


Abstract
The Biomolecular Interaction Network Database (BIND; http://binddb.org) is a database designed to store full descriptions of interactions, molecular complexes and pathways. Development of the BIND 2.0 data model has led to the incorporation of virtually all components of molecular mechanisms including interactions between any two molecules composed of proteins, nucleic acids and small molecules. Chemical reactions, photochemical activation and conformational changes can also be described. Everything from small molecule biochemistry to signal transduction is abstracted in such a way that graph theory methods may be applied for data mining. The database can be used to study networks of interactions, to map pathways across taxonomic branches and to generate information for kinetic simulations. BIND anticipates the coming large influx of interaction information from high-throughput proteomics efforts including detailed information about post-translational modifications from mass spectrometry. Version 2.0 of the BIND data model is discussed as well as implementation, content and the open nature of the BIND project. The BIND data specification is available as ASN.1 and XML DTD.
INTRODUCTION
The Biomolecular Interaction Network Database (BIND) has been designed to store information about biomolecular interactions, molecular complexes and pathways in a computer readable form. This type of data is typically stored as written English text in traditional journal publications and in PubMed, where it is difficult to mine. Because of technological advances and heightened interest, the field of proteomics is generating increasing amounts of scientific data on molecular interactions, pathways and post-translational modification of proteins. Proteomics techniques that generate large amounts of data include high-throughput two-hybrid studies and mass spectrometry (1). The genome era has taught us that it is important to design and use effective tools for storing and managing data before they become too large. A concerted effort by the biological community is required now to prepare for the interaction information of the near future (2).
The BIND project encompasses a data specification, a database and associated data mining and visualization tools. Goals of the project are to be a public proteomics resource to the community at large and to become a platform for data mining and visualization of interaction information. We hope that BIND may help in understanding complex cell signaling networks that play an important role in a number of cellular processes, from development to disease (3).
THE BIND DATA SPECIFICATION
The BIND project is based on an extensive data specification written using the Abstract Syntax Notation.1 (ASN.1) data description language (http://asn1.elibel.tm.fr). ASN.1 is used by the US National Center for Biotechnology Information (NCBI) to store and describe almost all of the information accessible via Entrez (4). The BIND data specification inherits the NCBI ASN.1 standards for representing biological sequences, three-dimensional (3D) molecular structure and publications (5). Currently, no other single system of data description standards in the field of biology has as many mature object descriptions and is as complete as the NCBI Data Model standard written in ASN.1, ranging from maps and sequences to 3D molecular structure. Other standards efforts are underway using more modern approaches, such as CORBA (6) and XML (7), but do not currently cover this broad spectrum of object descriptors. It is important to note that it is the semantic structure of the BIND specification, not the description language it is written in, that is important. Both the specifications written in ASN.1 and all the data can readily be converted to XML using tools currently available from the NCBI.
The three main data types (database records) in BIND are interaction, molecular complex and pathway. An interaction record stores a description of the binding event between two objects, A and B, which are generally molecules. We define a molecular complex as a generally stable aggregate of molecules that have a function when linked together and are usually described as having sub-units. An example is the ribosome. Molecular complexes are stored as a set of interactions along with information such as the topology of the complex and the order that the interactions occur to form the complex. The complex can also be defined more loosely if its internal interactions are unknown. A pathway is defined as a group of molecules that are generally free from each other, but form a network of interactions usually to mediate some cellular function. Metabolic pathways are usually connected by a series of chemical actions and results of those actions, for the purpose of changing one molecular species into another. Cell signaling pathways are generally connected by binding events sometimes involving chemical actions (e.g. conformational changes or phosphorylation events), for the purpose of transducing information from one place to another. Pathways are stored as a collection of interactions along with information such as cell cycle and associated phenotypes, both normal and disease. Each main record type also stores associated publications, record authors and a flag to mark a record as private (non-exportable) in the context of a private satellite BIND database. At least one publication is required to define a record. A record collection description is also attached to each record to mark it as part of a specific set. This is useful for keeping track of imported records in a data warehouse situation.
Most of the biological information in BIND is stored in an interaction record. An object in BIND may be of type protein, DNA, RNA, small molecule, molecular complex or photon. The object may store its name, a list of name synonyms, its origin—whether natural or not, where it occurs in the cell, the cell stages in which it occurs and a sequence database reference to or a full instantiation of biological sequence and 3D structure. Storing the origin with the molecule allows the description of inter-species interactions. However, the policy of the database states that such cross-species interactions remain biologically relevant e.g. a virus–host protein–protein interaction. An interaction also stores a text description, cellular place of interaction, experimental conditions used to observe binding, a comment on evolutionarily conserved biological sequence, binding sites on A and B and how they are connected, chemical action including kinetic and thermodynamic data and chemical state of the molecules involved. The specification can store 3D structure level of detail in object representations, binding site and chemical action descriptions.
Version 1.0 of the BIND specification was finalized in June 1999 (8). Since then, further implementation of the BIND database, data record entry into the database, user feedback and discussion with other groups working on standard representations of biological function (2,9,10) have led to many improvements in version 2.0. Photon, as a type of object, is now included which allows photochemical activation to be described. Inter- and intra-molecular interaction can now be unambiguously differentiated. The description of cellular localization has been reexamined. A simple enumerated list of 13 general cellular locations has been replaced by a hierarchical description of locations in the cell that enumerates at least 182 locations in plant and animal cells. In addition, standard ontologies (10) can be referenced to indicate cellular locations more precisely. We have added to the experimental condition data type the ability to precisely describe the experimental form of an object in an interaction. So, if a protein–protein interaction has been experimentally modeled using a non-physiological protein, such as a HIS tagged protein or a similar molecule from another organism, the biological form of the protein is stored in the interaction, but the experimental form (a full NCBI bioseq object) may be stored with the experiment description. Also added is the ability to describe experiments using a profile matrix as described by PROSITE (http://www.expasy.ch/txt/profile.txt). This allows experiments that predict interactions to be stored, e.g. searching for transcription factor binding sites in a genome using a consensus sequence stored in a profile. Version 2.0 also makes it easier to describe interactions with specific sub-units of a molecular complex. Space has been allocated to define links from many data types in BIND to other databases, whether they store experimental method, phenotype or sequence information. An NCBI user-object, which can store arbitrary user-defined data, has been added to the publication data type so that private BIND databases may store any information, such as an SDS–PAGE picture, as experimental evidence for information in the system.
The minimum information required for an interaction record is a reference to another database for each molecule, such as GenBank if the molecule is a biological sequence, and a PubMed publication reference. All other information is optional.
POST-TRANSLATIONAL MODIFICATIONS
Mass spectrometry will provide much information about post-translationally modified proteins and how these post-translational modifications affect interactions. The current IUPAC nomenclature for amino acids of single or three letter codes is not sufficient for easily representing modified amino acids. We have developed an extension to the IUPAC amino acid codes using the infrastructure of the NCBI toolkit to represent 60 common naturally occurring post-translationally modified amino acids such as phospho-tyrosine, hydroxy-proline and hypusine (see Supplementary Material). Representative structures for each amino acid in both residue, N- and C-terminal forms (where appropriate) have been integrated into our own version of the NCBI8aa encoding rules and the amino acid structure look up table files for Cn3D (11). Classes of modifications that are represented include acetylation, amidation, formylation, hydroxylation, methylation, phosphorylation, palmitoylation, myristoylation and geranyl geranylation. Each modified amino acid has a standard symbol in this scheme. This extension allows us to represent the most commonly modified amino acids easily in a sequence code. For example, O4′-phospho-l-tyrosine is represented as [Y:po] which can be used to describe the phospho-peptide ligand of Grb2 as [Y:po]VNV (12). We will be extending this system to represent more amino acid modifications in the near future.
METHODS
The BIND database is implemented using an object-relational scheme. ASN.1 binary objects are stored with accompanying indexed accession numbers in a relational database using the CodeBase database C library (http://www.sequiter.com). All programs have been written using the NCBI Software Toolkit (http://www.ncbi.nlm.nih.gov/Toolbox/ and http://bioinfo.mshri.on.ca/tkcourse/) in ANSI C. The freely available NCBI toolkit provides automatic C/C++ code generation directly from an ASN.1 specification, for parsing and dealing with ASN.1 objects. Binary ASN.1 objects are compactly encoded and thus efficient to read, write and transmit. The combination of ASN.1 and automatically generated C code means that programs are rapidly developed to run very quickly across many platforms. The BIND data manager server can run on, and has been tested with Windows NT, IRIX, Linux, LinuxPPC, Solaris and HP-UX. The BIND data specification provides a basis for tools to be developed that will be able to communicate with each other and the database across platforms and networks with minimal effort via ASN.1 or XML object transmission.
The user interface to BIND is web based. Currently a data entry tool allows most data in the specification to be entered and changed. The database may be queried using text from any field, or directly by accession number. An integrated Java applet, as shown in Figure 1, has been written to visually navigate the database starting from any interaction. An online help guide is available via the help link on the Data Manager menu.
Figure 1.
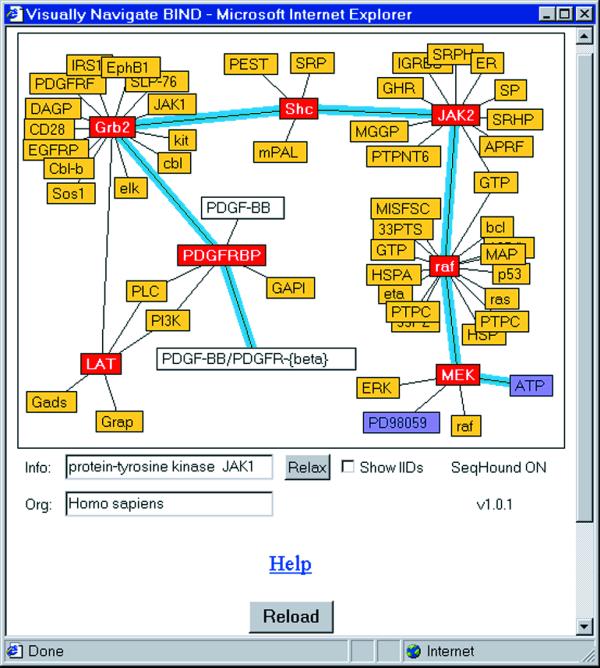
BIND Interaction Viewer Java applet showing how molecules can be connected in the database from molecular complex to small molecule. Yellow, protein; purple, small molecule; white, molecular complex; red, a square is fixed in place and will not be moved by the graph layout algorithm. This session was seeded by the interaction between human LAT and Grb2 proteins involved in cell signaling in the T-cell.
BIND has also been designed to function in a distributed manner. Multiple BIND databases may be set up, all using a common internet-based key-server to assign unique accession numbers. Collaborations are easy as information is efficiently shared.
DATA SUBMISSION
Data is entered into BIND either by manual or automatic methods. Expert curators on the BIND team are entering high quality records on a continuing basis. Users are encouraged to enter records into the database via the web-based system, or to contact the BIND staff if they have large data sets they want to process. A simple submission involves entering contact information (which only needs to be done the first time you submit to BIND), the PubMed identifier and two interacting molecules (which can easily be identified by their gi identifiers). Every record that is entered in this way will be validated by BIND indexers and by at least one other expert before it is made available in any public data release.
The GenBank policy on record ownership is followed as we hope that BIND becomes a primary public submission database for interaction, molecular complex and pathway data. Such a policy requires that the person who submits a record owns it and possesses the sole right to edit that record. Records in the public version of BIND are in the public domain.
Tools may also be written using the BIND API to import data from other sources. Such tools have been written to import information from the DIP database (13) and from recent yeast two-hybrid protein–protein interaction mapping projects (14,15). Databases that contain subsets of the interaction information that can be stored in BIND are increasing in number and are prime candidates for data import tools. In cases where such databases are free for academic use but are not allowed to be distributed by a third party, we will make import tools available.
THE OPEN NATURE OF BIND
BIND is meant to be an open effort to catalog molecular interactions, complexes and pathways. Not only are we releasing our own records into the public domain, but we will release all source code under the GNU general public license (GPL: http://www.fsf.org/) as soon as we have a fully tested API. We maintain copyright of the software, but the GPL allows anyone to freely distribute and modify the software source code provided they make their changes available under the GPL. Anyone may then install a copy of BIND on a private web server.
It is our strong belief that standard methods used in a community increase productivity and progress. We propose the BIND specification as an open standard for describing, storing and exchanging biomolecular interaction data in the scientific community.
Currently, we are making a 1.0 data release available that contains over 1000 interaction records, six pathways and 40 molecular complexes at ftp://ftp.mshri.on.ca/pub/BIND/DB/ in both XML and ASN.1. It has been estimated that there are 2–10 protein–protein interactions per protein in a cell (16). This estimate does not include other types of interactions such as protein–small molecule, of which there are undoubtedly at least as many. For example, this means that the ∼900 Saccharomyces cerevisiae interactions so far in BIND may represent ∼1–7% of the total protein–protein interactions in yeast. It is clear to us that yeast will be the first completely understood organism given the high-throughput experiments currently being undertaken in laboratories around the world.
FUTURE DIRECTIONS
Now that BIND has a stable data specification, a firm record base and a data release we are developing data mining methods for homologous interaction network finding, for finding pathways and for comparing interactions and scoring their similarity (analogous to BLAST for sequences). The data specification abstracts cellular interactions as a computer science concept of a graph, thus tools from the field of graph theory can be applied to data mining. A similarity algorithm can be used by the homologous interaction network algorithm and to create neighbor tables to deal with redundancy in the database. It is also possible that novel drug targets may be found by examining highly connected nodes in an interaction network (17). We plan to examine automatic data record generation directly from experimental sources, such as mass spectrometric data. Since BIND can contain information on the cellular place of all involved components of interaction networks and associated kinetics and thermodynamics data, models of cellular processes can be generated automatically for input into kinetics modeling software such as the Virtual Cell (18). We are also investigating the possibility of representing genetic interactions.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
Many thanks to Jim Ostell and Denis Vakatov at the NCBI for making the ASN.1 to XML conversion tools available. We are grateful to colleagues at the Samuel Lunenfeld Research Institute for helpful input. Katerina Michalickova developed the Seqhound database system. Van Le and Howard Feldman assisted in the post-translational amino acid modification work. BIND has been supported by an MRC (CIHR) grant to C.W.V.H. and T.P., by grants from Connaught (Aventis), the Canadian Foundation for Innovation, the Ontario R and D Challenge Fund. Special thanks to MDS Sciex and MDS Protana who have agreed to allow us to release BIND under the GNU Public License.
References
- 1.Mendelsohn A.R. and Brent,R. (1999) Protein interaction methods–toward an endgame. Science, 284, 1948–1950. [DOI] [PubMed] [Google Scholar]
- 2.Cassman M., Hunter,T. and Pawson,T. (2000) Proteins suggest form of their own database. Nature, 403, 591–592. [DOI] [PubMed] [Google Scholar]
- 3.Pawson T. (1995) Protein modules and signalling networks. Nature, 373, 573–580. [DOI] [PubMed] [Google Scholar]
- 4.Wheeler D.L., Chappey,C., Lash,A.E., Leipe,D.D., Madden,T.L., Schuler,G.D., Tatusova,T.A. and Rapp,B.A. (2000) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res., 28, 10–14. Updated article in this issue: Nucleic Acids Res. (2001), 29, 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ostell J. and Kans,J.A. (1998) In Baxevanis,A.D. and Ouellette,B.F. (eds), Bioinformatics, A Practical Guide to the Analysis of Genes and Proteins. John Wiley and Sons, New York, NY. Vol. 39, pp. 121–144.
- 6. Object Management Group (1996) CORBA Architecture and Specifications. OMG Publications, Needham, MA.
- 7.Fenyo D. (1999) The Biopolymer Markup Language. Bioinformatics ., 15, 339–340. [DOI] [PubMed] [Google Scholar]
- 8.Bader G.D. and Hogue,C.W. (2000) BIND-a data specification for storing and describing biomolecular interactions, molecular complexes and pathways. Bioinformatics, 16, 465–477. [DOI] [PubMed] [Google Scholar]
- 9.Karp P.D. (2000) An ontology for biological function based on molecular interactions. Bioinformatics, 16, 269–285. [DOI] [PubMed] [Google Scholar]
- 10.Ashburner M., Ball,C.A., Blake,J.A., Botstein,D., Butler,H., Cherry,J.M., Davis,A.P., Dolinski,K., Dwight,S.S., Eppig,J.T. et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hogue C.W. (1997) Cn3D: a new generation of three-dimensional molecular structure viewer. Trends Biochem. Sci., 22, 314–316. [DOI] [PubMed] [Google Scholar]
- 12.Salcini A.E., McGlade,J., Pelicci,G., Nicoletti,I., Pawson,T. and Pelicci,P.G. (1994) Formation of Shc-Grb2 complexes is necessary to induce neoplastic transformation by overexpression of Shc proteins. Oncogene, 9, 2827–2836. [PubMed] [Google Scholar]
- 13.Xenarios I., Rice,D.W., Salwinski,L., Baron,M.K., Marcotte,E.M. and Eisenberg,D. (2000) DIP: the database of interacting proteins. Nucleic Acids Res., 28, 289–291. Updated article in this issue: Nucleic Acids Res. (2001), 29, 239–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uetz P., Giot,L., Cagney,G., Mansfield,T.A., Judson,R.S., Knight,J.R., Lockshon,D., Narayan,V., Srinivasan,M., Pochart,P. et al. (2000) A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature, 403, 623–627. [DOI] [PubMed] [Google Scholar]
- 15.Ito T., Tashiro,K., Muta,S., Ozawa,R., Chiba,T., Nishizawa,M., Yamamoto,K., Kuhara,S. and Sakaki,Y. (1997) Toward a protein-protein interaction map of the budding yeast: A comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc. Natl Acad. Sci. USA, 97, 1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marcotte E.M., Pellegrini,M., Ng,H.L., Rice,D.W., Yeates,T.O. and Eisenberg,D. (1999) Detecting protein function and protein-protein interactions from genome sequences. Science, 285, 751–753. [DOI] [PubMed] [Google Scholar]
- 17.Albert R., Jeong,H. and Barabasi,A.L. (2000) Error and attack tolerance of complex networks. Nature, 406, 378–382. [DOI] [PubMed] [Google Scholar]
- 18.Schaff J. and Loew,L.M. (1999) The virtual cell. Pac. Symp. Biocomput., 228–239 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}