

Abstract
Interleukin-12 (IL-12) gene electrotransfer (GET) delivery is highly effective in inducing long-term, complete regression in mouse and human melanoma and other solid tumors. Therapeutic efficacy is enhanced by immune checkpoint inhibitors, and the combination of IL-12 plasmid GET (pIL-12 GET) and anti-programmed cell death protein 1 (PD-1) monoclonal antibodies has reached clinical trials. In this study, we designed peptides and plasmids encoding the mouse homologs of the pembrolizumab and nivolumab programmed cell death 1 ligand 1 (PD-L1) binding regions. We hypothesized that intratumor autocrine/paracrine peptide expression would block PD-1/PD-L1 binding and provide cancer patients with an effective and cost-efficient treatment alternative. We demonstrated that the mouse homolog to pembrolizumab was effective at blocking PD-1/PD-L1 in vitro. After intratumor plasmid delivery, both peptides bound PD-L1 on tumor cells. We established that plasmid DNA delivery to tumors in vivo or to tumor cells in vitro upregulated several immune modulators and PD-L1 mRNA and protein, potentiating this therapy. Finally, we tested the combination of pIL-12 GET therapy and peptide plasmids. We determined that pIL-12 GET therapeutic efficacy could be enhanced by combination with the plasmid encoding the pembrolizumab mouse homolog.
Keywords: MT: Delivery Strategies, interleukin 12, immune checkpoint inhibitor, gene electrotransfer
Graphical abstract
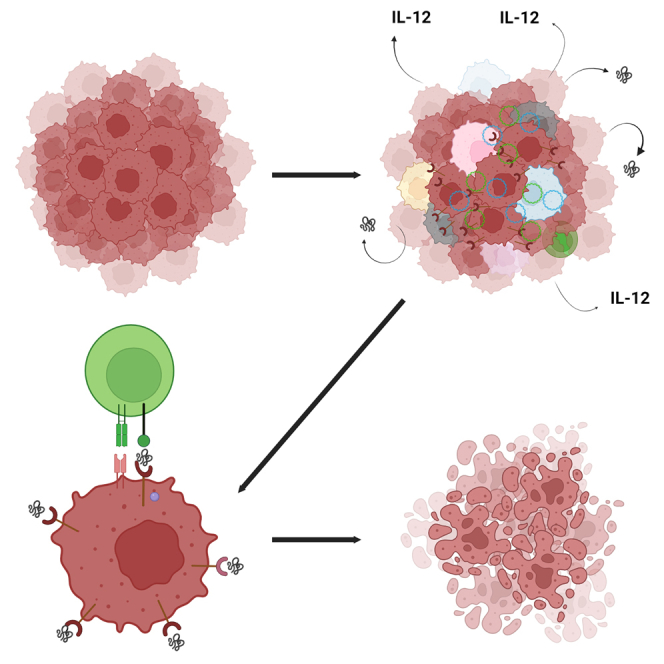
In this study, Heller and colleagues determined that the antitumor effect of IL-12 plasmid electrotransfer in mouse melanomas could be enhanced by the addition of a plasmid encoding a mouse homolog of the PD1P. The peptide blocked PD-1/PD-L1 binding, theoretically acting as an autocrine/paracrine checkpoint inhibitor.
Introduction
The immune checkpoint inhibitors (ICIs) pembrolizumab (Keytruda, Merck & Co.)1 and nivolumab (Opdivo, Bristol Myers Squibb)2 are approved by the US Food and Drug Administration for the treatment of several cancers, including melanoma. These therapies can be a significant financial burden to patients, leading to disparities in cancer treatment.3,4,5
ICI toxicity is associated with off-target activation of the immune system.6 Several immune-related adverse events are associated with ICI monoclonal antibody treatment. Melanoma therapy is most closely associated with dermatologic, hepatic, gastrointestinal, and endocrine adverse events.7,8,9
Our previous work determined that gene expression after intratumor interleukin-12 plasmid (pIL-12) GET modified the tumor microenvironment (TME) and induced a systemic cellular immune response, producing a significant antitumor effect on both primary tumors and metastases in the B16-F10 melanoma model.10,11,12,13,14,15 This work led to multiple clinical trials using pIL-12 GET, now termed tavokinogene telseplasmid electroporation (Tavo; OncoSec Medical Inc.) as a monotherapy.16,17,18,19 Clinical studies confirmed that intratumor pIL-12 GET increased lymphocytic infiltrate, decreased immune suppressive cells, and elevated checkpoints leading to a hot TME and potentiating ICI efficacy.18,19
The programmed cell death protein 1 (PD-1) epitopes recognized by pembrolizumab and nivolumab have been identified.20,21,22 We hypothesized that the expression of these peptides in the TME after plasmid gene electrotransfer (GET) could inhibit PD-1/programmed cell death 1 ligand 1 (PD-L1) binding. We, therefore, synthesized mammalian expression plasmids encoding these epitopes. Systemic antibody development would be hindered by the poor immunogenicity of peptides.23 The lack of antibody production could reduce associated systemic toxicities. In this study, we tested intratumor pIL-12 GET combined with plasmids encoding PD-1 epitopes as a potential alternative to combination with traditional checkpoint inhibitor therapy in the B16-F10 mouse melanoma model.
Results
PD-1 peptide expression and binding
Mouse-specific peptides corresponding with the nivolumab binding site (PD1N) and pembrolizumab binding site (PD1P) within human PD-1 were designed by alignment with mouse sequences using CLUSTAL Omega24 (Figure 1A) and commercially synthesized. PD1P but not PD1N significantly (p < 0.0001) inhibited mouse PD-1 detection by ELISA (Figure 1B). Plasmids pPD1P and pPD1N were developed based on the peptide sequences.
Figure 1.

Design and testing of PD-1 peptides
(A) CLUSTAL Omega24 multiple sequence alignment of human PD1 (NP_005009) and mouse PD1 (NP_032824). The binding sites of nivolumab and pembrolizumab are shown. Corresponding mouse peptide sequences are highlighted: PD1N (purple), PD1P (blue). (B) Inhibition of PD-1 ELISA by synthetic peptides. Mean ± standard deviation, ∗∗∗∗p < 0.0001.
To develop a gene-therapy based on this concept, we assessed the intratumor expression of the peptides encoded by the pPD1P and pPD1N plasmids after intratumor GET. Melanoma tumor cells endogenously express PD-L1 but not PD-1, while CD45+/CD8a+ T cells may endogenously express PD-1 but not PD-L1. We used these concepts to evaluate binding of plasmid-encoded PD-1 peptides to tumor cell PD-L1. Using immunohistochemistry and flow cytometry, we expected to detect PD-1 on some T cells based on their potential for endogenous expression of PD-1. We also assayed for PD-1 peptide binding PD-L1 on melanoma (Melan-A+) cells, which would be an indication that the expressed PD-1 peptide bound to PD-L1 expressed on the Melan-A+ tumor cells.
As expected, immunohistochemistry of control tumors confirmed the presence of PD-1 positive CD8a+ T cells, but no colocalization of PD-1 with Melan-A was observed (Figure 2A). Forty-eight hours after transfection with the PD-1 peptide plasmids, PD-1 was detected on both CD8a+ T cells and Melan-A+ tumor cells. This observation implicated the expression and binding of the encoded PD1N (Figure 2B) and PD1P (Figure 2C) peptides to PD-L1 on melanoma cells.
Figure 2.

PD-1 peptides bind B16-F10 cells in vivo 2 days after plasmid GET delivery
pPD1N and pPD1P were delivered to tumors using the EP2 pulse protocol. Representative composite images of (A) control tumors, (B) pPD1N GET tumors, and (C) pPD1P GET tumors are shown. Melan-A, Red; CD8a; cyan; PD-1, yellow. White arrows indicate an example of colocalization of PD-1 and CD8a; green arrows indicate an example of colocalization of PD-1 and Melan-A. Scale bar, 20 μm. Images of individual fluorescent channels are available in Figure S1. (D) Quantification of in vivo cell binding of PD1 peptides after intratumor pPD1N or pPD1P GET by flow cytometry. Mean ± standard deviation, ∗p < 0.05, ∗∗∗p < 0.001 with respect to control. Representative histograms from control tumors: (E) CD45+/PD-1+ cells, (F) CD45−/PD-1+ cells; representative histograms from pPD1N GET tumors: (G) CD45+/PD-1+ cells, (H) CD45−/PD-1+ cells; representative histograms from pPD1P GET tumors: (I) CD45+/PD-1+ cells, (J) CD45−/PD-1+ cells. Individual histograms are available in Figure S2.
Flow cytometry confirmed intratumor PD-1 binding (Figure 2D) to both CD45+ leukocytes and CD45-tumor cells. Figures 2E–2J shows the individual representative histograms of each experimental group. No changes in PD-1 levels were detected on CD45+ leukocytes from tumors after pPD1N or pPD1P GET, indicating PD-1 expression in T cells was unchanged. PD-1 levels increased on CD45-melanoma cells from approximately 1.3% in the control group to 7.3% (p < 0.05) after pPD1N GET and 12.7% (p < 0.001) after pPD1P GET. These results confirmed intratumor plasmid GET produced peptides capable of binding PD-L1 on melanoma cells.
Upregulation of PD-L1 expression in B16-F10 melanoma cells and tumors after DNA electrotransfer
PD-L1 (Cd274) RNA expression in melanoma tumors was minimally upregulated by pulse application, but upregulated 8-fold after plasmid DNA (pDNA) electrotransfer when compared with control tumors (Figure 3). Several cytokine mRNAs known to drive Cd274 expression, including interferon gamma Ifng, Il1b Il10, and Il6,25,26,27,28 were significantly regulated in several experimental groups. Ifng RNA was the sole cytokine upregulated nearly 5-fold by DNA injection, but was upregulated minimally by pulse application and pDNA electrotransfer. Il1b and Il10 were similarly regulated, approximately 2-fold after pulse application, increasing to 3-fold and 9-fold, respectively, after pDNA electrotransfer. Finally, Il6 RNA was upregulated more than 42-fold solely in the pDNA electrotransfer group. This significant regulation may indicate this cytokine is primarily responsible for Cd274 regulation in these tumors (Figure 3).
Figure 3.

PD-L1-related RNA signaling is regulated 4 h after vector pDNA electrotransfer (ET)
gWiz Blank was delivered using the EP1 pulse protocol to B16F10 tumors. The heatmap shows differentially expressed genes based on false discovery rate, ∗q < 0.05, ∗∗q < 0.01, ∗∗∗q < 0.001 with respect to control.
Protein expression experiments were performed in B16-F10 cells in vitro to confirm the observed changes in gene expression. A significant increase in the expression of PD-L1 on the B16-F10 cell surface was detected after DNA electrotransfer by flow cytometry (Figure 4A). While control cells minimally expressed PD-L1, the level of expression increased to 16% after pulse application (p < 0.05), then to nearly 50% after DNA electrotransfer (p < 0.001). Cytokine secretion from melanoma cells was quantified using a multiplex bead array 4 h after DNA electrotransfer (Figure 4B). At this time point, IL-1β and IFN-γ protein regulation was not detected. Interestingly, while mRNA expression in tumors was upregulated, IL-10 protein was downregulated 3-fold (p < 0.05) in cells after pDNA exposure alone; this regulation was 1.5-fold (p < 0.05) when combined with pulse delivery. Finally, IL-6 protein was upregulated approximately 20-fold (p < 0.01) compared with the control in the group receiving DNA electrotransfer, supporting the concept that this cytokine may be primarily responsible for PD-L1 regulation.
Figure 4.

PD-L1-related protein signaling is regulated 4 h after vector pDNA electrotransfer
gWiz Blank was delivered using the EP1 pulse protocol to B16F10 cells. (A) Expression of PD-L1 on the cell surface. (B) Cytokine production. Mean ± standard deviation, ∗p < 0.05, ∗∗∗p < 0.001 with respect to control.
Therapeutic efficacy of combination therapy in the B16-F10 mouse melanoma model
After plasmid delivery, PD-1 peptide/PD-L1 binding was observed in vivo (Figure 2). DNA electrotransfer upregulated PD-L1 mRNA expression in vivo (Figure 3), which was confirmed by PD-L1 protein expression in vitro (Figure 4A). It is established that pIL-12 GET potentiates checkpoint inhibitor therapy.15,17 Therefore, the potential therapeutic efficacy of this combined approach was assessed in the B16-F10 mouse melanoma model. To facilitate this combination, we used delivery protocols designed to achieve specific relative expression levels. Since the concept being tested is that the PD-1 peptide binds to upregulated PD-L1, we initially reasoned the best approach would be to utilize a pulse protocol (EP2) that typically leads to higher expression.13 For the plasmid encoding IL-12, we used a pulse protocol (EP1), which leads to moderate expression. This moderate expression induces the appropriate changes in the TME to result in a robust anti-tumor immune response.10,11,13,18,29,30 We delivered pIL-12 1 day before the peptide plasmids to enable TME changes to occur prior to delivery of PD-1. In additional experiments, we further explored this delivery approach by also delivering the pIL-12 with EP2.
We did not observe toxicity as indicated by weight loss (data not shown), coat appearance, general condition, and discomfort in any group or experiment. In the initial experiment, delivery of pVAX1 and pPD1N using EP2 had no significant effect on survival, while PD1P produced a minimal but significant effect (p = 0.0116) (Figure 5A). Delivery of pIL-12 using the EP1 pulse protocol significantly increased survival compared with mice with untreated tumors alone (p < 0.0001) or in combination with the vector plasmid, pVAX1 (p < 0.0001), pPD1P (p < 0.0001), and pPD1N (p < 0.0001) (Figure 5B).
Figure 5.

Delivery of plasmids encoding PD-1 peptides potentiates IL-12 efficacy in vivo
(A) PD1P, PD1N, and pVAX1 GET delivery as a sole therapy using the EP2 pulse protocol as described in Table 1. (B) Survival after pIL12 GET using pulse protocol EP1, n = 10 per group. (C) Survival after pIL12 GET using pulse protocol EP2, n = 20 per group. Significance was determined by Mantel-Cox log rank test followed by multiple comparisons of Kaplan-Meier curves using the Bonferroni method. For each test, a p value of <0.05 was considered statistically significant. ∗p < 0.05; ∗∗∗p < 0.001 respect to control tumors.
In evaluating this approach for potential translation, the pIL-12 delivery protocol was modified to correspond with the delivery protocol for the other plasmids (EP2). As expected, the higher pIL-12 expression associated with EP2 delivery significantly increased survival compared with mice with untreated tumors alone (p < 0.0001) (Figure 5C). While the addition of pVAX1 or pPD1N delivery produced no additional benefit, the addition of pPD1P increased survival minimally but significantly over pIL-12 delivery alone (p = 0.0135).
Discussion
In this study, we demonstrated that a mouse-specific synthetic peptide based on PD1P but not PD1N successfully inhibited PD-1/PD-L1 binding in vitro. Tumor cells transfected with peptide plasmids secreted these peptides, which subsequently bound PD-L1 on melanoma tumor cells in vivo. RNA and protein analyses demonstrated a potentiating effect of plasmid delivery by the upregulation of PD-L1 on tumor cells due to cytokine induction. Finally, we demonstrated that intratumor delivery of the PD1P-encoding plasmid increased the efficacy of pIL-12 GET therapy.
A synthetic peptide encoding PD1P but not PD1N inhibited PD-1 detection by ELISA (Figure 1). These peptides correspond with different regions of the PD-1 protein. Nivolumab binds an N-terminal loop not involved in the recognition of PD-L1.22 Pembrolizumab binds a longer region that overlaps the PD-1/PD-L1 binding site.20,21 Direct interference may be more likely to inhibit binding. These factors may influence the differential effectiveness in the inhibition of the in vitro assay, which was mirrored in vivo. Transfection of tumors resulted in autocrine/paracrine binding of each peptide to melanoma cells (Figure 2).
Tumor PD-L1 expression is a predictive biomarker for the efficacy of anti-PD-1/PD-L1 therapy.31,32 It is important to note that PD-L1 expression can be upregulated by anti-cancer therapies that induce intratumor expression of inflammatory molecules, such as chemotherapy, radiotherapy, and biological therapies.33,34 The activity of anti-PD-1 antibodies depends on PD-L1 expression. For tumors that do not express PD-L1, an elevation of expression is necessary for checkpoint activity. We found that empty vector plasmid GET significantly increased tumor PD-L1 RNA signaling pathways (Figure 3). Expression of the PD-L1 protein significantly increase in B16-F10 cells (Figure 4A). Although we detected this upregulation after vector delivery, this observation generally supports a previous study where increased PD-L1 expression was observed in hepatoma cells and tumors after exposure to a CCL21-encoding plasmid.35
PD-L1 expression can be driven by several factors, including inflammatory immune modulators such as IL-6, IL-10, IL-1β, and IFN-γ.25,26,27,28 We previously demonstrated that DNA electrotransfer into B16-F10 melanoma tumors induced upregulation of the mRNAs and proteins of many immune modulators, including IL-6,36,37 potentially due to the activation of cytosolic DNA-specific pattern recognition receptors.38,39 IL-6, IL-10, IL-1β, and IFN-γ mRNAs were upregulated after vector plasmid GET (Figure 3). While several immune modulators were upregulated 4 h after vector plasmid GET, the elevated upregulation of IL-6 implicates this protein driving PD-L1 expression (Figure 4B).
The efficacy of pIL-12 GET is well-established in the B16-F10 mouse melanoma model10,11,12,13,14 and in metastatic melanoma clinical trials.16,17,18,19 To enhance pIL-12 GET efficacy, we combined the upregulation of PD-L1 expression in response to DNA electrotransfer with the inhibition of PD-1/PD-L1 binding using plasmid-encoded peptides. In this study, the antitumor effects of pIL-12 GET delivered using EP2 were significantly enhanced by the addition of a plasmid encoding a mouse homolog of the pembrolizumab but not the PD1N (Figure 5C). This enhancement was not seen when pIL-12 GET was delivered with EP1. This may be because the high response to pIL-12 GET alone could not be enhanced with peptide expression. In a previous study, repeated injection of a plasmid encoding the complete PD-1 sequence was tested in intramuscular murine hepatomas.35 In this study, initial tumor growth was suppressed in 36% of mice; combination with a plasmid encoding CCL21 increased growth suppression to 57%. This study differed in tumor model and site, plasmid design, treatment regimen, and in the fact that we tested therapeutic efficacy rather than tumor growth suppression.
Localized intratumor delivery of IL-12 plasmid by electroporation leads to systemic responses and an abscopal effect.40 In our study, while pIL-12 GET was highly effective therapeutically, efficacy benefited from the addition of the plasmid encoding the PD1P peptide. Future studies will explore this approach in a multi-tumor or metastatic model to determine if these local effects can induce a systemic response.
Materials and methods
Peptides and plasmids
The binding site of pembrolizumab overlaps the PD-1/PD-L1 binding site,20,21 amino acids 74–99 of PD-1 (UniProt accession number: Q15116). Nivolumab primarily binds to amino acids 25–34, an N-terminal loop that is not involved in recognition of PD-L1.22 We performed an alignment between human and mouse (UniProt accession number: Q02242) PD-1 to find the homologous mouse sequence for each binding site (Figure 1A). Mouse-specific peptides PD1P (NQTEKQAAFCNGLSQPVQDARFQIIQ, 2,935.29 g/mol, 96.50% purity by high-performance liquid chromatography [HPLC]) and PD1N (LEVPNGPWRS, 1,154.3 g/mol, 98.91% purity by HPLC) were synthesized (ABI Scientific) and suspended in PBS. The corresponding DNA sequences were inserted between EcoRI and NotI in a mammalian expression plasmid (pVAX1, Thermo Fisher Scientific) in-frame behind an IgK leader sequence (5′-ATGGAGACAGACACACTCCTGCTATGGGTACTGCTGCTCTGGGTTCCAGGTTCCACTGGTGAC-3′) to ensure secretion of each peptide after transfection to create plasmids pPD1P and pPD1N. These plasmids were commercially prepared (Azenta Life SciencesUSA) and suspended in sterile H2O. Plasmids pUMVC3-mIL12 (University of Michigan Biomedical Research Core Facilities) and empty vectors pVAX1 and gWiz Blank (Aldevron LLC) were commercially prepared (Aldevron LLC) and suspended in sterile saline. Endotoxin levels were confirmed to be <0.1 EU/μg.
Binding inhibition
A Mouse PD-1 ELISA kit (Abcam) was used to quantify PD-1/PD-L1 binding inhibition. Briefly, the antibody cocktail was spiked with 1 mM synthetic PD1P, PD1N, or PBS diluent and incubated for 1 h at room temperature. The ELISA was then performed per manufacturer’s instruction using the 2,000 pg/mL standard as a positive control.
Electrotransfer
Two pulse protocols were used in this study (Table 1). EP1 produces moderate transgene expression and has been used in multiple clinical trials.16,17,18,19 EP2 produces higher expression,13 but has not yet reached clinical trials. Pulses were delivered using a legacy model ECM 830 Square Wave Electroporation System (BTX Harvard Apparatus).
Table 1.
Pulse protocols for plasmid electrotransfer
| Protocol | Pulse number | Pulse length (ms)a | Pulse intensity (V/cm)b | Pulse frequency (hertz) | Electrode |
|---|---|---|---|---|---|
| EP1 | 6 | 0.1 | 1,300 | 1 or 4 | 6-needle array |
| EP2 | 10 | 5 | 600 | 1 | caliper |
millisecond.
Volts/centimeter.
Cell lines and transfection
B16-F10 mouse melanoma cells (CRL-6475, American Type Culture Collection) were cultured in McCoy’s medium (Corning, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin-streptomycin in a 5% CO2 humidified incubator at 37°C. Cells were suspended to 2 × 107/mL in complete medium; groups were spiked with sterile physiological saline or 0.8 mg/mL gWiz Blank (Aldevron). In addition to the control group, the experimental groups included exposure to pulses alone by the application of pulse protocol EP1 at a frequency of 4 Hz (Table 1), plasmid suspension, and plasmid suspension followed by pulse application. Cells were then transferred to six-well plates and incubated for 24 h.
Cell flow cytometry
Transfected B16-F10 cells were harvested 24 h post-transfection. PD-L1 expression of DAPI (Akoya Biosciences) stained cells was quantified with an anti-PDL1 monoclonal antibody (Biolegend) via flow cytometry (CytoFLEX, Beckman). FlowJo analysis software (BD Biosciences) was used to determine total number of cells expressing PD-L1.
In vivo RNA expression
The animal experiments and oligonucleotide array dataset used in this analysis were previously described.37 All procedures were approved by the University of South Florida Institutional Animal Care and Use Committee. Briefly, 106 melanoma cells in 50 μL phosphate buffered saline were injected subcutaneously in the left flank of female 7- to 8-week-old C57Bl/6 mice (Jackson Laboratories) and allowed to grow for 6–8 to a diameter of approximately 4–6 mm. Mice were anesthetized by inhalation of 2% isoflurane in oxygen, tumors were injected with 50 μg gWiz Blank in 25 μL saline and pulse protocol EP1 at a frequency of 4 Hz was applied with a legacy model ECM 830 Square Wave Electroporation System (BTX Harvard Apparatus) (EP1). After 4 h, the mice were humanely euthanized and the tumors were removed and processed for gene expression analysis.37 The selection conditions of a significantly changed gene expression were based on a fold difference higher than absolute 2 and q-value after false discovery rate correction of less than 0.05 were analyzed using the DAVID (Database for Annotation, Visualization and Integrated Discovery) bioinformatics tool.41,42,43 Raw data and DAVID output files can be downloaded at https://www.mdpi.com/article/10.3390/pharmaceutics14102097/s1.37
In vitro protein expression
Proteins secreted by 106 transfected B16-F10 cells into 1 mL medium were quantified 4 h after pDNA electrotransfer using a multiplex panel (Millipore) for IL-6, IL-10, IL-1β, IFN-γ per the manufacturer’s instructions on a MAGPIX System (Luminex).
Tumor immunohistochemistry
Tumor induction was performed as described in the In vivo RNA expression section. Once tumors reached 4–6 mm in diameter, mice were anesthetized then treated via intratumor injection of pPD1N or pPD1P (100 μg/50 μL), followed by GET using EP2 (Table 1). Forty-eight hours after treatment, the mice were humanely euthanized, and the tumors removed. Paraffin-embedded sections (5 μm) were stained via immunohistochemistry (IHC) using Opal (520, 570, 690) Multiplex IHC kits (Akoya Biosciences) as directed by the manufacturer. Primary antibodies used include rabbit anti-mouse Melan-A (polyclonal antibody, Proteintech), rat anti-mouse CD8a (4SM15, Ebioscience), rabbit anti-mouse PD-1 (EPR20665, Abcam) using. Secondary antibodies used include horseradish peroxidase (HRP) goat anti-rat IgG (Poly4054, Biolegend) and HRP goat anti-rabbit IgG H&L (polyclonal antibody, Abcam). Sections were imaged using the PhenoImager Fusion platform (Akoya Biosciences) at a 40× (0.25 μm) resolution using Fusion software version 2.2.0 (Akoya Biosciences). Spectral unmixing, background autoflourescence removal, and image finalization were performed using Phenochart version 2.0.1 (Akoya Biosciences), Inform version 3.0 (Akoya Biosciences), and Qupath version 0.5.0.44
Tumor flow cytometry
Forty-eight hours after delivery of pPD1N or pPD1P, B16-F10 tumors were dissociated using a gentleMACS Octo Dissociator (Miltenyi Biotec) per the manufacturer’s instructions. Cells were stained for CD45 (Biolegend) and PD-1 (Miltenyi Biotec) and analyzed by flow cytometry (FACSCanto II, BD Biosciences).
Therapeutic efficacy
Under anesthesia, all plasmids were injected into the tumor at a volume of 50 μL when tumors reached 4–6 mm in diameter. pIL-12 was injected at a concentration of 1 mg/mL and pPD1N, pPD1P and pVAX1 were at a concentration of 2 mg/mL. After injection, GET was administered to the tumors. For mice receiving injections of pPD1N, pPD1P and pVAX1, the GET protocol was EP2 (Table 1). For mice receiving injections of pIL-12, the GET protocol for the initial experiment was EP1 (Table 1). In a subsequent experiment, mice also received pIL-12 using EP2. In each experiment, mice were administered pIL-12 GET on days 1, 5, and 8 and on days 2, 6, and 9 mice were administered pPD1N, pPD1P, and pVAX1 GET.
Statistical evaluation
Statistical analyses was carried out using GraphPad Prism 9.1.0. For binding inhibition assays and protein expression assays, significance was determined by a one-way ANOVA followed by a Tukey-Kramer post-test. The significance of survival was determined by an overall Mantel-Cox log rank test followed by multiple comparisons of Kaplan-Meier curves using the Bonferroni method. For each test, a p value of <0.05 was considered statistically significant.
Data and code availability
The data presented in this study are available on request from the corresponding authors.
Acknowledgments
The research reported in this publication was supported in part by the National Cancer Institute at the National Institutes of Health under award number R01CA196796 (L.C.H.) and award number R01CA186730 (R.H.) and by the Department of Medical Engineering. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funders. The funders had no role in study design, collection of data, decision to publish, or in preparation of this manuscript.
Author contributions
L.C.H.: Conceptualization, funding acquisition, methodology, project administration, supervision, visualization, writing – original draft preparation, writing – review & editing. G.S.: Formal analysis, investigation, methodology, visualization, writing – review & editing. A.S.C.: Formal analysis, investigation, visualization, methodology, writing – review & editing. J.Singh: Formal analysis, investigation, methodology, visualization, writing – review & editing. S.M.: Formal analysis, investigation, methodology, visualization, writing – review & editing. J.Synowiec: Formal Analysis, investigation, methodology, visualization, writing – review & editing. R.H.: conceptualization, funding acquisition, investigation, methodology, project administration, supervision, writing – review & editing.
Declaration of interests
L.H. and R.H. are inventors on patents that cover the technology that was used in the work reported in this manuscript. In addition, R.H. owns stock and stock options in Inovio Pharmaceutical, Inc.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2024.102267.
Contributor Information
Loree C. Heller, Email: lheller@usf.edu.
Richard Heller, Email: rheller@usf.edu.
Supplemental information
References
- 1.Robert C., Ribas A., Wolchok J.D., Hodi F.S., Hamid O., Kefford R., Weber J.S., Joshua A.M., Hwu W.J., Gangadhar T.C., et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384:1109–1117. doi: 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- 2.Robert C., Long G.V., Brady B., Dutriaux C., Maio M., Mortier L., Hassel J.C., Rutkowski P., McNeil C., Kalinka-Warzocha E., et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015;372:320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 3.Gilligan A.M., Alberts D.S., Roe D.J., Skrepnek G.H. Death or Debt? National Estimates of Financial Toxicity in Persons with Newly-Diagnosed Cancer. Am. J. Med. 2018;131:1187–1199.e5. doi: 10.1016/j.amjmed.2018.05.020. [DOI] [PubMed] [Google Scholar]
- 4.Cutler D.M. Early Returns From the Era of Precision Medicine. JAMA. 2020;323:109–110. doi: 10.1001/jama.2019.20659. [DOI] [PubMed] [Google Scholar]
- 5.Abuali I., Patel S., Kiel L., Meza K., Florez N. Disparities in cancer care-A call to action. Cancer Cell. 2023;41:1–4. doi: 10.1016/j.ccell.2022.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Martins F., Sofiya L., Sykiotis G.P., Lamine F., Maillard M., Fraga M., Shabafrouz K., Ribi C., Cairoli A., Guex-Crosier Y., et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019;16:563–580. doi: 10.1038/s41571-019-0218-0. [DOI] [PubMed] [Google Scholar]
- 7.Khoja L., Day D., Wei-Wu Chen T., Siu L.L., Hansen A.R. Tumour- and class-specific patterns of immune-related adverse events of immune checkpoint inhibitors: a systematic review. Ann. Oncol. 2017;28:2377–2385. doi: 10.1093/annonc/mdx286. [DOI] [PubMed] [Google Scholar]
- 8.Das S., Johnson D.B. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer. 2019;7:306. doi: 10.1186/s40425-019-0805-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Darnell E.P., Mooradian M.J., Baruch E.N., Yilmaz M., Reynolds K.L. Immune-Related Adverse Events (irAEs): Diagnosis, Management, and Clinical Pearls. Curr. Oncol. Rep. 2020;22:39. doi: 10.1007/s11912-020-0897-9. [DOI] [PubMed] [Google Scholar]
- 10.Lucas M.L., Heller L., Coppola D., Heller R. IL-12 plasmid delivery by in vivo electroporation for the successful treatment of established subcutaneous B16.F10 melanoma. Mol. Ther. 2002;5:668–675. doi: 10.1006/mthe.2002.0601. [DOI] [PubMed] [Google Scholar]
- 11.Lucas M.L., Heller R. IL-12 gene therapy using an electrically mediated nonviral approach reduces metastatic growth of melanoma. DNA Cell Biol. 2003;22:755–763. doi: 10.1089/104454903322624966. [DOI] [PubMed] [Google Scholar]
- 12.Heller L., Merkler K., Westover J., Cruz Y., Coppola D., Benson K., Daud A., Heller R. Evaluation of toxicity following electrically mediated interleukin-12 gene delivery in a B16 mouse melanoma model. Clin. Cancer Res. 2006;12:3177–3183. doi: 10.1158/1078-0432.CCR-05-2727. [DOI] [PubMed] [Google Scholar]
- 13.Shirley S.A., Lundberg C.G., Li F., Burcus N., Heller R. Controlled gene delivery can enhance therapeutic outcome for cancer immune therapy for melanoma. Curr. Gene Ther. 2015;15:32–43. doi: 10.2174/1566523214666141121111630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi G., Edelblute C., Arpag S., Lundberg C., Heller R. IL-12 Gene Electrotransfer Triggers a Change in Immune Response within Mouse Tumors. Cancers. 2018;10:498. doi: 10.3390/cancers10120498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi G., Scott M., Mangiamele C.G., Heller R. Modification of the Tumor Microenvironment Enhances Anti-PD-1 Immunotherapy in Metastatic Melanoma. Pharmaceutics. 2022;14:2429. doi: 10.3390/pharmaceutics14112429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daud A.I., DeConti R.C., Andrews S., Urbas P., Riker A.I., Sondak V.K., Munster P.N., Sullivan D.M., Ugen K.E., Messina J.L., Heller R. Phase I Trial of Interleukin-12 Plasmid Electroporation in Patients With Metastatic Melanoma. J. Clin. Oncol. 2008;26:5896–5903. doi: 10.1200/JCO.2007.15.6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Algazi A.P., Twitty C.G., Tsai K.K., Le M., Pierce R., Browning E., Hermiz R., Canton D.A., Bannavong D., Oglesby A., et al. Phase II Trial of IL-12 Plasmid Transfection and PD-1 Blockade in Immunologically Quiescent Melanoma. Clin. Cancer Res. 2020;26:2827–2837. doi: 10.1158/1078-0432.CCR-19-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Algazi A., Bhatia S., Agarwala S., Molina M., Lewis K., Faries M., Fong L., Levine L.P., Franco M., Oglesby A., et al. Intratumoral delivery of tavokinogene telseplasmid yields systemic immune responses in metastatic melanoma patients. Ann. Oncol. 2020;31:532–540. doi: 10.1016/j.annonc.2019.12.008. [DOI] [PubMed] [Google Scholar]
- 19.Greaney S.K., Algazi A.P., Tsai K.K., Takamura K.T., Chen L., Twitty C.G., Zhang L., Paciorek A., Pierce R.H., Le M.H., et al. Intratumoral Plasmid IL12 Electroporation Therapy in Patients with Advanced Melanoma Induces Systemic and Intratumoral T-cell Responses. Cancer Immunol. Res. 2020;8:246–254. doi: 10.1158/2326-6066.CIR-19-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horita S., Nomura Y., Sato Y., Shimamura T., Iwata S., Nomura N. High-resolution crystal structure of the therapeutic antibody pembrolizumab bound to the human PD-1. Sci. Rep. 2016;6 doi: 10.1038/srep35297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Na Z., Yeo S.P., Bharath S.R., Bowler M.W., Balikci E., Wang C.I., Song H. Structural basis for blocking PD-1-mediated immune suppression by therapeutic antibody pembrolizumab. Cell Res. 2017;27:147–150. doi: 10.1038/cr.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan S., Zhang H., Chai Y., Song H., Tong Z., Wang Q., Qi J., Wong G., Zhu X., Liu W.J., et al. An unexpected N-terminal loop in PD-1 dominates binding by nivolumab. Nat. Commun. 2017;8 doi: 10.1038/ncomms14369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trier N., Hansen P., Houen G. Peptides, Antibodies, Peptide Antibodies and More. Int. J. Mol. Sci. 2019;20:6289. doi: 10.3390/ijms20246289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Soding J., et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun C., Mezzadra R., Schumacher T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity. 2018;48:434–452. doi: 10.1016/j.immuni.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munir S., Lundsager M.T., Jorgensen M.A., Hansen M., Petersen T.H., Bonefeld C.M., Friese C., Met O., Straten P.T., Andersen M.H. Inflammation induced PD-L1-specific T cells. Cell Stress. 2019;3:319–327. doi: 10.15698/cst2019.10.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yi M., Niu M., Xu L., Luo S., Wu K. Regulation of PD-L1 expression in the tumor microenvironment. J. Hematol. Oncol. 2021;14:10. doi: 10.1186/s13045-020-01027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zerdes I., Matikas A., Bergh J., Rassidakis G.Z., Foukakis T. Genetic, transcriptional and post-translational regulation of the programmed death protein ligand 1 in cancer: biology and clinical correlations. Oncogene. 2018;37:4639–4661. doi: 10.1038/s41388-018-0303-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Canton D.A., Shirley S., Wright J., Connolly R., Burkart C., Mukhopadhyay A., Twitty C., Qattan K.E., Campbell J.S., Le M.H., et al. Melanoma treatment with intratumoral electroporation of tavokinogene telseplasmid (pIL-12, tavokinogene telseplasmid) Immunotherapy. 2017;9:1309–1321. doi: 10.2217/imt-2017-0096. [DOI] [PubMed] [Google Scholar]
- 30.Bhatia S., Longino N.V., Miller N.J., Kulikauskas R., Iyer J.G., Ibrani D., Blom A., Byrd D.R., Parvathaneni U., Twitty C.G., et al. Intratumoral Delivery of Plasmid IL12 Via Electroporation Leads to Regression of Injected and Noninjected Tumors in Merkel Cell Carcinoma. Clin. Cancer Res. 2020;26:598–607. doi: 10.1158/1078-0432.CCR-19-0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel S.P., Kurzrock R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Therapeut. 2015;14:847–856. doi: 10.1158/1535-7163.MCT-14-0983. [DOI] [PubMed] [Google Scholar]
- 32.Michot J.M., Bigenwald C., Champiat S., Collins M., Carbonnel F., Postel-Vinay S., Berdelou A., Varga A., Bahleda R., Hollebecque A., et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur. J. Cancer. 2016;54:139–148. doi: 10.1016/j.ejca.2015.11.016. [DOI] [PubMed] [Google Scholar]
- 33.Galluzzi L., Humeau J., Buque A., Zitvogel L., Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2020;17:725–741. doi: 10.1038/s41571-020-0413-z. [DOI] [PubMed] [Google Scholar]
- 34.Yi M., Zheng X., Niu M., Zhu S., Ge H., Wu K. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol. Cancer. 2022;21:28. doi: 10.1186/s12943-021-01489-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He Y.F., Zhang G.M., Wang X.H., Zhang H., Yuan Y., Li D., Feng Z.H. Blocking programmed death-1 ligand-PD-1 interactions by local gene therapy results in enhancement of antitumor effect of secondary lymphoid tissue chemokine. J. Immunol. 2004;173:4919–4928. doi: 10.4049/jimmunol.173.8.4919. [DOI] [PubMed] [Google Scholar]
- 36.Heller L.C., Cruz Y.L., Ferraro B., Yang H., Heller R. Plasmid injection and application of electric pulses alter endogenous mRNA and protein expression in B16.F10 mouse melanomas. Cancer Gene Ther. 2010;17:864–871. doi: 10.1038/cgt.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhandary M., Sales Conniff A., Miranda K., Heller L.C. Acute Effects of Intratumor DNA Electrotransfer. Pharmaceutics. 2022;14:2097. doi: 10.3390/pharmaceutics14102097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Znidar K., Bosnjak M., Cemazar M., Heller L.C. Cytosolic DNA Sensor Upregulation Accompanies DNA Electrotransfer in B16.F10 Melanoma Cells. Nucleic Acids. 2016;5 doi: 10.1038/mtna.2016.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bosnjak M., Jesenko T., Kamensek U., Sersa G., Lavrencak J., Heller L., Cemazar M. Electrotransfer of Different Control Plasmids Elicits Different Antitumor Effectiveness in B16.F10 Melanoma. Cancers. 2018;10:37. doi: 10.3390/cancers10020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen K.G., Vrabel M.R., Mantooth S.M., Hopkins J.J., Wagner E.S., Gabaldon T.A., Zaharoff D.A. Localized Interleukin-12 for Cancer Immunotherapy. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.575597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dennis G., Jr., Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 42.Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 43.Huang da W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bankhead P., Loughrey M.B., Fernandez J.A., Dombrowski Y., McArt D.G., Dunne P.D., McQuaid S., Gray R.T., Murray L.J., Coleman H.G., et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented in this study are available on request from the corresponding authors.