar]
- 13.Dvir A., Peterson,S.R., Knuth,M.W., Lu,H. and Dynan,W.S. (1992) Ku autoantigen is the regulatory component of a template-associated protein kinase that phosphorylates RNA polymerase II. Proc. Natl Acad. Sci. USA, 89, 11920–11924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gottlieb T.M. and Jackson,S.P. (1993) The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell, 72, 131–142. [DOI] [PubMed] [Google Scholar]
- 15.Suwa A., Hirakata,M., Takeda,Y., Jesch,S.A., Mimori,T. and Hardin,J.A. (1994) DNA-dependent protein kinase (Ku protein-p350 complex) assembles on double-stranded DNA. Proc. Natl Acad. Sci. USA, 91, 6904–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McElhinny S.A.N., Snowden,C.M., McCarville,J. and Ramsden,D.A. (2000) Ku recruits the XRCC4-Ligase IV complex to DNA ends. Mol. Cell. Biol., 20, 2996–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teo S.H. and Jackson,S.P. (2000) Lif1p targets the DNA ligase Lig4p to sites of DNA double-strand breaks. Curr. Biol., 10, 165–168. [DOI] [PubMed] [Google Scholar]
- 18.Li Z., Otevrei,T., Gao,Y., Cheng,H.-L., Seed,B., Stamato,T.D., Taccioli,G.E. and Alt,F.W. (1995) The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell, 83, 1079–1089. [DOI] [PubMed] [Google Scholar]
- 19.Gao Y., Frank,K.M., Dikkes,P., Fujiwara,Y., Seidl,K.J., Sekiguchi,J.M., Rathbun,G.A., Swat,W., Wang,J., Bronson,R., Malynn,B.A., Bryans,M., Zhu,C., Chaudhui,J., Davidson,L., Ferrini,R., Stamato,T., Orkin,S.H., Greenberg,M.E. and Alt,F.W. (1998) A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell, 95, 891–902. [DOI] [PubMed] [Google Scholar]
- 20.Critchlow S.E., Bowater,R.P. and Jackson,S.P. (1997) Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr. Biol., 7, 588–598. [DOI] [PubMed] [Google Scholar]
- 21.Grawunder U., Wilm,M., Wu,X., Kulesza,P., Wilson,T.E., Mann,M. and Lieber,M.R. (1997) Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature, 388, 492–495. [DOI] [PubMed] [Google Scholar]
- 22.Grawunder U., Zimmer,D., Fugmann,S., Schwarz,K. and Lieber,M.R. (1998) DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol. Cell, 2, 477–484. [DOI] [PubMed] [Google Scholar]
- 23.Wang H., Zeng,Z.C., Perrault,A.R., Cheng,X., Qin,W. and Iliakis,G. (2001) Genetic evidence for the involvement of DNA ligase IV in the DNA-PK-dependent pathway of non-homologous end joining in mammalian cells. Nucleic Acids Res., 29, 1653–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ouyang H., Nussenzweig,A., Kurimasa,A., Soares,V.C., Li,X., Cordon-Cardo,C., Li,W.H., Cheong,N., Nussenzweig,M., Iliakis,G., Chen,D. and Li,G. (1997) Ku70 is required for DNA repair but not for T cell antigen receptor gene recombination in vivo. J. Exp. Med., 15, 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li G.C., Ouyang,H., Li,X., Nagasawa,H., Little,J.B., Chen,D.J., Ling,C.C., Fuks,Z. and Cordon-Cardo,C. (1998) Ku70: a candidate tumor suppressor gene for murine T cell lymphoma. Mol. Cell, 2, 1–8. [DOI] [PubMed] [Google Scholar]
- 26.Nussenzweig A., Chen,C., da Costa Soares,V., Sanchez,M., Sokol,K., Nussenzweig,M.C. and Li,G.C. (1996) Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature, 382, 551–555. [DOI] [PubMed] [Google Scholar]
- 27.Zhu C., Bogue,M.A., Lim,D.-S., Hasty,P. and Roth,D.B. (1996) Ku86-deficient mice exhibit severe combined immunodeficiency and defective processing of V(D)J recombination intermediates. Cell, 86, 379–389. [DOI] [PubMed] [Google Scholar]
- 28.Jhappan C., Morse,H.C., Fleischmann,R.D., Gottesman,M.M. and Merlino,G. (1997) DNA-PKcs: a T-cell tumor suppressor encoded at the mouse scid locus. Nature Genet., 17, 483–486. [DOI] [PubMed] [Google Scholar]
- 29.Bogue M., Jhappan,C. and Roth,D.B. (1998) Analysis of variable (diversity) joining recombination in DNA-dependent protein kinase (DNA-PK)-deficient mice reveals DNA-PK-independent pathways for both signal and coding joint formation. Proc. Natl Acad. Sci. USA, 95, 15559–15564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao Y., Chaudhuri,J., Zhu,C., Davidson,L., Weaver,D.T. and Alt,F.W. (1998) A targeted DNA-PKcs-null mutation reveals DNA-PK-independent functions for Ku in V(D)J recombination. Immunity, 9, 367–376. [DOI] [PubMed] [Google Scholar]
- 31.Taccioli G.E., Amatucci,A.G., Beamish,H.J., Gell,D., Xiang,X.H., Torres Arzayus,M.I., Priestley,A., Jackson,S.P., Rothstein,A.M., Jeggo,P.A. and Herrera,V.L.M. (1998) Targeted disruption of the catalytic subunit of the DNA-PK gene in mice confers severe combined immunodeficiency and radiosensitivity. Immunity, 9, 355–366. [DOI] [PubMed] [Google Scholar]
- 32.Vogel H., Lim,D.-S., Karsenty,G., Finegold,M. and Hasty,P. (1999) Deletion of Ku86 causes early onset of senescence in mice. Proc. Natl Acad. Sci. USA, 96, 10770–10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karanjawala Z.E., Grawunder,U., Hsieh,C.L. and Lieber,M.R. (1999) The nonhomologous DNA end joining pathway is important for chromosome stability in primary fibroblasts. Curr. Biol., 9, 1501–1504. [DOI] [PubMed] [Google Scholar]
- 34.Difilippantonio M.J., Zhu,J., Chen,H.T., Meffre,E., Nussenzweig,M.C., Max,E.E., Ried,T. and Nussenzweig,A. (2000) DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature, 404, 510–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.d‘Adda di Fagagna F., Hande,M.P., Tong,W.-M., Roth,D.B., Lansdorp,P.M., Wang,Z.-Q. and Jackson,S.P. (2001) Effects of DNA nonhomologous end-joining factors on telomere length and chromosomal stability in mammalian cells. Curr. Biol., 11, 1192–1196. [DOI] [PubMed] [Google Scholar]
- 36.Bailey S.M., Meyne,J., Chen,D.J., Kurimasa,A., Li,G.C., Lehnert,B.E. and Goodwin,E.H. (1999) DNA double-strand break repair proteins are required to cap the ends of mammalian chromosomes. Proc. Natl Acad. Sci. USA, 96, 14899–14904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsu H.L., Gilley,D., Galande,S.A., Hande,M.P., Allen,B., Kim,S.H., Li,G.C., Campisi,J., Kohwi-Shigematsu,T. and Chen,D.J. (2000) Ku acts in a unique way at the mammalian telomere to prevent end joining. Genes Dev., 14, 2807–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Samper E., Goytisolo,F.A., Slijepcevic,P., van Buul,P.P.W. and Blasco,M.A. (2000) Mammalian Ku86 protein prevents telomeric fusions independently of the length of TTAGGG repeats and the G-strand overhang. EMBO Rep., 1, 244–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li G., Nelsen,C. and Hendrickson,E.A. (2002) Ku86 is essential in human somatic cells. Proc. Natl Acad. Sci. USA, 99, 832–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Myung K., He,D.M., Lee,S.E. and Hendrickson,E.A. (1997) KARP-1: a novel leucine zipper protein expressed from the Ku86 autoantigen locus is implicated in the control of DNA-dependent protein kinase activity. EMBO J., 16, 3172–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Myung K., Braastad,C., He,D.M. and Hendrickson,E.A. (1998) KARP-1 is induced by DNA damage in a p53- and ataxia telangiectasia mutated-dependent fashion. Proc. Natl Acad. Sci. USA, 95, 7664–7669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jongmans W., Artuso,M., Vuillaume,M., Bresil,H., Jackson,S.P. and Hall,J. (1996) The role of ataxia telangiectasia and the DNA-dependent protein kinase in the p53-mediated cellular response to ionising radiation. Oncogene, 13, 1133–1138. [PubMed] [Google Scholar]
- 43.Lee S.E., Mitchell,R.A., Cheng,A. and Hendrickson,E.A. (1997) Evidence for DNA-PK dependent and independent DNA double strand break repair pathways in mammalian cells as a function of the cell cycle. Mol. Cell. Biol., 17, 1425–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ludwig D.L., Chen,F., Peterson,S.R., Nussenzweig,A., Li,G.C. and Chen,D.J. (1997) Ku80 gene expression is Sp1-dependent and sensitive to CpG methylation within a novel cis element. Gene, 199, 181–194. [DOI] [PubMed] [Google Scholar]
- 45.Smith T.M., Lee,M.K., Szabo,C.I., Jerome,N., McEuen,M., Taylor,M., Hood,L. and King,M.C. (1996) Complete genomic sequence and analysis of 117 kb of human DNA containing the gene BRCA1. Genome Res., 6, 1029–1049. [DOI] [PubMed] [Google Scholar]
- 46.Xu C.F., Brown,M.A., Nicolai,H., Chambers,J.A., Griffiths,B.L. and Solomon,E. (1997) Isolation and characterisation of the NBR2 gene which lies head to head with the human BRCA1 gene. Hum. Mol. Genet., 6, 1057–1062. [DOI] [PubMed] [Google Scholar]
- 47.Magdinier F., Billard,L.M., Wittmann,G., Frappart,L., Benchaib,M., Lenoir,G.M., Guerin,J.F. and Dante,R. (2000) Regional methylation of the 5′ end CpG island of BRCA1 is associated with reduced gene expression in human somatic cells. FASEB J., 14, 1585–1594. [DOI] [PubMed] [Google Scholar]
- 48.Connelly M.A., Zhang,H., Kieleczawa,J. and Anderson,C.W. (1998) The promoters for human DNA-PKcs (PRKDC) and MCM4: divergently transcribed genes located at chromosome 8 band q11. Genomics, 47, 71–83. [DOI] [PubMed] [Google Scholar]
- 49.Byrd P.J., Cooper,P.R., Stankovic,T., Kullar,H.S., Watts,G.D., Robinson,P.J. and Taylor,M.R. (1996) A gene transcribed from the bidirectional ATM promoter coding for a serine rich protein: amino acid sequence, structure and expression studies. Hum. Mol. Genet., 5, 1785–1791. [DOI] [PubMed] [Google Scholar]
- 50.Imai T., Yamauchi,M., Seki,N., Sugawara,T., Saito,T., Matsuda,Y., Ito,H., Nagase,T., Nomura,N. and Hori,T. (1996) Identification and characterization of a new gene physically linked to the ATM gene. Genome Res., 6, 439–447. [DOI] [PubMed] [Google Scholar]
- 51.Waldman T., Kinzler,K.W. and Vogelstein,B. (1995) p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res., 55, 5187–5190. [PubMed] [Google Scholar]
- 52.Bunz F., Dutriaux,A., Lengauer,C., Waldman,T., Zhou,S., Brown,J.P., Sedivy,J.M., Kinzler,K.W. and Vogelstein,B. (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science, 282, 1497–1501. [DOI] [PubMed] [Google Scholar]
- 53.Koi M., Umar,A., Chauhan,D.P., Cherian,S.P., Carethers,J.M., Kunkel,T.A. and Boland,C.R. (1994) Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N′-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res., 54, 4308–4312. [PubMed] [Google Scholar]
- 54.Zaret K.S. and Yamamoto,K.R. (1984) Reversible and persistent changes in chromatin structure accompany activation of a glucocorticoid-dependent enhancer element. Cell, 38, 29–38. [DOI] [PubMed] [Google Scholar]
- 55.Liu J.-K., Bergman,Y. and Zaret,K.S. (1988) The mouse albumin promoter and a distal upstream site are simultaneously DNase I hypersensitive in liver chromatin and bind similar liver-abundant factors in vivo. Genes Dev., 2, 528–541. [DOI] [PubMed] [Google Scholar]
- 56.Frohman M.A., Dush,M.K. and Martin,G.R. (1988) Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc. Natl Acad. Sci. USA, 85, 8998–9002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hecht A., Strahl-Bolsinger,S. and Grunstein,M. (1996) Spreading of transcriptional repressor SIR3 from telomeric heterochromatin. Nature, 383, 92–96. [DOI] [PubMed] [Google Scholar]
- 58.Pinkert C.A., Ornitz,D.M., Brinster,R.L. and Palmiter,R.D. (1987) An albumin enhancer located 10 kb upstream functions along with its promoter to direct efficient, liver-specific expression in transgenic mice. Genes Dev., 1, 268–276. [DOI] [PubMed] [Google Scholar]
- 59.Felsenfeld G., Boyes,J., Chung,J., Clark,D. and Studitsky,V. (1996) Chromatin structure and gene expression. Proc. Natl Acad. Sci. USA, 93, 9384–9388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bird A.P. (1987) CpG islands as gene markers in the vertebrate nucleus. Trends Genet., 3, 342–347. [Google Scholar]
- 61.Antequera F. and Bird,A. (1993) Number of CpG islands and genes in human and mouse. Proc. Natl Acad. Sci. USA, 90, 11995–11999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baylin S.B., Esteller,M., Rountree,M.R., Bachman,K.E., Schuebel,K. and Herman,J.G. (2001) Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum. Mol. Genet., 10, 687–692. [DOI] [PubMed] [Google Scholar]
- 63.Jones P.A. (2001) Cancer. Death and methylation. Nature, 409, 141–144. [DOI] [PubMed] [Google Scholar]
- 64.Das A.K., Uhler,M.D. and Hajra,A.K. (2000) Molecular cloning and expression of mammalian peroxisomal trans-2-enoyl-coenzyme A reductase cDNAs. J. Biol. Chem., 275, 24333–24340. [DOI] [PubMed] [Google Scholar]
- 65.Lo K. and Smale,S.T. (1996) Generality of a functional initiator consensus sequence. Gene, 182, 13–22. [DOI] [PubMed] [Google Scholar]
- 66.Javahery R., Khachi,A., Lo,K., Zenzie-Gregory,B. and Smale,S.T. (1994) DNA sequence requirements for transcriptional initiator activity in mammalian cells. Mol. Cell. Biol., 14, 116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Szak S.T., Mays,D. and Pietenpol,J.A. (2001) Kinetics of p53 binding to promoter sites in vivo. Mol. Cell. Biol., 21, 3375–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.El-Deiry W.S., Kern,S.E., Pietenpol,J.A., Kinzler,K.W. and Vogelstein,B. (1992) Definition of a consensus binding site for p53. Nature Genet., 1, 45–49. [DOI] [PubMed] [Google Scholar]
- 69.Wang Y., Schwedes,J.F., Parks,D., Mann,K. and Tegtmeyer,P. (1995) Interaction of p53 with its consensus DNA-binding site. Mol. Cell. Biol., 15, 2157–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hecht A., Strahl-Bolsinger,S. and Grunstein,M. (1999) Mapping DNA interaction sites of chromosomal proteins. Crosslinking studies in yeast. Methods Mol. Biol., 119, 469–479. [DOI] [PubMed] [Google Scholar]
- 71.Papadopoulos N., Nicolaides,N.C., Liu,B., Parsons,R., Lengauer,C., Palombo,F., D‘Arrigo,A., Markowitz,S., Willson,J.K., Kinzler,K.W., Jiicny,J. and Vogelstein,B. (1995) Mutations of GTBP in genetically unstable cells. Science, 268, 1915–1917. [DOI] [PubMed] [Google Scholar]
- 72.Aebi S., Kurdi-Haidar,B., Gordon,R., Cenni,B., Zheng,H., Fink,D., Christen,R.D., Boland,C.R., Koi,M., Fishel,R. and Howell,S.B. (1996) Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res., 56, 3087–3090. [PubMed] [Google Scholar]
- 73.Banin S., Moyal,L., Shieh,S.-Y., Taya,Y., Anderson,C.W., Chessa,L., Smorodinsky,N.I., Prives,C., Reiss,Y., Shiloh,Y. and Ziv,Y. (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science, 281, 1674–1677. [DOI] [PubMed] [Google Scholar]
- 74.Canman C.E., Lim,D.-S., Cimprich,K.A., Taya,Y., Tamai,K., Sakaguchi,K., Appella,E., Kastan,M.B. and Siliciano,J.D. (1998) Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science, 281, 1677–1679. [DOI] [PubMed] [Google Scholar]
- 75.Sakaguchi K., Herrera,J.E., Saito,S., Miki,T., Bustin,M., Vassilev,A., Anderson,C.W. and Appella,E. (1998) DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev., 12, 2831–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu L., Scolnick,D.M., Trievel,R.C., Zhang,H.B., Marmorstein,R., Halazonetis,T.D. and Berger,S.L. (1999) p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol., 19, 1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ito A., Lai,C.H., Zhao,X., Saito,S., Hamilton,M.H., Appella,E. and Yao,T.P. (2001) p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J., 20, 1331–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Takenaka I., Morin,F., Seizinger,B.R. and Kley,N. (1995) Regulation of the sequence-specific DNA binding function of p53 by protein kinase C and protein phosphatases. J. Biol. Chem., 270, 5405–5411. [DOI] [PubMed] [Google Scholar]
- 79.Rogakou E.P., Pilch,D.R., Orr,A.H., Ivanova,V.S. and Bonner,W.M. (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem., 273, 5858–5868. [DOI] [PubMed] [Google Scholar]
- 80.Li Y., DeFatta,R., Anthony,C., Sunavala,G. and De Benedetti,A. (2001) A translationally regulated Tousled kinase phosphorylates histone H3 and confers radioresistance when overexpressed. Oncogene, 20, 726–738. [DOI] [PubMed] [Google Scholar]
- 81.Lis J. and Wu,C. (1993) Protein traffic on the heat shock promoter: parking, stalling and trucking along. Cell, 74, 1–4. [DOI] [PubMed] [Google Scholar]
- 82.Espinosa J.M. and Emerson,B.M. (2001) Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol. Cell, 8, 57–69. [DOI] [PubMed] [Google Scholar]
- 83.Lavia P., Macleod,D. and Bird,A. (1987) Coincident start sites for divergent transcripts at a randomly selected CpG-rich island of mouse. EMBO J., 6, 2773–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hentschel C.C. and Birnstiel,M.L. (1981) The organization and expression of histone gene families. Cell, 25, 301–313. [DOI] [PubMed] [Google Scholar]
- 85.Gavalas A., Dixon,J.E., Brayton,K.A. and Zalkin,H. (1993) Coexpression of two closely linked avian genes for purine nucleotide synthesis from a bidirectional promoter. Mol. Cell. Biol., 13, 4784–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gavalas A. and Zalkin,H. (1995) Analysis of the chicken GPAT/AIRC bidirectional promoter for de novo purine nucleotide synthesis. J. Biol. Chem., 270, 2403–2410. [DOI] [PubMed] [Google Scholar]
- 87.Linton J.P., Yen,J.Y., Selby,E., Chen,Z., Chinsky,J.M., Liu,K., Kellems,R.E. and Crouse,G.F. (1989) Dual bidirectional promoters at the mouse dhfr locus: cloning and characterization of two mRNA classes of the divergently transcribed Rep-1 gene. Mol. Cell. Biol., 9, 3058–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gessler M. and Bruns,G.A. (1993) Sequence of the WT1 upstream region including the Wit-1 gene. Genomics, 17, 499–501. [DOI] [PubMed] [Google Scholar]
- 89.Gowen L.C., Avrutskaya,A.V., Latour,A.M., Koller,B.H. and Leadon,S.A. (1998) BRCA1 required for transcription-coupled repair of oxidative DNA damage. Science, 281, 1009–1012. [DOI] [PubMed] [Google Scholar]