

Abstract
Background & Aims:
Hepatocellular carcinoma (HCC), a leading cause of cancer-related death, is associated with viral hepatitis, non-alcoholic steatohepatitis (NASH), and alcohol-related steatohepatitis, all of which trigger endoplasmic reticulum (ER) stress, hepatocyte death, inflammation, and compensatory proliferation. Using ER stress-prone MUP-uPA mice, we established that ER stress and hypernutrition cooperate to cause NASH and HCC, but the contribution of individual stress effectors, such as activating transcription factor 4 (ATF4), to HCC and their underlying mechanisms of action remained unknown.
Methods:
Hepatocyte-specific ATF4-deficient MUP-uPA mice (MUP-uPA/Atf4Δhep) and control MUP-uPA/Atf4F/F mice were fed a high-fat diet to induce NASH-related HCC, and Atf4F/F and Atf4Δhep mice were injected with diethylnitrosamine to model carcinogen-induced HCC. Histological, biochemical, and RNA-sequencing analyses were performed to identify and define the role of ATF4-induced solute carrier family 7a member 11 (SLC7A11) expression in hepatocarcinogenesis. Reconstitution of SLC7A11 in ATF4-deficient primary hepatocytes and mouse livers was used to study its effects on ferroptosis and HCC development.
Results:
Hepatocyte ATF4 ablation inhibited hepatic steatosis, but increased susceptibility to ferroptosis, resulting in accelerated HCC development. Although ATF4 activates numerous genes, ferroptosis susceptibility and hepatocarcinogenesis were reversed by ectopic expression of a single ATF4 target, Slc7a11, coding for a subunit of the cystine/glutamate antiporter xCT, which is needed for glutathione synthesis. A ferroptosis inhibitor also reduced liver damage and inflammation. ATF4 and SLC7A11 amounts were positively correlated in human HCC and livers of patients with NASH.
Conclusions:
Despite ATF4 being upregulated in established HCC, it serves an important protective function in normal hepatocytes. By maintaining glutathione production, ATF4 inhibits ferroptosis-dependent inflammatory cell death, which is known to promote compensatory proliferation and hepatocarcinogenesis. Ferroptosis inhibitors or ATF4 activators may also blunt HCC onset.
Keywords: HCC, Steatohepatitis, ER stress, ATF4, SLC7A11, NRF2, Ferroptosis
Graphical Abstract
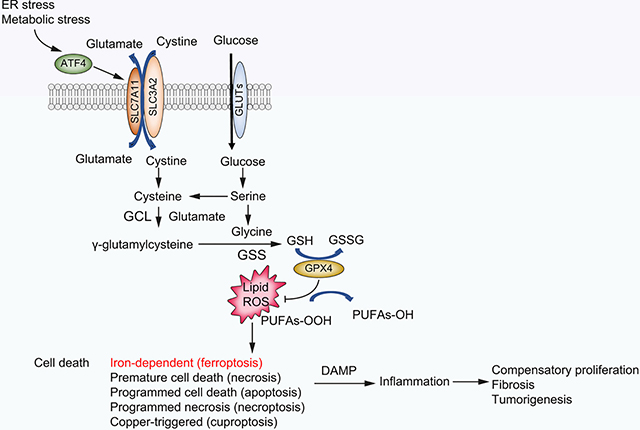
Introduction
Liver cancer is the third leading cause of cancer death and the sixth most common cancer type worldwide,1 whose prevalence constantly increases, paralleling the global rise in obesity, type 2 diabetes, metabolic syndrome, and excessive alcohol intake.2,3 Hepatocellular carcinoma (HCC), the main primary liver cancer, is associated with multiple aetiologies, including HBV and HCV infections, non-alcoholic (NAFLD), alcoholic (AFLD), and toxicant-associated (TAFLD) fatty liver diseases, which cause endoplasmic reticulum (ER) stress, liver damage, hepatocyte death, and inflammation. The spectrum of fatty liver disease ranges from simple steatosis without liver damage to steatohepatitis (non-alcoholic steatohepatitis [NASH], alcoholic steatohepatitis [ASH], or toxicant-associated steatohepatitis [TASH] according to their main cause), fibrosis, cirrhosis, and eventually HCC. Despite tight associations with well-defined risk factors, HCC is often diagnosed at an advanced stage, when the main therapeutic options are surgical and radiological ablations and localised chemotherapy. Currently, there are no curative treatments for late-stage HCC, whose median overall survival remains at about 1 year.4 Despite advances in NAFLD/NASH, AFLD/ASH, and TAFLD/TASH diagnosis and aetio-logical understanding, the mechanisms that govern progression from steatohepatitis to HCC remain elusive. Oxidative stress, ER stress, cell death, mitochondrial dysfunction, insulin resistance, inflammation, and gut dysbiosis were shown to be involved in steatohepatitis and its progression to HCC.2,3,5–7 We established the importance of excessive hepatocyte death, necroinflammation, and compensatory proliferation in HCC development.5,7,8 Nonetheless, the type of cell death promoting inflammation and HCC is contentious, with different studies suggesting it could be necrosis, apoptosis, or necroptosis.9–11 Ferroptosis is an iron-dependent cell death type that is often accompanied by lipid peroxidation caused by depletion of reduced glutathione (GSH), decreased glutathione peroxidase 4 (GPX4) activity, or increased lipid peroxidation.12 Ferroptosis is negatively regulated by solute carrier family 7a member 11 (SLC7A11), GPX4, and GSH. In previous studies conducted with hepatocyte-specific IKKβ knockout mice (IkkbΔhep), which are highly susceptible to diethylnitrosamine (DEN)-induced hepatocarcinogenesis, we observed increased lipid peroxidation and hepatocyte death, which were reversed by feeding the mice with N-acetyl cysteine (NAC), a precursor for GSH synthesis.8 A key regulator of ferroptosis is transcription factor nuclear factor erythroid 2-related factor 2 (NRF2), whose activation in HCC and elsewhere is promoted by SQSTM1/p62,5 which is encoded by an IKKβ/NF-κB-inducible gene.13 NRF2 induces SLC7A11 transcription, which is often elevated in human cancers and accompanied by decreased ferroptosis, increased tumour growth, and drug resistance.14,15 Ferroptosis may also be involved in liver injury and inflammation,16 but its role in hepatocarcinogenesis is unknown. Here we explored the role of the ER stress mediator activating transcription factor 4 (ATF4) in DEN- and high-fat diet (HFD)-induced HCC in major urinary protein (MUP)–urokinase-type plasminogen activator (uPA) mice, which express the secreted protein uPA from the MUP promoter. MUP-uPA mice undergo transient hepatic ER stress early in life owing to elevated uPA expression, which is sustained by HFD feeding.7 Using chemical chaperones, we demonstrated that NASH development in these mice depends on ER stress.
Protein misfolding after overexpression of a secreted protein triggers the unfolded protein response (UPR), which promotes stress adaptation.17 There are three primary UPR effectors: inositol-requiring enzyme 1 (IRE1), which controls expression of X-box-binding protein 1 (XBP1); activating transcription factor 6 (ATF6); and pancreatic ER-regulating kinase (PERK), which phosphorylates eukaryotic translation initiation factor 2 subunit α (eIF2α) to induce ATF4 translation. ATF4 activates numerous target genes, whose products mitigate stress or induce cell death if the stress cannot be resolved.17 UPR activation is often observed in cancer cells and thought to provide them with a survival advantage.17,18 Hepatocyte-specific IRE1a ablation decreases DEN-induced HCC and blunts acceleration of HCC progression by HFD,19 although it increases acute liver injury and hepatic steatosis.20,21 In contrast, XBP1 loss promotes tumorigenesis in mouse models of intestinal cancer,22 and hepatocyte-specific XBP1 ablation enhanced liver injury and fibrosis but attenuated steatosis in a high-fat/sugar diet-induced NASH model.23 ATF6 activates transformation-associated genes during hepatocarcinogenesis,24 whereas PERK promotes MYC-driven cell transformation and autophagy.25 ATF4 activates genes involved in redox homoeostasis, amino acid (AA) metabolism, protein synthesis, apoptosis, and autophagy,17,26 and was extensively studied for its roles in glucose, lipid, and cholesterol metabolism.27–29 Systemic ATF4 ablation attenuates fructose-induced hyper-triglyceridaemia and hepatic steatosis,30,31 in part through regulation of fibroblast growth factor 21 (FGF21) expression.32 In other studies, ATF4 ablation inhibited hepatic steatosis through different mechanisms, including expression of N-nicotinamide methyltransferase (NNMT)33, tribbles homologue 3 (TRB3),34 or nuclear respiratory factor 1 (NRF1)–mitochondrial transcription factor A (TFAM) signalling.35 ATF4 expression is low in healthy cells but is often elevated in cancer, where it promotes cell survival and tumour growth by inducing genes involved in AA metabolism and oxidant defenses.26,36,37 To test the role of ATF4 in hepatocarcinogenesis, we generated Atf4Δhep mice lacking hepatocyte ATF4. Despite its documented ability to support hepatic steatosis, ATF4 ablation enhanced DEN-induced tumorigenesis and NASH to HCC progression in HFD-fed MUP-uPA mice. Notably, the HCC suppressive activity of ATF4 was mainly caused by expression of a single target, SLC7A11, the small subunit of the cystine/glutamate antiporter (xCT), which is needed for GSH synthesis. Ectopic SLC7A11 expression suppressed HCC development in Atf4Δhep mice and provided protection from ferroptosis, suggesting that ferroptosis contributes to the HCC-promoting necroinflammatory response and that ferroptosis inhibitors may prevent NASH to HCC progression.
Materials and methods
Mouse models
Atf4F/F mice, a generous gift from Dr Christopher Adams (University of Iowa, Iowa City, IA, USA),38 were intercrossed with Alb-Cre mice (C57BL/6, Jackson Laboratory, Ellsworth, ME, USA) to generate Atf4F/F-Alb-Cre (Atf4Δhep) mice. Atf4Δhep mice were crossbred with MUP-uPA mice7 to obtain MUP-uPA/Atf4F/F and MUP-uPA/Atf4Δhep mice. B6.129X1-Nfe2l2tm1Ywk/J (Nrf2−/−) mice were described.39 All mouse lines were either on a pure C57BL/6 genetic background or crossed into it for at least 10 generations. The NASH and HCC model: 6-week-old (wo) MUP-uPA/Atf4F/F and MUP-uPA/Atf4Δhep mice were fed with low-fat diet (LFD) (composed of 12% fat, 23% protein, and 65% carbohydrates based on caloric content) or HFD (consisting of 59% fat, 15% protein, and 26% carbohydrates based on caloric content; Bio-Serv, Flemington, NJ, USA) and sacrificed when 4, 6, and 10 months old (mo) for the pathological analysis. The LFD-fed DEN-induced HCC model: DEN (25 mg/kg, Millipore Sigma N0258, Saint Louis, MO, USA) was injected i.p. into 14-day-old mice, and samples were collected from the LFD-fed mice when 5 and 10 mo. The HFD-fed DEN-induced HCC model: mice were placed on HFD when 3 mo, followed by DEN (80 mg/kg) administration when 4 mo, and water with 0.05% phenobarbital (PB; Sigma, P5178) when 5 mo. The mice were sacrificed and analysed when 10 mo. Animal experiments were performed in accordance with NIH guidelines for the use and care of live animals and approved by the University of California San Diego Institutional Animal Care and Use Committee, S00218. Experiments done at Shanghai University of Traditional Chinese Medicine were performed in accordance with the guidelines of the Ministry of Science and Research of China and approved by the Animal Experimental Ethics Committee of Shanghai University of Traditional Chinese Medicine.
RNA-seq data processing and analysis
Total liver RNAs were extracted from overnight fasted 6-wo Atf4F/F, Atf4Δhep, MUP-uPA/Atf4F/F, and MUP-uPA/Atf4Δhep mice using TRIzol® (Life Technologies, 15596–018, Carlsbad, CA, USA) according to the manufacturer’s instructions and was sequenced on the Illumina NovaSeq 6000 sequencer (2×150 bp read length, San Diego, CA, USA). Raw paired-end reads were trimmed and quality controlled by SeqPrep (https://github.com/jstjohn/SeqPrep) and Sickle (https://github.com/najoshi/sickle) with default parameters. Then the clean reads were separately aligned to a reference genome with orientation mode using HISAT2 (http://ccb.jhu.edu/software/hisat2/index.shtml) software. The mapped reads for each sample were assembled by StringTie (https://ccb.jhu.edu/software/stringtie/index.shtml?t=example) in a reference-based approach. RSEM (http://deweylab.biostat.wisc.edu/rsem/) was used to quantify gene abundance, and differential gene expression analysis was performed using DESeq2/DEGseq/EdgeR. Gene ontology (GO) functional enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were carried out using DAVID Bioinformatics Resources 6.8 (https://david.ncifcrf.gov/). For gene set enrichment analysis (GSEA), the gene expression matrix was pooled from gene expression estimates from StringTie output and processed with mouse-ENSEMBL-gene IDs with 1,000 permutations using a t test metric for gene ranking (https://www.gsea-msigdb.org/gsea/index.jsp). Enrichment was tested using default v5.2 MSigDb gene sets. Mouse RNA sequencing (RNA-seq) data have been deposited into the NCBI Gene Expression Omnibus (accession GSE191115).
Quantification and statistical analysis
Data are shown as mean ± SD as indicated. Statistical significance was determined using the two-tailed Student’s t test, and p values lower than 0.05 were considered statistically significant (*p <0.05, **p <0.01, ***p <0.001). GraphPad Prism (Boston, MA, USA) was used for statistical analysis and graphing.
Other details can be found in the Supplementary CTAT table and Supplementary information.
Results
ATF4 ablation increases uPA-induced liver injury without an effect on UPR activation
As previously found,7 5–6-wo MUP-uPA mice, whose liver uPA expression had peaked, exhibited elevated expression of ER stress markers, including endoplasmic reticulum protein 5 (ERP5), 78-kDa glucose-regulated protein (GRP78/BIP), C/EBP-homologous protein (CHOP), ATF4, and phosphorylated eIF2α (P-eIF2α) (Fig. 1A and Fig. S1A). To study the role of ATF4 in NASH and HCC development, we generated Atf4Δhep mice and crossbred them with MUP-uPA mice to obtain MUP-uPA/Atf4Δhep mice. Compared with livers of MUP-uPA/Atf4F/F mice, MUP-uPA/Atf4Δhep livers had a rough appearance with surface nodules (Fig. 1B) and elevated serum alanine aminotransferase (ALT) (Fig. 1C), suggesting enhanced liver injury. By contrast, livers of Atf4Δhep mice had normal appearance, and serum ALT was normal, too (Fig. 1B and C). In situ hybridisation analysis of Atf4 mRNA showed elevated expression in MUP-uPA/Atf4F/F hepatocytes, and efficient loss of the Atf4 mRNA signal was observed in both Atf4Δhep and MUP-uPA/Atf4Δhep mice (Fig. S1B and C), consistent with absence of the floxed allele in isolated hepatocytes (Fig. S1D). Histology revealed hepatocyte damage in 6-wo MUP-uPA/Atf4F/F mice accompanied by immune infiltration that was exacerbated in MUP-uPA/Atf4Δhep mice (Fig. 1D). Sox9 and F4/80 antibody staining were higher in MUP-uPA/Atf4Δhep livers, indicating injury-associated ductular reaction and enhanced macrophage infiltration (Fig. 1D). Notably, immunoblotting (IB) of liver lysates indicated that ATF4 ablation had little or no effect on the ER stress markers, TRB3, GRP78, CHOP, and P-eIF2α (Fig. 1E). The ATF6 and IRE1-Xbp1 branches of the UPR were not affected either based on expression of ATF6 target genes, ER degradation-enhancing alpha-mannosidase-like protein 2 (EDEM2) and GRP78 (Fig. S1E), and similar amounts of spliced Xbp1 (Xbp1s) mRNA in 6-wo MUP-uPA/Atf4F/F and MUP-uPA/Atf4Δhep mice (Fig. S1F), suggesting that the UPR in MUP-uPA mice is activated independently of ATF4. Interestingly, 4-hydroxynonenal (4HNE) staining, a marker of lipid peroxidation and ferroptosis, was elevated in the MUP-uPA/Atf4Δhep liver (Fig. 1D) and the reduced GSH to oxidised GSH (GSSG) ratio was decreased (Fig. 1F), suggesting oxidative stress and liver injury. These findings resemble observations made in IkkbΔhep mice, in which HCC development was suppressed by NAC, a GSH precursor.8 Quantitative reverse-transcription PCR (qRT-PCR) analysis of MUP-uPA/Atf4Δhep livers showed increased expression of F4/80, IL-1α, and C-X-C motif chemokine ligand 2 (CXCL2) mRNAs (Fig. S1G), indicating that ATF4-deficient hepatocytes are more susceptible to ER stress-related necroinflammatory injury, normally suppressed by ATF4, which plays an important role in maintaining redox homoeostasis.
Fig. 1. Hepatocyte-specific ATF4 ablation disrupts redox homoeostasis and exacerbates liver injury in MUP-uPA mice.
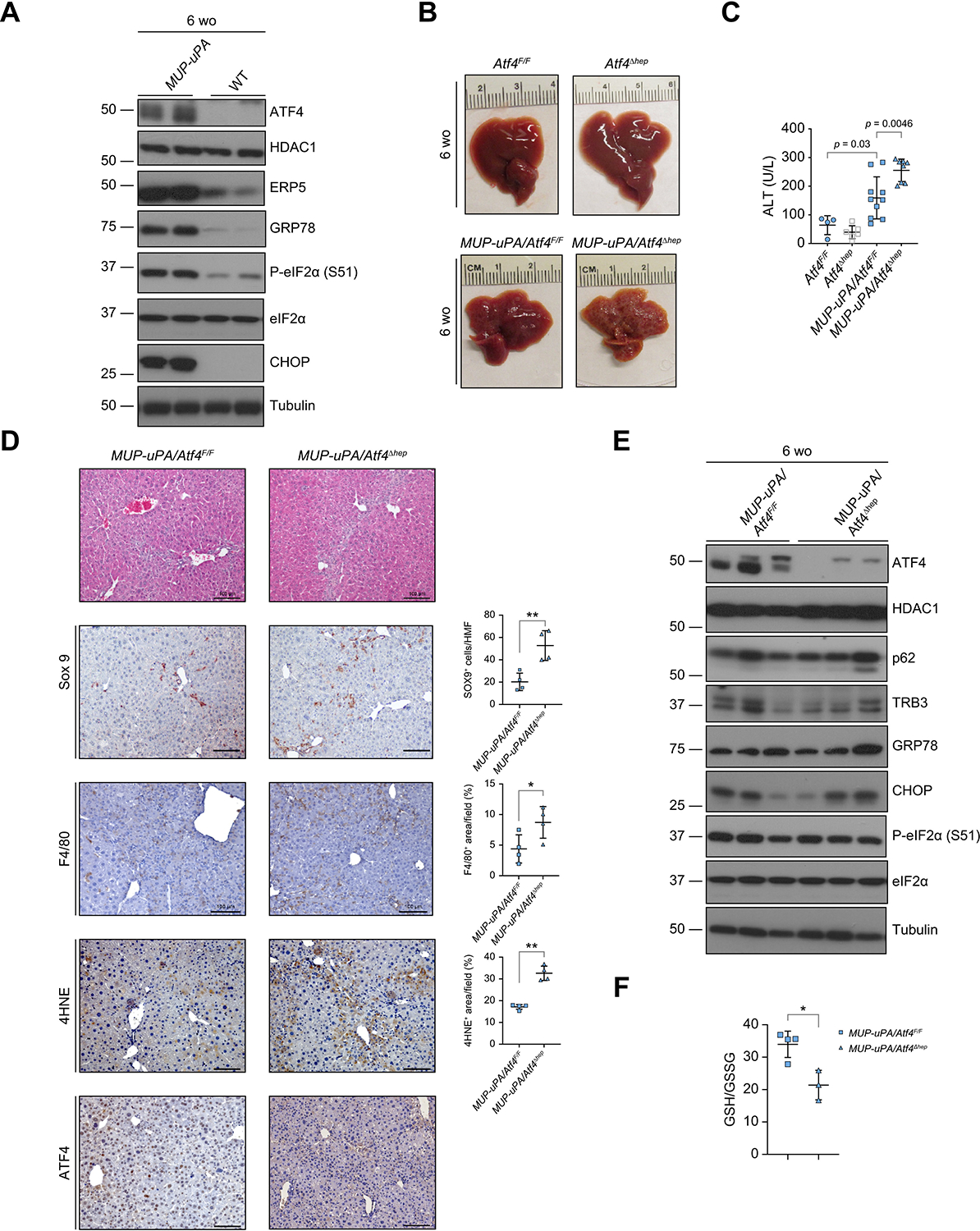
(A) Representative IB analysis of 6-wo BL6 (WT) and MUP-uPA mice. (B) Gross liver morphology of 6-wo Atf4F/F, Atf4Δhep, MUP-uPA/Atf4F/F, and MUP-uPA/Atf4Δhep mice. (C) Serum ALT in above mice. Mean ± SD (n = 4–10/group). Value of p indicates significance level, Student’s t test. (D) H&E staining and IHC analysis of 6-wo WT, MUP-uPA/Atf4F/F, and MUP-uPA/Atf4Δhep mice. Staining was quantitated as the number of positive cells per HMF or positively stained area per field, as indicated. Mean ± SD (n = 4/group). *p <0.05; **p <0.01 (Student’s t test). Scale bars, 100 lm. (E) IB analysis of 6-wo MUP-uPA/Atf4F/F and MUP-uPA/Atf4Δhep mice. (F) GSH:GSSG ratio in livers of indicated 6-wo mice. Mean ± SD (n = 3–4/group). *p = 0.012 (Student’s t test). 4HNE, 4-hydroxynonenal; ATF4, activating transcription factor 4; CHOP, C/EBP-homologous protein; eIF2α, eukaryotic translation initiation factor 2 subunit α; ERP5, endoplasmic reticulum protein 5; GRP78, 78-kDa glucose-regulated protein; GSH, glutathione; GSSG, oxidised glutathione; HDAC1, histone deacetylase 1; HMF, high magnification field; IB, immunoblotting; IHC, immunohistochemistry; MUP-uPA, major urinary protein promoter–urokinase plasminogen activator; P-eIF2α, phosphorylated eIF2α; TRB3, tribbles homologue 3; wo, week old; WT, wild-type.
ATF4 ablation attenuates target gene expression
Young MUP-uPA hepatocytes are ER-stressed, undergoing cell death and self-renewal.7,40 We used qRT-PCR to examine how ATF4 ablation affects target gene expression. MUP-uPA/Atf4F/F livers exhibited elevated expression of phosphoglycerate dehydrogenase (PHGDH), phosphoserine aminotransferase 1 (PSAT1), phosphoserine phosphatase (PSPH), cystathionine gamma-lyase (CTH), and methenyltetrahydrofolate dehydrogenase 2 (MTHFD2) mRNAs relative to wild-type (WT) livers (Fig. 2A). The products of these mRNAs catalyse serine and cysteine synthesis as well as one carbon metabolism/folate cycle (Fig. 2B). ATF4 ablation reduced these mRNAs (Fig. 2A). MUP-uPA/Atf4F/F livers also showed elevated expression of mRNAs for asparagine synthetase (ASNS), cysteinyl-tRNA synthetase (CARS), mitochondrial phosphoenolpyruvate carboxykinase (PCK2), and glutathione-specific gamma-glutamylcyclotransferase 1 (CHAC1), enzymes involved in AA metabolism, gluconoeogenesis, and GSH utilisation, all of which were reduced in MUP-uPA/Atf4Δhep livers (Fig. 2A). Decreased gene expression was not caused by reduced uPA expression or less ER stress in MUP-uPA/Atf4Δhep livers (Fig. 2A). Atf4 mRNA was higher in MUP-uPA/Atf4F/F livers compared with WT livers.
Fig. 2. ATF4 ablation disrupts expression of genes involved in amino acid and one-carbon metabolism.
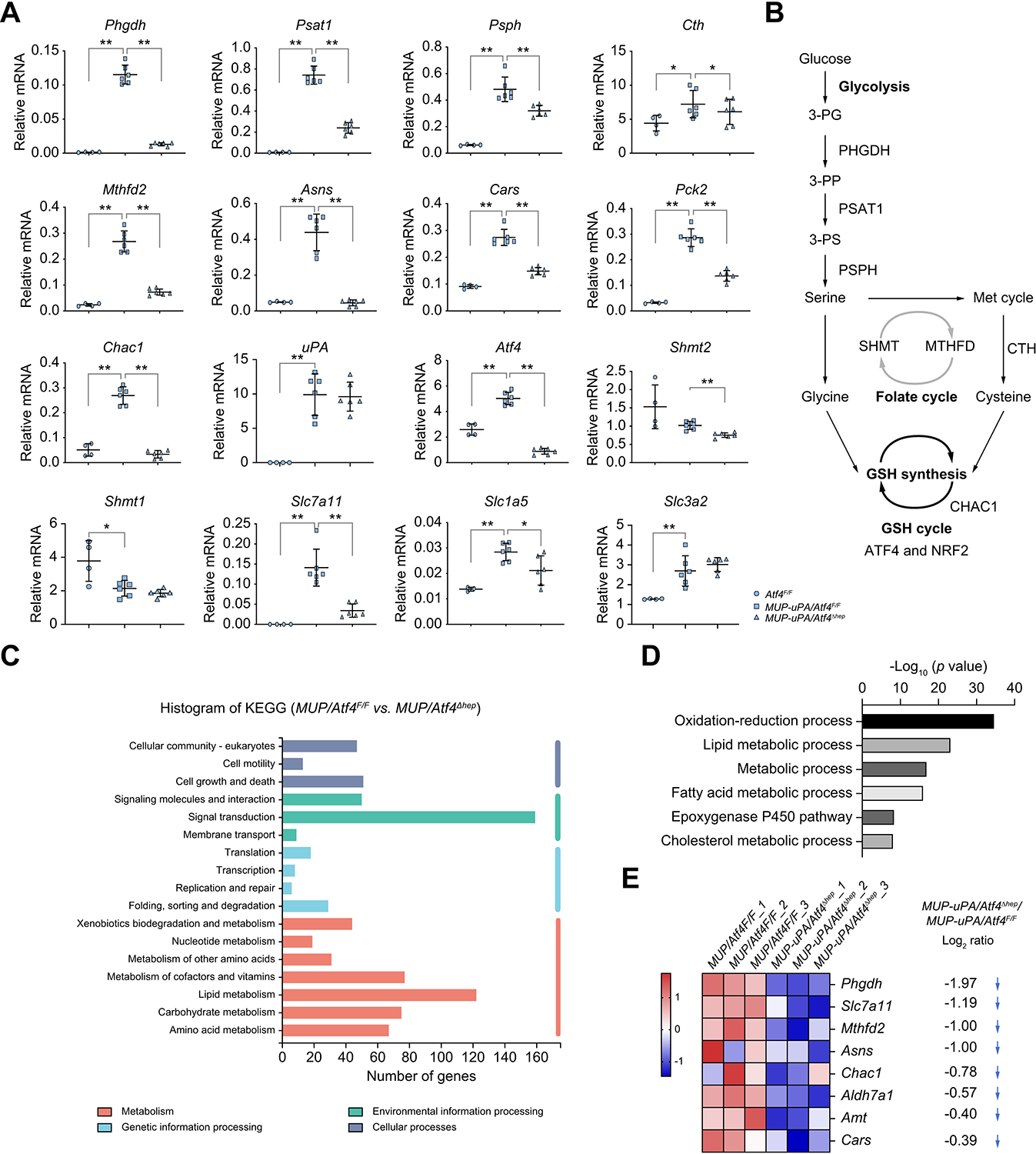
(A) qRT-PCR analysis of liver mRNAs from 6-wo Atf4F/F, MUP-uPA/Atf4F/F, and MUP-uPA/Atf4Δhep mice. Mean ± SD (n = 3–6/group). *p <0.05; **p <0.01 (Student’s t test). (B) Diagram showing metabolic pathways with key enzymes involved in serine, glycine, cysteine, and GSH metabolism as well as the folate and methionine cycles. (C) KEGG pathway analysis of genes differentially represented in RNA-seq data from livers of overnight fasted 6-wo MUP-uPA/Atf4Δhep and MUP-uPA/Atf4F/F mice (n = 3). (D) DAVID-based GO analysis of differentially expressed genes in above RNA-seq data (n = 3). (E) Heat map representation of significantly downregulated genes in MUP-uPA/Atf4Δhep relative to MUP-uPA/Atf4F/F livers (n = 3) associated with ‘amino acid metabolism’ including Log2FC value. ATF4, activating transcription factor 4; CHAC1, glutathione specific gamma-glutamylcyclotransferase 1; CTH, cystathionine gamma-lyase; GO, gene ontology; GSH, glutathione; KEGG, Kyoto Encyclopedia of Genes and Genomes; Log2FC, log2 fold-change; MTHFD2, methenyltetrahydrofolate dehydrogenase 2; MUP-uPA, major urinary protein promoter–urokinase plasminogen activator; NRF2, nuclear factor erythroid 2-related factor 2; PHGDH, phosphoglycerate dehydrogenase; PSAT1, phosphoserine aminotransferase 1; PSPH, phosphoserine phosphatase; qRT-PCR, quantitative reverse-transcription PCR; RNA-seq, RNA sequencing; SHMT, serine hydroxymethyltransferase; 3-PG, 3-phosphoglycerate; 3-PP, 3-phosphohydroxypyruvate; 3-PS, 3-phosphoserine; wo, week old.
GSH, whose amounts were reduced in MUP-uPA/Atf4Δhep livers, is a key antioxidant and redox regulator.41,42 De novo GSH synthesis is mediated via a two-step biosynthetic process catalysed by glutamate–cysteine ligase (GCL) and glutathione synthase (GSS). mRNAs for enzymes involved in glycine, cysteine, and glutamate synthesis were reduced on ATF4 ablation (Fig. 2A), which did not significantly affect mRNAs for glutaminolysis enzymes, glutaminase liver isoform (GSL2), and glutamate dehydrogenase 1 (GLUD1) (Fig. S2A). ATF4 ablation reduced mRNA for serine hydroxymethyltransferase 2 (SHMT2), which catalyses serine and glycine interconversion, but not for SHMT1 (Fig. 2A). Cysteine is the limiting GSH precursor, and its uptake is mediated by xCT.12,42 Liver-specific ATF4 ablation reduced expression of mRNAs for SLC7A11 and SLC1A5, which encodes the neutral AA transporter B (ASCT2) for uptake of alanine, serine, cysteine, and threonine, but had little effect on SLC1A4/ASCT1 mRNA amounts (Fig. 2A and Fig. S2A). ATF4 deletion also reduced the mRNAs for the large neutral AA transporter small subunit 1 (SLC7A5/4F2LC), but not for the heavy subunit of the large neutral AA transporter (SLC3A2/4F2HC), and seryl-tRNA synthetase (SARS) (Fig. 2A and Fig. S2A).
KEGG pathway analysis of RNA-Seq data collected from overnight-fasted 6-wo MUP-uPA/Atf4F/F and MUP-uPA/Atf4Δhep livers revealed that differentially expressed genes involved in lipid metabolism, metabolism of cofactors and vitamins, carbohydrate metabolism, and AA metabolism were the top four differentially expressed metabolic groups (Fig. 2C). DAVID-based GO analysis showed that top four significant changes in biological processes upon ATF4 ablation were oxidation–reduction process, lipid metabolic process, metabolic process, and fatty acid metabolic process (Fig. 2D). GSEA indicated that ATF4 ablation reduced expression of genes involved in AA metabolism (Fig. S2B). Heat map depiction showed significantly downregulated ‘AA metabolism’-related genes, including those encoding PHGDH, SLC7A11, MTHFD2, ASNS, glutathione-specific gamma-glutamylcyclotransferase 1 (CHAC1), alpha-aminoadipic semialdehyde dehydrogenase (ALDH7A1), aminomethyltransferase (AMT), and CARS in MUP-uPA/Atf4Δhep relative to MUP-uPA/Atf4F/F livers (Fig. 2E). By contrast, the Atf4F/F and Atf4Δhep liver transcriptomes were barely different (Fig. S2C and D), correlating with low ATF4 expression in non-stressed livers. These data explain how ATF4 ablation decreases the GSH:GSSG ratio.
The major transcriptional regulator of redox homoeostasis is NRF2.41,43 ER stress and oxidative stress are closely linked,43 and MUP-uPA/Atf4F/F mice showed elevated expression of numerous NRF2 target genes encoding SLC7A11, SLC1A4, SLC7A5, haem oxygenase 1 (HO1), NAD(P)H quinone dehydrogenase 1 (NQO1), and GSS (Fig. 2A, and Fig. S2A and E). ATF4 ablation had no effect on HO1, NQO1, GSS, GCLC, and GCLM mRNAs, while reducing SLC7A11, SLC7A5, and superoxide dismutase 2 (SOD2) mRNA amounts (Fig. 2A, and Fig. S2A and E), suggesting that some NRF2 targets also respond to ATF4. By contrast, ATF4 ablation increased expression of the pro-apoptotic mRNAs for Bcl-2-associated X protein (BAX) and death receptor 5 (DR5) but decreased expression of p53 upregulated modulator of apoptosis (PUMA) and had no effect on expression of BH3-interacting domain death agonist (BID), tumour necrosis factor receptor 1 (TNFR1), tumour necrosis factor receptor superfamily member 6 (TNFRSF6/FAS), and CHOP mRNAs (Fig. S2F). These results suggest that ATF4 ablation may contribute to apoptosis under stress.
Reduced SLC7A11 expression increases susceptibility to ferroptosis
The MUP-uPA/Atf4Δhep liver exhibited features of ferroptosis that could be attributed to reduced expression of xCT, a key regulator of ferroptosis.12 To examine whether ATF4 ablation increased susceptibility to ferroptosis and other forms of cell death, we cultured Atf4F/F and ATF4-deficient (Atf4Δ) hepatocytes and treated them with different cell death inducers, including palmitic acid (PA), tunicamycin (TM), DEN, the ferroptosis inducer RAS-selective lethal 3 (RSL3), and the proteasome inhibitor MG132. Although Atf4Δ hepatocytes grew as well as Atf4F/F hepatocytes under basal conditions, they exhibited higher cell death rates in response to drug treatment and had lower intracellular GSH (Fig. 3A and B, and Fig. S3A). NRF2-deficient (Nfe2l2−/−) hepatocytes were also more susceptible to RSL3-induced cell death than WT hepatocytes (Fig. S3B). Although most Atf4Δ hepatocytes died after incubation with low RSL3 concentrations, most Atf4F/F hepatocytes remained viable, but when RSL3 amounts were high, both Atf4F/F and Atf4Δ hepatocytes died (Fig. 3C–E and Fig. S3C). The ferroptosis inhibitor ferrostatin-1 (FER-1) completely inhibited RSL3-induced Atf4F/F hepatocyte death, but the caspase inhibitor Z-VAD or the necroptosis inhibitor necrostatin-1 (NEC-1) were ineffective (Fig. 3D). Full protection of Atf4Δ hepatocytes was seen in cells incubated with a low RSL3 concentration (Fig. S3D). Transmission electron microscopy (TEM) showed a typical feature of ferroptosis, smaller mitochondria, present in RSL3-treated hepatocytes, especially in the absence of ATF4 (Fig. S3E), consistent with previous reports.12,16 RSL3-induced ferroptosis of Atf4-deficient hepatocytes was also inhibited by the iron chelator deferoxamine (DFO) (Fig. S3F). β-Mercaptoethanol (β-Me), however, had little effect on Atf4F/F hepatocytes, consistent with previous reports,12,44 although it slightly inhibited ferroptosis in Atf4Δ hepatocytes (Fig. S3F). Furthermore, FER-1 partly inhibited the DEN-induced cell death of Atf4Δ hepatocytes (Fig. S3G), suggesting that DEN triggers different forms of cell death, including ferroptosis.
Fig. 3. ATF4-deficient hepatocytes are ferroptosis susceptible.
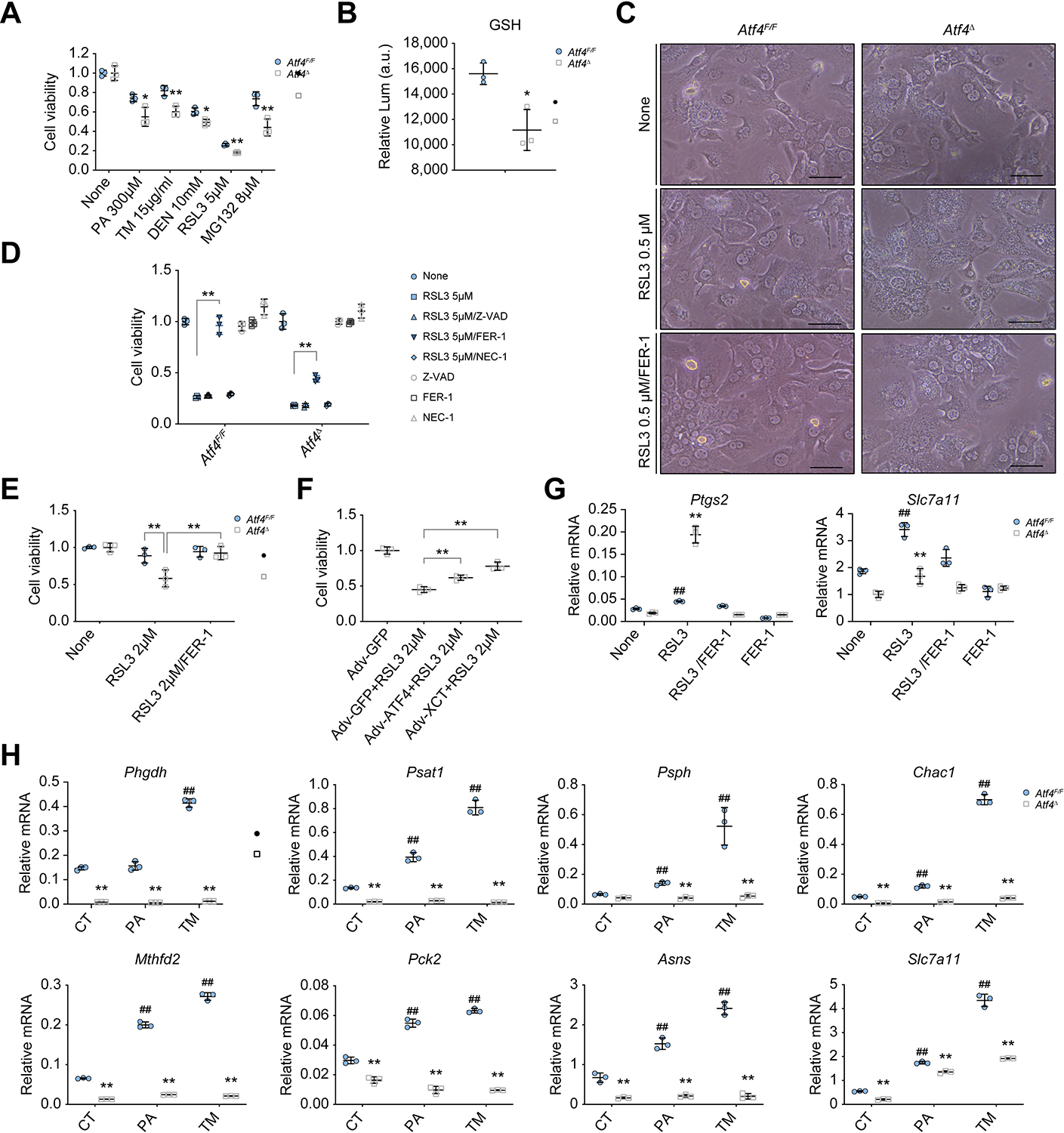
(A) Viability of Atf4F/F and Atf4Δ hepatocytes exposed to different cell death inducers. *p <0.05; **p <0.01 (vs. Atf4F/F, Student’s t test). (B) Relative GSH levels in above hepatocytes. (C) Representative images showing morphology of Atf4F/F and Atf4Δ hepatocytes treated with vehicle control (NONE) or 0.5 μM RSL3 in the absence or presence of 10 μM FER-1 for 10 h. Scale bars, 50 μm. (D) Viability of above hepatocytes treated without or with 5 μM RSL3 in the absence or presence of 50 μM Z-VAD, 10 μM FER-1, or 10 μM NEC-1 for 10 h. (E) Viability of Atf4F/F and Atf4Δ hepatocytes treated without or with 2 μM RSL3 or 2 μM RSL3 plus 10 μM FER-1 for 10 h. (F) Viability of Atf4Δ hepatocytes transduced with Adv-GFP, Adv-ATF4, or Adv-xCT 24 h before culturing in the presence of 2 μM RSL3, as indicated. **p <0.01 (Student’s t test). (G) qRT-PCR analysis of Ptgs2 and Slc7a11 mRNAs in Atf4F/F and Atf4Δ hepatocytes treated with NONE, 0.5 μM RSL3, 10 μM FER-1, or a 0.5 μM RSL3 plus 10 μM FER-1 combo for 8 h. **p <0.01 (vs. Atf4F/F, Student’s t test). ##p <0.01 (vs. NONE of Atf4F/F, Student’s t test). (H) qRT-PCR analysis of mRNA expression in Atf4F/F and Atf4Δ hepatocytes treated with NONE, 300 μM PA, or 4 μg/ml TM for 10 h. Mean ± SD (n = 3/group). **p <0.01 (vs. Atf4F/F, Student’s t test). ##p <0.01 (vs. CT of Atf4F/F, Student’s t test). Adv, adenovirus; AFT4, activating transcription factor 4; CT, Control; DEN, diethylnitrosamine; FER-1, ferrostatin-1; GSH, reduced glutathione; Lum, luminescence; NEC-1, necrostatin-1; PA, palmitic acid; RSL3, RAS-selective lethal 3; TM, tunicamycin.
To determine whether elevated ferroptosis susceptibility of Atf4Δ hepatocytes is caused by their SLC7A11 deficiency, we used adenovirus (Adv)-mediated transduction to express GFP, ATF4, or SLC7A11. Reconstitution of either ATF4 or SLC7A11 provided protection against RSL3-mediated ferroptosis, with SLC7A11 being more efficient owing to its higher expression from the Adv promoter than from the ATF4-activated endogenous promoter (Fig. 3F and Fig. S3H). RSL3-mediated oxidative stress led to NRF2 activation and induction of the NRF2 target prostaglandin G/H synthase 2 (PTGS2), a ferroptosis marker.16,45 Accordingly, ATF4 ablation increased PTGS2 and decreased SLC7A11 mRNA expression in RSL3-treated cells (Fig. 3G). These results suggest that ATF4 provides protection from ferroptosis by inducing SLC7A11 expression. PA or TM treatment of Atf4F/F and Atf4Δ hepatocytes resulted in strong induction of PHGDH, PSAT1, PSPH, CHAC1, MTHFD2, PCK2, ASNS, SLC7A11, FGF21, SLC7A5, and SLC1A4 mRNAs in Atf4F/F hepatocytes, although Atf4Δ hepatocytes were barely or modestly responsive (Fig. 3H and Fig. S3I). These results are consistent with those observed in MUP-uPA/Atf4F/F and MUP-uPA/Atf4Δhep mice.
ATF4 and NRF2 coordinately control SLC7A11 expression
NRF2 activates the antioxidant response by binding to antioxidant response elements (AREs) in its target promoters,46 whereas ATF4 controls AA metabolism by binding to AA response elements (AAREs).36 Previous studies in cancer cells showed some overlap between NRF2 and ATF4 target genes, including CHOP/Ddit347,48 and HO1.49 The Slc7a11 promoter contains one ARE and two AAREs.50,51 To understand how ATF4 and NRF2 regulate SLC7A11 expression in non-transformed hepatocytes, we generated a series of ARE and AARE deletion mutations in the Slc7a11 promoter fused to a luciferase reporter and examined their response to cotransfected ATF4 and NRF2 expression vectors (Fig. 4A). Either ATF4 or NRF2 stimulated luciferase expression, and the two combined gave the highest luciferase expression (Fig. 4B–E). Deletion of either the ARE or AAREs decreased luciferase expression to various degrees, and deletion of both elements completely blocked luciferase expression (Fig. 4B–E).
Fig. 4. ATF4 and NRF2 coordinately regulate Slc7a11 expression.
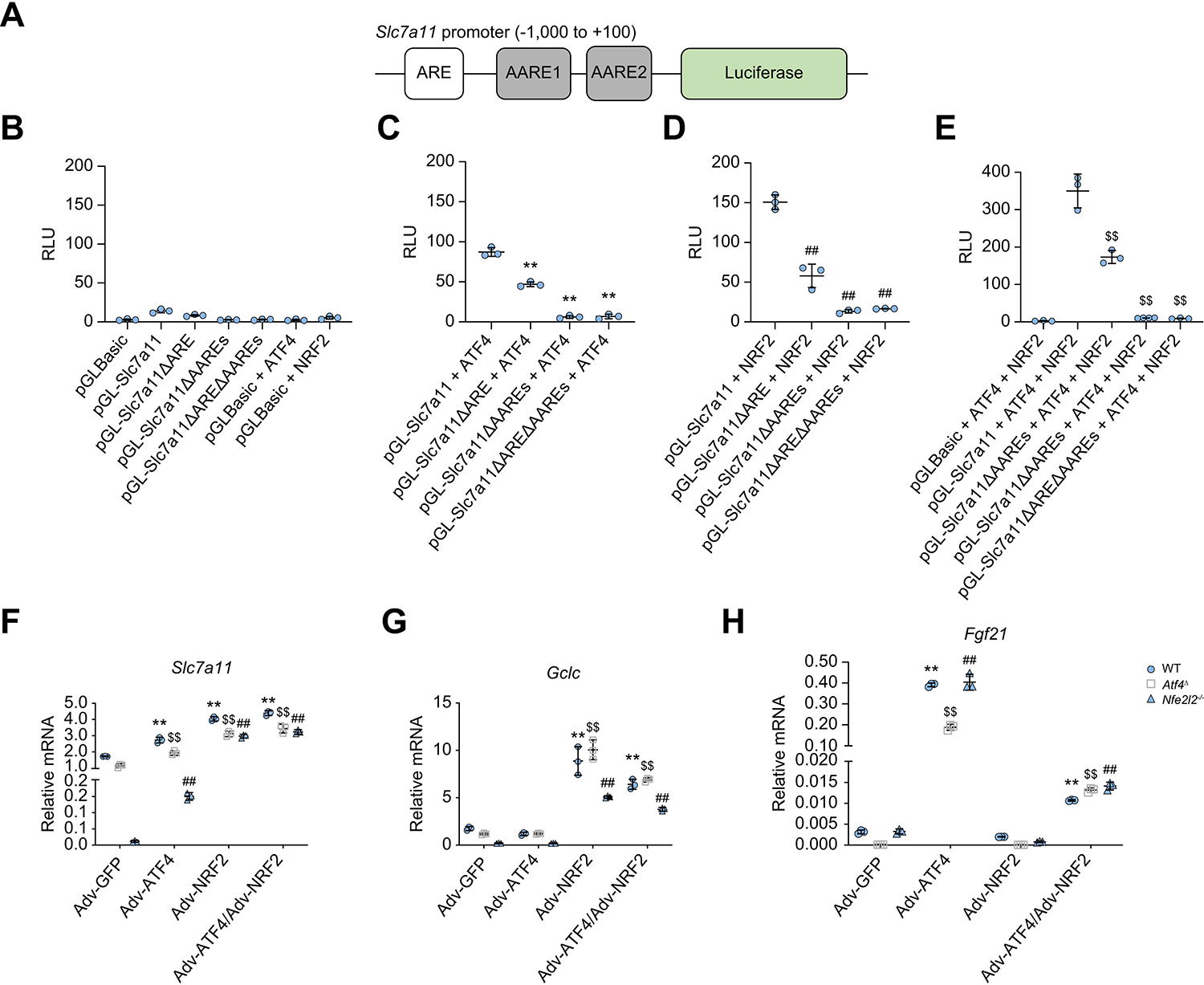
(A) Schematic representation of the mouse Slc7a11 gene promoter with the upstream ARE and the two AAREs (AARE-1 and AARE-2) indicated by open and solid rectangles, respectively, fused to a luciferase reporter. (B–E) Luciferase assay measuring Slc7a11 promoter activity before and after ARE and AARE deletion (ΔARE, ΔAAREs, and ΔAREΔAAREs) in the absence (B) or presence of ATF4 (C), NRF2 (D), and ATF4 + NRF2 (E). HEK293 cells were cotransfected with reporter plasmids (pGLBasic, pGL-Slc7a11, and its variants) in combination with pAd-track-NRF2 and/or ATF4 expression vectors, and a control Renilla luciferase vector PRL-TK. After 24 h, luciferase activities were measured and normalised to Renilla luciferase activity and presented as RLU (mean ± SD from three independent experiments). **p <0.01 (vs. pGL-Slc7a11 + ATF4); ##p <0.01 (vs. pGL-Slc7a11 + NRF2); $$p <0.01 (vs. pGL-Slc7a11 + ATF4 + NRF2), Student’s t test. (F–H) qRT-PCR analysis of Slc7a11 (F), Gclc (G), and Fgf21 (H) mRNAs in WT, Atf4Δ, and Nfe2l2−/− hepatocytes transduced with Adv-GFP, Adv-ATF4, Adv-NRF2, or Adv-ATF4 + NRF2. Mean ± SD (n = 3/group). **p <0.01 (vs. Adv-GFP of WT hepatocytes); $$p <0.01 (vs. Adv-GFP of Atf4Δ hepatocytes); ##p <0.01 (vs. Adv-GFP of Nfe2l2−/− hepatocytes), Student’s t test. AARE, amino acid response element; Adv, adenovirus; ARE, antioxidant response element; ATF4, activating transcription factor 4; NRF2, nuclear factor erythroid 2-related factor 2; qRT-PCR, quantitative reverse-transcription PCR; RLU, relative luminescence unit; SLC7A11, solute carrier family 7a member 11; WT, wild-type.
We reconstituted WT, Atf4Δ, and Nfe2l2−/− hepatocytes with Adv-GFP, Adv-ATF4, Adv-NRF2, or Adv-ATF4 + NRF2 and analysed target gene expression (Fig. S4A). Relative to Adv-GFP, either Adv-ATF4 or Adv-NRF2 induced Slc7a11 mRNA in WT and Atf4Δ hepatocytes, but the response to Adv-ATF4 was suppressed in Nfe2l2−/− hepatocytes, a defect that was overcome by Adv-NRF2 infection (Fig. 4F). This suggests that although NRF2 is a potent Slc7a11 inducer, maximal gene induction depends on ATF4, but without NRF2, ATF4 does not induce Slc7a11 mRNA. By contrast, ATF4 was not needed for induction of the typical NRF2 target genes Gclc and Ptgs2 (Fig. 4G and Fig. S4B). The typical ATF4 target gene Fgf21 was responsive to Adv-ATF4 but not to Adv-NRF2, which attenuated the response to ATF4 (Fig. 4H). Congruently, NRF2 ablation had no effect on Fgf21 induction by Adv-ATF4.
ATF4 ablation increases NASH severity and HCC burden in MUP-uPA mice
MUP-uPA/Atf4F/F and MUP-uPA/Atf4Δhep mice were kept on either LFD or HFD to examine NASH development and HCC progression. Livers of LFD-fed 4-mo MUP-uPA/Atf4F/F mice had a normal appearance, but those of MUP-uPA/Atf4Δhep mice retained a rough appearance, exhibited regenerative nodules, and had elevated serum ALT (Fig. 5A and B), indicating more liver damage. Histological analysis revealed more liver fibrosis, proliferating hepatocytes, lipid peroxidation, and cell death in MUP-uPA/Atf4Δhep relative to MUP-uPA/Atf4F/F mice (Fig. S5A). MUP-uPA/Atf4Δhep livers showed higher expression of Il6, Ly6g, and Dr5 mRNAs, suggesting increased monocyte, granulocyte, and neutrophil content (Fig. S5B). MUP-uPA/Atf4Δhep livers showed large reduction in Atf4 and Fgf21 mRNAs, but no effect on Tnf mRNA (Fig. S5B).
Fig. 5. Hepatocyte-specific ATF4 ablation increases lipid peroxidation, liver damage, inflammation, and HCC in HFD-fed MUP-uPA mice.
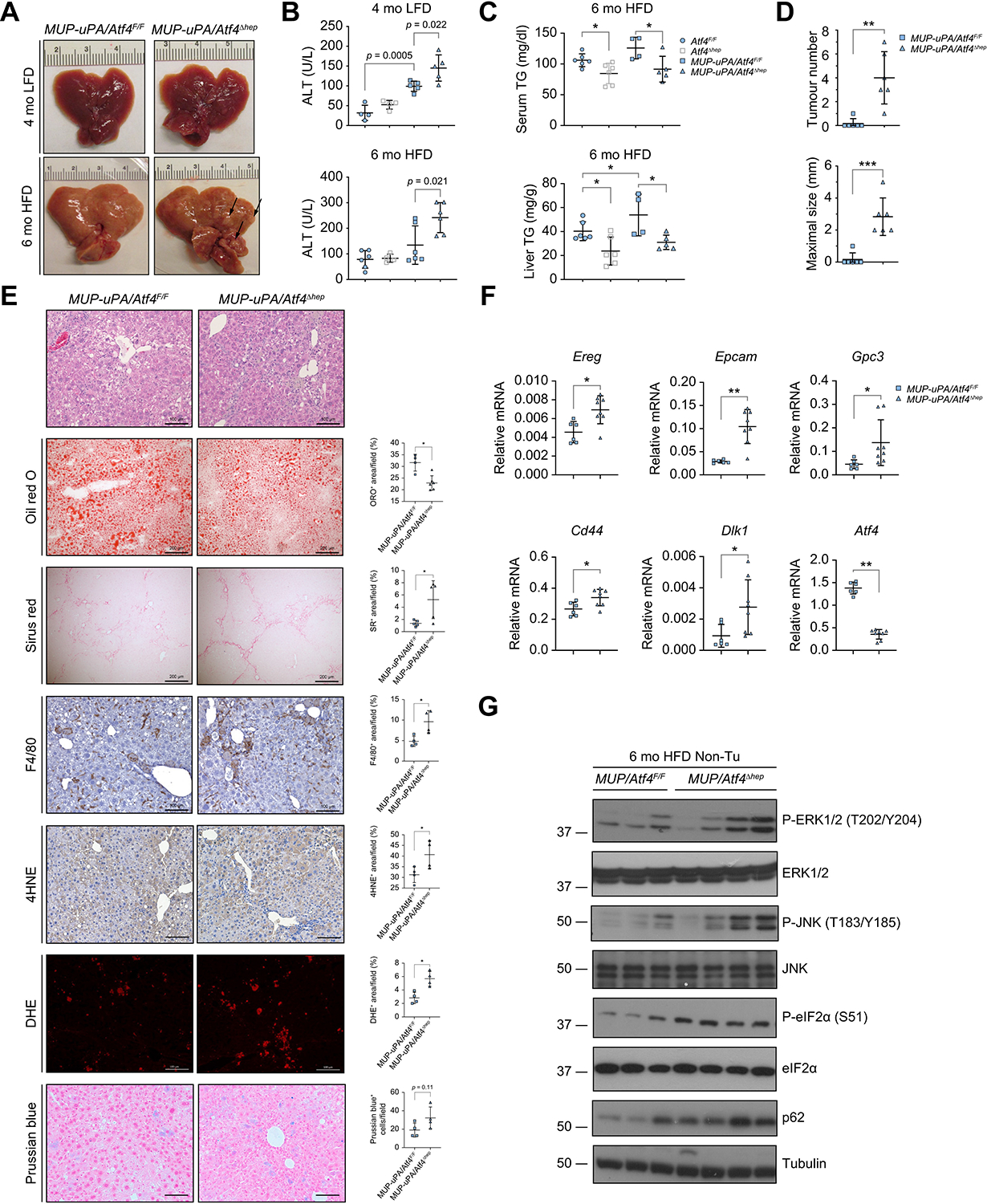
(A) Gross liver morphology of 4-mo LFD-fed and 6-mo HFD-fed MUP-uPA/Atf4F/F and MUP-uPA/Atf4Δhep mice. Arrows indicate tumours. (B) Serum ALT in 4-mo LFD-fed and 6-mo HFD-fed Atf4F/F, Atf4Δhep, MUP-uPA/Atf4F/F, and MUP-uPA/Atf4Δhep mice. Mean ± SD (n = 4–6/group). Value of p indicates significance level, Student’s t test. (C) Serum and liver TG in 6-mo HFD-fed Atf4F/F, Atf4Δhep, MUP-uPA/Atf4F/F, and MUP-uPA/Atf4Δhep mice. Mean ± SD (n = 4–6/group). *p <0.05 (Student’s t test). (D) Tumour numbers and maximal tumour sizes in livers of 6-mo HFD-fed MUP-uPA/Atf4F/F and MUP-uPA/Atf4Δhep mice. Mean ± SD (n = 6/group). **p <0.01; ***p <0.001 (Student’s t test). (E) H&E, Oil red O, Sirius Red, F4/80, 4HNE, DHE, and Prussian blue staining of liver sections from above mice. Staining was quantitated as the number of positive cells per HMF or positively stained area per field, as indicated. Scale bars, 100 μm for H&E, IHC, DHE, and Prussian blue staining; 200 μm for Oil Red O and Sirius Red staining. Mean ± SD (n = 6/group). *p <0.05 (Student’s t test). (F) qRT-PCR analysis of HCC markers in above mice. Mean ± SD (n = 6–8/group). *p <0.05; **p <0.01 (Student’s t test). (G) Representative IB analysis of Non-Tu liver tissues of above mice (n = 3–4/group). 4HNE, 4-hydroxynonenal; ALT, alanine aminotransferase; ATF4, activating transcription factor 4; DHE, dihydroethidium; eIF2α, eukaryotic translation initiation factor 2 subunit α; ERK1/2, extracellular signal-regulated kinase 1/2; HCC, hepatocellular carcinoma; HFD, high fat diet; HMF, high magnification field; IHC, immunohistochemistry; JNK, Jun N-terminal kinase; LFD, low-fat diet; mo, month old; MUP-uPA, major urinary protein promoter–urokinase plasminogen activator; Non-Tu, non-tumours; PB+, Prussian blue positive; P-ERK1/2, phosphorylated ERK1/2; P-eIF2α, phosphorylated eIF2α; P-JNK, phosphorylated JNK; qRT-PCR, quantitative reverse-transcription PCR; TG, triglycerides.
Similar to MUP-uPA mice,7 HFD-fed 6-mo MUP-uPA/Atf4F/F mice displayed typical NASH signs, including immune infiltration and fibrosis, which were more pronounced in MUP-uPA/Atf4Δhep mice, that also presented with liver tumour nodules and high circulating ALT (Fig. 5A–E). However, ATF4 ablation decreased serum triglyceride (TG) and total cholesterol (T-Chol), and liver TG, but not liver T-Chol (Fig. 5C and Fig. S5C). Notably, lipid droplet accumulation in MUP-uPA/Atf4Δhep livers was reduced relative to MUP-uPA/Atf4F/F livers (Fig. 5E), a situation typical of advanced human NASH.3,52 ATF4 ablation also accelerated and augmented HCC development, which was not apparent in MUP-uPA/Atf4F/F mice of a similar age (Fig. 5D), and increased lipid peroxidation (Fig. 5E). Prussian blue staining showed the presence of liver ferric iron, which contributes to ferroptosis induction, in both livers, which was not affected by ATF4 ablation. qRT-PCR analysis revealed increased expression of mRNAs coding for the fibrogenic cytokine transforming growth factor beta (TGFβ) and several fibrosis markers as well as inflammatory cytokines and chemokines, tumour necrosis factor (TNF), IL-1β, C–C motif chemokine ligand 2 (CCL2), CCL19, and IL-23A in the MUP-uPA/Atf4Δhep livers (Fig. S5D and E). Consistent with HCC development, MUP-uPA/Atf4Δhep livers exhibited elevated expression of mRNAs for HCC markers, epiregulin (EREG), epithelial cell adhesion molecule (EPCAM), glypican-3 (GPC3), CD44, and protein delta homologue 1 (DLK1) (Fig. 5F). ATF4 deficiency increased phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2) at threonine 202 and tyrosine 204, Jun N-terminal kinase (JNK) at threonine 183 and tyrosine 185, and eIF2α at serine 51, as well as p62 expression (Fig. 5G), which supports HCC development in MUP-uPA mice.5 Expression and phosphorylation of signal transducer and activator of transcription 3 (STAT3), AKT, and JNK, but not of ERK, were also increased in MUP-uPA/Atf4Δhep tumours isolated from 10-mo mice (Fig. S5F). MUP-uPA/Atf4Δhep HCCs were all ATF4 deficient (Fig. S5G), indicating they originated from ATF4-deficient hepatocytes. These data indicate that despite the reduction in hepatic steatosis, ATF4 deletion accelerates NASH pathogenesis and NASH to HCC progression.
ATF4 ablation enhances DEN-induced liver injury and tumorigenesis
DEN is a hepatotoxin and carcinogen that activates liver UPR signalling and increases ATF4 expression during HCC development.53 To examine the effect of ATF4 ablation on chemical liver injury and carcinogenesis, we challenged 3-mo Atf4F/F and Atf4Δhep mice with high-dose DEN (100 mg/kg). DEN is metabolically activated by cytochrome P450 2E1 (CYP2E1) in pericentral zone 3 hepatocytes.5,54 Accordingly, 24 h after DEN administration, Atf4F/F livers exhibited pericentral damage, whereas Atf4Δhep livers showed broader damage extending from zone 3 to zone 2; increased proliferating cell nuclear antigen (PCNA), F4/80, TUNEL, phosphorylated (γ) histone H2AX, and 4HNE staining; and elevated serum ALT (Fig. S6A–C). ATF4 ablation, however, did not affect CYP2E1 expression (Fig. S6A, D and E), suggesting that enhanced liver damage in Atf4Δhep mice is not caused by altered DEN metabolism.
To induce HCC development, 2-wo mice were given a lower DEN dose (25 mg/kg).5 Atf4F/F mice exhibited macroscopic tumours only after 10 months, whereas 5-mo Atf4Δhep mice showed small tumours, and when 10 mo, they showed higher tumour burden than Atf4F/F mice (Fig. 6A). Liver damage, tumour number, and size were higher in Atf4Δhep mice (Fig. 6B and C) and so was their liver to body weight ratio (Fig. S6F). The non-tumour portion of Atf4Δhep livers showed elevated cyclin-D1 (CCND1), mouse double minute 2 homologue (MDM2), connective tissue growth factor (CTGF), and DR5 mRNAs, but reduced FGF21 mRNA (Fig. 6D). Histological analysis showed that ATF4 ablation enhanced formation of nodular cancer foci positive for the HCC markers GPC3 and alpha-foetoprotein (AFP) (Fig. S7A). ATF4-deficient HCCs were stained more intensely for GPC3, AFP, and 4HNE (Fig. 6E). p38, STAT3, ERK1/2, and JNK phosphorylation were generally higher in ATF4-deficient tumours, which expressed more EREG but less FGF21 mRNA (Fig. 6F and G).
Fig. 6. ATF4 ablation accelerates DEN-induced HCC onset and burden.
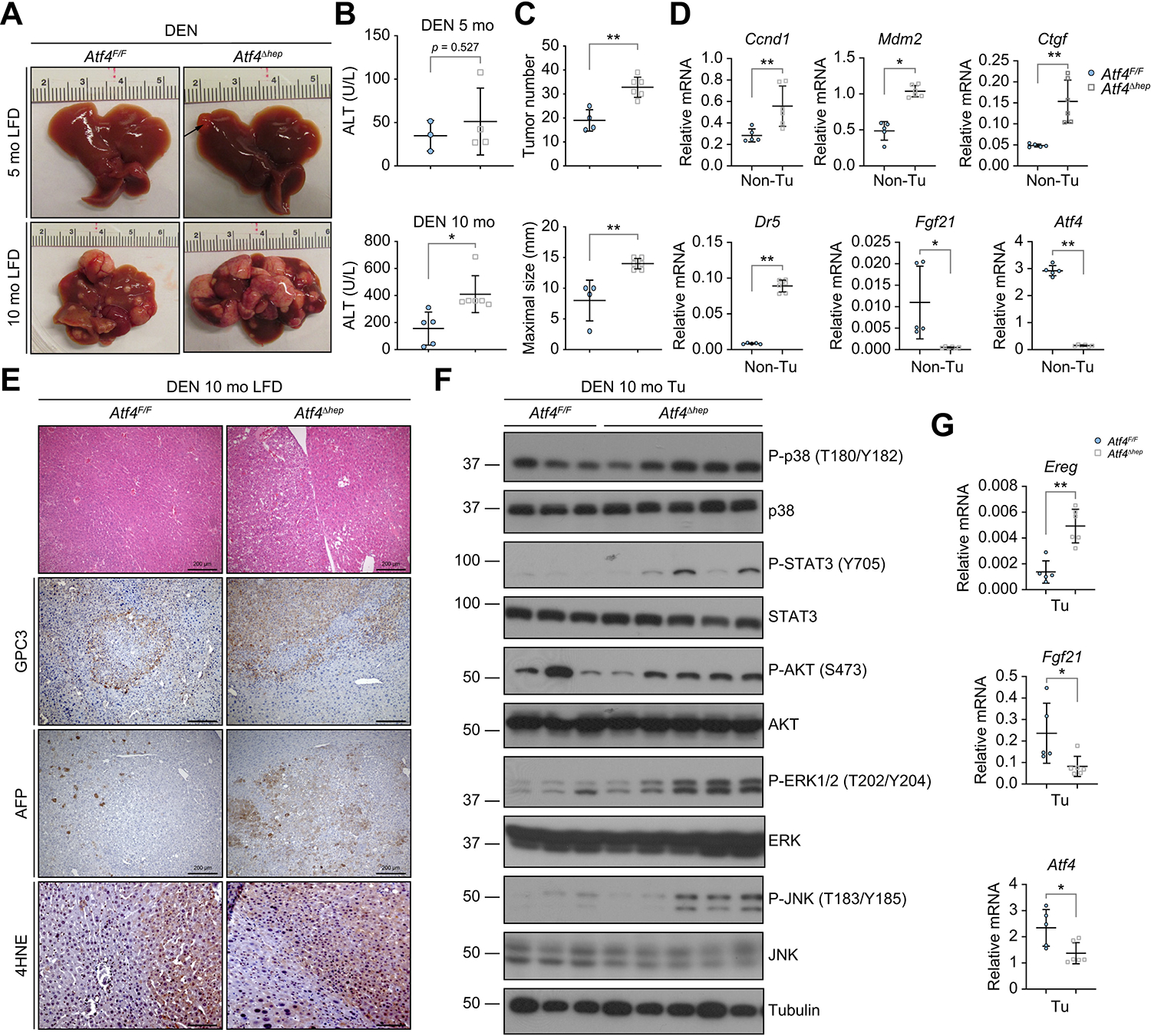
(A) Gross liver morphology of DEN-treated 5- and 10-mo LFD-fed Atf4F/F and Atf4Δhep mice. (B) Serum ALT in DEN-treated 5-mo (top) and 10-mo (bottom) Atf4F/F and Atf4Δhep mice. Mean ± SD (n = 3–6/group). *p <0.05; **p <0.01 (Student’s t test). (C) Liver tumour number (top) and maximal tumour size (bottom) in above 10-mo mice. Mean ± SD (n = 4–6/group). **p <0.01 (Student’s t test). (D) qRT-PCR analysis of mRNAs in the Non-Tu region of above 5-mo mice. Mean ± SD (n = 5–6/group). *p <0.05; **p <0.01 (Student’s t test). (E) H&E, GPC3, AFP, and 4HNE staining of liver sections from above 10-mo mice (n = 6/group). Scale bars, 200 μm for H&E, GPC3, and AFP staining; 100 μm for 4HNE staining. (F) IB analysis of tumours from above 10-mo mice (n = 3–5/group). (G) qRT-PCR analysis of tumour mRNAs from livers of above mice. Mean ± SD (n = 5–6/group). *p <0.05; **p <0.01 (Student’s t test). 4HNE, 4-hydroxynonenal; ALT, alanine aminotransferase; ATF4, activating transcription factor 4; AFP, alpha-foetoprotein; DEN, diethylnitrosamine; ERK1/2, extracellular signal-regulated kinase 1/2; GPC3, glypican 3; IB, immunoblotting; JNK, Jun N-terminal kinase; LFD, low-fat diet; mo, month old; Non-Tu, non-tumour; P-AKT, phosphorylated AKT; P-ERK1/2, phosphorylated ERK1/2; P-JNK, phosphorylated JNK; P-STAT3, phosphorylated STAT3; qRT-PCR, quantitative reverse-transcription PCR; STAT3, signal transducer and activator of transcription 3; Tu, tumour.
Restoration of Slc7a11/xCT expression inhibits liver tumorigenesis
To investigate whether SLC7A11 reconstitution can prevent the enhancement of liver damage and tumour formation in ATF4-deficient mice, we used adeno-associated virus 8 (AAV8) to transduce SLC7A11, whose N-terminus was fused to mCherry (AAV8-xCT), or mCherry (AAV8-mCherry), into livers of 8-wo MUP-uPA/Atf4F/F and MUP-uPA/Atf4Δhep mice (Fig. S7B). We first checked liver damage in 3.5-mo LFD-fed MUP-uPA/Atf4Δhep mice and found that AAV8-xCT-transduced MUP-uPA/Atf4Δhep livers had smooth appearances and fewer macroscopic regenerative nodules than AAV8-mCherry-transduced livers (Fig. 7A). AAV8-mCherry and AAV8-xCT were both well expressed in the MUP-uPA/Atf4Δhep livers. Histological analysis revealed less inflammation and lipid peroxidation in AAV-xCT-transduced livers compared with AAV8-mCherry-transduced MUP-uPA/Atf4Δhep livers (Fig. 7A), as well as lower serum ALT (Fig. 7B). IB and qRT-PCR confirmed exogenous xCT expression in the AAV8-xCT-transduced livers (Fig. 7C and D) and lower expression of mRNAs for the fibrosis markers collagen IV α1 (COL4A1) and tissue inhibitor of metalloproteinase 1 (TIMP1), and TNF (Fig. 7D). Administration of a low dose of FER-1 every other day for 2 weeks to 6-wo MUP-uPA/Atf4Δhep mice reduced liver inflammation (F4/80 staining) (Fig. 7E). AAV8-xCT transduction of MUP-uPA/Atf4Δhep mice reduced liver tumour size relative to the effect of AAV8-mCherry transduction, although its effect on tumour number was not significant (Fig. 7F and G). The negligible effect on tumour initiation might be as a result of either a small sample size or delayed xCT expression or action. To overcome this limitation, we used a model of delayed HCC induction, combining DEN administration to adult mice with HFD feeding and PB administration in the drinking water.55 Atf4F/F and Atf4Δhep mice subjected to this mode of HCC induction were first injected AAV8-mCherry or AAV8-xCT when 8 wo, followed by HFD feeding when 3 mo, DEN (80 mg/kg) administration when 4 mo, and water with 0.05% PB when 5 mo, and analysed for tumour burden when 10 mo (Fig. S7B). As in young DEN-challenged mice (Fig. 6A), ATF4 ablation enhanced HCC development, and compared with AAV8-mCherry transduction, AAV8-xCT transduction significantly reduced both tumour number and tumour size (Fig. S7C and D). These results indicate that the most critical ATF4 target for suppression of liver damage, inflammation, and tumorigenesis is the small xCT subunit.
Fig. 7. xCT reconstitution attenuates liver injury and reduces HCC burden.
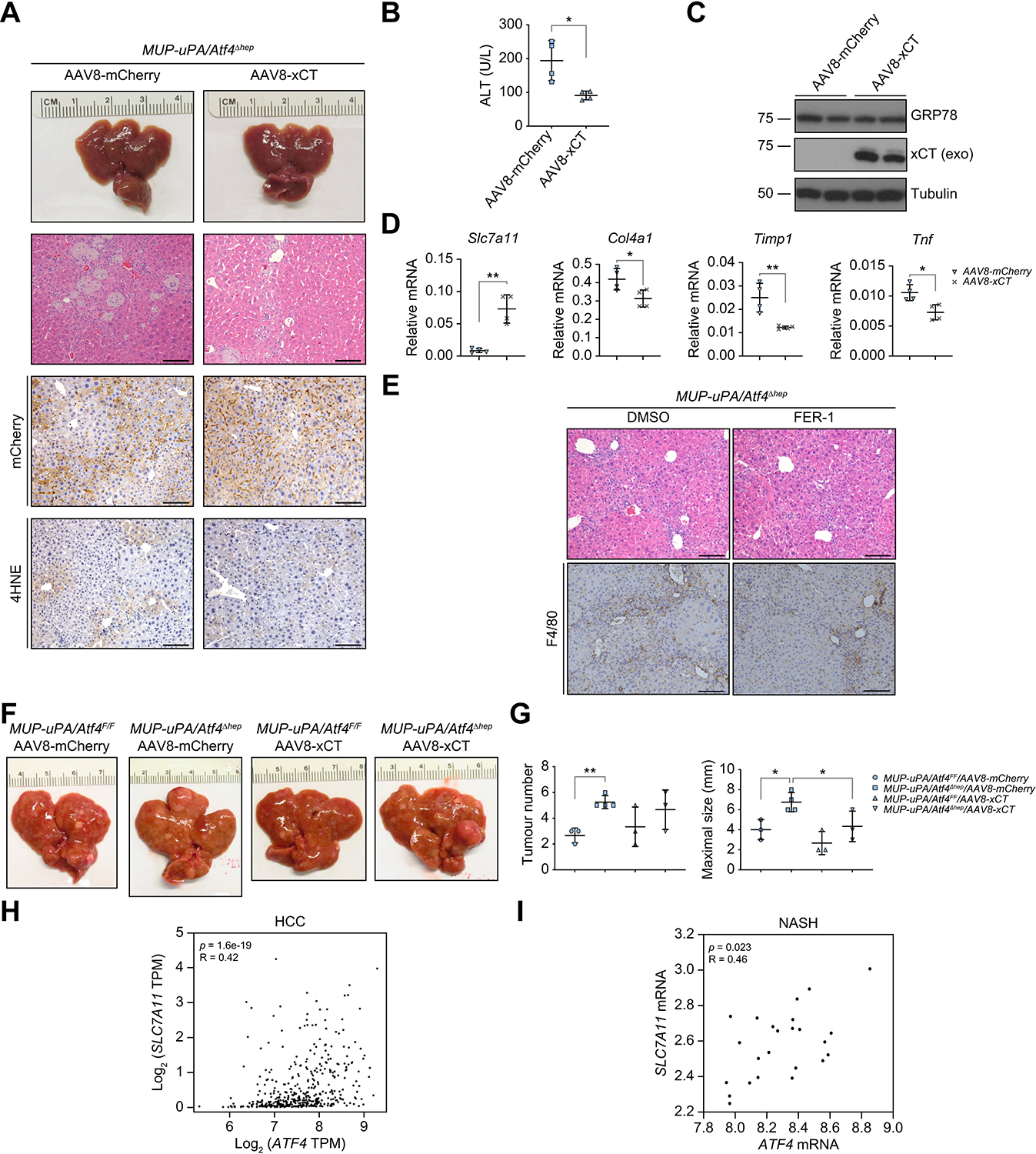
(A) Gross liver morphology, H&E staining, and IHC analysis of 3.5-mo LFD-fed MUP-uPA/Atf4Δhep mice injected with AAV8-mCherry or AAV8-xCT at 8 wo (n = 4/group). Scale bars, 100 μm. (B) Serum ALT in above mice. Mean ± SD (n = 4/group). *p <0.05 (Student’s t test). (C) IB analysis of liver lysates from above mice. Exo indicates exogenous mCherry-xCT fusion protein. (D) qRT-PCR analysis of liver mRNAs in above mice. Mean ± SD (n = 4/group). *p <0.05; **p <0.01 (Student’s t test). (E) H&E and F4/80 staining of 2-mo LFD-fed MUP-uPA/Atf4Δhep mice i.p. injected with vehicle (DMSO) or 1 mg/kg FER-1 every other day for 2 weeks (n = 3/group). Scale bars, 100 μm. (F and G) Gross liver morphology (F) and tumour burden (G) in 10-mo HFD-fed MUP-uPA/Atf4F/F and MUP-uPA/Atf4Δhep mice injected with AAV8-mCherry or AAV8-xCT when 8 wo. Mean ± SD (n = 3–4/group). *p <0.05; **p <0.01 (Student’s t test). (H and I) Correlation analysis of ATF4 and SLC7A11 expression. Spearman’s analysis of ATF4 and SLC7A11 mRNA amounts in human TCGA HCC datasets (H) and in patients with NASH (I) (data extracted from GSE61260). 4HNE, 4-hydroxynonenal; AAV8, adeno-associated virus 8; ALT, alanine aminotransferase; ATF4, activating transcription factor 4; FER-1, ferrostatin-1; GRP78, 78-kDa glucose-regulated protein; HCC, hepatocellular carcinoma; HFD, high-fat diet; IB, immunoblotting; IHC, immunohistochemistry; LFD, low-fat diet; MUP-uPA, major urinary protein promoter–urokinase plasminogen activator; qRT-PCR, quantitative reverse-transcription PCR; SLC7A11, solute carrier family 7a member 11; TCGA, The Cancer Genome Atlas; TPM, transcripts per million; wo, week old.
Discussion
Normal hepatocytes barely turnover, with virtually no cell death or proliferation.56 However, the liver retains the ability to quickly regenerate in response to extensive hepatocellular death caused by viral, toxic, metabolic, or autoimmune insults. This so-called compensatory proliferation contributes to oncogenic transformation, and elevated circulating aminotransferases, markers for hepatocyte death, are highly predictive of HCC development in patients.57,58 Different forms of hepatocyte death were suggested to contribute to the cancer-promoting necroinflammatory response, including apoptosis, necrosis, and necroptosis.11,59,60 As shown above, a predominant driver of HCC-promoting necroinflammation in NASH-afflicted MUP-uPA mice is ATF4–SLC7A11-suppressible ferroptosis. Although DEN-induced liver injury is complex and involves several distinct modes of cell death, ferroptosis also appears to be a predominant cause of liver injury in mice challenged with DEN in combination with HFD and PB. SLC7A11/xCT, which determines the intracellular concentration of GSH by importing cystine and exporting glutamate at a 1:1 ratio in an ATP-dependent manner, is a key regulator of ferroptosis.12,42 We demonstrate that ATF4-deficient hepatocytes and livers are sensitised to ER stress-induced cell death and exhibit a substantially lower GSH:GSSG ratio and elevated lipid peroxidation, classical hallmarks of ferroptosis that can be attributed to SLC7A11 deficiency. Of note, despite being tumour suppressive in other cell types and tissues, hepatocyte cell death, which is inhibited by NF-κB, is tumour-promoting in the liver.8 Even in this early study, published before the discovery of ferroptosis,12 it was clear that the liver-damaging and HCC-promoting effect of IKKβ ablation was fully reversed by an NAC-containing diet, which restored GSH levels and prevented oxidative stress and lipid peroxidation.8 Likewise, in the present study, liver damage and HCC promotion were suppressed by forced SLC7A11 expression, although the hepatocytes remained ATF4 deficient. Hepatocyte-specific ablation of the antiapoptotic proteins myeloid cell leukemia-1 and B-cell lymphoma-extra large also increased the rate of apoptosis and led to spontaneous HCC development.61,62 It will be interesting to determine whether ferroptosis inhibitors also suppress liver damage and hepatocarcinogenesis in these mice.
Previous studies had shown that ferroptosis is mainly regulated by NRF2, which controls expression of SLC7A11 and GPX4, as well as GSH-synthesising enzymes. Ferroptosis was implicated in drug-induced liver injury (DILI), ischaemia–reperfusion injury, and chronic liver disease with one or several features of GSH depletion, iron overload, and reduced SLC7A11 and GPX4 expression.16 Although the role of ferroptosis in NAFLD progression and carcinogen-induced HCC has not been established, 4-HNE staining was used as an oxidative stress marker in patients with NASH.63 Ferroptosis, however, was shown to trigger inflammation in a mouse steatohepatitis model induced by a choline-deficient, ethionine-supplemented diet.63 F-box and leucine-rich repeat protein 5 (FBXL5) regulates iron homoeostasis, and FBXL5-deficient mice exhibit features of ferroptosis, including liver iron overload, oxidative stress, liver injury, and steatohepatitis, and, similar to Atf4Δhep mice, are more susceptible to DEN-induced HCC.63 However, in all of these cases, it is not clear whether all hepatocytes die by ferroptosis or whether other forms of cell death, including necrosis, apoptosis, cuproptosis, and necroptosis, also take place.11,42,60,64 Despite these uncertainties, it is important to test whether FER-1 and other newly developed ferroptosis inhibitors can reduce liver injury and HCC susceptibility in NASH, ASH, DILI, α1 anti-trypsin deficiency, and iron overload diseases. Given that SLC7A11 is often highly expressed in different cancers, its overexpression can inhibit reactive oxygen species-induced ferroptosis, promote tumour growth, and confer drug resistance.14,15,65,66 ATF4 expression is also upregulated in different cancers to promote cell survival and tumour growth.36,37 Of note, ATF4 and SLC7A11 levels are positively correlated in human HCC (Fig. 7H), and ATF4-deficient HCC cells are more sensitive to ferroptosis (Fig. S7E). In human NASH specimens, ATF4 and SLC7A11 are also correlated (Fig. 7I), supporting our finding that inhibition of ferroptosis by the ATF4–SLC7A1 axis suppresses hepatocarcinogenesis in NASH-afflicted MUP-uPA mice (Fig. S7F). It is therefore plausible that ferroptosis inhibitors may be effective in preventing the progression of chronic liver diseases to HCC, although their effect on established HCC remains to be determined. The timing and duration of ferroptosis inhibition are important and should be optimised for achieving a maximal cancer preventive effect.
ATF4 is a key downstream effector of the ER and integrated stress responses, which are triggered by nutrient deprivation, viral infection, and oxidative stress.67 However, under persistent stress or when adaptation is insufficient, ATF4 promotes cell death.68 Of note, liver-specific ATF4 ablation inhibits hepatic steatosis,30,31,33–35 which could be a protective response that prevents further progression to steatohepatitis. Indeed, loss of ATF4 enhances liver damage.29 Unlike HFD-fed BL6 mice, which develop simple or benign steatosis, HFD-fed MUP-uPA mice progress to steatohepatitis, liver damage, and fibrosis owing to ER stress-driven hepatocyte death.7 The results described above suggest that ATF4 attenuates the progression from simple steatosis to NASH and HCC by suppressing ferroptosis, thereby blocking liver damage and necroinflammation. Consistent with previous publications,30,31,34 hepatocyte ATF4 ablation inhibited hepatic steatosis, reinforcing the notion that hepatic steatosis per se is not damaging and does not increase cancer risk unless it is accompanied by liver damage.
Conclusions
Our study suggests that ferroptosis, which is inhibited by the ATF4–SLC7A11 axis, may be the most relevant form of hepatocyte death that leads to HCC-accelerating inflammation and compensatory proliferation. Ferroptosis inhibitors or ATF4 activators may be useful in preventing NASH development and its progression to HCC.
Supplementary Material
Acknowledgements
We thank Eric P. Sandgren (University of Wisconsin-Madison, WI, USA) for MUP-uPA mice; Kang Zhang (University of California San Diego, CA, USA) for AAV8 vectors and advice on AAV generation; Dennis Petersen (The University of Colorado Denver, CO, USA) for the 4HNE antibody; Jonathan H. Lin (Stanford University, CA, USA), Tatiana Kisseleva (University of California San Diego, CA, USA), and David A. Brenner (University of California San Diego, CA, USA) for suggestions and constructive criticism; Karin laboratory members for helpful discussions; and eBioscience, Cell Signaling Technologies, Santa Cruz Technologies, and Promega for gifts of reagents. We also thank Lin Yuan, Na Hu, and Yi Jiang at the Electron Microscopy Core Facility of Shanghai University of Traditional Chinese Medicine for technical help.
Financial support
This research was supported by the Superfund Basic Research Program (P42-ES010337), NIH (P01-DK098108, 5R01DK120714-02, R01-CA118165, R37-AI043477, P01-CA128814, R01-CA211794, R01-CA198103, R01-DK103185, and R01-DK113171) and a C3 Pedal the Cause grant to MK, who holds the Ben and Wanda Hildyard Chair for Mitochondrial and Metabolic Diseases; FH was supported by the National Natural Science Foundation of China (82172947) and the Eli Lilly LIFA programme.
Abbreviations
- 4HNE
4-hydroxynonenal
- β-Me
β-mercaptoethanol
- AA
amino acid
- AARE
AA response element
- AAV8
adeno-associated virus 8
- Adv
adenovirus
- AFLD
alcoholic fatty liver disease
- AFP
alpha-foetoprotein
- ALDH7A1
alpha-aminoadipic semialdehyde dehydrogenase
- ALT
alanine aminotransferase
- AMT
aminomethyltransferase
- ARE
antioxidant response element
- ASCT2
alanine, serine, cysteine, threonine transporter 2
- ASH
alcoholic steatohepatitis
- ASNS
asparagine synthetase
- ATF4
activating transcription factor 4
- ATF6
activating transcription factor 6
- BAX
Bcl-2-associated X protein
- BID
BH3-interacting domain death agonist
- CARS
cysteinyl-tRNA synthetase
- CCL2
C–C motif chemokine ligand 2
- CCND1
cyclin-D1
- CHAC1
glutathione-specific gamma-glutamylcyclotransferase 1
- CHOP
C/EBP-homologous protein
- COL4A1
collagen IV α1
- CTGF
connective tissue growth factor
- CTH
cystathionine gamma-lyase
- CXCL2
C-X-C motif chemokine ligand 2
- CYP2E1
cytochrome P450 2E1
- DEN
diethylnitrosamine
- DFO
deferoxamine
- DILI
drug-induced liver injury
- DLK1
protein delta homologue 1
- DR5
death receptor 5
- EDEM2
ER degradation-enhancing alpha-mannosidase-like protein 2
- eIF2α
eukaryotic translation initiation factor 2 subunit α
- EPCAM
epithelial cell adhesion molecule
- ER
endoplasmic reticulum
- EREG
epiregulin
- ERK1/2
extracellular signal-regulated kinase ½
- ERP5
endoplasmic reticulum protein 5
- FBXL5
F-box and leucine-rich repeat protein 5
- FER-1
ferrostatin-1
- FGF21
fibroblast growth factor 21
- GCL
glutamate–cysteine ligase
- GLUD1
glutamate dehydrogenase 1
- GO
gene ontology
- GPC3
glypican-3
- GPX4
glutathione peroxidase 4
- GRP78
78-kDa glucose-regulated protein
- GSH
glutathione
- GSL2
glutaminase liver isoform
- GSS
glutathione synthase
- GSSG
oxidised glutathione
- HCC
hepatocellular carcinoma
- HFD
high-fat diet
- HO1
haem oxygenase 1
- IB
immunoblotting
- JNK
Jun N-terminal kinase
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LFD
low-fat diet
- MDM2
mouse double minute 2 homologue
- mo
month old
- MTHFD2
methenyltetrahydrofolate dehydrogenase 2
- MUP
major urinary protein
- NAC
N-acetyl cysteine
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- NEC-1
necrostatin-1
- NNMT
N-nicotinamide methyltransferase
- NQO1
NAD(P)H quinone dehydrogenase 1
- NRF1
nuclear respiratory factor 1
- NRF2
nuclear factor erythroid 2-related factor 2
- P-eIF2α
phosphorylated eIF2α
- PA
palmitic acid
- PB
phenobarbital
- PCK2
mitochondrial phosphoenolpyruvate carboxykinase
- PCNA
proliferating cell nuclear antigen
- PERK
pancreatic ER-regulating kinase
- PHGDH
phosphoglycerate dehydrogenase
- PSAT1
phosphoserine aminotransferase 1
- PSPH
phosphoserine phosphatase
- PTGS2
prostaglandin G/H synthase 2
- PUMA
p53 upregulated modulator of apoptosis
- qRT-PCR
quantitative reverse-transcription PCR
- RNA-seq
RNA sequencing
- RSL3
RAS-selective lethal 3
- SARS
seryl-tRNA synthetase
- SHMT2
serine hydroxymethyltransferase 2
- SLC7A11
solute carrier family 7a member 11
- SOD2
superoxide dismutase 2
- STAT3
signal transducer and activator of transcription 3
- T-Chol
total cholesterol
- TAFLD
toxicant-associated fatty liver disease
- TASH
toxicant-associated steatohepatitis
- TEM
transmission electron microscopy
- TFAM
mitochondrial transcription factor A
- TG
triglycerides
- TGFβ
transforming growth factor beta
- TIMP1
tissue inhibitor of metalloproteinase 1
- TM
tunicamycin
- TNF
tumour necrosis factor
- TNFR1
tumour necrosis factor receptor 1
- TNFRSF6
tumour necrosis factor receptor superfamily member 6
- TRB3
tribbles homologue 3
- uPA
urokinase-type plasminogen activator
- UPR
unfolded protein response
- wo
week old
- WT
wild-type
- XBP1
X-box-binding protein 1.
Footnotes
Conflicts of interest
BY is a full-time employee of Eli Lilly and Company. FH was partly supported by the Eli Lilly LIFA programme. The remaining authors declare no competing interests.
Please refer to the accompanying ICMJE disclosure forms for further details.
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jhep.2023.03.016.
Data availability statement
The data associated with this paper and further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, MK (karinoffice@ucsd.edu).
References
- [1].Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021;71:209–249. [DOI] [PubMed] [Google Scholar]
- [2].Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol 2019;16:411–428. [DOI] [PubMed] [Google Scholar]
- [3].Kim JY, He F, Karin M. From liver fat to cancer: perils of the Western diet. Cancers 2021;13:1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol 2018;15:599–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Umemura A, He F, Taniguchi K, Nakagawa H, Yamachika S, Font-Burgada J, et al. p62, upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell 2016;29:935–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021;184:2537–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014;26:331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKb couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005;121:977–990. [DOI] [PubMed] [Google Scholar]
- [9].Schneider AT, Gautheron J, Feoktistova M, Roderburg C, Loosen SH, Roy S, et al. RIPK1 suppresses a TRAF2-dependent pathway to liver cancer. Cancer Cell 2017;31:94–109. [DOI] [PubMed] [Google Scholar]
- [10].Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol 2018;15:738–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology 2014;147:765–783.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012;149:1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. NF-jB restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016;164:896–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015;520:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hu K, Li K, Lv J, Feng J, Chen J, Wu H, et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J Clin Invest 2020;130:1752–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang H, An P, Xie E, Wu Q, Fang X, Gao H, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017;66:449–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cel Biol 2020;21:421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer 2014;14:581–597. [DOI] [PubMed] [Google Scholar]
- [19].Wu Y, Shan B, Dai J, Xia Z, Cai J, Chen T, et al. Dual role for inositol-requiring enzyme 1ɑ in promoting the development of hepatocellular carcinoma during diet-induced obesity in mice. Hepatology 2018;68:533–546. [DOI] [PubMed] [Google Scholar]
- [20].Zhang K, Wang S, Malhotra J, Hassler JR, Back SH, Wang G, et al. The unfolded protein response transducer IRE1ɑ prevents ER stress-induced hepatic steatosis. EMBO J 2011;30:1357–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang JM, Qiu Y, Yang Z, Kim H, Qian Q, Sun Q, et al. IRE1ɑ prevents hepatic steatosis by processing and promoting the degradation of select microRNAs. Sci Signal 2018;11:eaao4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Niederreiter L, Fritz TM, Adolph TE, Krismer AM, Offner FA, Tschurtschenthaler M, et al. ER stress transcription factor Xbp1 suppresses intestinal tumorigenesis and directs intestinal stem cells. J Exp Med 2013;210:2041–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu X, Henkel AS, LeCuyer BE, Schipma MJ, Anderson KA, Green RM. Hepatocyte X-box binding protein 1 deficiency increases liver injury in mice fed a high-fat/sugar diet. Am J Physiol Gastrointest Liver Physiol 2015;309:G965–G974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Arai M, Kondoh N, Imazeki N, Hada A, Hatsuse K, Kimura F, et al. Transformation-associated gene regulation by ATF6ɑ during hepatocarcinogenesis. FEBS Lett 2006;580:184–190. [DOI] [PubMed] [Google Scholar]
- [25].Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest 2012;122:4621–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cel 2003;11:619–633. [DOI] [PubMed] [Google Scholar]
- [27].Seo J, Fortuno ES 3rd, Suh JM, Stenesen D, Tang W, Parks EJ, et al. Atf4 regulates obesity, glucose homeostasis, and energy expenditure. Diabetes 2009;58:2565–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wang C, Huang Z, Du Y, Cheng Y, Chen S, Guo F. ATF4 regulates lipid metabolism and thermogenesis. Cell Res 2010;20:174–184. [DOI] [PubMed] [Google Scholar]
- [29].Fusakio ME, Willy JA, Wang Y, Mirek ET, Al Baghdadi RJ, Adams CM, et al. Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Mol Biol Cel 2016;27:1536–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Xiao G, Zhang T, Yu S, Lee S, Calabuig-Navarro V, Yamauchi J, et al. ATF4 protein deficiency protects against high fructose-induced hyper-triglyceridemia in mice. J Biol Chem 2013;288:25350–25361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li H, Meng Q, Xiao F, Chen S, Du Y, Yu J, et al. ATF4 deficiency protects mice from high-carbohydrate-diet-induced liver steatosis. Biochem J 2011;438:283–289. [DOI] [PubMed] [Google Scholar]
- [32].Yueh MF, He F, Chen C, Vu C, Tripathi A, Knight R, et al. Triclosan leads to dysregulation of the metabolic regulator FGF21 exacerbating high fat diet-induced nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A 2020;117:31259–31266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Song Q, Chen Y, Wang J, Hao L, Huang C, Griffiths A, et al. ER stress-induced upregulation of NNMT contributes to alcohol-related fatty liver development. J Hepatol 2020;73:783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li K, Xiao Y, Yu J, Xia T, Liu B, Guo Y, et al. Liver-specific gene inactivation of the transcription factor ATF4 alleviates alcoholic liver steatosis in mice. J Biol Chem 2016;291:18536–18546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hao L, Zhong W, Dong H, Guo W, Sun X, Zhang W, et al. ATF4 activation promotes hepatic mitochondrial dysfunction by repressing NRF1-TFAM signalling in alcoholic steatohepatitis. Gut 2021;70:1933–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, et al. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J 2010;29:2082–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tameire F, Verginadis II, Leli NM, Polte C, Conn CS, Ojha R, et al. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat Cel Biol 2019;21:889–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ebert SM, Dyle MC, Kunkel SD, Bullard SA, Bongers KS, Fox DK, et al. Stress-induced skeletal muscle Gadd45a expression reprograms myonuclei and causes muscle atrophy. J Biol Chem 2012;287:27290–27301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A 1996;93:13943–13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sandgren EP, Palmiter RD, Heckel JL, Daugherty CC, Brinster RL, Degen JL. Complete hepatic regeneration after somatic deletion of an albumin-plasminogen activator transgene. Cell 1991;66:245–256. [DOI] [PubMed] [Google Scholar]
- [41].He F, Antonucci L, Yamachika S, Zhang Z, Taniguchi K, Umemura A, et al. NRF2 activates growth factor genes and downstream AKT signaling to induce mouse and human hepatomegaly. J Hepatol 2020;72:1182–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Stockwell BR. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022;185:2401–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal 2014;21:396–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014;3:e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shin D, Kim EH, Lee J, Roh JL. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic Biol Med 2018;129:454–462. [DOI] [PubMed] [Google Scholar]
- [46].Yamamoto M, Kensler TW, Motohashi H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev 2018;98:1169–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol 2002;318:1351–1365. [DOI] [PubMed] [Google Scholar]
- [48].Zong ZH, Du ZX, Li N, Li C, Zhang Q, Liu BQ, et al. Implication of Nrf2 and ATF4 in differential induction of CHOP by proteasome inhibition in thyroid cancer cells. Biochim Biophys Acta 2012;1823:1395–1404. [DOI] [PubMed] [Google Scholar]
- [49].He CH, Gong P, Hu B, Stewart D, Choi ME, Choi AM, et al. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J Biol Chem 2001;276:20858–20865. [DOI] [PubMed] [Google Scholar]
- [50].Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, et al. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem 2002;277:44765–44771. [DOI] [PubMed] [Google Scholar]
- [51].Ye P, Mimura J, Okada T, Sato H, Liu T, Maruyama A, et al. Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol Cel Biol 2014;34:3421–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Asgharpour A, Cazanave SC, Pacana T, Seneshaw M, Vincent R, Banini BA, et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J Hepatol 2016;65:579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Vandewynckel YP, Laukens D, Bogaerts E, Paridaens A, Van den Bussche A, Verhelst X, et al. Modulation of the unfolded protein response impedes tumor cell adaptation to proteotoxic stress: a PERK for hepatocellular carcinoma therapy. Hepatol Int 2015;9:93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Halpern KB, Shenhav R, Matcovitch-Natan O, Toth B, Lemze D, Golan M, et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 2017;542:352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010;140:197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Benedetti A, Jézéquel AM, Orlandi F. Preferential distribution of apoptotic bodies in acinar zone 3 of normal human and rat liver. J Hepatol 1988;7:319–324. [DOI] [PubMed] [Google Scholar]
- [57].Yang HI, Yuen MF, Chan HL, Han KH, Chen PJ, Kim DY, et al. Risk estimation for hepatocellular carcinoma in chronic hepatitis B (REACH-B): development and validation of a predictive score. Lancet Oncol 2011;12:568–574. [DOI] [PubMed] [Google Scholar]
- [58].Wen CP, Lin J, Yang YC, Tsai MK, Tsao CK, Etzel C, et al. Hepatocellular carcinoma risk prediction model for the general population: the predictive power of transaminases. J Natl Cancer Inst 2012;104:1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Seehawer M, Heinzmann F, D’Artista L, Harbig J, Roux PF, Hoenicke L, et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature 2018;562:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Nanji AA, Hiller-Sturmhöfel S. Apoptosis and necrosis: two types of cell death in alcoholic liver disease. Alcohol Health Res World 1997;21:325–330. [PMC free article] [PubMed] [Google Scholar]
- [61].Weber A, Boger R, Vick B, Urbanik T, Haybaeck J, Zoller S, et al. Hepatocyte-specific deletion of the antiapoptotic protein myeloid cell leukemia-1 triggers proliferation and hepatocarcinogenesis in mice. Hepatology 2010;51:1226–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hikita H, Kodama T, Shimizu S, Li W, Shigekawa M, Tanaka S, et al. Bak deficiency inhibits liver carcinogenesis: a causal link between apoptosis and carcinogenesis. J Hepatol 2012;57:92–100. [DOI] [PubMed] [Google Scholar]
- [63].Tsurusaki S, Tsuchiya Y, Koumura T, Nakasone M, Sakamoto T, Matsuoka M, et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis 2019;10:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022;375:1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gao R, Kalathur RKR, Coto-Llerena M, Ercan C, Buechel D, Shuang S, et al. YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis. EMBO Mol Med 2021;13:e14351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016;63:173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Costa-Mattioli M, Walter P. The integrated stress response: from mechanism to disease. Science 2020;368:eaat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cel Biol 2013;15:481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data associated with this paper and further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, MK (karinoffice@ucsd.edu).