

Abstract
Myotonic dystrophy (DM) is a multisystemic disorder caused by microsatellite expansion mutations in two unrelated genes leading to similar, yet distinct, diseases. DM disease presentation is highly variable and distinguished by differences in age-of-onset and symptom severity. In the most severe form, DM presents with congenital onset and profound developmental defects. At the molecular level, DM pathogenesis is characterized by a toxic RNA gain-of-function mechanism that involves the transcription of noncoding microsatellite expansions. These mutant RNAs disrupt key cellular pathways, including RNA processing, localization, and translation. In DM, these toxic RNA effects are predominantly mediated through the modulation of the muscleblind-like and CUGBP and ETR-3-like factor families of RNA binding proteins (RBPs). Dysfunction of these RBPs results in widespread RNA processing defects culminating in the expression of developmentally inappropriate protein isoforms in adult tissues. The tissue that is the focus of this review, skeletal muscle, is particularly sensitive to mutant RNA-responsive perturbations, as patients display a variety of developmental, structural, and functional defects in muscle. Here, we provide a comprehensive overview of DM1 and DM2 clinical presentation and pathology as well as the underlying cellular and molecular defects associated with DM disease onset and progression. Additionally, fundamental aspects of skeletal muscle development altered in DM are highlighted together with ongoing and potential therapeutic avenues to treat this muscular dystrophy.
Introduction
Myotonic dystrophy (dystrophia myotonica, DM) is a dominantly inherited and highly variable disease that affects nearly every organ system in the body (154). There are two types of DM defined by genetic etiology. DM type 1 (DM1) is caused by a CTG expansion (CTGexp) in the 3’ untranslated region (UTR) of dystrophia myotonica protein kinase (DMPK), while a CCTG expansion (CCTGexp) in the first intron of cellular nucleic acid binding protein (CNBP) leads to DM type 2 (DM2) (47, 231). In contrast to DM2, DM1 also occurs as a congenital disease (CDM) due to maternal transmission of exceptionally large (typically > 1000) CTGexp DMPK mutations (257). In fact, a variety of DM clinical symptoms are distinguished by their age-of-onset, systems involvement, and presentation (257). Skeletal muscle involvement is one of the most striking clinical manifestations of DM patients. Patients present with myotonia (delay in muscle relaxation following contraction), weakness of limb and facial musculature, and progressive adult-onset muscle wasting. Underscoring the dramatic variability of this disease, skeletal muscle development defects, including hypotonia (low basal muscle tone), are a characteristic neonatal feature of CDM together with perinatal mortality associated with respiratory insufficiency and swallowing difficulties. Cardiac and neurological dysfunctions are other prominent features of DM and contribute to patient mortality and diminished quality of life, respectively.
The molecular basis of DM pathogenesis has been the subject of intense investigation since the discoveries of the DM1 and DM2 mutations. Today, the prevailing pathomechanism is transcription across C(C)TGexp tracts produces toxic C(C)UGexp RNAs that disrupt the normal functions of effector proteins, most notably members of the muscleblind-like (MBNL) and CUGBP and ETR-3-like factor (CELF) families of RNA binding proteins (RBPs) (135, 319, 359). MBNL and CELF proteins are involved in diverse RNA processing steps, including alternative splicing (AS), alternative cleavage and polyadenylation (APA), mRNA stability, RNA localization, mRNA translation, and microRNA (miRNA) biogenesis (26, 203, 322, 428, 429). Interrogation of these MBNL- and CELF-responsive activities, and their misregulation in DM, has elucidated links between specific RNA processing events and patient symptoms and enhanced our understanding of the roles of certain RBPs in the developmental regulation of RNA processing. Current efforts are focused on comprehensive surveys to characterize the extent of RNA misprocessing events within the DM transcriptome. Many of the cellular pathways highlighted in these studies have known roles in skeletal muscle development and maintenance, and a common theme is the retention of developmentally immature RNA processing patterns in adult tissues.
In this review, we provide a comprehensive survey of DM skeletal muscle pathophysiology and highlight seminal studies that led to relatively rapid progress from the identification of the causative mutations to the development, and current implementation, of rationally designed therapeutics. We begin with a brief historical overview of DM research followed by a discussion of the clinical and histological hallmarks of DM skeletal muscle. Next, we examine working hypotheses and molecular models of DM pathogenesis leading to skeletal muscle dysfunctions that have been garnered through in vitro, in vivo, and in silico analyses. Finally, we summarize current and potential therapeutic interventions and conclude by addressing emerging and largely unresolved questions remaining in the DM field.
Historical Perspective
Disease characterization to causative mutation
In 1909, Hans Steinert described patients presenting with myotonia (Fig. 1) and progressive muscle wasting coupled with multisystem involvement, which led to the initial designation of this disorder as Steinert’s disease (332,381). Despite the autosomal dominant inheritance pattern, the variability of Steinert’s disease obscured its genetic etiology for decades. This was due in part because, unlike disorders associated with direct links between protein loss-of-function and disruption of tissue homeostasis (e.g., dystrophin/DMD mutations and Duchenne muscular dystrophy or DMD), consolidating the molecular mechanisms governing the pleiotropy of Steinert’s disease was, and remains, a complex task. Furthermore, the observation of increased symptom severity and decreased age-of-onset in successive generations of affected families, or genetic anticipation, underscored the complex and variable nature of this disease. Once dismissed as ascertainment bias, the molecular mechanism underlying anticipation was later identified following the sequencing of the disease-linked DMPK mutant gene (117, 150, 151). Steinert’s disease is one of the most striking examples of anticipation. For example, a mutation-harboring grandparent may be largely asymptomatic while her daughter presents with adult-onset disease that is diagnosed following the birth of her severely affected congenital infant, the proband. Indeed, the birth of a child with CDM is a common impetus for evaluation of affected families (251, 399). As studies of Steinert’s disease continued throughout the 20th century, the disorder was eventually renamed myotonic dystrophy, or DM, after its hallmark muscle symptoms.
Figure 1.
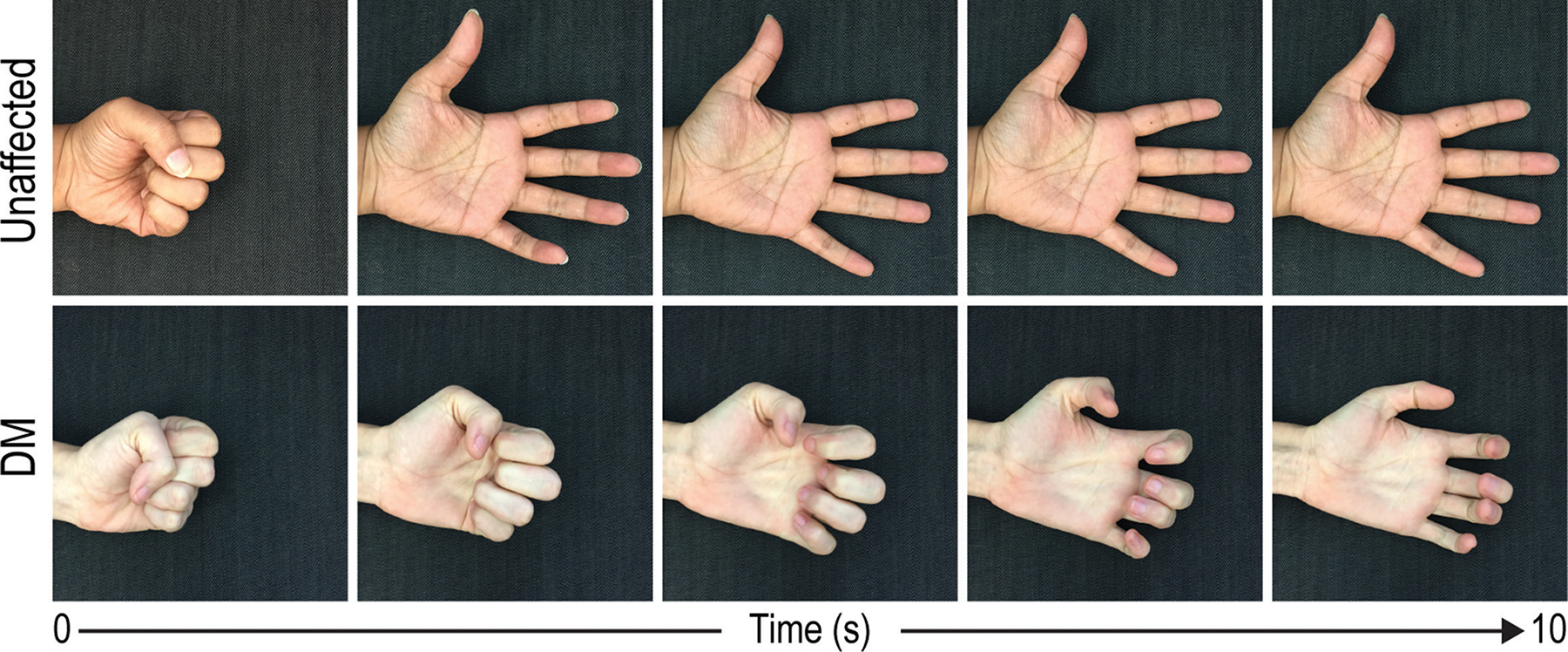
Myotonia is a characteristic skeletal muscle feature of DM patients. In unaffected individuals, grip relaxation is unencumbered and accompanied by muscle repolarization to resting potential (upper panels). For DM patients, loss of ion homeostasis results in delayed relaxation (lower panels).
The genetic basis of this classical form of DM was revealed in 1992, with the identification of a CTGexp in the 3’ UTR of DMPK located on chromosome 19q13.3 (Fig. 2) (47, 55, 150, 241). While unaffected individuals possess between 5 and 37 DMPK CTG repeats, disease symptoms emerge when the CTGexp surpasses 50 repeats. Thus, DM became the third disease associated with unstable nucleotide expansions after fragile X syndrome (FXS) and X-linked spinal-bulbar muscular atrophy (or Kennedy’s disease). Beyond a disease-specific threshold, these repeat tracts are unstable and prone to intergenerational and somatic expansions. Importantly, the discovery of microsatellite expansions provided a mechanistic foundation for understanding genetic anticipation that is associated with multiple microsatellite expansion disorders including FXS, Huntington disease (HD), and several types of spinocerebellar ataxia (SCA) (117, 206, 212). In DM, the earlier age-of-onset and increased severity of symptoms generally correlates with CTGexp repeat number (Fig. 3). The repeat expansions associated with clinically defined DM manifestations range from mild/asymptomatic (~50–<150), classic (~50–<1000) to congenital (>1000) (Fig. 4) (257).
Figure 2.
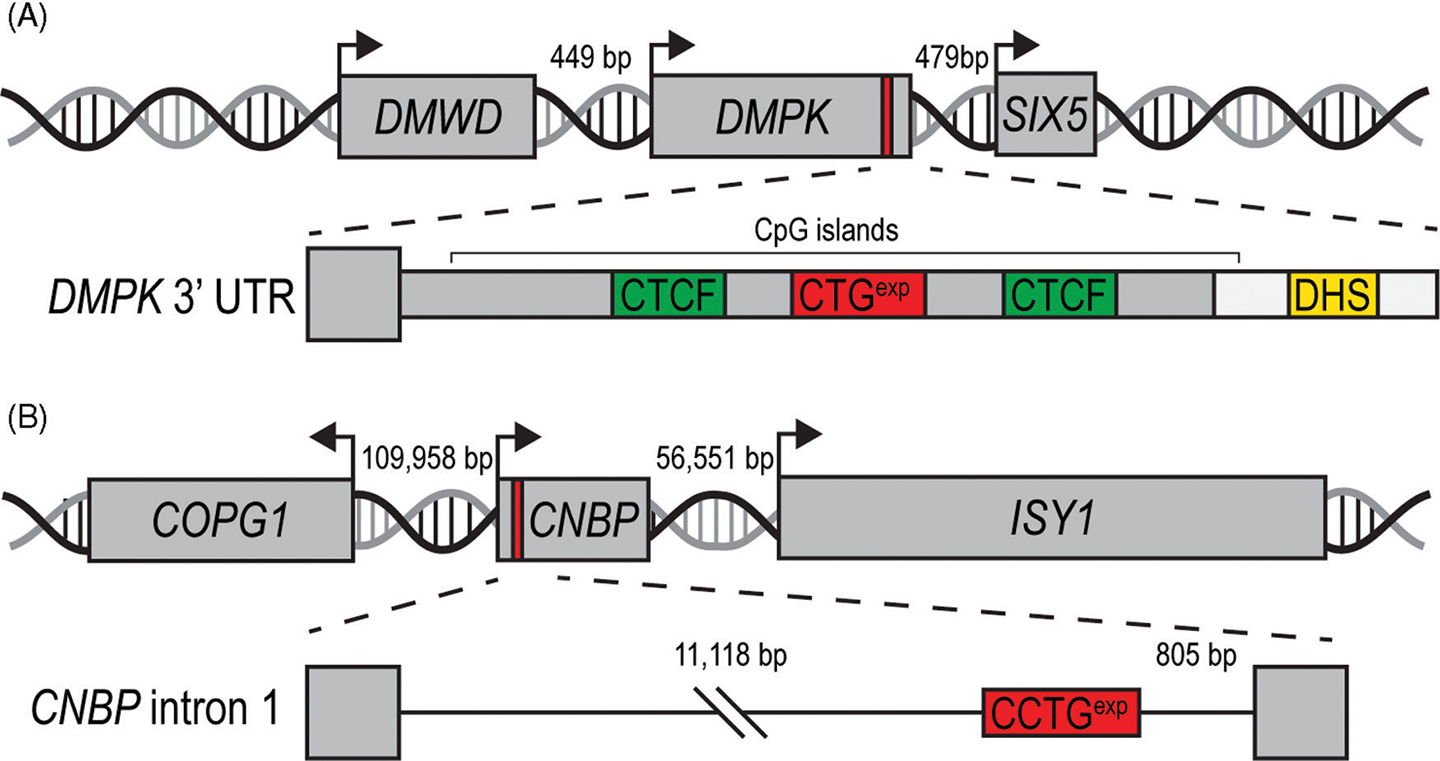
DM1- and DM2-associated gene loci. (A) The DMPK CTGexp (red box) is located in the 3’ UTR and is adjacent to two closely neighboring genes, DMWD and SIX5 (arrows indicate transcription start sites). CTCF binding sites (green boxes) flank the CTGexp along with a downstream DNase hypersensitivity site (DHS, yellow box). These elements may regulate the epigenetic features of this locus. (B) The DM2-associated CCTGexp (red box) is located in the first intron of CNBP. Neighboring genes are distal to this locus and may not be affected by this microsatellite expansion.
Figure 3.
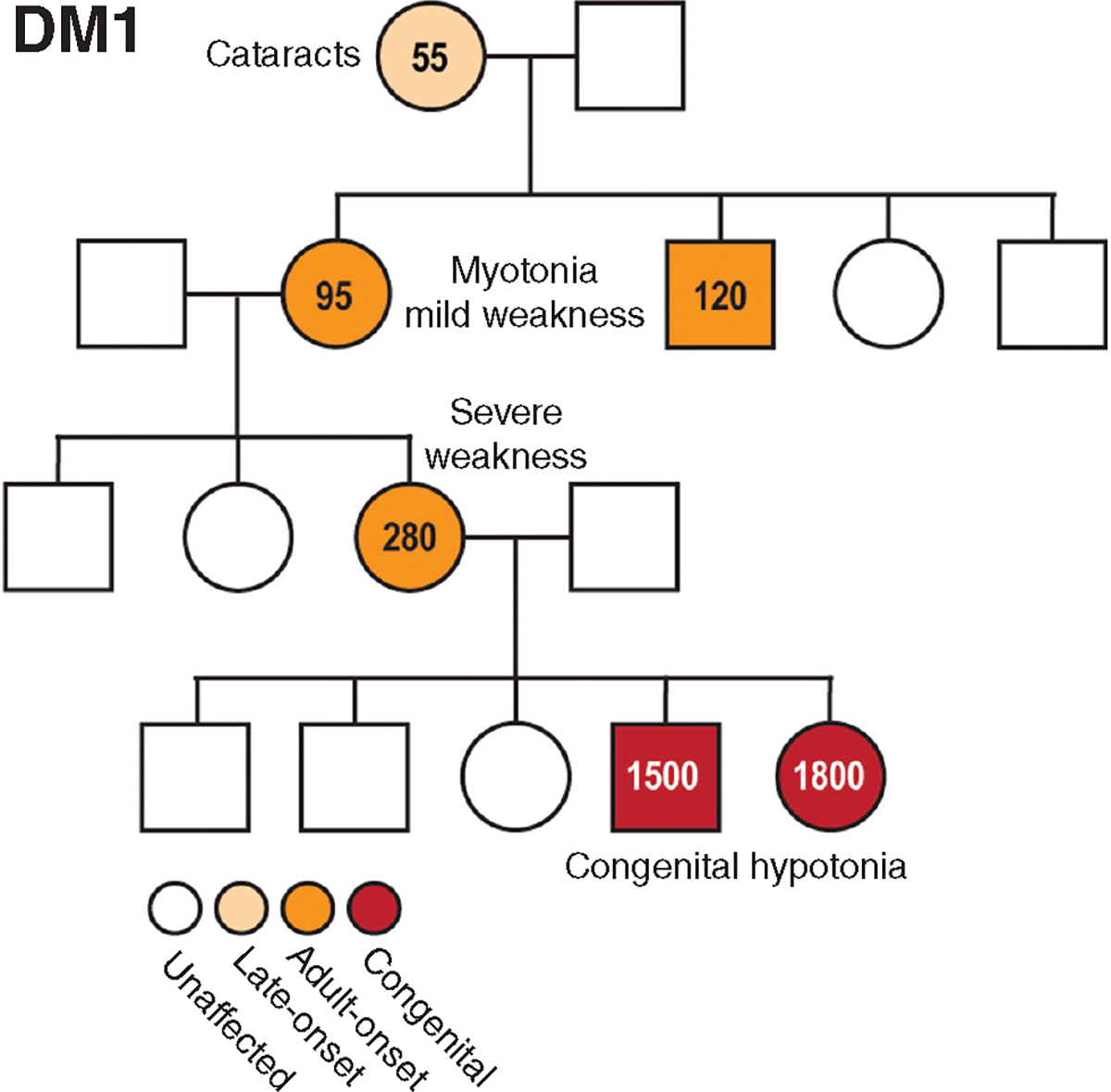
DM1 pedigree highlights genetic anticipation. Hypothetical pedigree of a DM1 family with males (boxes) and females (circles) and mutant allele CTG repeat lengths indicated.
Figure 4.
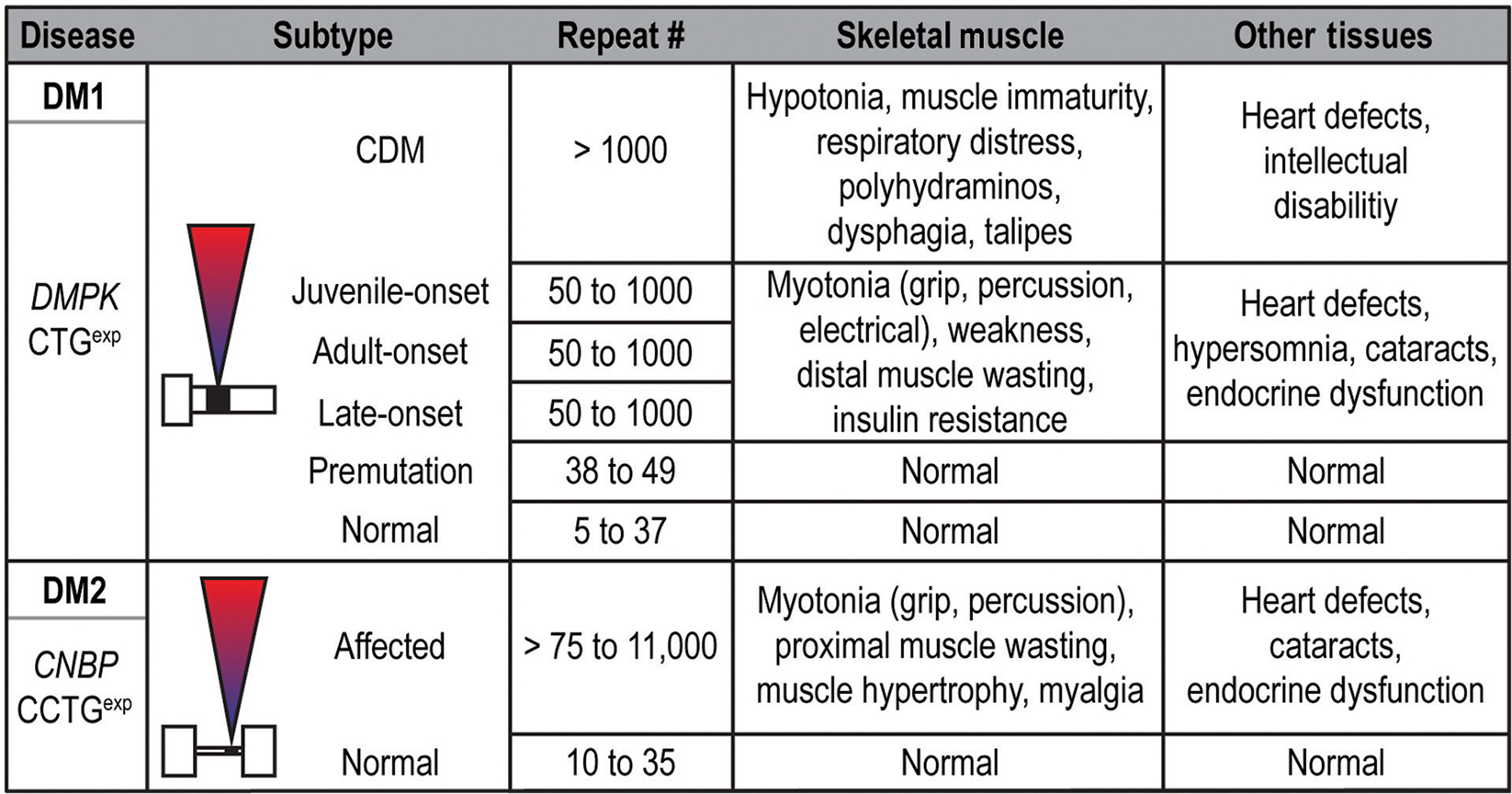
Clinical manifestations and disease stages in DM1, DM2, and CDM. In DM1, a variety of clinically defined subtypes are listed along with associated symptoms. While juvenile-, adult- and late-onset DM1 are all listed with 50 to 1000 repeats, earlier age-of-onset and exacerbated disease severity typically correlate with increased CTGexp size in DM1. This correlation is not as marked for DM2.
Following the identification of the DMPK-linked CTGexp mutation, reports emerged of patients presenting with DM-like symptoms who tested within the normal CTGexp range and showed preferential proximal, rather than distal, muscle involvement (331, 400). This disorder was originally termed proximal myotonic myopathy (PROMM). In the late 1990s, the casual mutation was linked to chromosomal region 3q21 and later revealed to be a CCTGexp in the first intron of CNBP (originally termed zinc finger 9, ZNF9) (Fig. 2) (231, 321). Shortly afterward, the DMPK-linked disease was renamed DM type 1 (DM1) and PROMM was designated DM type 2 (DM2) based on their phenotypic similarity, yet distinct etiology and presentation. In the context of DM2, normal individuals have < 30 CCTG repeats while disease manifestations have been observed in patients with as few as 55 CCTGexp repeats (231). In general, DM2 is later onset and less severe compared to DM1 and a congenital form of DM2 has not been reported (Fig. 4). Additionally, the prevalence of DM1 (1 in ~8000) is greater and more widespread than DM2 (~3% of DM cases worldwide) except in some regions of Northern Europe where the epidemiology is more comparable (254, 415). However, underdiagnosis of DM2 may be prevalent since it is often a late-onset disease and therefore confounded by the normal aging process (320).
Mutations to disease models
DM-associated C(C)TGexp mutations are located outside conventional protein-coding regions of the genome (Fig. 2), leading to a fundamental question: how do DNA simple sequence repeats in noncoding regions result in disease? Both forms of DM are inherited in an autosomal dominant pattern and DMPK and CNBP missense mutations have not been linked to either DM1 or DM2 arguing against a loss-of-function model. The similarity between DM1 and DM2 suggested shared pathogenic mechanisms might exist.
Two initial models emerged to reconcile these observations. Reduced expression of mutation harboring (DMPK, CNBP), and/or flanking (SIX5, DMWD for DM1), genes could result in haploinsufficiency. Alternatively, or in addition to haploinsufficiency, transcription across C(C)TGexp regions could generate toxic gain-of-function RNAs. The haploinsufficiency model garnered initial support since reduced DMPK mRNA and protein levels were reported in adult DM1 tissues (120). However, these results remain controversial, as other groups reported decreased, similar, or increased levels of DMPK mRNA and protein (59, 340, 432). Discrepancies in reported DMPK mRNA levels are likely due to the methodologies utilized for RNA isolation and analysis. While transcripts from the normal DMPK allele are readily purified using phenol-chloroform-based extraction procedures, expanded transcripts remain insoluble and are lost during purification (85, 147). The use of cesium chloride gradient-based purification techniques circumvented this issue and revealed normal steady-state levels of mutant DMPK transcripts (85). Nonetheless, CUGexp-containing DMPK transcripts may be susceptible to reduced translation. Therefore, Dmpk knockout (KO) mice were generated to test the hypothesis that DMPK depletion contributes to disease (Table 1) (179, 327). While homozygous Dmpk KO animals display mild phenotypes, heterozygous Dmpk KOs, a true model of haploinsufficiency, do not recapitulate features of DM arguing against a substantial role for DMPK loss-of-function. Additionally, the DM2 mutation is in a different gene on another chromosome, which suggests that the haploinsufficiency model fails to explain aspects of pathogenesis common between DM1 and DM2.
Table 1.
DM Mouse Models
| Mouse model | Rationale | Features and contributions | Limitations | References |
|---|---|---|---|---|
|
| ||||
| Dmpk knockout | DMPK haploinsufficiency in DM1. | Established that DM1 is not primarily caused by DMPK haploinsufficiency. | Conflicting reports on impacts of Dmpk ablation in mice. |
Reddy et al., 1996. Carrell et al., 2016. |
| Six5 knockout | SIX5 haploinsufficiency in DM1. | Established that DM1 is not caused by SIX5 haploinsufficiency. | Animals develop nuclear cataracts, different from the subcapsular cataracts in DM1. |
Klesert et al., 2000. Sarkar et al., 2000. Wakimoto et al., 2002. Sarkar et al., 2004. |
| Cnbp knockout | CNBP haploinsufficiency in DM2. | Heterozygous mice present myotonia, cardiac conduction defects, cataracts, and myopathic features. | Conflicting reports of CNBP downregulation in DM2. |
Chen et al. 2003. Chen et al., 2007. Margolis et al., 2006. Raheem et al., 2010. |
| Mbnl1 knockout | Mbnl1 loss-of-function. | Presents with myotonia, aberrant splicing, centralized myonuclei, and subcapsular cataracts. | Not a C(C)TGexp model so may not address some repeat-induced pathomechanisms including RAN translation, RNAi and CELF stabilization. Consitutive loss of both MBNL1 and 2 is embryonic lethal. |
Kanadia et al., 2003. Lee et al., 2013. |
| Mbnl2 knockout | Mbnl2 loss-of-function | Develops DM-relevant learning deficits and REM sleep disturbances | Charizanis et al., 2012. | |
| Mbnl3 knockout | Mbnl3 loss-of-function. | Absence of nuclear isoform impairs muscle regeneration | Poulos et al., 2013. | |
| CELF transgenic | Overexpression of CELF in skeletal muscle or heart. | Presents molecular, histological and physiological features of DM1. | Premature lethality. Limited applications for therapeutic development. |
Timchenko et al., 2004. Ward et al., 2010. Koshelev et al., 2010. |
| Dmt | First DM1 transgenic model. Transgene comprises ~ 162 CTG repeats and ~750 bp of flanking DNA from the DMPK 3’ UTR without any promoter element or coding sequence. | Occurrence of somatic and intergenerational instability. | Absence of promoter elements prevents transgene expression, limiting the applicability of this model to studies on repeat instability. |
Monckton et al., 1997. Fortune et al., 2000. |
| HSA LR | Transgenic overexpression of CTG250 within the human skeletal actin (HSA) 3’ UTR. | Established the RNA gain-of-function mechanism underlying DM1. Shows RNA foci and splicing alterations. Widely used for therapeutic development. | Transgene is expressed only in postdevelopmental skeletal muscle tissue at levels much higher than the endogenous Dmpk gene. Absence of muscle weakness or wasting. No effect on CELF levels. | Mankodi et al., 2000. |
| DM300 | Transgenic line carrying a 45 kb region from the DM1 locus including a mutant DMPK gene with ~300 CTG repeats. | Occurrence of somatic and intergenerational repeat instability. Myotonia and histological changes. | Variable expression levels. Mild phenotype. |
Seznec et al., 2000. Seznec et al., 2001. |
| DMSXL | Transgenic line derived from the DM300 model. Contains > 1000 CTG repeats. | Splicing changes in the central nervous system and skeletal muscle of homozygous mice. | Derived from a single founder. Low DMPK expression. Requires homozygosity to model a dominant disease. |
Gomes-Pereira et al., 2007. Huguet et al., 2012. |
| EpA960 | Tissue-specific overexpression of 960 interrupted CTG repeats within exon 15 of DMPK. | Suggested that CELF1 upregulation is dependent on the DMPK genomic context. | Interrupted repeat tract. Heart-specific model displayed premature death. |
Wang et al., 2007. Orengo et al., 2008. |
| TRE-EGFP-CTG5 and CTG200 | Transgenic line with inducible expression of DMPK 3’ UTR carrying 5 or 200 CTG repeats. | Dox-induced transgene activation leads to myotonia and cardiac defects. Dox withdrawal reverts these phenotypes. | Overexpression of a nonpathogenic repeat, CTG5, results in DM1 features. | Mahadevan et al., 2006. |
| Humanized Dmpk CTG84 knock-in | Transgenic line in which the genomic fragment between exons 13–15 from the endogenous Dmpk gene was replaced by the orthologous human fragment including 84 CTG repeats. | Somatic instability blocked in an Msh3-deficient background and increased in an Msh6-deficient background. | Absence of published data addressing molecular, histological or physiological phenotypes from the expression of humanized mutant Dmpk alleles. | van den Broek et al., 2002. |
As early as 1995, several hypotheses were proposed that DM1 was caused by a dominant-negative RNA gain-of-function mechanism (432). One study suggested that poly(A)+ RNA accumulation was blocked in trans by the DMPK expansion allele transcript but this finding was later refuted (432). However, another observation was made while tracking mutant DMPK transcript distribution in patient-derived cells. Using RNA fluorescence in situ hybridization, mutant DMPK transcripts were observed as punctate aggregates, or RNA foci, in the nuclei of DM1 patient-derived cells and tissues (Fig. 5) (85, 391). While unexpanded transcripts are efficiently exported to the cytoplasm, mutant DMPK transcripts are almost completely retained in the nucleus (85, 147). Strikingly, intranuclear CCUGexp RNA foci were also observed in DM2 patient-derived cells and tissues (Fig. 5) (231). These observations provided the first evidence for a shared mechanistic link between DM1 and DM2 and sparked considerable interest concerning the potential toxicity of these RNA aggregates.
Figure 5.
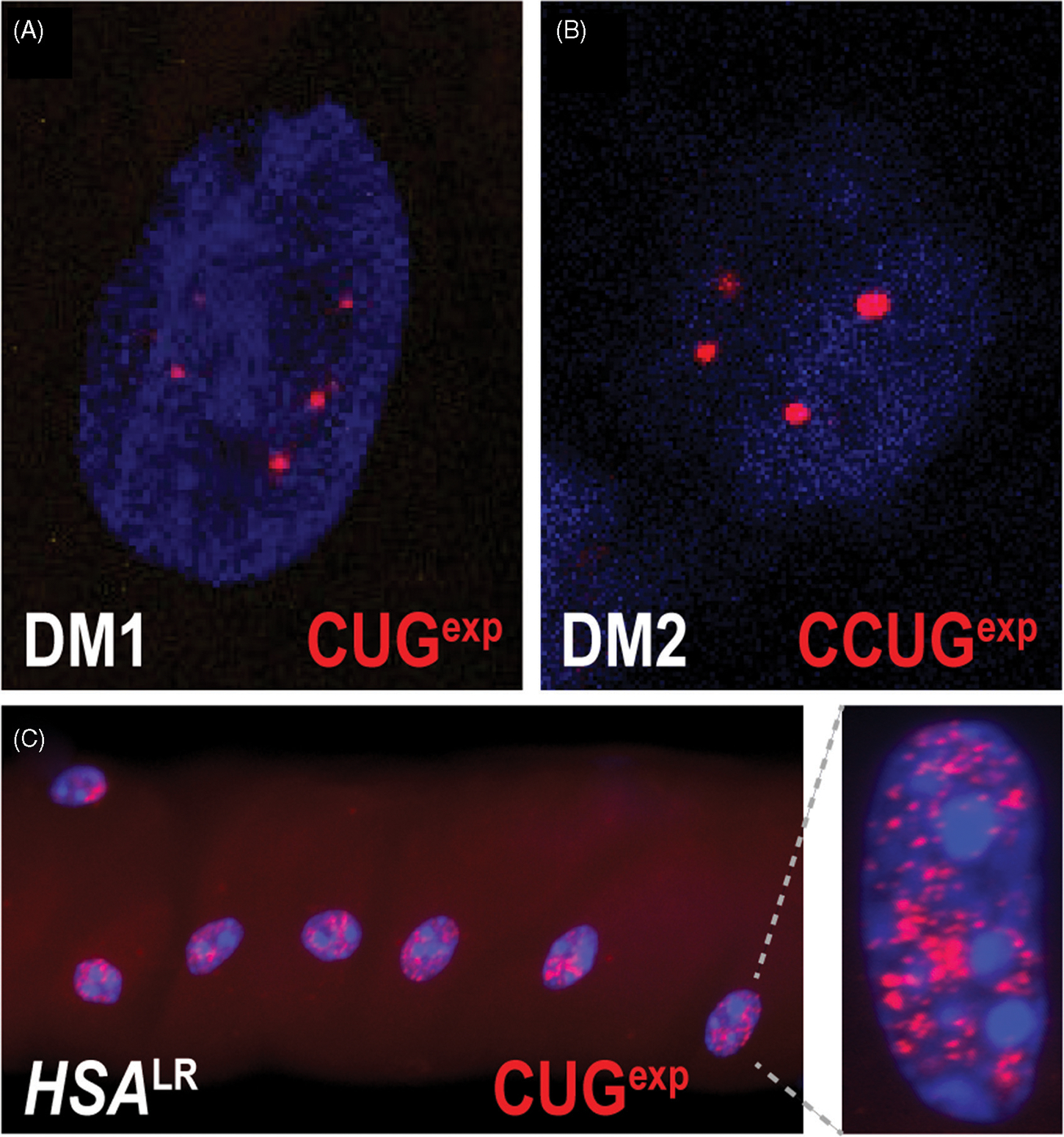
RNA foci in myotonic dystrophy. ((A) and (B)) Fluorescently labelled (CAG)10 or (CAGG)10 oligonucleotide probes hybridize to DMPK CUGexp transcripts in DM1 (A) or CNBP CCUGexp in DM2 (B), and reveal a punctate intranuclear staining pattern. These observations support the hypothesis that these mutant RNA transcripts are blocked for nucleocytoplasmic export and could exert toxicity in the nucleus. (C) Nuclear foci are abundant in myofibers isolated from the HSALR mouse DM1 model.
The RNA gain-of-function model was also tested using mouse models (Table 1). Early attempts to study the effects of CTGexp tracts in vivo utilized mice containing a DM1 DMPK transgene (265). While these animals showed a variety of phenotypes, it was unclear if the CUGexp RNAs were sufficient to generate disease independent of the DMPK gene context. To test this possibility, a transgenic mouse model was generated that expressed either a CTG5 (HSASR) or a CTG250 (HSALR) expansion, inserted into the 3’ UTR of a human skeletal actin transgene (245). Only the HSALR transgenic model recapitulated aspects of DM-associated myopathy, including myotonia, centralized myonuclei, and nuclear RNA foci (Fig. 5). Importantly, the severity of these features correlated with the degree of transgene expression demonstrating that the repeat expansion was toxic at the RNA, or a downstream, level. This study provided the first conclusive evidence that CUGexp RNAs could exert their toxic effect independent of gene context.
The remaining unresolved link for the RNA gain-of-function hypothesis was to determine the molecular pathways downstream of CUGexp expression that are adversely affected in disease and how CUGexp RNAs disrupt cellular homeostasis. Our group proposed a protein sequestration model in which CUGexp RNAs recruited, and subsequently sequestered, cellular factors with a high affinity for expanded CUG repeats. As an initial step to characterize these factors, in vitro pull-down assays were performed with (CUG)8 and these studies led to the identification of CUG-binding protein 1, CUGBP1 (currently CELF1) (406). However, further characterization of CELF1 did not support the hypothesis that this protein is a sequestered factor: (1) CELF1 binding to CUGexp RNA was not proportional to repeat number; (2) CELF1 failed to colocalize with CUGexp RNAs in RNA foci; and (3) CELF1 steady-state levels were upregulated in patient-derived cells (307, 406). Thus, the question of whether sequestered factors existed remained unresolved. To address this concern, an alternative experimental approach was tested that involved UV-crosslinking of proteins to radiolabeled RNAs following in vitro RNA processing reactions in HeLa cell nuclear extracts. These studies identified proteins homologous to Drosophila muscleblind (Mbl) (263). Importantly, these human proteins, termed MBNL, bound to CUGexp RNAs in a length-dependent manner, suggesting elevated sequestration as repeat tracts increase. Furthermore, all three human MBNL paralogs interact with CUGexp and CCUGexp RNAs in vitro and patient nuclei in vivo (105, 106, 247, 263, 386).
Another proposed, but not mutually exclusive, mechanism for C(C)UGexp toxicity in DM is a noncanonical form of protein translation termed repeat associated non-ATG (RAN) translation, which was originally discovered during studies on SCA8 pathomechanisms (78,463). DMPK, similar to other genes affected by microsatellite repeat expansions, is bidirectionally transcribed (27) so sense CTG repeats have the potential to encode polyleucine (polyLeu), polyalanine (polyAla), and polycysteine (polyCys) while the antisense CAG strand would translate into polyglutamine (poly-Gln), polyAla, and polyserine (polySer). Of these potential RAN products, only polyGln has been observed in DM1 myoblasts and skeletal muscle although this polyGln accumulation is more prevalent in blood cells (463).
Theories to therapies
Since both MBNL and CELF proteins were implicated in DM pathogenesis, understanding their normal cellular functions was the next critical step into understanding the molecular events misregulated in disease. Early work on CELF1 demonstrated that it was an AS factor that regulated human cardiac troponin T (cTNT) pre-mRNA splicing (307). Subsequent studies demonstrated that missplicing of the muscle chloride channel, CLCN1, was also responsive to CELF1. This missplicing event leads to nonsense-mediated decay of CLCN1 mRNA, which results in the myotonia, or muscle hyperexcitability, observed in DM. Subsequently, MBNL proteins were also shown to regulate the AS of gene transcripts misregulated in DM1 and DM2 (164, 189). DM-relevant missplicing also occurs in both HSALR transgenic and Mbnl1 KO mouse models and both animal models develop myotonia (189, 245). Currently, many RNA missplicing events have been identified in patient samples and animal models (Table 2) (67, 246, 272). Many of the affected pre-mRNAs (e.g., CLCN1, TNNT3, and INSR) are functionally related to known aspects of DM skeletal myopathy including myotonia, muscle weakness, and insulin insensitivity. A recurring theme from studying these splicing patterns is the retention of fetal exons in mature tissues and the antagonistic roles of MBNL and CELF proteins for some RNA targets (230). For example, CLCN1 exon 7a inclusion, which is the predominant pattern in embryonic and neonatal tissue of the developing mouse, leads to transcript degradation and possibly production of a CLCN1 C-terminal truncated protein (Fig. 6) (67, 246). As the muscle matures, CLCN1 exon 7a is increasingly excluded from the mRNA, facilitating increased CLCN1 RNA stability and downstream translation. For this event, CELF1 and MBNL1 promote inclusion and exclusion, respectively, leading to the aberrant retention of CLCN1 exon 7a in DM skeletal muscle when the activities of these RBPs are disrupted. In agreement with studies in the mouse, CLCN1 is lost from DM patient tissues and is associated with myotonia.
Table 2.
Missplicing Events Associated with DM Disease Symptoms
| Gene | Shift in DM | Protein function | Molecular and physiological consequence | References |
|---|---|---|---|---|
|
| ||||
| CLCN1 | Increased inclusion of exon 7a | Major skeletal muscle chloride channel responsible for transmembrane chloride conductance | Nonsense-mediated decay of transcript and loss of CLCN1 protein; myotonia |
Mankodi et al., 2002. Charlet et al., 2002. Wheeler et al., 2009. |
| CACNA1S | Decreased inclusion of exon 29 | CaV1.1 calcium voltage-gated calcium channel involved in excitation-contraction coupling | Increase CaV1.1 conductance and voltage sensitivity; in mature muscle fibers enhances electrically evoked Ca++ release; muscle weakness | Tang et al., 2012. |
| SCN5A | Increased inclusion of exon 6a and decreased inclusion of exon 6b | Cardiac sodium channel responsible for cardiomyocyte excitability and normal cardiac-conduction system function | Reduced excitability; heart arrhythmia, cardiac conduction delay | Freyermuth et al. 2016. |
| BIN1 | Decreased inclusion of exon 11 | Regulates T-tubule biogenesis | Lack of phosphatidylinositol 5-phosphate-binding and membrane-tubulating activities, altered T-tubules; muscle weakness | Fugier et al. 2011. |
| DMD | Decreased inclusion of exon 78 | Large structural and signaling protein linking the actin cytoskeletal to the extracellular matrix through the DAG complex | The dystrophin C-terminus is switched from an adult structure of a 13 aa beta-sheet into an embryonic 31 aa amphipathic alpha-helix. Abnormal dystrophin activity; muscle weakness and progressive atrophy | Rau et al. 2015. |
| INSR | Decreased inclusion of exon 11 (increased IR-A isoform) | Insulin receptor that mediates signal transduction involved in glucose storage and handling | Higher affinity for insulin; faster internalization and recycling time; lower signaling capacity | Savkur et al. 2001. |
| PKM | Increased inclusion of exon 10 (increased PKM2 isoform) | Pyruvate kinase M catalyzes phosphate group transfer reactions from phosphoenolpyruvate to ADP in glycolysis | Allosterically regulated isoform with reduced oxygen and increased glucose consumption; abnormal regulation of glucose metabolism in muscle; compromised glucose homeostasis; contributes to type 1 myofiber atrophy | Gao et al. 2013. |
| MBNL1, MBNL2 | Increased inclusion of exon 54nt, exon 36nt, and exon 95nt | RNA binding proteins that regulate diverse RNA processing activities | Increased nuclear localization and RNA splicing and polyadenylation activity |
Sznajder et al. 2016. Kino et al. 2015. Tran et al. 2010. Yuna et al. 2007. |
Figure 6.
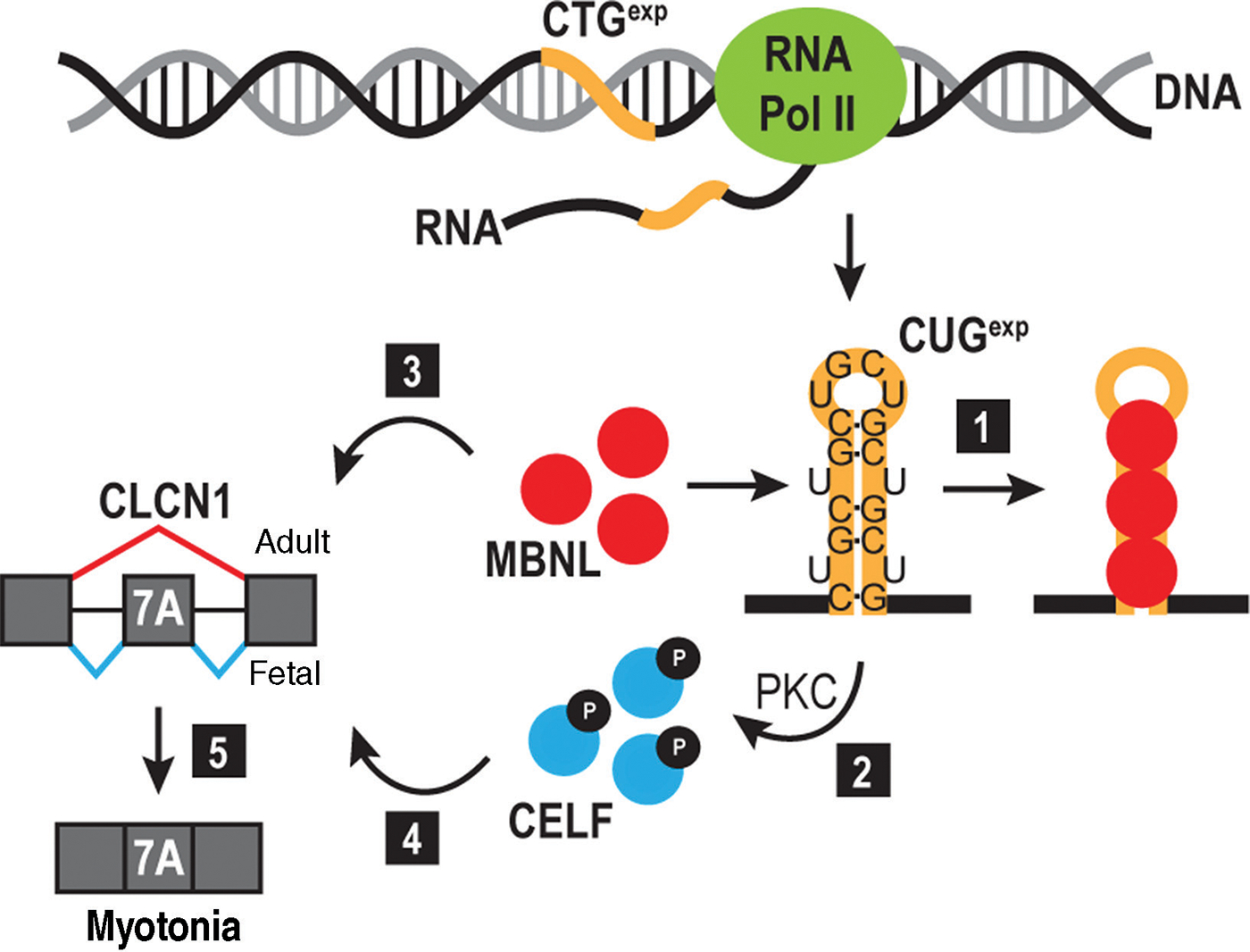
RNA toxicity model. Expression of the DMPK 3’ UTR CTGexp (orange line) produces a CUGexp RNA that sequesters MBNL proteins (red circles) (1) and triggers protein kinase C (PKC)-mediated CELF1 hyperphosphorylation (2) leading to an increase in its steady-state level. CELF and MBNL are antagonistic regulators of alternative splicing with MBNL promoting adult (3), and CELF favoring fetal (4), splicing isoforms. MBNL sequestration by CUGexp, in addition to CELF stabilization, leads to an imbalance in alternative splicing and emergence of fetal isoforms in adult tissues. In DM, this cascade leads to inclusion of exon 7A in CLCN1 mRNA, generating a fetal transcript that is degraded by nonsense-mediated decay. The absence of CLCN1 in the muscle membrane results in myotonia (5).
The advent of high-throughput transcript analysis technologies, such as microarrays and RNA-seq, have uncovered many RNA processing defects in DM. Furthermore, the use of crosslinking/immunoprecipitation and high-throughput sequencing (CLIP-seq) revealed direct MBNL and CELF binding targets, which has led to the identification of direct drivers of myopathy and other disease manifestations versus RNA processing errors resulting from generalized cellular dysfunction. Beyond a spliceopathy, DM is also a multifaceted RNA processing disorder as novel functions of MBNL and CELF proteins have emerged, including roles in alternative polyadenylation, RNA localization, miRNA biogenesis, RNA turnover, and control of translation. Today, unraveling the relative involvement of the complex mechanisms contributing to DM is an ongoing area of research. Identification of disease mediators is providing opportunities to design rationale therapeutics, such as antisense olignonucleotides (ASOs) targeting toxic RNA (438) or the use of pharmaceuticals to ameliorate symptoms (234).
DM Clinical Presentation
DM is a multisystemic muscular dystrophy affecting nearly every organ system of the body. A striking aspect of the DM1 phenotype results from skeletal muscle dysfunction, including myotonia, weakness, wasting, and myalgia (muscle pain) (Fig. 4) (332). However, DM is a truly multisystemic disorder with dysfunction of the cardiovascular system (arrhythmias, conduction blocks, cardiogenic syncope, and hypotension), respiratory system (respiratory muscle weakness, aspiration, and sleep apnea), gastrointestinal track (dysphagia, reflux, dyspepsia, choleostasis, constipation, diarrhea), central nervous system (hypersomnia, intellectual disability, executive dysfunction, peripheral neuropathy, and behavioral, emotional, and social difficulties), eye (particulate cataracts, ptosis, retinopathy, and ocular hypotension), endocrine system (insulin resistance/diabetes and metabolic syndrome), immune system (hypogammaglobulinemia), liver (steatosis and cirrhosis), reproductive system (testicular atrophy, female infertility, pregnancy, and neonatal complications), premature frontal balding, and pilomatrixoma (253,257,332,399). Many of these dysfunctions are more prominent in DM1 than DM2, and for overlapping features (e.g., myotonia), the severity is typically greatest in DM1. On the other hand, DM2 patients are more affected by myalgia (257).
Within DM1, subtypes are clinically defined based on age-of-onset and symptoms that correlate with repeat copy number: (1) late/asymptomatic; (2) adult/classic; (3) juvenile/childhood; and (4) congenital (257). In particular, CDM is often considered a unique disorder despite its shared genetic etiology to DM1 due to the exceptionally large CTGexp alleles (>750–1000), prenatal onset, and the unique constellation of symptoms, which include reduced fetal movement, polyhydramnios, hypotonia, respiratory distress, talipes, hydrocephalus, arthrogryposis, and intellectual disability (399). There are no genetically or clinically defined DM2 subtypes.
The birth of a CDM infant is often the impetus for evaluation and diagnosis of DM1 affected families. In cases where individuals pursue an independent clinical evaluation, hypersomnia is typically the motivating complaint and often occurs prior to adult-onset myopathy. Interestingly, many patients are unaware of their myotonia prior to clinical evaluation. In all forms of DM, cataracts are an early prognostic event particularly for late-onset/asymptomatic cases of DM1. Molecular diagnosis has almost eliminated the need for diagnostic muscle biopsies and is the only definitive test to diagnose DM and distinguish between DM1- and DM2-linked mutations. While it is not clinically utilized for diagnosis, DM skeletal muscle histopathology is sufficiently characteristic to identify the disorder as DM and differentiate between DM1 and DM2 (discussed below). Due to the later onset and variably muted phenotype of DM2, there is underdiagnosis of affected individuals even for those carrying large CCTGexp mutations.
Hypersomnia and behavioral changes typically precede skeletal muscle involvement and are among the most substantial features affecting patient quality of life (213, 214). As progressive muscle weakness and wasting emerge, additional complications arise such as gait abnormalities and difficulties performing tasks requiring fine dexterity. Altogether, these dysfunctions can be physically and socially disabling, and many DM patients have trouble securing employment. Premature mortality, typically occurring within the fourth decade of life, is most frequently associated with cardiac dysfunction and/or severe skeletal wasting leading to respiratory insufficiency (420). Many aspects of DM1 skeletal muscle dysfunction similar to age-related sarcopenia, suggesting DM1 is a progeroid-like syndrome (242, 253). For DM2, myalgia is one of the primary complaints, but life span is not significantly reduced. While DM is a multisystemic disorder, we will focus on skeletal muscle involvement for the remainder of this review. In the current section, we will provide a clinical perspective of the core DM skeletal muscle manifestations (myotonia, weakness, and wasting) and a discussion of events unique to CDM skeletal myopathy.
Myotonia
Following excitation of skeletal muscle by lower motor neurons, a variety of voltage-dependent, ion, and mechanosensory channels stimulate the release of calcium from extra- and intracellular stores. These events couple neuron-stimulated excitation to uniform and robust contraction of sarcomeres, a process termed excitation-contraction (EC) coupling. Following contraction, repolarization of the sarcolemmal membrane potential is required to allow stabilization of homeostatic calcium gradients and muscle relaxation. This process is also regulated by channel-mediated ion redistribution.
Myotonia is an abnormal delay in muscle relaxation following contraction, and occurs in the tongue, jaw, feet, and hand musculature of DM patients (25, 171, 434). While myotonia is widespread, it is initially assessed by grip myotonia—an abnormal delay in extending fingers after forming a fist (Fig. 1) (171). A more reliable diagnosis of myotonia is achieved by stimulating the thenar eminence (25). This is termed percussion myotonia and can be observed in the absence of grip myotonia (25). To obtain quantitative measurements of electrical myotonia, electromyography (EMG) is performed. Insertion of concentric needle electrodes elicits depolarization of the sarcolemma and results in measurable action potential number, duration, amplitude, and qualities (e.g., waxing and waning) (309). As electrical myotonia can be observed independent of grip and percussion myotonia, EMG is the most sensitive measurement of myotonia (18, 238, 434). Additionally, outputs from EMG can be useful in differentiating between myotonic versus other (e.g., inflammatory) myopathies (128, 160).
DM is one of several myotonic disorders including myotonia congenita, paramyotonia congenita, and hyperkalemic periodic paralysis (238,384). While these diseases result from mutations in specific ion channels, DM-associated myotonia results from aberrant CLCN1 pre-mRNA splicing. As noted above, this missplicing destabilizes the transcript leading to its degradation and loss of CLCN1 from the sarcolemma. Because CLCN1 acts as a major regulator of chloride flux in mature muscle, its loss results in muscle hyperexcitability.
EMG-measured myotonic discharges are generally of greater frequency and amplitude in DM1 than DM2, and the distribution of myotonia differs in DM types with distal muscle groups more affected in DM1 while proximal muscle problems are more prominent in DM2 (160, 233). Additionally, DM2 action potentials are characterized almost exclusively by a waning characteristic (233, 452). Interestingly, in cases of pronounced myotonia in DM2 patients, there is often a cosegregating mutation in the CLCN1 protein-coding region or sodium channel, voltage gated, type IV alpha subunit (SCN4A) (52, 385).
Chloride-mediated myotonic disorders, such as DM, display ameliorated severity following repeated contraction-relaxation episodes, known as the warm-up phenomenon (346). Consequently, patients have the greatest degree of difficulty initiating movement with improvements over time. DM patients are spared of sensitivity to cold climates, a feature frequently observed in sodium-mediated myotonic disorders (115, 128). Although the consequences of myotonia are most evident while performing tasks requiring fine motor skills, health may be compromised if severity reaches a debilitating threshold in bulbar muscles important for chewing and swallowing. Furthermore, myotonia in diaphragm and intercostal muscles may contribute to respiratory complications and life-threatening myotonia can be induced by the use of neuromuscular blockers during surgery. DM is one of the few dystrophic myotonic disorders, and as the disease progresses, myotonia typically becomes undetectable as muscle atrophy and weakness become more prevalent (378).
Muscle weakness
The contraction of muscle generates force, the magnitude of which is dictated by factors including myofiber number, size, and fiber type. Additionally, the efficiency of EC coupling and integrity of the contractile apparatus within a myofiber are critical components influencing muscle strength. Muscle is a highly plastic organ, and physical training can increase its size through hyperplastic and hypertrophic mechanisms culminating in a greater force potential. A balance of anabolic and catabolic pathways governs this plasticity with predominant catabolic activity contributing to muscle weakness in muscle disuse atrophy, aging, and disease. Furthermore, myofiber loss causes debilitating weakness. While these are examples of weakness subsequent to loss of myofiber size or number, defects in the contractile apparatus and/or architecture of the costamere also compromise muscle strength.
The assessment of muscle strength in a sensitive, quantitative, and reproducible manner is a nontrivial task. This is a particularly important consideration as clinic-to-clinic variability can lead to technical artifacts, hindering development of reliable data useful as clinical trial outcome measures. Hand-held dynamometry devices are a commonly utilized method to assess grip strength and meet the needs of quantitative output, reproducibility, and ease of use (377). However, for many muscle groups, the slow progressivity of weakness poses practical concerns for using traditional strength measurement scales (295, 441). The measurement of handgrip strength has been proposed as a possible exception, as changes can be detected within 6 months for most patients, a reasonable time scale to test therapeutic efficacy (441). These findings have been supported by a retrospective analysis of 204 DM1 patients, and both studies highlight the slow progressivity of weakness in the majority of DM muscle groups (45).
DM patients are affected by progressive muscle weakness with early signs of onset in the face, neck, ankles, and hands (238, 441). While this progressivity is slow, it can be debilitating in some instances (146, 279). Interestingly, weakness typically manifests secondary to myotonia, suggesting a temporal hierarchy in disease symptom emergence (238). Facial muscle weakness contributes to difficulties chewing and swallowing, ptosis, and the drooping appearance of the face. Weakness of the leg musculature impairs ambulation and is associated with difficulties lifting the foot, termed foot drop. Gait abnormalities and reduced muscle force in the legs correlates with increased propensity for falls in more severely affected DM patients (146). Late stage weakness in bulbar and respiratory (diaphragm and intercostal) muscles increase patient morbidity (238, 257).
As with myotonia, muscle weakness is more severe in DM1 than DM2, and DM1 shows a preferential involvement of distal rather than proximal muscles (257). Interestingly, DM1 is unique in the distal, rather than proximal, involvement seen in most myopathies including DM2, suggesting a particular sensitivity of muscles such as the tibialis anterior (TA) to CUGexp-mediated toxicity (441).
Muscle wasting
The normal development, function, and maintenance of muscle requires a complex interplay between transcriptional, co-/posttranscriptional, and downstream pathways (89). In general, a rigid network of extracellular support and a variety of membrane repair mechanisms help to maintain the structural integrity of muscle. Furthermore, resident skeletal muscle stem, or satellite, cells (SCs) provide muscle with a high regenerative capacity and also contribute to hypertrophy (98). In response to activity such as exercise, a variety of endocrine signals and mechanosensors stimulate growth receptors that activate anabolic signaling cascades, most notably the Akt/mTOR pathway (40). Conversely, muscle wasting, or atrophy, is the decline of muscle mass resulting from excessive catabolic activity and commonly occurs because of disuse, inadequate innervation, aging, and disease (39,80). Loss of muscle bulk can result from a decrease in individual myofiber size and/or a reduction in total myofiber number. Progressive muscle wasting leads to a dramatic decrease in body weight, muscle weakness, disability, and when severe, poses a formidable health risk. For example, cancer-induced muscle loss, or cachexia, is the proximal cause of death of many cancer patients (461).
Early signs of wasting in DM1 are seen in facial muscles (e.g., temporalis and masseter) and distal limbs. In late-stage disease, proximal muscles are similarly affected. DM2 patients experience preferential proximal muscle wasting and can unexpectedly present with hypertrophy of some distal muscles such as the gastrocnemius. While there is a scarcity of quantitative data regarding DM muscle wasting, qualitative assessment of patient muscle biopsies reveals features of degenerative disease and will be discussed in more detail below (423). Beyond myofiber wasting, satellite cell dysfunction may be compromised in DM (243, 244). While this has yet to be explicitly studied, the complex nature of muscle wasting in DM is likely to be explained by a combination of systemic pathology, defects inherent to myofiber function, increased proliferative burden of satellite cells, and ultimately exhaustion of the satellite cell population. As discussed below, many aspects of DM wasting resemble age-related sarcopenia (253).
Developmental abnormalities in CDM
While CDM is also caused by CTGexp mutations in the DMPK 3’ UTR, the exceptionally large repeat copy number (typically > 1000), unique constellation of symptoms, and prenatal onset is often the basis for its classification as a clinically distinct disorder (162, 256, 257). This is similar to the repeat-length-based criteria distinguishing fragile X-associated tremor/ataxia syndrome (FXTAS) and FXS (144).
Another critical distinction between DM1 and CDM is the fact that CDM patients inherit highly expanded alleles, resulting in the present of large CTGexp mutations throughout embryogenesis. As such, several symptoms emerge in utero including reduced fetal movement, polyhydramnios, talipes, and borderline ventriculomegaly (399, 456). Polyhydramnios is likely reflective of myogenic defects impairing the ability of fetal swallowing (148, 355, 456). Perinatally, CDM infants present with hypotonia, poor suckling due to bilateral facial weakness, dysphagia, and respiratory insufficiency necessitating supportive ventilation (100). Hypotonia is one of the most visually striking feature of newborn CDM infants, and affects posture and movement leading to the hallmark “floppy baby” appearance (38, 301). Assisted feeding is necessary for the majority of CDM infants (57).
Diaphragm and intercostal muscle weakness contributes to respiratory insufficiency and is the greatest source of mortality (58, 326, 339). Perinatal asphyxia correlates with reduced APGAR score and measures of neurological function in later years of life, suggesting a contribution of muscle weakness to other phenotypes (390). CDM infants will show gradual improvement but display reduced motor milestones and eventually develop symptoms associated with childhood onset DM1 (100,399). Intellectual disability including autism spectrum disorder is a prominent feature (100,102). Although clinical and electrical myotonia eventually emerges, it is absent in CDM neonates likely due to the dispensable role of CLCN1 in immature muscle. Interestingly, many CDM symptoms are similar to those seen in other congenital myopathies. This observation suggests that different genetic defects affect developmental myogenesis to culminate in similar clinical presentations (175, 328).
Skeletal Muscle Architecture in DM
A skeletal muscle is organized into highly interconnected and organized structural units—an organizational pattern important for proper function (Fig. 7). Histological and ultrastructural examinations of diseased skeletal muscle offer insights into the degree of tissue and cellular pathology and even provides clues into the nature of molecular dysfunction. For example, disrupted transverse tubules morphology observed via electron microscopy is suggestive of defective EC coupling, calcium handling, and potential underlying dysfunction of dihydropyridine receptors.
Figure 7.
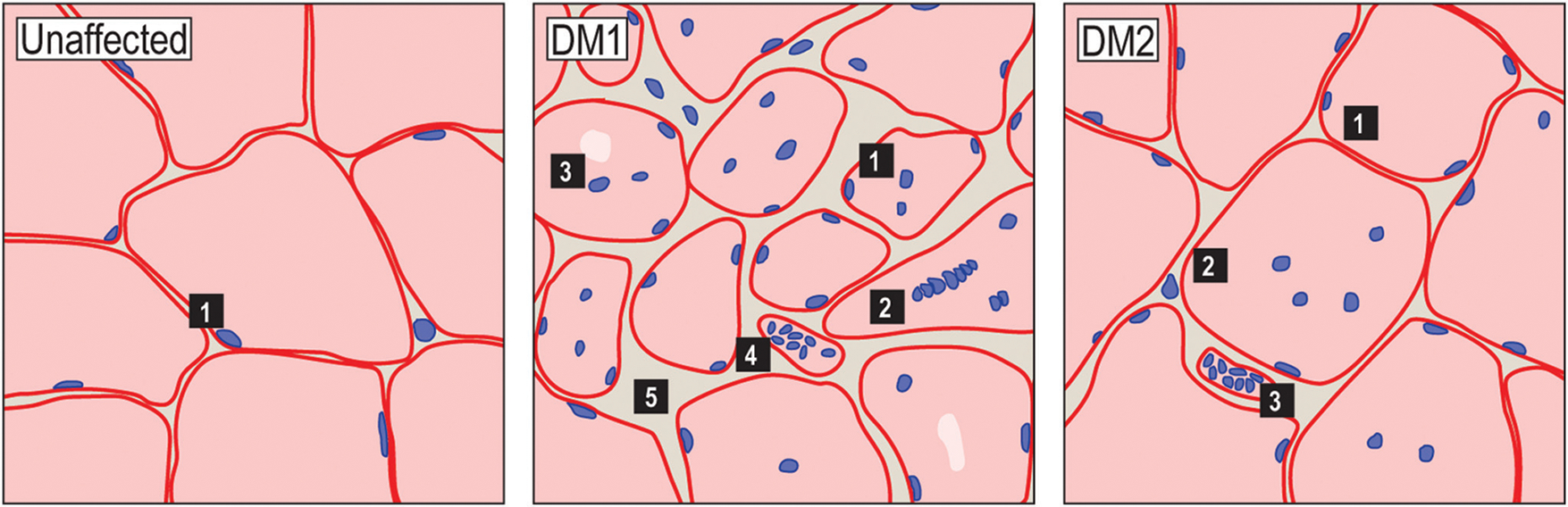
Histological features of DM1 and DM2 skeletal muscle. Schematic representations of H&E-stained skeletal muscle cross-sections from unaffected (left), DM1 (center), and DM2 (right) patients depicting common histological features (images available at http://neuromuscular.wustl.edu/pathol/). Typically, myofibers are uniform in size and have subsarcolemmal myonuclei (left panel). In DM1, histopathological features include central myonuclei, myofiber size variability, pyknotic nuclear clumps and fibrosis. Other features include type I fiber atrophy, irregular nuclei shape, and acid phosphatase stained granules and several of these features roughly correlate with disease severity and progression. In DM2, these histopathological features are generally less pronounced and may include some variability in fiber size, internal myonuclei, and pyknotic nuclear clumps. Acid phosphatase positive granules are also observed in DM2.
An individual muscle is comprised of many muscle fascicles—bundles of myofibers surrounded by a connective tissue layer termed the perimysium. Within a single muscle fascicle are the muscle cells themselves, the myofibers, that contain the functional units of muscle contraction (sarcomeres) comprised of the thin (actin) and thick (myosin) filaments. Mature myofibers contain hundreds of myonuclei located beneath the sarcolemma. The positioning of organelles in a myofiber is important to avoid obstructing the sarcomere, thus allowing uniform and undisturbed contraction. While observation of typical muscle cross-sections reveals striking homogeneity, differences in fibers can be observed using a variety of staining methods to identify biochemical fiber types within a muscle, of which, there are four major categories—type I, IIA, IIB, and IIX—largely defined based on their relative sarcomeric myosin heavy chain composition (354). The relative proportion of these varies between muscle groups and dictates aspects of muscle performance. For example, TA is mostly a type II fast-twitch muscle, with a greater force potential than type I muscles. On the other hand, predominant type I slow-twitch muscles (e.g., soleus) are better suited for endurance tasks such as maintaining posture. Fiber-type patterning is first established during embryogenesis by homeoproteins such as SIX1 and SIX4, but has substantial postnatal plasticity based on hormones, innervation, electrical stimulation, and activity (141, 177, 330). Training regimes stimulate fiber-type transitions, typically in a predictable manner: I ↔ IIA ↔ IIX ↔ IIB (304). Type IIB fibers typically have the greatest cross-sectional area, providing the greatest force capacity. Fiber types also dramatically differ in their metabolic signature with type I being the most oxidative and type IIB the most glycolytic. Importantly, these differences provide the foundation for histochemical staining procedures allowing for the identification of different fiber types within a muscle cross-section (354).
DM1 and DM2 muscle structure profile
Haemotoxylin and eosin staining, a common technique for analysis of patient skeletal muscle biopsies, reveals several hallmark characteristics of DM histopathology, including myofiber atrophy and centralized nuclei (Fig. 7). The histological hallmarks of DM patient biopsies are sufficient to distinguish DM from other myopathies and even differentiate between DM1 and DM2. For example, disease specific biochemical fiber-type histopathology is predictive of DM1 versus DM2 (308). While genetic analysis is currently the only supported diagnostic measure, technical challenges in genetic testing for DM2 have resulted in a greater need for diagnostic biopsy. Thus, histological analysis of DM2 skeletal muscle is more common than in DM1.
In DM2, there is an increase in type II fibers and fiber atrophy appears greater in these compared to type I fibers (270, 423). In DM1, there is preferential type I fiber atrophy (423). Given electrical activity can stimulate fiber-type transitions, it is possible that myotonic discharges in DM influence fiber types. Fiber atrophy is preferential for distal muscle in DM1 and proximal muscle in DM2. Furthermore, within DM1 biopsies, type I fibers show preferential atrophy while type II fibers are more affected in DM2 (270). In DM2, pyknotic nuclear clumps—a marker of late atrophic myofibers—are observed.
The subsarcolemma positioning of myonuclei is a hallmark of mature muscle. Typically, myonuclei are regularly spaced within a myofiber and deviation from this normal organization patterns impairs muscle function (48). In several myopathic disorders and age-related sarcopenia, nuclei are mislocalized in the center of a myofiber upon histological examination. This is considered a marker of active regeneration and is associated with several congenital myopathies (182,183). Beyond a marker for myopathy, central nuclei may directly contribute to muscle dysfunction by disrupting sarcomere organization and affect myofibril contraction (113). Several regulators of myonuclear positioning are emerging and their contribution to centralized nuclei in DM has not yet been characterized (114, 260). While central nuclei are observed in both DM1 and DM2, DM2 patients tend to show a preference for central nucleation in type II fibers (308). However, as with other measures of DM histopathology, the frequency of central nuclei appears greater in DM1 (356,423). While less studied, other features of DM muscle histopathology include split fibers, ring fibers, late-stage fibrosis and steatosis, nuclear chains, and large/irregularly shaped myonuclei (133, 423).
CDM muscle
CDM patients display a variety of in utero and perinatal phenotypes, indicative of disrupted muscle development, and histopathological features throughout embryogenesis and postnatal life. Indeed, CDM muscle contains an increased proportion of immature myotubes, small muscle fascicles, central nuclei, myofiber size variability, and fiber-type disproportion (14, 108, 174, 193, 342, 343, 352, 369, 411).
One of the first studies of CDM histopathology reported a reduction in IIB fibers and atrophy of type I fibers, similar to results obtained from adult DM1 biopsies (14). These authors suggested the involvement of dysfunctional motor neurons in these phenotypes, similar to congenital fiber-type disproportion disease (14). While abnormal motor endings have been observed in CDM, other groups have suggested normal innervation patterns in CDM (108, 343). Given the establishment and maintenance of mature neuromuscular junctions is dependent on both motor neuronand muscle-intrinsic mechanisms, both cell populations may play a role in disease (447). CDM infants are often born prematurely and muscles at 27, 34, and 37 weeks gestational age have been studied, revealing morphological and histochemical markers of fiber immaturity (342). Some patients displayed differences in satellite cell number and all three patients showed evidence of increased lysosome accumulation—a marker of fiber necrosis (342). Unfortunately, studies of CDM histopathology are often limited based on small sample numbers and lack of appropriate controls. Therefore, the current data regarding characteristics of CDM muscle should be interpreted with caution. This scarcity of data highlights the need for more thorough analysis of CDM patient skeletal muscle as well as the generation of animal models that can address the fundamental basis of myogenic defects in CDM. Although a variety of staining methods have revealed aspects of DM histopathology, additional work utilizing immunolabeling techniques should provide information regarding important myogenic cell populations, such as satellite cells, which have been proposed as being dysfunctional in CDM (122). Furthermore, cell-cell and cell-matrix interactions are essential for muscle development and may play a role in CDM manifestations (61).
Molecular Mechanisms Involved in DM Pathogenesis
In the previous sections, we provided an introduction into the DM field and highlighted key clinical and histological presentations of DM patients. Complex molecular mechanisms underlie these phenotypes and, in many cases, aspects of the disease are still under investigation. In the remainder of this review, we provide detailed information regarding the molecular pathogenesis of DM and how particular events relate to disease. In this section, we first discuss aspects of DM molecular pathology that are likely at play in any given cell type. We then focus our attention on studies exploring aspects of muscle development, function, and maintenance and the model organisms used to understand these processes. Given the prevailing view that disruption of RNA processing pathways is a central pathomechanism in DM, particular attention is given to these regulatory networks.
A fundamental question in understanding DM pathogenesis is how simple repetitive CTG and CCTG sequence motifs in noncoding regions of the genome give rise to the variety of features described above. The complexity of this question is underscored by several observations. First, DM is a multisystemic disease with nearly every organ system in the body affected to some degree. Second, given the location of the DMPK and CNBP C(C)TGexp repeats in noncoding regions, it is unlikely DMPK or CNBP gain-of-function mediated through alterations in amino acid sequence or loss-of-function resulting from frame-shifting would contribute to disease. Third, the striking genetic anticipation of DM1 suggests a repeat toxicity dose effect. Finally, and perhaps most intriguing, the partial phenocopy between DM1 and DM2 hints at some overlapping disease mechanism.
Two main pathogenic mechanisms have been proposed. First, C(C)TGexp mutations may alter the expression patterns of DMPK, CNBP, or neighboring genes through epigenetic mechanisms. Second, DM is an RNA-mediated disease and C(C)UGexp RNAs are toxic through the modulation of downstream effectors. As explained below, the latter RNA gain-of-function mechanism has emerged as the predominant contributor to molecular toxicity. However, cooperativity may also exist between these mechanisms, particularly in the case of CDM where highly expanded repeats are present throughout embryogenesis. In this section, we will survey the fundamental principles of genome-, transcript-, and effector-level mechanisms of DM molecular pathology as they pertain to any tissue. In the next section, we will address molecular dysfunction as it relates to specific defects in skeletal muscle.
Genome level
DM is one of over two dozen microsatellite expansion disorders, and lessons learned from these other diseases can inform mechanistic theories of DM pathogenesis. For example, FXS is caused by a CGG microsatellite expansion in the 5’ UTR region of the FMR1 gene and results in intellectual disability, psychiatric dysfunction, and other neurological impairments (21). CGGexp modulates epigenetic modification of this locus, including altered methylation and histone modifications, ultimately silencing FMR1 transcription and eliminating the production of FMRP protein—a key regulator of local translation in neurons (21). CTGexp elements influence nucleosome positioning, suggesting transcriptional dysregulation of the DMPK locus may also be involved in DM pathogenesis (424). Indeed, increased nucleosome occupancy was preferentially observed on mutant DMPK alleles and appeared to correlate with repeat number, suggesting transcriptional disruption of the DMPK locus may be involved (433). The DMPK 3’ UTR contains a DNase I hypersensitivity site in the wild-type allele, but shows resistance to DNase I cleavage in DM1-patient-derived cells and tissues, indicative of heterochromatin (287). Furthermore, CpG-islands are located upstream and downstream of the CTGexp that are unmethylated in normal adults and most adult DM1 patients, but show frequent hypermethylation in CDM (380). This result indicates a unique contribution of highly expanded DMPK alleles to certain epigenetic modifications and also provides one of the first molecular distinctions between adult-onset DM1 and CDM. The spread of heterochromatin at this locus may be restricted by two CTCF-dependent insulator regions flanking the CTGexp (74, 112). While DMPK hypermethylation is postulated to disrupt CTCF-binding and chromatin condensation near the CTGexp, CTCF occupancy at this locus appears to be methylation-insensitive (74, 235, 451).
The DMPK CTGexp mutation resides in a gene-rich region. The dystrophia myotonica WD repeat-containing protein (DMWD) and sine oculis homeobox homolog 5 (SIX5) are upstream and downstream of DMPK, respectively, and reside within an ~30 kb window. Both of these genes are expressed in developing and mature muscle. The DMPK 3’ UTR overlaps with putative SIX5 promoter elements and the DNase I hypersensitivity site lost in DM1 serves as an enhancer element (200). SIX5 expression is reduced in DM1 patient-derived fibroblasts, myoblasts, muscle, and heart tissue (200, 402). This reduction is allele-specific and correlates with repeat number (200, 402). Additionally, DMPK RNA and protein levels were originally reported as being decreased in DM1 patients (120). As DM is inherited in an autosomal dominant fashion, this would support a haploinsufficiency model (120). While this model is not expected to agree with the anticipation observed in DM, it is possible that repeat length-dependent increases in epigenetic changes would result in a step-wise decrease in RNA and protein production of DM1-linked genes. However, hypermethylation of the DMPK CTGexp proximal locus does not correlate with repeat length (235). Rather, the hypermethylation status appears specific to CDM and suggests the presence of pathogenic allele sizes during development triggers the establishment of epigenetic changes that cannot be recapitulated during postnatal repeat expansion (235). Recent work using a large cohort of patient blood, chorionic villus, and human embryonic stem cell (ESC) samples confirms hypermethylation adjacent to the DMPK CTGexp is a prominent, and unique, feature of CDM (24). Additionally, the authors report that methylation upstream of the CTGexp is unique to maternally derived germ cells, which may explain the transmission bias of CDM alleles from mothers (24). In male spermatogonia, methylation-induced reduction in SIX5 expression may result in loss of these cells, and this may explain the exceedingly rare paternal transmission pattern. In agreement, Six5 KO mice display male infertility associated with impaired post-natal spermatogenesis (351). Because germ cells are haploid, CTGexp-induced gene silencing would be expected to be more deleterious than in diploid cell populations.
The most compelling evidence against a DM haploinsufficiency model comes from a variety of heterozygous and homozygous mouse KO studies (Table 1). Heterozygous Six5 KO mice are normal and do not support a model whereby partial loss of SIX5 recapitulates DM phenotypes (199). While homozygous Six5 KO mice develop cataracts, they do not resemble the subcapsular particulate cataracts present in DM patients (199). Heterozygous Six5 KOs may display subtle cardiac abnormalities, but a direct link to DM pathology is unclear (427). To date, reduced SIX5 protein levels have not been demonstrated in DM tissues. However, as mentioned above, reductions in SIX5 levels may be restricted to, and particularly toxic in, haploid cells such as male germ cells.
Although mechanistically unexplored, the CTGexp may affect the expression of DMPK itself. Furthermore, the presence of 3’ UTR CUGexp tracts could theoretically disrupt other aspects of DMPK RNA metabolism such as nuclear export and/or translational efficiency. To test the contribution of DMPK loss-of-function to DM1, heterozygous and homozygous Dmpk KO mice were generated (327). Heterozygous KO mice are overtly normal, with no reported overt abnormalities or decreased life expectancy (327). Later studies reported cardiac abnormalities but recent work suggests normal cardiac and muscle function in various DMPK-depleted mouse models (30, 62). Homozygous KO mice were originally described as having mild, late-onset myopathy but again, recent studies do not support these original observations (62, 179, 327). Along with a lack of robust DM-relevant phenotypes in mouse KO studies, the original evidence of reduced DMPK mRNA and protein levels is controversial and may be associated with technical artifacts including RNA purification techniques and quality of anti-DMPK antibodies (85, 124, 147, 215, 240, 432). As mentioned above, use of cesium-chloride gradient RNA purification strategies recovers normal DMPK mRNA levels in DM1 samples compared to controls (85). While DMWD loss-of-function studies have not been thoroughly conducted, DMWD mRNA levels are not reduced in patient-derived fibroblasts (147). Overall, these results fail to support a major contribution of SIX5, DMPK, or DMWD haploinsufficiency to DM1.
Unlike DM1, the CCTGexp in DM2 is located far from neighboring genes, suggesting no gene besides CNBP would be susceptible to haploinsufficiency in DM2. CNBP protein is localized to the nuclei of embryonic mouse tissues where it promotes cell proliferation partly through transcriptional activation of c-MYC (68, 365). Although controversial, studies have shown CCTGexp-associated decreases in CNBP expression levels (316,345). Homozygous Cnbp KO mice are embryonic lethal, and present with dramatic developmental abnormalities including largely absent forebrain and craniofacial defects (Table 1) (68). Interestingly, Cnbp heterozygous KO mice recapitulate electrical myotonia, cardiac conduction defects, cataracts, and myopathic features (69). CNBP is typically expressed at high levels in skeletal muscle and is localized to Z-lines (316). In DM2, the CNBP expression pattern may be altered and may affect the translation of CNBP-target RNAs, several of which have known roles in muscle function (172,316). A key distinction between DM1- and DM2-associated microsatellite expansions is the location of the repeat. In DM1, the CTGexp in the 3’ UTR of DMPK is preserved after mRNA processing, yielding capped and polyadenylated transcripts. Other pathogenic microsatellites, such as the CTGexp in Fuchs endothelial corneal dystrophy (FECD) (442), the GGGGCCexp in C9-ALS/FTD or the CCTGexp in DM2, reside in introns of TCF4, C9orf72, and CNBP, respectively, and therefore are expected to be completely spliced out of the final mRNA population. In this context, the spliced intron would be less stable and the mature mRNA would be spared from downstream disruption. The expected processing pattern (i.e., intron 1 splicing) of mutant CNBP transcripts is supported by early studies (249), in contrast to recent findings in C9-ALS/FTD, in which the GGGGCCexp led to intron retention in patient lymphoblasts (275).
Knowledge regarding the normal functions of the DMPK, SIX5, and CNBP proteins is important, as this information may provide insights into the molecular defects associated with their possible loss-of-function in patient tissue and animal models. DMPK is a serine/threonine kinase and is localized to the nuclear envelope of HeLa and C2C12 cells (153, 185). Overexpression or knockdown is associated with altered laminin protein levels and localization in these cells and disruption of myotube formation in C2C12 cells (152, 153). DMPK may also support resistance to reactive oxygen species (ROS) and antagonize ROS-induced cell death (292). CNBP is a single-stranded RBP and DNA binding protein and has been suggested to bind to genes associated with Wnt signaling pathways (248). SIX5 is a homeodomain protein, a protein family essential for embryonic development, and its misregulation would be expected to exacerbate myopathy (46).
Transcript level
Transcription across C(C)TGexp DNA generates C(C)UGexp-containing RNAs. Early studies proposed that CUGexp RNAs contribute to DM1 (63, 405, 432). According to this model, highly expanded C(C)UGexp would disrupt cellular pathways leading to disease manifestations possibly via some RNA gain-of-function mechanism.
In the context of DM1, mutant DMPK transcripts are expressed and undergo normal pre-mRNA processing (i.e., 5’ cap addition, splicing, and polyadenylation), but these mRNAs are selectively retained in the nucleus of DM1 muscle and fibroblast cell lines (85, 147). Retention is length-dependent, as CUG80 repeats are more often found in the cytoplasmic fraction than CUG400 repeat-containing transcripts (147). Beyond nuclear retention, CUGexp RNAs are localized as punctate inclusions, or RNA foci, that also increase in DM1 myoblast cell lines containing longer repeats (391). As discussed more below, RNA foci are complex structures comprised of C(C)UGexp RNA and RBPs which can be compact and crowded structures depending on mutant RNA repeat length and copy number as well as the total amount of MBNL available to bind (386). Recently, CUGexp RNA has been observed undergoing phase transitions to form viscous, gel-like structures in vitro, and these structures can merge, divide, or completely dissolve over time (178, 386).
A variety of transgenic mouse lines expressing CUGexp transcripts support a role for toxic RNAs eliciting disease symptoms. One transgenic mouse model, generated using an ~45 kb human mutant transgene containing a CTG repeat expansion, underwent intergenerational expansion to yield a variety of large repeat mice (Table 1) (137). DM300 mice, a derivative of the original line, recapitulate features of DM1 and those with larger (1200–1800) repeats, show a more severe phenotype (132, 361, 362). However, the expression of these transgenes is low, likely below the normal levels observed in affected DM1 tissues, and therefore underestimates the contribution of CUGexp RNA to disease progression. A direct role for CUGexp RNAs in disease progression is most convincingly demonstrated in a mouse model expressing CUG250 transcripts under the control of a human skeletal actin promoter (HSALR mice) (245). These mice develop centralized nuclei, myotonia, and CUGexp RNA foci that correlate with transgene expression level (245). Importantly, these observations reveal CUGexp RNAs exert toxicity and recapitulate aspects of the disease independent of gene context. A variety of additional repeat mouse models have confirmed and extended these findings, many of which show muscle atrophy, myotonia, and heart defects (284).
Nuclear retention of mutant DMPK and CNBP transcripts is not the consequence of disrupted pre-mRNA processing (147, 249). Furthermore, the punctate localization of RNA foci suggests the involvement of coalescing factors recruited to these transcripts (261, 406). The first of these was identified as CELF1/CUGBP1 (406), but despite its interaction with CUGexp RNAs in vitro CELF1 does not colocalize with RNA foci in patient tissues, suggesting that its sequestration does not occur in vivo. On the other hand, other CUGexp binding proteins, most notably members of the MBNL family colocalize with RNA foci and directly interact with C(C)UGexp RNAs in vivo (105, 106, 134, 263, 386).
Effector level
Modulation of CELF and MBNL activities is the most significant and widely supported cause of DM pathogenesis (73, 221, 311). In vitro, CELF proteins preferentially interact with CUGexp or UG-enriched RNAs (110,396,406). However, CELF1 does not colocalize with RNA foci in vivo (105, 106). Instead, CELF1 levels are increased in DM1 skeletal and cardiac muscle (188, 307, 407). CUGexp RNAs can directly stimulate CELF1 increases through PKC-dependent hyperphosphorylation and stabilization of CELF1 protein (403) and transgenic DMPK CTG960 mice, which display cardiac abnormalities and increased CELF1 levels, show amelioration of symptoms and reduced CELF1 protein following PKC inhibition (211, 431). Other regulators of CELF1 steady-state levels include GSK3β, cyclin D3-CDK4, and calcineurin (180,324). CELF1 levels are also increased in regenerating and denervated muscle fibers (285, 392). Since features of regenerative myogenesis occur in DM1 muscle, is the increase in CELF1 levels simply a consequence? In support of a direct role, CELF1 levels are increased in the CTG960 mouse model prior to the onset of overt histopathology (284). Immunofluorescent labelling of CELF1 also revealed increased levels in mature, nonregenerative myonuclei of this mouse model, implicating CELF1 upregulation as a contributor to DM1 pathology (285). Additionally, CELF1 repression ameliorates myopathy in DM1 models (29).
MBNL proteins are orthologs of Drosophila muscleblind (Mbl) and in mammals, three MBNL paralogs exist, MBNL1, MBNL2, and MBNL3 (294). All three mammalian paralogs bind to CUGexp RNAs in vitro and colocalize with RNA foci in vivo (105, 106, 263, 386). MBNL proteins associate with CUGexp RNAs by binding GC steps interrupted by unpaired pyrimidines (DM1, U-U mismatches in CUG repeats; DM2, C-U/U-C in CCUG) and are stabilized via homotypic interactions mediated in their C-terminal domains (454). All MBNL paralogs bind CUG repeats in vitro with very high affinity and with even higher affinity to RNA fragments containing CCUG repeats (203, 386). This feature, together with higher expression of the CNBP gene, should evoke stronger MBNL-dependent spliceopathy in DM2. However, MBNL sequestration is likely to be limited by rapid turnover of spliced CNBP intron 1 (249). In contrast to CELF, in vitro MBNL binding is proportional to CUG repeat length (263). MBNL1 knockdown reduces the number RNA foci in DM1 patient cells (84) and MBNL overexpression increases the number and size of RNA foci in vitro (386). In adult muscle, MBNL1 is highly expressed and predominantly localized throughout the nucleoplasm. In the presence of C(C)UGexp RNAs, MBNL1 is redistributed to RNA foci where it is thought to be functionally inactivated. When CUGexp size is small, or in cell populations where DMPK transcript copy number is low, excess MBNL leads to saturation of binding sites in foci and MBNL can be rapidly exchanged between foci and the nucleoplasm (165, 315, 386). In these instances, MBNL functional inactivation and target missplicing are relatively low. In instances where CUGexp size increases (e.g., inter- or intergenerational repeat expansions), or in tissues with high DMPK expression, MBNL proteins are effectively titrated from the nucleoplasm and tend to circulate within foci between available binding sites (386). In agreement with this model, many DM-relevant splicing events show strong dose-response relationships between nuclear MBNL concentration and target exon inclusion levels (425). Furthermore, using a metric of inferred MBNL concentration based on >40 validated splicing events, the severity of spliceopathy in DM1 muscle can be accurately predicted (425). Together, these data are consistent with the model that increased sequestration of MBNL leads to progressive severity within DM patient populations and when critical thresholds are reached, may explain the distinguishing pathogenic features of presymptomatic, adult, juvenile, and congenital forms of DM1. In support of a direct role of MBNL in DM pathogenesis, AAV-mediated overexpression of MBNL1 in HSALR muscle ameliorates myotonia, restores CLCN1 protein levels, and corrects missplicing (190). In a separate approach, a transgenic MBNL1 overexpression mouse corrects DM-associated pathology when bred to HSALR mice (64). Furthermore, Mbnl1 KO mice recapitulate several DM associated phenotypes including myotonia, subcapsular cataracts, and histopathology (189). MBNL1 protein levels increase in postnatal muscle whereas CELF1 levels typically decline >10-fold, expression patterns that reinforce the pathological nature of their misregulation in DM (188). As discussed in more detail below, this pattern agrees with functional antagonism between members of these protein families.
Beyond MBNL proteins, other constituents of RNA foci are described (306). However direct versus indirect binding events need to be carefully distinguished to determine the proximal contributors to disease rather than proteins associated with the RBPs that are directly bound to C(C)UGexp RNAs. For example, hnRNP H normally functions in coordination with MBNL1 and CELF1 to regulate AS (296). Increased concentration of MBNL proteins in RNA foci could lead to an increase in colocalizing hnRNP H. Additionally, an important pathogenic hallmark of MBNL sequestration in DM is its depletion from the nucleoplasm in conjunction with colocalization with RNA foci. This suggests a high degree of functional inactivation for MBNL that may not be the case for abundant nuclear proteins such as hnRNPs. While colocalization with RNA foci is considered an important hallmark of RBP inactivation in DM, a variety of RBPs interact with CUGexp RNAs in vitro and are misregulated in DM patient samples and mouse models. For example, Staufen1 (STAU1) is increased in DM1 patient skeletal muscle as well as HSALR and other DM1 mouse models, and increased STAU1 activity is associated with myopathic phenotypes in transgenic mice (83). While STAU1 interacts with CUGexp RNAs in vitro, it does not colocalize with RNA foci in vivo, but may be associated with nuclear export of single CUGexp RNA molecules (325). In agreement with the model that STAU1 interacts with nonfoci associated DMPK transcripts, increased STAU1 does not affect the number of foci in cell models or the association of MBNL1 with these structures (325). Interestingly, STAU1 also modulates the splicing of several DM-relevant transcripts including INSR, CLCN1, and many others, which may modify disease progression in DM (43). While STAU1 does not modulate RNA foci abundance, two other C(C)UGexp-interacting RBPs, DDX5 and DDX6, reduce foci accumulation and rescue missplicing of some targets in DM cell models (181, 305). Furthermore, muscle histopathology is reduced in HSALR mice following DDX5 overexpression (181). As DDX5 and DDX6 are RNA helicases, it has been suggested that their activity unwinds structured CUG RNAs, making them more susceptible to turnover. As RNA foci are increasingly appreciated as complex structures, it is likely a variety of RBPs remodel these structures in a step-wise manner (178, 386).
Models and Modulators of DM Myopathy
In the previous sections, we discussed features of DM skeletal muscle functional and structural pathology followed by an overview of molecular mechanisms whereby C(C)TGexp mutations elicit abnormal cellular responses. RNA toxicity mediated through the modulation of MBNL and CELF activities has emerged as the prominent contributor to disease pathogenesis. Although reduced expression of C(C)TGexp-linked loci may partially contribute to disease, in the remainder of this review, we will primarily focus on RNA-toxicity-associated events with discussion of alternative hypotheses where appropriate.
The goal of dissecting molecular lesions downstream of C(C)UGexp RNA toxicity is twofold: (1) comprehensively reveal the extent of cellular dysfunction in each affected cell type; (2) establish links between molecular dysfunction to patient symptoms with the hope of identifying avenues for therapeutic intervention. The latter objective has been achieved and is most thoroughly exemplified by CLCN1 splicing errors eliciting myotonia. However, the molecular basis of other symptoms, such as impaired myogenesis in CDM and adult muscle wasting in DM1, has proven more elusive. Answers to these unresolved questions may lie in identifying subtle changes in several components of complex pathways. To address this, the use of high-throughput and increasingly unbiased sequencing technologies is lending a comprehensive and detailed view of disease relevant pathways. Indeed, the scope and resolution of RNA-seq afforded by increased sequencing depth and read length is revealing the dramatic extent of RNA processing errors in DM patients and animal models. Many of these changes have likely been overlooked using less-sensitive and lower-throughput experimental strategies, underscoring the importance of developing and utilizing new technologies for interrogating patient transcriptomes. While generating data is one step, deconvoluting the drivers of myopathy from passenger events is a complex task. To this end, the use of sophisticated computational and statistical tools is allowing the dissection of complex pathways and providing resources to develop global views of dysfunction. Below, we focus on three aspects of muscle biology disrupted in DM: development, function, and maintenance. We discuss key models used to understand these pathways, RNA processing networks associated with each, and where possible, specific events linked to dysfunction.
Muscle development
The development of muscle, myogenesis, is a multistep differentiation process whereby muscle precursor cells (MPCs) differentiate into mature, contractile myofibers (Fig. 8) (28,455). Two major forms of myogenesis occur in vivo, developmental and regenerative myogenesis, with many common cellular intermediates and molecular requirements (7, 28). While there are differences between periods of myogenesis, a common cellular prerequisite for any stage is the myoblast. Myoblasts undergo extensive proliferation prior to an initial differentiation event into a myocyte, a migratory cell with enhanced cell adhesion properties (2, 3). Myoblasts fuse to generate new myofibers, or fuse with preexisting myotubes to increase muscle bulk. In Drosophila, specialized fusion competent myoblasts initiate this process, although a homologous cell type has not yet been identified in vertebrates (313). Prior to fusion, myoblasts exit the cell cycle and commit to myogenic differentiation. Hundreds of cell fusion events eventually give rise to multinucleated myofibers, which then undergo a variety of maturation processes. For example, biogenesis of sarcomeres provides the contractile properties of muscle and coincides with relocalization of myonuclei to the cell periphery (113, 204).
Figure 8.
Expression patterns of DM-associated transcripts throughout myogenesis. As muscle precursor cells differentiate and mature into adult myofibers, the expression of DMPK (grey) increases transiently. MBNL1 (red) levels increase steadily as muscle develops while MBNL2 (blue) levels remain relatively constant. Both MBNL3 (green) and CELF1 (purple) are associated with early muscle precursors and other embryonic cell populations. The relative expression level of these genes in quiescent satellite cells is currently unknown. While CNBP (not shown) is highly expressed in proliferative cell populations, its relative expression in various myogenic cells is unclear.
In the mouse, developmental myogenesis occurs in successive waves. Myofibers first appear during embryonic, or primary myogenesis, beginning on embryonic day 11 (E11) followed by fetal or secondary myogenesis between E15.5 and E17.5. During developmental myogenesis, MPCs originate from the dermomyotome, a compartment of mesodermal cells adjacent to the notochord and neural tube of postgastrulation embryos. The cells migrate into the developing limb bud and establish the initial framework of skeletal muscle. By birth, muscle patterning is already established and myofibers mature through a combination of hyperplastic, cell-fusion, and hypertrophic processes (155). Postnatally, myofiber growth is largely dictated by hypertrophic mechanisms highlighted by increased sarcomeric protein production (101, 347). However, resident muscle stem cells, or satellite cells, can be activated by various stimuli, including muscle injury, to initiate a postnatal myogenic program. These cells also contribute to the basal maintenance of noninjured muscle and their decline is linked to age-, or disease-, related loss in muscle mass (194).
The molecular control of myogenesis has been most thoroughly studied in the context of transcriptional programs mediated by myogenic regulatory factors (MRFs) whose expression oscillates throughout myogenesis to control a finely tuned gene expression profile (28, 49). The MRFs are a family of four basic helix loop helix transcription factors, MyoD, Myf5, myogenin, and MRF4. The essential, combinatorial, and lineage specific contribution to myogenesis played by MRFs has been demonstrated using a variety of murine KO models. Myf5 and MyoD play redundant roles in early stages of the myogenic program followed by myogenin and Mrf4-responsive transcriptional programs driving myofiber maturation. Upstream of these, another set of transcription factors, the paired-box homeodomain proteins Pax3 and Pax7 control myogenic specification of early precursor cells and contribute to the satellite cell quiescence-activation axis.
Beyond transcriptional control of gene expression, co-/posttranscriptional mechanisms regulate myogenesis. Indeed, the degree of AS in the regulation of gene expression was first appreciated in skeletal muscle (232). Splicing patterns change throughout muscle differentiation and regions surrounding alternative exons are enriched for several well-characterized RBPs including CELF, MBNL, and RBFOX families (36, 413, 414). Furthermore, targeted mRNA decay controls transcript levels and helps to maintain satellite cell quiescence (107, 156). RNAs, such as miRNAs and lncRNAs are also emerging as another layer of myogenic control (277).
In the context of DM, studies of developmental myogenic defects are most applicable to CDM as overt neonatal musculature defects go undetected in adult-onset forms of DM1 and DM2. Several CDM manifestations suggest myogenic defects, including reduced fetal movement in utero, polyhydraminos (increased amniotic fluid related to insufficient swallowing by the developing child) and talipes (foot deformities). Given bone and skeletal muscle development are intrinsically linked, it is possible myogenic defects contribute to talipes (197). As discussed above, muscle biopsy reveals immature myotubes containing centralized nuclei, disorganized fibers, and small muscle fascicles (108). Small myofiber size suggests a growth deficiency mediated, in part, by reduced myoblast fusion during embryogenesis. Importantly, DMPK mRNA and protein increases during induction of human myogenesis in vitro, and DMPK protein levels decrease soon after birth (Fig. 8) (123). Interestingly, we have found that several genes associated with NMJ development and innervation show similar expression patterns during in vitro myogenesis, suggesting a potential role for DMPK in the regulation of innervation.
Myoblasts isolated from fetal CDM skeletal muscle display several markers of impaired function including reduced doubling time, lifespan, and fusion potential (122). These results are supported by other studies that demonstrated p16-induced senescence of CDM-derived satellite cells (34, 398). In vitro-derived CDM myotubes express immature myosin heavy chain isoforms (122). Of note, DMPK expression increases in differentiating myoblasts, suggesting that enhanced CUGexp-mediated toxicity exists during myogenesis (397). Based on ChIP-seq data, this Dmpk expression pattern appears to be regulated by core MRFs (e.g., MYOD and MYOG) during intermediate stages of C2C12 differentiation (397). RNA foci are detected in DM1, DM2, and CDM-derived myoblasts and the DMPK CTGexp is unstable in CDM-derived myoblasts, with a tendency toward expansions (122, 300). Furthermore, MBNLs colocalize with nuclear CUGexp RNA foci in patient-derived or model myogenic cell lines, suggesting the DM RNA gain-of-function model holds true in these cell populations (105, 106, 263). Myogenic defects are also observed in DM1, but not DM2, patient-derived myoblasts (298). This raises two important questions. First, given DMPK CTGexp expansions predominantly occur in postmitotic tissues in adult-onset DM1, is developmental myogenesis normal, but regenerative myogenesis impaired, in DM1? However, this is a difficult question to address in patients, given the challenges associated with acquiring myoblasts from presymptomatic DM1 patients as this necessitates prior knowledge that patients will eventually manifest disease. Second, the lack of in vitro myogenic defects in DM2 myoblasts suggests one possible mechanism for lack of congenital disease in DM2. Of note, CELF1 is upregulated in DM1 but not DM2 myoblasts, and there is evidence that CCUGexp RNAs modulate CELF1 levels in other systems (298). However, RNA foci are observed in DM2 myoblasts with colocalization of MBNL1, snRNPs and hnRNPs, but not RNAPII, SC35, CStF, or PML, suggesting the formation of these foci occurs post-/cotranscriptionally (300). PLCβ1, a promyogenic factor, is upregulated in both DM1 and DM2, perhaps to provide functional compensation to DM MPCs (103). Another RBP, hnRNP H, is also upregulated in DM1 myoblasts, colocalizes with RNA foci, and antagonizes adult pattern insulin receptor splicing (296). ZNF37A, a transcription factor that is downregulated in DM1 myoblasts, is associated with myogenic defects and its expression is responsive to CELF1 levels (127). Unfortunately, studies of patient-derived cells are confounded by technical limitations including disease severity at the time of isolation, culturing conditions, use of primary/immortalized cell lines, and number of in vitro population doublings. Indeed, there are conflicting reports of increased and decreased numbers of satellite cells in CDM muscle biopsies (342, 343). The use of animal and cell models provides a partial solution and allows more detailed mechanistic studies.
In the mouse, Dmpk expression is first observed in the developing somites along the anteroposterior axis of E10.5 and E11.5 embryos (191). Somites originate between E8 and E9.5 in the mouse embryo, and contain some of the first myogenic lineage-committed cell populations (170). While DMPK has not been localized to subsomitic compartments, such as the dermomyotome, its substantial overlap using in situ hybridization techniques is striking and suggestive of early deficiencies in the myogenic program (51, 170). This suggests that the toxic effects of CUGexp RNA expression occur at the beginning of myogenic specification. In agreement with the observed DMPK localization, DMPK regulatory regions display skeletal muscle enriched enhancer activity. Transgenic mice expressing a GFP-Dmpk 3’ UTR reporter construct under its native regulatory elements shows expression beginning in somites and in a variety of embryonic muscle groups (382, 383). Cnbp is expressed in embryonic and adult mouse tissues (68, 365). Cnbp RNA is detectable by northern blot as early as E7 and is expressed throughout embryogenesis (365). In situ labeling reveals staining in a variety of pregastrulation tissues, and the signal becomes increasingly enriched in the midbrain and forelimbs by E11.5 (365).
Expression of the human DMPK CTGexp 3’ UTR with as few as 57 CTG repeats impairs C2C12 in vitro myogenesis (11). Surprisingly, in vitro myogenesis is also impaired by a human DMPK 3’ UTR construct that contains only 5 CTG repeats (31). This activity was mapped to a region 5’ to the DMPK 3’ UTR CTGexp and is associated with increased CELF1 activity (9,31,341). This result underscores the importance of CTGexp gene context in the emergence of certain aspects of disease. Of course, the presence of a phenotype in CTG5 experiments is unexpected given this repeat size is within the normal range in DM patients, but may reflect toxicity resulting from overexpression of small repeats. Furthermore, additional CTGexp proximal cis-elements in the DMPK may exert some form of toxicity when these RNAs accumulate in the nucleus. In agreement, cultured myoblasts from transgenic mice overexpressing an unexpanded DMPK 3’ UTR display fusion defects and increased steady-state levels of CELF1 (274). Transgenic mice expressing CUG11 RNAs in the context of the DMPK 3’ UTR also show signs of myopathy in adult muscle and myogenic defects in cultured myoblasts (382). This myopathy is exacerbated in CUG91 RNAs expressed in the same context, suggesting that while CUGexp RNAs are inherently toxic, their context within the DMPK 3’ UTR is necessary to recapitulate all aspects of toxicity (382). Interestingly, while myogenic defects are observed in this mouse beginning at primary myogenesis, somite architecture appears normal, providing clues that pathology begins largely at the stage of myoblast differentiation (382). Conclusive evidence for inherent toxicity of CUGexp RNAs was shown using the HSA CTGexp mouse model (245). While control, CTG5 mice are normal, mice with CTG>200 (HSALR) display adult-onset phenotypes including myotonia, RNA foci, MBNL sequestration, and RNA missplicing. Interestingly, these animals do not present with overt early developmental phenotypes despite high transgene expression. CELF levels are not upregulated in HSALR mice, which may be due to a lack of DMPK cis-elements required for CELF increase. Expression of DMPK under the correct regulatory regions is also likely to play an essential role in disease manifestations. Indeed, primary myoblasts isolated from HSALR muscle, where the CTGexp is driven by the α-actin promoter, fail to accumulate RNA foci although these foci are induced following myogenic differentiation (our unpublished data). Mouse models constitutively overexpressing DMPK 3’ UTR CUGexp RNAs in a variety of tissues display embryonic lethality, while skeletal muscle conditional models display severe muscular atrophy and myotonia (284). Conditional expression of this same construct in heart results in early mortality due to arrhythmia and cardiomyopathy, recapitulating the other major cause of DM patient mortality (430). Both of these models display increased CELF1 levels, MBNL sequestration in RNA foci, and misregulation of many AS events (284). Interestingly, several of these splicing changes are not observed in HSALR mice, reinforcing the importance of CUGexp RNA context and expression pattern. The inherent toxicity of CUGexp RNAs has also been shown in zebrafish utilizing direct injection of CUG91 RNAs into the nervous system and skeletal muscle of embryos (410).
In mice, expression of Mbnl1 overlaps with Dmpk in E10.5 and E11.5 somites and developing limb buds (191). Temporal expression patterns reveal an intriguing correlation between Mbnl expression and events associated with myogenesis. The expression of Mbnl1, Mbnl2, and Mbnl3 peaks between E13.5–15.5, E17.5–18.5, and E11.5–15.5, respectively, in the developing mouse (191). Interestingly, the observed Mbnl3 peak overlaps with a hyperplastic phase (E11-E15) of muscle development while Mbnl1 peak expression occurs during a time of terminal myofiber differentiation (E12-E17). While Mbnl1 and Mbnl2 are both expressed during embryogenesis and in mature tissues, MBNL3 and CELF1 levels decline as cells differentiate (Fig. 8) (188). This in vivo observation is recapitulated in C2C12 myoblasts since MBNL1 and MBNL2 protein levels remain consistent throughout C2C12 differentiation while MBNL3 is lost soon after the induction of differentiation (Fig. 8) (312). MBNL3 is undetectable in adult skeletal muscle, but strikingly reemerges following muscle injury with particularly high levels during active myoblast proliferation (312).
In Drosophila, depletion of muscleblind (Mbl) proteins results in dysfunctional sarcomerogenesis due to impaired maturation of Z-lines (16). MBNL proteins regulate other developmental processes. For example, MBNL1 and MBNL2 regulate early differentiation events in embryonic stem cells (ESCs) and increased expression of MBNL1 and MBNL2 promote mature splicing events that activate ES cell commitment (149, 373, 419). In the heart, loss of MBNL1 disrupts normal valve development (81). On the other hand, CELF activity is typically most prominent in immature cells, reinforcing the antagonism observed between MBNL and CELF proteins.
One of the most thoroughly studied DM-relevant modulator of myogenesis is MBNL3. While MBNL3 is not detectable in mature skeletal muscle, or PAX7+-satellite cells associated with single isolated myofibers, two isoforms—37 and 28 kDa—of this protein are observed in C2C12 and primary mouse myoblasts (312, 397). Steady-state levels of these proteins are rapidly reduced upon induction of C2C12 myoblast differentiation (312). On the other hand, MBNL1 and MBNL2 protein levels remain relatively constant throughout C2C12 differentiation (226, 252, 312). This suggests a specific role of MBNL3 in the control of early myogenic events—a hypothesis supported by an enrichment of MBNL3 target RNAs in pathways associated with cell cycle and myoblast fusion (3, 312). MBNL3 was originally identified as a negative regulator of myogenesis using a variety of knockdown and overexpression models (223–225, 225, 375). One study identified disrupted MYOD-dependent transcriptional networks and decreased total MYOD protein levels following MBNL3 overexpression in C2C12 cells, indicative of impaired myogenesis (225). Particularly, several differentiation associated gene expression markers are downregulated following MBNL3 overexpression in C2C12 cells (224). Currently, there is no evidence supporting direct MBNL3 binding to MYOD transcripts, including a lack of MBNL3 CLIP clusters (312). MBNL3 supports inclusion of the Mef2 β-exon, coding for a region of the Mef2 transactivation domain, through binding of a region in intron 7 (224). Loss of this inclusion is correlated with delayed myogenesis in C2C12 myoblasts and can be rescued by overexpression of Mef2 β-exon containing constructs (224). Mef2 cooperates with MyoD to regulate early myogenic events and the β-exon inclusion isoform of Mef2 proteins results in an increased activation of target genes (224, 462). This missplicing event was also observed in DM patient skeletal muscle and cardiac tissue (224). Interestingly, Mef2 transcriptional networks are also disrupted in DM patient cardiac tissue and this has been explained by alterations in Mef2-responsive miRNAs (187). In contrast to data supporting MBNL3 as an antagonist of myogenesis, later studies suggested that siRNA-mediated knockdown of MBNL3 in C2C12 myoblasts delays in vitro myogenesis (312). While seemingly in conflict, other myogenic regulators are known to carefully balance myoblast differentiation dynamics in a manner similar to MBNL3. For example, constitutive activation or KO of beta-catenin disrupts myogenesis and is suggested to maintain a balance between cell proliferation and differentiation (336). Our group generated an Mbnl3 isoform KO (Mbnl3ΔE2/Y) mouse, which displays age-dependent regeneration defects but not overt developmental defects (312). Other studies utilizing Mbnl3ΔE2/Y KOs found similar age-dependent defects, but no developmental abnormalities (75, 76). Furthermore, the transcriptome of Mbnl3ΔE2/Y muscle, as surveyed by RNA-seq analysis of E15 forelimb, is largely normal with no overt splicing changes and only subtle gene expression changes (312). Since MBNL3 binds predominantly to 3’ UTRs, it is possible that this protein regulates APA or translation, mechanisms not explored in our original study (26, 76). Given the other prominent 28 kD MBNL3 isoform is upregulated and redistributed to the nucleus in Mbnl3ΔE2/Y mice, these events may act as a compensatory mechanism that masks potential phenotypes (312). Recently, our group has generated an Mbnl3 KO model (3KO) lacking both MBNL3 37 kDa and 28 kDa protein isoforms, and we have identified myogenic abnormalities in primary myoblasts isolated from these animals (397). These defects include failure to form mature, multinucleated myotubes in vitro following induction of differentiation as well as morphological abnormalities in cell spreading (397). Interestingly, recent work has described MBNL3 as a regulator of tumorigenesis partly through regulation of Paxillin (PXN) antisense transcripts. PXN protein is a component of focal adhesion complexes of nonstriated cells and dysregulation of its activity, or other cell adhesion components, may be linked to adherence defects in 3KO myoblasts. Another surprising feature of 3KO myoblasts is the emergence of adult pattern exon utilization in many DM-relevant transcripts including Bin1, Mef2d, and Neb (397). Because loss of MBNL activity typically results in reversion to developmentally immature splicing patterns, these observations were unexpected, and while they remain to be mechanistically explained, the relative splicing strength of MBNL paralogs may be linked (386).
The activity of MBNL1 and MBNL2 during mouse muscle development has not been extensively studied, but combined loss of these proteins results in embryonic lethality near E16.5, a period associated with terminal stages of primary myogenesis (227). While High-throughput sequencing-crosslinking immunoprecipitation (HITS-CLIP) data suggest that MBNL1 is predominantly an intronic binding protein in C2C12 myoblasts, it is primarily a 3’ UTR binding protein in adult mouse skeletal muscle (428). This differential binding may be mediated, in part, by highly dynamic MBNL localization patterns during myogenic differentiation (166, 252). MBNL binding to target RNAs in C2C12 myoblasts regulates global AS regulation and mRNA localization that is conserved between mouse and fruit fly (428). Mbnl1 KO mice do not display overt myogenic defects but this is likely due to compensatory upregulation and functional compensation provided by MBNL2 (227, 428). While MBNL2 loss alone has no obvious effects on muscle performance in mouse models, morpholino-mediated loss of MBNL2 in zebrafish results in paralysis and musculature defects (239). Further work should more closely analyze aspects of neonatal myopathy in Mbnl KO lines, particularly in Mbnl1 and Mbnl2 compound KO models.
CELF upregulation has also been implicated in developmental abnormalities (163, 409). Two transgenic CELF1 overexpression lines were generated and showed dramatic developmental delays correlating with the expression level of the transgene (409). Mice with the highest transgene expression (approximately eightfold) died at birth and showed muscle histopathology (409). Interestingly, p21 is upregulated in these mice, which suggests premature cell-cycle arrest in myoblasts (409). To evaluate the tissue specificity of CELF1 overexpression, cardiac and skeletal muscle conditional transgenic CELF1 lines were generated producing approximately fourfold to sixfold higher CELF1 levels than endogenous (163). As with other CELF1 overexpression studies, developmental abnormalities were observed in this mouse including failure to thrive, histopathology and splicing abnormalities (163).
The cellular composition of skeletal muscle is complex and includes many nonmyogenic cell populations: lower motor neurons, junctional Schwann cells, fibroblasts, monocytes, pericytes, and capillary-associated cells (28). Sustained capacity for adult regenerative myogenesis is dependent on many of these, as satellite cells reside in a highly specialized niche important for quiescence and activation (267). Furthermore, nonmyogenic cell populations such as pericytes, have been shown to possess myogenic potential (91). During developmental myogenesis, cross-talk between motor neurons and myofibers regulates normal formation of both, and disruption is associated with impaired development and disease (87, 192). While these nonmyogenic cell populations are understudied in the context of DM, recent studies have suggested their importance, as MBNL1 regulates myofibroblast development and fibrotic responses (86). One possible avenue to circumvent the challenges of these rare cell populations in patients is using induced pluripotent stem cells (iPSCs) generated from DM1 patient fibroblasts that have already been utilized in other studies (448, 449). In animal models, synaptogenesis is impaired in a CTGexp mouse with cooccurring neuronal RNA foci, behavioral abnormalities, and disrupted vesicular trafficking proteins (161). Although early studies suggested normal lower motor neuron morphology in CDM infants, CDM infants are hypotonic, a symptom typically associated with neurologic involvement (38). Using human ESC derived neuronal cultures, two SLITRK paralogs were found to be misspliced in DM1 and associated with defective neurite outgrowth and establishment of neuromuscular junctions in muscle coculture experiments (250). Mbnl homologs in C. elegans have also been shown to regulate synapse formation, highlighting conserved roles across species (374). These data invite the hypothesis that similar abnormalities exist in motor neurons throughout developmental myogenesis.
Developmental myogenesis is clearly affected in CDM, and likely during regenerative myogenesis in adult-onset DM (10). Many DM splicing biomarkers have been described, and it will be important to address if similar or unique changes are observed in CDM (272, 425). While many normal splicing transitions begin postnatally, there is evidence for groups of splicing transitions beginning in utero (188). In agreement, missplicing of INSR has been reported in several CDM quadriceps samples obtained from aborted or still-born fetuses (123). BIN1 exon 11 missplicing has been thoroughly described as contributing to myopathy and CDM-patient-derived cells display missplicing of this exon (121). Importantly, BIN1 mutations are associated with centronuclear myopathy, a congenital muscle disease (104). Furthermore, a muscle-specific isoform of myotubularin-related 1 (MTMR1) increases upon induction of myogenesis and is misspliced in CDM and DM1 (53). A homolog of MTMR1, MTM1, is mutated in X-linked myotubular myopathy, a congenital myopathy with striking similarities to CDM including generalized muscle weakness, hypotonia, and perinatal lethality (218). MTMR1 is more frequently misspliced in DM1 than DM2, and correlates with age-of-onset in DM1 patients (349). Importantly, misregulation of this exon is observed during in vitro myogenesis of patient-derived cells, confirming aberrant processing during myogenesis (349). Understanding the extent of RNA misprocessing in CDM tissues will provide valuable insights into disease pathomechanisms, and generate valuable biomarkers. Moreover, studies of altered RNA processing during myogenesis are increasing our basic understanding of posttranscriptional regulation of development, and this will provide useful information for other common and rare developmental myopathies (273). To address these last two points, transcriptome-wide analysis of CDM muscle samples, and identified hundreds of misregulated AS and polyadenylation events present as early as 3 months of age (397). Interestingly, most CDM missplicing events are also disrupted in DM1 patient muscle, but are more severely misregulated in the former (397, 425). Furthermore, many CDM-relevant exons appear to undergo developmental RNA isoform transitions that are completed by birth, suggesting that disruption of these events in utero would impair normal myogenic differentiation necessary to sustain life (397). In agreement, mouse MBNL loss-of-function in utero results in congenital spliceopathy and gene expression abnormalities characteristic of CDM as well as frequent perinatal lethality, respiratory distress, muscle histopathology, and failure to thrive in compound KO models (397).
Muscle function
Diverse functional properties are provided by skeletal muscle including thermoregulation, posture, respiration, metabolic function such as glucose storage, and most fundamentally, movement (119). In addition to its role in mobility, muscle is increasingly understood as an endocrine organ producing muscle-intrinsic cytokines or myokines (440).
The basic functional unit of muscle contraction is the sarcomere, a repeating unit of myosin, actin, and associated regulatory proteins positioned along the length of a myofiber (173). Mutations in a variety of sarcomeric proteins contribute to both adult-onset and congenital myopathies (173,282,317). The sarcomere is linked to the muscle membrane through additional structural proteins and ultimately connected to an intricate structural network in the extracellular matrix (ECM), which also plays essential roles in muscle contraction, signaling, and other aspects of muscle function (129). Again, several mutations in components of these structures are causative of human disease, most notably DMD mutations and DMD disease (129, 258).
Contraction is governed by events linking neurotransmission to activation of myofibril cross-bridge cycling. Electrical stimuli transmitted through motor neurons culminate with the release of acetylcholine, an excitatory neurotransmitter, at the neuromuscular junction. These stimulatory molecules traverse synaptic junctions and bind to acetycholine receptors embedded within the postsynaptic sarcolemmal membrane. Transmission of the endplate potential leads to an initial influx of excitatory sodium ions leading to a robust, uniform contraction of muscle. The resulting endplate potential stimulates a variety of calcium channels allowing the influx of calcium from extracellular and intracellular storage compartments. The binding of calcium to regulatory units of the sarcomeric thin filaments allows for ATP-dependent contraction prior to repolarization of muscle fibers and rest (56, 136, 216). While this scheme is generally true for all striated muscle, distinct functional requirements by certain fiber types (e.g., type I, IIA, etc.) or developmental time periods require unique specializations. For example, many developmental and myofiber specific titin isoforms exist and are controlled through AS (142, 288). Modulation of muscle RBPs or alterations in specific exon usage patterns, disrupts the structure and function of muscle (130, 297). Some RBPs, even those within the same family, appear to have importance in the function and maintenance of mature muscle, but may be dispensable for myogenesis. For instance, RBFOX2 controls early splicing transitions essential for myoblast fusion although depletion of RBFOX1 does not disrupt in vitro myogenesis and global splicing networks are largely unperturbed (370). However, conditional deletion of RBFOX1 in adult muscle results in histopathology, calcium mishandling, weakness, and spliceopathy (297). Despite >70% deletion of RBFOX1 in satellite cells in this same model, regenerative myogenesis is not affected (297). While the remaining RBFOX1-positive satellite cells may compensate, these data support the dispensable role for RBFOX1 in early myogenic cells.
The most profound functional defects associated with DM skeletal muscle are myotonia and weakness. As described above, skeletal muscle contraction is activated by excitatory neurotransmitters originating from lower motor neurons and disruption in excitatory signaling or muscle denervation can disrupt muscle function (192). While early stage DM1 patients are spared substantial muscle weakness, they present with a marked reduction in reflexes and this is suggestive of lower motor neuron impairment (338). Several of these patients also showed signs of reduction in several parameters of motor neuron electrophysiology (338). While the structural integrity of DM1 motor neurons has not been explored in detail, there are reports of hyperproliferation of synapses in DM1 patient-derived spinal cord neurons and decreased axon myelination in phrenic nerves (289). Other studies have reported normal innervation in a cohort of DM1 patients in contrast to markedly disrupted innervation in ALS (95). With this in mind, the transcriptome of DM1 and DM2 skeletal muscle is highly disrupted and this misregulation greatly contributes to muscle-intrinsic pathogenesis (210). High-throughput expression profile techniques, including microarray and RNA-seq, suggest hundreds of misregulated splice patterns are present in DM1 skeletal muscle, with over 20 events correlating with markers of disease severity, including muscle weakness (272, 425). Given DM1 and DM2 patients show differences in affected distal versus proximal muscle groups, it will be important to understand the degree of these splicing changes in different muscles. Furthermore, type I and type II fibers are differentially susceptible in DM1 and DM2, respectively. The use of laser capture microdissection technology has allowed the study of splicing changes and their effects in specific fiber types of DM1 and DM2 patients (348). While this study found no significant differences in splicing of INSR between fiber types, other RNA processing changes may be fiber specific (348). Of note, many splicing changes observed in DM1 are observed in other myopathies, and direct links between specific splicing changes and patient symptoms must be experimentally validated.
Several transgenic and other forms of DM mouse models have been generated that possess DM-relevant functional deficits (131, 367, 435). HSALR mice develop myotonia at approximately 4 weeks of age, due to Clcn1 pre-mRNA missplicing (245). As with DM patients, myotonia is first present in the absence of overt muscle wasting and histopathology. While subtle markers of histopathology (e.g., some central nuclei) are present at later stages in this mouse model, the lack of overt muscle wasting is surprising, particularly given the transgene has been measured as being >1000-fold higher than endogenous Dmpk (138). In fact, this study compared transgene expression levels in several mouse models, including DM500, DMSXL and Tg26, and found the expression level of the HSALR transgene to be higher than all other models (138). These data suggest the spatiotemporal expression pattern of CUGexp RNAs and their sequence context are important for these phenotypes. The HSA transgene is not expressed in NMJ-associated nuclei, whereas human DMPK is expressed and RNA foci are observed in these subjunctional nuclei (Fig. 9) (437). In addition, RNA foci and MBNL sequestration are observed in lower motor neurons of human spinal cord (437). Given the importance of NMJ maintenance in muscle function, these expression differences may be critical in understanding DM pathology (37). Other RNA misprocessing events are also present in the HSALR mouse, such as APA abnormalities (26). However, the lack of overt muscle weakness in the HSALR model suggests that the APA changes present in this mouse are not sufficient to generate these phenotypes.
Figure 9.

RNA foci in HSALR myofibers. A nonuniform distribution of RNA foci-positive (red) and negative (white arrows) nuclei (blue, DAPI) is present in HSALR myofibers. Foci-negative nuclei are likely satellite cells, subjunctional myonuclei, or nuclei from other myofiber-associated cells. This is the expression pattern generated by the HSA promoter, so expression of DMPK CUGexp RNAs in these nuclei may contribute to disease progression in DM1 patients.
Transgenic CTGexp mice containing additional human DMPK elements, overcome the limitations of restricted spatiotemporal expression. DM300 mice recapitulate myotonia and progressive muscle weakness (361, 421). At 5 months of age, no difference is observed between DM300 and wild-type littermates, but hindlimb muscle strength is reduced ~30% at 10 months, suggesting the accumulation of dysfunction over time (421). This weakness is recapitulated in various isolated muscle groups (421). Components of the ubiquitin-proteasome pathway are increased in these same mice, but a cause/effect relationship has not been established (421). Metabolic disruptions are also present in DM300 mice and correlate with the severity of INSR exon 11 missplicing (140). As skeletal muscle is the body’s largest reservoir for glucose, defects in glucose storage may be associated with the insulin resistance in DM (268). DMSXL mice, a derivative of the DM300 line, display growth abnormalities as early as 4 weeks when bred to homozygosity, but do not display this phenotype in hemizygotes likely due to low transgene expression levels (132). Only two DM-associated splicing targets were tested in this study and only one, Insr, showed missplicing (132). Large CTGexp mice from this line, including DM600 and DMSXL, that survive into adulthood display respiratory dysfunction, diaphragm histopathology and RNA foci in the diaphragm and phrenic nerve (290, 291). These results are important given diaphragm weakness and respiratory distress are major causes of morbidity and mortality in DM1 and CDM. Another derivative of these lines, with approximately 550 repeats displays skeletal muscle weakness and atrophy along with activation of the proteolytic Fbox32-ubiquitin pathway (421). EpA960 mice allow for conditional cardiac or skeletal muscle expression of 960 interrupted CUGexp RNAs (430). Upon transgene induction, mice develop cardiac or skeletal muscle abnormalities secondary to Tnnt2 missplicing and CELF1 upregulation occur as early as 12 to 24 and 6 hours postinduction, respectively (430). Importantly, these time-course data link these molecular changes as occurring prior to the onset of overt symptoms.
MBNL1 proteins are major contributors to adult-pattern CLCN1 splicing in human, and thus, Mbnl1 KO mice display Clcn1 missplicing, loss of membrane-associated CLCN1 protein, and myotonia (189). Mbnl2 KO mice do not recapitulate these phenotypes, and this suggests a particularly important role for MBNL1 in the mature skeletal muscle (66). While its loss is not sufficient to generate overt myopathy in adult mice, MBNL2 is upregulated in Mbnl1 KO muscle, its relative nuclear abundance increases, and it provides functional compensation (227). While constitutive Mbnl1; Mbnl2 compound KO (DKO) mice are embryonic lethal, skeletal muscle conditional Mbnl1; Mbnl2 DKO mice develop exacerbated myotonia and missplicing (227). In addition, these mice develop dramatic muscle wasting (discussed below) (227).
While linking specific RNA processing events to a given clinical symptom is difficult, a well-established link between CLCN1 splicing and myotonia exemplifies the greatest success toward this goal (67, 246). On the other hand, muscle weakness can be due to a variety of events, making a direct one gene-one symptom link more challenging. While defective EC coupling and/or inherent defects in the contractile apparatus are expected to contribute to the DM muscle weakness, the major, and life-threatening, contributor to muscle weakness is the age-associated loss in muscle mass, or muscle wasting, observed in late-stage DM patients.
Muscle maintenance
Skeletal muscle wasting, or atrophy, is the loss of muscle mass and occurs in response to aging, disuse, and disease. The maintenance of muscle is dependent on the longevity of healthy myofibers and a muscle’s inherent potential for self-repair. Typically, muscle atrophy is not associated with overt loss in the number of myofibers, but rather the degradation of proteins, particularly sarcomere-associated components, within myofibers. Myofibrillar components comprise 70% to 85% of muscle protein and are the primary target of catabolic ubiquitin-proteasome pathways (80, 89). Catabolism-dependent muscle wasting is often seen in age-related atrophy (sarcopenia) or cancer-induced atrophy (cachexia) (6, 42). Satellite cells also contribute to steady-state myofiber maintenance and regenerative myogenesis following injury. Importantly, satellite cell numbers decline as a function of age and this reduction is associated with sarcopenia. In cases of overt myofiber loss, a critical threshold of calcium mishandling is typically the major contributor to necrotic pathway activation in disease such as DMD (54). The downstream effectors of this calcium mishandling are typically calcium-dependent calpains as well as activation of ER stress response pathways (44,54). In these cases, a fibrotic and inflammatory response typically cooccurs.
Skeletal muscle atrophy accounts for approximately 60% of DM patient mortality, followed by cardiac-related mortality (157, 344). Given the lack of inflammatory cell invasion and fibrotic deposits in DM1, muscle wasting is likely due to loss of muscle mass independent of myofiber loss. In DMD, disruption of the sarcolemma is the prominent contributor to the calcium-mediated necrosis discussed above. In contrast, sarcolemmal integrity in DM and a variety of mouse models appears normal (133). Furthermore, elevation of serum creatine kinase, a marker of membrane permeability, is typically absent or unremarkable in most DM patients (133, 158, 159). While some studies have reported apoptotic markers in DM muscle, the contribution of apoptosis to muscle wasting is controversial as the proportion of lost nuclei is small compared to the number of nuclei found in mature myofibers (236). Many aspects of DM muscle wasting are similar to sarcopenia and this suggests that DM is a progeroid-type disorder (8). Muscle wasting in DM may result from activation of catabolic pathways downstream of a primary pathology such as missplicing, or from the direct role of DM-associated factors in translation (404). Indeed, both MBNL and CELF proteins have been implicated in direct or indirect control of mRNA translational efficiency. As previously discussed, missplicing of genes associated with other myopathies is prevalent in DM skeletal muscle tissues. The challenges of muscle maintenance in DM are compounded by the increased burden of age-associated increases CTG copy number in these muscle fibers (12, 266, 400, 444). The late stage onset of muscle wasting is likely linked to these increases in CTGexp size.
Satellite cells are one of the major contributors to muscle repair in the context of disease (363). During normal development, a subset of Pax3/7 specific myogenic cells avoid MRF expression into adulthood and give rise to a subset of cells with a high degree of regeneration potential. Satellite cells are situated between the basal lamina and sarcolemma and are quiescent until activated by mechanical disruption or signaling events. Under normal circumstances of activation from quiescence, these cells undergo asymmetric cell divisions to yield cells that differentiate into myoblasts and cells that maintain the stem cell pool (283). Unfortunately, in situ studies of satellite cells in DM are limited and in general, challenging to study in the context of any muscle disease. However, ex vivo analysis suggests several aspects of satellite cells are dysfunctional in DM1 (398). For example, most studies of patient-derived cells suggest they undergo premature senescence that is independent of telomere shortening, but rather, may be associated with premature p16 activation (34). This suggests alterations of specific intracellular pathways. CELF1 is known to bind to p21 mRNA and provides one possible explanation (408). In contrast, telomere shortening might impair the regenerative capacity of DM2 MPCs (329). Beyond satellite cells, a variety of nonmyogenic cells also contribute to muscle homeostasis and regeneration, but their contribution to DM pathomechanisms is largely unexplored. One notable exception is the contribution of MBNL1 to myofibroblast differentiation, but these cells have not yet been studied in a clinical context (86).
In mice, overexpression of wild-type human DMPK RNA results in centralized nuclei and type I fiber atrophy (274). These results reinforce the concept that DMPK cis-elements enhance the inherent toxicity of CUGexp RNAs (9,382). While DMPK levels are not increased in DM1 patients, one possibility is that nuclear retention of CUGexp-containing RNAs increases the local concentration of DMPK transcripts in the nucleus, effectively mimicking DMPK increased steady-state levels. As the CTGexp is in the DMPK 3’UTR and full-length DMPK transcripts are retained in DM muscle nuclei, retention of CUGexp RNAs inevitably results in an increase in the remaining DMPK mRNA sequence. Notably, HSALR mice do not exhibit overt muscle wasting, but rather show evidence of myofiber hypertrophy, which may be related to increased calcineurin signaling in adult animals (324). It is possible that the lack of native CUGexp RNA flanking sequence or the normal DMPK spatiotemporal expression pattern may contribute to this absent phenotype.
Muscle-specific, combined loss of MBNL1 and MBNL2 results in dramatic adult-onset muscle wasting, with nearly 100% of fibers containing one, or multiple, centralized nuclei (227). Both quadriceps and TA muscles contain a mixture of type I and type II fibers, and both display muscle wasting in compound Mbnl1; Mbnl2 DKO mice, invoking a contribution of MBNL proteins to maintenance of myofibers implicated in both DM1 and DM2 (227). Many splicing events known to contribute to muscle weakness and wasting, such as Bin1 exon 11 and Cacna1s exon 29, are severely misspliced in these animals and may represent underlying contributors to this pathology (227). However, as these markers were assessed in end-stage animals, earlier time points should be studied to determine if these events are simply secondary to muscle regeneration (Fig. 10) (19, 285).
Figure 10.
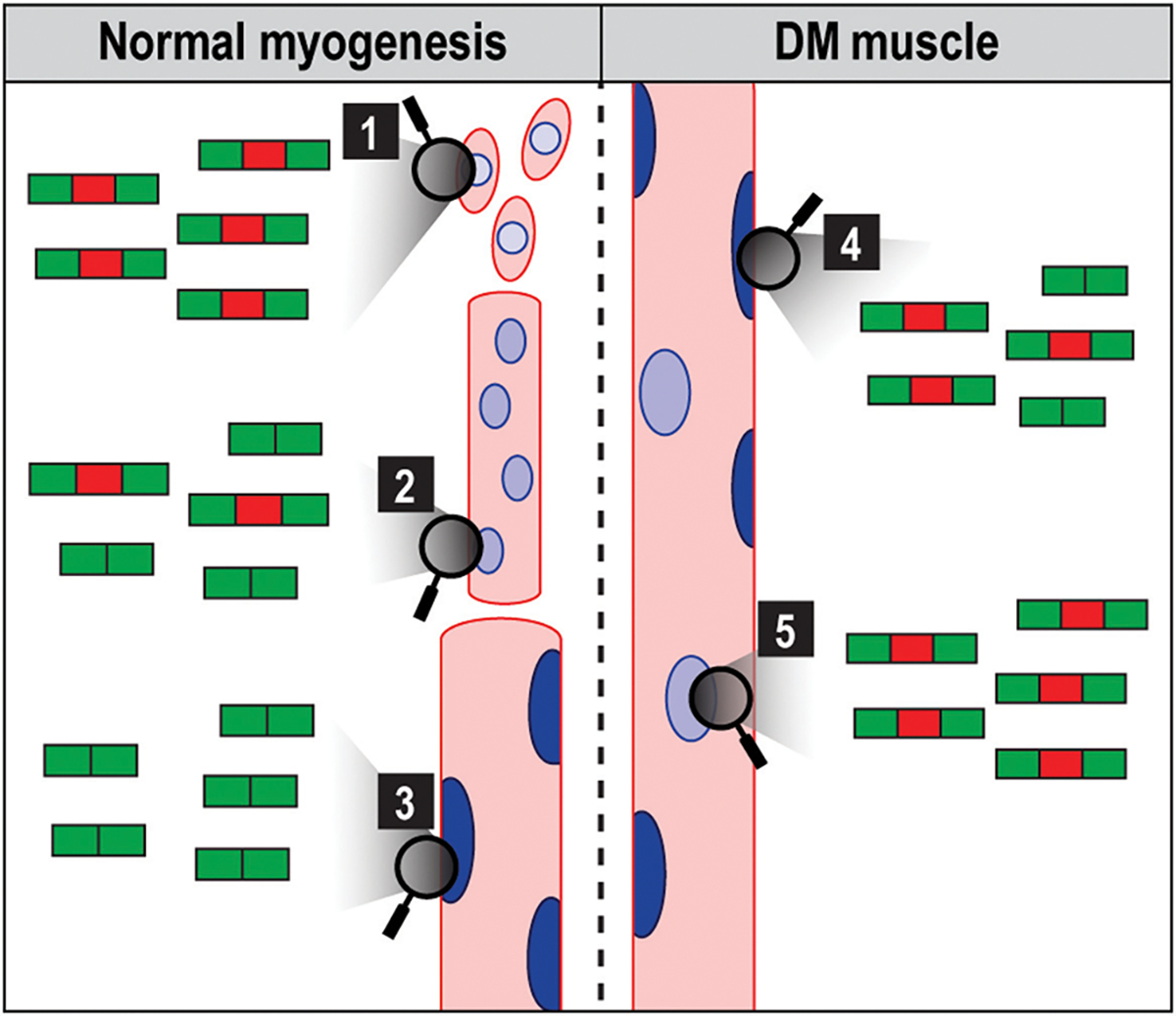
RNA splicing in unaffected and DM muscles. In unaffected adults, C(C)UG repeat number is in the nonpathogenic range and adult/mature RNA isoforms (red exon exclusion) are expressed (3). During injury-induced regeneration, fetal RNA isoform (red exon inclusion (1) and (2)) expression patterns are recapitulated. In DM, C(C)UGexp RNA expression inhibits MBNL splicing activity by sequestration leading to fetal/immature isoform reexpression in mature myofibers (4), which is also accompanied by elevated regeneration indicated by centralized myonuclei (5).
Overall, a variety of model organisms recapitulate hallmark features of DM myopathy. Importantly, DM-relevant RNA processing networks are dysregulated in these organisms including AS, APA, and RNA localization. Certain biological processes are enriched in datasets of misregulated RNA processing in DM, including components of the ECM and channels important for ion homeostasis, many of which are implicated in other channelopathies (17,96). Next, we discuss RNA processing events of particular interest based on their known association with DM phenotypes, common misregulation in DM patients and several models or involvement in other myopathies.
Potential Links: Genes and Phenotypes
Profiling of the DM skeletal muscle transcriptome has revealed misprocessing of hundreds of transcripts (26, 121, 202, 272, 281, 302, 428). Some of these abnormal splicing events might contribute to dysfunctional contractile and metabolic properties of DM skeletal muscle as exemplified by CLCN1 missplicing and links to myotonia. Importantly, spliceopathy is highly variable between patients. Affected individuals differ in the total number of genes affected, particular exons disrupted and the degree of missplicing (272, 425). Some of these differences may relate to disease severity and progression. Indeed, the degree of missplicing for BIN1 exon 11, NFIX exon 7, DTNA exons 11/12a, and other splicing events correlates with ankle dorsiflexion weakness in a cohort of DM1 patients (272). Separating disease-initiating from secondary misprocessing events is a topic of active investigation. KO studies provide information regarding the critical role of DM-relevant genes in the context of muscle development, function, and maintenance, but these studies fall short of providing information regarding the contribution of structural or regulatory changes induced by missplicing. Deciphering the specific role of differential exon inclusion requires targeted disruption of splice site selection, which may be accomplished using ASOs (121,323). Many misprocessing events are reproducibly identified in studies of DM patients and animal models, suggesting these are important events to consider. Furthermore, pathway analysis of groups of genes often reveals common themes in EC coupling, sarcomeric proteins and regulation of ion homeostasis. In this section, we will discuss some of these events as they relate to various aspects of muscle biology. While not an exhaustive list, these represent common themes in DM pathogenesis and are probably important contributors to myopathy. Where possible, we will discuss our current understanding of how DM-associated misprocessing of these RNAs have been experimentally validated to contribute to myopathy.
Migration, adhesion, and fusion in muscle development and DM
MPCs, including satellite cells, myoblasts, and myocytes, are a migratory population of cells. During developmental myogenesis, MPCs migrate from the dermomyotome into the developing limb bud prior to extensive proliferation and fusion to generate myofibers. During regenerative myogenesis, quiescent satellite cells are activated and migrate to points of injury (357). Satellite cells also move rapidly along isolated myofibers grown in culture (368). MPC intrinsic and extrinsic components are critical for migration and MPCs connect to a variety of collagens, laminins, and fibronectin of the ECM and transmembrane proteoglycans of the myofiber. MPC attachment to ECM components is largely mediated through transmembrane alpha and beta integrins, which are linked to a dense array of intermembrane proteins linked to actin filaments (Fig. 11). Collectively, these structures are known as focal adhesions, and these structures provide many signaling functions in addition to their roles in motility (264). These attachments provide traction points and act as “molecular feet” during migration. Focal adhesions are typically found in the posterior region of a cells leading edge and anterior to this is a branching array of actin protrusions forming lamellipodia and filopodia. These structures effectively push the membrane forward and are stabilized or recycled by a variety of actin binding proteins. As cells move, ECM components are also cleared, often by secreted metalloproteases, at the leading edge of migrating MPCs to provide space for movement (278). Extracellular chemoattractant molecules, such as IL-4, HGF, and PDGF, promote directional cell migration (3). Repulsive signals have also been described, such as ephrins embedded in the membrane of healthy myofibers which may promote MPC migration away from points where repair is not necessary (376). When two MPCs come in contact, or a single MPC contacts a potential site for fusion with a preformed myofiber, cell-cell adherens junctions form. These structures are similar to focal adhesions, and even share many of the same components, but are distinct in many ways, including adherens junction specific proteins, the posttranslational modification status of shared components and the fact that MPC adherens junctions establish the initial contact points necessary for cell fusion. A variety of signaling and accessory molecules such as β-catenin, FERMT2 (also known as kindlin-2) and a variety of small G-proteins are localized to MPC adherens junctions and are thought to be involved in signal transduction events necessary to initiate fusion (3, 94, 418). Upon induction of fusion, actin is rearranged at the sites of fusion, forming a dense network parallel to the cell membrane (97). The formation of this structure is dependent on nonmuscle myosin IIa (MYH9), an actin binding motor protein (97).
Figure 11.
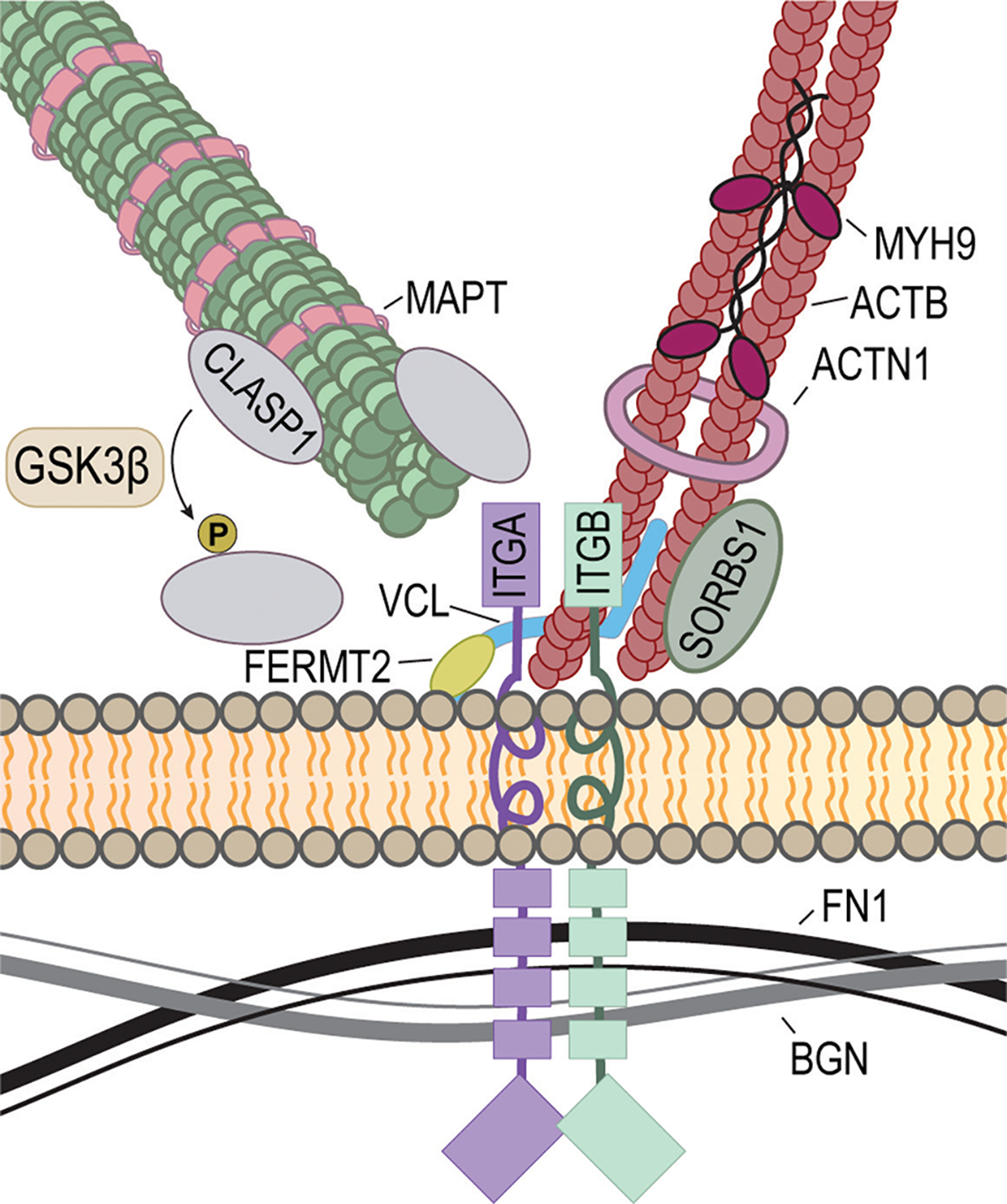
DM-associated components of focal adhesions. A schematic of a focal adhesion is shown along with some associated components implicated in DM.
In DM muscle, transcriptome studies have largely focused on patient biopsy/autopsy skeletal muscle or in vitro differentiated myotubes (121, 272, 425). Therefore, transcripts important for MPC migration and fusion have not been specifically studied in the context of DM. However, many of the gene products disrupted in adult DM myofibers are utilized by MPCs for controlling early myogenic events. Fortunately, this provides a priori knowledge as to which transcripts are likely disrupted during these early stages of myogenesis. For example, many of the large array of actin-associated protein-coding transcripts are also expressed in MPCs and control focal adhesions, filopodia extension and cell-cell adherens junctions. Furthermore, CLIP-seq studies have identified the binding profile of MBNL1, MBNL3, and CELF1 in C2C12 myoblasts, providing a catalogue of potential pathways misregulated in DM patient MPCs. Among these pathways are processes associated with membrane dynamics, cell adhesion, cell-cell contact, and actin reorganization (312, 428). While the functional consequences of MBNL binding to these transcripts have not yet been explored in detail, prior studies suggest roles in localization and/or transcript stability (252, 428). These observations are supported by cytoplasmic MBNL1 and MBNL3 localization, particularly during the reestablishment of cell-matrix contacts following trypsinization and replating of C2C12 myoblasts (166, 312, 428).
In the following sections, we discuss a selection of gene transcripts implicated in DM myopathy grouped by functional pathways important for MPCs, contraction/muscle structure, and modulators of gene expression.
Cell adhesion and ECM components
PDLIMs
PDZ and LIM domain (PDLIM) proteins are a large family of cytoskeleton-associated proteins important for stabilizing and/or redistributing F-actin through associations with alpha-actinin and other actin binding proteins (416). Additionally, PDLIMs modulate protein phosphorylation and function via kinase activity found in their LIM domains (13). In the context of cell-migration, PDLIMs are known to phosphorylate cofilin, another actin mediator important for directional cell migration. Cofilin is localized to the leading edge of migratory cells and, when activated, severs actin filaments at the F-actin pointed end. This facilitates rapid depolymerization of F-actin into G-actin which can then be reincorporated into the barbed end of growing actin filaments—a process termed treadmilling. Loss of one PDLIM family member, PDLIM7, disrupts cell motility and is sufficient to cause perinatal lethality in mice (207,394). In addition to the multiple PDLIM paralogs found in mammals, a variety of splice isoforms exists, some of which regulate the abundance of certain PDLIM paralogs. For example, a splice isoform of PDLIM4 accumulates during stress that generates an unstable, and rapidly degraded protein isoform and despite its short half-life, this PDLIM4 isoform impairs cell migration (143). In DM, several PDLIM family members are misprocessed including PDLIM3, PDLIM5, PDLIM6 (also known as LDB3), and PDLIM7 (272, 425). While the function of these various splice isoforms has not been totally explored, LDB3 has received some attention (see discussion below).
CAPZB
Capping actin protein of muscle Z-line beta (CAPZB) subunit is a subunit of an F-actin cap binding complex. As with PDLIM, CAPZB is associated with actin filaments at the Z-line of mature muscle, but also participates in the regulation of actin dynamics during cell migration where it blocks the incorporation of actin at the barbed end (92). Propolymerization factors, such as cofilin, compete with CAPZB for barbed end interaction, and changes in local pH are speculated to shift the balance of interacting proteins (92). Interestingly, AS may adjust the pH sensitivity of actin binding proteins (92). In a cohort of patients presenting with hypotonia, micrognathia, and other developmental abnormalities, a chromosome 1 and 13 translocation, t(1;13)(p36.13;q12.11), has been identified that disrupted CAPZB expression (269). In agreement with a role in development, capzb is expressed throughout embryogenesis in zebrafish and loss-of-function mutants display disrupted muscle structure (269). When stimulated by force, local CAPZB levels increase to support focal adhesion maturation (209), and CAPZB loss-of-function impairs cell migration and filopodia morphology (371). CAPZB exon 8 is abnormally excluded in DM1 and DM2 patients, and missplicing is slightly more severe in DM1 patients (60, 188). Transgenic CELF1 overexpression mice also show missplicing of CAPZB exon 8 in developing heart and this event is not disrupted in Mbnl1 KO mice (188). While this may represent an MBNL-independent splice event, functional compensation provided by MBNL2 may mask the contribution of MBNL loss-of-function (227). However, alterations in CELF1 and MBNL activities in DM skeletal muscle likely contribute to CAPZB exon 8 missplicing and may affect its ability to regulate actin polymerization in migratory cells and stabilize actin in mature muscle.
CLASP
Cytoplasmic linker associated protein 1 (CLASP1) is a microtubule-associated protein (MAP) involved in stabilizing microtubule plus ends along with a variety of other MAPs (168). In migratory fibroblasts, CLASPs are localized to the distal portion of microtubules at the cells leading edge where they are believed to stabilize microtubules via their associated with CLIP proteins (5). CLASP exon 19 is misspliced in DM1 patients and displays a broad range of missplicing that correlates with disease severity (425). While this exon has not been tested in current DM mouse models, another CLASP exon (exon 25a in mice), is disrupted in Mbnl1 KO mice (457). The function of this exon has not been explored in detail, but its loss is expected to affect microtubule dynamics. Interestingly, microtubules are critical regulators of focal adhesion disassembly in migratory cells (379). Furthermore, microtubules are also believed to provide a conduit for MMP-containing vesicle trafficking to cell-ECM junction, allowing ECM remodeling during migration (379, 389).
ITGA3
Integrin subunit alpha 3 (ITGA3) is an integral membrane protein enriched at focal adhesions—sites of tight cell-ECM connection. The integrin protein family forms heterodimeric integrin-alpha and integrin-beta complexes that provide an essential link between the intracellular actin cytoskeleton and the ECM. Beyond mechanical support, integrins provide signaling roles, often mediated through a variety of intermembrane, integrin-associated proteins, such as focal adhesion kinase (FAK). Deletion of Itga3 disrupts myogenesis as early as the myoblast fusion stage in vitro (50). MBNL2/MBL directly interacts with the ITGA3 3’ UTR and controls its localization to focal adhesions (4). This localization likely depends on the known ACACCC zip-code localization motif originally described as an essential motif for ACTB RNA localization. Interestingly, MBNL and CELF proteins also bind to the Actb 3’ UTR in C2C12 myoblasts, suggesting a global role in trafficking RNAs important for association with focal adhesions (312, 428, 429). In agreement, there is striking colocalization of MBNL2 with activated FAK in A549 cells, suggesting MBNL2, and associated RNAs, are actively shuttled to these regions (4). ITGA3 is the first transmembrane protein-coding transcript shown to be localized to a specific subcellular region, and it has been suggested that this allows for local translation at these junctions (4). If this is the case, disruption of MBNL-mediated ITGA3 localization by C(C)UGexp RNAs would be expected to inhibit mature focal adhesion formation and myoblast fusion.
ITGB1
Integrin subunit beta 1 (ITGB1) is another major structural unit of integrin-associated adhesion complexes. As with Itga3, loss of Itgb1 expression disrupts myoblast differentiation as well as sarcomerogenesis (358). Additionally, ITGB1 cooperates with FGF2, to maintain the satellite cell niche and decline of this activity is observed in satellite cells (SCs) from aged mice (335). While the localization of Itgb1 has not been studied, this RNA is a target of all three MBNL proteins in mouse embryonic fibroblasts (MEFs) (26). MBNLs bind slightly upstream of the distal Itgb1 PAS and MBNL loss results in preferential proximal PAS usage, suggesting MBNL proteins promote expression of the full-length Itgb1 3’ UTR (26). The truncated Itgb1 3’ UTR isoform lacks motifs for the HuR RNA-stabilizing protein so the mRNA half-life may be reduced. In agreement, single-nucleotide mutagenesis of MBNL binding sites in the Itgb1 3’ UTR is sufficient to increase proximal PAS usage and lead to reduced protein production in luciferase reporter constructs (26). As with ITGA3, disrupted MBNL activity in the context of DM1 may lead to reduced ITGB1 protein levels and impaired cell-cell and cell-ECM adhesion complexes.
FN1
Fibronectin 1 (FN1) is a secreted component of the ECM. FN1, along with other extracellular proteins, provides scaffolding essential for developmental myogenesis, muscle patterning, and maintenance of the adult satellite cell. Loss of FN1 contributes to satellite cell depletion and age-associated muscle wasting (237). Fn1 mRNA is another 3’ UTR target of MBNL1 and MBNL3 in C2C12 myoblasts (312, 428) and siRNA-mediated knockdown of Mbnl1 and Mbnl2 in C2C12 myoblasts results in relocalization of FN1 away from the cell membrane toward the insoluble fraction of cell lysates (428). Using secreted luciferase reporters linked to the Fn1 3’ UTR, the expression of CUGexp RNAs is sufficient to decrease protein secretion (428). Importantly, this activity was rescued following MBNL1 overexpression, which indicates that MBNL-mediated localization of RNAs is important for secretion (428). Beyond localization, Fn1 and many other ECM-protein encoding transcripts are misspliced in DM1 animal models (96). Altogether, these results suggest that the ECM is compromised in DM1.
Myofiber structural proteins
DMD
Dystrophin (DMD) is the largest human gene and is composed of 79 exons encoding the 427 kDa dystrophin protein (Fig. 12) (35). Dystrophin is a component of a large dystrophin-associated glycoprotein complex that links the cytoskeleton and ECM of muscle and provides a scaffold for force transmission during muscle contraction, as well as transduction of extracellular-mediated signals to the muscle cytoskeleton (35) and DMD frame-shift and truncation mutations cause DMD. The DMD pre-mRNA splicing pattern is regulated during development and involves exons 71 to 74 and 78 and the splicing pattern of all these exons is altered in DM1 muscles (271). However, only missplicing of exon 78 has pathological implications because mice lacking exons 71 to 74 are normal (82). Aberrant exclusion of DMD exon 78 in adults shifts the open reading frame and alters the DMD C-terminus with a hydrophilic and positively charged β-sheet domain exchanged by a negatively charged amphipathic α-helix. This missplicing event compromises muscle fiber organization during contraction (323). Conditional Mbnl1; Mbnl2 KO mice display aberrant splicing of DMD exon 78 (227). In addition to its function in mature myofibers, an unexpected role of DMD in satellite cells has emerged in the control of cell polarity and satellite cell fate decisions (99). It is possible that DMD exon 78 missplicing might lead to disruption of the activation-quiescence axis in DM satellite cells.
Figure 12.
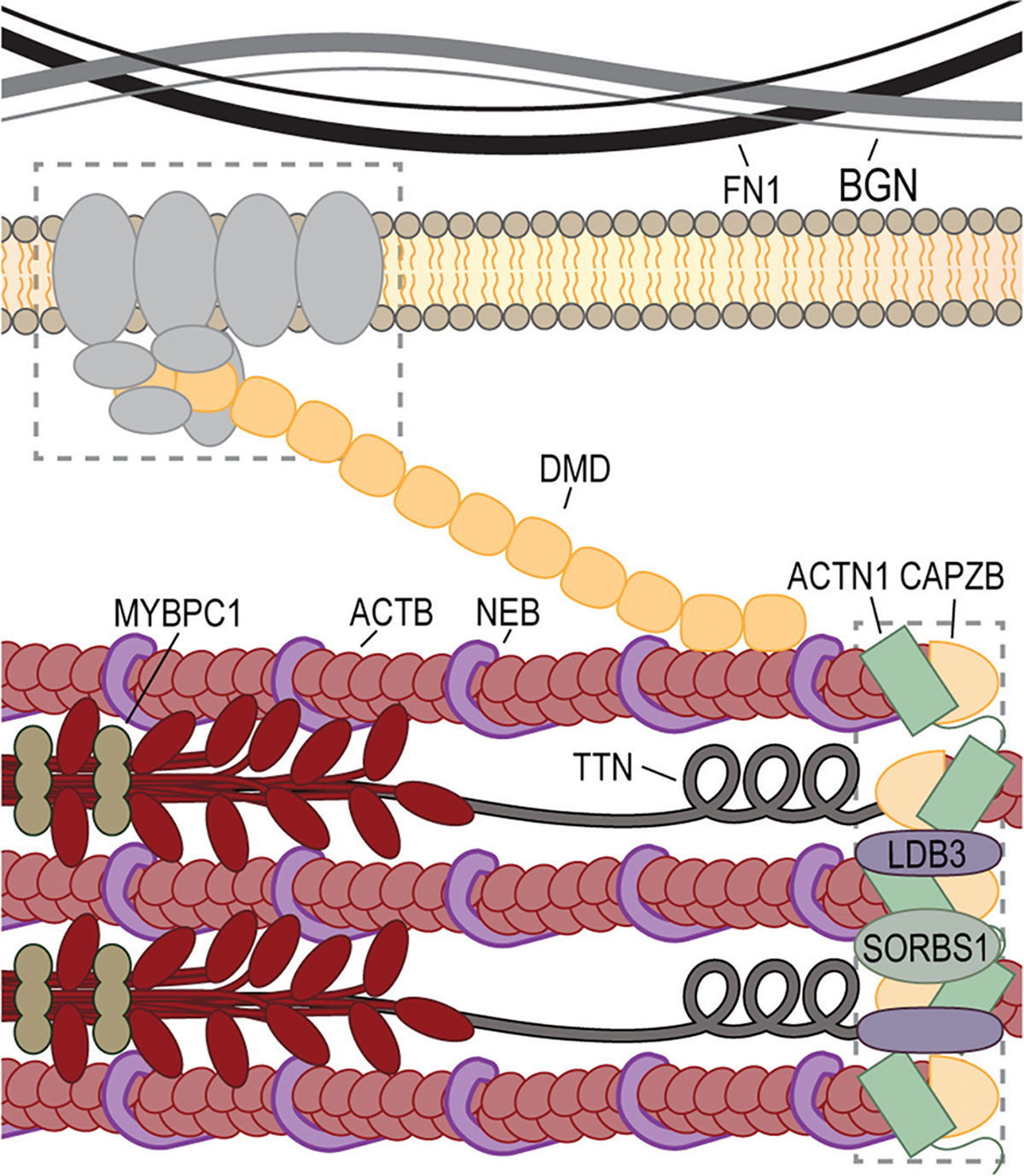
DM-associated contractile and structural proteins. A schematic of a sarcomere is shown along with the DMD-mediated link to the sarcolemma. Gray boxes are shown outlining the dystrophin-associated glycoprotein complex (left) and the muscle Z-line (right).
LDB3
Lim domain binding 3 (LDB3) is a PDLIM family member that is highly expressed in skeletal muscle. In mature myofibers, LDB3 localizes to Z-lines and interacts with alpha-actinin, an actin stabilizing protein, through its PDZ domain (109). Several LDB3 alternative splice isoforms exist, some of which lack LIM domains containing kinase activity involved in signal transduction (109). Various LDB3 loss-of-function mutations have been identified in humans presenting with dilated cardiomyopathy (15) or distal myopathy (70). In the mouse, deletion of the Ldb3 gene results in perinatal lethality (460). Interestingly, sarcomerogenesis and Z-line formation are not overtly disrupted in this animal, suggesting LDB3 is important for the maintenance of muscle rather than development (460). Interestingly, some cases of cardiomyopathy are associated with LDB3 mutations that disrupt its interaction with PKC (15). In the context of DM1, LDB3 exon 11 is abnormally included in mature skeletal muscle and in cells transfected with CTGexp constructs (450). The emergence of an abnormal protein isoform was confirmed via western blot in this study (450). Furthermore, the LDB3 exon 11 containing protein isoform exhibits reduced binding affinity for PKC proteins (450). It is possible that this reduced affinity partly contributes to PKC hyperactivation in DM skeletal muscle, and provides a possible link for CELF1 increase. Furthermore, while the ability LDB3 exon 11 containing isoforms to properly maintain Z-lines has not been studied, it is possible that missplicing of exon 11 also directly contributes to muscle weakness in DM.
ABLIM1
Actin binding LIM protein 1 (ABLIM1) is highly expressed in skeletal and cardiac muscle (333). In cardiac muscle, ABLIM1 localizes to Z-lines where it likely interacts with other actin binding proteins via its LIM domain to stabilize actin filaments (333). However, the exact function of this protein has not been explored in detail. ABLIM exon 11 is fully excluded in fetal skeletal muscle and inclusion gradually increases during postnatal muscle development (281). In a cohort DM1, exon 11 is completely excluded in the majority DM1 patients and in HSALR skeletal muscle (281). This results in an in-frame loss of 28 amino acids downstream of one of ABLIM1’s LIM domains and is speculated to disrupt protein-protein interactions (281). Given exon 11 inclusion is specific to cardiac and skeletal muscle, this is expected to disrupt muscle-specific processes (281). Using an exon 11 minigene, it was shown MBNL and CELF proteins directly regulate this splice event in an antagonistic manner—MBNL and CELF promote inclusion and exclusion, respectively (281). This suggests missplicing of this exon is directly responsive to MBNL and CELF dysregulation and may affect sarcomere function in DM. In agreement, overexpression of CUGexp reduced exon 11 inclusion in C2C12 cells (281).
TNNT3
Troponin T type 3 (TNNT3) is a fast-twitch skeletal muscle troponin subunit. Binding of calcium to regulatory domains within the troponin C subunit of the troponin trimer transmits conformational changes to tropomyosin leading to its displacement from myosin binding sites. Troponin T is the specific subunit that transmits the conformational change. Several TNNT3 exons are alternatively spliced, contributing to subtle protein differences throughout development. In DM, TNNT3 shows premature inclusion of a fetal (F) exon, and fetal exon inclusion has been reported to be more common in DM2 than DM1 (422). An orthologous exon is misspliced in Mbnl1 KO mice suggesting disruption of MBNL activity in DM contributes increased fetal exon inclusion (189). As with other structural proteins disrupted in DM, the contribution of this missplicing event to disease progression requires further investigation.
Myofiber ion homeostasis and EC coupling
BIN1
Bridging integrator 1 (BIN1) protein is expressed in many tissues and functions as a regulator of actin and membrane dynamics (176). As nascent myotubes mature into terminally differentiated, functional myofibers, specialized structures including the sarcoplasmic reticulum (SR), transverse tubules, and myofibrillar components develop. In the mouse, these structures are first observed beginning with the SR at E14, followed by the first observable transverse tubules at E15 (388). BIN1 is an essential mediator of transverse tubule invagination and development (219). BIN1 exon 11, encoding the protein’s phosphoinositide-binding domain, is abnormally excluded in CDM patient-derived cells and DM1 skeletal muscle (121, 272). ASO-mediated exclusion of BIN1 exon 11 results in defective transverse tubule biogenesis and is sufficient to cause muscle weakness in mice (121). Interestingly, alterations in BIN1 are associated with several forms of human disease including both congenital (276) and adult-onset (41) forms of centronuclear myopathy. In the brain, altered CpG methylation upstream of BIN1 is associated with Alzheimer’s disease progression (88) and increased BIN1 expression is a modifier of Tau pathology (65). This suggests that BIN1 misregulation may also contribute to central nervous system defects observed in DM patients.
CACNA1S
Calcium voltage-gated channel subunit alpha 1S (CACNA1S) a multipass, transmembrane calcium ion channel located in the transverse tubules of skeletal muscle. CACNA1S is found only in skeletal muscle, where it participates in EC coupling (23). In DM1 and DM2, skipping of CACNA1S exon 29 increases with more severe exclusion in DM1 (393). This exon is undergoes a developmental splice transition between E16 and P10 in the mouse (393) and this may provide important functional characteristics for postnatal muscle. CACNA1S exon 29 encodes 19 extracellular amino acids (23), and a direct contribution of their loss to DM1 myopathy is supported by several lines of evidence: (1) the absence of this splicing shift in another muscular dystrophy, Facioscapulohumeral muscular dystrophy (FSHD); (2) a correlation between this splicing defect and TA muscle weakness; and (3) the appearance of a myopathy in wild-type mice followed morpholino-mediated exon 29 exclusion (393). Moreover, Mbnl1; Mbnl2 muscle-specific DKO mice show a significant shift in Cacna1s splicing, supporting the combinatorial role of multiple MBNL proteins in DM pathogenesis (227, 393). CELF1 overexpression also promotes the exon 29 exclusion isoform, reinforcing the antagonistic relationship observed between MBNL and CELF regulated events (393). Missplicing of CACNA1S represents a common theme is DM spliceopathy, where many affected genes have known roles in ion homeostasis and are implicated in other channelopathies (17). In agreement with a loss of proper calcium homeostasis in DM muscle, increased cytoplasmic calcium levels were identified in DM patients with a greater affect in DM1 than DM2 (350). These levels correlated with the degree of splicing changes observed in DM muscle (350). Importantly, these splicing defects may have consequences beyond contraction defects, and may play a role in myofiber death in DM (see below).
ATP2A1
ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 1 (ATP2A1) (also known as SERCA1) is a transmembrane calcium reuptake pump located in the SR (303). Following myofiber excitation, calcium is released from the SR via the RYR1 channel, and ATP2A1 regulates the reuptake of calcium and supports relaxation following contraction. In DM, ATP2A1 exon 22 is abnormally excluded, resulting in truncation of a C-terminal domain and a portion of the mRNA 3’ UTR. ATP2A1 exon 22 is also misspliced in a variety of mouse models including HSALR, Mbnl1 KO, and CELF1 overexpression models. Importantly, overexpression of MBNL1 in HSALR skeletal muscle rescues this splicing defect and validates MBNL as an important mediator of this splicing event (190). The exon 22 lacking protein, termed ATP2A1b, is highly expressed in DM1 muscle and may be particularly enriched in type I myofibers (459). Missplicing of ATP2A1 is also believed to contribute to altered calcium homeostasis in cultured DM myotubes (350). In agreement, the calcium reuptake activity of ATP2A1a (exon 22 containing isoform) is approximately double that of ATP2A1b when expressed in cultured myotubes (459). Beyond DM, mutations in ATP2A1 cause Brody disease, another disorder characterized by delayed muscle relaxation (280). Interestingly, this delay in relaxation is not myotonia, as action potentials are not present during relaxation (139). Excess intracellular calcium levels can cause ER stress, mitochondrial dysfunction, and the activation of calcium-dependent proteases (44, 54). Underscoring the importance of calcium regulation is the contribution excess intracellular calcium plays in myofiber necrosis in DMD (54). ATP2A1 is one of three SERCA pumps expressed in muscle. Another, ATP2A2, is also misspliced in DM1 skeletal muscle and displays altered functional properties (459).
Myofiber metabolism and protein homeostasis
INSR
Insulin receptor (INSR, also known as IR) is a transsarcolemmal receptor that responds to the ligand insulin. INSR is a tyrosine kinase receptor that regulates glucose metabolism (184) and upon ligand binding, autophosphorylation promotes intracellular signaling events that lead to activation of gene expression, protein synthesis, and glucose metabolism (79). Skeletal muscle is the body’s largest storage compartment for glucose and IR is a critical mediator. Normally, INSR undergoes a developmentally regulated splicing event with alternative exon 11 encoding a portion of the C-terminal domain (360).
IR missplicing generates an isoform with less affinity for insulin, IR-A (− exon 11) rather than IR-B (+ exon 11), and it has been suggested that this leads to increased blood glucose levels in DM patients (353). This splicing event is controlled by MBNL and CELF proteins and is also misregulated in HSALR muscle (84). The IR-A isoform also shows a greater affinity for IGF-II (another IR ligand) compared to IGF-I with implications for hypertrophic muscle growth. Upon binding of IGFs to INSR, key components of the Akt/mTOR pathway are upregulated, notably mTOR and GSK3β, while the atrophy-promoting transcription factor FOXO is downregulated. Interestingly, GSK3β, a modulator of mTOR anabolic activity, is increased in HSALR skeletal muscle and DM1 muscle cultures (180). Alternating GSK3β levels may be one therapeutic strategy to correct muscle weakness in DM patients (436). As with other RNA processing events, differences in the extent of IR missplicing exist between DM1 and DM2 (348).
PKM
Pyruvate kinase is a glycolytic enzyme produced as two major isoforms, PKM1 and PKM2, that differ in their metabolic activities and expression profile. For example, PKM2 is typically expressed in cells with high metabolic demands such as proliferating cells and is also an important modulator of tumor progression (77). PKM isoform choice is dictated by an AS event regulating mutually exlcusive exons 9 and 10. Alternative splice choice results in PKM1 (+ exon 10) and PKM2 (+ exon 9) differing in a 22 amino acid C-terminal domain that dictates whether the protein is constitutively (PKM1) or allosterically active (PKM2). As skeletal muscle differentiates, a developmental switch occurs from PKM2 in MPCs to PKM1 in myofibers (126). In DM1 patients, PKM2 expression is increased and correlates with altered glucose metabolism and type I fiber atrophy (126). Using ASO morpholinos to specifically disrupt this splice choice in otherwise healthy muscle results in an increase in glycolytic metabolism (126). This exon is specifically regulated by CELF1 overexpression, but not MBNL1 knockdown (126). Importantly, PKM2 reexpression is highly enriched in type I fibers and may explain fiber-specific muscle atrophy in DM1 (126).
Myofiber gene expression and RNA processing
NFIX
Nuclear factor IX (NFIX) is a transcription factor expressed in a variety of tissues, and in skeletal muscle it regulates an embryonic to fetal myogenic gene expression program (259). While NFIX is nearly undetectable in embryonic muscle and myoblasts, its expression greatly increases during fetal myogenesis (259). In fetal muscle, NFIX cooperates with transcriptional repressors to prevent the expression of embryonic genes, such as Nfat2c, another transcription factor important to activating embryonic genes (259). Skeletal muscle ablation of Nfix is sufficient to cause defective myoblast fusion and sarcomereogenesis (259). Additionally, Nfix regulates regenerative myogenesis partly through the control of myostatin expression (334). NFIX exon 7 is misspliced in DM patients, HSALR mice, and Mbnl1 KO mice (96, 227, 272). While the functional consequences of this splicing abnormality have not been explored, increased exon 7 inclusion in DM may disrupt the association of NFIX with other transcriptional coactivators and result in disruption of prenatal myogenic gene expression. This would be expected to contribute to disrupted developmental myogenesis in CDM and adult regeneration deficiencies in adult DM. Interestingly, NFIX increases the expression of some genes (e.g., Bgn) (259) that are targets of MBNL proteins (428), suggesting cooperativity between multiple layers of gene expression dysfunction in DM.
MEF2D
Myocyte enhancer factor 2D (MEF2D) is a transcriptional coactivator and regulates several aspects of muscle differentiation (310). The splicing of MEF2D is regulated by several RBPs, including MBNL and RBFOX proteins (224, 337, 370). In DM, two MEF2D missplicing events are present: (1) β exon; (2) a change in mutually exclusive α1 and α2 exon usage. The Mef2d β-exon is excluded following overexpression of CUGexp RNAs in C2C12 cells, suggesting this splicing event is susceptible to misregulation in DM (224). Furthermore, MEF2D β-exon inclusion is significantly reduced in DM1 skeletal muscle relative to controls (224). MEF2D cooperates with MYOD to regulate early myogenic events and the β-exon inclusion isoform of MEF2D protein results in an increased activation of target genes (222, 224, 310, 462). Strikingly, overexpression of the myofiber-specific MEF2D isoform (α1 and β exon positive) is sufficient to rescue myogenic defects in fusion-deficient RBFOX2 depleted cells (370). These data support a critical role for correct MEF2D splicing in muscle development. Other MEF proteins are disrupted in DM1, such as MEF2A, which shows reduced mRNA and protein levels in DM1 heart (187). Alterations in MEF2-regulated mRNA and miRNA pathways are also observed in DM1 heart (187), a finding consistent with another study demonstrating altered miR-1 processing and heart defects in DM1 (322). Interestingly, MEF2 proteins are activated in a mouse model of Becker syndrome, a nondystrophic congenital myotonia disorder (446). The activity of MEF2 proteins increases in response to increased intracellular calcium, and promotes fiber-type specific gene expression programs (445). It will be interesting to test if altered MEF2 activity in DM modulates fiber-type specific pathologies. Importantly, missplicing of MEF2 proteins is observed in other neuromuscular disorders, and has been suggested to be a compensatory, rather than pathologic, mechanism (20).
MBNL
While MBNL proteins have been extensively discussed above, their RNA processing is also misregulated in DM patients and would be expected to influence their roles in pre-mRNA AS and polyadenylation (26, 189, 428), mRNA stability (252, 429), RNA localization (428) and miRNA-1 biogenesis (Fig. 13) (111, 322). The three MBNL paralogs possess inherent functional differences, which are further modulated by presence of variable amino acid sequences encoded by alternative exons (386). MBNL1 exon 5, also referred to as exon 7, is alternatively spliced and modulates subcellular localization (412). In DM, this exon is preferentially included compared to controls and may act as a compensatory mechanism to increase the abundance of nuclear MBNL. However, the presence of this sequence decreases the splicing activity of MBNLs and that may be related to differences in MBNL homotypic interactions (386, 412).
Figure 13.
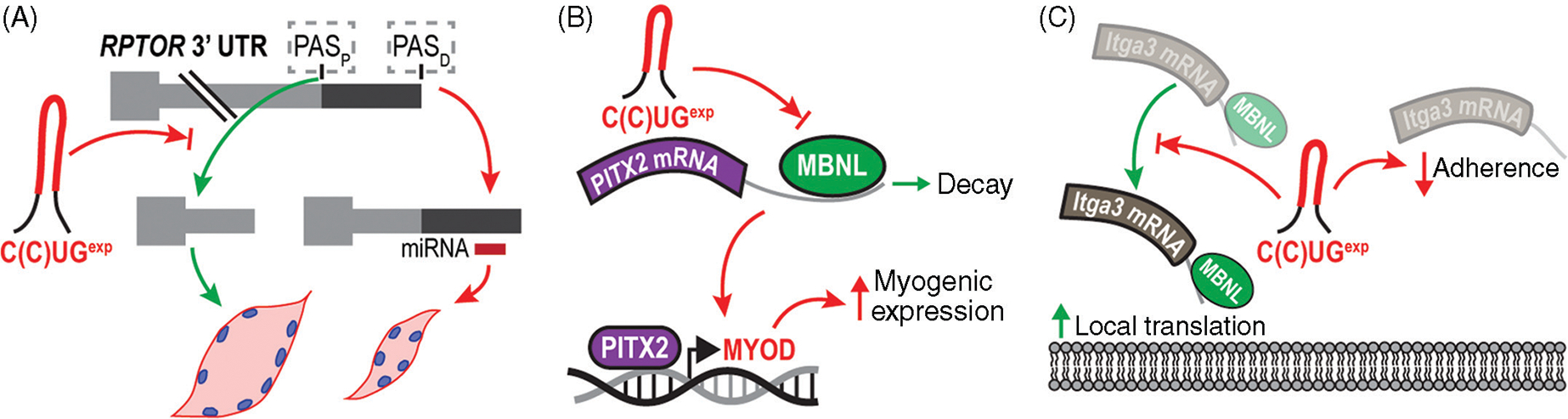
Additional RNA processing events implicated in DM. (A) RPTOR polyadenylation site (PAS) selection (PASP, proximal PAS; PASD, distal PAS) is altered in DM1 by CUGexp RNA and perhaps CCUGexp RNAs (red hairpin) in DM2. Increased PASD utilization may contribute to muscle wasting in DM because the increased 3’ UTR length allows regulation by miRNAs (red box) (24). (B) MBNL1 contributes to PITX2 mRNA (purple box) decay (green arrow), and C(C)UGexp-associated blocking of MBNL increases PITX2-mediated myogenic gene expression (244). (C) MBNL2/MLP1 has also been proposed to regulate ITGA3 mRNA localization to focal adhesions, presumably to allow local translation at these sites. Disruption of this activity in DM has been proposed to affect cell adherence (4).
Additional DM Pathomechanisms
RNA missplicing is a global phenomenon in DM1 and DM2 (302, 425) muscle, and we have largely focused on its role as a driver of disease symptoms. However, the direct role of spliceopathy in disease progression has been called into question due to the common occurrence of DM-relevant missplicing events in a variety of neuromuscular disorders (19). Given these observations, and the fact that many missplicing events are associated with muscle degeneration-regeneration (285), aspects of spliceopathy may be secondary to muscle pathology rather than a driver of disease. Furthermore, additional molecular pathomechanisms are clearly present. For example, bidirectional transcription of CTGexp alleles results in the production of CUG-CAG repeat structures and associated downregulation of endogenous CUG/CAG containing transcripts via an RNAi-like pathway in fly models (453). It is possible similar mechanisms occur in DM-affected tissues, as SIX5 antisense transcripts can extend through CTG repeats (74). However, the expression of DMPK antisense transcripts and the abundance of antisense RNA foci appear to be very low (262). Another class of regulatory RNAs, namely miRNAs, are also disrupted in DM Drosophila models, with levels of important myogenic miRNAs such as miR-1 and miR-7 reduced and conserved in DM-patient-derived fibroblast and myoblast cell lines with associated increases in several downstream target RNAs (111). In this study, expression of CUGexp RNAs was sufficient to cause these changes and was linked to alterations in Drosophila Mbl availability and control of miR-1 and miR-7 biogenesis (111). Interestingly, while miRNA networks are also misregulated in DM1 heart, these alterations are not linked to MBNL loss- or CELF gain-of-function, but rather dysregulation of MEF2 transcriptional networks (187). Heart-specific CUGexp RNA overexpression in the EpA960 mouse model is sufficient to induce these changes, MEF2A protein levels are significantly reduced in DM1 patient heart tissue, and exogenous expression of MEF2C in a DM1 cardiac cell model rescues MEF2 associated miRNA and mRNA expression (187). In addition to MEF2-transcriptional networks, other transcriptional regulators are affected in DM1 mouse models. For example, the abundance and nuclear localization of NFATc1 is increased in HSALR muscle and is coincident with increased levels of transcriptional targets (324). Interestingly, calcineurin (a phosphatase that increases the activity of NFATc1) is also increased in the HSALR mouse model as well as DMD muscle and is thought to act as a compensatory response to calcium mishomeostasis in both (324).
The distinction between RNA and protein toxicity is blurred in the case of another emerging DM-associated pathology, RAN translation. RAN translation was originally identified in the neurological disease SCA type 8 (SCA8) and a mouse model of this disease (463), but is increasingly appreciated in several microsatellite disorders including FXTAS, HD, and C9orf72-linked ALS/FTD (78, 463). In the case of HD-associated RAN peptides, a direct toxic role has been revealed using glial and neuronal cell models (22). Furthermore, antisense transcription across C9orf72-linked G4C2-repeat results in the accumulation of G2C4-associated RAN proteins in patient brain that have been demonstrated to exert toxicity in cell model systems (464). In the context of DM1, polyglutamine RAN proteins have been identified in the muscle of DMSXL and DM55 mouse models as well as in patient myoblast cell lines (463). Since, bidirectional transcription can occur across CUGexp tracts in DM1, both CUG and CAG RNAs might escape into the cytoplasm resulting in the synthesis of up to six RAN peptides. For DM2, the identification of RAN proteins is most prevalent in CNS tissues and has not yet been demonstrated in muscle tissues.
Therapeutic Interventions
The efficacy of therapeutic approaches requires the development of measurable outcomes that must be carefully defined. Studies in the past 25 years have linked some DM disease features to specific RNA processing events (e.g., myotonia and insulin insensitivity caused by CLCN1 and INSR missplicing, respectively). These splicing biomarkers are important tools to determine the therapeutic success in mouse models, but the requirement for biopsies from affected tissues undermines their feasibility to evaluate treatment efficacy in clinical trials. In contrast, less invasive approaches can be used to monitor muscle strength and myotonia. Using newly developed equipment to monitor muscle strength, a longitudinal study in the DMSXL mouse has identified outcome measures for preclinical assessments (90). Likewise, myotonia can be monitored qualitatively through grip and percussion tests or more quantitatively using EMG. Altogether, these outcome measures have provided reasonable avenues to develop and evaluate multiple strategies for targeted intervention.
While the ideal therapy for DM would be the correction of expanded repeats to a nonpathogenic size, this approach requires further advancements in targeted gene editing as well as a change in societal attitudes toward the use of these technologies in a clinical setting. Alternative strategies have been proposed, including the use of ASOs that target CUGexp transcripts and prevent MBNL sequestration or induce transcript degradation. This approach has been successful in reverting phenotypes in animal models, and holds great promise in clinical trials. Several other avenues have been explored, including the use of small molecules designed to bind repeats and release MBNL or block DMPK transcription. In this section, we will discuss these and other potential therapeutic interventions for DM, including their technological limitations and unresolved questions.
Gene editing
Correction of the expanded repeat in the DMPK or CNBP genes to a nonpathogenic size has been a major objective since the discovery of the mutations underlying DM1 and DM2 (47, 150, 231, 241). For many years, this site-specific modification in the human genome remained technically challenging, mostly due to the substantial complexity and inefficiency of gene-editing approaches. Recent additions to the genome engineering toolbox such as zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and the revolutionary CRISPR/Cas system have provided efficient strategies to accomplish the goal of targeted gene editing (Fig. 14) (93). Currently in Phase II clinical trials, ZFNs were used to KO the CCR5 receptor from autologous hematopoietic stem cells, making them resistant to HIV (395). Similarly, the CRISPR/Cas system has been recently used in a mouse model of DMD, providing a successful approach to remove the mutant exon 23 from the DMD gene, producing an in-frame mRNA and a truncated but functional DMD protein (387).
Figure 14.
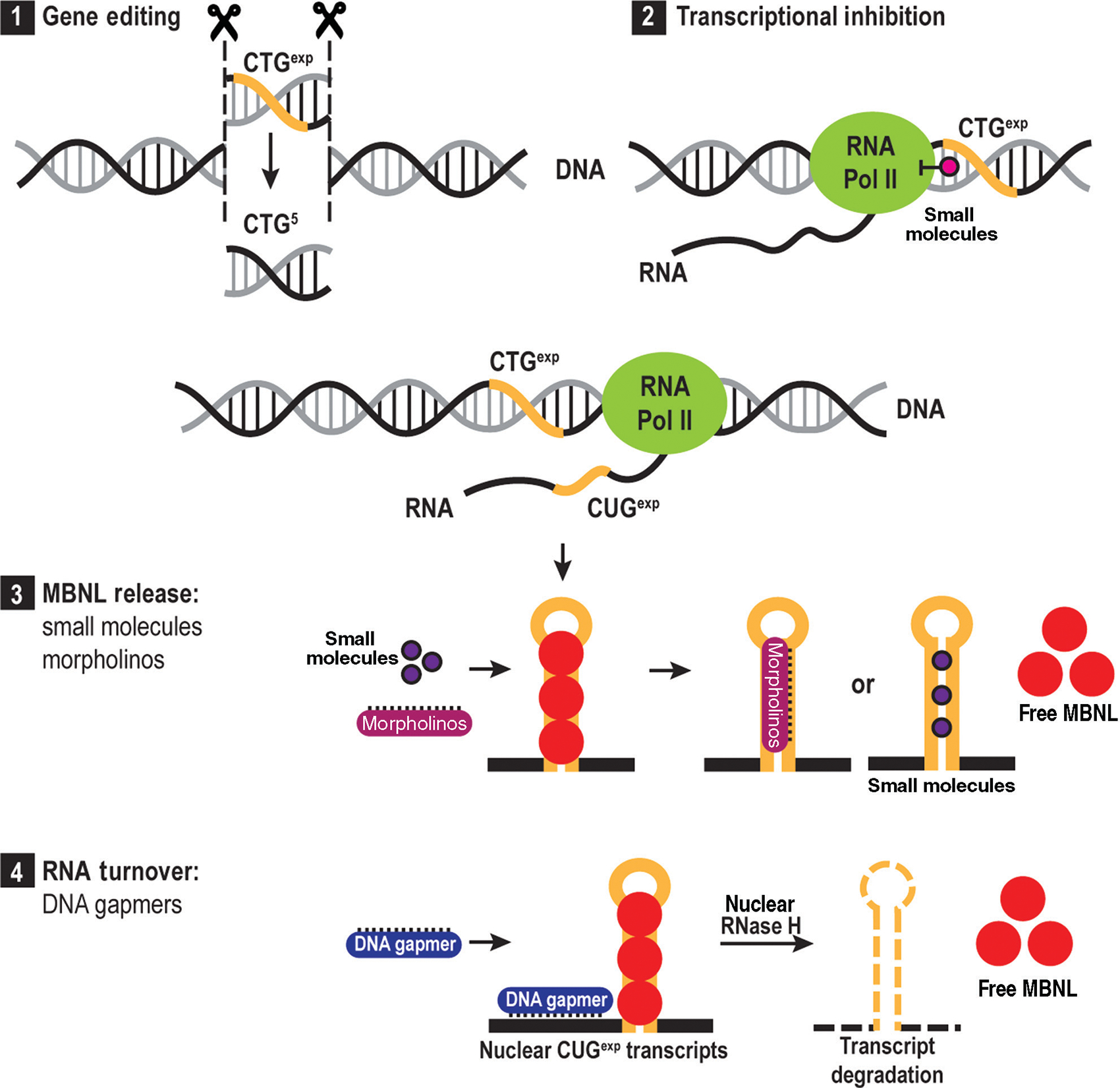
Therapeutic interventions. Proposed avenues for therapeutic intervention in DM, including: (1) gene editing of the expanded repeats to a nonpathogenic size; (2) use of small molecules that intercalate into GC-rich DNA and arrest the elongating RNA polymerase II; (3) use of small molecules or morpholinos that displace or sterically inhibit MBNL binding; (4) use of DNA antisense oligonucleotide (ASO) gapmers that bind to mutant transcripts and trigger their degradation by RNase H.
In DM, TALENs have been used to correct patient-derived stem cells by insertion of a polyadenylation signal upstream of the CTG repeats (125, 448). While this strategy does not remove the repeats from the DMPK gene, it results in the premature termination of DMPK transcription, preventing the production of mutant transcripts that accumulate in RNA foci, and restoring normal splicing patterns in neural progenitor cells. Similarly, expanded CTG repeats can be corrected to a nonpathogenic size or the DMPK gene can be knocked out, despite the existence of conflicting reports regarding the effects of Dmpk ablation in mice (62, 327).
A recurrent concern with any gene editing strategy is the potential for off-target, or nonspecific, DNA cleavage. To enhance specificity, Cas9 nucleases, which alone are capable of generating a DNA double-stranded break, can be replaced by a pair of nickases, enzymes that can only cut one of the DNA strands (364). As a result, the generation of a double-stranded break now requires the simultaneous and proximal binding of two different nickases, which in turn dramatically reduces the occurrence of off-target DNA cleavage events. Additionally, these nucleases can be engineered for enhanced specificity. For example, the widely used Cas9 nuclease from Staphylococcus pyogenes (SpCas9) has been modified to alter some hydrogen bond-forming amino acid residues resulting in a Cas9 enzyme with nearly undetectable off-target effects (198).
The largest disadvantage of gene-editing applications for DM1 and DM2 is the multisystemic nature of these diseases. Systemic delivery is very challenging and the idea of correcting all affected tissues is not viable with the current technological repertoire. Local delivery of nucleases to skeletal muscle is possible with adeno-associated virus (AAV) vectors but the cargo is limited to ~4.5 kb so AAVs are not suitable to deliver the relatively large (~4.2 kb) SpCas9 and its guide RNA. As an alternative, smaller, albeit less active, nucleases have been utilized, including the ~3 kb Staphylococcus aureus Cas9 (SaCas) that is currently being used in multiple preclinical studies (318, 387).
Antisense oligonucleotides
Currently, ASO technology is the most promising therapeutic avenue for DM. ASOs are short nucleic acid fragments whose sequence is complementary to a target RNA of interest. Depending on the specific chemistry used, ASOs can bind to their RNA target and sterically prevent the association of transacting factors, such as MBNL, or bind and activate targeted degradation of mutant transcripts. The first scenario relies on ASOs such as phosphorodiamidate morpholino oligomers or all-lock-nucleic acid oligomers, which are composed of a short stretch of pure CA(G)G repeats that bind to expanded C(C)UG transcripts without triggering RNA degradation. This enables the release of sequestered MBNL proteins, restoring their function within the cell (439,443). In the second scenario, chimeric ASOs have been developed including DNA gapmers, which consist of 7 to 10 phosphorothioate DNA nucleotides flanked by modified RNA bases. Upon entry into the cell nucleus, these ASOs bind to their target RNA sequences and the subsequent DNA:RNA hybrid is recognized by endogenous RNase H resulting in the degradation of the RNA portion of the duplex (220). Because C(C)UGexp RNAs are largely retained in the nucleus, this increases the potency of therapeutic compounds that require endogenous intranuclear factors such as RNase H. In fact, variations in many factors such as C(C)UGexp repeat length, C(C)UGexp expression level, and available intranuclear MBNL produce a dynamic environment that has important implications for therapeutic intervention (401). For example, under conditions of C(C)UGexp RNA excess relative to MBNL, increased MBNL occupancy within RNA foci may reduce the movement of foci-associated MBNL into the nucleoplasm resulting in increased molecular crowding with implications in preventing the availability of C(C)UGexp to small molecules or ASOs (386,401). As hundreds of nuclei are present in muscle fibers, it is possible that different disease progression states exist in distinct nuclei within a single muscle cell. Beyond targeting toxic C(C)UGexp RNAs, ASOs can modulate downstream effectors by targeting RNA processing events underlying specific DM symptoms, such as CLCN1 missplicing (201). Similar approaches have been successfully used to promote exon skipping in Becker and Duchenne muscular dystrophies (1) or enhanced SMN2 exon 7 inclusion in spinal muscular atrophy (169). Intramuscular delivery of ASOs has been shown to be effective (220), despite the fact that membrane integrity is not significantly compromised in DM (133).
Systemic distribution of ASOs is challenging, but studies have shown it can be improved by peptide-linkage strategies (229) and liposomal delivery followed by ultrasound exposure (201). Changes in ASO chemical composition affect tissue permeability, distribution, stability, affinity for repeats, and the mechanism of action. For instance, modified 2’-O-methoxyethyl (MOE) flanking gapmers increase ASO bioavailability, resulting in a remarkable reversion of ~85% of the splicing changes in the HSALR mouse model (438).
Following their marked success in preclinical studies, ASOs designed to degrade mutant DMPK transcripts entered a Phase I/II clinical trial in December 2014. In early 2017, this trial was discontinued due to inadequate efficacy in muscle tissue although safety was not an issue. Modifications in ASO composition are promising strategies to overcome this issue and future preclinical studies and therapeutic trials will inform us regarding the success of these structural changes, including their impact on efficacy, tissue distribution, and long-term turnover. Importantly, long-term studies are required to address several safety issues concerning ASOs, including their specificity and toxicity.
The concept of toxicity is limited by the assay and thus ASO off-target effects must be carefully monitored using different experimental approaches. For example, the current generation of DNA gapmers for DM1 is designed to preferentially degrade mutant DMPK transcripts, due to their nuclear retention and RNase H susceptibility, but transcript levels from the normal allele are also affected and depletion of this kinase may adversely affect the regulation of its phosphorylation targets. For DM2, Cnbp is an essential gene in mice so gapmers targeting human CNBP exonic regions may lead to deleterious consequences. An alternative is to target the CCUGexp region directly, but other genes containing CCUG repeats may be affected so global gene expression studies should also be performed to address this issue. Additionally, the downstream consequences of morpholinos aimed to release MBNL proteins from toxic transcripts must be evaluated. Once free from MBNL, toxic transcripts might be exported to the cytoplasm and trigger RAN translation or RNAi pathway alterations (111,205,208). Despite their occurrence in DM, the specific contribution of these pathways for DM disease pathogenesis remains unclear.
Small molecules
The knowledge gathered from structural studies on expanded C(C)UG RNAs and their interaction with MBNL proteins has paved another avenue for therapeutic intervention: the use of small molecules that displace or sterically prevent MBNL binding. Designed to specifically bind C(C)UG repeats, small molecules are promising therapeutic candidates due to their lower production costs and potential for increased tissue permeability. To date, several compounds have been proposed, including Ligand 1 (145), Hoechst 33258 (314), lomofungin and dilomofungin (167), and others (71, 72, 293). While the majority of these compounds specifically bind to CUG repeats, some target CCUG expansions as well (72, 228).
Another promising class of small molecules consists of transcriptional inhibitors that intercalate into GC-rich DNA, arresting elongating RNA polymerase II (118). These compounds are mostly chemotherapeutics with high affinity and specificity for CUG repeats at low dosages. For example, actinomycin D (ActD), an FDA-approved drug used to treat cancers at dosages between 0.015 and 0.045 mg/kg, was deployed to reduce CUG expression at a human equivalent dosage of 0.002 mg/kg and rescue splicing at 0.02 mg/kg in the HSALR mouse model (366). Nevertheless, the ability of ActD to induce global transcriptional inhibition is a potential drawback and its long-term side effects are unknown. Currently, our repertoire of small molecules is expanding, primarily supported by high-throughput screens that address the impact of these compounds on RNA foci formation, splicing rescue, MBNL1 and CELF1 localization and function (195).
Additional strategies
Several additional approaches have been proposed as potential therapeutic interventions for DM. For example, the utilization of mexiletine has been shown to ameliorate myotonia (234) and the use of steroids such as dehydroepiandrosterone (DHEA) to treat muscle weakness (299). Exercise has been suggested as another therapy (196), as well as the implantation of automatic defibrillators to prevent sudden death, despite the risks of exposing DM patients to generalized anesthesia (32, 426).
At the cellular and molecular levels, multiple additional strategies have also been explored. Artificial site-specific RNA endonucleases have been used to degrade expanded CUG transcripts (458). In patient-derived cells, engineered human U7 small nuclear RNAs containing a CAG repeat were utilized to selectively degrade mutant DMPK transcripts (116). Complete DMPK knockdown has also been suggested (217, 372), as well as MBNL1 overexpression, despite the potential detrimental effects of these strategies in humans (190).
Conclusion
In this review, we have summarized key concepts of skeletal myopathy in DM. However, DM1, DM2, and CDM are multisystemic diseases and require a multifaceted approach to pinpoint the key drivers of pathology in the various affected tissues. We believe the use of increasingly sophisticated animal models will support these studies. Current generations of DM modeling studies have revealed recurrent themes in DM pathomechanisms, but no animal model recapitulates the complete spectrum of DM disease. While differences between mouse and humans may prevent this, attempts to generate these models have largely been stifled by technical barriers. First, the generation of expanded DNA repeats is limited by the capacity of E. coli to harbor large expansions and poses a barrier to traditional cloning strategies (33). Techniques such as rolling circle amplification have been utilized to circumvent this, allowing for the generation of CTG repeats well beyond the CDM threshold of ~1000 (286). While theoretically possible, it remains to be seen if similar successes can be achieved with CCTG expansions. Even with DNA templates of the appropriate length, introduction into the mouse genome is challenging particularly for targeted knockins. The use of traditional mouse ESC targeting strategies have not yielded models with large expansions possibly due to toxicity of these lengthy repeats in these cell populations or the inability of endogenous mouse loci to harbor large expansions. To date, CTG<100 is the largest repeat knocked into the mouse Dmpk locus (417). The use of newer technologies, such as CRISPR/Cas9, may provide a valuable resource in the generation of a new generation of knockin models. Furthermore, while many current transgenic models of DM are available, few insertions have been mapped and will be important to ensure off-target effects are not contributing to phenotypes observed in these animals. Given CRISPR/Cas9 is also susceptible to off target effects, careful mapping and characterization of mutations should be performed in conjunction with efforts to phenotype models.
Another largely unanswered question in the field is the mechanistic basis of congenital DM. One key distinction between DM1 and CDM is the presence of highly expanded CTGexp repeats throughout embryonic development in CDM due to inheritance of large alleles. Similar to adult-onset DM1 where RNA alternative processing is linked to disease manifestations, the presence of highly expanded CUGexp RNAs during embryonic development would be expected to affect RNA isoform transitions important for the tissue morphogenesis (186). In agreement with the RNA toxicity model holding true during tissue development, CUGexp RNA foci are highly abundant in embryonic and fetal cardiac, skeletal muscle, and brain tissues expressing CUGexp RNA (262). These data suggest MBNL loss-of-function contributes to CDM disease, a hypothesis supported by our recent findings of congenital myopathy in mouse Mbnl KO models (397). Beyond inherent toxicity throughout embryonic development, the dynamics of repeat instability may differ in developing tissues compared to postmitotic tissues (255). Along with these, the lack of congenital forms of DM2 suggests fundamental differences in the ability of CNBP-linked CCTGexp mechanisms to elicit developmental defects. Differences in the spatiotemporal expression and steady-state transcript levels of CCUGexp versus CUGexp containing transcripts may underlie this difference, but this question warrants further investigation.
Acknowledgements
The authors thank J. Bubenik for reviewing the manuscript and members of E. Wang’s lab for lending a hand. Research studies performed in the Swanson lab are supported by grants from the NIH (NS058901, NS98819) and the MDA (RG480539). The authors also thank the Myotonic Dystrophy Foundation and Wyck Foundation for support to Ł.J. Sznajder.
References
- 1.Aartsma-Rus A, van Ommen GJ. Antisense-mediated exon skipping: A versatile tool with therapeutic and research applications. RNA 13: 1609–1624, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abmayr SM, Balagopalan L, Galletta BJ, Hong SJ. Cell and molecular biology of myoblast fusion. Int Rev Cytol 225: 33–89, 2003. [DOI] [PubMed] [Google Scholar]
- 3.Abmayr SM, Pavlath GK. Myoblast fusion: Lessons from flies and mice. Development 139: 641–656, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adereth Y, Dammai V, Kose N, Li R, Hsu T. RNA-dependent integrin alpha3 protein localization regulated by the Muscleblind-like protein MLP1. Nat Cell Biol 7: 1240–1247, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akhmanova A, Hoogenraad CC, Drabek K, Stepanova T, Dortland B, Verkerk T, Vermeulen W, Burgering BM, De Zeeuw CI, Grosveld F, Galjart N. Clasps are CLIP-115 and -170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell 104: 923–935, 2001. [DOI] [PubMed] [Google Scholar]
- 6.Ali S, Garcia JM. Sarcopenia, cachexia and aging: Diagnosis, mechanisms and therapeutic options: A mini-review. Gerontology 60: 294–305, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Almada AE, Wagers AJ. Molecular circuitry of stem cell fate in skeletal muscle regeneration, ageing and disease. Nat Rev Mol Cell Biol 17: 267–279, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alway SE, Myers MJ, Mohamed JS. Regulation of satellite cell function in sarcopenia. Front Aging Neurosci 6: 246, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amack JD, Mahadevan MS. The myotonic dystrophy expanded CUG repeat tract is necessary but not sufficient to disrupt C2C12 myoblast differentiation. Hum Mol Genet 10: 1879–1887, 2001. [DOI] [PubMed] [Google Scholar]
- 10.Amack JD, Mahadevan MS. Myogenic defects in myotonic dystrophy. Dev Biol 265: 294–301, 2004. [DOI] [PubMed] [Google Scholar]
- 11.Amack JD, Paguio AP, Mahadevan MS. Cis and trans effects of the myotonic dystrophy (DM) mutation in a cell culture model. Hum Mol Genet 8: 1975–1984, 1999. [DOI] [PubMed] [Google Scholar]
- 12.Anvret M, Ahlberg G, Grandell U, Hedberg B, Johnson K, Edstrom L. Larger expansions of the CTG repeat in muscle compared to lymphocytes from patients with myotonic dystrophy. Hum Mol Genet 2: 1397–1400, 1993. [DOI] [PubMed] [Google Scholar]
- 13.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 393: 805–809, 1998. [DOI] [PubMed] [Google Scholar]
- 14.Argov Z, Gardner-Medwin D, Johnson MA, Mastaglia FL. Congenital myotonic dystrophy: Fiber type abnormalities in two cases. Arch Neurol 37: 693–696, 1980. [DOI] [PubMed] [Google Scholar]
- 15.Arimura T, Hayashi T, Terada H, Lee SY, Zhou Q, Takahashi M, Ueda K, Nouchi T, Hohda S, Shibutani M, Hirose M, Chen J, Park JE, Yasunami M, Hayashi H, Kimura A. A Cypher/ZASP mutation associated with dilated cardiomyopathy alters the binding affinity to protein kinase C. J Biol Chem 279: 6746–6752, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Artero R, Prokop A, Paricio N, Begemann G, Pueyo I, Mlodzik M, Perez-Alonso M, Baylies MK. The muscleblind gene participates in the organization of Z-bands and epidermal attachments of Drosophila muscles and is regulated by Dmef2. Dev Biol 195: 131–143, 1998. [DOI] [PubMed] [Google Scholar]
- 17.Ashcroft FM. From molecule to malady. Nature 440: 440–447, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Ashizawa T, Sarkar PS. Myotonic dystrophy types 1 and 2. Handb Clin Neurol 101: 193–237, 2011. [DOI] [PubMed] [Google Scholar]
- 19.Bachinski LL, Baggerly KA, Neubauer VL, Nixon TJ, Raheem O, Sirito M, Unruh AK, Zhang J, Nagarajan L, Timchenko LT, Bassez G, Eymard B, Gamez J, Ashizawa T, Mendell JR, Udd B, Krahe R. Most expression and splicing changes in myotonic dystrophy type 1 and type 2 skeletal muscle are shared with other muscular dystrophies. Neuromuscul Disord 24: 227–240, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bachinski LL, Sirito M, Bohme M, Baggerly KA, Udd B, Krahe R. Altered MEF2 isoforms in myotonic dystrophy and other neuromuscular disorders. Muscle Nerve 42: 856–863, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bagni C, Tassone F, Neri G, Hagerman R. Fragile X syndrome: Causes, diagnosis, mechanisms, and therapeutics. J Clin Invest 122: 4314–4322, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banez-Coronel M, Porta S, Kagerbauer B, Mateu-Huertas E, Pantano L, Ferrer I, Guzman M, Estivill X, Marti E. A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet 8: e1002481, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bannister RA, Beam KG. Ca(V)1.1: The atypical prototypical voltage-gated Ca(2)(+) channel. Biochim Biophys Acta 1828: 1587–1597, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barbe L, Lanni S, Lopez-Castel A, Franck S, Spits C, Keymolen K, Seneca S, Tome S, Miron I, Letourneau J, Liang M, Choufani S, Weksberg R, Wilson MD, Sedlacek Z, Gagnon C, Musova Z, Chitayat D, Shannon P, Mathieu J, Sermon K, Pearson CE. CpG methylation, a parent-of-origin effect for maternal-biased transmission of congenital myotonic dystrophy. Am J Hum Genet 100: 488–505, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barroso FA, Nogues MA. Images in clinical medicine. Percussion myotonia. N Engl J Med 360: e13, 2009. [DOI] [PubMed] [Google Scholar]
- 26.Batra R, Charizanis K, Manchanda M, Mohan A, Li M, Finn DJ, Goodwin M, Zhang C, Sobczak K, Thornton CA, Swanson MS. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol Cell 56: 311–322, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Batra R, Charizanis K, Swanson MS. Partners in crime: Bidirectional transcription in unstable microsatellite disease. Hum Mol Genet 19: R77–R82, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bentzinger CF, Wang YX, Rudnicki MA. Building muscle: Molecular regulation of myogenesis. Cold Spring Harb Perspect Biol 4: pii: a008342, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berger DS, Ladd AN. Repression of nuclear CELF activity can rescue CELF-regulated alternative splicing defects in skeletal muscle models of myotonic dystrophy. PLoS Curr 4: RRN1305, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berul CI, Maguire CT, Aronovitz MJ, Greenwood J, Miller C, Gehrmann J, Housman D, Mendelsohn ME, Reddy S. DMPK dosage alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model. J Clin Invest 103: R1–R7, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhagavati S, Shafiq SA, Xu W. (CTG)n repeats markedly inhibit differentiation of the C2C12 myoblast cell line: implications for congenital myotonic dystrophy. Biochim Biophys Acta 1453: 221–229, 1999. [DOI] [PubMed] [Google Scholar]
- 32.Bhakta D, Shen C, Kron J, Epstein AE, Pascuzzi RM, Groh WJ. Pacemaker and implantable cardioverter-defibrillator use in a US myotonic dystrophy type 1 population. J Cardiovasc Electrophysiol 22: 1369–1375, 2011. [DOI] [PubMed] [Google Scholar]
- 33.Bichara M, Wagner J, Lambert IB. Mechanisms of tandem repeat instability in bacteria. Mutat Res 598: 144–163, 2006. [DOI] [PubMed] [Google Scholar]
- 34.Bigot A, Klein AF, Gasnier E, Jacquemin V, Ravassard P, Butler-Browne G, Mouly V, Furling D. Large CTG repeats trigger p16-dependent premature senescence in myotonic dystrophy type 1 muscle precursor cells. Am J Pathol 174: 1435–1442, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev 82: 291–329, 2002. [DOI] [PubMed] [Google Scholar]
- 36.Bland CS, Wang ET, Vu A, David MP, Castle JC, Johnson JM, Burge CB, Cooper TA. Global regulation of alternative splicing during myogenic differentiation. Nucleic Acids Res 38: 7651–7664, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bloch-Gallego E Mechanisms controlling neuromuscular junction stability. Cell Mol Life Sci 72: 1029–1043, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bodensteiner JB. The evaluation of the hypotonic infant. Semin Pediatr Neurol 15: 10–20, 2008. [DOI] [PubMed] [Google Scholar]
- 39.Bodine SC. Disuse-induced muscle wasting. Int J Biochem Cell Biol 45: 2200–2208, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3: 1014–1019, 2001. [DOI] [PubMed] [Google Scholar]
- 41.Bohm J, Biancalana V, Malfatti E, Dondaine N, Koch C, Vasli N, Kress W, Strittmatter M, Taratuto AL, Gonorazky H, Laforet P, Maisonobe T, Olive M, Gonzalez-Mera L, Fardeau M, Carriere N, Clavelou P, Eymard B, Bitoun M, Rendu J, Faure J, Weis J, Mandel JL, Romero NB, Laporte J. Adult-onset autosomal dominant centronuclear myopathy due to BIN1 mutations. Brain 137: 3160–3170, 2014. [DOI] [PubMed] [Google Scholar]
- 42.Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 6: 25–39, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bondy-Chorney E, Crawford Parks TE, Ravel-Chapuis A, Klinck R, Rocheleau L, Pelchat M, Chabot B, Jasmin BJ, Cote J. Staufen1 regulates multiple alternative splicing events either positively or negatively in DM1 indicating its role as a disease modifier. PLoS Genet 12: e1005827, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Botta A, Malena A, Loro E, Del Moro G, Suman M, Pantic B, Szabadkai G, Vergani L. Altered Ca2+ homeostasis and endoplasmic reticulum stress in myotonic dystrophy type 1 muscle cells. Genes (Basel) 4: 275–292, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bouchard JP, Cossette L, Bassez G, Puymirat J. Natural history of skeletal muscle involvement in myotonic dystrophy type 1: A retrospective study in 204 cases. J Neurol 262: 285–293, 2015. [DOI] [PubMed] [Google Scholar]
- 46.Braun T, Gautel M. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat Rev Mol Cell Biol 12: 349–361, 2011. [DOI] [PubMed] [Google Scholar]
- 47.Brook JD, Mccurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, Sohn R, Zemelman B, Snell RG, Rundle SA, Crow S, Davies J, Shelbourne P, Buxton J, Jones C, Juvonen V, Johnson K, Harper PS, Shaw DJ, Housman DE. Molecular-basis of myotonic-dystrophy: Expansion of a trinucleotide (Ctg) tepeat at the 3’ end of a transcript encoding a protein-kinase family member. Cell 68: 799–808, 1992. [DOI] [PubMed] [Google Scholar]
- 48.Bruusgaard JC, Liestol K, Ekmark M, Kollstad K, Gundersen K. Number and spatial distribution of nuclei in the muscle fibres of normal mice studied in vivo. J Physiol 551: 467–478, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bryson-Richardson RJ, Currie PD. The genetics of vertebrate myogenesis. Nat Rev Genet 9: 632–646, 2008. [DOI] [PubMed] [Google Scholar]
- 50.Brzoska E, Bello V, Darribere T, Moraczewski J. Integrin alpha3 subunit participates in myoblast adhesion and fusion in vitro. Differentiation 74: 105–118, 2006. [DOI] [PubMed] [Google Scholar]
- 51.Buckingham M, Bajard L, Chang T, Daubas P, Hadchouel J, Meilhac S, Montarras D, Rocancourt D, Relaix F. The formation of skeletal muscle: From somite to limb. J Anat 202: 59–68, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bugiardini E, Rivolta I, Binda A, Soriano Caminero A, Cirillo F, Cinti A, Giovannoni R, Botta A, Cardani R, Wicklund MP, Meola G. SCN4A mutation as modifying factor of myotonic dystrophy type 2 phenotype. Neuromuscul Disord 25: 301–307, 2015. [DOI] [PubMed] [Google Scholar]
- 53.Buj-Bello A, Furling D, Tronchère H, Laporte J, Lerouge T, Butler-Browne GS, Mandel J-L. Muscle-specific alternative splicing of myotubularin-related 1 gene is impaired in DM1 muscle cells. Hum Mol Genet 11: 2297–2307, 2002. [DOI] [PubMed] [Google Scholar]
- 54.Burr AR, Molkentin JD. Genetic evidence in the mouse solidifies the calcium hypothesis of myofiber death in muscular dystrophy. Cell Death Differ 22: 1402–1412, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buxton J, Shelbourne P, Davies J, Jones C, Van Tongeren T, Aslanidis C, de Jong P, Jansen G, Anvret M, Riley B, et al. Detection of an unstable fragment of DNA specific to individuals with myotonic dystrophy. Nature 355: 547–548, 1992. [DOI] [PubMed] [Google Scholar]
- 56.Calderon JC, Bolanos P, Caputo C. The excitation-contraction coupling mechanism in skeletal muscle. Biophys Rev 6: 133–160, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Campbell C, Levin S, Siu VM, Venance S, Jacob P. Congenital myotonic dystrophy: Canadian population-based surveillance study. J Pediatr 163: 120–125 e121–123, 2013. [DOI] [PubMed] [Google Scholar]
- 58.Campbell C, Sherlock R, Jacob P, Blayney M. Congenital myotonic dystrophy: Assisted ventilation duration and outcome. Pediatrics 113: 811–816, 2004. [DOI] [PubMed] [Google Scholar]
- 59.Carango P, Noble JE, Marks HG, Funanage VL. Absence of myotonic dystrophy protein kinase (DMPK) mRNA as a result of a triplet repeat expansion in myotonic dystrophy. Genomics 18: 340–348, 1993. [DOI] [PubMed] [Google Scholar]
- 60.Cardani R, Bugiardini E, Renna LV, Rossi G, Colombo G, Valaperta R, Novelli G, Botta A, Meola G. Overexpression of CUGBP1 in skeletal muscle from adult classic myotonic dystrophy type 1 but not from myotonic dystrophy type 2. PLoS One 8: e83777, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carmignac V, Durbeej M. Cell-matrix interactions in muscle disease. J Pathol 226: 200–218, 2012. [DOI] [PubMed] [Google Scholar]
- 62.Carrell ST, Carrell EM, Auerbach D, Pandey SK, Bennett CF, Dirksen RT, Thornton CA. Dmpk gene deletion or antisense knockdown does not compromise cardiac or skeletal muscle function in mice. Hum Mol Genet 25: 4328–4338, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Caskey CT, Swanson MS, Timchenko LT. Myotonic dystrophy: Discussion of molecular mechanism. Cold Spring Harb Symp Quant Biol 61: 607–614, 1996. [PubMed] [Google Scholar]
- 64.Chamberlain CM, Ranum LP. Mouse model of muscleblind-like 1 overexpression: Skeletal muscle effects and therapeutic promise. Hum Mol Genet 21: 4645–4654, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chapuis J, Hansmannel F, Gistelinck M, Mounier A, Van Cauwenberghe C, Kolen KV, Geller F, Sottejeau Y, Harold D, Dourlen P, Grenier-Boley B, Kamatani Y, Delepine B, Demiautte F, Zelenika D, Zommer N, Hamdane M, Bellenguez C, Dartigues JF, Hauw JJ, Letronne F, Ayral AM, Sleegers K, Schellens A, Broeck LV, Engelborghs S, De Deyn PP, Vandenberghe R, O’Donovan M, Owen M, Epelbaum J, Mercken M, Karran E, Bantscheff M, Drewes G, Joberty G, Campion D, Octave JN, Berr C, Lathrop M, Callaerts P, Mann D, Williams J, Buee L, Dewachter I, Van Broeckhoven C, Amouyel P, Moechars D, Dermaut B, Lambert JC, consortium G. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry 18: 1225–1234, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, Yuan Y, Shiue L, Cline M, Scotti MM, Xia G, Kumar A, Ashizawa T, Clark HB, Kimura T, Takahashi MP, Fujimura H, Jinnai K, Yoshikawa H, Gomes-Pereira M, Gourdon G, Sakai N, Nishino S, Foster TC, Ares M Jr., Darnell RB, Swanson MS. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 75: 437–450, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Charlet-B N, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell 10: 45–53, 2002. [DOI] [PubMed] [Google Scholar]
- 68.Chen W, Liang YQ, Deng WJ, Shimizu K, Ashique AM, Li E, Li YP. The zinc-finger protein CNBP is required for forebrain formation in the mouse. Development 130: 1367–1379, 2003. [DOI] [PubMed] [Google Scholar]
- 69.Chen W, Wang Y, Abe Y, Cheney L, Udd B, Li YP. Haploinsuffciency for Znf9 in Znf9+/− mice is associated with multiorgan abnormalities resembling myotonic dystrophy. J Mol Biol 368: 8–17, 2007. [DOI] [PubMed] [Google Scholar]
- 70.Cheng H, Zheng M, Peter AK, Kimura K, Li X, Ouyang K, Shen T, Cui L, Frank D, Dalton ND, Gu Y, Frey N, Peterson KL, Evans SM, Knowlton KU, Sheikh F, Chen J. Selective deletion of long but not short Cypher isoforms leads to late-onset dilated cardiomyopathy. Hum Mol Genet 20: 1751–1762, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Childs-Disney JL, Stepniak-Konieczna E, Tran T, Yildirim I, Park H, Chen CZ, Hoskins J, Southall N, Marugan JJ, Patnaik S, Zheng W, Austin CP, Schatz GC, Sobczak K, Thornton CA, Disney MD. Induction and reversal of myotonic dystrophy type 1 pre-mRNA splicing defects by small molecules. Nat Commun 4: 2044, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Childs-Disney JL, Yildirim I, Park H, Lohman JR, Guan L, Tran T, Sarkar P, Schatz GC, Disney MD. Structure of the myotonic dystrophy type 2 RNA and designed small molecules that reduce toxicity. ACS Chem Biol 9: 538–550, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cho DH, Tapscott SJ. Myotonic dystrophy: Emerging mechanisms for DM1 and DM2. Biochim Biophys Acta 1772: 195–204, 2007. [DOI] [PubMed] [Google Scholar]
- 74.Cho DH, Thienes CP, Mahoney SE, Analau E, Filippova GN, Tapscott SJ. Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol Cell 20: 483–489, 2005. [DOI] [PubMed] [Google Scholar]
- 75.Choi J, Dixon DM, Dansithong W, Abdallah WF, Roos KP, Jordan MC, Trac B, Lee HS, Comai L, Reddy S. Muscleblind-like 3 deficit results in a spectrum of age-associated pathologies observed in myotonic dystrophy. Sci Rep 6: 30999, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Choi J, Personius KE, DiFranco M, Dansithong W, Yu C, Srivastava S, Dixon DM, Bhatt DB, Comai L, Vergara JL, Reddy S. Muscleblind-Like 1 and muscleblind-like 3 depletion synergistically enhances myotonia by altering Clc-1 RNA translation. EBioMedicine 2: 1034–1047, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452: 230–233, 2008. [DOI] [PubMed] [Google Scholar]
- 78.Cleary JD, Ranum LP. Repeat associated non-ATG (RAN) translation: New starts in microsatellite expansion disorders. Curr Opin Genet Dev 26: 6–15, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cohen P The twentieth century struggle to decipher insulin signalling. Nat Rev Mol Cell Biol 7: 867–873, 2006. [DOI] [PubMed] [Google Scholar]
- 80.Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat Rev Drug Discov 14: 58–74, 2015. [DOI] [PubMed] [Google Scholar]
- 81.Coram RJ, Stillwagon SJ, Guggilam A, Jenkins MW, Swanson MS, Ladd AN. Muscleblind-like 1 is required for normal heart valve development in vivo. BMC Dev Biol 15: 36, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Crawford GE, Faulkner JA, Crosbie RH, Campbell KP, Froehner SC, Chamberlain JS. Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain. J Cell Biol 150: 1399–1410, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Crawford Parks TE, Ravel-Chapuis A, Bondy-Chorney E, Renaud JM, Cote J, Jasmin BJ. Muscle-specific expression of the RNA-binding protein Staufen1 induces progressive skeletal muscle atrophy via regulation of phosphatase tensin homolog. Hum Mol Genet 26: 1821–1838, 2017. [DOI] [PubMed] [Google Scholar]
- 84.Dansithong W, Paul S, Comai L, Reddy S. MBNL1 is the primary determinant of focus formation and aberrant insulin receptor splicing in DM1. J Biol Chem 280: 5773–5780, 2005. [DOI] [PubMed] [Google Scholar]
- 85.Davis BM, McCurrach ME, Taneja KL, Singer RH, Housman DE. Expansion of a CUG trinucleotide repeat in the 3’ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci U S A 94: 7388–7393, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Davis J, Salomonis N, Ghearing N, Lin SC, Kwong JQ, Mohan A, Swanson MS, Molkentin JD. MBNL1-mediated regulation of differentiation RNAs promotes myofibroblast transformation and the fibrotic response. Nat Commun 6: 10084, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Davis-Dusenbery BN, Williams LA, Klim JR, Eggan K. How to make spinal motor neurons. Development 141: 491–501, 2014. [DOI] [PubMed] [Google Scholar]
- 88.De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L, Eaton ML, Keenan BT, Ernst J, McCabe C, Tang A, Raj T, Replogle J, Brodeur W, Gabriel S, Chai HS, Younkin C, Younkin SG, Zou F, Szyf M, Epstein CB, Schneider JA, Bernstein BE, Meissner A, Ertekin-Taner N, Chibnik LB, Kellis M, Mill J, Bennett DA. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci 17: 1156–1163, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.de Rezende Pinto WB, de Souza PV, Oliveira AS. Normal muscle structure, growth, development, and regeneration. Curr Rev Musculoskelet Med 8: 176–181, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Decostre V, Vignaud A, Matot B, Huguet A, Ledoux I, Bertil E, Gjata B, Carlier PG, Gourdon G, Hogrel JY. Longitudinal in vivo muscle function analysis of the DMSXL mouse model of myotonic dystrophy type 1. Neuromuscul Disord 23: 1016–1025, 2013. [DOI] [PubMed] [Google Scholar]
- 91.Dellavalle A, Sampaolesi M, Tonlorenzi R, Tagliafico E, Sacchetti B, Perani L, Innocenzi A, Galvez BG, Messina G, Morosetti R, Li S, Belicchi M, Peretti G, Chamberlain JS, Wright WE, Torrente Y, Ferrari S, Bianco P, Cossu G. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat Cell Biol 9: 255–267, 2007. [DOI] [PubMed] [Google Scholar]
- 92.dos Remedios CG, Chhabra D, Kekic M, Dedova IV, Tsubakihara M, Berry DA, Nosworthy NJ. Actin binding proteins: Regulation of cytoskeletal microfilaments. Physiol Rev 83: 433–473, 2003. [DOI] [PubMed] [Google Scholar]
- 93.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346: 1258096, 2014. [DOI] [PubMed] [Google Scholar]
- 94.Dowling JJ, Vreede AP, Kim S, Golden J, Feldman EL. Kindlin-2 is required for myocyte elongation and is essential for myogenesis. BMC Cell Biol 9: 36, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Drachman DB, Fambrough DM. Are muscle fibers denervated in myotonic dystrophy? Arch Neurol 33: 485–488, 1976. [DOI] [PubMed] [Google Scholar]
- 96.Du H, Cline MS, Osborne RJ, Tuttle DL, Clark TA, Donohue JP, Hall MP, Shiue L, Swanson MS, Thornton CA, Ares M Jr. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat Struct Mol Biol 17: 187–193, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Duan R, Gallagher PJ. Dependence of myoblast fusion on a cortical actin wall and nonmuscle myosin IIA. Dev Biol 325: 374–385, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dumont NA, Bentzinger CF, Sincennes MC, Rudnicki MA. Satellite cells and skeletal muscle regeneration. Compr Physiol 5: 1027–1059, 2015. [DOI] [PubMed] [Google Scholar]
- 99.Dumont NA, Wang YX, von Maltzahn J, Pasut A, Bentzinger CF, Brun CE, Rudnicki MA. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat Med 21: 1455–1463, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Echenne B, Rideau A, Roubertie A, Sebire G, Rivier F, Lemieux B. Myotonic dystrophy type I in childhood long-term evolution in patients surviving the neonatal period. Eur J Paediatr Neurol 12: 210–223, 2008. [DOI] [PubMed] [Google Scholar]
- 101.Egerman MA, Glass DJ. Signaling pathways controlling skeletal muscle mass. Crit Rev Biochem Mol Biol 49: 59–68, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ekstrom AB, Hakenas-Plate L, Samuelsson L, Tulinius M, Wentz E. Autism spectrum conditions in myotonic dystrophy type 1: A study on 57 individuals with congenital and childhood forms. Am J Med Genet B Neuropsychiatr Genet 147B: 918–926, 2008. [DOI] [PubMed] [Google Scholar]
- 103.Faenza I, Blalock W, Bavelloni A, Schoser B, Fiume R, Pacella S, Piazzi M, D’Angelo A, Cocco L. A role for PLCbeta1 in myotonic dystrophies type 1 and 2. FASEB J 26: 3042–3048, 2012. [DOI] [PubMed] [Google Scholar]
- 104.Falcone S, Roman W, Hnia K, Gache V, Didier N, Laine J, Aurade F, Marty I, Nishino I, Charlet-Berguerand N, Romero NB, Marazzi G, Sassoon D, Laporte J, Gomes ER. N-WASP is required for amphiphysin-2/BIN1-dependent nuclear positioning and triad organization in skeletal muscle and is involved in the pathophysiology of centronuclear myopathy. EMBO Mol Med 6: 1455–1475, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fardaei M, Larkin K, Brook JD, Hamshere MG. In vivo co-localisation of MBNL protein with DMPK expanded-repeat transcripts. Nucleic Acids Res 29: 2766–2771, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fardaei M, Rogers MT, Thorpe HM, Larkin K, Hamshere MG, Harper PS, Brook JD. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum Mol Genet 11: 805–814, 2002. [DOI] [PubMed] [Google Scholar]
- 107.Farina NH, Hausburg M, Betta ND, Pulliam C, Srivastava D, Cornelison D, Olwin BB. A role for RNA post-transcriptional regulation in satellite cell activation. Skelet Muscle 2: 21, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Farkas-Bargeton E, Barbet JP, Dancea S, Wehrle R, Checouri A, Dulac O. Immaturity of muscle fibers in the congenital form of myotonic dystrophy: Its consequences and its origin. J Neurol Sci 83: 145–159, 1988. [DOI] [PubMed] [Google Scholar]
- 109.Faulkner G, Pallavicini A, Formentin E, Comelli A, Ievolella C, Trevisan S, Bortoletto G, Scannapieco P, Salamon M, Mouly V, Valle G, Lanfranchi G. ZASP: A new Z-band alternatively spliced PDZ-motif protein. J Cell Biol 146: 465–475, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Faustino NA, Cooper TA. Identification of putative new splicing targets for ETR-3 using sequences identified by systematic evolution of ligands by exponential enrichment. Mol Cell Biol 25: 879–887, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fernandez-Costa JM, Garcia-Lopez A, Zuniga S, Fernandez-Pedrosa V, Felipo-Benavent A, Mata M, Jaka O, Aiastui A, Hernandez-Torres F, Aguado B, Perez-Alonso M, Vilchez JJ, Lopez de Munain A, Artero RD. Expanded CTG repeats trigger miRNA alterations in Drosophila that are conserved in myotonic dystrophy type 1 patients. Hum Mol Genet 22: 704–716, 2013. [DOI] [PubMed] [Google Scholar]
- 112.Filippova GN, Thienes CP, Penn BH, Cho DH, Hu YJ, Moore JM, Klesert TR, Lobanenkov VV, Tapscott SJ. CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nat Genet 28: 335–343, 2001. [DOI] [PubMed] [Google Scholar]
- 113.Folker ES, Baylies MK. Nuclear positioning in muscle development and disease. Front Physiol 4: 363, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Folker ES, Schulman VK, Baylies MK. Translocating myonuclei have distinct leading and lagging edges that require kinesin and dynein. Development 141: 355–366, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fournier E, Viala K, Gervais H, Sternberg D, Arzel-Hézode M, Laforêt P, Eymard B, Tabti N, Willer J-C, Vial C, Fontaine B. Cold extends electromyography distinction between ion channel mutations causing myotonia. Ann Neurol 60: 356–365, 2006. [DOI] [PubMed] [Google Scholar]
- 116.Francois V, Klein AF, Beley C, Jollet A, Lemercier C, Garcia L, Furling D. Selective silencing of mutated mRNAs in DM1 by using modified hU7-snRNAs. Nat Struct Mol Biol 18: 85–87, 2011. [DOI] [PubMed] [Google Scholar]
- 117.Friedman JE. Anticipation in hereditary disease: The history of a biomedical concept. Hum Genet 130: 705–714, 2011. [DOI] [PubMed] [Google Scholar]
- 118.Fromaget M, Cook PR. Photobleaching reveals complex effects of inhibitors on transcribing RNA polymerase II in living cells. Exp Cell Res 313: 3026–3033, 2007. [DOI] [PubMed] [Google Scholar]
- 119.Frontera WR, Ochala J. Skeletal muscle: A brief review of structure and function. Calcif Tissue Int 96: 183–195, 2015. [DOI] [PubMed] [Google Scholar]
- 120.Fu YH, Friedman DL, Richards S, Pearlman JA, Gibbs RA, Pizzuti A, Ashizawa T, Perryman MB, Scarlato G, Fenwick RG Jr., et al. Decreased expression of myotonin-protein kinase messenger RNA and protein in adult form of myotonic dystrophy. Science 260: 235–238, 1993. [DOI] [PubMed] [Google Scholar]
- 121.Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, Tosch V, Vignaud A, Ferry A, Messaddeq N, Kokunai Y, Tsuburaya R, de la Grange P, Dembele D, Francois V, Precigout G, Boulade-Ladame C, Hummel MC, Lopez de Munain A, Sergeant N, Laquerriere A, Thibault C, Deryckere F, Auboeuf D, Garcia L, Zimmermann P, Udd B, Schoser B, Takahashi MP, Nishino I, Bassez G, Laporte J, Furling D, Charlet-Berguerand N. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med 17: 720–725, 2011. [DOI] [PubMed] [Google Scholar]
- 122.Furling D, Coiffier L, Mouly V, Barbet JP, St Guily JL, Taneja K, Gourdon G, Junien C, Butler-Browne GS. Defective satellite cells in congenital myotonic dystrophy. Hum Mol Genet 10: 2079–2087, 2001. [DOI] [PubMed] [Google Scholar]
- 123.Furling D, Doucet G, Langlois MA, Timchenko L, Belanger E, Cossette L, Puymirat J. Viral vector producing antisense RNA restores myotonic dystrophy myoblast functions. Gene Ther 10: 795–802, 2003. [DOI] [PubMed] [Google Scholar]
- 124.Furling D, Lemieux D, Taneja K, Puymirat J. Decreased levels of myotonic dystrophy protein kinase (DMPK) and delayed differentiation in human myotonic dystrophy myoblasts. Neuromuscul Disord 11: 728–735, 2001. [DOI] [PubMed] [Google Scholar]
- 125.Gao Y, Guo X, Santostefano K, Wang Y, Reid T, Zeng D, Terada N, Ashizawa T, Xia G. Genome therapy of myotonic dystrophy type 1 iPS cells for development of autologous stem cell therapy. Mol Ther 24: 1378–1387, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gao Z, Cooper TA. Reexpression of pyruvate kinase M2 in type 1 myofibers correlates with altered glucose metabolism in myotonic dystrophy. Proc Natl Acad Sci U S A 110: 13570–13575, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gauthier M, Marteyn A, Denis JA, Cailleret M, Giraud-Triboult K, Aubert S, Lecuyer C, Marie J, Furling D, Vernet R, Yanguas C, Baldeschi C, Pietu G, Peschanski M, Martinat C. A defective Krab-domain zinc-finger transcription factor contributes to altered myogenesis in myotonic dystrophy type 1. Hum Mol Genet 22: 5188–5198, 2013. [DOI] [PubMed] [Google Scholar]
- 128.Ghosh PS, Sorenson EJ. Use of clinical and electrical myotonia to differentiate childhood myopathies. J Child Neurol 30: 1300–1306, 2015. [DOI] [PubMed] [Google Scholar]
- 129.Gillies AR, Lieber RL. Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 44: 318–331, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Giudice J, Xia Z, Li W, Cooper TA. Neonatal cardiac dysfunction and transcriptome changes caused by the absence of Celf1. Sci Rep 6: 35550, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gomes-Pereira M, Cooper TA, Gourdon G. Myotonic dystrophy mouse models: Towards rational therapy development. Trends Mol Med 17: 506–517, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gomes-Pereira M, Foiry L, Nicole A, Huguet A, Junien C, Munnich A, Gourdon G. CTG trinucleotide repeat “big jumps”: Large expansions, small mice. PLoS Genet 3: e52, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gonzalez-Barriga A, Kranzen J, Croes HJ, Bijl S, van den Broek WJ, van Kessel ID, van Engelen BG, van Deutekom JC, Wieringa B, Mulders SA, Wansink DG. Cell membrane integrity in myotonic dystrophy type 1: Implications for therapy. PLoS One 10: e0121556, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Goodwin M, Mohan A, Batra R, Lee KY, Charizanis K, Fernandez Gomez FJ, Eddarkaoui S, Sergeant N, Buee L, Kimura T, Clark HB, Dalton J, Takamura K, Weyn-Vanhentenryck SM, Zhang C, Reid T, Ranum LP, Day JW, Swanson MS. MBNL sequestration by toxic RNAs and RNA misprocessing in the myotonic dystrophy brain. Cell Rep 12: 1159–1168, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Goodwin M, Swanson MS. RNA-binding protein misregulation in microsatellite expansion disorders. Adv Exp Med Biol 825: 353–388, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev 80: 853–924, 2000. [DOI] [PubMed] [Google Scholar]
- 137.Gourdon G, Radvanyi F, Lia AS, Duros C, Blanche M, Abitbol M, Junien C, Hofmann-Radvanyi H. Moderate intergenerational and somatic instability of a 55-CTG repeat in transgenic mice. Nat Genet 15: 190–192, 1997. [DOI] [PubMed] [Google Scholar]
- 138.Gudde AE, Gonzalez-Barriga A, van den Broek WJ, Wieringa B, Wansink DG. A low absolute number of expanded transcripts is involved in myotonic dystrophy type 1 manifestation in muscle. Hum Mol Genet 25: 1648–1662, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Guglielmi V, Vattemi G, Gualandi F, Voermans NC, Marini M, Scotton C, Pegoraro E, Oosterhof A, Kosa M, Zador E, Valente EM, De Grandis D, Neri M, Codemo V, Novelli A, van Kuppevelt TH, Dallapiccola B, van Engelen BG, Ferlini A, Tomelleri G. SERCA1 protein expression in muscle of patients with Brody disease and Brody syndrome and in cultured human muscle fibers. Mol Genet Metab 110: 162–169, 2013. [DOI] [PubMed] [Google Scholar]
- 140.Guiraud-Dogan C, Huguet A, Gomes-Pereira M, Brisson E, Bassez G, Junien C, Gourdon G. DM1 CTG expansions affect insulin receptor isoforms expression in various tissues of transgenic mice. Biochim Biophys Acta 1772: 1183–1191, 2007. [DOI] [PubMed] [Google Scholar]
- 141.Gundersen K Excitation-transcription coupling in skeletal muscle: The molecular pathways of exercise. Biol Rev Camb Philos Soc 86: 564–600, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Guo W, Bharmal SJ, Esbona K, Greaser ML. Titin diversity: Alternative splicing gone wild. J Biomed Biotechnol 2010: 753675, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Guryanova OA, Drazba JA, Frolova EI, Chumakov PM. Actin cytoskeleton remodeling by the alternatively spliced isoform of PDLIM4/RIL protein. J Biol Chem 286: 26849–26859, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hagerman PJ, Hagerman RJ. Fragile X-associated tremor/ataxia syndrome (FXTAS). Ment Retard Dev Disabil Res Rev 10: 25–30, 2004. [DOI] [PubMed] [Google Scholar]
- 145.Haghighat Jahromi A, Honda M, Zimmerman SC, Spies M. Single-molecule study of the CUG repeat-MBNL1 interaction and its inhibition by small molecules. Nucleic Acids Res 41: 6687–6697, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hammaren E, Kjellby-Wendt G, Lindberg C. Muscle force, balance and falls in muscular impaired individuals with myotonic dystrophy type 1: A five-year prospective cohort study. Neuromuscul Disord 25: 141–148, 2015. [DOI] [PubMed] [Google Scholar]
- 147.Hamshere MG, Newman EE, Alwazzan M, Athwal BS, Brook JD. Transcriptional abnormality in myotonic dystrophy affects DMPK but not neighboring genes. Proc Natl Acad Sci U S A 94: 7394–7399, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hamza A, Herr D, Solomayer EF, Meyberg-Solomayer G. Polyhydramnios: Causes, diagnosis and therapy. Geburtshilfe Frauenheilkd 73: 1241–1246, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Han H, Irimia M, Ross PJ, Sung HK, Alipanahi B, David L, Golipour A, Gabut M, Michael IP, Nachman EN, Wang E, Trcka D, Thompson T, O’Hanlon D, Slobodeniuc V, Barbosa-Morais NL, Burge CB, Moffat J, Frey BJ, Nagy A, Ellis J, Wrana JL, Blencowe BJ. MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature 498: 241–245, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Harley HG, Brook JD, Rundle SA, Crow S, Reardon W, Buckler AJ, Harper PS, Housman DE, Shaw DJ. Expansion of an unstable DNA region and phenotypic variation in myotonic dystrophy. Nature 355: 545–546, 1992. [DOI] [PubMed] [Google Scholar]
- 151.Harley HG, Rundle SA, MacMillan JC, Myring J, Brook JD, Crow S, Reardon W, Fenton I, Shaw DJ, Harper PS. Size of the unstable CTG repeat sequence in relation to phenotype and parental transmission in myotonic dystrophy. Am J Hum Genet 52: 1164–1174, 1993. [PMC free article] [PubMed] [Google Scholar]
- 152.Harmon EB, Harmon ML, Larsen TD, Paulson AF, Perryman MB. Myotonic dystrophy protein kinase is expressed in embryonic myocytes and is required for myotube formation. Dev Dyn 237: 2353–2366, 2008. [DOI] [PubMed] [Google Scholar]
- 153.Harmon EB, Harmon ML, Larsen TD, Yang J, Glasford JW, Perryman MB. Myotonic dystrophy protein kinase is critical for nuclear envelope integrity. J Biol Chem 286: 40296–40306, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Harper PS. Major Problems in Neurology: Myotonic Dystrophy (3rd ed.). London: WB Saunders, 2001. [Google Scholar]
- 155.Hasson P “Soft” tissue patterning: Muscles and tendons of the limb take their form. Dev Dyn 240: 1100–1107, 2011. [DOI] [PubMed] [Google Scholar]
- 156.Hausburg MA, Doles JD, Clement SL, Cadwallader AB, Hall MN, Blackshear PJ, Lykke-Andersen J, Olwin BB. Post-transcriptional regulation of satellite cell quiescence by TTP-mediated mRNA decay. Elife 4: e03390, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Heatwole C, Bode R, Johnson NE, Dekdebrun J, Dilek N, Eichinger K, Hilbert JE, Logigian E, Luebbe E, Martens W, McDermott MP, Pandya S, Puwanant A, Rothrock N, Thornton C, Vickrey BG, Victorson D, Moxley RT III. Myotonic dystrophy health index: Correlations with clinical tests and patient function. Muscle Nerve 53: 183–190, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Heatwole C, Johnson N, Goldberg B, Martens W, Moxley R III. Laboratory abnormalities in patients with myotonic dystrophy type 2. Arch Neurol 68: 1180–1184, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Heatwole CR, Miller J, Martens B, Moxley RT III. Laboratory abnormalities in ambulatory patients with myotonic dystrophy type 1. Arch Neurol 63: 1149–1153, 2006. [DOI] [PubMed] [Google Scholar]
- 160.Hehir MK, Logigian EL. Electrodiagnosis of myotonic disorders. Phys Med Rehabil Clin N Am 24: 209–220, 2013. [DOI] [PubMed] [Google Scholar]
- 161.Hernandez-Hernandez O, Guiraud-Dogan C, Sicot G, Huguet A, Luilier S, Steidl E, Saenger S, Marciniak E, Obriot H, Chevarin C, Nicole A, Revillod L, Charizanis K, Lee KY, Suzuki Y, Kimura T, Matsuura T, Cisneros B, Swanson MS, Trovero F, Buisson B, Bizot JC, Hamon M, Humez S, Bassez G, Metzger F, Buee L, Munnich A, Sergeant N, Gourdon G, Gomes-Pereira M. Myotonic dystrophy CTG expansion affects synaptic vesicle proteins, neurotransmission and mouse behaviour. Brain 136: 957–970, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ho G, Cardamone M, Farrar M. Congenital and childhood myotonic dystrophy: Current aspects of disease and future directions. World J Clin Pediatr 4: 66–80, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ho TH, Bundman D, Armstrong DL, Cooper TA. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum Mol Genet 14: 1539–1547, 2005. [DOI] [PubMed] [Google Scholar]
- 164.Ho TH, Charlet BN, Poulos MG, Singh G, Swanson MS, Cooper TA. Muscleblind proteins regulate alternative splicing. EMBO J 23: 3103–3112, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ho TH, Savkur RS, Poulos MG, Mancini MA, Swanson MS, Cooper TA. Colocalization of muscleblind with RNA foci is separable from mis-regulation of alternative splicing in myotonic dystrophy. J Cell Sci 118: 2923–2933, 2005. [DOI] [PubMed] [Google Scholar]
- 166.Holt I, Jacquemin V, Fardaei M, Sewry CA, Butler-Browne GS, Furling D, Brook JD, Morris GE. Muscleblind-like proteins: Similarities and differences in normal and myotonic dystrophy muscle. Am J Pathol 174: 216–227, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hoskins JW, Ofori LO, Chen CZ, Kumar A, Sobczak K, Nakamori M, Southall N, Patnaik S, Marugan JJ, Zheng W, Austin CP, Disney MD, Miller BL, Thornton CA. Lomofungin and dilomofungin: Inhibitors of MBNL1-CUG RNA binding with distinct cellular effects. Nucleic Acids Res 42: 6591–6602, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Howard J, Hyman AA. Dynamics and mechanics of the microtubule plus end. Nature 422: 753–758, 2003. [DOI] [PubMed] [Google Scholar]
- 169.Hua Y, Vickers TA, Baker BF, Bennett CF, Krainer AR. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol 5: e73, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hubaud A, Pourquie O. Signalling dynamics in vertebrate segmentation. Nat Rev Mol Cell Biol 15: 709–721, 2014. [DOI] [PubMed] [Google Scholar]
- 171.Hughes BN, Hogue JS, Hsieh DT. Grip and percussion myotonia in myotonic dystrophy type 1. J Pediatr 164: 1234–1234 e1231, 2014. [DOI] [PubMed] [Google Scholar]
- 172.Huichalaf C, Schoser B, Schneider-Gold C, Jin B, Sarkar P, Timchenko L. Reduction of the rate of protein translation in patients with myotonic dystrophy 2. J Neurosci 29: 9042–9049, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hwang PM, Sykes BD. Targeting the sarcomere to correct muscle function. Nat Rev Drug Discov 14: 313–328, 2015. [DOI] [PubMed] [Google Scholar]
- 174.Iannaccone ST, Bove KE, Vogler C, Azzarelli B, Muller J. Muscle maturation delay in infantile myotonic dystrophy. Arch Pathol Lab Med 110: 405–411, 1986. [PubMed] [Google Scholar]
- 175.Iannaccone ST, Castro D. Congenital muscular dystrophies and congenital myopathies. Continuum (Minneap Minn) 19: 1509–1534, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Itoh T, Erdmann KS, Roux A, Habermann B, Werner H, De Camilli P. Dynamin and the actin cytoskeleton cooperatively regulate plasma membrane invagination by BAR and F-BAR proteins. Dev Cell 9: 791–804, 2005. [DOI] [PubMed] [Google Scholar]
- 177.Jackson HE, Ingham PW. Control of muscle fibre-type diversity during embryonic development: The zebrafish paradigm. Mech Dev 130: 447–457, 2013. [DOI] [PubMed] [Google Scholar]
- 178.Jain A, Vale RD. RNA phase transitions in repeat expansion disorders. Nature 546: 243–247, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Jansen G, Groenen PJ, Bachner D, Jap PH, Coerwinkel M, Oerlemans F, van den Broek W, Gohlsch B, Pette D, Plomp JJ, Molenaar PC, Nederhoff MG, van Echteld CJ, Dekker M, Berns A, Hameister H, Wieringa B. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat Genet 13: 316–324, 1996. [DOI] [PubMed] [Google Scholar]
- 180.Jones K, Wei C, Iakova P, Bugiardini E, Schneider-Gold C, Meola G, Woodgett J, Killian J, Timchenko NA, Timchenko LT. GSK3beta mediates muscle pathology in myotonic dystrophy. J Clin Invest 122: 4461–4472, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Jones K, Wei C, Schoser B, Meola G, Timchenko N, Timchenko L. Reduction of toxic RNAs in myotonic dystrophies type 1 and type 2 by the RNA helicase p68/DDX5. Proc Natl Acad Sci U S A 112: 8041–8045, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Jungbluth H, Gautel M. Pathogenic mechanisms in centronuclear myopathies. Front Aging Neurosci 6: 339, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Jungbluth H, Voermans NC. Congenital myopathies: Not only a paediatric topic. Curr Opin Neurol 29: 642–650, 2016. [DOI] [PubMed] [Google Scholar]
- 184.Kahn CR, White MF. The insulin receptor and the molecular mechanism of insulin action. J Clin Invest 82: 1151–1156, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kaliman P, Catalucci D, Lam JT, Kondo R, Gutierrez JC, Reddy S, Palacin M, Zorzano A, Chien KR, Ruiz-Lozano P. Myotonic dystrophy protein kinase phosphorylates phospholamban and regulates calcium uptake in cardiomyocyte sarcoplasmic reticulum. J Biol Chem 280: 8016–8021, 2005. [DOI] [PubMed] [Google Scholar]
- 186.Kalsotra A, Cooper TA. Functional consequences of developmentally regulated alternative splicing. Nat Rev Genet 12: 715–729, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kalsotra A, Singh RK, Gurha P, Ward AJ, Creighton CJ, Cooper TA. The Mef2 transcription network is disrupted in myotonic dystrophy heart tissue, dramatically altering miRNA and mRNA expression. Cell Rep 6: 336–345, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, Cooper TA. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc Natl Acad Sci USA 105: 20333–20338, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS. A muscleblind knockout model for myotonic dystrophy. Science 302: 1978–1980, 2003. [DOI] [PubMed] [Google Scholar]
- 190.Kanadia RN, Shin J, Yuan Y, Beattie SG, Wheeler TM, Thornton CA, Swanson MS. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc Natl Acad Sci U S A 103: 11748–11753, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kanadia RN, Urbinati CR, Crusselle VJ, Luo D, Lee YJ, Harrison JK, Oh SP, Swanson MS. Developmental expression of mouse muscleblind genes Mbnl1, Mbnl2 and Mbnl3. Gene Expr Patterns 3: 459–462, 2003. [DOI] [PubMed] [Google Scholar]
- 192.Kanning KC, Kaplan A, Henderson CE. Motor neuron diversity in development and disease. Annu Rev Neurosci 33: 409–440, 2010. [DOI] [PubMed] [Google Scholar]
- 193.Karpati G, Carpenter S, Watters GV, Eisen AA, Andermann F. Infantile myotonic dystrophy. Histochemical and electron microscopic features in skeletal muscle. Neurology 23: 1066–1077, 1973. [DOI] [PubMed] [Google Scholar]
- 194.Keefe AC, Lawson JA, Flygare SD, Fox ZD, Colasanto MP, Mathew SJ, Yandell M, Kardon G. Muscle stem cells contribute to myofibres in sedentary adult mice. Nat Commun 6: 7087, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ketley A, Chen CZ, Li X, Arya S, Robinson TE, Granados-Riveron J, Udosen I, Morris GE, Holt I, Furling D, Chaouch S, Haworth B, Southall N, Shinn P, Zheng W, Austin CP, Hayes CJ, Brook JD. High-content screening identifies small molecules that remove nuclear foci, affect MBNL distribution and CELF1 protein levels via a PKC-independent pathway in myotonic dystrophy cell lines. Hum Mol Genet 23: 1551–1562, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kierkegaard M, Harms-Ringdahl K, Edstrom L, Widen Holmqvist L, Tollback A. Feasibility and effects of a physical exercise programme in adults with myotonic dystrophy type 1: A randomized controlled pilot study. J Rehabil Med 43: 695–702, 2011. [DOI] [PubMed] [Google Scholar]
- 197.Kindler JM, Lewis RD, Hamrick MW. Skeletal muscle and pediatric bone development. Curr Opin Endocrinol Diabetes Obes 22: 467–474, 2015. [DOI] [PubMed] [Google Scholar]
- 198.Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529: 490–495, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Klesert TR, Cho DH, Clark JI, Maylie J, Adelman J, Snider L, Yuen EC, Soriano P, Tapscott SJ. Mice deficient in Six5 develop cataracts: Implications for myotonic dystrophy. Nat Genet 25: 105–109, 2000. [DOI] [PubMed] [Google Scholar]
- 200.Klesert TR, Otten AD, Bird TD, Tapscott SJ. Trinucleotide repeat expansion at the myotonic dystrophy locus reduces expression of DMAHP. Nat Genet 16: 402–406, 1997. [DOI] [PubMed] [Google Scholar]
- 201.Koebis M, Kiyatake T, Yamaura H, Nagano K, Higashihara M, Sonoo M, Hayashi Y, Negishi Y, Endo-Takahashi Y, Yanagihara D, Matsuda R, Takahashi MP, Nishino I, Ishiura S. Ultrasound-enhanced delivery of morpholino with bubble liposomes ameliorates the myotonia of myotonic dystrophy model mice. Sci Rep 3: 2242, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Koebis M, Ohsawa N, Kino Y, Sasagawa N, Nishino I, Ishiura S. Alternative splicing of myomesin 1 gene is aberrantly regulated in myotonic dystrophy type 1. Genes Cells 16: 961–972, 2011. [DOI] [PubMed] [Google Scholar]
- 203.Konieczny P, Stepniak-Konieczna E, Sobczak K. MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Res 42: 10873–10887, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Kontrogianni-Konstantopoulos A, Ackermann MA, Bowman AL, Yap SV, Bloch RJ. Muscle giants: Molecular scaffolds in sarcomerogenesis. Physiol Rev 89: 1217–1267, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Koscianska E, Witkos TM, Kozlowska E, Wojciechowska M, Krzyzosiak WJ. Cooperation meets competition in microRNA-mediated DMPK transcript regulation. Nucleic Acids Res 43: 9500–9518, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Koshy BT, Zoghbi HY. The CAG/polyglutamine tract diseases: Gene products and molecular pathogenesis. Brain Pathol 7: 927–942, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Krcmery J, Gupta R, Sadleir RW, Ahrens MJ, Misener S, Kamide C, Fitchev P, Losordo DW, Crawford SE, Simon HG. Loss of the cytoskeletal protein Pdlim7 predisposes mice to heart defects and hemostatic dysfunction. PLoS One 8: e80809, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Krol J, Fiszer A, Mykowska A, Sobczak K, de Mezer M, Krzyzosiak WJ. Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol Cell 25: 575–586, 2007. [DOI] [PubMed] [Google Scholar]
- 209.Kuo JC. Mechanotransduction at focal adhesions: Integrating cytoskeletal mechanics in migrating cells. J Cell Mol Med 17: 704–712, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kuyumcu-Martinez NM, Cooper TA. Misregulation of alternative splicing causes pathogenesis in myotonic dystrophy. Prog Mol Subcell Biol 44: 133–159, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state in levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell 28: 68–78, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.La Spada AR, Paulson HL, Fischbeck KH. Trinucleotide repeat expansion in neurological disease. Ann Neurol 36: 814–822, 1994. [DOI] [PubMed] [Google Scholar]
- 213.Laberge L, Begin P, Montplaisir J, Mathieu J. Sleep complaints in patients with myotonic dystrophy. J Sleep Res 13: 95–100, 2004. [DOI] [PubMed] [Google Scholar]
- 214.Laberge L, Gagnon C, Dauvilliers Y. Daytime sleepiness and myotonic dystrophy. Curr Neurol Neurosci Rep 13: 340, 2013. [DOI] [PubMed] [Google Scholar]
- 215.Lam LT, Pham YCN, Man NT, Morris GE. Characterization of a monoclonal antibody panel shows that the myotonic dystrophy protein kinase, DMPK, is expressed almost exclusively in muscle and heart. Hum Mol Genet 9: 2167–2173, 2000. [DOI] [PubMed] [Google Scholar]
- 216.Lamb GD. Excitation-contraction coupling in skeletal muscle: Comparisons with cardiac muscle. Clin Exp Pharmacol Physiol 27: 216–224, 2000. [DOI] [PubMed] [Google Scholar]
- 217.Langlois MA, Boniface C, Wang G, Alluin J, Salvaterra PM, Puymirat J, Rossi JJ, Lee NS. Cytoplasmic and nuclear retained DMPK mRNAs are targets for RNA interference in myotonic dystrophy cells. J Biol Chem 280: 16949–16954, 2005. [DOI] [PubMed] [Google Scholar]
- 218.Laporte J, Biancalana V, Tanner SM, Kress W, Schneider V, Wallgren-Pettersson C, Herger F, Buj-Bello A, Blondeau F, Liechti-Gallati S, Mandel JL. MTM1 mutations in X-linked myotubular myopathy. Hum Mutat 15: 393–409, 2000. [DOI] [PubMed] [Google Scholar]
- 219.Lee E, Marcucci M, Daniell L, Pypaert M, Weisz OA, Ochoa GC, Farsad K, Wenk MR, De Camilli P. Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science 297: 1193–1196, 2002. [DOI] [PubMed] [Google Scholar]
- 220.Lee JE, Bennett CF, Cooper TA. RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc Natl Acad Sci U S A 109: 4221–4226, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Lee JE, Cooper TA. Pathogenic mechanisms of myotonic dystrophy. Biochem Soc Trans 37: 1281–1286, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Lee K-SS, Smith K, Amieux PS, Wang EH. MBNL3/CHCR prevents myogenic differentiation by inhibiting MyoD-dependent gene transcription. Differentiation 76: 299–309, 2008. [DOI] [PubMed] [Google Scholar]
- 223.Lee K-SS, Squillace RM, Wang EH. Expression pattern of muscleblind-like proteins differs in differentiating myoblasts. Biochem Biophys Res Commun 361: 151–155, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Lee KS, Cao Y, Witwicka HE, Tom S, Tapscott SJ, Wang EH. RNA-binding protein muscleblind-like 3 (MBNL3) disrupts myocyte enhancer factor 2 (Mef2) {beta}-exon splicing. J Biol Chem 285: 33779–33787, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Lee KS, Smith K, Amieux PS, Wang EH. MBNL3/CHCR prevents myogenic differentiation by inhibiting MyoD-dependent gene transcription. Differentiation 76: 299–309, 2008. [DOI] [PubMed] [Google Scholar]
- 226.Lee KS, Squillace RM, Wang EH. Expression pattern of muscleblind-like proteins differs in differentiating myoblasts. Biochem Biophys Res Commun 361: 151–155, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Lee KY, Li M, Manchanda M, Batra R, Charizanis K, Mohan A, Warren SA, Chamberlain CM, Finn D, Hong H, Ashraf H, Kasahara H, Ranum LP, Swanson MS. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol Med 5: 1887–1900, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Lee MM, Pushechnikov A, Disney MD. Rational and modular design of potent ligands targeting the RNA that causes myotonic dystrophy 2. ACS Chem Biol 4: 345–355, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Leger AJ, Mosquea LM, Clayton NP, Wu IH, Weeden T, Nelson CA, Phillips L, Roberts E, Piepenhagen PA, Cheng SH, Wentworth BM. Systemic delivery of a peptide-linked morpholino oligonucleotide neutralizes mutant RNA toxicity in a mouse model of myotonic dystrophy. Nucleic Acid Ther 23: 109–117, 2013. [DOI] [PubMed] [Google Scholar]
- 230.Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, Thornton CA. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet 15: 2087–2097, 2006. [DOI] [PubMed] [Google Scholar]
- 231.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293: 864–867, 2001. [DOI] [PubMed] [Google Scholar]
- 232.Llorian M, Smith CW. Decoding muscle alternative splicing. Curr Opin Genet Dev 21: 380–387, 2011. [DOI] [PubMed] [Google Scholar]
- 233.Logigian EL, Ciafaloni E, Quinn LC, Dilek N, Pandya S, Moxley RT III, Thornton CA. Severity, type, and distribution of myotonic discharges are different in type 1 and type 2 myotonic dystrophy. Muscle Nerve 35: 479–485, 2007. [DOI] [PubMed] [Google Scholar]
- 234.Logigian EL, Martens WB, Moxley RTt, McDermott MP, Dilek N, Wiegner AW, Pearson AT, Barbieri CA, Annis CL, Thornton CA, Moxley RT III. Mexiletine is an effective antimyotonia treatment in myotonic dystrophy type 1. Neurology 74: 1441–1448, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Lopez Castel A, Nakamori M, Tome S, Chitayat D, Gourdon G, Thornton CA, Pearson CE. Expanded CTG repeat demarcates a boundary for abnormal CpG methylation in myotonic dystrophy patient tissues. Hum Mol Genet 20: 1–15, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Loro E, Rinaldi F, Malena A, Masiero E, Novelli G, Angelini C, Romeo V, Sandri M, Botta A, Vergani L. Normal myogenesis and increased apoptosis in myotonic dystrophy type-1 muscle cells. Cell Death Differ 17: 1315–1324, 2010. [DOI] [PubMed] [Google Scholar]
- 237.Lukjanenko L, Jung MJ, Hegde N, Perruisseau-Carrier C, Migliavacca E, Rozo M, Karaz S, Jacot G, Schmidt M, Li L, Metairon S, Raymond F, Lee U, Sizzano F, Wilson DH, Dumont NA, Palini A, Fassler R, Steiner P, Descombes P, Rudnicki MA, Fan CM, von Maltzahn J, Feige JN, Bentzinger CF. Loss of fibronectin from the aged stem cell niche affects the regenerative capacity of skeletal muscle in mice. Nat Med 22: 897–905, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Machuca-Tzili L, Brook D, Hilton-Jones D. Clinical and molecular aspects of the myotonic dystrophies: A review. Muscle Nerve 32: 1–18, 2005. [DOI] [PubMed] [Google Scholar]
- 239.Machuca-Tzili LE, Buxton S, Thorpe A, Timson CM, Wigmore P, Luther PK, Brook JD. Zebrafish deficient for muscleblind-like 2 exhibit features of myotonic dystrophy. Dis Model Mech 4: 381–392, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Maeda M, Taft CS, Bush EW, Holder E, Bailey WM, Neville H, Perryman MB, Bies RD. Identification, tissue-specific expression, and subcellular localization of the 80- and 71-kDa forms of myotonic dystrophy kinase protein. J Biol Chem 270: 20246–20249, 1995. [DOI] [PubMed] [Google Scholar]
- 241.Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J, O’Hoy K, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3’ untranslated region of the gene. Science 255: 1253–1255, 1992. [DOI] [PubMed] [Google Scholar]
- 242.Malatesta M Skeletal muscle features in myotonic dystrophy and sarcopenia: Do similar nuclear mechanisms lead to skeletal muscle wasting? Eur J Histochem 56: e36, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Malatesta M, Cardani R, Pellicciari C, Meola G. RNA transcription and maturation in skeletal muscle cells are similarly impaired in myotonic dystrophy and sarcopenia: The ultrastructural evidence. Front Aging Neurosci 6: 196, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Malatesta M, Giagnacovo M, Cardani R, Meola G, Pellicciari C. Human myoblasts from skeletal muscle biopsies: In vitro culture preparations for morphological and cytochemical analyses at light and electron microscopy. Methods Mol Biol 976: 67–79, 2013. [DOI] [PubMed] [Google Scholar]
- 245.Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton CA. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 289: 1769–1773, 2000. [DOI] [PubMed] [Google Scholar]
- 246.Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT, Cannon SC, Thornton CA. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell 10: 35–44, 2002. [DOI] [PubMed] [Google Scholar]
- 247.Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, Krym M, Henderson D, Schalling M, Swanson MS, Thornton CA. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet 10: 2165–2170, 2001. [DOI] [PubMed] [Google Scholar]
- 248.Margarit E, Armas P, Garcia Siburu N, Calcaterra NB. CNBP modulates the transcription of Wnt signaling pathway components. Biochim Biophys Acta 1839: 1151–1160, 2014. [DOI] [PubMed] [Google Scholar]
- 249.Margolis JM, Schoser BG, Moseley ML, Day JW, Ranum LP. DM2 intronic expansions: Evidence for CCUG accumulation without flanking sequence or effects on ZNF9 mRNA processing or protein expression. Hum Mol Genet 15: 1808–1815, 2006. [DOI] [PubMed] [Google Scholar]
- 250.Marteyn A, Maury Y, Gauthier MM, Lecuyer C, Vernet R, Denis JA, Pietu G, Peschanski M, Martinat C. Mutant human embryonic stem cells reveal neurite and synapse formation defects in type 1 myotonic dystrophy. Cell Stem Cell 8: 434–444, 2011. [DOI] [PubMed] [Google Scholar]
- 251.Martorell L, Cobo AM, Baiget M, Naudo M, Poza JJ, Parra J. Prenatal diagnosis in myotonic dystrophy type 1. Thirteen years of experience: Implications for reproductive counselling in DM1 families. Prenat Diagn 27: 68–72, 2007. [DOI] [PubMed] [Google Scholar]
- 252.Masuda A, Andersen HS, Doktor TK, Okamoto T, Ito M, Andresen BS, Ohno K. CUGBP1 and MBNL1 preferentially bind to 3’ UTRs and facilitate mRNA decay. Sci Rep 2: 209, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Mateos-Aierdi AJ, Goicoechea M, Aiastui A, Fernandez-Torron R, Garcia-Puga M, Matheu A, Lopez de Munain A. Muscle wasting in myotonic dystrophies: A model of premature aging. Front Aging Neurosci 7: 125, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Mathieu J, Prevost C. Epidemiological surveillance of myotonic dystrophy type 1: A 25-year population-based study. Neuromuscul Disord 22: 974–979, 2012. [DOI] [PubMed] [Google Scholar]
- 255.McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet 11: 786–799, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Meola G Clinical aspects, molecular pathomechanisms and management of myotonic dystrophies. Acta Myol 32: 154–165, 2013. [PMC free article] [PubMed] [Google Scholar]
- 257.Meola G, Cardani R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim Biophys Acta 1852: 594–606, 2015. [DOI] [PubMed] [Google Scholar]
- 258.Mercuri E, Muntoni F. Muscular dystrophies. Lancet 381: 845–860, 2013. [DOI] [PubMed] [Google Scholar]
- 259.Messina G, Biressi S, Monteverde S, Magli A, Cassano M, Perani L, Roncaglia E, Tagliafico E, Starnes L, Campbell CE, Grossi M, Goldhamer DJ, Gronostajski RM, Cossu G. Nfix regulates fetal-specific transcription in developing skeletal muscle. Cell 140: 554–566, 2010. [DOI] [PubMed] [Google Scholar]
- 260.Metzger T, Gache V, Xu M, Cadot B, Folker ES, Richardson BE, Gomes ER, Baylies MK. MAP and kinesin-dependent nuclear positioning is required for skeletal muscle function. Nature 484: 120–124, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Michalowski S, Miller JW, Urbinati CR, Paliouras M, Swanson MS, Griffith J. Visualization of double-stranded RNAs from the myotonic dystrophy protein kinase gene and interactions with CUG-binding protein. Nucleic Acids Res 27: 3534–3542, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Michel L, Huguet-Lachon A, Gourdon G. Sense and antisense DMPK RNA foci accumulate in DM1 tissues during development. PLoS One 10: e0137620, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J 19: 4439–4448, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: In command and control of cell motility. Nat Rev Mol Cell Biol 6: 56–68, 2005. [DOI] [PubMed] [Google Scholar]
- 265.Monckton DG, Coolbaugh MI, Ashizawa KT, Siciliano MJ, Caskey CT. Hypermutable myotonic dystrophy CTG repeats in transgenic mice. Nat Genet 15: 193–196, 1997. [DOI] [PubMed] [Google Scholar]
- 266.Monckton DG, Wong LJ, Ashizawa T, Caskey CT. Somatic mosaicism, germline expansions, germline reversions and intergenerational reductions in myotonic dystrophy males: Small pool PCR analyses. Hum Mol Genet 4: 1–8, 1995. [DOI] [PubMed] [Google Scholar]
- 267.Morrison SJ, Spradling AC. Stem cells and niches: Mechanisms that promote stem cell maintenance throughout life. Cell 132: 598–611, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Moxley RT, Corbett AJ, Minaker KL, Rowe JW. Whole body insulin resistance in myotonic dystrophy. Ann Neurol 15: 157–162, 1984. [DOI] [PubMed] [Google Scholar]
- 269.Mukherjee K, Ishii K, Pillalamarri V, Kammin T, Atkin JF, Hickey SE, Xi QJ, Zepeda CJ, Gusella JF, Talkowski ME, Morton CC, Maas RL, Liao EC. Actin capping protein CAPZB regulates cell morphology, differentiation, and neural crest migration in craniofacial morphogenesisdagger. Hum Mol Genet 25: 1255–1270, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Nadaj-Pakleza A, Lusakowska A, Sulek-Piatkowska A, Krysa W, Rajkiewicz M, Kwiecinski H, Kaminska A. Muscle pathology in myotonic dystrophy: Light and electron microscopic investigation in eighteen patients. Folia Morphol (Warsz) 70: 121–129, 2011. [PubMed] [Google Scholar]
- 271.Nakamori M, Kimura T, Fujimura H, Takahashi MP, Sakoda S. Altered mRNA splicing of dystrophin in type 1 myotonic dystrophy. Muscle Nerve 36: 251–257, 2007. [DOI] [PubMed] [Google Scholar]
- 272.Nakamori M, Sobczak K, Puwanant A, Welle S, Eichinger K, Pandya S, Dekdebrun J, Heatwole CR, McDermott MP, Chen T, Cline M, Tawil R, Osborne RJ, Wheeler TM, Swanson MS, Moxley RT III, Thornton CA. Splicing biomarkers of disease severity in myotonic dystrophy. Ann Neurol 74: 862–872, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Nance JR, Dowling JJ, Gibbs EM, Bonnemann CG . Congenital myopathies: An update. Curr Neurol Neurosci Rep 12: 165–174, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Narang Monica A, Waring James D, Sabourin Luc A, Rajcan-Separovic E, Parry D, Jirik F, Korneluk Robert G. Skeletal myopathy in mice over-expressing the human myotonic dystrophy protein kinase (DMPK) gene. Gene Funct Dis 1: 134–144, 2000. [Google Scholar]
- 275.Niblock M, Smith BN, Lee YB, Sardone V, Topp S, Troakes C, Al-Sarraj S, Leblond CS, Dion PA, Rouleau GA, Shaw CE, Gallo JM. Retention of hexanucleotide repeat-containing intron in C9orf72 mRNA: Implications for the pathogenesis of ALS/FTD. Acta Neuropathol Commun 4: 18, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Nicot AS, Toussaint A, Tosch V, Kretz C, Wallgren-Pettersson C, Iwarsson E, Kingston H, Garnier JM, Biancalana V, Oldfors A, Mandel JL, Laporte J. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat Genet 39: 1134–1139, 2007. [DOI] [PubMed] [Google Scholar]
- 277.Nie M, Deng ZL, Liu J, Wang DZ. Noncoding RNAs, emerging regulators of skeletal muscle development and diseases. Biomed Res Int 2015: 676575, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Nishimura T, Nakamura K, Kishioka Y, Kato-Mori Y, Wakamatsu J, Hattori A. Inhibition of matrix metalloproteinases suppresses the migration of skeletal muscle cells. J Muscle Res Cell Motil 29: 37–44, 2008. [DOI] [PubMed] [Google Scholar]
- 279.Nitz JC, Burns YR, Jackson RV. A longitudinal physical profile assessment of skeletal muscle manifestations in myotonic dystrophy. Clin Rehabil 13: 64–73, 1999. [DOI] [PubMed] [Google Scholar]
- 280.Odermatt A, Taschner PE, Khanna VK, Busch HF, Karpati G, Jablecki CK, Breuning MH, MacLennan DH. Mutations in the gene-encoding SERCA1, the fast-twitch skeletal muscle sarcoplasmic reticulum Ca2+ ATPase, are associated with Brody disease. Nat Genet 14: 191–194, 1996. [DOI] [PubMed] [Google Scholar]
- 281.Ohsawa N, Koebis M, Mitsuhashi H, Nishino I, Ishiura S. ABLIM1 splicing is abnormal in skeletal muscle of patients with DM1 and regulated by MBNL, CELF and PTBP1. Genes Cells 20: 121–134, 2015. [DOI] [PubMed] [Google Scholar]
- 282.Ohtsuki I, Morimoto S. Troponin: Regulatory function and disorders. Biochem Biophys Res Commun 369: 62–73, 2008. [DOI] [PubMed] [Google Scholar]
- 283.Ono Y, Urata Y, Goto S, Nakagawa S, Humbert PO, Li TS, Zammit PS. Muscle stem cell fate is controlled by the cell-polarity protein Scrib. Cell Rep 10: 1135–1148, 2015. [DOI] [PubMed] [Google Scholar]
- 284.Orengo JP, Chambon P, Metzger D, Mosier DR, Snipes GJ, Cooper TA. Expanded CTG repeats within the DMPK 3’ UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. Proc Natl Acad Sci U S A 105: 2646–2651, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Orengo JP, Ward AJ, Cooper TA. Alternative splicing dysregulation secondary to skeletal muscle regeneration. Ann Neurol 69: 681–690, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Osborne RJ, Thornton CA. Cell-free cloning of highly expanded CTG repeats by amplification of dimerized expanded repeats. Nucleic Acids Res 36: e24, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Otten AD, Tapscott SJ. Triplet repeat expansion in myotonic dystrophy alters the adjacent chromatin structure. Proc Natl Acad Sci U S A 92: 5465–5469, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Ottenheijm CA, Knottnerus AM, Buck D, Luo X, Greer K, Hoying A, Labeit S, Granzier H. Tuning passive mechanics through differential splicing of titin during skeletal muscle development. Biophys J 97: 2277–2286, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Panaite PA, Gantelet E, Kraftsik R, Gourdon G, Kuntzer T, Barakat-Walter I. Myotonic dystrophy transgenic mice exhibit pathologic abnormalities in diaphragm neuromuscular junctions and phrenic nerves. J Neuropathol Exp Neurol 67: 763–772, 2008. [DOI] [PubMed] [Google Scholar]
- 290.Panaite PA, Kuntzer T, Gourdon G, Barakat-Walter I. Respiratory failure in a mouse model of myotonic dystrophy does not correlate with the CTG repeat length. Respir Physiol Neurobiol 189: 22–26, 2013. [DOI] [PubMed] [Google Scholar]
- 291.Panaite PA, Kuntzer T, Gourdon G, Lobrinus JA, Barakat-Walter I. Functional and histopathological identification of the respiratory failure in a DMSXL transgenic mouse model of myotonic dystrophy. Dis Model Mech 6: 622–631, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Pantic B, Trevisan E, Citta A, Rigobello MP, Marin O, Bernardi P, Salvatori S, Rasola A. Myotonic dystrophy protein kinase (DMPK) prevents ROS-induced cell death by assembling a hexokinase II-Src complex on the mitochondrial surface. Cell Death Dis 4: e858, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Parkesh R, Childs-Disney JL, Nakamori M, Kumar A, Wang E, Wang T, Hoskins J, Tran T, Housman D, Thornton CA, Disney MD. Design of a bioactive small molecule that targets the myotonic dystrophy type 1 RNA via an RNA motif-ligand database and chemical similarity searching. J Am Chem Soc 134: 4731–4742, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Pascual M, Vicente M, Monferrer L, Artero R. The Muscleblind family of proteins: An emerging class of regulators of developmentally programmed alternative splicing. Differentiation 74: 65–80, 2006. [DOI] [PubMed] [Google Scholar]
- 295.Paternostro-Sluga T, Grim-Stieger M, Posch M, Schuhfried O, Vacariu G, Mittermaier C, Bittner C, Fialka-Moser V. Reliability and validity of the Medical Research Council (MRC) scale and a modified scale for testing muscle strength in patients with radial palsy. J Rehabil Med 40: 665–671, 2008. [DOI] [PubMed] [Google Scholar]
- 296.Paul S, Dansithong W, Kim D, Rossi J, Webster NJ, Comai L, Reddy S. Interaction of muscleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. EMBO J 25: 4271–4283, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Pedrotti S, Giudice J, Dagnino-Acosta A, Knoblauch M, Singh RK, Hanna A, Mo Q, Hicks J, Hamilton S, Cooper TA. The RNA-binding protein Rbfox1 regulates splicing required for skeletal muscle structure and function. Hum Mol Genet 24: 2360–2374, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Pelletier R, Hamel F, Beaulieu D, Patry L, Haineault C, Tarnopolsky M, Schoser B, Puymirat J. Absence of a differentiation defect in muscle satellite cells from DM2 patients. Neurobiol Dis 36: 181–190, 2009. [DOI] [PubMed] [Google Scholar]
- 299.Penisson-Besnier I, Devillers M, Porcher R, Orlikowski D, Doppler V, Desnuelle C, Ferrer X, Bes MC, Bouhour F, Tranchant C, Lagrange E, Vershueren A, Uzenot D, Cintas P, Sole G, Hogrel JY, Laforet P, Vial C, Vila AL, Sacconi S, Pouget J, Eymard B, Chevret S, Annane D. Dehydroepiandrosterone for myotonic dystrophy type 1. Neurology 71: 407–412, 2008. [DOI] [PubMed] [Google Scholar]
- 300.Perdoni F, Malatesta M, Cardani R, Giagnacovo M, Mancinelli E, Meola G, Pellicciari C. RNA/MBNL1-containing foci in myoblast nuclei from patients affected by myotonic dystrophy type 2: An immunocytochemical study. Eur J Histochem 53: 151–158, 2009. [DOI] [PubMed] [Google Scholar]
- 301.Peredo DE, Hannibal MC. The floppy infant: Evaluation of hypotonia. Pediatr Rev 30: e66–e76, 2009. [DOI] [PubMed] [Google Scholar]
- 302.Perfetti A, Greco S, Fasanaro P, Bugiardini E, Cardani R, Garcia-Manteiga JM, Riba M, Cittaro D, Stupka E, Meola G, Martelli F. Genome wide identification of aberrant alternative splicing events in myotonic dystrophy type 2. PLoS One 9: e93983, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Periasamy M, Kalyanasundaram A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 35: 430–442, 2007. [DOI] [PubMed] [Google Scholar]
- 304.Pette D, Staron RS. Myosin isoforms, muscle fiber types, and transitions. Microsc Res Tech 50: 500–509, 2000. [DOI] [PubMed] [Google Scholar]
- 305.Pettersson OJ, Aagaard L, Andrejeva D, Thomsen R, Jensen TG, Damgaard CK. DDX6 regulates sequestered nuclear CUG-expanded DMPK-mRNA in dystrophia myotonica type 1. Nucleic Acids Res 42: 7186–7200, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Pettersson OJ, Aagaard L, Jensen TG, Damgaard CK. Molecular mechanisms in DM1: A focus on foci. Nucleic Acids Res 43: 2433–2441, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 280: 737–741, 1998. [DOI] [PubMed] [Google Scholar]
- 308.Pisani V, Panico MB, Terracciano C, Bonifazi E, Meola G, Novelli G, Bernardi G, Angelini C, Massa R. Preferential central nucleation of type 2 myofibers is an invariable feature of myotonic dystrophy type 2. Muscle Nerve 38: 1405–1411, 2008. [DOI] [PubMed] [Google Scholar]
- 309.Pitt M Update in electromyography. Curr Opin Pediatr 25: 676–681, 2013. [DOI] [PubMed] [Google Scholar]
- 310.Potthoff MJ, Wu H, Arnold MA, Shelton JM, Backs J, McAnally J, Richardson JA, Bassel-Duby R, Olson EN. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J Clin Invest 117: 2459–2467, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Poulos MG, Batra R, Charizanis K, Swanson MS. Developments in RNA splicing and disease. Cold Spring Harb Perspect Biol 3: a000778, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Poulos MG, Batra R, Li M, Yuan Y, Zhang C, Darnell RB, Swanson MS. Progressive impairment of muscle regeneration in muscleblind-like 3 isoform knockout mice. Hum Mol Genet 22: 3547–3558, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Powell GT, Wright GJ. Do muscle founder cells exist in vertebrates? Trends Cell Biol 22: 391–396, 2012. [DOI] [PubMed] [Google Scholar]
- 314.Pushechnikov A, Lee MM, Childs-Disney JL, Sobczak K, French JM, Thornton CA, Disney MD. Rational design of ligands targeting triplet repeating transcripts that cause RNA dominant disease: Application to myotonic muscular dystrophy type 1 and spinocerebellar ataxia type 3. J Am Chem Soc 131: 9767–9779, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Querido E, Gallardo F, Beaudoin M, Menard C, Chartrand P. Stochastic and reversible aggregation of mRNA with expanded CUG-triplet repeats. J Cell Sci 124: 1703–1714, 2011. [DOI] [PubMed] [Google Scholar]
- 316.Raheem O, Olufemi SE, Bachinski LL, Vihola A, Sirito M, Holmlund-Hampf J, Haapasalo H, Li YP, Udd B, Krahe R. Mutant (CCTG)n expansion causes abnormal expression of zinc finger protein 9 (ZNF9) in myotonic dystrophy type 2. Am J Pathol 177: 3025–3036, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Rahimov F, Kunkel LM. The cell biology of disease: Cellular and molecular mechanisms underlying muscular dystrophy. J Cell Biol 201: 499–510, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520: 186–191, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Ranum LP, Cooper TA. RNA-mediated neuromuscular disorders. Annu Rev Neurosci 29: 259–277, 2006. [DOI] [PubMed] [Google Scholar]
- 320.Ranum LP, Day JW. Myotonic dystrophy: RNA pathogenesis comes into focus. Am J Hum Genet 74: 793–804, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Ranum LP, Rasmussen PF, Benzow KA, Koob MD, Day JW. Genetic mapping of a second myotonic dystrophy locus. Nat Genet 19: 196–198, 1998. [DOI] [PubMed] [Google Scholar]
- 322.Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B, Dembele D, Gourdon G, Nicole A, Duboc D, Wahbi K, Day JW, Fujimura H, Takahashi MP, Auboeuf D, Dreumont N, Furling D, Charlet-Berguerand N. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol 18: 840–845, 2011. [DOI] [PubMed] [Google Scholar]
- 323.Rau F, Laine J, Ramanoudjame L, Ferry A, Arandel L, Delalande O, Jollet A, Dingli F, Lee KY, Peccate C, Lorain S, Kabashi E, Athanasopoulos T, Koo T, Loew D, Swanson MS, Le Rumeur E, Dickson G, Allamand V, Marie J, Furling D. Abnormal splicing switch of DMD’s penultimate exon compromises muscle fibre maintenance in myotonic dystrophy. Nat Commun 6: 7205, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Ravel-Chapuis A, Belanger G, Cote J, Michel RN, Jasmin BJ. Misregulation of calcium-handling proteins promotes hyperactivation of calcineurin-NFAT signaling in skeletal muscle of DM1 mice. Hum Mol Genet 26: 2192–2206, 2017. [DOI] [PubMed] [Google Scholar]
- 325.Ravel-Chapuis A, Belanger G, Yadava RS, Mahadevan MS, DesGroseillers L, Cote J, Jasmin BJ. The RNA-binding protein Staufen1 is increased in DM1 skeletal muscle and promotes alternative pre-mRNA splicing. J Cell Biol 196: 699–712, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Reardon W, Newcombe R, Fenton I, Sibert J, Harper PS. The natural history of congenital myotonic dystrophy: Mortality and long term clinical aspects. Arch Dis Child 68: 177–181, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Reddy S, Smith DB, Rich MM, Leferovich JM, Reilly P, Davis BM, Tran K, Rayburn H, Bronson R, Cros D, Balice-Gordon RJ, Housman D. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat Genet 13: 325–335, 1996. [DOI] [PubMed] [Google Scholar]
- 328.Reed UC. Congenital muscular dystrophy. Part I: A review of phenotypical and diagnostic aspects. Arq Neuropsiquiatr 67: 144–168, 2009. [DOI] [PubMed] [Google Scholar]
- 329.Renna LV, Cardani R, Botta A, Rossi G, Fossati B, Costa E, Meola G. Premature senescence in primary muscle cultures of myotonic dystrophy type 2 is not associated with p16 induction. Eur J Histochem 58: 2444, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Richard AF, Demignon J, Sakakibara I, Pujol J, Favier M, Strochlic L, Le Grand F, Sgarioto N, Guernec A, Schmitt A, Cagnard N, Huang R, Legay C, Guillet-Deniau I, Maire P. Genesis of muscle fiber-type diversity during mouse embryogenesis relies on Six1 and Six4 gene expression. Dev Biol 359: 303–320, 2011. [DOI] [PubMed] [Google Scholar]
- 331.Ricker K, Koch MC, Lehmann-Horn F, Pongratz D, Speich N, Reiners K, Schneider C, Moxley RT III. Proximal myotonic myopathy. Clinical features of a multisystem disorder similar to myotonic dystrophy. Arch Neurol 52: 25–31, 1995. [DOI] [PubMed] [Google Scholar]
- 332.Romeo V Myotonic dystrophy type 1 or Steinert’s disease. Adv Exp Med Biol 724: 239–257, 2012. [DOI] [PubMed] [Google Scholar]
- 333.Roof DJ, Hayes A, Adamian M, Chishti AH, Li T. Molecular characterization of abLIM, a novel actin-binding and double zinc finger protein. J Cell Biol 138: 575–588, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Rossi G, Antonini S, Bonfanti C, Monteverde S, Vezzali C, Tajbakhsh S, Cossu G, Messina G. Nfix regulates temporal progression of muscle regeneration through modulation of myostatin expression. Cell Rep 14: 2238–2249, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Rozo M, Li L, Fan CM. Targeting beta1-integrin signaling enhances regeneration in aged and dystrophic muscle in mice. Nat Med 22: 889–896, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Rudolf A, Schirwis E, Giordani L, Parisi A, Lepper C, Taketo MM, Le Grand F. Beta-catenin activation in muscle progenitor cells regulates tissue repair. Cell Rep 15: 1277–1290, 2016. [DOI] [PubMed] [Google Scholar]
- 337.Runfola V, Sebastian S, Dilworth FJ, Gabellini D. Rbfox proteins regulate tissue-specific alternative splicing of Mef2D required for muscle differentiation. J Cell Sci 128: 631–637, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Russo LS. Altered motor neuron excitability in myotonic dystrophy. Electromyogr Clin Neurophysiol 31: 461–466, 1991. [PubMed] [Google Scholar]
- 339.Rutherford MA, Heckmatt JZ, Dubowitz V. Congenital myotonic-dystrophy—Respiratory-function at birth determines survival. Arch Dis Child 64: 191–195, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Sabouri LA, Mahadevan MS, Narang M, Lee DS, Surh LC, Korneluk RG. Effect of the myotonic dystrophy (DM) mutation on mRNA levels of the DM gene. Nat Genet 4: 233–238, 1993. [DOI] [PubMed] [Google Scholar]
- 341.Sabourin LA, Tamai K, Narang MA, Korneluk RG. Overexpression of 3’-untranslated region of the myotonic dystrophy kinase cDNA inhibits myoblast differentiation in vitro. J Biol Chem 272: 29626–29635, 1997. [DOI] [PubMed] [Google Scholar]
- 342.Sahgal V, Bernes S, Sahgal S, Lischwey C, Subramani V. Skeletal muscle in preterm infants with congenital myotonic dystrophy. Morphologic and histochemical study. J Neurol Sci 59: 47–55, 1983. [DOI] [PubMed] [Google Scholar]
- 343.Sahgal V, Sahgal S, Bernes S, Subramani V. Ultrastructure of muscle spindle in congenital myotonic dystrophy. A study of preterm infant muscle spindles. Acta Neuropathol 61: 207–213, 1983. [DOI] [PubMed] [Google Scholar]
- 344.Salehi LB, Bonifazi E, Stasio ED, Gennarelli M, Botta A, Vallo L, Iraci R, Massa R, Antonini G, Angelini C, Novelli G. Risk prediction for clinical phenotype in myotonic dystrophy type 1: Data from 2,650 patients. Genet Test 11: 84–90, 2007. [DOI] [PubMed] [Google Scholar]
- 345.Salisbury E, Schoser B, Schneider-Gold C, Wang GL, Huichalaf C, Jin B, Sirito M, Sarkar P, Krahe R, Timchenko NA, Timchenko LT. Expression of RNA CCUG repeats dysregulates translation and degradation of proteins in myotonic dystrophy 2 patients. Am J Pathol 175: 748–762, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Sander HW, Tavoulareas GP, Quinto CM, Menkes DL, Chokroverty S. The exercise test distinguishes proximal myotonic myopathy from myotonic dystrophy. Muscle Nerve 20: 235–237, 1997. [DOI] [PubMed] [Google Scholar]
- 347.Sandri M Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 23: 160–170, 2008. [DOI] [PubMed] [Google Scholar]
- 348.Santoro M, Masciullo M, Bonvissuto D, Bianchi ML, Michetti F, Silvestri G. Alternative splicing of human insulin receptor gene (INSR) in type I and type II skeletal muscle fibers of patients with myotonic dystrophy type 1 and type 2. Mol Cell Biochem 380: 259–265, 2013. [DOI] [PubMed] [Google Scholar]
- 349.Santoro M, Modoni A, Masciullo M, Gidaro T, Broccolini A, Ricci E, Tonali PA, Silvestri G. Analysis of MTMR1 expression and correlation with muscle pathological features in juvenile/adult onset myotonic dystrophy type 1 (DM1) and in myotonic dystrophy type 2 (DM2). Exp Mol Pathol 89: 158–168, 2010. [DOI] [PubMed] [Google Scholar]
- 350.Santoro M, Piacentini R, Masciullo M, Bianchi ML, Modoni A, Podda MV, Ricci E, Silvestri G, Grassi C. Alternative splicing alterations of Ca2+ handling genes are associated with Ca2+ signal dysregulation in myotonic dystrophy type 1 (DM1) and type 2 (DM2) myotubes. Neuropathol Appl Neurobiol 40: 464–476, 2014. [DOI] [PubMed] [Google Scholar]
- 351.Sarkar PS, Paul S, Han J, Reddy S. Six5 is required for spermatogenic cell survival and spermiogenesis. Hum Mol Genet 13: 1421–1431, 2004. [DOI] [PubMed] [Google Scholar]
- 352.Sarnat HB, Silbert SW. Maturational arrest of fetal muscle in neonatal myotonic dystrophy. A pathologic study of four cases. Arch Neurol 33: 466–474, 1976. [DOI] [PubMed] [Google Scholar]
- 353.Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet 29: 40–47, 2001. [DOI] [PubMed] [Google Scholar]
- 354.Schiaffino S, Reggiani C. Fiber types in mammalian skeletal muscles. Physiol Rev 91: 1447–1531, 2011. [DOI] [PubMed] [Google Scholar]
- 355.Schild RL, Plath H, Hofstaetter C, Brenner R, Mann E, Mundegar RR, Steinbach P, Hansmann M. Polyhydramnios: An association with congenital myotonic dystrophy. J Obstet Gynaecol 18: 484–485, 1998. [DOI] [PubMed] [Google Scholar]
- 356.Schoser BG, Schneider-Gold C, Kress W, Goebel HH, Reilich P, Koch MC, Pongratz DE, Toyka KV, Lochmuller H, Ricker K. Muscle pathology in 57 patients with myotonic dystrophy type 2. Muscle Nerve 29: 275–281, 2004. [DOI] [PubMed] [Google Scholar]
- 357.Schultz E, Jaryszak DL, Valliere CR. Response of satellite cells to focal skeletal muscle injury. Muscle Nerve 8: 217–222, 1985. [DOI] [PubMed] [Google Scholar]
- 358.Schwander M, Leu M, Stumm M, Dorchies OM, Ruegg UT, Schittny J, Muller U. Beta1 integrins regulate myoblast fusion and sarcomere assembly. Dev Cell 4: 673–685, 2003. [DOI] [PubMed] [Google Scholar]
- 359.Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet 17: 19–32, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Seino S, Bell GI. Alternative splicing of human insulin receptor messenger RNA. Biochem Biophys Res Commun 159: 312–316, 1989. [DOI] [PubMed] [Google Scholar]
- 361.Seznec H, Agbulut O, Sergeant N, Savouret C, Ghestem A, Tabti N, Willer JC, Ourth L, Duros C, Brisson E, Fouquet C, Butler-Browne G, Delacourte A, Junien C, Gourdon G. Mice transgenic for the human myotonic dystrophy region with expanded CTG repeats display muscular and brain abnormalities. Hum Mol Genet 10: 2717–2726, 2001. [DOI] [PubMed] [Google Scholar]
- 362.Seznec H, Lia-Baldini AS, Duros C, Fouquet C, Lacroix C, Hofmann-Radvanyi H, Junien C, Gourdon G. Transgenic mice carrying large human genomic sequences with expanded CTG repeat mimic closely the DM CTG repeat intergenerational and somatic instability. Hum Mol Genet 9: 1185–1194, 2000. [DOI] [PubMed] [Google Scholar]
- 363.Shadrach JL, Wagers AJ. Stem cells for skeletal muscle repair. Philos Trans R Soc Lond B Biol Sci 366: 2297–2306, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, Wang L, Hodgkins A, Iyer V, Huang X, Skarnes WC. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods 11: 399–402, 2014. [DOI] [PubMed] [Google Scholar]
- 365.Shimizu K, Chen W, Ashique AM, Moroi R, Li YP. Molecular cloning, developmental expression, promoter analysis and functional characterization of the mouse CNBP gene. Gene 307: 51–62, 2003. [DOI] [PubMed] [Google Scholar]
- 366.Siboni RB, Nakamori M, Wagner SD, Struck AJ, Coonrod LA, Harriott SA, Cass DM, Tanner MK, Berglund JA. Actinomycin D specifically reduces expanded CUG repeat RNA in myotonic dystrophy models. Cell Rep 13: 2386–2394, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Sicot G, Gomes-Pereira M. RNA toxicity in human disease and animal models: From the uncovering of a new mechanism to the development of promising therapies. Biochim Biophys Acta 1832: 1390–1409, 2013. [DOI] [PubMed] [Google Scholar]
- 368.Siegel AL, Atchison K, Fisher KE, Davis GE, Cornelison DD. 3D timelapse analysis of muscle satellite cell motility. Stem Cells 27: 2527–2538, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Silver MM, Vilos GA, Silver MD, Shaheed WS, Turner KL. Morphologic and morphometric analyses of muscle in the neonatal myotonic dystrophy syndrome. Hum Pathol 15: 1171–1182, 1984. [DOI] [PubMed] [Google Scholar]
- 370.Singh RK, Xia Z, Bland CS, Kalsotra A, Scavuzzo MA, Curk T, Ule J, Li W, Cooper TA. Rbfox2-coordinated alternative splicing of Mef2d and Rock2 controls myoblast fusion during myogenesis. Mol Cell 55: 592–603, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Sinnar SA, Antoku S, Saffin JM, Cooper JA, Halpain S. Capping protein is essential for cell migration in vivo and for filopodial morphology and dynamics. Mol Biol Cell 25: 2152–2160, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Sobczak K, Wheeler TM, Wang W, Thornton CA. RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy. Mol Ther 21: 380–387, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Solana J, Irimia M, Ayoub S, Orejuela MR, Zywitza V, Jens M, Tapial J, Ray D, Morris Q, Hughes TR, Blencowe BJ, Rajewsky N. Conserved functional antagonism of CELF and MBNL proteins controls stem cell-specific alternative splicing in planarians. Elife 5: pii: e16797, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Spilker KA, Wang GJ, Tugizova MS, Shen K. Caenorhabditis elegans muscleblind homolog mbl-1 functions in neurons to regulate synapse formation. Neural Dev 7: 7, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Squillace RM, Chenault DM, Wang EH. Inhibition of muscle differentiation by the novel muscleblind-related protein CHCR. Dev Biol 250: 218–230, 2002. [DOI] [PubMed] [Google Scholar]
- 376.Stark DA, Karvas RM, Siegel AL, Cornelison DD. Eph/ephrin interactions modulate muscle satellite cell motility and patterning. Development 138: 5279–5289, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Stark T, Walker B, Phillips JK, Fejer R, Beck R. Hand-held dynamometry correlation with the gold standard isokinetic dynamometry: A systematic review. PM R 3: 472–479, 2011. [DOI] [PubMed] [Google Scholar]
- 378.Statland JM, Barohn RJ. Muscle channelopathies: The nondystrophic myotonias and periodic paralyses. Continuum (Minneap Minn) 19: 1598–1614, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Stehbens S, Wittmann T. Targeting and transport: How microtubules control focal adhesion dynamics. J Cell Biol 198: 481–489, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Steinbach P, Glaser D, Vogel W, Wolf M, Schwemmle S. The DMPK gene of severely affected myotonic dystrophy patients is hypermethylated proximal to the largely expanded CTG repeat. Am J Hum Genet 62: 278–285, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Steinberg H, Wagner A. [Hans Steinert: 100 years of myotonic dystrophy]. Nervenarzt 79: 961–962, 965–970, 2008. [DOI] [PubMed] [Google Scholar]
- 382.Storbeck CJ, Drmanic S, Daniel K, Waring JD, Jirik FR, Parry DJ, Ahmed N, Sabourin LA, Ikeda JE, Korneluk RG. Inhibition of myogenesis in transgenic mice expressing the human DMPK 3’-UTR. Hum Mol Genet 13: 589–600, 2004. [DOI] [PubMed] [Google Scholar]
- 383.Storbeck CJ, Sabourin LA, Waring JD, Korneluk RG. Definition of regulatory sequence elements in the promoter region and the first intron of the myotonic dystrophy protein kinase gene. J Biol Chem 273: 9139–9147, 1998. [DOI] [PubMed] [Google Scholar]
- 384.Suetterlin K, Mannikko R, Hanna MG. Muscle channelopathies: Recent advances in genetics, pathophysiology and therapy. Curr Opin Neurol 27: 583–590, 2014. [DOI] [PubMed] [Google Scholar]
- 385.Suominen T, Schoser B, Raheem O, Auvinen S, Walter M, Krahe R, Lochmuller H, Kress W, Udd B. High frequency of co-segregating CLCN1 mutations among myotonic dystrophy type 2 patients from Finland and Germany. J Neurol 255: 1731–1736, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Sznajder LJ, Michalak M, Taylor K, Cywoniuk P, Kabza M, Wojtkowiak-Szlachcic A, Matloka M, Konieczny P, Sobczak K. Mechanistic determinants of MBNL activity. Nucleic Acids Res 44: 10326–10342, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Tabebordbar M, Zhu K, Cheng JK, Chew WL, Widrick JJ, Yan WX, Maesner C, Wu EY, Xiao R, Ran FA, Cong L, Zhang F, Vandenberghe LH, Church GM, Wagers AJ. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351: 407–411, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Takekura H, Flucher BE, Franzini-Armstrong C. Sequential docking, molecular differentiation, and positioning of T-Tubule/SR junctions in developing mouse skeletal muscle. Dev Biol 239: 204–214, 2001. [DOI] [PubMed] [Google Scholar]
- 389.Takino T, Watanabe Y, Matsui M, Miyamori H, Kudo T, Seiki M, Sato H. Membrane-type 1 matrix metalloproteinase modulates focal adhesion stability and cell migration. Exp Cell Res 312: 1381–1389, 2006. [DOI] [PubMed] [Google Scholar]
- 390.Tanabe Y, Iai M, Tamai K, Fujimoto N, Sugita K. Neuroradiological findings in children with congenital myotonic dystrophy. Acta Paediatr 81: 613–617, 1992. [DOI] [PubMed] [Google Scholar]
- 391.Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J Cell Biol 128: 995–1002, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Tang Y, Wang H, Wei B, Guo Y, Gu L, Yang Z, Zhang Q, Wu Y, Yuan Q, Zhao G, Ji G. CUG-BP1 regulates RyR1 ASI alternative splicing in skeletal muscle atrophy. Sci Rep 5: 16083, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Tang ZZ, Yarotskyy V, Wei L, Sobczak K, Nakamori M, Eichinger K, Moxley RT, Dirksen RT, Thornton CA. Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of Ca(V)1.1 calcium channel. Hum Mol Genet 21: 1312–1324, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.te Velthuis AJW, Bagowski CP. PDZ and LIM domain-encoding genes: molecular interactions and their role in development. ScientificWorld-Journal 7: 1470–1492, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, Spratt SK, Surosky RT, Giedlin MA, Nichol G, Holmes MC, Gregory PD, Ando DG, Kalos M, Collman RG, Binder-Scholl G, Plesa G, Hwang WT, Levine BL, June CH. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N EnglJ Med 370: 901–910, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Teplova M, Song J, Gaw HY, Teplov A, Patel DJ. Structural insights into RNA recognition by the alternate-splicing regulator CUG-binding protein 1. Structure 18: 1364–1377, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Thomas JD, Sznajder ŁJ, Bardhi O, Aslam FN, Anastasiadis ZP, Scotti MM, Nishino I, Nakamori M, Wang ET, Swanson MS. Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy. Genes Dev 31: 1122–1133, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Thornell LE, Lindstom M, Renault V, Klein A, Mouly V, Ansved T, Butler-Browne G, Furling D. Satellite cell dysfunction contributes to the progressive muscle atrophy in myotonic dystrophy type 1. Neuropathol Appl Neurobiol 35: 603–613, 2009. [DOI] [PubMed] [Google Scholar]
- 399.Thornton CA. Myotonic dystrophy. Neurol Clin 32: 705–719, viii, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Thornton CA, Griggs RC, Moxley RT III. Myotonic dystrophy with no trinucleotide repeat expansion. Ann Neurol 35: 269–272, 1994. [DOI] [PubMed] [Google Scholar]
- 401.Thornton CA, Wang E, Carrell EM. Myotonic dystrophy: Approach to therapy. Curr Opin Genet Dev 44: 135–140, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Thornton CA, Wymer JP, Simmons Z, McClain C, Moxley RT III. Expansion of the myotonic dystrophy CTG repeat reduces expression of the flanking DMAHP gene. Nat Genet 16: 407–409, 1997. [DOI] [PubMed] [Google Scholar]
- 403.Tian B, White RJ, Xia TB, Welle S, Turner DH, Mathews MB, Thornton CA. Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA 6: 79–87, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Timchenko L Molecular mechanisms of muscle atrophy in myotonic dystrophies. Int J Biochem Cell Biol 45: 2280–2287, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Timchenko LT, Miller JW, Timchenko NA, DeVore DR, Datar KV, Lin L, Roberts R, Caskey CT, Swanson MS. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res 24: 4407–4414, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Timchenko LT, Timchenko NA, Caskey CT, Roberts R. Novel proteins with binding specificity for DNA CTG repeats and RNA CUG repeats: Implications for myotonic dystrophy. Hum Mol Genet 5: 115–121, 1996. [DOI] [PubMed] [Google Scholar]
- 407.Timchenko NA, Cai ZJ, Welm AL, Reddy S, Ashizawa T, Timchenko LT. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J Biol Chem 276: 7820–7826, 2001. [DOI] [PubMed] [Google Scholar]
- 408.Timchenko NA, Iakova P, Cai ZJ, Smith JR, Timchenko LT. Molecular basis for impaired muscle differentiation in myotonic dystrophy. Mol Cell Biol 21: 6927–6938, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Timchenko NA, Patel R, Iakova P, Cai ZJ, Quan L, Timchenko LT. Overexpression of CUG triplet repeat-binding protein, CUGBP1, in mice inhibits myogenesis. J Biol Chem 279: 13129–13139, 2004. [DOI] [PubMed] [Google Scholar]
- 410.Todd PK, Ackall FY, Hur J, Sharma K, Paulson HL, Dowling JJ. Transcriptional changes and developmental abnormalities in a zebrafish model of myotonic dystrophy type 1. Dis Model Mech 7: 143–155, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Tominaga K, Hayashi YK, Goto K, Minami N, Noguchi S, Nonaka I, Miki T, Nishino I. Congenital myotonic dystrophy can show congenital fiber type disproportion pathology. Acta Neuropathol 119: 481–486, 2010. [DOI] [PubMed] [Google Scholar]
- 412.Tran H, Gourrier N, Lemercier-Neuillet C, Dhaenens CM, Vautrin A, Fernandez-Gomez FJ, Arandel L, Carpentier C, Obriot H, Eddarkaoui S, Delattre L, Van Brussels E, Holt I, Morris GE, Sablonniere B, Buee L, Charlet-Berguerand N, Schraen-Maschke S, Furling D, Behm-Ansmant I, Branlant C, Caillet-Boudin ML, Sergeant N. Analysis of exonic regions involved in nuclear localization, splicing activity, and dimerization of muscleblind-like-1 isoforms. J Biol Chem 286: 16435–16446, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32: 381–386, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Udd B, Krahe R. The myotonic dystrophies: Molecular, clinical, and therapeutic challenges. Lancet Neurol 11: 891–905, 2012. [DOI] [PubMed] [Google Scholar]
- 416.Vallenius T, Scharm B, Vesikansa A, Luukko K, Schafer R, Makela TP. The PDZ-LIM protein RIL modulates actin stress fiber turnover and enhances the association of alpha-actinin with F-actin. Exp Cell Res 293: 117–128, 2004. [DOI] [PubMed] [Google Scholar]
- 417.van den Broek WJAA. Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum Mol Genet 11: 191–198, 2002. [DOI] [PubMed] [Google Scholar]
- 418.Vasyutina E, Martarelli B, Brakebusch C, Wende H, Birchmeier C. The small G-proteins Rac1 and Cdc42 are essential for myoblast fusion in the mouse. Proc Natl Acad Sci U S A 106: 8935–8940, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Venables JP, Lapasset L, Gadea G, Fort P, Klinck R, Irimia M, Vignal E, Thibault P, Prinos P, Chabot B, Abou Elela S, Roux P, Lemaitre JM, Tazi J. MBNL1 and RBFOX2 cooperate to establish a splicing programme involved in pluripotent stem cell differentiation. Nat Commun 4: 2480, 2013. [DOI] [PubMed] [Google Scholar]
- 420.Verhaert D, Richards K, Rafael-Fortney JA, Raman SV. Cardiac involvement in patients with muscular dystrophies: Magnetic resonance imaging phenotype and genotypic considerations. Circ Cardiovasc Imaging 4: 67–76, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Vignaud A, Ferry A, Huguet A, Baraibar M, Trollet C, Hyzewicz J, Butler-Browne G, Puymirat J, Gourdon G, Furling D. Progressive skeletal muscle weakness in transgenic mice expressing CTG expansions is associated with the activation of the ubiquitin-proteasome pathway. Neuromuscul Disord 20: 319–325, 2010. [DOI] [PubMed] [Google Scholar]
- 422.Vihola A, Bachinski LL, Sirito M, Olufemi SE, Hajibashi S, Baggerly KA, Raheem O, Haapasalo H, Suominen T, Holmlund-Hampf J, Paetau A, Cardani R, Meola G, Kalimo H, Edstrom L, Krahe R, Udd B. Differences in aberrant expression and splicing of sarcomeric proteins in the myotonic dystrophies DM1 and DM2. Acta Neuropathol 119: 465–479, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Vihola A, Bassez G, Meola G, Zhang S, Haapasalo H, Paetau A, Mancinelli E, Rouche A, Hogrel JY, Laforet P, Maisonobe T, Pellissier JF, Krahe R, Eymard B, Udd B. Histopathological differences of myotonic dystrophy type 1 (DM1) and PROMM/DM2. Neurology 60: 1854–1857, 2003. [DOI] [PubMed] [Google Scholar]
- 424.Volle CB, Delaney S. CAG/CTG repeats alter the affinity for the histone core and the positioning of DNA in the nucleosome. Biochemistry 51: 9814–9825, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Wagner SD, Struck AJ, Gupta R, Farnsworth DR, Mahady AE, Eichinger K, Thornton CA, Wang ET, Berglund JA. Dose-dependent regulation of alternative splicing by MBNL proteins reveals biomarkers for myotonic dystrophy. PLoS Genet 12: e1006316, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Wahbi K, Meune C, Porcher R, Becane HM, Lazarus A, Laforet P, Stojkovic T, Behin A, Radvanyi-Hoffmann H, Eymard B, Duboc D. Electrophysiological study with prophylactic pacing and survival in adults with myotonic dystrophy and conduction system disease. JAMA 307: 1292–1301, 2012. [DOI] [PubMed] [Google Scholar]
- 427.Wakimoto H, Maguire CT, Sherwood MC, Vargas MM, Sarkar PS, Han J, Reddy S, Berul CI. Characterization of cardiac conduction system abnormalities in mice with targeted disruption of Six5 gene. J Interv Card Electrophysiol 7: 127–135, 2002. [DOI] [PubMed] [Google Scholar]
- 428.Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S, Lecuyer E, Burge CB. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 150: 710–724, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Wang ET, Ward AJ, Cherone J, Wang TT, Giudice J, Treacy D, Freese P, Lambert NJ, Saxena T, Cooper TA, Burge CB. Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res 25(6):858–871, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Wang GS, Kearney DL, De Biasi M, Taffet G, Cooper TA. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J Clin Invest 117: 2802–2811, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Wang GS, Kuyumcu-Martinez MN, Sarma S, Mathur N, Wehrens XH, Cooper TA. PKC inhibition ameliorates the cardiac phenotype in a mouse model of myotonic dystrophy type 1. J Clin Invest 119: 3797–3806, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Wang J, Pegoraro E, Menegazzo E, Gennarelli M, Hoop RC, Angelini C, Hoffman EP. Myotonic dystrophy: Evidence for a possible dominant-negative RNA mutation. Hum Mol Genet 4: 599–606, 1995. [DOI] [PubMed] [Google Scholar]
- 433.Wang YH, Amirhaeri S, Kang S, Wells RD, Griffith JD. Preferential nucleosome assembly at DNA triplet repeats from the myotonic-dystrophy gene. Science 265: 669–671, 1994. [DOI] [PubMed] [Google Scholar]
- 434.Wang ZJ, Huang XS. Images in clinical medicine. Myotonia of the tongue. N Engl J Med 365: e32, 2011. [DOI] [PubMed] [Google Scholar]
- 435.Wansink DG, Wieringa B. Transgenic mouse models for myotonic dystrophy type 1 (DM1). Cytogenet Genome Res 100: 230–242, 2003. [DOI] [PubMed] [Google Scholar]
- 436.Wei C, Jones K, Timchenko NA, Timchenko L. GSK3beta is a new therapeutic target for myotonic dystrophy type 1. Rare Dis 1: e26555, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Wheeler TM, Krym MC, Thornton CA. Ribonuclear foci at the neuromuscular junction in myotonic dystrophy type 1. Neuromuscul Disord 17: 242–247, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Wheeler TM, Leger AJ, Pandey SK, MacLeod AR, Nakamori M, Cheng SH, Wentworth BM, Bennett CF, Thornton CA. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 488: 111–115, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT, Thornton CA. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 325: 336–339, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Whitham M, Febbraio MA. The ever-expanding myokinome: Discovery challenges and therapeutic implications. Nat Rev Drug Discov 15: 719–729, 2016. [DOI] [PubMed] [Google Scholar]
- 441.Whittaker RG, Ferenczi E, Hilton-Jones D. Myotonic dystrophy: Practical issues relating to assessment of strength. J Neurol Neurosurg Psychiatry 77: 1282–1283, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Wieben ED, Aleff RA, Tosakulwong N, Butz ML, Highsmith WE, Edwards AO, Baratz KH. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2–2) gene predicts Fuchs corneal dystrophy. PLoS One 7: e49083, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Wojtkowiak-Szlachcic A, Taylor K, Stepniak-Konieczna E, Sznajder LJ, Mykowska A, Sroka J, Thornton CA, Sobczak K. Short antisense-locked nucleic acids (all-LNAs) correct alternative splicing abnormalities in myotonic dystrophy. Nucleic Acids Res 43: 3318–3331, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Wong LJC, Ashizawa T, Monckton DG, Caskey CT, Richards CS. Somatic heterogeneity of the Ctg repeat in myotonic-dystrophy is age and size-dependent. Am J Hum Genet 56: 114–122, 1995. [PMC free article] [PubMed] [Google Scholar]
- 445.Wu H, Naya FJ, McKinsey TA, Mercer B, Shelton JM, Chin ER, Simard AR, Michel RN, Bassel-Duby R, Olson EN, Williams RS. MEF2 responds to multiple calcium-regulated signals in the control of skeletal muscle fiber type. EMBO J 19: 1963–1973, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Wu H, Olson EN. Activation of the MEF2 transcription factor in skeletal muscles from myotonic mice. J Clin Invest 109: 1327–1333, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Wu H, Xiong WC, Mei L. To build a synapse: Signaling pathways in neuromuscular junction assembly. Development 137: 1017–1033, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Xia G, Gao Y, Jin S, Subramony SH, Terada N, Ranum LP, Swanson MS, Ashizawa T. Genome modification leads to phenotype reversal in human myotonic dystrophy type 1 induced pluripotent stem cell-derived neural stem cells. Stem Cells 33: 1829–1838, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Xia G, Santostefano KE, Goodwin M, Liu J, Subramony SH, Swanson MS, Terada N, Ashizawa T. Generation of neural cells from DM1 induced pluripotent stem cells as cellular model for the study of central nervous system neuropathogenesis. Cell Reprogram 15: 166–177, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Yamashita Y, Matsuura T, Kurosaki T, Amakusa Y, Kinoshita M, Ibi T, Sahashi K, Ohno K. LDB3 splicing abnormalities are specific to skeletal muscles of patients with myotonic dystrophy type 1 and alter its PKC binding affinity. Neurobiol Dis 69: 200–205, 2014. [DOI] [PubMed] [Google Scholar]
- 451.Yanovsky-Dagan S, Avitzour M, Altarescu G, Renbaum P, Eldar-Geva T, Schonberger O, Mitrani-Rosenbaum S, Levy-Lahad E, Birnbaum RY, Gepstein L, Epsztejn-Litman S, Eiges R. Uncovering the role of hypermethylation by CTG expansion in myotonic dystrophy type 1 using mutant human embryonic stem cells. Stem Cell Reports 5: 221–231, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Young NP, Daube JR, Sorenson EJ, Milone M. Absent, unrecognized, and minimal myotonic discharges in myotonic dystrophy type 2. Muscle Nerve 41: 758–762, 2010. [DOI] [PubMed] [Google Scholar]
- 453.Yu Z, Teng X, Bonini NM. Triplet repeat-derived siRNAs enhance RNA-mediated toxicity in a Drosophila model for myotonic dystrophy. PLoS Genet 7: e1001340, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Yuan Y, Compton SA, Sobczak K, Stenberg MG, Thornton CA, Griffith JD, Swanson MS. Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res 35: 5474–5486, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Yusuf F, Brand-Saberi B. Myogenesis and muscle regeneration. Histochem Cell Biol 138: 187–199, 2012. [DOI] [PubMed] [Google Scholar]
- 456.Zaki M, Boyd PA, Impey L, Roberts A, Chamberlain P. Congenital myotonic dystrophy: Prenatal ultrasound findings and pregnancy outcome. Ultrasound Obstet Gynecol 29: 284–288, 2007. [DOI] [PubMed] [Google Scholar]
- 457.Zhang C, Lee KY, Swanson MS, Darnell RB. Prediction of clustered RNA-binding protein motif sites in the mammalian genome. Nucleic Acids Res 41: 6793–6807, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Zhang WJ, Wang Y, Dong SY, Choudhury R, Jin YF, Wang ZF. Treatment of type 1 myotonic dystrophy by engineering site-specific RNA endonucleases that target (CUG)(n) repeats. Mol Ther 22: 312–320, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Zhao Y, Ogawa H, Yonekura S, Mitsuhashi H, Mitsuhashi S, Nishino I, Toyoshima C, Ishiura S. Functional analysis of SERCA1b, a highly expressed SERCA1 variant in myotonic dystrophy type 1 muscle. Biochim Biophys Acta 1852: 2042–2047, 2015. [DOI] [PubMed] [Google Scholar]
- 460.Zhou Q, Chu PH, Huang C, Cheng CF, Martone ME, Knoll G, Shelton GD, Evans S, Chen J. Ablation of Cypher, a PDZ-LIM domain Z-line protein, causes a severe form of congenital myopathy. J Cell Biol 155: 605–612, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, Lacey DL, Goldberg AL, Han HQ. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142: 531–543, 2010. [DOI] [PubMed] [Google Scholar]
- 462.Zhu B, Ramachandran B, Gulick T. Alternative pre-mRNA splicing governs expression of a conserved acidic transactivation domain in myocyte enhancer factor 2 factors of striated muscle and brain. J Biol Chem 280: 28749–28760, 2005. [DOI] [PubMed] [Google Scholar]
- 463.Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LP. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A 108: 260–265, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, Ostrow LW, Rothstein JD, Troncoso JC, Ranum LP. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A 110: E4968–E4977, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]