

Abstract
患儿,男,7月龄,表现为重度全面发育落后、难治性癫痫、肌张力降低、眼球震颤、眼距宽、鼻梁塌陷、上唇外翻、高腭弓和隐睾,基因检测发现EEF1A2基因存在c.364G>A(p.E122K)新生杂合错义变异,最终该患儿确诊为EEF1A2基因变异致常染色体显性遗传发育性癫痫性脑病33型。该病例报道提示,对不明原因婴儿期起病的重度-极重度全面发育落后/智力障碍、难治性癫痫患儿,尤其是存在肌张力低下、语言缺失、颅面部畸形者,应考虑EEF1A2基因变异可能,应尽早完善遗传学检测协助诊断。
Keywords: 全面发育落后, 智力障碍, 癫痫, EEF1A2基因, 婴儿
Abstract
A boy, aged 7 months, presented with severe global developmental delay (GDD), refractory epilepsy, hypotonia, nystagmus, ocular hypertelorism, a broad nasal bridge, everted upper lip, a high palatal arch, and cryptorchidism. Genetic testing revealed a de novo heterozygous missense mutation of c.364G>A(p.E122K) in the EEF1A2 gene, and finally the boy was diagnosed with autosomal dominant developmental and epileptic encephalopathy 33 caused by the EEF1A2 gene mutation. This case report suggests that for children with unexplained infancy-onset severe to profound GDD/intellectual disability and refractory epilepsy, genetic testing for EEF1A2 gene mutations should be considered. This is particularly important for those exhibiting hypotonia, nonverbal communication, and craniofacial deformities, to facilitate a confirmed diagnosis.
Keywords: Global developmental delay, Intellectual disability, Epilepsy, EEF1A2 gene, Infant
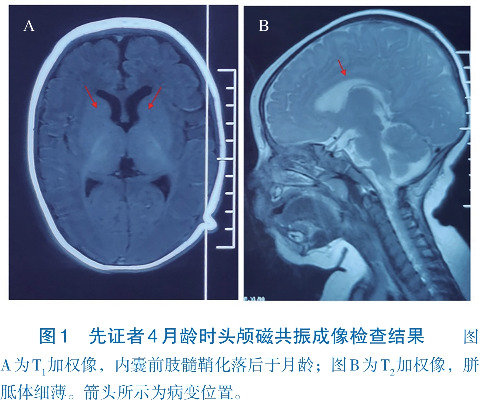
患儿,男,7月龄,因发育落后、抽搐4个月就诊。患儿3月龄不能抬头,并出现抽搐,表现为大叫一声后,意识丧失,双眼上翻,双手握拳,面色口唇发绀,持续数十秒,发作时测体温37.5℃,当日共发作3次,未予特殊处理。4月龄再次出现抽搐,发作形式及持续时间与之前相同,遂至当地医院住院,完善4 h脑电图示多量广泛尖波、棘波、多棘波阵发。头颅磁共振成像示内囊前肢髓鞘化落后于月龄,胼胝体细薄(图1)。诊断为“癫痫、发育落后”,予左乙拉西坦(每日40 mg/kg)治疗,仍有反复发作,1~2次/周。4个月20 d加用托吡酯(每日3 mg/kg),发作改善不明显。6月龄出现发呆愣神,双眼凝视,呼之不应。7月龄出现间断双侧眼球水平震颤,抬头仍不稳,不会独坐、翻身、爬,不能“咿呀”发音。为进一步诊治,来我院就诊。既往有反复呼吸道感染病史。患儿系第2胎第2产,胎龄39周,因“疤痕子宫”剖宫产娩出,无抢救窒息史,出生体重3.1 kg,生后混合喂养。母亲孕期身体健康,否认毒物、放射线接触史。父母身体健康,否认近亲婚配,有一同母异父哥哥,身体健康,否认家族有神经精神疾病或遗传病史。
体格检查:体重10 kg(P 75~P 90),身长79 cm(>P 97),头围45 cm(P 75)。前囟平软,约0.5 cm×0.5 cm。神志清楚,反应迟钝,追光、追物、追声欠佳,偶可逗笑,双眼球间断有水平震颤,不能抓物,眼距宽,鼻梁塌陷、上唇外翻、高腭弓。心肺腹检查未见异常。四肢肌力正常,四肢肌张力稍降低,腱反射未引出,病理征阴性,手掌、脚掌脱皮。双侧阴囊未触及睾丸。
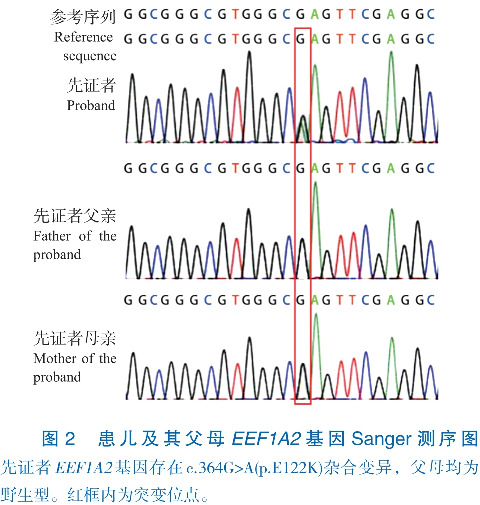
辅助检查:入院后血常规、尿常规、肝功能、肾功能、心肌酶、电解质、血脂、动脉血气分析、血乳酸、血糖、血氨基酸和酰基肉碱筛查未见异常。7月龄4 h视频脑电图示弥漫性大量2~5 Hz混合活动低-中波幅,发作间期睡眠期广泛性棘慢波/尖慢波。家系全外显子组测序示EEF1A2基因4号外显子存在c.364G>A(p.E122K)(NM_001958.5)杂合错义变异。Sanger测序示患儿父母该位点均为野生型,提示该变异为新生变异(图2)。本例患儿是第364位核苷酸G突变为A,导致其编码的蛋白第122位谷氨酸变成赖氨酸,依据美国医学遗传学与基因组学学会变异分类标准与指南[1],该变异评定为致病性变异(PS1+PS2+PS3+PM1+PM2)。
结合临床特点及辅助检查,最终该患儿诊断为:(1)常染色体显性遗传发育性癫痫性脑病33型(developmental and epileptic encephalopathy 33, DEE33);(2)隐睾。将患儿托吡酯加量至5 mg/(kg·d),发作减少,7~10 d发作1次。1岁1月龄发作频率增加,4 h视频脑电图示清醒期、睡眠期双侧后头部为主多灶性或广泛性尖波、棘波、棘慢波/尖慢波、多棘慢波,清醒期全面性发作、肌阵挛发作,加用丙戊酸钠(每日20 mg/kg),仍有反复发作,表现为全身抖动一下,持续2~3 s,每小时发作数次。1岁3月龄加用氯巴占(每日10 mg),左乙拉西坦减量,发作减少,每日发作数次,表现为白天入睡前出现轻微点头。随访至1岁8个月,患儿对外界反应好转,追光、追物可,能“咿呀”发音,会无意识发“baba、mama”音,不能听懂简单指令,抬头稳,不能翻身、独坐、独走,持物差,四肢肌张力偏低,腱反射未引出,病理征阴性。双侧阴囊未触及睾丸。
讨论:EEF1A2基因定位于人类染色体20q13.3,编码真核细胞翻译延长因子1α2(eukaryotic translation elongation factor 1, alpha-2, eEF1A2)。EEF1A2是常染色体显性遗传DEE33和常染色体显性遗传智力障碍38型的致病基因。真核细胞翻译延长因子1α有两种同分异构体,即eEF1A1和eEF1A2,分别由EEF1A1和EEF1A2基因编码,其通过促进氨基酰tRNA与核糖体A位点的结合,对蛋白质合成过程新生多肽的延伸发挥重要作用[2]。eEF1A1在各种组织中广泛表达;eEF1A2在脑、心肌、骨骼肌中特异表达[3]。eEF1A1在整个发育过程中均广泛表达,但出生后在神经元、骨骼和心脏中表达量下降,最终被eEF1A2完全取代[3],这个过程在小鼠出生后21 d左右完成。eEF1A1和eEF1A2的互斥表达与Eef1a2基因敲除小鼠断奶前表现正常,约生后21 d开始出现震颤、共济失调、伴或不伴自发性癫痫发作,28 d内死亡相符[4]。
2012年de Ligt等[5]首次报道1例EEF1A2基因错义变异导致重度智力障碍(intellectual disability, ID)和癫痫患者。迄今为止,英文文献共报道41例EEF1A2基因变异患者[6-18],未见相关中文病例报道。包括本文病例,共42例EEF1A2基因变异患者,男性17例,女性23例,2例未描述性别,报道时年龄为1岁3个月至32岁[6-18]。EEF1A2基因共有18种不同变异位点,均为错义变异,无移码、剪接、无义等功能缺失变异[6-18]。这种独特的基因变异谱提示,EEF1A2基因杂合功能丧失性变异可能导致人类胚胎致死,或者人类可以耐受EEF1A2基因杂合功能丧失性变异,也提示EEF1A2基因杂合错义变异可能是通过功能获得或显性负效应致病。18种EEF1A2基因变异散布在整个基因的编码序列中,无明确的基因型-表型相关性[6-18]。42例EEF1A2基因变异患者均有全面发育落后(global developmental delay, GDD)/ID,以重度-极重度为主,多数患者存在语言缺失、难治性癫痫、肌张力降低、运动障碍、面部外观异常、小头畸形等[6-18]。该病预后差,多数癫痫发作难以控制,已报道4例死亡病例,2例死于神经退行性病变[7],2例死于心力衰竭[6],因此EEF1A2基因变异患者除了神经功能评估,还应进行心脏功能评估。本例患儿以重度GDD、语言缺失、早发难治性癫痫、四肢肌张力低下、眼球震颤、面部外观异常和隐睾为主要表现,EEF1A2基因存在c.364G>A(p.E122K)新生杂合错义变异,该变异已在8例患者中报道[7-9,12,16]。文献报道的8例p.E122K变异患者均存在中度-重度GDD/ID和癫痫发作,7例语言缺失,5例肌张力低下,6例颅面部外观异常[7-9,12,16]。本例患儿临床表现与文献报道的8例p.E122K变异患者临床表型相符,因此,结合临床表现及基因检测结果最终确诊为常染色体显性遗传DEE33。
综上所述,EEF1A2基因变异患者临床特点为婴儿期起病,重度-极重度GDD/ID、语言缺失、难治性癫痫、肌张力减低、颅面畸形等。该病预后差,可因心力衰竭和神经退行性病变导致早期死亡,因此EEF1A2基因变异患者都应进行神经功能和心脏功能评估。
基金资助
国家自然科学基金(82201316)。
利益冲突声明
所有作者声明无利益冲突。
作者贡献
贺海兰负责文章设计和撰写;林雪芹、王晓乐、彭盼、肖慧负责病例收集;尹飞、彭镜负责病例筛选和审核。
参 考 文 献
- 1. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17(5): 405-424. DOI: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McLachlan F, Sires AM, Abbott CM. The role of translation elongation factor eEF1 subunits in neurodevelopmental disorders[J]. Hum Mutat, 2019, 40(2): 131-141. DOI: 10.1002/humu.23677. [DOI] [PubMed] [Google Scholar]
- 3. Chambers DM, Peters J, Abbott CM. The lethal mutation of the mouse wasted (wst) is a deletion that abolishes expression of a tissue-specific isoform of translation elongation factor 1alpha, encoded by the Eef1a2 gene[J]. Proc Natl Acad Sci U S A, 1998, 95(8): 4463-4468. DOI: 10.1073/pnas.95.8.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Newbery HJ, Gillingwater TH, Dharmasaroja P, et al. Progressive loss of motor neuron function in wasted mice: effects of a spontaneous null mutation in the gene for the eEF1A2 translation factor[J]. J Neuropathol Exp Neurol, 2005, 64(4): 295-303. DOI: 10.1093/jnen/64.4.295. [DOI] [PubMed] [Google Scholar]
- 5. de Ligt J, Willemsen MH, van Bon BW, et al. Diagnostic exome sequencing in persons with severe intellectual disability[J]. N Engl J Med, 2012, 367(20): 1921-1929. DOI: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 6. Cao S, Smith LL, Padilla-Lopez SR, et al. Homozygous EEF1A2 mutation causes dilated cardiomyopathy, failure to thrive, global developmental delay, epilepsy and early death[J]. Hum Mol Genet, 2017, 26(18): 3545-3552. DOI: 10.1093/hmg/ddx239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carvill GL, Helbig KL, Myers CT, et al. Damaging de novo missense variants in EEF1A2 lead to a developmental and degenerative epileptic-dyskinetic encephalopathy[J]. Hum Mutat, 2020, 41(7): 1263-1279. DOI: 10.1002/humu.24015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Rinaldis M, Giorda R, Trabacca A. Mild epileptic phenotype associates with de novo eef1a2 mutation: case report and review[J]. Brain Dev, 2020, 42(1): 77-82. DOI: 10.1016/j.braindev.2019.08.001. [DOI] [PubMed] [Google Scholar]
- 9. Inui T, Kobayashi S, Ashikari Y, et al. Two cases of early-onset myoclonic seizures with continuous parietal delta activity caused by EEF1A2 mutations[J]. Brain Dev, 2016, 38(5): 520-524. DOI: 10.1016/j.braindev.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 10. Kaneko M, Rosser T, Raca G. Dilated cardiomyopathy in a patient with autosomal dominant EEF1A2-related neurodevelopmental disorder[J]. Eur J Med Genet, 2021, 64(1): 104121. DOI: 10.1016/j.ejmg.2020.104121. [DOI] [PubMed] [Google Scholar]
- 11. Kaur S, Van Bergen NJ, Gold WA, et al. Whole exome sequencing reveals a de novo missense variant in EEF1A2 in a Rett syndrome-like patient[J]. Clin Case Rep, 2019, 7(12): 2476-2482. DOI: 10.1002/ccr3.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lam WW, Millichap JJ, Soares DC, et al. Novel de novo EEF1A2 missense mutations causing epilepsy and intellectual disability[J]. Mol Genet Genomic Med, 2016, 4(4): 465-474. DOI: 10.1002/mgg3.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lance EI, Kronenbuerger M, Cohen JS, et al. Successful treatment of choreo-athetotic movements in a patient with an EEF1A2 gene variant[J]. SAGE Open Med Case Rep, 2018, 6: 2050313X18807622. DOI: 10.1177/2050313X18807622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Long K, Wang H, Song Z, et al. EEF1A2 mutations in epileptic encephalopathy/intellectual disability: understanding the potential mechanism of phenotypic variation[J]. Epilepsy Behav, 2020, 105: 106955. DOI: 10.1016/j.yebeh.2020.106955. [DOI] [PubMed] [Google Scholar]
- 15. Lopes F, Barbosa M, Ameur A, et al. Identification of novel genetic causes of Rett syndrome-like phenotypes[J]. J Med Genet, 2016, 53(3): 190-199. DOI: 10.1136/jmedgenet-2015-103568. [DOI] [PubMed] [Google Scholar]
- 16. Nakajima J, Okamoto N, Tohyama J, et al. De novo EEF1A2 mutations in patients with characteristic facial features, intellectual disability, autistic behaviors and epilepsy[J]. Clin Genet, 2015, 87(4): 356-361. DOI: 10.1111/cge.12394. [DOI] [PubMed] [Google Scholar]
- 17. Veeramah KR, Johnstone L, Karafet TM, et al. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies[J]. Epilepsia, 2013, 54(7): 1270-1281. DOI: 10.1111/epi.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vogt LM, Lorenzo M, Prendergast D B, et al. EEF1A2 pathogenic variant presenting in an infant with failure to thrive and frequent apneas requiring respiratory support[J]. Am J Med Genet A, 2022, 188(10): 3106-3109. DOI: 10.1002/ajmg.a.62932. [DOI] [PubMed] [Google Scholar]