

Summary
Patients with long-standing inflammatory bowel disease (IBD) face an increased risk of developing colitis-associated cancer (CAC). Although IBD-induced prolonged inflammation seems to be involved in CAC pathogenesis, the specific molecular changes that contribute remain unknown. Here, we applied digital spatial RNA profiling, RNAscope, and imaging mass cytometry to examine paired uninflamed, inflamed, and early dysplastic mucosa of patients with IBD. We observed robust type 3 (IL-17) responses during inflammation, accompanied by elevated JAK-STAT signaling and phosphorylated STAT3 (P-STAT3) levels, with both inflamed and dysplastic mucosa displaying immune cell activation. Higher stromal P-STAT3 was detected in uninflamed and inflamed mucosa of patients who eventually developed dysplasia. CD8a+ T cells did not infiltrate inflamed or dysplastic epithelial regions in these patients, while control patients showed elevated CD8a in inflamed mucosa. Our study reveals distinct inflammatory patterns throughout CAC development, marked by an activated IL-17 pathway, engaged STAT3, and diminished cytotoxic T cell infiltration.
Subject areas: disease, biopsy sample, components of the immune system, bioinformatics, expression study, transcriptomics
Graphical abstract
Highlights
-
•
Immune-epithelial crosstalk changes during inflammation and dysplasia development
-
•
Type 3 (IL-17) and cytotoxic immune responses increase in inflammation and dysplasia
-
•
CD8+ T cells localize more distantly from the epithelium during inflammation
-
•
Elevated IL-17A and P-STAT3 mark inflamed mucosa from patients with dysplasia
Disease; Biopsy sample; Components of the immune system; Bioinformatics; Expression study; Transcriptomics.
Introduction
Colitis-associated colorectal cancer (CAC) is a well-known complication of long-standing inflammatory bowel disease (IBD), with standardized incidence ratios ranging from 1.7 up to 7.2 in high-risk groups.1,2 Despite this high rate of incidence, molecular mechanisms underlying the pathogenesis of CAC are incompletely understood. Several lines of evidence suggest that inflammation predisposes patients to colitis-associated dysplasia.1 For example, proinflammatory cytokines, such as interleukin-6 (IL-6) and IL-23, were reported to have a role in this process.3,4 Additionally, reduction of inflammation through genetic manipulation of key inflammatory pathways, such as nuclear factor κB (NF-κB) activation, has been shown to suppress colitis-associated tumorigenesis.5 Taken together, these studies underscore the importance of specific inflammatory pathways in inflammation-driven cancer; however, specific molecular changes that drive this process in patients have not been characterized.
Previous genetic studies revealed that most of the early colitis-associated dysplastic lesions feature loss-of-function mutations in tumor suppressor gene P53.6,7 Furthermore, chromosomal instability has been shown to progress from low-grade to high-grade dysplasia in this setting.8 In neighboring non-dysplastic tissue, an increased level of a variety of somatic single-nucleotide alterations in genes involved in IL-17 and Toll-like receptor signaling has also been detected, indicating that mutations accumulate under chronic inflammatory conditions.9 Of note, single-nucleotide polymorphisms in inflammation response genes, such as IL23R and NOD2/CARD15, are associated with IBD,9,10,11,12,13 but the involvement of these inflammatory pathways in CAC development is presently unclear. Moreover, the majority of insights into the changes that accompany CAC development have been obtained using mouse models, which imperfectly capture human physiology.
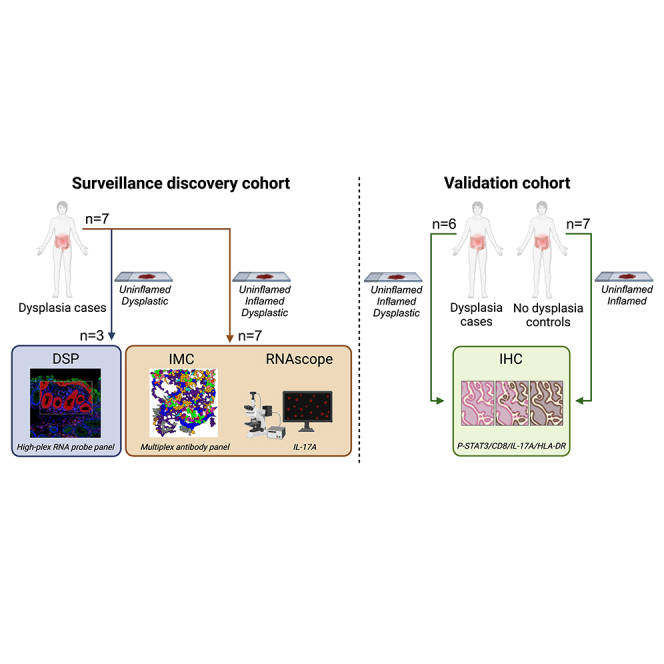
Here, we describe initial analysis of genetic and molecular signatures of colitis-associated tumorigenesis in humans enabled by access to a unique patient cohort composed of patients with long-standing colitis (>9 years) enlisted in the Utrecht IBD Surveillance Study. We used digital spatial RNA profiling (DSP), RNAscope, and imaging mass cytometry (IMC) and analyzed paired uninflamed, inflamed, and early dysplastic mucosa obtained from patient surveillance endoscopies. Next, we compared mucosal samples from patients that developed dysplasia (cases) and patients who did not (controls) to identify the most significant differences between the two groups. This allowed us to pinpoint IL-17 pathway and JAK-STAT3 signaling pathway activation and diminished cytotoxic T cell mucosal infiltration as distinguishing features of CAC development. Overall, this improved understanding of the underlying pathophysiological mechanisms in patients at risk for CAC development could contribute to more effective surveillance protocols and treatment strategies.
Results
Inflammation and cancer pathways are upregulated in dysplastic mucosa
To explore the inflammatory landscape in the mucosa of patients at risk for CAC, we compared dysplastic colonic mucosa from 3 patients with long-standing colitis (>9 years) enrolled in the Utrecht IBD Surveillance Study with matched uninflamed mucosa (mucosa with no signs of ongoing chronic or active inflammation) (Tables 1 and 2). Since dysplasia mostly comprises a relatively small region, we used selected, pathologist-annotated regions within tissue sections and performed DSP of immune cells (CD45+) and epithelial cells (panCK+) (Figures 1A and S1A).14 Gene expression profiles of epithelial and immune cells are clearly separated in uniform manifold approximation and projection (UMAP) (Figure S1B) and unsupervised clustering analyses (Figure S1C).
Table 1.
Patient characteristics
| Patient ID | Diagnosis | Sex | Agea | Disease duration (years)a | Montreal class | Maximal extent of inflammation |
IBD medicationa | |
|---|---|---|---|---|---|---|---|---|
| endoscopic | histological | |||||||
| 1 | CD | M | 68–72 | 45–48 | A2L3B1 | Pancolitis | pancolitis | AZA |
| 2 | CD | M | 44–52 | 36–44 | A1L3B2 | Pancolitis | pancolitis | AZA |
| 3 | UC | M | 28–34 | 9–15 | E3 | Pancolitis | left-sided colitis | 6-MP |
| 4 | CD | M | 49–51 | 10–11 | A2L2B1 | left-sided colitis | pancolitis | 5-ASA, AZA |
| 5 | CD | M | 55–62 | 30–37 | A2L3B1 | >50% | pancolitis | 5-ASA |
| 6 | UC | F | 50–53 | 15–19 | E3 | extensive colitis | extensive colitis | 5-ASA, AZA |
| 7 | IC + PSC | M | 44–47 | 24–26 | E3 | Pancolitis | pancolitis | 5-ASA |
CD, Crohn’s disease; UC, ulcerative colitis; IC, indeterminate colitis; PSC, primary sclerosing cholangitis; AZA, azathioprine (6-MP prodrug); 6-MP, 6-mercaptopurine; 5-ASA, 5-aminosalicylates.
at the time of sample collection.
Table 2.
Biopsy characteristics of GeoMx digital spatial profiling
| Patient ID | Status | nROI |
|---|---|---|
| 1 | Uninflamed | 3 |
| 1 | Uninflamed | 3 |
| 1 | Uninflamed | 3 |
| 1 | Dysplastic | 2 |
| 4 | Uninflamed | 3 |
| 4 | Dysplastic | 2 |
| 6 | Uninflamed | 3 |
| 6 | Uninflamed | 3 |
| 6 | Dysplastic | 2 |
Figure 1.

Digital spatial RNA profiling reveals elevated inflammatory signals in colitis-associated dysplasia
Formalin-fixed paraffin embedded mucosal biopsies (3 dysplastic and 7 paired non-dysplastic, uninflamed tissues) were included from 3 patients enrolled in surveillance.
(A) Masks for epithelium (panCK+) and immune cells (CD45+) were generated, for 3 regions per biopsy. Scale bars represent 1 mm.
(B) Epithelial and (C) immune cell gene expression-based pathway analysis of uninflamed versus dysplastic biopsies. Top 20 of KEGG human pathway enrichment, most significant processes are highlighted in red, and the less significant processes are highlighted in blue according to −log10(FDR). Dot size indicates number of genes involved: larger dots in the graph correspond to a greater number of genes.
(D) Volcano plots showing differential gene expression with genes enriched in uninflamed (left) or in dysplastic tissue (right). Significance is indicated by color: p < 0.05 (light blue), FDR<0.05 (dark blue), FDR<0.001 (orange, and names indicated).
(E) Gene expression level of IL-17 family (type 3 inflammation) cytokines compared between dysplastic and uninflamed epithelial regions and immune cells. Displayed are IL-17A, IL-17F, and IL-21. Student’s t test was performed. p values are indicated in the graphs. See also Tables S1A and S1B and Figure S1.
Pathway analysis revealed significant upregulation of inflammation-related pathways (systemic lupus erythematosus, NET formation, cytokine-cytokine receptor interaction, and chemokine signaling) and cancer-related pathways (transcriptional misregulation in cancer, PI3K-AKT signaling pathway, and viral carcinogenesis) in the dysplastic epithelium (Figure 1B). Immune cells displayed significant upregulation of the cytokine-cytokine receptor interaction pathway, as well as other pathways related to inflammation (complement and coagulation cascades, systemic lupus erythematosus) and cancer (choline metabolism in cancer, and transcriptional misregulation in cancer) (Figure 1C). Gene expression analysis of the epithelium showed significantly reduced expression of multiple histone genes (H2, H3, and H4) in dysplasia, consistent with the upregulation of “systemic lupus erythematosus” and “NET formation” Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways since histones play a major role in both pathways (Figures 1D and S1A).15,16 The cytokine-cytokine receptor pathways were upregulated as indicated by increased levels of IL-3, IL-5, IL-16, IL-19, IFNW1, IFNAR2, IL-7R, and IL-22RA1, and notably three IL-17 pathway cytokines (IL-17A, IL-17F, and IL-21), suggesting a role for type 3 inflammation (Figure 1E; Table S1A). We also observed that chemokines CCL2, CCL16, CCL19, CXCL9, and CXCL14 were upregulated in dysplasia (Table S1A). Analysis of the immune cell compartment showed no significant differentially expressed genes overall (FDR<0.05), possibly due to the large diversity of cells within this compartment and the bulk level analysis due to the use of the DSP method (Figure 1D; Table S1B). Taken together, the DSP results indicate that inflammation-related pathways were upregulated in the dysplastic epithelium, as evidenced by the presence of increased levels of cytokines, cytokine receptors, and chemokines, especially those involved in type 3 inflammation.
Dysplastic epithelium shows low inflammatory signaling and MHC-II expression
To explore the changes of immune cell subsets and the inflammatory interactions during dysplasia development, we selected a set of longitudinally collected colon biopsies from 7 patients with IBD for IMC analysis, including consecutive sections of the tissue samples used for DSP analysis (Table 1). Selection of these colonic site-matched biopsies was based on the presence of inflammation (uninflamed: no signs of ongoing chronic or active inflammation; inflamed: active and/or chronic inflammation) and on the presence or absence of dysplasia (dysplastic). Again, regions of inflamed and dysplastic mucosa within tissue sections we chose for analysis were annotated by a pathologist (Figure S2A; Table S2). To assess cellular and inflammatory changes in the tissue samples, we applied the multiplex imaging pipeline “MATISSE,”17,18 specifically designed for multiplex single-cell protein expression analysis of epithelial, immune, and stromal cell subsets in tissue biopsies (Figure 2A). This method combines fluorescent microscopy (DAPI), with a 33-plex panel for IMC. The antibody panel (Table S3) contains custom-conjugated antibodies, which are all validated with IMC (Figures S2B and S2C).
Figure 2.

The epithelial compartment displays lower inflammatory signaling and HLA-DR-DP-DQ expression in dysplasia
(A) Overview of the experimental design including sample selection, tissue staining, and downstream computational protein expression and spatial analysis. 48 tissue biopsies were included from 7 patients with IBD, of >3 separately timed biopsies. Tissue sections were labeled, and regions of interest (ROIs) were imaged and segmented using the MATISSE pipeline.
(B) Hierarchical clustering of median expression data of all cells in the epithelial compartment per biopsy. Disease states and individual patients are color coded. Relative expression level is indicated by color intensity in the heatmap.
(C) Boxplot showing median expression of KI67, cleaved caspase-3, P-STAT3 in tissues annotated as different disease states (uninflamed, inflamed, and dysplastic). Black dots indicate the median expression per biopsy. Colored points represent the mean expression of all biopsies included for each individual patient. Kruskal-Wallis statistical test was performed, with Dunn’s post-test and Bonferroni multiple testing correction. p values are indicated in the graphs.
(D) Boxplot showing median of the nuclear/cellular ratio of NF-κB. ANOVA statistical test was performed, with Tukey HSD post-test. p values are indicated in the graphs.
(E) Boxplot showing median expression of HLA-DR-DP-DQ. Kruskal-Wallis statistical test was performed, with Dunn’s post-test and Bonferroni multiple testing correction. p values are indicated in the graphs.
(F) Representative image of HLA-DR-DP-DQ staining; the epithelial compartment border is indicated with a purple outline. Scale bars represent 50 μm. See also Tables S2 and S3 and Figure S2.
First, we evaluated the epithelial cell compartment and assessed proliferation (KI67), and active signal transduction of multiple pathways, including JAK-STAT (phosphorylated STAT3, P-STAT3), apoptosis (cleaved caspase-3), NF-κB (nuclear NF-κB-P65 localization), PI3K (P-AKT, P-S6), and Ras-MAPK signaling (P-ERK). Hierarchical clustering of median expression levels per image revealed a cluster containing most dysplastic samples, and a cluster containing most of the inflamed samples, except for 2 samples that co-clustered with uninflamed mucosa samples and 1 sample of dysplastic mucosa that appears not to be correlated with other samples (Figures 2B and S2E). Apoptosis (cleaved caspase-3) was found to be significantly decreased in dysplastic mucosa compared to uninflamed mucosa, while proliferation (KI67) did not vary in the different states (Figures 2C and S2D). Inflamed and uninflamed mucosa do not differ with respect to apoptosis and proliferation. P-STAT3 levels were higher in inflamed epithelium as compared to uninflamed and dysplastic epithelium, but this was not significant (p = 0.05 compared to uninflamed, and p = 0.07 compared to dysplastic) (Figure 2C). NF-κB activation, defined by nuclear localization ratio, decreased significantly in both inflamed epithelium and in dysplasia, suggesting an overall reduction of inflammatory signaling in dysplastic cells (Figures 2D and S2F). Other included signaling markers were not significantly different between uninflamed, inflamed, and dysplastic tissues (Figure S2G).
The major histocompatibility complex-II (MHC-II) expressed on murine intestinal epithelium has been demonstrated to present antigens to CD4+ T helper cells.19 We showed that HLA-DR-DP-DQ, which is part of the MHC-II complex, is expressed at high levels in inflamed epithelium, while dysplastic epithelium shows lower HLA-DR-DP-DQ expression, suggesting reduced antigen-presenting capacity (Figures 2E and 2F). Together, dysplastic epithelium displays reduced apoptosis compared to inflamed and uninflamed mucosa. While inflamed epithelium shows high P-STAT3 and MHC-II levels, NF-κB activation is reduced in both inflamed and dysplastic epithelium. In dysplastic mucosa, we found reduced STAT3 activation and MHC-II expression. These observations suggest a diminished inflammatory crosstalk between dysplastic epithelium and immune cells.
Lamina propria immune cells display active inflammatory responses in dysplasia
Next, we set out to identify changes in the immune cell compartment in the inflamed and dysplastic mucosa. First, we assessed global changes in expression of cytokines, checkpoint molecules, and active signaling in the lamina propria cells, where most immune cells reside. Hierarchical clustering of median cellular expression values per image revealed high levels of the inflammatory mediators IL-10, IL-17A, and granzyme B and low levels of IFNγ in inflamed and dysplastic regions (Figure 3A). In addition, the checkpoint molecules ICOS, CTLA-4, and PD1 are high in these settings. Moreover, inflamed and dysplastic tissues display high phosphorylation of STAT3, ERK, AKT, and S6, demonstrating active receptor and inflammatory signaling and a mild increase in proliferation (KI67). Other markers, such as nuclear NF-κB, cleaved caspase-3, and HLA-DR-DP-DQ did not differ between the dysplastic and uninflamed mucosa (Figures 3A and S3A). Next, we assessed expression levels of these proteins per patient and per disease stage. Expression of checkpoint molecules ICOS, CTLA-4, and PD1 was significantly higher in inflamed as compared to uninflamed conditions (Figures 3B and S3B). While ICOS shows a significantly higher expression in dysplasia, the expression of PD1 and CTLA-4 was more variable and showed no significant differences. HLA-DR-DP-DQ expression was similar in uninflamed and inflamed mucosal lamina propria cells, but its expression dropped in the dysplastic microenvironment (Figures 3C and S3C). Analysis of active signal transduction revealed significantly elevated levels of phosphorylated S6 in inflammation but not in dysplasia (p = 0.08) (Figure 3D). P-STAT3 was high in both inflammation and dysplasia, suggesting increased immune cell activation and inflammatory JAK-STAT signaling, while P-AKT and P-ERK levels showed no clear changes (Figures 3D and S3D). Furthermore, apoptosis (cleaved caspase-3) was high in the inflamed mucosa, but proliferation (KI67) did not change (Figure S3E). NF-κB signaling did not differ between uninflamed, inflamed, and dysplastic mucosa in lamina propria cells (Figure 3E). Taken together, our results highlight the differences between uninflamed, inflamed, and dysplastic mucosa in lamina propria cells, with both inflamed and dysplastic mucosa exhibiting clear signs of inflammatory response. However, we did not detect clear correlations between different states and key checkpoint molecules, cytokines, and active signaling.
Figure 3.

Lamina propria cells display elevated expression of immune checkpoints, cytokines and active signaling in inflammation, and early dysplasia
(A) Hierarchical clustering of median expression data of all cells in the lamina propria compartment per biopsy. Disease states and individual patients are color coded. Expression level is indicated by color intensity in the heatmap.
(B–D) Boxplots showing median expression of proteins on lamina propria cells in tissues annotated as different disease states (uninflamed, inflamed, and dysplastic). Median expression is displayed for (B) checkpoint molecules CTLA-4, ICOS, PD-1, (C) HLA-DR-DP-DQ, (D) phosphorylated S6 (P-S6), and STAT3 (P-STAT3). Kruskal-Wallis statistical test was performed, with Dunn’s post-test and Bonferroni multiple testing correction. p values are indicated in the graphs. Black dots indicate the median expression per biopsy. Colored points represent the mean expression of all biopsies included for each individual patient.
(E) Boxplot of the median nuclear/cellular localization ratio of NF-κB in lamina propria cells. ANOVA statistical test was performed, with Tukey HSD post-test. p values are indicated in the graphs.
(F) Median expression of granzyme B, Interferon gamma (IFNγ), interleukin-10 (IL-10), and IL-17A. Kruskal-Wallis statistical test was performed, with Dunn’s post-test and Bonferroni multiple testing correction. p values are indicated in the graphs. Black dots indicate the median expression per biopsy. Colored points represent the mean expression of all biopsies included for each individual patient.
(G) Representative IMC images are shown of granzyme B, and IL-17A in uninflamed, inflamed, and dysplastic tissue. Scale bar represents 50 μm.
(H) Percentage of cells in lamina propria with more than 5 IL-17A mRNA counts per cell. Healthy colon value was obtained from a tissue microarray (TMA) of healthy colon tissue. two-way ANOVA with main effects only was performed, with Tukey HSD post-test. Colored points represent the percentage of cells per tissue biopsy.
(I) Representative images of IL-17A mRNA expression in uninflamed, inflamed, and dysplastic tissue are shown. Scale bars represent 200 μm. See also Figure S3.
IL-17A is upregulated in inflammatory conditions
As described earlier, we observed that the overall production of secreted factors by lamina propria cells, such as granzyme B, IL-10, and IL-17A, increased significantly in inflamed tissue, but not consistently in dysplastic mucosa, and that IFNγ, the driver of type 1 inflammation, did not differ between disease states. Both type 1 and type 3 inflammation have been linked to IBD and targeting these pathways has been shown to be effective in its treatment.20,21 In murine models, type 3 inflammation promotes sporadic colorectal cancer (CRC).4 In agreement with this, we observed higher IL-17A levels in the inflamed pre-dysplastic tissue microenvironment (Figures 3F and 3G). Since DSP is limited to bulk expression data and does not detect significant differences in mRNA expression in total CD45+ immune cells, we extended our analysis using high-resolution single-cell RNAscope to quantify RNA expression in each lamina propria cell. IL-17A mRNA is indeed significantly upregulated in inflamed conditions (p = 0.02), with a similar trend in dysplasia (p = 0.08), characterized by a higher percentage of cells with more than five mRNA counts per cell compared to uninflamed conditions (Figures 3H, 3I, and S3F). In epithelium, we observe a high expression of IL-17A mRNA in all disease states although this is absent on a protein level (Figures 3G and S3G). Collectively, our data suggest that the main source of IL-17A is immune cells and that type 3 inflammation is the main form of inflammation in these patients.
The mucosal cellular composition changes during dysplasia development
To understand the contributions of specific cellular subsets to the observed differences in the inflammatory landscape, we performed unsupervised clustering on all segmented cells using Rphenograph,22 which resulted in 18 distinct clusters (Figure 4A). Clusters were annotated based on the expression of protein markers (Figure S4A), and proportional frequencies of cells are displayed per overall disease stage (Figure 4B) and per patient (Figure 4C). Overall, dysplastic tissue showed higher proportions of epithelial cells, although this difference was not significant. Of the myeloid immune cell-containing clusters, the “T cell macrophage & CD16+” cluster was found to be reduced in the dysplastic mucosa compared to inflamed mucosa. Clusters “IL-17” and “IL-17 epithelial” displayed significantly increased frequencies in inflamed mucosa compared to uninflamed mucosa. All other clusters did not show significant changes between disease stages (Figure 4C). We also mapped different cell clusters onto the tissue, to visualize the spatial distribution (Figure 4D). Together, we show that the (epithelial) IL-17 frequencies are increased in inflamed tissue, while the frequency of the “T cell macrophage & CD16+” cluster is reduced in the setting of dysplasia. We observed no significant changes in frequencies of T cell clusters. We also note that hierarchical clustering reveals the coinciding presence of cell phenoclusters, but cannot fully distinguish between disease stages, and shows interpatient variability (Figure S4B).
Figure 4.

Changed cellular composition of the tissue during dysplasia development
(A) UMAP displaying all identified cells clustered into cell subsets using Rphenograph.
(B) Bar graph showing cellular proportions of each cell type cluster.
(C) Boxplots showing proportions of cell types per tissue biopsy analyzed in the annotated disease states. Each dot represents the result of individual biopsies. ANOVA statistical test was performed, with Tukey HSD post-test. p values were adjusted for multiple comparisons with Bonferroni and indicated in the graphs.
(D) Representative images displaying localization of cells, colored by cluster. See also Figure S4.
T cells display inflammatory markers, elevated immune checkpoints, and reduced epithelial infiltration
Next, we evaluated changes in the inflammatory signatures of T cell subsets, such as cytokine production and expression of immune checkpoints. First, we identified CD4+ T helper cells, CD4+CD8+ double-positive cells (DP T cells), and CD8+ cytotoxic T cells (Figure S5A); of note, the proportion of DP T cells may have been relatively overestimated due to spatial signal overlap of membranous CD4 and CD8a in some cases. Proportional analysis revealed that overall frequencies of T cell subsets do not differ significantly between uninflamed, inflamed, and dysplastic tissues (Figures 5A and S5F). Expression of FOXP3, the memory T cell marker CD45RO, and the proliferation marker KI67 did not differ in the three groups (Figures S5B and S5C). Expression of checkpoint CTLA-4 was significantly higher in the inflamed and dysplastic lamina propria, while ICOS was only increased in inflamed tissue (Figures 5B and S5E). No significant differences were observed in overall PD1 expression (Figure 5B), which was similar in different T cell subsets (Figure S5C). Signal transduction (P-S6) in T helper cells increased in inflamed and dysplastic tissue, while P-ERK remained unchanged (Figure S5D).
Figure 5.

Inflamed mucosa shows elevated Th17 and cytotoxic T cells, which avoid epithelium in dysplasia
(A) Proportions of CD4+ T helper cells, CD4+CD8+ (DP) T cells, and CD8+ cytotoxic T cells per patient. ANOVA statistical test was performed, with Tukey HSD post-test. p values are indicated in the graphs.
(B) Expression of checkpoint molecules in T helper cells (CD4+ T cells). Individual datapoints represent median expression of all cells, and colored dots represent mean of all biopsies included per patient. Kruskal-Wallis statistical test was performed, with Dunn’s post-test and Bonferroni multiple testing correction. p values are indicated in the graphs.
(C) Production of cytokines in T helper cells. Kruskal-Wallis statistical test was performed, with Dunn’s post-test and Bonferroni multiple testing correction. p values are indicated in the graphs.
(D) Granzyme B expression in DP T cells and cytotoxic T cells. Kruskal-Wallis statistical test was performed, with Dunn’s post-test and Bonferroni multiple testing correction. p values are indicated in the graphs.
(E) Median epithelial distance (μm) of CD8+ cytotoxic T cells per disease state. Distance was clipped to 75 μm. Kruskal-Wallis statistical test was performed, with Dunn’s post-test and Bonferroni multiple testing correction. p values are indicated in the graphs.
(F) Analysis of localization of CD8+ cytotoxic T cells in the tissue, quantified as distance from the epithelium. Bins indicate “intra-epithelial” (≤0 μm), “proximal” (1–25 μm), and “distal” (>25 μm). Distance was clipped to 75 μm. ANOVA statistical test was performed, with Tukey HSD post-test. p values are indicated in the graphs. See also Figure S5.
Next, we assessed cytokine production, and showed that while IFNγ and IL-10 expression in T helper cells shows no significant differences, IL-17A expression in T helper cells is significantly higher in inflamed but not in dysplastic tissue (Figures 5C and S5F). While granzyme B expression was higher in the inflamed and dysplastic lamina propria (Figure 3F), granzyme B expression in DP T cells and cytotoxic CD8+ T cells was similar between conditions (Figure 5D).
To assess potential interactions between T cells and the epithelium, we calculated the median distance between T cell subsets and the epithelium. There were no significant differences in median epithelial distance between disease states of CD4+ and CD4+CD8+ T cells (Figures S5G and S5H). Cytotoxic T cells displayed a divergent localization pattern: the median distance to the epithelium was relatively low in uninflamed and dysplastic tissue, compared to inflamed tissue (Figure 5E). Infiltration of cytotoxic T cells has been associated with reduced progression of sporadic CRC.23 Moreover, while stromal localization of CD8+ T cells is increased in microsatellite instability (MSI) metastatic CRC, CD8+ T cells localize within the epithelium in MSI non-metastatic tumors.24 To explore the infiltration of cytotoxic T cells in the epithelium, we separated the cells in three distance bins24: “intra-epithelial” (≤0 μm), “proximal” (1–25 μm), and “distal” (>25 μm). Intra-epithelial CD8+ T cell frequency was significantly reduced in the inflamed mucosa, accompanied by a significant increase in distal CD8+ T cell frequency, while in dysplastic epithelium intra-epithelial CD8+ T cell infiltration showed a near-significant reduction (p = 0.0578), and proximal CD8+ T cell frequency a near-significant increase (p = 0.0563). These results indicate reduced infiltration of dysplastic epithelium by cytotoxic CD8+ T cells in inflammation and dysplasia, but less defined as has been described in fully developed (metastatic) tumors in CRC, which contain large areas of stroma (Figure 5F). Taken together, our analyses performed at the level of T cells subsets show that T cells expressed higher levels of inflammatory markers and checkpoint molecules in inflamed and dysplastic mucosa and exhibit distinct localization patterns, especially with respect to reduced epithelial infiltration during inflammation.
Elevated P-STAT3 in inflamed mucosa associates with dysplasia development
To confirm the altered expression of P-STAT3, HLA-DR-DP-DQ, CD8a, and IL-17A in inflamed mucosa of patients who eventually developed dysplasia, we selected 14 new patients from the Utrecht Surveillance Study of whom 7 developed dysplasia (cases). The other 7 matched patients did not develop dysplastic lesions and served as controls (Table S4). One dysplasia case was excluded because the biopsies did not meet quality requirements. Using immunohistochemistry (IHC), the expression of proteins was evaluated in paired tissue sections of uninflamed and inflamed mucosa at baseline for each patient. In controls, the number of HLA-DR-DP-DQ+ cells was higher in inflamed compared to uninflamed epithelium but this was not significant in uninflamed versus inflamed tissue of dysplasia cases (Figures S6A and S6B). However, a significant decrease in HLA-DR-DP-DQ+ cells was observed in the dysplastic epithelium of dysplasia cases, confirming IMC results (Figure S6C).
Next, we investigated STAT3 activation by IHC and confirmed that P-STAT3 expression, here quantified as proportion of P-STAT3+ cells, was significantly higher in inflamed compared to paired uninflamed epithelium of dysplasia cases (Figures 6A and 6B), with a similar but not significant trend in the lamina propria (p = 0.07) (Figure 6C). Of note, proportions of P-STAT3+ cells did not change in inflamed versus paired uninflamed epithelium of controls. We did not observe significant changes of P-STAT3+ cells in dysplastic tissue, only in (pre-dysplastic) inflamed conditions (Figure S6D). These findings imply that increased P-STAT3 expression in the inflamed epithelium could be relevant for dysplasia development.
Figure 6.

Elevated IL-17A and P-STAT3 in inflamed mucosa associates with dysplasia development
(A) Representative images of P-STAT3 expression in uninflamed and inflamed tissues of controls (no dysplasia), and dysplasia cases (dysplasia). Scale bars represent 50 μm.
(B) Single-cell analysis of P-STAT3 expression, displayed as the proportion of P-STAT3+ cells in the epithelium in uninflamed and inflamed tissues of dysplasia cases and controls. Paired t test was performed. p values are indicated in the graphs.
(C) Single-cell analysis of P-STAT3 expression, displayed as the proportion of P-STAT3+ cells in the lamina propria in uninflamed and inflamed tissues of dysplasia cases and controls. Paired t test was performed. p values are indicated in the graphs.
(D) Representative images of CD8a expression in uninflamed and inflamed tissues of dysplasia cases and controls. Scale bars represent 50 μm.
(E) Single-cell analysis of CD8a expression, depicted as proportion of CD8a+ cells in the epithelium in uninflamed and inflamed tissues of dysplasia cases and controls. Paired t test was performed. p values are indicated in the graphs.
(F) Single-cell analysis of CD8a expression, depicted as proportion of CD8a+ cells in the lamina propria in uninflamed and inflamed tissues of dysplasia cases and controls. Paired t test was performed. p values are indicated in the graphs.
(G) Single-cell analysis of CD8a expression, depicted as proportion of CD8a+ cells in uninflamed, inflamed, and dysplastic epithelium of dysplasia cases. Kruskal-Wallis statistical test was performed, with Dunn’s post-test. p values are indicated in the graphs. A representative image of CD8a expression in dysplastic tissue is shown. Scale bar represents 50 μm.
(H) Single-cell analysis of IL-17A expression, depicted as proportion of IL-17A+ cells in uninflamed and inflamed lamina propria of dysplasia cases and controls. Paired t test was performed. p values are indicated in the graphs. See also Tables S4 and S5 and Figure S6.
Next, we assessed whether the presence of cytotoxic T cells was relevant for the development of dysplasia and showed that the proportion of CD8a+ cells is significantly higher in inflamed lamina propria of controls but is similar in uninflamed and inflamed mucosa of dysplasia cases (Figures 6D–6F), confirming IMC results. Moreover, there was a significant reduction of CD8a+ cells in the dysplastic epithelium of dysplasia cases, which was not observed in the dysplastic lamina propria (Figures 6G and S6E), confirming reduced cytotoxic T cell infiltration of dysplastic epithelium. As we demonstrated that IL-17A mRNA and protein levels are high in the inflamed lamina propria of patients with dysplasia (Figures 3F–3I), we examined their status in this new patient cohort. We showed that IL-17A+ cells tended to be elevated in the inflamed lamina propria of dysplasia cases (p = 0.07), but not in controls (Figures 6H and S6H). Since IL-17A can also be secreted, we next analyzed the annotated region as an area. In the inflamed area, IL-17A was significantly increased in the lamina propria of dysplasia cases but not in controls (Figure S6F). There was no significant difference in the dysplastic lamina propria of dysplasia cases, confirming upregulated IL-17A protein specifically in inflammation as observed in our IMC analyses (Figure S6G). Together, we demonstrate increased proportions of P-STAT3+ cells in epithelium and higher total levels of IL-17A in inflamed lamina propria of dysplasia cases, while only controls showed increased CD8a+ cells.
Discussion
In this study, we explored the immunological landscape in the uninflamed, inflamed, and dysplastic epithelium and lamina propria of patients with IBD. Our comprehensive approach, which combined bulk and single-cell data at the mRNA and protein levels, revealed distinct inflammatory gene expression patterns, cell compositions, and inflammatory profiles under different conditions we studied. We observed that type 3 (IL-17) inflammatory responses increase during inflammation and persist in dysplasia, as well as P-STAT3 levels. Furthermore, dysplasia was associated with decrease in macrophages and elevated checkpoint molecule expression on T helper cells. Spatial analysis indicated that cytotoxic CD8a+ T cells infiltrate the epithelium in uninflamed tissue, but not in inflammation and dysplasia. Subsequently, we studied pre-dysplastic changes in a second surveillance patient cohort and found increased P-STAT3+ epithelial cells but no changes in lamina propria CD8a+ cells in inflamed as compared to uninflamed tissue. Control patients who did not develop dysplasia had higher proportions of CD8a+ lamina propria cells but no changes in P-STAT3+ cells in inflamed tissue compared to paired uninflamed tissue.
In our high-resolution multiplex IMC analyses, we observed reduced immune-epithelial interactions in dysplasia, characterized by reduced epithelial CD8+ cytotoxic T cell infiltration and downregulation of epithelial HLA-DR-DP-DQ expression, an essential component of MHC-II, leading to reduced antigen presentation of epithelial cells to CD4+ T helper cells. Increased expression of MHC-II components has been consistently observed in intestinal tissues of patients with active IBD,25,26,27 similar to our results, both in inflamed tissue from dysplasia cases and controls. Expression of MHC-II by intestinal stem cells is essential for inflammatory T cell responses and epithelial differentiation in experimental infection models,19 and reduced MHC-II expression is related to dietary and microbiome changes.26 Additionally, in a mouse model of sporadic CRC, MHC-II deletion in intestinal stem cells promotes intestinal tumor growth,26 showing that the regulation of T helper activation via epithelial MHC-II plays a role in inflammation and tumor development. This suggests that MHC-II downregulation occurs as an anti-inflammatory mechanism in dysplastic epithelium.
Although CD8+ T cell frequencies did not change, our spatial analysis showed lower epithelial CD8+ cytotoxic T cell infiltration in inflamed and dysplastic epithelium, which underscores the reduced immune-epithelial interaction in patients with dysplasia. Consistently, the CD8a IHC signal did not change in inflamed mucosa from dysplasia cases but, in fact, was higher in inflamed mucosa from controls. Together, this suggests a role for CD8+ T cell presence and epithelial infiltration in dysplasia development. This is in line with observations in sporadic CRC tissue, where reduced infiltration of cytotoxic T cells is associated with tumor progression.23,24
Different proinflammatory pathways, such as NF-κB, IL-6/STAT3, and IL-23/IL-17, may contribute to a tumor-supporting microenvironment resulting in CAC.28 Our studies demonstrate an increased type 3 (IL-17) response in inflamed and dysplastic conditions as we observed elevated levels of IL-17A, IL-17F, IL-22 mRNA, and IL-17A mRNA in lamina propria. Type 3 responses have previously been associated with IBD inflammation and cancer development. Additionally, in mouse models for sporadic CRC, IL-23 produced by myeloid cells enhanced tumor growth and progression,4 and IL-17RA signaling reduced the production of the T cell-recruiting chemokines CXCL-9 and -10 by the tumor.29 Of note, the accumulation of mutations in IL-17 signaling-related genes was observed in the epithelium of patients with ulcerative colitis (UC) and concomitant CAC, genes that are infrequently affected in sporadic CRC.9 Together, type 3 inflammatory responses may promote tumor development in both a paracrine fashion within the epithelium and via modulation of epithelium-immune inflammatory crosstalk. Remarkably, IL-17 blockade therapy has shown promise in dermatology and rheumatology, but exacerbated disease in IBD,30 suggesting that IL-17 signaling has a role in the maintenance of the gut barrier. Additionally, murine models showed that signaling via IL-17A and the IL-17 receptor A (IL-17RA) promotes secretory cell differentiation and regeneration, of which the latter may be involved in dysplasia development.31 Although the exact role of IL-17 signaling in IBD and CAC development may depend on a specific IL-17 family member and receptors expressed,32 blocking mediators of the IL-17 pathway, such as IL-23, could be beneficial for patients with long-standing colitis and may prevent dysplasia.
Another important observation that we made is that inflamed tissue of patients that developed dysplasia has higher P-STAT3+ cell proportions and total expression levels, which is in contrast to patients who did not develop dysplasia. This suggests that JAK-STAT3 upregulation indicates a pre-dysplastic inflammatory condition. In line with this, colonic crypts of Stat3ΔIEC mice show decreased proliferation and increased apoptosis after DSS exposure and develop less and smaller adenomas compared to wild-type mice.3 Moreover, the inhibition of JAK-STAT3 signaling in CRC cells induced apoptosis and cell-cycle arrest,33 suggesting that JAK-STAT3 signaling is essential for epithelial cell survival and proliferation, thereby driving dysplasia. A previous study showed increased numbers of epithelial P-STAT3-positive cells during CAC progression; P-STAT3 was increased in active UC, low-grade dysplasia, high-grade dysplasia, and CAC compared to inactive UC.34 However, we show that epithelial P-STAT3 in established dysplasia is lower compared to inflamed tissue, implying that the early dysplastic epithelium becomes less susceptible to specific inflammatory signals that promote JAK-STAT3 signaling (e.g., IL-6).35 Targeting the JAK-STAT3 pathway with JAK inhibitors may therefore be both beneficial for the treatment of IBD36 and for preventing dysplasia development.
Since JAK-STAT3 signaling is mainly driven by cytokines and cytokine receptors, this suggests that specific inflammatory profiles in IBD can promote risk of dysplasia development. How and whether elevated type 3 inflammatory responses are related to increased P-STAT3 levels in patients at risk is presently unclear. Our data suggest that drugs targeting specific inflammatory pathways, such as IL-23 receptor blockade or JAK inhibitors, may be beneficial for the prevention of CAC. A better understanding of the functional implications of our findings may provide guiding for a safe and effective treatment of IBD while preventing dysplasia development.
Together, our analyses demonstrate changes in immune-epithelial crosstalk during chronic inflammation and dysplasia development in the colonic mucosa of patients with long-standing IBD. Moreover, we identified specific inflammatory signatures in the inflamed mucosa of patients who developed dysplastic lesions over time, most notably elevated P-STAT3, accompanied by elevated IL17A levels, and unchanged CD8a expression. Future studies should investigate the potential predictive value of this signature for dysplasia development in a large cohort. Moreover, future research focused on the role of these inflammatory pathways in colitis-associated tumor development will provide insights into its pathophysiology and might open new avenues for prevention and treatment of this dreaded complication of IBD.
Limitations of the study
One limitation of this study is the small sample size used. This is a unique longitudinal cohort with paired samples, and we only included patients that were not treated with biologicals before, to limit variation. Hence, we were limited to this sample size. Still, some variation between patients occurs due to different treatments (AZA, 6MP, and 5-ASA) and variable disease duration, and 1 patient with primary sclerosing cholangitis colitis was included. Moreover, while this study focused on cellular composition and inflammatory pathways, other factors such as bacterial infection and oxidative stress could play a role in inflammation-induced DNA damage and cancer development, as has been demonstrated in murine models.37
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CTLA4 (clone CAL49) | Abcam | Cat# ab237712; RRID: AB_2905652 |
| Goat anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody | Invitrogen | Cat# A16112; RRID: AB_2534785 |
| E-Cadherin (clone 24E10) | Cell Signaling Technology | Cat# CST3195BF; RRID: AB_2728822 |
| CD20 (clone H1) | BD Biosciences | Cat# 555677; RRID: AB_396030 |
| Non-phospho (active) beta catenin (clone D13A1) | Cell Signaling Technology | Cat# CST8814BF; RRID: AB_11127203 |
| PanKeratin (clone C11) | Cell Signaling Technology | Cat# CST4545BF; RRID: AB_490860 |
| IFN-γ (clone D3H2) | Cell Signaling Technology | Cat# CST8455BF; RRID: AB_2797644 |
| CD45RO (clone UCHL1) | Cell Signaling Technology | Cat# CST55618BF; RRID: AB_2799491 |
| Akt (pan) (clone 40D4) | Cell Signaling Technology | Cat# CST2920BF; RRID: AB_1147620 |
| HLA-DR+DP + DQ (clone CR3/43) | Abcam | Cat# ab7856; RRID: AB_306142 |
| FOXP3 (clone 236A/E7) | Abcam | Cat# ab96048; RRID: AB_10861686 |
| CD4 (clone EPR6855) | Abcam | Cat# ab181724; RRID: AB_2864377 |
| pSTAT3 (Tyr705) (clone D3A7) | Cell Signaling Technology | Cat# CST9145BF; RRID: AB_2491009 |
| IL-10 (polyclonal) | R&D Systems | Cat# AF-217-NA; RRID: AB_354401 |
| CD45 (clone D9M8I) | Cell Signaling Technology | Cat# CST13917; RRID: AB_2750898 |
| CD8a (clone C8/144B) | Thermo Fisher Scientific | Cat# 14-0085-82; RRID: AB_11150240 |
| ICOS (clone D1K2T) | Cell Signaling Technology | Cat# CST89601BF; RRID: AB_2800142 |
| pS6 (Ser235/236) (clone D57.2.2E) | Cell Signaling Technology | Cat# CST4858BF; RRID: AB_916156 |
| PD1 (clone EPR4877(2)) | Abcam | Cat# ab186928; RRID: AB_2894896 |
| NFkB (clone L8F6) | Cell Signaling Technology | Cat# CST6956BF; RRID: AB_2797704 |
| IL-17a (polyclonal) | R&D Systems | Cat# AF-317-NA; RRID: AB_354463 |
| Ki-67 (clone B56) | BD Biosciences | Cat# 550609; RRID: AB_393778 |
| Granzyme B (clone D6E9W) | Cell Signaling Technology | Cat# CST46890BF; RRID: AB_2799313 |
| CD3 (polyclonal) | Dako | Cat# A045229-2; RRID: AB_2335677 |
| pERK (Thr202/Tyr204) (clone D13.14.4E) | Cell Signaling Technology | Cat# CST4370BF; RRID: AB_2315112 |
| Cleaved Caspase-3 (clone 5A1E) | Cell Signaling Technology | Cat# CST9664BF; RRID: AB_2070042 |
| ERK (Pan) (clone 137F5) | Cell Signaling Technology | Cat# CST4695BF; RRID: AB_390779 |
| pAKT (Ser473) (clone D9E) | Cell Signaling Technology | Cat# CST4060BF; RRID: AB_2315049 |
| Histone H3 (clone D1H2) | Cell Signaling Technology | Cat# CST4499BF; RRID: AB_10544537 |
| aSMA (clone 1A4) | Thermo Fisher Scientific | Cat# 14-9760-82; RRID: AB_2572996 |
| CD16 (clone EPR16784) | Abcam | Cat# ab215977; RRID: AB_2877105 |
| CD68 (clone KP1) | Thermo Fisher Scientific | Cat# 14-0688-82; RRID: AB_11151139 |
| CD8a (clone C8/144B) | Cell Signaling Technology | Cat# 70306; RRID: AB_2799781 |
| BrightVision poly HRP-Anti-Rabbit IgG | ImmunoLogic | Cat#: DPVR55HRP; RRID: AB_2915958 |
| Human TruStain FcX (Fc receptor blocking solution) | BioLegend | Cat# 422302; RRID: AB_2818986 |
| Biological samples | ||
| Formalin-fixed colon biopsies from patients with ulcerative colitis | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Cell-ID Intercalator-Ir | Fluidigm | Cat# 201192B; RRID: N/A |
| DAPI (40,6-Diamindino-20-Phenyindole, Dihydrochloride) | Sigma-Aldrich | Cat# D9542; CAS: 28718-90-3 |
| ProLong™ Gold Antifade Mountant | Thermo Fisher Scientific | Cat# P36930; RRID: N/A |
| PAP pen for immunostaining (5 mm tip) | Sigma-Aldrich | Cat# Z377821 |
| Xylene | Klinipath | Cat# 4055–9005; Cas 1330-20-7 |
| Ethanol absolute ≥99% | Klinipath | Cat# 4099–9005; Cas 64-17-5 |
| Hydrogen peroxide solution | Merck Life science | Cat# 216763; Cas 7722-84-1 |
| Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA) | Sigma-Aldrich | Cat# 27285; Cas 6381-92-6 |
| 2-Amino-2-methyl-1,3-propanediol (Tris base) | Sigma-Aldrich | Cat# 10708976001; Cas 77-86-1 |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | Cat# A9647; Cas 9048-46-8 |
| Sodium Chloride (NaCl) | Sigma-Aldrich | Cat# 31434; Cas 7647-14-5 |
| Tween 20 | Sigma-Aldrich | Cat# P9416; Cas 9005-64-5 |
| Hydrochloric acid fuming 37% | Sigma-Aldrich | Cat# 100317; Cas 7647-01-0 |
| Sodium hydroxide | Sigma-Aldrich | Cat# 106498; Cas 1310-73-2 |
| 3,3-Diaminobenzidine tetrahydrochloride hydrate | Sigma-Aldrich | Cat# D5637; Cas 868272-85-9 |
| Citric acid monohydrate | Sigma-Aldrich | Cat# 33114; Cas 5949-29-1 |
| Sodium phosphate dibasic dihydrate | Honeywell | Cat# 30435; Cas 10028-24-7 |
| Hematoxylin Solution, Gill No. 3 | Sigma-Aldrich | Cat# GHS316 |
| ClearVue Mountant XYL | Thermo Fisher Scientific | Cat# 4212; Cas 1330-20-7 |
| Critical commercial assays | ||
| RNAscope™ Multiplex Fluorescent Reagent Kit v2 | ACD Bio Techne | Cat# 323100; RRID: N/A |
| RNAscope™ Probe- Hs-IL17A | ACD Bio Techne | Cat# 310931; RRID: N/A |
| Opal 650 Reagent Pack | Akoya Biosciences | Cat# FP1496001KT; RRID: N/A |
| RNAscope™ Multiplex Fluorescent Detection Kit v2 | ACD Bio Techne | Cat# 323110; RRID: N/A |
| Deposited data | ||
| Cell-segmented raw imaging mass cytometry data | This study | Dataverse: https://doi.org/10.34894/LXHZQF |
| Software and algorithms | ||
| MATISSE pipeline | Baars et al.17; Krijgsman et al.18 | https://github.com/VercoulenLab/MATISSE-Pipeline; RRID: N/A |
| Fiji (ImageJ) (version 2.9.0) | Schindelin et al.38 | https://github.com/fiji/fiji; RRID: SCR_002285 |
| QuPath | Bankhead et al.39 | https://qupath.github.io/; RRID: SCR_018257 |
| HALO™ Multiplex IHC module | Indica Labs | https://www.indicalab.com/halo/; RRID: SCR_018350 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yvonne Vercoulen (Y.Vercoulen@umcutrecht.nl).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
All single-cell segmented raw IMC data derived from participants have been deposited on Dataverse, accessible via https://doi.org/10.34894/LXHZQF
-
•
All code used for DSP and IMC data processing and analysis have been deposited on Dataverse, accessible via https://doi.org/10.34894/LXHZQF
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Study participants
Formalin-fixed paraffin embedded (FFPE) tissue blocks of colonic biopsies were obtained from patients with inflammatory bowel disease (IBD), enrolled in the Utrecht IBD Surveillance Study. Samples were only included if biopsies of the compared disease states were available from the same segment of the colon per individual patient. Biopsies taken during or less than 3 months before stopping systemic or local corticosteroid use were excluded. Montreal classes were retrieved from endoscopy reports or retrospectively assessed from endoscopic images. Detailed patient characteristics are provided in Tables 1 and S4. Median age was 51,5 years (50–70) and 50 years (31–70), with 1/3 (33.3%) and 1/7 (14.3%) females, for DSP and IMC, respectively. Median age was 53.75 years (31,5–69,5) and 60 years (34,5–70), with 1/7 (14.3%) and 4/7 (57.1%) females, for IHC with dysplasia and without dysplasia, respectively. No data on participants’ race, ethnicity or ancestry were collected as part of this study and neither were these data available from routine care. This study was approved by the medical ethical board of the UMC Utrecht (METC protocol #11–050/E, biobank protocols #18–413). Written informed consent was obtained from all patients according to the Declaration of Helsinki.
Method details
GeoMX digital spatial profiling
Paired tissue samples from uninflamed colon and dysplastic colon were included from 3 patients. Nine freshly cut serial tissue sections of 4 μm were incubated with the human Whole Transcriptome Atlas panel and subsequently stained with antibodies for pan-cytokeratin (AE1+AE3, Novus Biologicals), CD45 (2B11+PD7/26, Novus Biologicals) and nuclear SYTO13 (Nanostring, 121303303) according to the manufacturer’s protocol.14 After loading and scanning of the slides on the Geomx instrument, 27 regions of interest were drawn, and segmented into masks for epithelium (pan-cytokeratin, pan-CK), and immune cells (CD45) in the Nanostring Geomx software.
Functional enrichment analysis
To evaluate functional pathways of genes involved in epithelium and CD45+ cells, gene enrichment analysis was performed, using Kyoto Encyclopedia of Genes and Genomes (KEGG) from human pathways. Network representation from the gene enrichment analysis was performed using ShinyGO v0.77.40 Statistical significance was obtained by calculating false discovery rate which was set to 0.05.
Imaging mass cytometry (IMC)
Sample inclusion
Forty-eight tissue biopsies from 7 patients with IBD, taken at 1–3 consecutive colonoscopies and including at least 3 different disease states were selected. Additionally, tissue microarray (TMA) sections of tissue rest-material were included from healthy and diseased control tissues according to the no-objection-agreement, approved by the UMC Utrecht biobank committee (protocol #18–222). Before and during colonoscopy, all included patients underwent similar treatment regimen comprised of 5-aminosalicylates and/or thiopurines. None of the patients had been exposed to biologicals previously.
Region selection and annotations
Regions of interest were selected by a gastrointestinal pathology expert based on serial H&E-stained sections. Selection criteria included tissue quality, longitudinal crypts (at least 4 in field of view (FOV)), and absence of tertiary lymphoid structure (TLS). In addition, the inflammatory and/or dysplastic state of the tissue was determined by expert pathologists (Table S2). The latter was based on pathological and clinical assessment and defined as follows.
-
i)
Uninflamed: no signs of ongoing chronic or active inflammation.
-
ii)
Inflamed: chronic and/or actively inflamed mucosa with basal lymphoplasmacytosis, an increased density of inflammatory cells in the lamina propria and presence of neutrophils in the epithelium (the latter indicating active inflammation).
-
iii)
Dysplastic: pseudostratified epithelium, crowded, elongated and hyperchromatic nuclei with preserved nuclear polarity and lack of surface maturation.
All selected regions had to contain 4 to 10 epithelial crypts and had to span the mucosa from lumen to muscularis mucosae.
Immunofluorescence and IMC
Tissue slides were stained with DAPI as described in the MATISSE protocol.17,18 DAPI images were generated using the Zeiss AxioImager Z1 stand microscope. Objective: 20x dry, NA 0.5 (Zeiss 420350). Software: ZEN Pro (2.6.76).
IMC was performed as previously reported,17,18 with a few deviations.
-
i)
Samples were kept dry after the staining protocol (no mounting).
-
ii)
Antibodies were newly conjugated and titrated (Table S3), and CTLA-4 staining was performed in 2 steps, using a primary and metal-conjugated secondary antibody.
Image processing and single-cell segmentation
Single-cell segmentation was performed as previously described,18 with a few deviations.
-
i)
The proportion of DAPI & IMC images annotated: random selection of 15% of image regions of which a random 10% area of FOV was annotated.
-
ii)
DAPI images were normalized by quantiles preceding pixel classification workflow.
The following IMC channels for pixel classification in Ilastik were employed: DNA-intercalator (Ir193), Histone H3, aSMA (In115), E-Cadherin, Beta Catenin, Pan Keratin, CD45, CD45RO, CD16, HLA DR-DP-DQ, CD68, CD3, CD8a, CD4, CD103, CD20, IL-17A.
For single-cell segmentation the following classes were included in training: membranes immune-cells, membranes epithelium, nuclei immune-cells, nuclei epithelium, and background pixels. Feature selection settings: Gaussian blur (σ 0.3, 0.7, 1.0, 1.6, 3.5), Laplacian of Gaussian (σ 0.3, 0.7, 1.0, 1.6, 3.5), Gaussian Gradient Magnitude (σ 0.3, 0.7, 1.0, 1.6, 3.5), Difference of Gaussians (σ 0.3, 0.7, 1.0, 1.6, 3.5), Structure Tensor Eigenvalues (σ 0.3, 0.7, 1.0, 1.6, 3.5), Hessian of Gaussian Eigenvalues (σ 0.3, 0.7, 1.0, 1.6, 3.5).
For tissue compartment segmentation the following classes were included in training: background, epithelium, stroma, muscularis mucosae. Feature selection settings: Gaussian blur (σ 1.6, 3.5, 5), Hessian of Gaussian Eigenvalues (σ 1.6, 3.5, 5).
High-intensity pixel noise artifacts in at least three sequential channels were identified with a pixel classification workflow in Ilastik.
IMC channels used to perform pixel classification: DNA-intercalator, Histone H3, aSMA, E-Cadherin, Beta Catenin, Pan Keratin, CD45, CD45RO, CD16, HLA DR-DP-DQ, CD68, CD3, CD8a, CD4, CD103, CD20, IL-17A. Classes included in training: noise, signal. Feature selection settings: Gaussian blur (σ 0.3, 1.0).
Single-cell data
Remove noise
Data cleanup was performed by excluding image regions with >10% of cells overlap with noisy regions, based on noise probability map (2 ROIs were excluded) and cells overlapping with noisy regions.
Normalization
Single cell expression values were log1p transformed. Scaled expression value was calculated per channel as median expression/median expression all images.
Cell-type annotation
Intensity rescaling/standardization was performed per channel using scale function = (x-mean(x))/sd(x). Rphenocluster22 was used in R version 4.1.2 with the following parameters: number of nearest neighbors k = 30, channels = aSMA, Pan Keratin, E-Cadherin, CD16, CD20, CD3, CD45, CD68, IL-17a. Clusters were annotated based on marker expression and, when annotated as the same cell type, merged in further analysis. Dimensionality reduction was performed using UMAP (n_neighbors = 15, min-dist = 0.1).
Spatial analysis
The cell-epithelial distance was calculated between cell-centroid and epithelial outline. Pearson correlation analysis was performed using CytoMAP41 in MATLAB version 2020b. Acceptance or avoidance of cells expressing specific markers was shown by Pearson correlation coefficient of cells residing within 10 μm radius (neighbors).
RNA in situ hybridization (ISH)
The RNAscope Multiplex Fluorescent V2 Assay kit (ACD Bio Techne) was used for RNA ISH on 4 μm serial FFPE colonic tissue sections. Following the manufacturers protocol, sections were blocked with H2O2, targets were retrieved in a decloaking chamber for 15 min, and proteases present in the tissue were digested. Colonic tissue sections were hybridized with the RNAscope Probe- Hs-IL-17A (ACD Bio Techne) and Opal 650 (Akoya Biosciences) was assigned to this probe. After hybridization, the slides were counterstained with DAPI and mounted in ProLong Gold Antifade Mountant (Invitrogen).
Z-stacks of regions of interest were obtained using a water immersion 20X objective on a Zeiss Cell Observer microscope. Obtained images were stacked, stitched, and adjusted using Fiji ImageJ software.38 Single cell analysis was performed in QuPath39; a selection of epithelial and lamina propria regions were annotated blindly followed by DAPI single cell detection. Finally, IL-17A mRNA counts per cell were measured.
Immunohistochemistry
FFPE tissue blocks of colonic biopsies were sliced into 4 μm sections. Obtained slides were dewaxed, rehydrated, and endogenous peroxidase activity was blocked with 3,5% (v/v) H2O2 for 15 min. Masked antigens were retrieved by incubation with pre-heated Tris-EDTA (pH9.0) for 30 min. After cooling and washing, tissue sections were further blocked using 3% BSA and Fc blockers (Human TruStain FcX, BioLegend). Finally, slides were incubated overnight at 4°C in a humidified chamber with primary antibodies (Table S5) diluted in 0.5% BSA in TBST. The following day, sections were washed three times and incubated with an HRP-conjugated secondary antibody for 1 h. After washing with TBS, sections were developed with 3,3′-Diaminobenzidine (DAB) chromogen for 10 min at room temperature. Tissue sections were counterstained with hematoxylin before dehydration and mounting.
Images were visualized and captured using the NanoZoomerXR whole slide scanner (Hamamatsu) at 40X magnification, with a resolution of 0.25 μm/pixel. Epithelium, lamina propria, background, and debris were dissected using DenseNet classification in HALO image analysis software (Indica Labs). Subsequently, single cells were detected using the HALO Multiplex IHC module and DAB positive cells were quantified in both epithelium and lamina propria of annotated regions. Finally, the percentages of DAB positive cells were plotted.
Patient and public involvement statement
Patients were not involved in design of this study. We have established routes for dissemination with the Dutch patient society representatives of Crohn & Colitis NL, via newsletters and social media.
Quantification and statistical analysis
IMC
Quantitative data are shown as independent data points with median and Box-Whisker. Analyses of statistical significance for expression values were performed using the Kruskal-Wallis test (non-parametric). Pairwise statistical testing between groups was performed by Dunn’s test, the Bonferroni correction was used for multiple testing correction. Adjusted p-values are shown in graphs.
Analyses of statistical significance for abundance and nuclear translocation were performed by ANOVA (parametric). Pairwise statistical testing was performed by Tukey HSD. Adjusted p-values are shown in graphs. p-values <0.05 were considered statistically significant.
DSP
Raw counts from each gene in each segment were extracted from the NanoString GeoMx NGS processing pipeline. For each region, the expected background was estimated with the mean of the 139 negative control probes. Segment data were normalized with the Q3 normalization method. Two of the 54 segments failed QC and were discarded. For each segment and each gene, differential expression for uninflamed vs. dysplastic was evaluated with a linear mixed model (Tables S1A and S1B).
Acknowledgments
The authors wish to thank the support staff of the pathology tissue facility and Domenico Castigliego for technical support, the Utrecht IBD research group and the Center for Molecular Medicine community for discussions, the CMM ICT team for excellent ICT support, Pascalle Hemelop and Livio Kleij for lab support, and Anouk Wijnands for discussions and advice. We acknowledge the Utrecht Sequencing Facility (USEQ) for providing nanostring Spatial Omics and/or sequencing service and data. USEQ is subsidized by the University Medical Center Utrecht and The Netherlands X-omics Initiative (NWO project 184.034.019).
This work has been supported by a grant from cancergenomicscenter.nl NWO Gravitation 024.001.028, a grant from WKZ research Foundation project TREAT-PID to Y.V., and the Gastrostart award from NVGE to B.O. and Y.V. Funders had no role in study design, data acquisition, analysis, and interpretation. This work is dedicated to the memory of Neeraj Sinha.
Author contributions
M.J.D.B., E.F., and N.S. contributed equally to this paper. Y.V. and B.O. drafted the study concept. M.J.D.B., N.S., J.J.M.t.L., J.R.t.H., M.M.L., G.J.A.O., B.O., and Y.V. contributed to the design of the study. M.J.D.B., E.F., N.S., M.A., and S.v.D. acquired and analyzed data. All authors contributed to data interpretation. M.J.D.B., N.S., E.F., and Y.V. prepared the first draft of the manuscript. Y.V. acquired funding. All authors revised and approved the final version of the manuscript.
Declaration of interests
B.O. received research grants from AbbVie, Celltrion, Ferring, Takeda, Galapagos, and Pfizer and is a member of the advisory boards of Cablon, Pfizer, BMS, Janssen, MSD, Takeda, and Galapagos. Y.V. received a Public Private Partnership grant from Health Holland (#TKI2017), with TigaTx BV and received a research grant from Galapagos.
Published: July 20, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.110550.
Supplemental information
References
- 1.Beaugerie L., Itzkowitz S.H. Cancers Complicating Inflammatory Bowel Disease. N. Engl. J. Med. 2015;372:1441–1452. doi: 10.1056/NEJMra1403718. [DOI] [PubMed] [Google Scholar]
- 2.Lutgens M.W.M.D., van Oijen M.G.H., van der Heijden G.J.M.G., Vleggaar F.P., Siersema P.D., Oldenburg B. Declining Risk of Colorectal Cancer in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2013;19:789–799. doi: 10.1097/MIB.0b013e31828029c0. [DOI] [PubMed] [Google Scholar]
- 3.Grivennikov S., Karin E., Terzic J., Mucida D., Yu G.-Y., Vallabhapurapu S., Scheller J., Rose-John S., Cheroutre H., Eckmann L., Karin M. IL-6 and Stat3 Are Required for Survival of Intestinal Epithelial Cells and Development of Colitis-Associated Cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grivennikov S.I., Wang K., Mucida D., Stewart C.A., Schnabl B., Jauch D., Taniguchi K., Yu G.-Y., Österreicher C.H., Hung K.E., et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491:254–258. doi: 10.1038/nature11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greten F.R., Eckmann L., Greten T.F., Park J.M., Li Z.-W., Egan L.J., Kagnoff M.F., Karin M. IKKβ Links Inflammation and Tumorigenesis in a Mouse Model of Colitis-Associated Cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 6.Burmer G.C., Rabinovitch P.S., Haggitt R.C., Crispin D.A., Brentnall T.A., Kolli V.R., Stevens A.C., Rubin C.E. Neoplastic progression in ulcerative colitis: Histology, DNA content, and loss of a p53 allele. Gastroenterology. 1992;103:1602–1610. doi: 10.1016/0016-5085(92)91184-6. [DOI] [PubMed] [Google Scholar]
- 7.Hussain S.P., Amstad P., Raja K., Ambs S., Nagashima M., Bennett W.P., Shields P.G., Ham A.-J., Swenberg J.A., Marrogi A.J., Harris C.C. Increased p53 Mutation Load in Noncancerous Colon Tissue from Ulcerative Colitis: A Cancer-prone Chronic Inflammatory Disease. Cancer Res. 2000;60:3333–3337. [PubMed] [Google Scholar]
- 8.Baker A.-M., Cross W., Curtius K., Al Bakir I., Choi C.-H.R., Davis H.L., Temko D., Biswas S., Martinez P., Williams M.J., et al. Evolutionary history of human colitis-associated colorectal cancer. Gut. 2019;68:985–995. doi: 10.1136/gutjnl-2018-316191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nanki K., Fujii M., Shimokawa M., Matano M., Nishikori S., Date S., Takano A., Toshimitsu K., Ohta Y., Takahashi S., et al. Somatic inflammatory gene mutations in human ulcerative colitis epithelium. Nature. 2020;577:254–259. doi: 10.1038/s41586-019-1844-5. [DOI] [PubMed] [Google Scholar]
- 10.Duerr R.H., Taylor K.D., Brant S.R., Rioux J.D., Silverberg M.S., Daly M.J., Steinhart A.H., Abraham C., Regueiro M., Griffiths A., et al. A Genome-Wide Association Study Identifies IL23R as an Inflammatory Bowel Disease Gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olafsson S., McIntyre R.E., Coorens T., Butler T., Jung H., Robinson P.S., Lee-Six H., Sanders M.A., Arestang K., Dawson C., et al. Somatic Evolution in Non-neoplastic IBD-Affected Colon. Cell. 2020;182:672–684.e11. doi: 10.1016/j.cell.2020.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baars M.J.D., Douma T., Simeonov D.R., Myers D.R., Kulhanek K., Banerjee S., Zwakenberg S., Baltissen M.P., Amini M., de Roock S., et al. Dysregulated RASGRP1 expression through RUNX1 mediated transcription promotes autoimmunity. Eur. J. Immunol. 2021;51:471–482. doi: 10.1002/eji.201948451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hugot J.-P., Laurent-Puig P., Gower-Rousseau C., Olson J.M., Lee J.C., Beaugerie L., Naom I., Dupas J.-L., Van Gossum A., Groupe d’Etude Thérapeutique des Af Mapping of a susceptibility locus for Crohn’s disease on chromosome 16. Nature. 1996;379:821–823. doi: 10.1038/379821a0. [DOI] [PubMed] [Google Scholar]
- 14.Merritt C.R., Ong G.T., Church S.E., Barker K., Danaher P., Geiss G., Hoang M., Jung J., Liang Y., McKay-Fleisch J., et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 2020;38:586–599. doi: 10.1038/s41587-020-0472-9. [DOI] [PubMed] [Google Scholar]
- 15.Liu C.L., Tangsombatvisit S., Rosenberg J.M., Mandelbaum G., Gillespie E.C., Gozani O.P., Alizadeh A.A., Utz P.J. Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res. Ther. 2012;14:R25. doi: 10.1186/ar3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pieterse E., Hofstra J., Berden J., Herrmann M., Dieker J., van der Vlag J. Acetylated histones contribute to the immunostimulatory potential of neutrophil extracellular traps in systemic lupus erythematosus. Clin. Exp. Immunol. 2015;179:68–74. doi: 10.1111/cei.12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baars M.J.D., Sinha N., Amini M., Pieterman-Bos A., van Dam S., Ganpat M.M.P., Laclé M.M., Oldenburg B., Vercoulen Y. MATISSE: a method for improved single cell segmentation in imaging mass cytometry. BMC Biol. 2021;19:99. doi: 10.1186/s12915-021-01043-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krijgsman D., Sinha N., Baars M.J.D., van Dam S., Amini M., Vercoulen Y. MATISSE: An analysis protocol for combining imaging mass cytometry with fluorescence microscopy to generate single-cell data. STAR Protoc. 2022;3 doi: 10.1016/j.xpro.2021.101034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biton M., Haber A.L., Rogel N., Burgin G., Beyaz S., Schnell A., Ashenberg O., Su C.-W., Smillie C., Shekhar K., et al. T Helper Cell Cytokines Modulate Intestinal Stem Cell Renewal and Differentiation. Cell. 2018;175:1307–1320.e22. doi: 10.1016/j.cell.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobayashi T., Okamoto S., Hisamatsu T., Kamada N., Chinen H., Saito R., Kitazume M.T., Nakazawa A., Sugita A., Koganei K., et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn’s disease. Gut. 2008;57:1682–1689. doi: 10.1136/gut.2007.135053. [DOI] [PubMed] [Google Scholar]
- 21.Moschen A.R., Tilg H., Raine T. IL-12, IL-23 and IL-17 in IBD: immunobiology and therapeutic targeting. Nat. Rev. Gastroenterol. Hepatol. 2019;16:185–196. doi: 10.1038/s41575-018-0084-8. [DOI] [PubMed] [Google Scholar]
- 22.Chen H. Rphenograph: R implementation of the PhenoGraph algorithm. R package v. 0.99 1. 2015. https://github.com/JinmiaoChenLab/Rphenograph
- 23.Mlecnik B., Bindea G., Angell H.K., Maby P., Angelova M., Tougeron D., Church S.E., Lafontaine L., Fischer M., Fredriksen T., et al. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity. 2016;44:698–711. doi: 10.1016/j.immuni.2016.02.025. [DOI] [PubMed] [Google Scholar]
- 24.Küçükköse E., Baars M.J.D., Amini M., Schraa S.J., Floor E., Bol G.M., Borel Rinkes I.H.M., Roodhart J.M.L., Koopman M., Laoukili J., et al. Stromal localization of inactive CD8+ T cells in metastatic mismatch repair deficient colorectal cancer. Br. J. Cancer. 2024;130:213–223. doi: 10.1038/s41416-023-02500-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wosen J.E., Mukhopadhyay D., Macaubas C., Mellins E.D. Epithelial MHC Class II Expression and Its Role in Antigen Presentation in the Gastrointestinal and Respiratory Tracts. Front. Immunol. 2018;9 doi: 10.3389/fimmu.2018.02144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beyaz S., Chung C., Mou H., Bauer-Rowe K.E., Xifaras M.E., Ergin I., Dohnalova L., Biton M., Shekhar K., Eskiocak O., et al. Dietary suppression of MHC class II expression in intestinal epithelial cells enhances intestinal tumorigenesis. Cell Stem Cell. 2021;28:1922–1935.e5. doi: 10.1016/j.stem.2021.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kondo A., Ma S., Lee M.Y.Y., Ortiz V., Traum D., Schug J., Wilkins B., Terry N.A., Lee H., Kaestner K.H. Highly Multiplexed Image Analysis of Intestinal Tissue Sections in Patients With Inflammatory Bowel Disease. Gastroenterology. 2021;161:1940–1952. doi: 10.1053/j.gastro.2021.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo C., Zhang H. The Role of Proinflammatory Pathways in the Pathogenesis of Colitis-Associated Colorectal Cancer. Mediators Inflamm. 2017;2017:5126048. doi: 10.1155/2017/5126048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen J., Ye X., Pitmon E., Lu M., Wan J., Jellison E.R., Adler A.J., Vella A.T., Wang K. IL-17 inhibits CXCL9/10-mediated recruitment of CD8+ cytotoxic T cells and regulatory T cells to colorectal tumors. J. Immunother. Cancer. 2019;7:324. doi: 10.1186/s40425-019-0757-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fauny M., Moulin D., D’Amico F., Netter P., Petitpain N., Arnone D., Jouzeau J.-Y., Loeuille D., Peyrin-Biroulet L. Paradoxical gastrointestinal effects of interleukin-17 blockers. Ann. Rheum. Dis. 2020;79:1132–1138. doi: 10.1136/annrheumdis-2020-217927. [DOI] [PubMed] [Google Scholar]
- 31.Lin X., Gaudino S.J., Jang K.K., Bahadur T., Singh A., Banerjee A., Beaupre M., Chu T., Wong H.T., Kim C.-K., et al. IL-17RA-signaling in Lgr5+ intestinal stem cells induces expression of transcription factor ATOH1 to promote secretory cell lineage commitment. Immunity. 2022;55:237–253.e8. doi: 10.1016/j.immuni.2021.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGeachy M.J., Cua D.J., Gaffen S.L. The IL-17 Family of Cytokines in Health and Disease. Immunity. 2019;50:892–906. doi: 10.1016/j.immuni.2019.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiong H., Zhang Z.-G., Tian X.-Q., Sun D.-F., Liang Q.-C., Zhang Y.-J., Lu R., Chen Y.-X., Fang J.-Y. Inhibition of JAK1, 2/STAT3 Signaling Induces Apoptosis, Cell Cycle Arrest, and Reduces Tumor Cell Invasion in Colorectal Cancer Cells. Neoplasia. 2008;10:287–297. doi: 10.1593/neo.07971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y., de Haar C., Chen M., Deuring J., Gerrits M.M., Smits R., Xia B., Kuipers E.J., van der Woude C.J. Disease-related expression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut. 2010;59:227–235. doi: 10.1136/gut.2009.184176. [DOI] [PubMed] [Google Scholar]
- 35.Mudter J., Neurath M.F. Il-6 signaling in inflammatory bowel disease: Pathophysiological role and clinical relevance. Inflamm. Bowel Dis. 2007;13:1016–1023. doi: 10.1002/ibd.20148. [DOI] [PubMed] [Google Scholar]
- 36.Salas A., Hernandez-Rocha C., Duijvestein M., Faubion W., McGovern D., Vermeire S., Vetrano S., Vande Casteele N. JAK–STAT pathway targeting for the treatment of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020;17:323–337. doi: 10.1038/s41575-020-0273-0. [DOI] [PubMed] [Google Scholar]
- 37.Irrazabal T., Thakur B.K., Kang M., Malaise Y., Streutker C., Wong E.O.Y., Copeland J., Gryfe R., Guttman D.S., Navarre W.W., Martin A. Limiting oxidative DNA damage reduces microbe-induced colitis-associated colorectal cancer. Nat. Commun. 2020;11:1802. doi: 10.1038/s41467-020-15549-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bankhead P., Loughrey M.B., Fernández J.A., Dombrowski Y., McArt D.G., Dunne P.D., McQuaid S., Gray R.T., Murray L.J., Coleman H.G., et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ge S.X., Jung D., Yao R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics. 2020;36:2628–2629. doi: 10.1093/bioinformatics/btz931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stoltzfus C.R., Filipek J., Gern B.H., Olin B.E., Leal J.M., Wu Y., Lyons-Cohen M.R., Huang J.Y., Paz-Stoltzfus C.L., Plumlee C.R., et al. CytoMAP: A Spatial Analysis Toolbox Reveals Features of Myeloid Cell Organization in Lymphoid Tissues. Cell Rep. 2020;31 doi: 10.1016/j.celrep.2020.107523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All single-cell segmented raw IMC data derived from participants have been deposited on Dataverse, accessible via https://doi.org/10.34894/LXHZQF
-
•
All code used for DSP and IMC data processing and analysis have been deposited on Dataverse, accessible via https://doi.org/10.34894/LXHZQF
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.