

Abstract
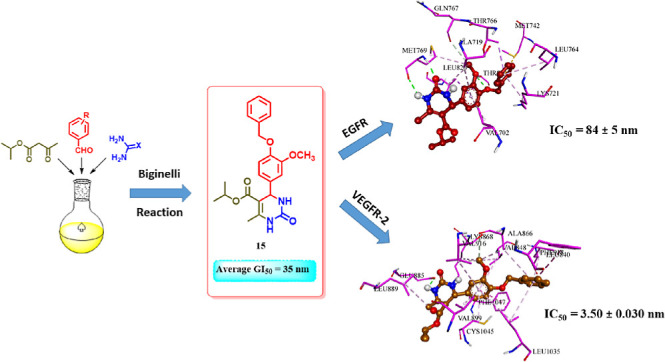
We developed and synthesized tetrahydropyrimidine derivatives as possible cytotoxic agents to inhibit EGFR and VEGFR-2 in the present study. Our study completely assesses the cytotoxic efficiency of pyrimidine-based derivatives 4–15 against various cancer cell lines, revealing derivatives 12 and 15 for their remarkable activity with GI50 values of 37 and 35 nM, respectively, when compared to the reference erlotinib (33 nM). In vitro enzyme assays showed that target compounds, particularly 12, 14, and 15, effectively inhibited EGFR and VEGFR-2. In vitro enzyme testing revealed that compound 15 was the most promising, with IC50 values of 84 and 3.50 nM for EGFR and VEGFR-2, respectively. Additionally, an in vitro assessment of the novel targets’ apoptotic potential revealed that both pro-apoptotic and antiapoptotic behaviors were promising, indicating that the apoptotic induction pathway is a strongly proposed action method for the newly developed targets. Finally, molecular docking experiments are elaborately discussed to corroborate the exact binding interactions of the most active hybrids 12 and 15 with the EGFR and VEGFR-2 active sites.
1. Introduction
Cancer ranks among the leading causes of mortality globally, affecting both developed and developing nations. As populations age and lifestyles evolve, the prevalence and fatality rates of cancer continue to rise each year. Projections indicate that by 2025, there will be over 20 million new cases.1,2 While surgical removal remains a viable treatment for some initial malignancies, its efficacy diminishes in advanced stages. Fortunately, chemotherapy and targeted therapeutic medications offer renewed optimism for cancer patients.3,4
Receptor tyrosine kinases (RTKs) are key in several physiological processes and signaling networks.5 RTKs are powerful oncoproteins that can induce angiogenesis, metastasis, and dysregulated cell proliferation when mutated or overexpressed;4,6 this makes them important targets for small molecule inhibitors in cancer treatment. Several RTK inhibitors have been discovered to have effective cytotoxic activity; some are in clinical studies or have been approved.7−10
EGFR, also known as epidermal growth factor receptor, is a specific type of membrane RTK that is found at higher levels than normal in several types of cancer. Because cancer progression is closely linked to EGFR tyrosine kinase signal transmission, inhibiting receptor activation can effectively halt tumor growth.11−13 VEGFR-2, also known as vascular endothelial growth factor receptor, is a RTK that has the ability to induce angiogenesis.14 VEGFR-2, a member of the VEGFR family, plays a crucial role in regulating the growth of blood vessels in tumors and is necessary for the formation of solid tumors. Blocking VEGFR-2 has been suggested as a viable strategy to prevent angiogenesis.15,16
It has been established that EGFR and VEGFR-2 are potential therapeutic targets in the fight against cancer. They play critical roles in signaling networks that control tumor cell angiogenesis, motility, differentiation, and proliferation.17−20 EGFR and VEGFR-2 commonly have overlapping downstream signaling pathways within a complex network of interconnected circuits. Suppressing EGFR can reduce VEGF production and inhibit the formation of new blood vessels. While also increasing VEGFR-2 expression, this can eventually lead to resistance to EGFR inhibitors.21,22 Consequently, the simultaneous inhibition of both EGFR and VEGFR-2 has emerged as a potent cancer treatment approach, yielding a synergistic effect.23−25
Multicomponent reactions (MCRs) have received much interest in the drug discovery process, particularly in developing tailored libraries with structural diversity.26 The current interest is developing innovative therapeutic candidates by MCRs for preclinical investigations. The cyclocondensation reaction was first reported by Pietro Biginelli in 1893. It involved using an acid catalyst and little heating to combine β-keto ester, aldehyde, and urea/thiourea dissolved in solvent ethanol. After cooling the reaction mixture, the resultant product precipitated and was identified as new 1,4-dihydropyrimidin-2-one.27 This one-pot MCR’s emphasis was expanded by altering the building elements to synthesize effective compounds for various medicinal uses. 1,4-Dihydropyrimidines are known to have various pharmacological actions, including anticancer activity.28 Because of the vast spectrum of medicinal characteristics, the pharmaceutical sector is interested in developing innovative dihydropyrimidines29−31
Naishima et al.28 recently published a study on 33 new Biginelli 1,4-dihydropyrimidines with anticancer activity as RTK inhibitors against breast cancer (MCF-7) cell line. Compound I (Figure 1) was the most potent derivative, significantly inhibiting the tyrosine kinases EGFR and human epidermal growth factor receptor 2 (HER2). In another study from our group,32 we describe the design, synthesis, and biological evaluation of a new class of dihydropyrimidine derivatives as apoptotic cytotoxic agents. Compound II (Figure 1) had the strongest cytotoxic effect when exposed to most cancer cells. The studied compound combined with 5-FU had significantly higher anticancer activity than 5-FU alone. Compound II enhanced pro-apoptotic markers (caspase 3, 8, 9, p53, cytochrome C, and BAX) while downregulating the antiapoptotic BCL-2. Furthermore, compound II caused cell cycle arrest at the PreG1 and G2/M phases in the HepG2 cell line.
Figure 1.

Structures of some dihydropyrimidine-based cytotoxic RTK inhibitors.
Recently, Ahmed et al.33 identified 18 new indolyl-dihydropyrimidine-based compounds as EGFR inhibitors with potential anticancer action. Compound III (Figure 1) had the most inhibitory profile against the cancer cell lines evaluated. Compound III has a lower IC50 value against EGFR (0.25 ± 0.01 μM) than erlotinib (0.30 ± 0.01 μM). When compared to the reference erlotinib, molecular docking analysis demonstrated that the dihydropyrimidine nucleus improves the binding within the active sites of EGFR by interacting with additional amino acids in these active sites.
In another study, Mostafa and Selim.34 developed a series of Biginelli hybrids with various heterocyclic moieties and tested 12 compounds against the NCI-60 cancer cell line panel. Compound IV (Figure 1), the most effective in the series, was tested as a potential inhibitor of the tyrosine kinases VEGFR-2 and mTOR. It showed strong inhibitory activity, with IC50 values of 1.97 and 0.64 μM, respectively. Additionally, compound IV boosted caspase-3 levels by 10-fold, whereas caspase-9 levels increased by around 100 fold. It also raised the percentage of A549 cells undergoing early apoptosis by 3.27% and late apoptosis by 3.31%.
Based on prior studies highlighting the tyrosine kinase inhibitory effects of dihydropyrimidine-based derivatives and our ongoing efforts to develop dual or multitargeted cytotoxic agents,35−42 the strategy of this work was centered on the design and synthesis of new cytotoxic compounds (4–15) based on dihydropyrimidine scaffold (Figure 2). Each of the newly synthesized analogues’ cytotoxic efficacy was assessed against four human cancer cell lines and one normal human cell line. In addition, the compounds with the highest cytotoxic activity were tested in vitro as apoptosis inducers and dual-EGFR and VEGFR-2 inhibitors. Finally, molecular docking against their respective molecular targets tested the most active variants.
Figure 2.

Structures of new targets 4–15.
2. Results and Discussion
2.1. Chemistry
The synthetic process for targets 4–15 is shown in Scheme 1. Commercially available isopropyl acetoacetate (2) was treated to the Biginelli reaction with various substituted benzaldehydes (1a–l) in the presence of urea or thiourea (3a or 3b) to produce pyrimidine-5-carboxylates 4–15. The best yields were obtained by refluxing in ethanol for 8 h with glacial acetic acid as a catalyst.
Scheme 1. Synthesis of Pyrimidine-5-Carboxylates 4–15.

The final compounds 4–15 were characterized using spectroscopic techniques such as 1H and 13C NMR and elemental analysis. The 1H NMR of the desired compounds showed two characteristic peaks appearing as two doublets at δ 0.99–1.07 and δ 1.15–1.19 ppm in addition to a multiplet peak at δ 4.80–4.88 ppm, which correspond to the isopropyl group. The ring closure was confirmed by the appearance of a doublet peak at δ 5.00–5.13 ppm, which resonated to the proton at the C4 position of the ring. The methyl group at C6 appeared as a singlet at δ 2.22–2.28 ppm. The two NH groups of the cyclic thiourea moiety in compounds 4–8 appeared as two weak singlets at δ 9.52–9.61 and δ 10.20–10.31 ppm, respectively, while they appeared at δ 7.57–7.69 and δ 9.06–9.14 ppm in compounds 9–15 containing a cyclic urea moiety. Methoxy groups on the C4 phenyl ring in compounds 4–6, 9–11, 14, and 15 appeared as intense singlet peaks at δ 3.63–3.74 ppm. Compound 15 showed a singlet peak at δ 5.05 ppm, which indicates the presence of the benzylic CH2 group. 13C NMR DEPTQ-135 spectra showed a peak at δ 17.05–18.18 ppm, corresponding to CH3 at C6 of the ring. The isopropyl group appeared as two adjacent peaks at δ 21.43–22.28 ppm and a peak at δ 66.19–67.35 ppm. The peak of C4 was found in the range of δ 53.54–54.51 ppm. The peaks of aromatic carbons of the C4 phenyl ring as well as C5 and C6 carbons were observed in the range of δ 99.50–157.85 ppm, while the carbonyl group of the ester moiety appeared at δ 164.68–165.40 ppm. Compounds 4–8 showed characteristic C2 thiocarbonyl peak at δ 173.83–174.62 ppm, while the C2 carbonyl group of compounds 9–15 displayed a signal at δ 152.11–152.66 ppm. Compound 15 peaked at δ 70.43 ppm, corresponding to the benzylic methylene group.
2.2. Biology
2.2.1. Assay of Cell Viability Effect
The human mammary gland epithelial (MCF-10A) normal cell line was employed to assess the impact of new targets 4–15 on viability.43,44 The MTT assay was employed to assess the cell viability of 4–15 following a four-day incubation period on MCF-10A cells. Table 1 indicates that none of the compounds analyzed exhibited cytotoxicity since all compounds showed over 87% cell viability at a concentration of 50 μM.
Table 1. Median Inhibitory Concentration (IC50) and Average IC50 (GI50) of Each Compound against the Four Cancer Cell Lines.
| comp | cell viability % | cytotoxic
activity IC50 ± SEM (nM) |
||||
|---|---|---|---|---|---|---|
| A-549 | MCF-7 | Panc-1 | HT-29 | average (GI50) | ||
| 4 | 91 | 47 ± 4 | 50 ± 5 | 48 ± 4 | 49 ± 4 | 49 |
| 5 | 89 | 67 ± 6 | 71 ± 6 | 68 ± 6 | 70 ± 6 | 68 |
| 6 | 90 | 42 ± 4 | 46 ± 4 | 44 ± 4 | 47 ± 4 | 45 |
| 7 | 93 | 70 ± 7 | 74 ± 7 | 72 ± 7 | 73 ± 7 | 71 |
| 8 | 92 | 64 ± 6 | 68 ± 6 | 67 ± 6 | 68 ± 6 | 67 |
| 9 | 90 | 74 ± 7 | 79 ± 7 | 75 ± 7 | 77 ± 7 | 76 |
| 10 | 89 | 52 ± 5 | 55 ± 5 | 52 ± 5 | 54 ± 5 | 53 |
| 11 | 92 | 56 ± 5 | 58 ± 5 | 56 ± 5 | 59 ± 5 | 57 |
| 12 | 87 | 36 ± 3 | 38 ± 3 | 36 ± 3 | 38 ± 3 | 37 |
| 13 | 90 | 62 ± 6 | 64 ± 6 | 61 ± 6 | 64 ± 6 | 62 |
| 14 | 89 | 40 ± 4 | 44 ± 4 | 42 ± 4 | 43 ± 4 | 41 |
| 15 | 91 | 33 ± 3 | 36 ± 3 | 34 ± 3 | 36 ± 3 | 35 |
| erlotinib | ND | 30 ± 3 | 40 ± 3 | 30 ± 3 | 30 ± 3 | 33 |
2.2.2. Cytotoxicity Assay
The cytotoxic effect of targets 4–15 on four human cancer cell lines (HT-29 for colon cancer, Panc-1 for pancreatic cancer, A-549 for lung cancer, and MCF-7 for breast cancer) was assessed using the MTT assay.45,46 Erlotinib was utilized as a reference.
Generally, targets 4–12 had significant cytotoxic efficacy, with GI50 values ranging from 35 to 76 nM when compared to the reference erlotinib (GI50 = 33 nM), and all tested compounds were more sensitive to the lung cancer (A-549) cell line than the other cell lines used. Additionally, 2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylates (9–15) are more reactive than 2-thioxo-congeners, 4–8.
Compounds 4, 6, 12, 14, and 15 had the highest cytotoxic activity, with GI50 values of 49, 45, 37, 41, and 35 nM, respectively, making compounds 12 and 15 equivalent to erlotinib. However, derivatives 12 and 15 outperformed erlotinib against the MCF-7 cancer cell line, with IC50 values of 38 and 36 nM, respectively, compared to erlotinib’s IC50 value of 40 nM.
Compound 15 (R = 4-O-benzyl-3-OMe, X = O) was the most effective derivative of all synthesized compounds, with a GI50 value of 35 nM, equaling erlotinib (GI50 = 33 nM) against the four cancer cell lines tested. The phenyl group substitution pattern at position four, as well as the type of substitution at position two, has a significant impact on the cytotoxic activity of compounds 4–12. For example, compound 9 (R = 4-OH-3-OMe, X = O), a debenzylated derivative, was found to have decreased cytotoxic efficacy. Compound 9 had a GI50 of 76 nM, 2.2 times lower than 15, indicating that the benzyl group at position 4 of the phenyl moiety is more tolerated for cytotoxic activity than the free hydroxyl group. Compound 14 (R = 3-OH-4-OMe, X = O) showed a GI50 value of 41 nM, making it 1.9 times more effective than compound 9, suggesting that the hydroxyl group was tolerated for cytotoxic activity at position 3 rather than 4.
Compound 12 (R = 3-OH, X = O) ranked second in activity with a GI50 value of 37 nM, comparable to compound 15. Replacing 3-OH in compound 12 with 4-OH as in compound 13 (R = 4-OH, X = O) resulted in a marketed decrease in cytotoxic action with a GI50 value of 62 nM, adding to the evidence that the hydroxyl group was more tolerated at position 3 rather than 4. Replacing the oxygen atom at position 2 with a sulfur atom resulted in a reported decrease in cytotoxic activity. Compound 7 (R = 3-OH, X = S), the sulfur derivative of compound 12, revealed a GI50 value of 71 nM, being 2-folds less potent than compound 12, showing that the oxygen atom at position 2 is more favored than the sulfur atom for the cytotoxic action.
Finally, compounds 10 (R = 3,4-di-OMe, X = O) and 11 (R = 3,4,5-tri-OMe, X = O) showed GI50 values of 53 and 57 nM, respectively, suggesting that the number of methoxy groups did not significantly affect the cytotoxic action. However, this rule does not apply to the sulfur derivatives 5 (R = 3,4-di-OMe, X = S) and 6 (R = 3,4,5-tri-OMe, X = S), where the trimethoxy derivative, compound 6, is approximately 1.5 times more potent than the dimethoxy derivative, compound 5.
2.2.3. EGFR Inhibitory Assay
The most potent cytotoxic derivatives, 6, 12, 14, and 15, were tested for their ability to inhibit EGFR using the EGFR-TK test.47,48 The results are shown in Table 2. The outcomes of this assay align with the findings of the cytotoxic assay, which demonstrated that compounds 12 (R = 3-OH, X = O) and 15 (R = 4-O-benzyl-3-OMe, X = O) were the most potent derivatives of EGFR inhibitors, with IC50 values of 87 ± 5 and 84 ± 5, respectively, which are equivalent to the reference drug erlotinib (IC50 = 80 ± 5). Compounds 12 and 15 were also the most potent derivatives with cytotoxic properties. Compounds 6 (R = 3,4,5-tri-OMe, X = S) and 14 (R = 3-OH-4-OMe, X = O) both showed notable anti-EGFR activity, with IC50 values of 98 ± 6 and 92 ± 5 nM. These findings suggest compounds 12 and 15 have promising EGFR inhibitory action and could be used as cytotoxic agents.
Table 2. IC50 Values of Compounds 6, 12, 14, and 15 against EGFR and VEGFR-2.
| compound | EGFR inhibition IC50 ± SEM (nM) | VEGFR-2 inhibition IC50 ± SEM (nM) |
|---|---|---|
| 6 | 98 ± 6 | 5.30 ± 0.050 |
| 12 | 87 ± 5 | 4.20 ± 0.040 |
| 14 | 92 ± 5 | 4.70 ± 0.040 |
| 15 | 84 ± 5 | 3.50 ± 0.030 |
| erlotinib | 80 ± 5 | |
| sorafenib | 0.17 ± 0.001 |
2.2.4. Assay for Inhibiting VEGFR-2
The inhibitory effects of compounds 6, 12, 14, and 15 on VEGFR-2 were evaluated using the kinase-glo-luminescent kinase assay,49 with sorafenib as the control drug. The results are shown as IC50 values in Table 2. The findings indicated that the compounds under investigation effectively inhibited VEGFR-2, with IC50 values ranging from 3.50 to 5.30 nM. In comparison, sorafenib had a lower IC50 value of 0.17 nM. Compounds 12 and 15 exhibited the highest potency, with IC50 values of 4.20 and 3.50 nM, respectively. The results indicate that compounds 12 and 15 exhibit potent cytotoxic activity and have the potential to function as dual inhibitors of EGFR and VEGFR-2.
2.2.5. Apoptosis Assays
Apoptosis is a programmed cell death via two primary processes.50 The intrinsic pathway is launched by internal events such as mitochondrial oxidative stress, while the extrinsic pathway is triggered by external influences such as tumor necrosis factor-α.51 Some antiapoptotic proteins, including BCL-2 and CK-18, regulate apoptosis. They inhibit apoptosis by deactivating pro-apoptotic proteins such as BAX, p53, caspase 3, and caspase 6.52 This age has piqued researchers’ interest in discovering new potent candidates capable of promoting apoptosis in malignant cells while having little effect on normal cells.
To assess their apoptotic inhibitory potential against A-549, three candidates (12, 14, and 15) were chosen. In vitro, quantitative assessments of pro-apoptotic proteins (p53, BAX, caspase 3, and caspase 6) and antiapoptotic proteins (BCL-2 and CK-18) were performed using a sandwiched immunoassay, as shown in Figure 3.
Figure 3.

Protein expression levels after treatment with 12, 14, and 15; (A) p53, (B) BAX, (C) caspase 3, (D) caspase 6, (E) BCL-2, and (F) CK 18. Values are represented as mean ± SD for three independent experiments.
Results revealed that all tested candidates (12, 14, and 15) significantly raised protein expression levels for pro-apoptotic indicators. In comparison to the control untreated cells, p53, BAX, caspase 3, and caspase 6 were elevated by (4.75, 13.95, 8.20, and 9.65), (3.10, 11.50, 7.40, and 8.50), and (5.90, 14.50, 8.70, and 9.80)-fold for 12, 14, and 15, respectively (Figure 3A–D). As a result, all the investigated compounds had very significant and comparable pro-apoptotic potentials, particularly 15, followed by 12 and 14.
In contrast, the treatment with tested compounds (12, 14, and 15) significantly reduced BCL-2 and CK-18 expression. BCL-2 and CK 18 protein expression levels were (0.60 and 0.55), (0.55 and 0.40), and (0.70 and 0.60)-folds in 12, 14, and 15 compared to the control untreated cells (Figure 3E,F). As a result, the pro-apoptotic and antiapoptotic behaviors were promising, showing that the apoptotic induction pathway is a highly proposed mode of action for the newly identified targets.
2.3. Molecular Docking Studies
Employing computational methodologies, we conducted molecular docking simulations of the most potent compounds, 12 and 15, against EGFR (PDB ID: 1M17)13 and VEGFR-2 (PDB ID: 4ASD).53 By focusing on these specific protein structures as docking targets, we aimed to elucidate the intricate interactions and binding affinities between these compounds and their respective receptor binding sites. Regarding EGFR, to verify the accuracy of our docking methodology, we performed a redocking experiment with the cocrystallized erlotinib in the EGFR binding site. Our results demonstrated that the redocked ligand exhibited a notable binding affinity of −8.18 kcal/mol, suggesting a strong interaction with the receptor. Furthermore, the root-mean-square deviation (RMSD) value of 1.12 Å between the experimental and redocked ligands indicates a close alignment, reinforcing the reliability of our docking approach. Superimposition of both redocked and cocrystallized erlotinib is depicted in Figure 4.
Figure 4.
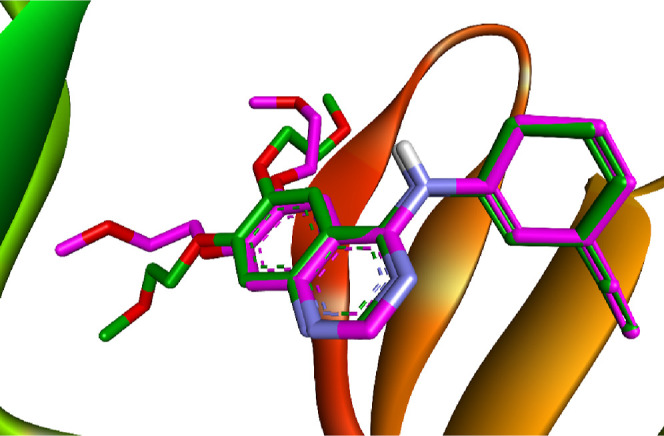
Comparison between the docked pose (green) and the X-ray cocrystallized structure (magenta) of erlotinib.
Furthermore, the most potent inhibitors, 12 and 15, were docked into the binding site of EGFR. Results showed that both compounds interacted strongly with the binding pocket with affinities of −5.6495 and −7.0684 kcal/mol, respectively. Analysis of the docking results of both compounds revealed intriguing insights. Specifically, the dihydropyrimidine ring in both compounds exhibited a critical hydrogen bonding interaction with the amino acid Met769, similar to that observed with the cocrystallized erlotinib. This shared interaction provides a rationale for the comparable potencies of compounds 12 and 15 with erlotinib toward EGFR. Compound 15 displayed an additional hydrogen bonding interaction with THR830, further enhancing its binding affinity. Moreover, the incorporation of the benzyl moiety in compound 15 facilitated numerous hydrophobic interactions with amino acids LYS721, THR766, and LEU764, effectively increasing binding and stabilizing the compound within the EGFR binding site (as depicted in Figures 5 and 6).
Figure 5.

Two-dimensional interactions of erlotinib and compounds 12 and 15 inside the EGFR binding pocket.
Figure 6.

Three-dimensional interactions of erlotinib and compounds 12 and 15 inside the EGFR binding pocket.
Similarly, the cocrystallized sorafenib was docked into the binding site of VEGFR-2 to assess the reliability of our docking protocol. The redocked sorafenib demonstrated an outstanding binding affinity of −10.1476 kcal/mol, underscoring a robust interaction within the VEGFR-2 binding site with an RMSD value of 0.1542 Å. Superimposition of both redocked and co-crystallized sorafenib is illustrated in Figure 7.
Figure 7.
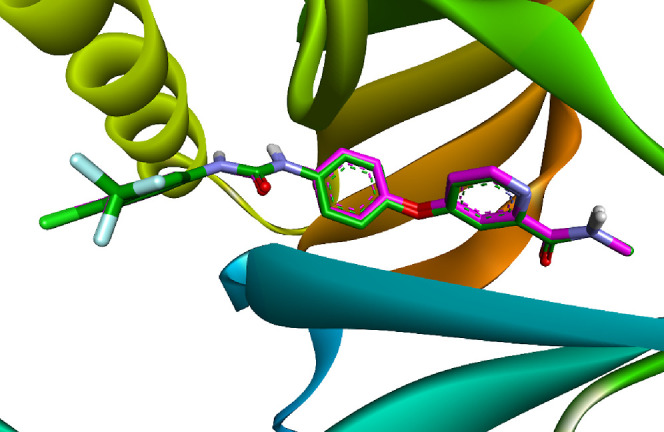
Comparison between the docked pose (green) and the X-ray cocrystallized structure (magenta) of sorafenib.
Moreover, docking the most potent inhibitors, compounds 12 and 15, into the VEGFR-2 binding site was conducted. The results revealed strong interactions of both compounds within the binding pocket, showcasing binding affinities of approximately −5.8777 and −8.2422 kcal/mol, respectively. Compound 15, with its cyclic urea moiety, not only established critical hydrogen bonding with Glu885 analogous to sorafenib but also anchored itself to the binding pocket by establishing hydrophobic interactions with various amino acids such as Val899, Leu889, Val916, Val848, and Leu1035, thereby reinforcing its affinity for the receptor pocket. Additionally, it engaged in pi–pi T-shaped interactions with Phe1047 akin to sorafenib. Compound 12 exhibited a similar interaction pattern to sorafenib and compound 15, except for engaging in a salt bridge with Glu885 through its urea moiety instead of a hydrogen bond, as seen in 15 and sorafenib (Figures 8 and 9).
Figure 8.

Two-dimensional interactions of sorafenib and compounds 12 and 15 inside the VEGFR-2 binding pocket.
Figure 9.

Three-dimensional interactions of sorafenib and compounds 12 and 15 inside the VEGFR-2 binding pocket.
2.4. Structure–Activity Relationship
Figure 10 illustrates the structure–activity relationship of compounds 4–15.
Figure 10.

Structure–activity relationship of compounds 4–15 as dual EGFR/VEGFR-2 inhibitors.
3. Conclusions
In the current study, we aim to achieve progress in cancer treatment by designing and synthesizing novel dihydropyrimidine-based compounds (4–15) as dual EGFR/VEGFR-2 inhibitors. Compounds 12 and 15 were discovered to be the most effective cytotoxic compounds, with significant EGFR and VEGFR-2 inhibition, revealing IC50 values of 84 and 3.5 nM, respectively. At 50 μM doses, 12 and 15 had no effect on nontumor cells MCF-10A, showing tumor-cell selectivity of these compounds. Apoptotic experiments show that compounds 12 and 15 can increase the levels of proapoptotic markers (p53, caspase 3, caspase 6, and BAX) while suppressing the antiapoptotic BCL-2 and CK-18. Molecular docking experiments have successfully revealed that compounds 12 and 15 have precise binding affinities with the EGFR and VEGFR-2 active sites. According to the data, 12 and 15 are promising dual EGFR and VEGFR-2 inhibitor candidates for the development of novel cancer therapies.
4. Experimental Section
General details: see Appendix A.
4.1. Chemistry
4.1.1. General Procedure for Synthesis of Isopropyl 6-Methyl-2-oxo/thioxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylates (4–15)
To a stirred solution of the appropriate aldehydes (1a–l) (2 mmol) and isopropyl acetoacetate (2) (2 mmol, 290 μL) in ethanol was added glacial acetic acid (0.5 mmol, 29 μL) and the mixture was stirred at room temperature for 1 h. Then, urea or thiourea (3a or 3b) (3 mmol) was added, and the obtained suspension was heated under reflux for 8 h. The reaction mixture was then poured on crushed ice and neutralized with sodium bicarbonate. The formed precipitate was filtered off and washed with distilled water, and the crude product was recrystallized from the suitable solvent.
4.1.2. Isopropyl 4-(4-Hydroxy-3-methoxyphenyl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4)
Yellow crystals (methanol) in (0.425 g, 63% yield), mp 190–195 °C; 1H NMR (400 MHz, DMSO-d6, δ): (ppm) 10.22 (s, 1H, N3-H), 9.53 (s, 1H, N1-H), 8.98 (s, 1H, OH), 6.77 (s, 1H, Ar-H), 6.72 (d, J = 8.0 Hz, 1H, Ar-H), 6.60 (d, J = 8.0 Hz, 1H, Ar-H), 5.06 (br s, 1H, C4-H), 4.89–4.80 (m, 1H, isopropyl-CH), 3.72 (s, 3H, OCH3), 2.27 (s, 3H, C6-CH3), 1.17 (d, J = 6.4 Hz, 3H, isopropyl-CH3), 1.03 (d, J = 6.4 Hz, 3H, isopropyl-CH3); 13C NMR (100 MHz, DMSO-d6, δ): (ppm): 174.56, 165.23, 147.81, 146.65, 144.76, 135.20, 119.17, 115.90, 111.62, 101.85, 67.23, 56.15, 54.25, 22.19, 21.95, 17.52; MS (ESI+) m/z 336.78 [M]+. Anal. Calcd for C16H20N2O4S (336.41): C, 57.31; H, 5.99; N, 8.33. Found: C, 57.41; H, 6.21; N, 8.44.
4.1.3. Isopropyl 4-(3,4-Dimethoxyphenyl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (5)
Yellow crystals (acetonitrile) in (0.295 g, 42% yield), mp 178–180 °C; 1H NMR (400 MHz, DMSO-d6, δ): (ppm): 10.26 (s, 1H, N3-H), 9.57 (s, 1H, N1-H), 6.92 (d, J = 8.4 Hz, 1H, Ar-H), 6.81 (s, 1H, Ar-H), 6.71 (d, J = 8.4 Hz, 1H, Ar-H), 5.11 (br s, 1H, C4-H), 4.90–4.80 (m, 1H, isopropyl-CH), 3.72 (s, 6H, 2× OCH3), 2.28 (s, 3H, C6-CH3), 1.17 (d, J = 6.4 Hz, 3H, isopropyl-CH3), 1.04 (d, J = 6.4 Hz, 3H, isopropyl-CH3); 13C NMR (100 MHz, DMSO-d6, δ): (ppm): 174.62, 165.19, 148.94, 148.78, 145.06, 136.57, 118.71, 112.30, 110.98, 101.63, 67.31, 56.01, 55.92, 54.10, 22.20, 21.97, 17.56; MS (ESI+) m/z 350.47 [M]+. Anal. Calcd for C17H22N2O4S (350.43): C, 58.27; H, 6.33; N, 7.99. Found: C, 58.39; H, 6.43; N, 8.14.
4.1.4. Isopropyl 6-Methyl-2-thioxo-4-(3,4,5-trimethoxyphenyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate (6)
White crystals (ethanol) in (0.573 g, 75% yield), mp 203–204 °C; 1H NMR (400 MHz, DMSO-d6, δ): (ppm): 10.31 (s, 1H, N3-H), 9.61 (s, 1H, N1-H), 6.51 (s, 2H, Ar-H), 5.13 (br s, 1H, C4-H), 4.93–4.84 (m, 1H, isopropyl-CH), 3.72 (s, 6H, 2× OCH3), 3.63 (s, 3H, OCH3), 2.28 (s, 3H, C6-CH3), 1.19 (d, J = 6.4 Hz, 3H, isopropyl-CH3), 1.07 (d, J = 6.4 Hz, 3H, isopropyl-CH3); 13C NMR (100 MHz, DMSO-d6, δ): (ppm): 174.50, 164.73, 152.82, 144.88, 139.19, 137.03, 103.52, 100.93, 66.93, 60.02, 55.82, 53.87, 21.73, 21.52, 17.13; MS (ESI+) m/z 380.93 [M]+. Anal. Calcd for C18H24N2O5S (380.46): C, 56.83; H, 6.36; N, 7.36. Found: C, 56.64; H, 6.54; N, 7.45.
4.1.5. Isopropyl 4-(3-Hydroxyphenyl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (7)
Brown crystals (acetonitrile) in (0.355 g, 58% yield), mp 210–211 °C; 1H NMR (400 MHz, DMSO-d6, δ): (ppm): 10.25 (s, 1H, N3-H), 9.57 (s, 1H, N1-H), 9.42 (s, 1H, OH), 7.11 (t, J = 8.4 Hz, 1H, Ar-H), 6.64 (d, J = 8.4 Hz, 3H, Ar-H), 5.06 (br s, 1H, C4-H), 4.88–4.79 (m, 1H, isopropyl-CH), 2.27 (s, 3H, C6-CH3), 1.17 (d, J = 6.4 Hz, 3H, isopropyl-CH3), 1.03 (d, J = 6.4 Hz, 3H, isopropyl-CH3); 13C NMR (100 MHz, DMSO-d6, δ): (ppm): 174.59, 165.14, 157.85, 145.42, 144.98, 129.87, 117.53, 115.03, 113.78, 101.57, 67.35, 54.51, 22.19, 21.89, 17.55; MS (ESI+) m/z 306.16 [M]+. Anal. Calcd for C15H18N2O3S (306.38): C, 58.80; H, 5.92; N, 9.14. Found: C, 58.91; H, 6.00; N, 9.28.
4.1.6. Isopropyl 4-(4-Hydroxyphenyl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (8)
Brown crystals (acetonitrile) in (0.369 g, 60% yield), mp 189–193 °C; 1H NMR (400 MHz, DMSO-d6, δ): (ppm): 10.20 (s, 1H, N3-H), 9.52 (s, 1H, N1-H), 9.39 (s, 1H, OH), 7.00 (d, J = 8.4 Hz, 2H, Ar-H), 6.70 (d, J = 8.4 Hz, 2H, Ar-H), 5.04 (br s, 1H, C4-H), 4.86–4.77 (m, 1H, isopropyl-CH), 2.27 (s, 3H, C6-CH3), 1.16 (d, J = 6.4 Hz, 3H, isopropyl-CH3), 1.00 (d, J = 6.4 Hz, 3H, isopropyl-CH3); 13C NMR (100 MHz, DMSO-d6, δ): (ppm): 173.83, 164.68, 156.87, 144.24, 134.25, 127.70, 115.07, 101.41, 66.72, 53.67, 21.72, 21.43, 17.05; MS (ESI+) m/z 306.20 [M]+. Anal. Calcd for C15H18N2O3S (306.38): C, 58.80; H, 5.92; N, 9.14. Found: C, 58.92; H, 6.12; N, 9.38.
4.1.7. Isopropyl 4-(4-Hydroxy-3-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (9)
White powder (ethanol) in (0.463 g, 60% yield), mp 197–198 °C; 1H NMR (400 MHz, DMSO-d6, δ): (ppm): 9.07 (s, 1H, N3-H), 8.88 (s, 1H, OH), 7.60 (s, 1H, N1-H), 6.78 (s, 1H, Ar-H), 6.70 (d, J = 8.0 Hz, 1H, Ar-H), 6.62 (d, J = 8.0 Hz, 1H, Ar-H), 5.04 (br s, 1H, C4-H), 4.87–4.77 (m, 1H, isopropyl-CH), 3.72 (s, 3H, OCH3), 2.23 (s, 3H, C6-CH3), 1.16 (d, J = 6.4 Hz, 3H, isopropyl-CH3), 1.02 (d, J = 6.4 Hz, 3H, isopropyl-CH3); 13C NMR (100 MHz, DMSO-d6, δ): (ppm): 164.97, 152.21, 147.62, 147.18, 145.75, 136.07, 118.40, 115.26, 110.93, 99.87, 66.24, 55.58, 53.64, 21.81, 21.56, 17.68; MS (ESI+) m/z 320.24 [M]+. Anal. Calcd for C16H20N2O5 (320.35): C, 59.99; H, 6.29; N, 8.74. Found: C, 60.16; H, 6.38; N, 8.92.
4.1.8. Isopropyl 4-(3,4-Dimethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (10)
White powder (acetonitrile) in (0.557 g, 83% yield), mp 215–218 °C; 1H NMR (400 MHz, DMSO-d6, δ): (ppm): 9.10 (s, 1H, N3-H), 7.64 (s, 1H, N1-H), 6.89 (d, J = 8.0 Hz, 1H, Ar-H), 6.83 (s, 1H, Ar-H), 6.73 (d, J = 8.0 Hz, 1H, Ar-H), 5.08 (br s, 1H, C4-H), 4.87–4.78 (m, 1H, isopropyl-CH), 3.71 (s, 6H, 2× OCH3), 2.23 (s, 3H, C6-CH3), 1.17 (d, J = 6.4 Hz, 3H, isopropyl-CH3), 1.02 (d, J = 6.4 Hz, 3H, isopropyl-CH3); 13C NMR (100 MHz, DMSO-d6, δ): (ppm): 165.06, 152.33, 148.48, 148.11, 147.93, 137.55, 118.10, 111.82, 110.59, 99.84, 66.45, 55.63, 55.52, 53.63, 21.89, 21.65, 17.78; MS (ESI+) m/z 334.95 [M]+. Anal. Calcd for C17H22N2O5 (334.37): C, 61.07; H, 6.63; N, 8.38. Found: C, 61.16; H, 6.75; N, 8.53.
4.1.9. Isopropyl 6-Methyl-2-oxo-4-(3,4,5-trimethoxyphenyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate (11)
White powder (ethanol) in (0.591 g, 81% yield), mp 165–170 °C; 1H NMR (400 MHz, DMSO-d6, δ): (ppm): 9.14 (s, 1H, N3-H), 7.69 (s, 1H, N1-H), 6.52 (s, 2H, Ar-H), 5.10 (br s, 1H, C4-H), 4.90–4.80 (m, 1H, isopropyl-CH), 3.72 (s, 6H, 2× OCH3), 3.63 (s, 3H, OCH3), 2.24 (s, 3H, C6-CH3), 1.17 (d, J = 6.4 Hz, 3H, isopropyl-CH3), 1.04 (d, J = 6.4 Hz, 3H, isopropyl-CH3); 13C NMR (100 MHz, DMSO-d6, δ): (ppm): 165.06, 152.80, 152.33, 148.27, 140.66, 136.86, 103.58, 99.50, 66.53, 60.12, 55.89, 54.01, 21.88, 21.64, 17.81; MS (ESI+) m/z 364.27 [M]+. Anal. Calcd for C18H24N2O6 (364.40): C, 59.33; H, 6.64; N, 7.69. Found: C, 59.51; H, 6.41; N, 7.91.
4.1.10. Isopropyl 4-(3-Hydroxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (12)
Brown powder (ethanol) in (0.406 g, 70% yield), mp 193–195 °C; 1H NMR (400 MHz, DMSO-d6, δ): (ppm): 9.34 (s, 1H, OH), 9.11 (s, 1H, N3-H), 7.66 (s, 1H, N1-H), 7.10 (t, J = 8.4 Hz, 1H, Ar-H), 6.69–6.61 (m, 3H, Ar-H), 5.05 (br s, 1H, C4-H), 4.87–4.78 (m, 1H, isopropyl-CH), 2.24 (s, 3H, C6-CH3), 1.18 (d, J = 6.4 Hz, 3H, isopropyl-CH3), 1.03 (d, J = 6.4 Hz, 3H, isopropyl-CH3); 13C NMR (100 MHz, DMSO-d6, δ): (ppm): 164.88, 157.30, 152.18, 147.79, 146.38, 129.19, 116.95, 114.12, 113.16, 99.68, 66.33, 53.95, 21.81, 21.49, 17.71; MS (ESI+) m/z 290.74 [M]+. Anal. Calcd for C15H18N2O4 (290.32): C, 62.06; H, 6.25; N, 9.65. Found: C, 62.31; H, 6.34; N, 9.91.
4.1.11. Isopropyl 4-(4-Hydroxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (13)
Yellow powder (acetonitrile) in (0.432 g, 74% yield), mp 186–189 °C [lit. 192–194 °C]; 1H NMR (400 MHz, DMSO-d6, δ): (ppm): 9.30 (s, 1H, OH), 9.06 (s, 1H, N3-H), 7.57 (s, 1H, N1-H), 7.02 (d, J = 8.0 Hz, 2H, Ar-H), 6.68 (d, J = 8.0 Hz, 2H, Ar-H), 5.02 (br s, 1H, C4-H), 4.84–4.75 (m, 1H, isopropyl-CH), 2.22 (s, 3H, C6-CH3), 1.15 (d, J = 6.4 Hz, 3H, isopropyl-CH3), 0.99 (d, J = 6.4 Hz, 3H, isopropyl-CH3); 13C NMR (100 MHz, DMSO-d6, δ): (ppm): 164.90, 156.49, 152.11, 147.47, 135.57, 127.45, 114.90, 100.01, 66.19, 53.54, 21.80, 21.50, 17.66; MS (ESI+) m/z 290.27 [M]+. Anal. Calcd for C15H18N2O4 (290.32): C, 62.06; H, 6.25; N, 9.65. Found: C, 61.85; H, 6.39; N, 9.77.
4.1.12. Isopropyl 4-(3-Hydroxy-4-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (14)
White powder (acetonitrile) in (0.492 g, 77% yield), mp 216–217 °C; 1H NMR (400 MHz, DMSO-d6, δ): (ppm): 9.07 (s, 1H, N3-H), 8.91 (s, 1H, OH), 7.59 (s, 1H, N1-H), 6.82 (d, J = 8.4 Hz, 1H, Ar-H), 6.69 (s, 1H, Ar-H), 6.61 (d, J = 8.4 Hz, 1H, Ar-H), 5.00 (br s, 1H, C4-H), 4.86–4.77 (m, 1H, isopropyl-CH), 3.72 (s, 3H, OCH3), 2.22 (s, 3H, C6-CH3), 1.16 (d, J = 6.4 Hz, 3H, isopropyl-CH3), 1.03 (d, J = 6.4 Hz, 3H, isopropyl-CH3); 13C NMR (100 MHz, DMSO-d6, δ): (ppm): 165.40, 152.66, 147.91, 147.30, 146.72, 138.20, 117.42, 114.16, 112.41, 100.47, 66.75, 56.13, 53.99, 22.28, 22.01, 18.15; MS (ESI+) m/z 320.70 [M]+. Anal. Calcd for C16H20N2O5 (320.35): C, 59.99; H, 6.29; N, 8.74. Found: C, 60.24; H, 6.38; N, 8.90.
4.1.13. Isopropyl 4-[4-(Benzyloxy)-3-methoxyphenyl]-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (15)
White crystals (dioxane) in (0.561 g, 68% yield), mp 229–232 °C; 1H NMR (400 MHz, DMSO-d6, δ): (ppm): 9.11 (s, 1H, N3-H), 7.65 (s, 1H, N1-H), 7.47–7.28 (m, 5H, Ar-H), 6.98 (d, J = 8.4 Hz, 1H, Ar-H), 6.87 (s, 1H, Ar-H), 6.71 (d, J = 8.4 Hz, 1H, Ar-H), 5.08 (br s, 1H, C4-H), 5.05 (s, 2H, benzylic-CH2), 4.88–4.78 (m, 1H, isopropyl-CH), 3.74 (s, 3H, OCH3), 2.24 (s, 3H, C6-CH3), 1.17 (d, J = 6.4 Hz, 3H, isopropyl-CH3), 1.02 (d, J = 6.4 Hz, 3H, isopropyl-CH3); 13C NMR (100 MHz, DMSO-d6, δ): (ppm): 165.40, 152.66, 149.28, 148.38, 147.44, 138.42, 137.64, 128.85, 128.26, 128.19, 118.44, 114.08, 111.29, 100.12, 70.43, 66.78, 56.02, 54.04, 22.28, 22.03, 18.18; MS (ESI+) m/z 410.59 [M]+. Anal. Calcd for C23H26N2O5 (410.47): C, 67.30; H, 6.38; N, 6.82. Found: C, 67.40; H, 6.64; N, 7.01.
4.2. Biology
4.2.1. Cell Viability Assay
The human mammary gland epithelial (MCF-10A) normal cell line was utilized to analyze the viability effect of new targets 4–15.43,44 After 4 days of incubation on MCF-10A cells, the cell viability of 4–15 was assessed using the MTT test. For more details, please see Appendix A (Supporting Information).
4.2.2. Cytotoxic Assay
Targets 4–15 were tested for cytotoxic effects against four human cancer cell lines using the MTT assay.45,46 Refer to Appendix A for more information.
4.2.3. Assay for EGFR Inhibitory Action
Using the EGFR-TK assay, the most effective cytotoxic compounds, 6, 12, 14, and 15, were examined for their capacity to inhibit EGFR.47,48 See Appendix A.
4.2.4. Assay for VEGFR-2 Inhibitory Action
Using the kinase-glo-luminescent kinase assay,49 compounds 6, 12, 14, and 15 were evaluated for their ability to inhibit VEGFR-2, with sorafenib as the control medication. Table 2 displays the results as IC50 values. See Appendix A.
4.2.5. Apoptosis Induction Assay
Three candidates (12, 14, and 15) were selected to test their apoptotic inhibitory potential against A-549. In vitro, quantitative assessments of pro-apoptotic proteins (p53, BAX, caspase 3, and caspase 6) and antiapoptotic proteins (BCL-2 and CK-18) were done using a sandwiched immunoassay.54 See Appendix A.
4.3. Docking Study
The structures of the human epidermal growth factor receptor (PDB code: 1M17) and human vascular endothelial growth factor receptor 2 (PDB code: 4ASD) were downloaded from the protein data bank.13,53 Chemical structures were drawn and optimized using the molecular editors Marvin Sketch and Avogadro. The preparation of both protein structures was performed using Autodock tools v1.5.7,55 involving the removal of cocrystallized water molecules and reference compounds (erlotinib and sorafenib), followed by the addition of Kollman charges and polar hydrogens. See Appendix A.
Acknowledgments
The authors acknowledge the support by Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP2024R3), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors also acknowledge support from the KIT-Publication Fund of the Karlsruhe Institute of Technology.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c01361.
1H NMR, 13C NMR spectra, and elemental analysis of compounds 4–15 and Appendix A (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ma J.; Jemal A.; Fedewa S. A.; Islami F.; Lichtenfeld J. L.; Wender R. C.; Cullen K. J.; Brawley O. W. The American Cancer Society 2035 challenge goal on cancer mortality reduction. Ca-Cancer J. Clin. 2019, 69 (5), 351–362. 10.3322/caac.21564. [DOI] [PubMed] [Google Scholar]
- Milroy M. J.Cancer statistics: Global and national. Quality Cancer Care: Survivorship Before, During and After Treatment; Springer, 2018; pp 29–35. [Google Scholar]
- Faisca Phillips A. M. Recent developments in anti-cancer drug research. Curr. Med. Chem. 2020, 26 (41), 7282–7284. 10.2174/092986732641200108093655. [DOI] [PubMed] [Google Scholar]
- Zhong L.; Li Y.; Xiong L.; Wang W.; Wu M.; Yuan T.; Yang W.; Tian C.; Miao Z.; Wang T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduction Targeted Ther. 2021, 6 (1), 201. 10.1038/s41392-021-00572-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batool Z.; Azfal A.; Liaquat L.; Sadir S.; Nisar R.; Inamullah A.; Ghalib A. U. F.; Haider S.. Receptor tyrosine kinases (RTKs): from biology to pathophysiology. Receptor Tyrosine Kinases in Neurodegenerative and Psychiatric Disorders; Elsevier, 2023; pp 117–185. [Google Scholar]
- Saraon P.; Pathmanathan S.; Snider J.; Lyakisheva A.; Wong V.; Stagljar I. Receptor tyrosine kinases and cancer: oncogenic mechanisms and therapeutic approaches. Oncogene 2021, 40 (24), 4079–4093. 10.1038/s41388-021-01841-2. [DOI] [PubMed] [Google Scholar]
- Sabbah M.; Najem A.; Krayem M.; Awada A.; Journe F.; Ghanem G. E. RTK inhibitors in melanoma: from bench to bedside. Cancers 2021, 13 (7), 1685. 10.3390/cancers13071685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colardo M.; Segatto M.; Di Bartolomeo S. Targeting RTK-PI3K-mTOR axis in gliomas: An update. Int. J. Mol. Sci. 2021, 22 (9), 4899. 10.3390/ijms22094899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird A. D.; Cherrington J. M. Small molecule tyrosine kinase inhibitors: clinical development of anticancer agents. Expert Opin. Invest. Drugs 2003, 12 (1), 51–64. 10.1517/13543784.12.1.51. [DOI] [PubMed] [Google Scholar]
- Pottier C.; Fresnais M.; Gilon M.; Jérusalem G.; Longuespée R.; Sounni N. E. Tyrosine kinase inhibitors in cancer: breakthrough and challenges of targeted therapy. Cancers 2020, 12 (3), 731. 10.3390/cancers12030731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L.; Fu L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm. Sin. B 2015, 5 (5), 390–401. 10.1016/j.apsb.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewale C.; Baradia D.; Vhora I.; Patil S.; Misra A. Epidermal growth factor receptor targeting in cancer: a review of trends and strategies. Biomaterials 2013, 34 (34), 8690–8707. 10.1016/j.biomaterials.2013.07.100. [DOI] [PubMed] [Google Scholar]
- Mohamed F. A.; Gomaa H. A.; Hendawy O.; Ali A. T.; Farghaly H. S.; Gouda A. M.; Abdelazeem A. H.; Abdelrahman M. H.; Trembleau L.; Youssif B. G. Design, synthesis, and biological evaluation of novel EGFR inhibitors containing 5-chloro-3-hydroxymethyl-indole-2-carboxamide scaffold with apoptotic antiproliferative activity. Bioorg. Chem. 2021, 112, 104960. 10.1016/j.bioorg.2021.104960. [DOI] [PubMed] [Google Scholar]
- Modi S. J.; Kulkarni V. M. Vascular endothelial growth factor receptor (VEGFR-2)/KDR inhibitors: medicinal chemistry perspective. Med. Drug Discovery 2019, 2, 100009. 10.1016/j.medidd.2019.100009. [DOI] [Google Scholar]
- Falcon B. L.; Chintharlapalli S.; Uhlik M. T.; Pytowski B. Antagonist antibodies to vascular endothelial growth factor receptor 2 (VEGFR-2) as anti-angiogenic agents. Pharmacol. Ther. 2016, 164, 204–225. 10.1016/j.pharmthera.2016.06.001. [DOI] [PubMed] [Google Scholar]
- Peng F.-W.; Liu D.-K.; Zhang Q.-W.; Xu Y.-G.; Shi L. VEGFR-2 inhibitors and the therapeutic applications thereof: a patent review (2012–2016). Expert Opin. Ther. Pat. 2017, 27 (9), 987–1004. 10.1080/13543776.2017.1344215. [DOI] [PubMed] [Google Scholar]
- Lemmon M. A.; Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141 (7), 1117–1134. 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J. J.-L. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J. Med. Chem. 2007, 50 (3), 409–424. 10.1021/jm0608107. [DOI] [PubMed] [Google Scholar]
- Wheler J. J.; Janku F.; Naing A.; Li Y.; Stephen B.; Zinner R.; Subbiah V.; Fu S.; Karp D.; Falchook G. S.; et al. TP53 alterations correlate with response to VEGF/VEGFR inhibitors: implications for targeted therapeutics. Mol. Cancer Ther. 2016, 15 (10), 2475–2485. 10.1158/1535-7163.mct-16-0196. [DOI] [PubMed] [Google Scholar]
- Xie C.; Wan X.; Quan H.; Zheng M.; Fu L.; Li Y.; Lou L. Preclinical characterization of anlotinib, a highly potent and selective vascular endothelial growth factor receptor-2 inhibitor. Cancer Sci. 2018, 109 (4), 1207–1219. 10.1111/cas.13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.-L.; Chen H.-H.; Zheng L.-L.; Sun L.-P.; Shi L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduction Targeted Ther. 2023, 8 (1), 198. 10.1038/s41392-023-01460-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S.; Zhang J.; Wang N.; Kong X.; Fu F.; Wang H.; Yao J. Design and discovery of quinazoline-and thiourea-containing sorafenib analogs as EGFR and VEGFR-2 dual TK inhibitors. Molecules 2018, 23 (1), 24. 10.3390/molecules23010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.-J.; Zhao H.-C.; Hou S.-J.; Zhang H.-J.; Cheng L.; Yuan S.; Zhang L.-R.; Song J.; Zhang S.-Y.; Chen S.-W. Recent development of multi-target VEGFR-2 inhibitors for the cancer therapy. Bioorg. Chem. 2023, 133, 106425. 10.1016/j.bioorg.2023.106425. [DOI] [PubMed] [Google Scholar]
- Farghaly T. A.; Al-Hasani W. A.; Abdulwahab H. G. An updated patent review of VEGFR-2 inhibitors (2017-present). Expert Opin. Ther. Pat. 2021, 31 (11), 989–1007. 10.1080/13543776.2021.1935872. [DOI] [PubMed] [Google Scholar]
- Zhang H.-Q.; Gong F.-H.; Li C.-G.; Zhang C.; Wang Y.-J.; Xu Y.-G.; Sun L.-P. Design and discovery of 4-anilinoquinazoline-acylamino derivatives as EGFR and VEGFR-2 dual TK inhibitors. Eur. J. Med. Chem. 2016, 109, 371–379. 10.1016/j.ejmech.2015.12.032. [DOI] [PubMed] [Google Scholar]
- Ganem B. Strategies for innovation in multicomponent reaction design. Acc. Chem. Res. 2009, 42 (3), 463–472. 10.1021/ar800214s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza R. O.; da Penha E. T.; Milagre H. M.; Garden S. J.; Esteves P. M.; Eberlin M. N.; Antunes O. A. The three-component Biginelli reaction: a combined experimental and theoretical mechanistic investigation. Chem.—Eur. J. 2009, 15 (38), 9799–9804. 10.1002/chem.200900470. [DOI] [PubMed] [Google Scholar]
- Naishima N. L.; Faizan S.; Raju R. M.; Sruthi A. S. V. L.; Ng V.; Sharma G. K.; Vasanth K. S.; Shivaraju V. K.; Ramu R.; Kumar B. P. Design, Synthesis, Analysis, Evaluation of Cytotoxicity Against MCF-7 Breast Cancer Cells, 3D QSAR Studies and EGFR, HER2 Inhibition Studies on Novel Biginelli 1, 4-Dihydropyrimidines. J. Mol. Struct. 2023, 1277, 134848. 10.1016/j.molstruc.2022.134848. [DOI] [Google Scholar]
- Faizan S.; Roohi T. F.; Raju R. M.; Sivamani Y.; Br P. K. A Century-Old One-Pot Multicomponent Biginelli Reaction Products Still Finds a Niche in Drug Discoveries: Synthesis, Mechanistic studies and Diverse Biological Activities of Dihydropyrimidines. J. Mol. Struct. 2023, 1291, 136020. 10.1016/j.molstruc.2023.136020. [DOI] [Google Scholar]
- Padmashali B.; Chidananda B. N.; Govindappa B.; Basavaraj S. M.; Chandrashekharappa S.; Venugopala K. N. Synthesis and characterization of novel 1, 6-dihydropyrimidine derivatives for their pharmacological properties. J. Appl. Pharm. Sci. 2019, 9 (5), 117–124. 10.7324/JAPS.2019.90517. [DOI] [Google Scholar]
- Nammalwar B.; Bunce R. A. Recent Advances in Pyrimidine-Based Drugs. Pharmaceuticals 2024, 17 (1), 104. 10.3390/ph17010104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim T.; Youssif B. G.; AL-Mahmoudy A.; Hassan A.; Taher E.; Tantawy M.; Abdel-Aal E.; Osman N. Novel Benzyloxyphenyl Pyrimidine-5-Carbonitrile Derivatives as Potential ApoptoticAntiproliferative Agents. Anti-Cancer Agents Med. Chem. 2022, 22 (5), 978–990. 10.2174/1871520621666210612043812. [DOI] [PubMed] [Google Scholar]
- Ahmed N. M.; Youns M. M.; Soltan M. K.; Said A. M. Design, Synthesis, Molecular Modeling and Antitumor Evaluation of Novel Indolyl-Pyrimidine Derivatives with EGFR Inhibitory Activity. Molecules 2021, 26 (7), 1838. 10.3390/molecules26071838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafa A. S.; Selim K. B. Synthesis and anticancer activity of new dihydropyrimidinone derivatives. Eur. J. Med. Chem. 2018, 156, 304–315. 10.1016/j.ejmech.2018.07.004. [DOI] [PubMed] [Google Scholar]
- Abou-Zied H. A.; Beshr E. A.; Gomaa H. A.; Mostafa Y. A.; Youssif B. G.; Hayallah A. M.; Abdel-Aziz M. Discovery of new cyanopyridine/chalcone hybrids as dual inhibitors of EGFR/BRAFV600E with promising antiproliferative properties. Arch. Pharm. 2023, 356 (4), 2200464. 10.1002/ardp.202200464. [DOI] [PubMed] [Google Scholar]
- Alshammari M. B.; Aly A. A.; Youssif B. G.; Bräse S.; Ahmad A.; Brown A. B.; Ibrahim M. A.; Mohamed A. H. Design and synthesis of new thiazolidinone/uracil derivatives as antiproliferative agents targeting EGFR and/or BRAFV600E. Front. Chem. 2022, 10, 1076383. 10.3389/fchem.2022.1076383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Wahaibi L. H.; Abou-Zied H. A.; Beshr E. A.; Youssif B. G.; Hayallah A. M.; Abdel-Aziz M. Design, Synthesis, Antiproliferative Actions, and DFT Studies of New Bis–Pyrazoline Derivatives as Dual EGFR/BRAFV600E Inhibitors. Int. J. Mol. Sci. 2023, 24 (10), 9104. 10.3390/ijms24109104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Wahaibi L. H.; Abou-Zied H. A.; Hisham M.; Beshr E. A.; Youssif B. G.; Bräse S.; Hayallah A. M.; Abdel-Aziz M. Design, Synthesis, and Biological Evaluation of Novel 3-Cyanopyridone/Pyrazoline Hybrids as Potential Apoptotic Antiproliferative Agents Targeting EGFR/BRAFV600E Inhibitory Pathways. Molecules 2023, 28 (18), 6586. 10.3390/molecules28186586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Wahaibi L. H.; El-Sheref E. M.; Hammouda M. M.; Youssif B. G. One-Pot Synthesis of 1-Thia-4-azaspiro[4.4/5]alkan-3-ones via Schiff Base: Design, Synthesis, and Apoptotic Antiproliferative Properties of Dual EGFR/BRAFV600E Inhibitors. Pharmaceuticals 2023, 16 (3), 467. 10.3390/ph16030467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Wahaibi L. H.; El-Sheref E. M.; Hassan A. A.; Bräse S.; Nieger M.; Youssif B. G.; Ibrahim M. A.; Tawfeek H. N. Synthesis and Structure Determination of Substituted Thiazole Derivatives as EGFR/BRAFV600E Dual Inhibitors Endowed with Antiproliferative Activity. Pharmaceuticals 2023, 16 (7), 1014. 10.3390/ph16071014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Wahaibi L. H.; Gouda A. M.; Abou-Ghadir O. F.; Salem O. I.; Ali A. T.; Farghaly H. S.; Abdelrahman M. H.; Trembleau L.; Abdu-Allah H. H.; Youssif B. G. Design and synthesis of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as antiproliferative EGFR and BRAFV600E dual inhibitors. Bioorg. Chem. 2020, 104, 104260. 10.1016/j.bioorg.2020.104260. [DOI] [PubMed] [Google Scholar]
- Al-Wahaibi L. H.; Mahmoud M. A.; Mostafa Y. A.; Raslan A. E.; Youssif B. G. Novel piperine-carboximidamide hybrids: design, synthesis, and antiproliferative activity via a multi-targeted inhibitory pathway. J. Enzyme Inhib. Med. Chem. 2023, 38 (1), 376–386. 10.1080/14756366.2022.2151593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekheimer R. A.; Allam S. M.; Al-Sheikh M. A.; Moustafa M. S.; Al-Mousawi S. M.; Mostafa Y. A.; Youssif B. G.; Gomaa H. A.; Hayallah A. M.; Abdelaziz M.; et al. Discovery of new pyrimido[5,4-c]quinolines as potential antiproliferative agents with multitarget actions: Rapid synthesis, docking, and ADME studies. Bioorg. Chem. 2022, 121, 105693. 10.1016/j.bioorg.2022.105693. [DOI] [PubMed] [Google Scholar]
- Mahmoud M. A.; Mohammed A. F.; Salem O. I.; Gomaa H. A.; Youssif B. G. New 1,3,4-oxadiazoles linked with the 1,2,3-triazole moiety as antiproliferative agents targeting the EGFR tyrosine kinase. Arch. Pharm. 2022, 355 (6), 2200009. 10.1002/ardp.202200009. [DOI] [PubMed] [Google Scholar]
- Hisham M.; Hassan H. A.; Gomaa H. A.; Youssif B. G.; Hayallah A. M.; Abdel-Aziz M. Structure-based design, synthesis and antiproliferative action of new quinazoline-4-one/chalcone hybrids as EGFR inhibitors. J. Mol. Struct. 2022, 1254, 132422. 10.1016/j.molstruc.2022.132422. [DOI] [Google Scholar]
- Hagar F. F.; Abbas S. H.; Abdelhamid D.; Gomaa H. A.; Youssif B. G.; Abdel-Aziz M. New 1,3,4-oxadiazole-chalcone/benzimidazole hybrids as potent antiproliferative agents. Arch. Pharm. 2023, 356 (2), 2200357. 10.1002/ardp.202200357. [DOI] [PubMed] [Google Scholar]
- Maghraby M.; Salem O.; Youssif B.; Sheha M. Design, synthesis, and modelling study of new 1,2,3-triazole/chalcone hybrids with antiproliferative action as epidermal growth factor receptor inhibitors. Chem. Biol. Drug Des. 2022, 101, 749–759. 10.1111/cbdd.14178. [DOI] [PubMed] [Google Scholar]
- Al-Wahaibi L. H.; Mostafa Y. A.; Abdelrahman M. H.; El-Bahrawy A. H.; Trembleau L.; Youssif B. G. Synthesis and Biological Evaluation of Indole-2-Carboxamides with Potent Apoptotic Antiproliferative Activity as EGFR/CDK2 Dual Inhibitors. Pharmaceuticals 2022, 15 (8), 1006. 10.3390/ph15081006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud M. A.; Mohammed A. F.; Salem O. I.; Rabea S. M.; Youssif B. G. Design, synthesis, and antiproliferative properties of new 1,2,3-triazole-carboximidamide derivatives as dual EGFR/VEGFR-2 inhibitors. J. Mol. Struct. 2023, 1282, 135165. 10.1016/j.molstruc.2023.135165. [DOI] [Google Scholar]
- Schultz D. R.; Harringto W. J. Jr.. Apoptosis: programmed cell death at a molecular level. Seminars in Arthritis and Rheumatism; Elsevier, 2003; pp 345–369. [DOI] [PubMed]
- Chota A.; George B. P.; Abrahamse H. Interactions of multidomain pro-apoptotic and anti-apoptotic proteins in cancer cell death. Oncotarget 2021, 12 (16), 1615–1626. 10.18632/oncotarget.28031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulukaya E.; Acilan C.; Yilmaz Y. Apoptosis: why and how does it occur in biology?. Cell Biochem. Funct. 2011, 29 (6), 468–480. 10.1002/cbf.1774. [DOI] [PubMed] [Google Scholar]
- McTigue M.; Murray B. W.; Chen J. H.; Deng Y.-L.; Solowiej J.; Kania R. S. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2012, 109 (45), 18281–18289. 10.1073/pnas.1207759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler J. E. Enzyme-linked immunosorbent assay. J. Immunoassay 2000, 21 (2–3), 165–209. 10.1080/01971520009349533. [DOI] [PubMed] [Google Scholar]
- Shaykoon M. S.; Marzouk A. A.; Soltan O. M.; Wanas A. S.; Radwan M. M.; Gouda A. M.; Youssif B. G.; Abdel-Aziz M. Design, synthesis and antitrypanosomal activity of heteroaryl-based 1, 2, 4-triazole and 1, 3, 4-oxadiazole derivatives. Bioorg. Chem. 2020, 100, 103933. 10.1016/j.bioorg.2020.103933. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.