

Abstract
Background
The BLAST algorithm compares biological sequences to one another in order to determine shared motifs and common ancestry. However, the comparison of all non-redundant (NR) sequences against all other NR sequences is a computationally intensive task. We developed NBLAST as a cluster computer implementation of the BLAST family of sequence comparison programs for the purpose of generating pre-computed BLAST alignments and neighbour lists of NR sequences.
Results
NBLAST performs the heuristic BLAST algorithm and generates an exhaustive database of alignments, but it only computes 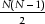 alignments (i.e. the upper triangle) of a possible N2 alignments, where N is the set of all sequences to be compared. A task-partitioning algorithm allows for cluster computing across all cluster nodes and the NBLAST master process produces a BLAST sequence alignment database and a list of sequence neighbours for each sequence record. The resulting sequence alignment and neighbour databases are used to serve the SeqHound query system through a C/C++ and PERL Application Programming Interface (API).
alignments (i.e. the upper triangle) of a possible N2 alignments, where N is the set of all sequences to be compared. A task-partitioning algorithm allows for cluster computing across all cluster nodes and the NBLAST master process produces a BLAST sequence alignment database and a list of sequence neighbours for each sequence record. The resulting sequence alignment and neighbour databases are used to serve the SeqHound query system through a C/C++ and PERL Application Programming Interface (API).
Conclusions
NBLAST offers a local alternative to the NCBI's remote Entrez system for pre-computed BLAST alignments and neighbour queries. On our 216-processor 450 MHz PIII cluster, NBLAST requires ~24 hrs to compute neighbours for 850000 proteins currently in the non-redundant protein database.
Background
Sequence comparison algorithms are widely used to search protein and DNA databases in reconstructing relationships, phylogenies and assigning gene/protein function. Several implementations have been developed to either increase the sensitivity of the search or to speed up the comparison. One implementation that is widely used to speed up similarity searching is the related sequences, or "neighbour" service of the National Centre for Biotechnology Information's (NCBI) Entrez system http://www.ncbi.nlm.nih.gov. The neighbour service is dependent on the fast, heuristic protein and DNA similarity algorithm in the BLAST program [1]. Upon retrieval of a sequence with the Entrez system, one is offered a choice to retrieve a set of similar sequences based on a table of pre-computed BLAST comparisons. Neighbour systems are effective speedups when a sequence is already known to reside in the database, as one does not have to re-compute the BLAST comparison.
Since the amount of time required for such a comparison is currently on the order of 5000 CPU hours, there is requirement for a supercomputer or cluster-computing version. NBLAST provides a platform for the parallel computing and storage of BLAST sequences in an all against all comparison and further provides the ability to generate sequence neighbours in a parallel fashion. We created NBLAST since to our knowledge no such implementation of BLAST exists outside NCBI to compute neighbours.
Results
NBLAST is a freely available multi-platform application, which was written in C using the NCBI Toolkit [2] and has been cross-compiled on Windows 98/ME/NT/2000, MacOsX, Linux, HP-UX, PA-RISC Linux, Compaq Tru64, IRIX, Solaris, QNX, FreeBSD and PowerPC-Linux operating systems. Source code and binaries are freely available at http://www.sourceforge.net/projects/slritools/. NBLAST uses a modified version of NCBI's blastall.c source file and can be compiled using definition flags into the NBLAST binary executable. NBLAST source files are divided into an application layer and a separate database layer, for maximal portability to other database systems. The NBLAST binary executables include the xBase file-compatible CodeBase (Sequiter Software Inc., http://www.sequiter.com), a lightweight, royalty-free, cross-platform database management system, to store sequence alignments and neighbour lists. CodeBase provides the output database format that are used to store the large alignment files which amount to about 70 GB of information for an e-value cut-off of 0.01.
The sequence database must be a FASTA file formatted with formatdb ftp://ftp.ncbi.nih.nlm.gov/ using the option to generate full indexes (-oT). NBLAST first iterates through the data file to parse out the GenInfo (GI) identifier, a unique identifier given for each sequence record deposited in the NCBI repository. NBLAST stores each GI along with a unique ordinal (ORD) number in the NBlastDB database. The ordinal numbers are hashed to store the BLAST comparisons in the BlastDB database.
NBLAST computes the minimal number of required comparisons for an all by all or otherwise specified range of BLAST sequence comparisons. The NBLAST application also includes a task-division algorithm to enable computer cluster or supercomputer computation of all or a user-specified range of sequences. For cluster computing, NBLAST integrates the MoBiDiCK API for easy deployment and status reporting of each node [3], but could be adapted to other cluster management software like Load Sharing Facility, LSF http://www.platform.com or Portable Batch System, PBS http://www.openpbs.org. A checkpoint restart function has been implemented so that the program can be interrupted at any point during the BLAST job and can be continued using a command line option from where it left off.
When the cluster nodes have completed their tasks, the databases must be moved to a master server. Once all the databases are located together, NBLAST can assemble the databases into a single database, and/or generate a neighbour list for each GI using a specified e-value cut-off. Both alignments and sequence neighbours are retrieved through API calls to SeqHound, a sequence database manager [4].
All against all comparison
An exhaustive analysis between all sequences (N) in a database would result in a total of N2 comparisons. However, the BLAST comparison of sequence A with sequence B is roughly equivalent to the comparison of sequence B with sequence A for significant hits, thus, just less than one half of the comparisons are redundant and only 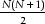 comparisons should be made. Moreover, we can ignore the identity comparison of sequence A with itself, thereby minimizing an exhaustive N2 comparison to comparisons.
comparisons should be made. Moreover, we can ignore the identity comparison of sequence A with itself, thereby minimizing an exhaustive N2 comparison to comparisons.
Partition algorithm and cluster computing
The largest problem facing the computation of all-against-all sequence alignments is the total time required for the comparison. A simple task-partitioning algorithm has been implemented to divide the database for equal number of comparisons according to the number of machines/processes working on the computation. The algorithm determines the number of rows each machine must work on and then assigns half of the rows from the upper half of the triangle and half from the lower half (Figure 1a). Each machine must be distributed a copy of the FASTA formatted database so that NBLAST may seek out the set of query and database sequences to perform the BLAST comparisons. The result is that each machine performs a similar number of comparisons over a comparable time frame. On our 108 node cluster computer, where each node possesses two 450 MHz PIII processors and 512 MB RAM, NBLAST requires ~24 hrs to compute neighbours for 850000 proteins from the non-redundant protein database.
Figure 1.

NBLAST task partitioning and unique identifier assignment scheme. (a) The division of the all-against-all (N2) sequence comparison using NBLAST is shown here for i = 4 computers. Each computer is allocated N/i rows from the sequence database, which are partitioned between the upper half and the lower half of the triangle. This results in an equivalent number of N2/(2*i) comparisons for each computer over a similar time frame. (b) The mapping function generates a unique identifier (UID) (in small boxes) for pairwise comparison between ordinal numbers (ORD1, ORD2) assigned to database records.
BLAST results, indexing and storage
The result of a BLAST comparison is called a SeqAlign. It is an ASN.1 structure http://www.ncbi.nlm.nih.gov/IEB/ToolBox/SDKDOCS/ASNLIB.HTML that describes the sequence alignment. NBLAST stores a modified SeqAlign that includes the query GI and subject GI along with several elements of important alignment information. The first is the bit score and the e-value. The bit score is a normalized score that represents the probability level for sequence comparisons that is independent of the scoring system used and provides an estimate of the search space required to find such an alignment by chance. The expected probability of the number of hits occurring by chance is called the e-value, and is generated from the bit score and the size of the database. Thus, as the database size increases, the e-value can be rapidly be regenerated from the bit score, without having to redo the comparison. The second part of the SeqAlign describes the alignment with the number and lengths of alignment segments, and their positions relative to the start of each sequence.
Indexing pairwise alignments uniquely in a database comparison involving large numbers of sequences growing as the square of the database size represents another problem we had to solve. We found that we were able to generate a unique identifier (UID) by means of a mapping function using the ordinal numbers of the two sequences for each pairwise comparison (Figure 1b). The 64-bit integer identifier UID is computed as follows: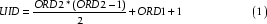
where ORD1 and ORD2 are 32 bit integers. By definition ORD1 < ORD2, so we can choose to set ORD1 = ORD2-1 and substitute into eq. 1: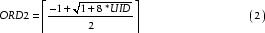
When ORD1 = ORD2-1 eq. 2 yields an exact integer, I, however for values of ORD1 in the interval [0,ORD2-2], the result will be a real number R, such that (I-R) < 1. Thus by applying the ceiling function we get the correct ORD2 for all values of ORD1. The ordinal number ORD1 is then easily obtained by rearrangement of eq. 1: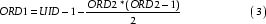
Another advantage of this method is that all generated UIDs are consecutive integers starting at 1 and require less space in ASCII encoded-form than full 64-bit keys.
Using simple shell scripts, we transfer the completed comparison databases (BlastDB) files from each node to a central server.
Neighbour generation
NBLAST iterates through the node-generated sequence comparison databases using a user-specified e-value threshold to generate the neighbour lists for each sequence record. The ASN.1 structure containing the neighbour lists has the query GI at the head and has its children as sorted GI/e-value pairs. Pre-computed CodeBase neighbour tables are available at ftp://ftp.mshri.on.ca/pub/NBLAST/.
Alignment and neighbour retrieval using the SeqHound API
NBLAST generates a table of computed sequence comparisons and sequence neighbours. C/C++/PERL functions to retrieve the ASN structures from the databases are incorporated into the SeqHound API [4]. Sequence alignments for a GI pair can be returned either as the NBLAST SeqAlign or NCBI SeqAlign, for processing and alignment visualization. Sequence neighbours to a query GI are returned in a dual array, called a FLinkSet, one for the subject GI and its corresponding e-value. Neighbour API calls include returning neighbours from a list of GIs or taxonomy id. Moreover, one can retrieve neighbours of neighbours for extended neighbour searching. Since NBLAST only parses out the first GI of every sequence record and SeqHound keeps track of redundant GIs, all valid NCBI GIs can be queried through SeqHound. Additional API calls integrate the sequence and structure databases, by querying the neighbour list for GIs for associated structures (neighbours with structures).
Daily update
A daily update scheme has been implemented to add the newest sequences from the non-redundant database and remove killed sequences from the list of BLAST comparisons and sequence neighbours. The update mechanism works on a single or multiple machines to BLAST new sequences, save the alignments and add to the neighbours list.
This program provides the framework for current sequence and structure studies and allows neighbour linking through SeqHound [4] and the BIND database [5] http://bioinfo.mshri.on.ca.
Contributor Information
Michel Dumontier, Email: micheld@mshri.on.ca.
Christopher WV Hogue, Email: hogue@mshri.on.ca.
Acknowledgments
We would like to thank our colleagues at the Samuel Lunenfeld Research Institute for their support in our work, especially Howard Feldman for deriving the mapping function, Gary Bader for his valuable input and Katerina Michalickova for developing the SeqHound database system. This research was supported by grants to C.W.V Hogue and M. Dumontier by the Natural Sciences and Engineering Research Council of Canada.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, DJ Lipman. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostell JM, Wheelan SJ, Kans JA. The NCBI data model. Methods Biochem Anal. 2001;43:19–43. doi: 10.1002/0471223921.ch2. [DOI] [PubMed] [Google Scholar]
- Dharsee M, Hogue CWV. 9th Heterogeneous Computing Workshop. IEEE Computer Society. 2000. pp. 323–335.
- Michalikova K, Bader GD, Dumontier M, Isserlin R, Hogue CWV. SeqHound biological sequence database system as a platform for bioinformatics research. in press . [DOI] [PMC free article] [PubMed]
- Bader GD, Donaldson I, Wolting C, Ouellette BF, Pawson T, Hogue CW. BIND – The Biomolecular Interaction Network Database. Nucleic Acids Res. 2001;29:242–5. doi: 10.1093/nar/29.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]