

Abstract
There is increasing interest in developing in-depth proteomic approaches for mapping tissue heterogeneity in a cell-type-specific manner to better understand and predict the function of complex biological systems such as human organs. Existing spatially resolved proteomics technologies cannot provide deep proteome coverage due to limited sensitivity and poor sample recovery. Herein, we seamlessly combined laser capture microdissection with a low-volume sample processing technology that includes a microfluidic device named microPOTS (microdroplet processing in one pot for trace samples), multiplexed isobaric labeling, and a nanoflow peptide fractionation approach. The integrated workflow allowed us to maximize proteome coverage of laser-isolated tissue samples containing nanogram levels of proteins. We demonstrated that the deep spatial proteomics platform can quantify more than 5000 unique proteins from a small-sized human pancreatic tissue pixel (∼60,000 μm2) and differentiate unique protein abundance patterns in pancreas. Furthermore, the use of the microPOTS chip eliminated the requirement for advanced microfabrication capabilities and specialized nanoliter liquid handling equipment, making it more accessible to proteomic laboratories.
Biological tissues contain a wide variety of cell types and unique microenvironments with distinct functions and interaction networks.1,2 Proteome analysis aims to better understand basic biology and disease processes in specific tissue types, as well as to identify novel disease biomarkers and potential therapeutic targets.3 However, widely used bulk analysis approaches average out localized biological processes and signaling across whole cell populations and as such, obscure cellular heterogeneity and spatial localization.4 Thus, examining cellular diversity and tissue heterogeneity while retaining spatial information is of great interest in biomedical research. Therefore, single-cell and spatial proteomic technologies are becoming increasingly important to understand and discern cellular heterogeneity.4
Recently, increasing attention has been focused on enabling spatially resolved proteomic profiling of low-input samples.5−13 A significant effort has been made to scale down bulk proteomics techniques to enable processing of mass-limited samples.14 Even though the various techniques use different cell isolation approaches and processing platforms,7,15−24 the common goal of them is to minimize sample loss and enhance proteome profiling of low-input samples. Miniaturization of tube-based bottom-up processing enabled innovative and sensitive technologies for proteome measurements of low cell numbers,20,21,23−27 yet the automation of manual workflows is still needed. On the other hand, droplet- or nanowell-based sample preparation techniques illustrate the power of automated sample processing miniaturization, yet they utilize technology that is not easily accessible to the broader research community. Therefore, we modified the previously developed processing platform called nanodroplet processing in one pot for trace samples (nanoPOTS), which demonstrated the ability to effectively analyze bottom-up proteomic samples with limited numbers of cells and even down to a single mammalian cell.1,10,18,28,29 We scaled up our nanoPOTS platform to microdroplet volume by designing larger-sized microwells,30 which can accommodate tissue sample processing from near single-cell to multicell levels in up to 2 μL processing volume. By developing a microvolume device that operates in low-microliter range and utilizes commercially available laboratory micropipettes, we overcame barriers to adoption that we faced with the nanoPOTS platform, such as the cleanroom usage for the chip fabrication and demand for a highly customized nanoliter liquid handling robot and highly skilled personnel to operate the specialized platform.30,31 The microdroplet processing in one pot for trace samples (microPOTS) technology has the potential to be affordable, accessible, and easy-to-use for a wide research community. Previously, the microPOTS technology was employed for the analysis of ∼25 cultured HeLa cells and ∼10 cells from mouse liver thin sections, where ∼1800 and 1200 unique proteins were identified, respectively, with high reproducibility.30 The microPOTS technology was also utilized to identify proteomic changes in ∼200 Barrett’s esophageal cells, where >1500 protein groups were confidently identified, achieving a high reproducibility with a Pearson’s correlation coefficient of R > 0.9 among replicates.31
To optimize the discovery potential and obtain mechanistic insights from spatial proteomics measurements, the depth of proteome coverage needs to be maximized to include low-abundance proteins. In-depth proteome profiling of low-input samples poses a significant challenge due to low sensitivity and throughput of analytical methods.32 Peptides are typically identified using a data-dependent acquisition label-free approach, which is biased toward selecting peptides with the strongest signal for fragmentation;33 hence, the low-abundance protein quantification is unreliable due to a small number of MS/MS spectra being identified.34 Although a data-independent acquisition label-free approach has shown promising results in detecting low-abundance peptides,27,35 its throughput lags behind multiplexing approaches.11 A widely used multiplexing approach designed to enable the identification and precise quantitation of peptides is tandem mass tag (TMT) isobaric labeling. Several variations on the standard TMT approach have been developed to improve the detection of less abundant proteins7,10 and post-translational modifications.5 Common to all those approaches is the addition of a carrier channel to the multiplex, which contains a mixture of cells or tissues that mimics the experimental samples with a ratio (ratio between the carrier channel and the sample channel) of 30–500-fold.10 Another approach that was recently developed that reduces sample complexity and hence enables a dramatic increase in proteome coverage and enhancement of MS detection sensitivity is a multidimensional liquid chromatography (LC) separation that includes nanoflow fractionation with an online concatenation process. Previously, a coverage of >6000 proteins has been obtained from ∼650 HeLa cells and 10 single human pancreatic islets (∼1000 cells) utilizing a nanowell-mediated two-dimensional (2D) LC approach.13 Our in-house-built nanoflow Fractionation and Automated Concatenation (nanoFAC) 2D LC platform enabled in-depth proteomic analysis of small-sized samples allowing comprehensive proteome characterization of >6600 proteins with only a 100 ng HeLa digest, equivalent to ∼500 HeLa cells.12
In this article, we addressed the difficulties of in-depth proteome profiling of low-input samples by advancing and integrating the existing technologies. We further enhanced our microPOTS technology by integrating the TMT carrier channel concept into the sample prep workflow, then combined it with our in-house-built nanoflow fractionation system to further improve the overall proteome coverage. This effort resulted in a unique platform that can provide deep proteomic profiling of micron-scale tissue samples. Successful implementation of our advanced microdroplet processing technology resulted in quantification of 53,710 unique peptide sequences that mapped to >5000 unique proteins from a 125,000 μm2 rat pancreas tissue voxel. Subsequently, we employed the same technology to image a specific tissue region of a human pancreatic tissue section to gain biological insights into the pancreatic islet microenvironment. Using the optimized microdroplet processing protocol with the TMT carrier concept, we were able to dramatically increase the sensitivity and depth of the proteome coverage. Hence, from a 60,000 μm2 laser-captured microdissected human pancreas tissue section, we identified 52,000 unique peptide sequences that map to >5500 unique proteins.
Experimental Section
Reagents and Chemicals
Polypropylene microwell chips with a 2.2 mm well diameter were manufactured on polypropylene substrates by Protolabs (Maple Plain, MN). LC–MS-grade water, formic acid (FA), iodoacetamide (IAA), triethylammonium bicarbonate (TEAB), TMT-10plex and TMT11–131C reagents, anhydrous acetonitrile, tris(2-carboxyethyl)phosphine hydrochloride (TCEP-HCl), and 50% hydroxylamine (HA) were purchased from Thermo Fisher Scientific (Waltham, MA). N-Dodecyl β-d-maltose (DDM), dimethyl sulfoxide (DMSO) (HPLC grade), phosphate-buffered saline (PBS), and Periodic Acid-Schiff (PAS) staining kit were purchased from Sigma-Aldrich (St. Louis, MO). Both Lys-C and trypsin were purchased from Promega (Madison, WI). Ethanol was purchased from Decon Laboratories (King of Prussia, PA).
Tissue Collection
To evaluate and demonstrate the utility of our enhanced microPOTS technology, we used rodent and human models of pancreas tissue. Sprague–Dawley rats (Crl/CD[SD]) were obtained from Charles River Laboratories and were 7–8 weeks of age when the pancreas was collected. Animals were euthanized by exsanguination while under isoflurane gas anesthesia, and the pancreas was collected immediately and snap-frozen in an optimal cutting temperature compound (OCT). For the alkylation optimization experiments, we used rat gastrocnemius muscle tissue from Sprague–Dawley rats housed at Pacific Northwest National Laboratory according to NIH and institutional guidelines for the use of laboratory animals. Dissected tissues were immediately plunge-frozen in LN2 and stored at −80 °C until use. All protocols for this study were reviewed and approved by the Institutional Animal Care and Use Committee of Battelle, Pacific Northwest Division. Human pancreas tissue that was used for microPOTS imaging application was obtained from a 17 year old male donor. The donor was selected based on our eligibility criterion (https://dx.doi.org/10.17504/protocols.io.yxmvmnye5g3p/v2). Organ recovery and tissue processing were performed per our standard protocol (https://dx.doi.org/10.17504/protocols.io.n2bvj6dnblk5/v1). Briefly, the pancreas was sliced into 0.5 cm-thick tissue segments, subdivided, and immediately frozen in carboxymethylcellulose (CMC, prepared per https://dx.doi.org/10.17504/protocols.io.br4fm8tn) in cryotray molds that were prechilled on dry ice/isopentane slurry. Frozen CMC tissue blocks were stored at −80 °C until sectioning.
Cryosectioning
OCT-embedded rat pancreas tissue was cryosectioned to 12 micrometer-thick sections using a cryostat and thaw-mounted onto polyethylene naphthalate (PEN) membrane slides. The OCT was removed from the tissue sections by immersing slides into 50% ethanol for 30 s followed by immersion into the gradient of ethanol solutions (70, 96, and 100% ethanol) for 30 s each change, to fix the tissue sections.
CMC-embedded human pancreas tissue was cut to ten micrometer-thick slices using a cryostat and collected on PEN membrane slides. Samples were washed with the gradient of ethanol solutions (70, 96, and 100% ethanol, respectively) for 30 s each change, to dehydrate the tissue sections and to remove embedding material.
Laser Capture Microdissection
Sample dissection and collection were completed using a PALM Technologies (Carl Zeiss MicroImaging, Munich, Germany) which contains a PALM MicroBeam and RoboStage designed for high-precision laser micromanipulation in the micrometer range and a PALM RoboMover that collects dissected samples directly into microwells of the microPOTS chip. For sample collection, microwells were preloaded with 2 μL of DMSO that served as a capturing medium for excised tissue sections.1
First, we used rat pancreatic tissue sections to assess our platform for deep spatial proteomics profiling in terms of robustness and reproducibility. We collected five replicates of each acinar and islet tissue, with a total tissue area of 125,000 μm2 per replicate, roughly 60 ng of proteins. For islet tissue replicates, this corresponds to 4–5 islet voxels, while acinar tissue replicates contained a single tissue voxel. In our experimental design, we also included a carrier sample of roughly equally distributed islet and acinar tissue volume, containing 16-fold more tissue material than in individual samples, which was approximately 2,000,000 μm2 of the total tissue area split among two microwells, each processed as an industrial carrier sample and later pooled.
For the microPOTS imaging experiment, we first stained a 10 μm-thick human pancreas section using the PAS staining kit, following the manufacturer’s protocol. Informed by the islet localizations from the serial PAS-stained section, a 3 × 3 grid was used to collect an islet and surrounding acinar tissue. The grid was positioned to capture the whole islet in one 200 × 300 μm pixel and the surrounding acinar tissue in the other 8 pixels. Voxels were dissected in grid mode and collected into corresponding microwells of the chip. One carrier sample was also obtained, containing a similar-sized islet and surrounding acinar tissue with a total area equivalent to the entire grid size, which was 540,000 μm2.
Proteomics Sample Processing in a Microdroplet
The whole sample prep workflow including the subsequent TMT labeling was carried out on-chip by a manual pipetting protocol. Evaporation was minimized through the combination of chip cooling during dispensing of reagent solutions and using a humidified chamber for the incubation steps, where the chip was previously sealed with a contactless cover and wrapped in aluminum foil.
Rat pancreas tissue voxels collected in the microPOTS chip were incubated at 75 °C for 1 h to dry out remaining DMSO solvent. After DMSO was completely evaporated, 2 μL of extraction buffer containing 0.1% DDM, 0.5 × PBS, 38 mM TEAB, and 1 mM TCEP was dispensed to each well of the chip, following incubation at 75 °C for an hour. Next, 0.5 μL of an IAA solution (25 mM IAA in 100 mM TEAB) was added to the samples, and the samples were allowed to incubate at room temperature for 30 min. All samples were subsequently digested by dispensing 0.5 μL of an enzyme mixture (10 ng of Lys-C and 40 ng of trypsin in 100 mM TEAB) and incubating at 37 °C for 10 h. TMT-10 and TMT-11 isobaric mass tag reagents were resuspended in anhydrous acetonitrile to a concentration of 6.4 μg/μL. For all samples, 1 μL of appropriate TMT tag was dispensed to the corresponding well. Since the carrier sample was equally distributed between two microwells, 1 μL of 126 TMT tag was dispensed to each microwell. The peptide–TMT mixtures were incubated for 1 h at room temperature, and the labeling reaction was quenched by adding 1 μL of 5% HA in 100 mM TEAB and incubated for 15 min at room temperature. Next, all samples were pooled together into one Eppendorf tube, brought up to the final 1% FA, then centrifuged at 12,300 rcf for 5 min at 25 °C to spin down the precipitate. The supernatant was then transferred to an autosampler vial, dried, and stored in −20 °C.
Human pancreas tissue samples, collected for the proteomics imaging experiment, were processed following the aforementioned protocol, with a slight modification of the alkylation step and TMT labeling strategy. Informed by the overalkylation of the previously processed rat pancreas samples, we used 0.5 μL of a 10 mM IAA solution in 100 mM TEAB to reach a final concentration of 2 mM IAA in the reaction. Following our experimental design, peptides were labeled using a TMT 11-plex, leaving the 130N channel empty and using the 131N channel for the carrier sample; the 128N channel was used for the islet voxel and the other 8 channels for the acinar tissue voxels.
Nanoflow LC Fractionation
Sample was resuspended in 62 μL of 0.1% FA in water. The first-dimension high-pH fractionation was performed off-line by loading 50 μL of the sample onto a precolumn (150 μm i.d., 5 cm length) using 0.1% FA in water at a flow rate of 9 μL/min for 9 min. The sample was then transferred on an LC column (75 μm i.d., 60 cm length). Precolumn and column were packed in-house with 5 and 3 μm Jupiter C18 packing material (300 Å pore size) (Phenomenex, Terrence, USA), respectively. An Ultimate 3000 RSLCnano system (Thermo Scientific) was used to deliver gradient flow to the LC column at a nanoflow rate of 300 nL/min. 10 mM ammonium formate (pH 9.5) in water was used as mobile phase A and acetonitrile as mobile phase B. The peptides were eluted using the following increasing gradients: from 1 to 8% of mobile phase B in 11 min and then to 12% mobile phase B in 18 min, followed by an increase to 30% mobile phase B in 55 min, then to 45% mobile phase B in 22 min, to 65% mobile phase B in 6 min, and finally to 95% mobile phase B in 5 min. Peptides eluted from the high-pH nano-LC separation were fractionated using a HTX PAL collect system into the vials preloaded with 25 μL of 0.1% FA in water, containing 0.01% (m/v) DDM. The PAL autosampler allowed us to automatically perform the concatenation by robotically moving the dispensing capillary among 12 collection vials; hence, a total of 96 fractions were concatenated into 12 fractions. The vials were stored at −20 °C until the following low-pH LC–MS/MS analysis.
LC–MS/MS Peptide Analyses
Fractionated samples were separated in the second dimension, using the Ultimate 3000 RSLCnano system (Thermo Scientific), by being injected fully into a 20 μL loop and loaded onto a precolumn using 0.1% FA in water at a flow rate of 7 μL/min for 5 min. A 3 cm-long Jupiter trapping column with 150 μm i.d. was in-house packed using a 5 μm C18 packing material (300 Å pore size, Phenomenex, Terrence, USA). The sample was then transferred on an LC column (1.7 μm Waters BEH 130 75 μm i.d. × 30 cm) that was heated at 45 °C. Chromatographic separation was performed at 200 nL/min using the following gradient: 1–8% (2.3–12.6 min), 8–25% (12.6–107 min), 25–35% (107.6–117.6), 35–75% (117–122.6 min), and 75–95% (122.6–125.9 min) of buffer B (0.1% FA in acetonitrile). Separated peptides were introduced to the ion source of a Q Exactive HF-X (Thermo Scientific) instrument, where high voltage (2200 V) was applied to generate electrospray and ionize peptides. The ion transfer tube was heated to 300 °C and the S-Lens RF level was set to 40. Similar acquisition parameters were used for analysis of the two different pancreas tissue models. A full MS scan of rat pancreas tissue sample fractions was acquired across a scan range of 300 to 1800 m/z at a resolution of 60,000, combined with a maximum injection time (IT) of 50 ms and automatic gain control (AGC) target value of 3 × 106. Sixteen data-dependent MS/MS scans were recorded per MS scan, at a resolving power of 60,000 combined with a maximum IT of 120 ms and AGC target value of 2 × 105, with an isolation window of 0.7 m/z. A dynamic exclusion time was set to 45 s to reduce the repeated selection of precursor ions. For the human pancreas tissue fractions, precursor ions from 300 to 1800 m/z were scanned with a mass resolution of 60,000 combined with an IT of 20 ms and (AGC) target of 3 × 106. The MS/MS spectra were acquired in data-dependent mode where the 12 most intense precursor ions were fragmented and recorded at a resolving power of 45,000 combined with a maximum IT of 100 ms and AGC target value of 1 × 105, with an isolation window of 0.7 m/z. A dynamic exclusion of 45 s was used.
Data Analysis
Thermo RAW files were processed using mzRefinery to correct for mass calibration errors.36 Spectra were then searched with MS-GF + v988137,38 to match against the Uniprot human database downloaded in March 2021 (20,371 proteins), combined with common contaminants (e.g., trypsin and keratin). A partially tryptic search was used with a ±20 ppm parent ion mass tolerance. TMT global proteomics data processing was performed as we previously described.39
Gene set enrichment analysis was performed on rat pancreas data using the gseGO40 function of the ClusterProfiler R package in conjunction with genome-wide annotations for rats (available from org.Rn.eg.db), with the Gene Ontology Biological Process41,42 as the reference database. To obtain the ranked list, we used the MSnSet.utils (https://rdrr.io/github/vladpetyuk/vp.misc/) and limma R43 package to compute a t-test comparison between acinar and islet samples, then filtered the original 5021 genes down to those with BH-adjusted p-values below 0.05, leaving 2339 differentially expressed genes. Finally, we ranked the differentially expressed genes according to the log-fold change of islet minus acinar, in descending order, to obtain the input for the GSEA.
Human pancreas data was visualized using a Python class designed for proteomics imaging data.
Data is available through MassIVE (https://massive.ucsd.edu), a full partner of ProteomeXchange, through the following database accession: MSV000093925; password: Nano4427.
Results and Discussion
MicroPOTS Platform for Deep Spatial Proteomics
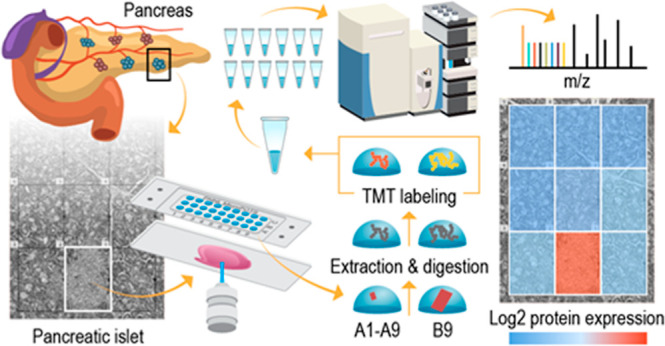
Figure 1 depicts the workflow for our deep spatial proteomics platform, which combines the laser capture microdissection (LCM)-based sample isolation with the microPOTS sample preparation approach for high sample recovery, a TMT carrier channel strategy, and a nanoscale fractionation system with automated concatenation to dramatically increase the depth of proteome coverage. This approach combines in-depth quantitative information on protein abundance with spatial context for untargeted investigations of biological tissues. The coupling of the microPOTS chip with LCM provides simple, robust, and efficient voxel collection. Using the chip with larger well diameters, such as microwell chips with a 2.2 mm well diameter, ensures a higher collection success rate allowing a high-precision capture of tissue voxels into the corresponding microwells, compared to 1.2 mm nanowell collection. Similar to the nanoPOTS chip, the microPOTS chip allows the direct visualization of the sample with an LCM microscope to confirm sample capture (Figure S1). In terms of sample processing, the microPOTS platform retains some advantages of the nanoPOTS platform, ensuring good digestion kinetics while reducing contaminants and side reactions. As opposed to our nanodroplet-based proteomic approach, which requires highly customized sample processing equipment, our microdroplet-based proteomic approach operates in the low-microliter range; hence, the samples can be processed manually using standard pipettes. As such, microPOTS is a low-cost technology, designed to be easily adopted by other research laboratories.
Figure 1.

Schematic overview of the deep spatial proteomics analysis pipeline.
Next, we utilized a multiplexing strategy by combining TMT isobaric labeling and a TMT carrier channel with microPOTS technology to improve proteome depth. Collection and labeling of large enough samples enabled us to utilize our custom-built nanoscale fractionation 2D LC system. The resolving power and overall peak capacity are dramatically increased by using 2D separation, which decreased sample complexity in each of the fractions and therefore enabled us to maximize proteome coverage of small-sized samples. In addition, 2D LC has been shown to significantly reduce ratio compression when using isobaric labeling approaches in complex samples.44
Implementation and Evaluation of Our Deep Spatial Proteomics Platform
To demonstrate that our recently developed platform for microscale proteomics can provide in-depth quantitative analyses of limited sample amounts, we employed it to compare proteome profiles of two distinct pancreatic tissue types. Exocrine pancreas tissue (acinar) and endocrine pancreas tissue (islet) were dissected and collected directly into a microPOTS chip following the experimental design detailed in the Experimental Section. To assess the robustness of our platform, we first looked at various analytical figures of merit. We found that the microPOTS sample processing approach provided great efficiency with a missed cleavage rate < 15%. TMT evaluation of the peptides labeled with TMT on the internal serine, threonine, and tyrosine residues revealed 1.3–6.8% of of peptides were over-labeled. peptide-spectrum match. Also, TMT labeling efficiency was evaluated by comparing the number of unlabeled peptides with the labeled peptides,45 and the labeling efficiency was >99%, demonstrating that the microPOTS platform provides as highly efficient TMT labeling as the nanoPOTS platform. Looking across the data set, the sample channels showed between 15- and 26-fold median abundance difference when compared to the carrier channel loaded with a 16-fold excess sample by area (Figure 2a). Additionally, we looked at missing data at the peptide and protein levels of the 11-plex set. Among the identified, >99% of proteins and >95% peptides were quantified without any missing values in the channels, demonstrating good data completeness achievable using TMT with a carrier channel, which is especially important considering that peptide loading amounts were at the low-μg level.
Figure 2.
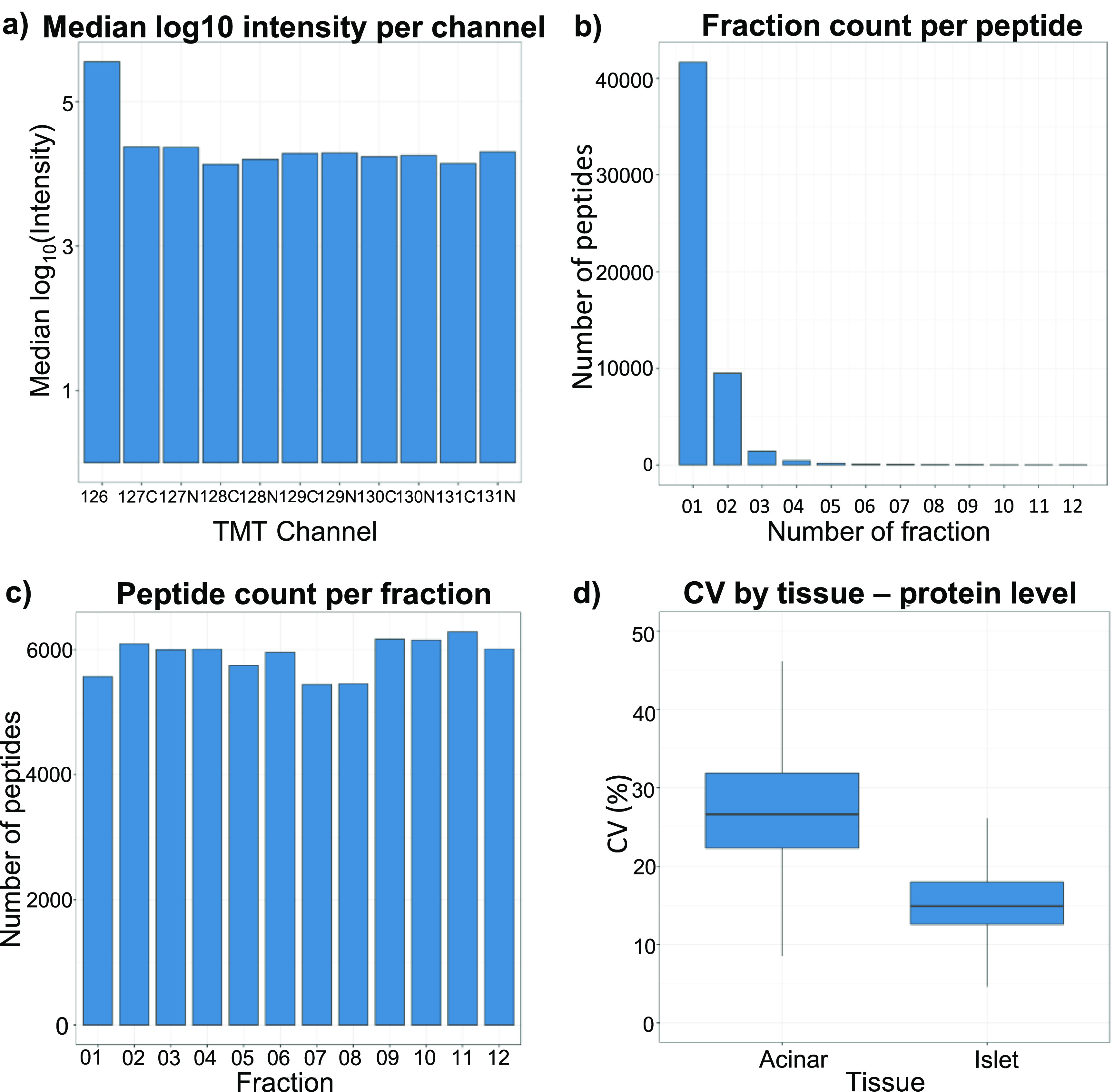
Evaluation of the deep spatial proteomics platform. (a) Median log10 intensities in the 11-plex set. (b) Fraction count in which a given peptide was detected. (c) Sampling depth (peptide count) for each fractionation. (d) Median CVs calculated across 5 replicates of each of the two pancreas functional units.
We also assessed the performance of our custom nanofractionation system. We identified 53,710 unique peptide sequences that correspond to 5202 unique proteins, whereas 78% of unique peptide identifications were found within a single fraction and 95% of peptides were found in two or fewer fractions, indicating high fractionation efficiency (Figure 2b). Peptides were also evenly distributed across the fractions, with each fraction yielding between 5300 and 6200 peptides, showing low fraction-to-fraction variation (Figure 2c). Furthermore, we computed the coefficient of variation (CV) for 5 islet and 5 acinar tissue replicates at the protein level (Figure 2d), and the results showed that variation among the islet replicates was comparatively small, median CV < 15%, indicating good reproducibility of the platform. We found that the CV for the acinar replicates, median CV 27%, was much higher relative to the islet. One potential reason for the different grouping of the acinar replicates and their high CV is that samples were collected from different tissue locations, indicating that the acinar tissue is not as homogeneous as we expected it to be, whereas each islet sample replicate consisted of 4–5 whole islets, creating a more homogeneous sample.
After assessing the microPOTS figures of merit, we compared proteomic profiles generated to evaluate the platform’s ability to accurately identify biological differences. Principal component (PC) analysis (PCA) was employed on the data from all 10 samples to visualize whether a clear separation exists between the islet and acinar proteomic profiles. This analysis clearly separated islet and acinar tissues forming two separate clusters in the first PC, accounting for 2/3 of the variability in the data set (Figure 3a). While the five islet replicates clustered closely together, the second PC separated acinar replicates into two groups, accounting for 10% of the variation. Next, we used linear models for microarray data to compare the two different tissue types. As expected, given the disparate functions of these tissue types, we found 2339 differentially expressed proteins. We then took the genes with an adjusted p-value below an FDR of 0.05; hierarchical clustering was used to identify differences in gene expression between the islet and acinar tissue types. The gene expression heat map, depicted in Figure S2, shows a distinct cluster of genes, indicating different biological functions of the two tissue types. A heat map of the top 50 clustered genes differentially expressed among the islet and acinar tissue is shown in Figure 3b. As expected, islet-specific endocrine hormones insulin and glucagon46 were upregulated in the islet samples as well as islet amyloid polypeptide.47 On the other hand, digestion enzymes chymotrypsin-like elastase family member 2A and pancreatic lipase-related protein 2 were upregulated in the acinar samples.46 Next, we linked overexpressed genes to biological processes (Figure 3c). Acinar tissue was enriched in biological processes, such as translation ribosomal assembly, digestion, and metabolic terms, which is in line with the previous publications,48,49 and islet samples overexpressed genes related to inter- and intracellular signaling, protein secretion, and hormone-level regulation, as reported previously.50
Figure 3.

(a) PCA showing sample variation in 2D PC space for islet and acinar proteomic profiles. (b) Heat map depicts hierarchical clustering of top 50 differently expressed genes in 2 functional units of the pancreas. (c) Gene set enrichment analysis for 2339 genes differentially expressed in endocrine and exocrine pancreas.
In-Depth Proteome Imaging of the Human Islet Microenvironment
Having established the quality of our microPOTS platform, we sought to demonstrate its utility as a proteome imaging platform. To do this, we imaged a region of pancreas tissue containing an islet and proximal acinar cells. We captured the entire islet in one pixel and acinar tissue in the other 8 pixels in order to investigate changes at the proteome level of the islet and the surrounding acinar microenvironment, Figure 4a. From 200 × 300 μm pancreas tissue, we reliably identified 52,000 unique peptide sequences that map to >5500 unique proteins. We then mapped known tissue-specific proteins to see whether the localization of these markers aligns with the functional role. As depicted in Figure 4b, endocrine hormones insulin and glucagon were predominantly present in the islet tissue region. On the other hand, digestive enzymes alpha-amylase 2B and pancreatic triacylglycerol lipase were highly abundant in the acinar tissue voxels of the imaged pancreas section (Figure 4c). Next, we leveraged our advanced microPOTS platform to look at the low-abundance proteins and gain molecular and spatial insights into distinct expressions of proteins throughout the imaged pancreas tissue. Syntaxin, which promotes fusion of insulin granules in pancreatic β cells,51 and Ca2+/calmodulin-dependent protein kinase type II subunit beta, which regulates insulin secretion and neurotransmitter release,52 were profiled with the higher abundance in the islet (Figure 4d). ERP27, a member of the protein disulfide isomerase family of endoplasmic reticulum proteins,53 showed high widespread expression in the imaged voxels related to acinar cells (Figure 4e). In addition to demonstrating changes in the proteome level between the islet and surrounding acinar tissue area, we sought to validate our profiling capability by looking at the proteins that are expected to be uniformly distributed across all the pixels. As depicted in Figure 4f, phosphatidylinositol 3-kinase catalytic subunit type 3 protein was efficiently profiled with medium abundance across all pixels, which is in line with the Human Protein Atlas54 reporting (https://www.proteinatlas.org/ENSG00000078142-PIK3C3/tissue/pancreas). This imaging experiment demonstrates the platform’s ability to provide proteomic profiles at a high spatial resolution, allowing us to image an important region of the pancreas with unprecedented depth of proteomic coverage. Further characterization of specific biological pathways that are uniquely expressed in pancreatic islet cells shows the ability of the microPOTS spatial proteomics platform to be exploited in a clinical setting.55
Figure 4.

Proteome imaging data obtained using our advanced microPOTS platform for deep spatial proteomics profiling. (a) Optical image of the 10 μm-thick PAS-stained pancreas section with the regions selected for LCM-proteome imaging. (b-f) Colored maps with scaled protein log2 abundance values (yellow–high and red–low), as examples of protein abundance changes across all 9 imaged pixels.
Conclusions
Here, we introduce a novel platform that combines LCM, microPOTS processing, multiplex labeling with a carrier channel, and nanoFAC to dramatically increase the sensitivity and depth of proteome coverage for small, spatially resolved, samples. We have demonstrated the performance and reproducibility of our platform by obtaining deep protein coverage and matching functional units of the pancreas with their protein profiles at a 200 μm spatial resolution.
Our advanced microPOTS technology can be applied to virtually any other tissue sample, and hence, it can be broadly applied across biomedical research. Microliter sample preparation technology can be performed with a micropipette without the requirement of a nanoliter liquid handling robot. Thus, this low-cost technology is easily implemented and adopted by other research laboratories. Furthermore, the nanoFAC system is assembled from commercially available instrumentation and parts. Currently, this technology uses a TMT 11-plex study design; future experiments can take advantage of TMT 18-plex reagents to further increase the throughput and proteome coverage.
Acknowledgments
This work was supported by the NIH HubMAP initiative grant to JL NIH UH3CA255132 and Pfizer Worldwide Research and Development. Much of this research was performed using the Environmental Molecular Sciences Laboratory, a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory (PNNL). PNNL is operated for the DOE by Battelle Memorial Institute under contract DE-AC05-76RLO1830. The authors thank graphic designer Nathan Johnson (PNNL) for preparing the figures.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.4c00523.
Images of the dissection and collection of pancreas tissue voxels into the microPOTS chip and heat map visualization of the distinct cluster of all significant genes, indicating different biological functions of the two tissue types (islet and acinar) (PDF)
Author Present Address
§ Department of Microchemistry, Proteomics, Lipidomics and Next Generation Sequencing, Genentech, 1 DNA Way, South San Francisco, 94080, United States
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Zhu Y.; Dou M.; Piehowski P. D.; Liang Y.; Wang F.; Chu R. K.; Chrisler W. B.; Smith J. N.; Schwarz K. C.; Shen Y.; et al. Spatially Resolved Proteome Mapping of Laser Capture Microdissected Tissue with Automated Sample Transfer to Nanodroplets. Mol. Cell. Proteomics 2018, 17 (9), 1864–1874. 10.1074/mcp.TIR118.000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y. H.; Wang X.; Huang P.; Tian R. Spatial proteomics for understanding the tissue microenvironment. Analyst 2021, 146 (12), 3777–3798. 10.1039/D1AN00472G. [DOI] [PubMed] [Google Scholar]
- Huang H. Z.; Shukla H.; Wu C.; Saxena S. Challenges and solutions in proteomics. Curr. Genomics 2007, 8 (1), 21–28. 10.2174/138920207780076910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L. W.; George J.; Wang J. Deep Profiling of Cellular Heterogeneity by Emerging Single-Cell Proteomic Technologies. Proteomics 2020, 20 (13), 1900226. 10.1002/pmic.201900226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi L.; Tsai C. F.; Dirice E.; Swensen A. C.; Chen J.; Shi T.; Gritsenko M. A.; Chu R. K.; Piehowski P. D.; Smith R. D.; et al. Boosting to Amplify Signal with Isobaric Labeling (BASIL) Strategy for Comprehensive Quantitative Phosphoproteomic Characterization of Small Populations of Cells. Anal. Chem. 2019, 91 (9), 5794–5801. 10.1021/acs.analchem.9b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C. F.; Zhao R.; Williams S. M.; Moore R. J.; Schultz K.; Chrisler W. B.; Pasa-Tolic L.; Rodland K. D.; Smith R. D.; Shi T.; et al. An Improved Boosting to Amplify Signal with Isobaric Labeling (iBASIL) Strategy for Precise Quantitative Single-cell Proteomics. Mol. Cell. Proteomics 2020, 19 (5), 828–838. 10.1074/mcp.RA119.001857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik B.; Levy E.; Harmange G.; Slavov N. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018, 19, 161. 10.1186/s13059-018-1547-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ctortecka C.; Stejskal K.; Krššáková G.; Mendjan S.; Mechtler K. Quantitative Accuracy and Precision in Multiplexed Single-Cell Proteomics. Anal. Chem. 2022, 94 (5), 2434–2443. 10.1021/acs.analchem.1c04174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung T. K.; Lee C. Y.; Bayer F. P.; McCoy A.; Kuster B.; Rose C. M. Defining the carrier proteome limit for single-cell proteomics. Nat. Methods 2021, 18 (1), 76–83. 10.1038/s41592-020-01002-5. [DOI] [PubMed] [Google Scholar]
- Dou M. W.; Clair G.; Tsai C. F.; Xu K.; Chrisler W. B.; Sontag R. L.; Zhao R.; Moore R. J.; Liu T.; Pasa-Tolic L.; et al. High-Throughput Single Cell Proteomics Enabled by Multiplex Isobaric Labeling in a Nanodroplet Sample Preparation Platform. Anal. Chem. 2019, 91 (20), 13119–13127. 10.1021/acs.analchem.9b03349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoof E. M.; Furtwängler B.; Üresin N.; Rapin N.; Savickas S.; Gentil C.; Lechman E.; Keller U. a. d.; Dick J. E.; Porse B. T. Quantitative single-cell proteomics as a tool to characterize cellular hierarchies. Nat. Commun. 2021, 12 (1), 3341. 10.1038/s41467-021-23667-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou M. W.; Tsai C. F.; Piehowski P. D.; Wang Y.; Fillmore T. L.; Zhao R.; Moore R. J.; Zhang P.; Qian W. J.; Smith R. D.; et al. Automated Nanoflow Two-Dimensional Reversed-Phase Liquid Chromatography System Enables In-Depth Proteome and Phosphoproteome Profiling of Nanoscale Samples. Anal. Chem. 2019, 91 (15), 9707–9715. 10.1021/acs.analchem.9b01248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou M. W.; Zhu Y.; Liyu A.; Liang Y.; Chen J.; Piehowski P. D.; Xu K.; Zhao R.; Moore R. J.; Atkinson M. A.; et al. Nanowell-mediated two-dimensional liquid chromatography enables deep proteome profiling of < 1000 mammalian cells. Chem. Sci. 2018, 9 (34), 6944–6951. 10.1039/C8SC02680G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupp-Sutton K. A.; Fang M. L.; Wu S. Separation methods in single-cell proteomics: RPLC or CE?. Int. J. Mass Spectrom. 2022, 481, 116920. 10.1016/j.ijms.2022.116920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leduc A.; Huffman R. G.; Cantlon J.; Khan S.; Slavov N. Exploring functional protein covariation across single cells using nPOP. Genome Biol. 2022, 23 (1), 261. 10.1186/s13059-022-02817-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z. Y.; Huang M.; Wang X. K.; Zhu Y.; Li J. S.; Wong C. C. L.; Fang Q. Nanoliter-Scale Oil-Air-Droplet Chip-Based Single Cell Proteomic Analysis. Anal. Chem. 2018, 90 (8), 5430–5438. 10.1021/acs.analchem.8b00661. [DOI] [PubMed] [Google Scholar]
- Woo J.; Williams S. M.; Markillie L. M.; Feng S.; Tsai C. F.; Aguilera-Vazquez V.; Sontag R. L.; Moore R. J.; Hu D.; Mehta H. S.; et al. High-throughput and high-efficiency sample preparation for single-cell proteomics using a nested nanowell chip. Nat. Commun. 2021, 12 (1), 6246. 10.1038/s41467-021-26514-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Piehowski P. D.; Zhao R.; Chen J.; Shen Y.; Moore R. J.; Shukla A. K.; Petyuk V. A.; Campbell-Thompson M.; Mathews C. E.; et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nat. Commun. 2018, 9, 882. 10.1038/s41467-018-03367-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ctortecka C.; Hartlmayr D.; Seth A.; Mendjan S.; Tourniaire G.; Udeshi N. D.; Carr S. A.; Mechtler K. An Automated Nanowell-Array Workflow for Quantitative Multiplexed Single-Cell Proteomics Sample Preparation at High Sensitivity. Mol. Cell. Proteomics 2023, 22 (12), 100665. 10.1016/j.mcpro.2023.100665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard-Banek C.; Li J.; Portero E. P.; Onjiko R. M.; Singer C. D.; Plotnick D. O.; Al Shabeeb R. Q.; Nemes P. In Vivo Subcellular Mass Spectrometry Enables Proteo-Metabolomic Single-Cell Systems Biology in a Chordate Embryo Developing to a Normally Behaving Tadpole (X. laevis)*. Angew. Chem., Int. Ed. 2021, 60 (23), 12852–12858. 10.1002/anie.202100923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard-Banek C.; Moody S. A.; Manzini M. C.; Nemes P. Microsampling Capillary Electrophoresis Mass Spectrometry Enables Single-Cell Proteomics in Complex Tissues: Developing Cell Clones in Live Xenopus laevis and Zebrafish Embryos. Anal. Chem. 2019, 91 (7), 4797–4805. 10.1021/acs.analchem.9b00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha-Shah A.; Esmaeili M.; Sidoli S.; Hwang H.; Yang J.; Klein P. S.; Garcia B. A. Single Cell Proteomics by Data-Independent Acquisition To Study Embryonic Asymmetry in Xenopus laevis. Anal. Chem. 2019, 91 (14), 8891–8899. 10.1021/acs.analchem.9b00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L. L.; Dubiak K. M.; Peuchen E. H.; Zhang Z.; Zhu G.; Huber P. W.; Dovichi N. J. Single Cell Proteomics Using Frog (Xenopus laevis) Blastomeres Isolated from Early Stage Embryos, Which Form a Geometric Progression in Protein Content. Anal. Chem. 2016, 88 (13), 6653–6657. 10.1021/acs.analchem.6b01921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard-Banek C.; Reddy S.; Moody S. A.; Nemes P. Label-free Quantification of Proteins in Single Embryonic Cells with Neural Fate in the Cleavage-Stage Frog (Xenopus laevis) Embryo using Capillary Electrophoresis Electrospray Ionization High-Resolution Mass Spectrometry (CE-ESI-HRMS). Mol. Cell. Proteomics 2016, 15 (8), 2756–2768. 10.1074/mcp.M115.057760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard-Banek C.; Moody S. A.; Nemes P. Single-Cell Mass Spectrometry for Discovery Proteomics: Quantifying Translational Cell Heterogeneity in the 16-Cell Frog (Xenopus) Embryo. Angew. Chem., Int. Ed. 2016, 55 (7), 2454–2458. 10.1002/anie.201510411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigdel T. K.; Piehowski P. D.; Roy S.; Liberto J.; Hansen J. R.; Swensen A. C.; Zhao R.; Zhu Y.; Rashmi P.; Schroeder A. Near-Single-Cell Proteomics Profiling of the Proximal Tubular and Glomerulus of the Normal Human Kidney. Front. Med. 2020, 7, 499. 10.3389/fmed.2020.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha-Shah A.; Esmaeili M.; Sidoli S.; Hwang H.; Yang J.; Klein P. S.; Garcia B. A. Single Cell Proteomics by Data-Independent Acquisition To Study Embryonic Asymmetry in Xenopus laevis. Anal. Chem. 2019, 91 (14), 8891–8899. 10.1021/acs.analchem.9b00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piehowski P. D.; Zhu Y.; Bramer L. M.; Stratton K. G.; Zhao R.; Orton D. J.; Moore R. J.; Yuan J.; Mitchell H. D.; Gao Y.; et al. Automated mass spectrometry imaging of over 2000 proteins from tissue sections at 100-mu m spatial resolution. Nat. Commun. 2020, 11 (1), 8. 10.1038/s41467-019-13858-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S. M.; Liyu A. V.; Tsai C. F.; Moore R. J.; Orton D. J.; Chrisler W. B.; Gaffrey M. J.; Liu T.; Smith R. D.; Kelly R. T.; et al. Automated Coupling of Nanodroplet Sample Preparation with Liquid Chromatography-Mass Spectrometry for High-Throughput Single-Cell Proteomics. Anal. Chem. 2020, 92 (15), 10588–10596. 10.1021/acs.analchem.0c01551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K. R.; Liang Y.; Piehowski P. D.; Dou M.; Schwarz K. C.; Zhao R.; Sontag R. L.; Moore R. J.; Zhu Y.; Kelly R. T. Benchtop-compatible sample processing workflow for proteome profiling of < 100 mammalian cells. Anal. Bioanal. Chem. 2019, 411 (19), 4587–4596. 10.1007/s00216-018-1493-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weke K.; Singh A.; Uwugiaren N.; Alfaro J. A.; Wang T.; Hupp T. R.; O’Neill J. R.; Vojtesek B.; Goodlett D. R.; Williams S. M.; et al. MicroPOTS Analysis of Barrett’s Esophageal Cell Line Models Identifies Proteomic Changes after Physiologic and Radiation Stress. J. Proteome Res. 2021, 20 (5), 2195–2205. 10.1021/acs.jproteome.0c00629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavov N. Single-cell protein analysis by mass spectrometry. Curr. Opin. Chem. Biol. 2021, 60, 1–9. 10.1016/j.cbpa.2020.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B.; Shi J.; Wang X.; Jiang H.; Zhu H. J. Label-free absolute protein quantification with data-independent acquisition. J. Proteonomics 2019, 200, 51–59. 10.1016/j.jprot.2019.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J. Y.; Dann G. P.; Liew C. W.; Smith R. D.; Kulkarni R. N.; Qian W. J. Unraveling pancreatic islet biology by quantitative proteomics. Expet Rev. Proteonomics 2011, 8 (4), 495–504. 10.1586/epr.11.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzinger M.; Müller E.; Dürnberger G.; Pichler P.; Mechtler K. Robust and Easy-to-Use One-Pot Workflow for Label-Free Single-Cell Proteomics. Anal. Chem. 2023, 95 (9), 4435–4445. 10.1021/acs.analchem.2c05022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons B. C.; Chambers M. C.; Monroe M. E.; Tabb D. L.; Payne S. H. Correcting systematic bias and instrument measurement drift with mzRefinery. Bioinformatics 2015, 31 (23), 3838–3840. 10.1093/bioinformatics/btv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Pevzner P. A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 2014, 5, 5277. 10.1038/ncomms6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Gupta N.; Pevzner P. A. Spectral probabilities and generating functions of tandem mass spectra: A strike against decoy databases. J. Proteome Res. 2008, 7 (8), 3354–3363. 10.1021/pr8001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosline S. J. C.; Tognon C.; Nestor M.; Joshi S.; Modak R.; Damnernsawad A.; Posso C.; Moon J.; Hansen J. R.; Hutchinson-Bunch C.; et al. Proteomic and phosphoproteomic measurements enhance ability to predict ex vivo drug response in AML. Clin. Proteomics 2022, 19 (1), 30. 10.1186/s12014-022-09367-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.; Hu E.; Xu S.; Chen M.; Guo P.; Dai Z.; Feng T.; Zhou L.; Tang W.; Zhan L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2 (3), 100141. 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M.; Ball C. A.; Blake J. A.; Botstein D.; Butler H.; Cherry J. M.; Davis A. P.; Dolinski K.; Dwight S. S.; Eppig J. T.; et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 2000, 25 (1), 25–29. 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon S.; Douglass E.; Good B. M.; Unni D. R.; Harris N. L.; Mungall C. J.; Basu S.; Chisholm R. L.; Dodson R. J.; Hartline E.; et al. The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021, 49 (D1), D325–D334. 10.1093/nar/gkaa1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M. E.; Phipson B.; Wu D.; Hu Y.; Law C. W.; Shi W.; Smyth G. K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43 (7), e47 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ow S. Y.; Salim M.; Noirel J.; Evans C.; Wright P. C. Minimising iTRAQ ratio compression through understanding LC-MS elution dependence and high-resolution HILIC fractionation. Proteomics 2011, 11 (11), 2341–2346. 10.1002/pmic.201000752. [DOI] [PubMed] [Google Scholar]
- Hutchinson-Bunch C.; Sanford J. A.; Hansen J. R.; Gritsenko M. A.; Rodland K. D.; Piehowski P. D.; Qian W. J.; Adkins J. N. Assessment of TMT Labeling Efficiency in Large-Scale Quantitative Proteomics: The Critical Effect of Sample pH. ACS Omega 2021, 6 (19), 12660–12666. 10.1021/acsomega.1c00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swensen A. C.; Veličković D.; Williams S. M.; Moore R. J.; Day L. Z.; Niessen S.; Hennessy S.; Posso C.; Monetti M.; Qian W. J.; et al. Proteomic Profiling of Intra-Islet Features Reveals Substructure-Specific Protein Signatures. Mol. Cell. Proteomics 2022, 21 (12), 100426. 10.1016/j.mcpro.2022.100426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark P.; Andersson A.; Westermark G. T. Islet Amyloid Polypeptide, Islet Amyloid, and Diabetes Mellitus. Physiol. Rev. 2011, 91 (3), 795–826. 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]
- Lugea A.; Waldron R. T.; Mareninova O. A.; Shalbueva N.; Deng N.; Su H. Y.; Thomas D. D.; Jones E. K.; Messenger S. W.; Yang J.; et al. Human Pancreatic Acinar Cells. Am. J. Pathol. 2017, 187 (12), 2726–2743. 10.1016/j.ajpath.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson A.; Hedin A.; Müller M.; Skog O.; Korsgren O. Transcriptional profiles of human islet and exocrine endothelial cells in subjects with or without impaired glucose metabolism. Sci. Rep. 2020, 10 (1), 22315. 10.1038/s41598-020-79313-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng X. W.; Chung Y. H.; Piston D. W. Intercellular Communication in the Islet of Langerhans in Health and Disease. Compr. Physiol. 2021, 11 (3), 2191–2225. 10.1002/cphy.c200026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunii M.; Ohara-Imaizumi M.; Takahashi N.; Kobayashi M.; Kawakami R.; Kondoh Y.; Shimizu T.; Simizu S.; Lin B.; Nunomura K.; et al. Opposing roles for SNAP23 in secretion in exocrine and endocrine pancreatic cells. J. Cell Biol. 2016, 215 (1), 121–138. 10.1083/jcb.201604030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos G. J. d.; Ferreira S. M.; Ortis F.; Rezende L. F.; Li C.; Naji A.; Carneiro E. M.; Kaestner K. H.; Boschero A. C. Metabolic memory of ß-cells controls insulin secretion and is mediated by CaMKIIa. Mol. Metabol. 2014, 3 (4), 484–489. 10.1016/j.molmet.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielsson A.; Pontén F.; Fagerberg L.; Hallström B. M.; Schwenk J. M.; Uhlén M.; Korsgren O.; Lindskog C. The Human Pancreas Proteome Defined by Transcriptomics and Antibody-Based Profiling. PLoS One 2014, 9 (12), e115421 10.1371/journal.pone.0115421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M.; Fagerberg L.; Hallström B. M.; Lindskog C.; Oksvold P.; Mardinoglu A.; Sivertsson Å.; Kampf C.; Sjöstedt E.; Asplund A. Tissue-based map of the human proteome. Science 2015, 347, 6220. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Gosline S. J. C.; Veličković M.; Pino J. C.; Day L. Z.; Attah I. K.; Swensen A. C.; Danna V.; Posso C.; Rodland K. D.; Chen J.; et al. Proteome Mapping of the Human Pancreatic Islet Microenvironment Reveals Endocrine- Exocrine Signaling Sphere of Influence. Mol. Cell. Proteomics 2023, 22 (8), 100592. 10.1016/j.mcpro.2023.100592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.