

Summary
Histone deacetylase 3 (HDAC3) is a crucial epigenetic modulator essential for various developmental and physiological functions. Although its dysfunction is increasingly recognized in abnormal phenotypes, to our knowledge, there have been no established reports of human diseases directly linked to HDAC3 dysfunction. Using trio exome sequencing and extensive phenotypic analysis, we correlated heterozygous de novo variants in HDAC3 with a neurodevelopmental disorder having variable clinical presentations, frequently associated with intellectual disability, developmental delay, epilepsy, and musculoskeletal abnormalities. In a cohort of six individuals, we identified missense variants in HDAC3 (c.277G>A [p.Asp93Asn], c.328G>A [p.Ala110Thr], c.601C>T [p.Pro201Ser], c. 797T>C [p.Leu266Ser], c.799G>A [p.Gly267Ser], and c.1075C>T [p.Arg359Cys]), all located in evolutionarily conserved sites and confirmed as de novo. Experimental studies identified defective deacetylation activity in the p.Asp93Asn, p.Pro201Ser, p.Leu266Ser, and p.Gly267Ser variants, positioned near the enzymatic pocket. In addition, proteomic analysis employing co-immunoprecipitation revealed that the disrupted interactions with molecules involved in the CoREST and NCoR complexes, particularly in the p.Ala110Thr variant, consist of a central pathogenic mechanism. Moreover, immunofluorescence analysis showed diminished nuclear to cytoplasmic fluorescence ratio in the p.Ala110Thr, p.Gly267Ser, and p.Arg359Cys variants, indicating impaired nuclear localization. Taken together, our study highlights that de novo missense variants in HDAC3 are associated with a broad spectrum of neurodevelopmental disorders, which emphasizes the complex role of HDAC3 in histone deacetylase activity, multi-protein complex interactions, and nuclear localization for proper physiological functions. These insights open new avenues for understanding the molecular mechanisms of HDAC3-related disorders and may inform future therapeutic strategies.
Keywords: histone deacetylase 3, CoREST, NCoR, neurodevelopmental disorder, epigenetics, exome sequencing, HDAC activity, proteomics, cellular mislocalization
Graphical abstract
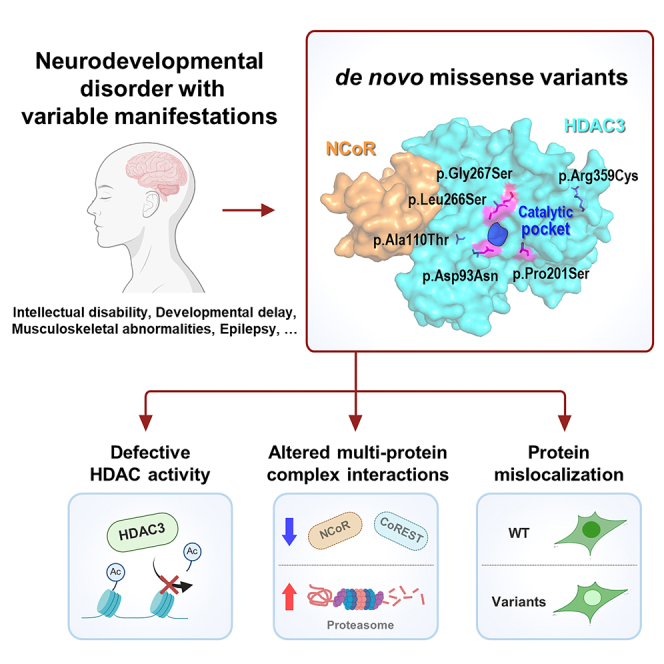
Yoon et al. identify de novo missense variants in HDAC3 that are associated with neurodevelopmental disorders with variable clinical features. Characterization of these variants focusing on their functions in histone deacetylation, multi-protein complex interactions, and nuclear localization highlights the multifaceted role of HDAC3 in human physiology and diseases.
Introduction
Epigenetic control of gene expression plays a pivotal role in numerous developmental processes. Among the major epigenetic modifications, histone acetylation is critical, regulated by two classes of proteins: histone acetyltransferases (HATs) and histone deacetylases (HDACs).1,2 HDACs remove acetyl groups from lysine (K) residues on histones (H) and are thereby often associated with inducing a closed chromatin structure and transcriptional repression.3,4,5 The deacetylation of histones H3K9 and H3K27 is particularly important, as these lysine residues can be methylated, facilitating chromatin compaction and gene silencing.6,7,8
There are 18 mammalian HDACs classified into four classes (I, II, III, and IV).9 HDAC1, HDAC2, HDAC3, and HDAC8 are class I HDACs, primarily nuclear enzymes with strong HDAC activity. These HDACs are often found in multi-protein complexes including the nuclear receptor co-repressor (NCoR) complex, co-repressor of repressor element 1 silencing transcription factor (CoREST) complex, and nucleosome remodeling and deacetylase (NuRD) complex.10 Notably, either NCoR1 or NCoR2 (also known as silencing mediator of retinoic acid and thyroid hormone receptor [SMRT]) is essential for HDAC activity.11,12,13 HDACs also interact with lysine-specific histone demethylase 1 (KDM1A; also known as LSD1), which is found in the CoREST complex. KDM1A is responsible for demethylating H3K4 and potentially H3K9 through REST corepressor 1/2 (RCOR1/2).14,15,16 HDACs also work cooperatively with chromatin remodelers, such as chromodomain helicase DNA binding proteins (CHD3, CHD4, and CHD5), by forming the NuRD complex.17
HDAC3, widely present in the cell nucleus and cytoplasm, is recognized as a crucial regulator of numerous developmental and physiological functions.18 Global deletion of Hdac3 in mice has resulted in embryonic lethality,19,20 suggesting that other HDACs cannot compensate for the loss of Hdac3 in early development. The conditional depletion of Hdac3 in mouse models has revealed profound impacts on metabolic and developmental functions in various tissues and organs, including the cerebral cortex and cerebellum,21,22,23 heart,20,24 liver,25,26 lung,27 skeletal bone and muscles,28,29 brown adipose tissue,30 and uterus.31 Particularly, the absence of Hdac3 in the forebrain and Purkinje neurons of the cerebellum has revealed a disrupted organization of neuronal and glial cell types in both the cortex and cerebellum, highlighting its essential role in regulating both neuronal and glial cell development.21 It is not only the deacetylation activity that is critical; the interaction with multi-protein complexes also plays a vital role in brain functions. For instance, NCoR complexes, comprising NCoR1 or NCoR2, are essential in regulating GABA signaling, thereby influencing memory and learning.32 Furthermore, HDAC3 has been implicated in Rett-like phenotypes through its interaction with methyl CpG binding protein 2 (MECP2). This interaction between MECP2 and HDAC3 contributes to the activation of forkhead box protein O3 (FOXO3), which in turn positively regulates a specific subset of neuronal genes playing crucial roles in cognition, sociability, and locomotor coordination.18,22
To date, several genes in the epigenetic machinery have been associated with Mendelian disorders, collectively termed chromatinopathies.33 Among HDACs, HDAC4 (MIM: 605314) and HDAC8 (MIM: 300269) have been identified as contributors to neurodevelopmental disorder with central hypotonia and dysmorphic facies (MIM: 619797) and Cornelia de Lange syndrome 5 (MIM: 300882), respectively.34,35 While previous studies have robustly supported the association between abnormal brain development and Hdac3 defects in mice, and although one individual has been reported,32 further evidence is needed to establish a clear gene-disease association and genotype-phenotype relationships in humans. To address this, we describe the phenotypes of six individuals with heterozygous de novo variants in HDAC3 (MIM: 605166) and characterize the functional impacts of these missense variants on HDAC activity, interactions with multi-protein complexes, and nuclear localization.
Subjects and methods
Subjects
Two individuals (individuals 1 and 2, see Table S1) were enrolled from the Rare Disease Center of Seoul National University Hospital (SNUH) cohort, Seoul, Republic of Korea.36 Three other individuals (individuals 3, 4, and 5) were ascertained from the Deciphering Developmental Disorders (DDD) study,37 while individual 6 was enrolled in the Boston Children’s Hospital (BCH) Rare Disease Cohorts Initiative.38 Appropriate informed consent was obtained from individuals in the SNUH, BCH, and DDD cohorts, including for the publication of all photographic material, if applicable. The study received approval from the Institutional Review Boards of the participating institutions (IRB no. SNUH 1406-081-588, BCH X10-04-0197) in compliance with the Declaration of Helsinki.
Sequencing analysis
Blood samples were collected from trios (proband and parents), and genomic DNA was subjected to Illumina short-read sequencing. Detailed methods for exome sequencing have been previously documented.39 Sequenced reads were aligned to the human reference genome hg38 and processed using the Exome Germline Single Sample pipeline (version 3.0.4), as provided by the WDL Analysis Research Pipelines (WARP; Broad Institute, MA, USA). Detected de novo variants in HDAC3 were subsequently validated through Sanger sequencing (Figure S1) with PCR experiments using the target-specific primers (Table S2).
Protein modeling
The protein structure of the NCoR2 DAD domain-HDAC3 complex was retrieved from the Protein DataBank (PDB: 4A69).40 Subsequent structural adjustment and three-dimensional visualization were performed using PyMOL (v.2.5.5; PyMOL Molecular Graphics System, Schrödinger Inc., New York, NY, USA).
Mutagenesis of HDAC3 variants
To generate HDAC3 variants, we performed PCR with mutagenic primers (Macrogen, Seoul, Korea; Table S2) using nPfu-Forte polymerase (Enzynomics, Daejeon, Korea). After the PCR reaction, PCR products were digested with DpnI restriction enzyme (Enzynomics, Daejeon, Korea) and incubated at 37°C for 1 h to remove the template DNA. The variant clones were then transformed into DH5α competent cells and were confirmed by Sanger sequencing (Figure S2B).
Purification of proteins using baculovirus expression system
To purify the complexes of the human NCoR1 DAD domain and HDAC3, 6 × His-tagged NCoR1 DAD domain and FLAG-tagged HDAC3 were cloned into the baculovirus expression vector, pFastBac1 (Invitrogen, Cat. 10-360-014). The NCoR1 DAD domain, HDAC3 wild type (WT), or variants were co-expressed in Sf9 cells (Life Technologies, Cat. 11496-015) through baculovirus infection (Figure S2C). After 64 h of infection, Sf9 cells were harvested in BC350 buffer (20 mM HEPES-NaCl at pH 7.8, 350 mM NaCl, 10% glycerol, 0.1% NP-40, and 1 mM EDTA) with protease inhibitors (1 mM phenylmethylsulfonyl fluoride [PMSF], 0.1 mM Benzamidine hydrochloride, 1.25 mg/mL Leupeptin, and 0.625 mg/mL Pepstatin A) and phosphatase inhibitors (20 mM NaF, 1 mM Na3VO4). Cells were lysed by sonication, and the WT or variant recombinants were purified by Ni-NTA Agarose (QIAGEN) and Anti-FLAG M2 Affinity Gel (Sigma). The NCoR1 DAD domain (Clone ID: KU017294) and HDAC3 plasmids (Clone ID: hMU006316) were provided by the Korea Human Gene Bank (Medical Genomics Research Center, KRIBB, Korea).
HDAC assay
HDAC assays were performed with HDAC buffer (10 mM Tris-HCl at pH 8.0, 150 mM NaCl, and 10% glycerol), recombinant H3K27-acetylated mononucleosomes or recombinant H3K4,9,14,18-/H4K5,8,12,16-acetylated mononucleosomes (EpiCypher), and recombinant human NCoR1 DAD domain-HDAC3 complexes. The reaction proceeded for 60 min at 30°C and stopped with the addition of 4 μL of sample buffer (50 mM Tris-HCl at pH 6.8, 2% sodium dodecyl sulfate [SDS], 6% glycerol, 0.004% bromophenol blue, and 5% β-mercaptoethanol). HDAC activities were measured with H3K27ac, H3ac, and H4ac antibodies (Table S3).
Cell culture, lentiviral production and transduction, and lysate preparation
Human embryonic kidney (HEK) 293T cells (Cat. CRL-3216) were obtained from the American Type Culture Collection (ATCC, USA). HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Biowest) supplemented with 5% (v/v) fetal bovine serum (FBS) and 1× Antibiotic-Antimycotic (Gibco) in a humidified atmosphere of 5% CO2 at 37°C. HDAC3 WT or variant plasmids were subcloned into the pLV-EF1α-IRES-Puro vector (Addgene, Cat. 85132) for lentiviral production. Lentiviral vectors containing HDAC3 WT or variants at a concentration of 10 μg each were co-transfected with 2.5 μg of pcREV, 3 μg of BH10, and 5 μg of vesicular stomatitis virus G (VSVG) packaging vector into HEK293T cells. The viral supernatant was harvested 48 h after transfection and used to infect target cells, which were plated in 6-well plates. Infected cells were selected with 2 μg/ml puromycin for 5 days, and the medium containing puromycin was refreshed daily. To prepare cell lysates, 8.8 × 106 cells were washed with ice-cold phosphate-buffered saline (PBS) and subsequently lysed using RIPA buffer (50 mM Tris-HCl at pH 8.0, 400 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS) along with protease inhibitors, as well as phosphatase inhibitors. The cells were subjected to sonication for lysis and subsequently clarified by centrifugation at 13,000 rpm at 4°C for 20 min. The total protein concentration was measured by Pierce BCA Assay Kit (Thermo Fisher Scientific).
Western blotting
Cell lysates were prepared by quantifying and normalizing protein concentrations, followed by denaturation of 10–15 μg of total protein in sample buffer (50 mM Tris-HCl, pH 6.8, 2% SDS, 6% glycerol, 0.004% bromophenol blue, and 5% β-mercaptoethanol) at 95°C for 5 min. The samples were then resolved by SDS-PAGE, running initially at 100 V for 15 min and subsequently at 120 V for 75 min. Proteins were transferred onto polyvinylidene fluoride (PVDF) membranes (Merck Millipore), which were subsequently blocked with 5% skim milk in TBS containing 0.1% Tween 20 (TBS-T) at room temperature (RT) for 1 h. The membranes were incubated overnight at 4°C with the primary antibodies. Following primary incubation, membranes were rinsed three times for 10 min each with TBS-T at RT. Next, the membranes were probed with horseradish peroxidase (HRP)-conjugated secondary antibodies at RT for 1 h. After secondary antibody incubation, the membranes underwent three additional 5-min washes with TBS-T. Detection was performed using an enhanced chemiluminescent substrate (Thermo Fisher Scientific). The antibodies utilized are specified in Table S3.
Co-immunoprecipitation (Co-IP)
A total of 1.5 mg of cell lysate was prepared with BC150 buffer composed of 20 mM HEPES-NaCl, pH 7.8, 150 mM NaCl, 10% glycerol, 0.1% NP-40, and 1 mM EDTA, supplemented with protease inhibitors and phosphatase inhibitors. The lysate was then incubated with 50 μL of Anti-FLAG M2 Affinity Gel (Sigma) at 4°C for 3 h. After incubation, the gel was washed three times with BC150 buffer at 4°C for 10 min per wash. Following the final wash, the complexes were eluted using 0.2 mg/ml 3 × FLAG peptide at 4°C for 1 h. The eluted proteins were resolved on SDS-PAGE gels, which were subsequently either silver stained or electro-transferred onto 0.45 μm PVDF membranes (Merck Millipore) for western blot analysis.
Silver staining
The SDS-PAGE gel was fixed using a fixative solution containing methanol. The gel was immersed in this solution at RT for 20 min; the procedure was repeated twice and subsequently rinsed twice with distilled water for 10 min each. The gel was then stained with a solution containing NaOH, ammonium hydroxide, and silver nitrate (Chemicals Duksan, Incheon, Korea) at RT for 15 min. Following the staining, the gel was rinsed twice with distilled water and developed in a solution of citric acid and formaldehyde at RT for a duration ranging from 2 to 20 min, depending on the desired intensity of staining.
Mass-spectrometry (MS) analysis
Proteomic analysis was performed through an optimized protocol integrating filter-aided sample preparation and StageTip desalting, as previously established.41 Protein samples were initially denatured using a buffer containing 2% SDS, 50 mM chloroacetamide, and 10 mM tris(2-carboxyethyl)phosphine hydrochloride in a 0.1 M Tris-HCl solution (pH 8.5). The mixture underwent reduction and alkylation processes at 95°C for 15 min. Digestion was facilitated overnight at 37°C with a trypsin/Lys-C mix at a 1:100 protease-to-protein mass ratio. Post-digestion, peptides were acidified with 10% trifluoroacetic acid and purified using StageTip columns, with styrene-divinylbenzene-reverse phase sulfonate as the adsorbent.
The peptides were then separated and analyzed on a Q-Exactive HF-X mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) coupled with an Ultimate 3000 RSLCnano system (Dionex, Sunnyvale, CA, USA).42 A comprehensive dual-column setup, comprising a C18 trapping column and an EASY-Spray C18 analytical column, was utilized for peptide separation. A 120-min gradient, ranging from 5% to 40% acetonitrile, was employed at a 300 nL/min flow rate. MS detection was operated in positive ion mode, performing an initial full scan across an m/z range of 300 to 1,800 at 60,000 resolutions, followed by MS/MS scans at 15,000 resolutions for the top 15 precursor ions, selected within a 1.6 m/z isolation window and fragmented with a normalized collision energy of 30%.
Data processing was conducted using MaxQuant (v.2.2.0.0; Max Planck Institute of Biochemistry, Munich, Germany),43 with MS/MS spectra matched against the Human UniProtKB/Swiss-Prot database (June 2023 release; Homo sapiens, 20,423 entries) complemented by four HDAC3 variant proteins and common contaminants. A 6-ppm tolerance was applied to the precursor ion, with 20 ppm for MS/MS ions. Fixed and variable modifications included carbamidomethylation of cysteine and, respectively, N-terminal acetylation and methionine oxidation. A stringent 1% false discovery rate was maintained for all peptide and protein identifications. Label-free quantification leveraged the intensity-based absolute quantification (iBAQ) algorithm,44 facilitating robust absolute quantification within the MaxQuant framework (Table S4).
Immunofluorescence staining
HEK293T cells were seeded onto Cell Culture Slide I (SPL, Gyeonggi-do, Korea). The cells were fixed with 4% paraformaldehyde (v/v) for 30 min and then permeabilized with 0.1% Triton X-100 for 20 min. Nonspecific antibody binding sites were blocked by incubation with 1% bovine serum albumin in PBS for 20 min. The cells were then incubated overnight at 4°C with a primary antibody specific to FLAG M2, followed by washing with PBS three times. An anti-mouse IgG-Alexa Fluor 488 secondary antibody was added to the cells and incubated for 2 h at RT. Cell culture slides were then washed and mounted by applying Antifade Mounting Medium (Vectashield). Fluorescence images were acquired using a confocal laser-scanning microscope and software (FV31S-SW v.2.3), with a 60× objective (Olympus FV3000, Tokyo, Japan). 4',6-diamidino-2-phenylindole (DAPI) staining was performed for nuclear staining.
Statistical analysis
Our analysis employed two-sample t tests to compare protein interaction alterations between HDAC3 WT and variant proteins. Significance was assigned to proteins displaying a minimum fold change (FC) of 1.5 coupled with a p value less than 0.05. We utilized Perseus software (v.2.0.11) for the statistical analysis of the MS data.45 To discern potential interaction partners, we processed the iBAQ intensities through a log2 transformation. Proteins were considered for further analysis if they exhibited at least two-thirds valid data points within each experimental condition. We addressed missing values using the deterministic minimal value (MinDet) imputation method,46 which aids in mitigating the bias that can arise from non-random data absence. ImageJ (v.1.54) was used for the quantification of western blot and analysis of immunofluorescent images. Statistical analysis was performed using GraphPad Prism (v.9.5.0).
Results
Clinical presentations in individuals with HDAC3 variants
We first identified two unrelated females with de novo variants in HDAC3 (GenBank: NM_003883.4) who were previously undiagnosed by exome sequencing and presented with neurodevelopmental problems with additional syndromic features among 2,510 individuals enrolled in the rare disease cohort.36 The identified variants, c.328G>A (p.Ala110Thr) and c.1075C>T (p.Arg359Cys), were validated through Sanger sequencing as depicted in Figure S1. Further analysis of publicly available databases revealed three additional heterozygous de novo HDAC3 variants, c.277G>A (p.Asp93Asn), c.797T>C (p.Leu266Ser), and c.799G>A (p.Gly267Ser), among 13,451 individuals enrolled in the DDD cohort.37 We also found a proband with c.601C>T (p.Pro201Ser) variant in an epilepsy cohort comprising 522 individuals.38 Consequently, six different missense variants were found, as outlined in Table 1; all variants occurred as de novo, were predicted to be functionally deleterious by multiple in silico tools (Table S1), and are located at evolutionarily conserved sites in vertebrates (Figure 1A), thus classified as likely pathogenic variants according to the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) criteria.47
Table 1.
Clinical findings in individuals with heterozygous de novo HDAC3 variants
| Proband | Individual 1 | Individual 2 | Individual 3 | Individual 4 | Individual 5 | Individual 6 |
|---|---|---|---|---|---|---|
| mRNA (GenBank: NM_003883.4) | c.328G>A | c.1075C>T | c.277G>A | c.797T>C | c.799G>A | c.601C>T |
| Protein (GenBank: NP_003874.2) | p.Ala110Thr | p.Arg359Cys | p.Asp93Asn | p.Leu266Ser | p.Gly267Ser | p.Pro201Ser |
| Ancestry | Asian | Asian | European | European | European | European |
| Sex | female | female | male | female | male | male |
| Age at last evaluation | 25 years | 5 years | 6 years | 12 years | 15 years | 12 years |
| Gestational age at delivery | full-term, cesarean delivery | 36 weeks, vaginal delivery | 40 weeks, vaginal delivery | not available | 35 weeks, vaginal delivery (dizygotic twin) | full-term, vaginal delivery |
| Birth weight | 1.98 kg | 2.10 kg | 3.30 kg | not available | 2.04 kg | 4.17 kg |
| Intellectual disability | severe | severe | moderate | mild | moderate | no |
| Neurodevelopmental delay | yes | yes | yes | yes | yes | no |
| Musculoskeletal abnormalities | scoliosis, varus of the proximal tibia, hand joint deformity | polydactyly, syndactyly | no | joint hypermobility | hand joint deformity | neonatal torticollis |
| Seizure | yes | no | yes | no | no | yes |
| Facial dysmorphism | yes | yes | no | no | yes | no |
| Brain MRI findings | no specific findings | no specific findings | subcortical heterotopia | focus of nodular heterotopia | mildly dilated vestibular ducts | no specific findings |
| Genitourinary anomaly | no | congenital hydronephrosis | phimosis | no | congenital unilateral right hydronephrosis with vesicoureteral reflux | no |
| Microcephaly | yes | yes | no | no | no | no |
| Hearing impairment | sensorineural type | no | no | no | mixed type | no |
| Failure to thrive | yes | yes | no | no | no | no |
| Congenital heart disease | yes | yes | no | no | no | no |
| Autistic behavior | yes | no | yes | no | no | no |
| Other findings | microtia | – | – | subcutaneous scalp arteriovenous malformation | type 1 diabetes, chronic acral warts | – |
Figure 1.

Locus conservation, phenotypic characteristics, and three-dimensional location on the protein structure of HDAC3 variants
(A) Genomic locations of identified variants. All variants are missense and situated in highly conserved regions across vertebrate species. The blue bar indicates the presence of the nuclear localization signal (NLS) domain.
(B) Phenotypic characteristics of individuals 1 and 2 carrying the p.Ala110Thr and p.Arg359Cys variants, respectively. Both females exhibited facial dysmorphism, profound microcephaly with no abnormal brain MRI findings, and skeletal abnormalities such as joint stiffness, scoliosis, or polysyndactyly (yellow triangle).
(C) Three-dimensional structure of the bound form of NCoR2 DAD domain-HDAC3 complex and variant locations on HDAC3 protein (modified from PDB: 4A69). This panel shows the three-dimensional positioning of HDAC3 variants on the protein structure, highlighting the responsible amino acid residues in magenta.
Individual 1, who harbors the p.Ala110Thr variant, has two older sisters with no reported developmental problems. She exhibited various syndromic features, including facial dysmorphism, profound primary microcephaly (Z score −7.0), microphthalmia, sensorineural hearing loss, and short stature. She also underwent cardiac surgery for an atrial septal defect. X-ray imaging revealed several skeletal anomalies, including early epiphyseal fusion of the middle phalanx of the fifth finger and scoliosis (Figure 1B, upper). Her growth was severely retarded, and she experienced generalized seizures; electroencephalogram (EEG) findings showed a few spikes or spike-and-wave discharges in the left frontal lobe. Moreover, she exhibited severe intellectual disability and autistic behaviors, including hand stereotypy, while no anatomical abnormalities were detected in brain magnetic resonance imaging (MRI) findings. Individual 2, with the p.Arg359Cys variant, was born as the third baby. The first and second children, who are dizygotic twins, had no medical issues. In contrast, she received neonatal intensive care after birth for more than a month due to laryngomalacia and feeding difficulties. This individual exhibited facial dysmorphism with broad thumbs and fingers; a small, retracted chin; low-set ears; and hypertrichosis (Figure 1B, lower). Rubinstein-Taybi syndrome (MIM: 180849) was clinically suspected, but the sequencing of CREBBP (MIM: 600140) and EP300 (MIM: 602700) failed to detect causative variants. She exhibited primary microcephaly (Z score −5.1), global developmental delay, short stature, and failure to thrive. Furthermore, congenital hydronephrosis and limb anomalies were observed, including complete syndactyly on the fourth web of the left hand, partial syndactyly on the third web of both hands, polysyndactyly on the left fifth toe, and bilateral triangular shape of the proximal phalanx on the toe. Brain MRI findings revealed no structural anomalies.
Similarly, individual 3, harboring the p.Asp93Asn variant, initially presented at 16 months of age with delayed speech and language development, delayed motor development, and phimosis. Over the following years, this individual additionally showed a moderate intellectual disability, autistic features, and epilepsy. A brain MRI study revealed subcortical heterotopia, with otherwise normal findings. Compared to the previous individuals, individual 4, possessing the p.Leu266Ser variant, exhibited relatively milder phenotypes, although the brain MRI also indicated nodular heterotopia. She exhibited mild intellectual disability, subcutaneous scalp arteriovenous malformation, and joint hypermobility. Individual 5, with the p.Gly267Ser variant, is a 15-year-old male and a fraternal twin, with his twin sibling being unaffected. He has presented multiple syndromic clinical features, including facial dysmorphism and mixed-type hearing loss, and he recently developed type 1 diabetes. He also exhibited renal pelviectasis both antenatally and postnatally and had unilateral hydronephrosis associated with vesicoureteral reflux, evolving into an atrophic non-functioning kidney. His brain MRIs revealed mildly dilated vestibular ducts. Lastly, individual 6 with the p.Pro201Ser variant exhibited mild or no notable abnormalities in developmental progress. He reported no perinatal issues except for neonatal torticollis. He is currently a 12-year-old boy with normal intelligence but has experienced two unprovoked seizures, one of which was noticed at the age of two. EEG monitoring has shown generalized spikes, indicating high-risk recurrent seizures.
Overall, the clinical presentations of individuals with de novo HDAC3 variants vary, and they were frequently associated with a wide range of neurodevelopmental conditions (Table 2). Intellectual disability and neurodevelopmental delay were the most common conditions (83.3%), followed by musculoskeletal abnormalities (66.7%). Additionally, seizure, facial dysmorphism, abnormalities of the genitourinary system, and brain imaging abnormalities were observed in half of this cohort. Other associated phenotypes include microcephaly, hearing impairments, congenital heart disease, and autistic behavior, each observed in two individuals. Furthermore, an endocrinological issue, such as type 2 diabetes, was noted in a single individual, indicating the systemic involvement of HDAC3-related disorders.
Table 2.
Summary of clinical features in this cohort
| Clinical findings (HPO identifier) | Frequency | % |
|---|---|---|
| Ancestry | 2 Asian, 4 European | – |
| Sex | 3 male, 3 female | – |
| De novo inheritance (HP:0025352) | 6/6 | 100.0 |
| Intellectual disability (HP:0001249) | 5/6 | 83.3 |
| Neurodevelopmental delay (HP:0012758) | 5/6 | 83.3 |
| Abnormality of the musculoskeletal system (HP:0033127) | 4/6 | 66.7 |
| Seizure (HP:0001250) | 3/6 | 50.0 |
| Abnormal facial shape (HP:0001999) | 3/6 | 50.0 |
| Brain imaging abnormality (HP:0410263) | 3/6 | 50.0 |
| Abnormality of the genitourinary system (HP:0000119) | 3/6 | 50.0 |
| Microcephaly (HP:0000252) | 2/6 | 33.3 |
| Hearing impairment (HP:0000365) | 2/6 | 33.3 |
| Failure to thrive (HP:0001508) | 2/6 | 33.3 |
| Abnormal heart morphology (HP:0001627) | 2/6 | 33.3 |
| Autistic behavior (HP:0000729) | 2/6 | 33.3 |
| Abnormality of the endocrine system (HP:0000818) | 1/6 | 16.7 |
Abbreviation: HPO, human phenotype ontology.
Changes in the HDAC activities
Next, we reviewed the published structure of the NCoR2 DAD-HDAC3 complex (PDB: 4A69) to determine the location of each variant (Figure 1C).40 This enabled us to categorize the variants based on their proximity to the catalytic pocket (His134 and His135) of HDAC3. The residues Asp93, Pro201, Leu266, and Gly267 were found near the catalytic pocket, whereas the residues Ala110 and Arg359 were positioned distant from the catalytic center. Following this classification, we investigated the impact of HDAC3 variants on HDAC activity in the cellular context by over-expressing either HDAC3 WT or four selected variants (p.Ala110Thr, p.Leu266Ser, p.Gly267Ser, and p.Arg359Cys) in HEK293T cells using lentivirus expression system (Figure 2A). Through the expression system, we noted that the protein accumulation of HDAC3 WT and its variant forms were comparable. Additionally, there were no significant alterations in the accumulation of NCoR1 and NCoR2, which are crucial proteins interacting with HDAC3 and necessary for its activity, across the samples. The p.Leu266Ser and p.Gly267Ser variants, which are located near the catalytic pocket, exhibited higher levels of acetylated histones (H3K9ac, H3K27ac, and H3ac) compared to the WT counterparts (Figure 2B). These findings suggest that the missense variants close to locating the catalytic pocket substantially impair HDAC activity. In contrast, the p.Ala110Thr and p.Arg359Cys variants did not exhibit noticeable defects in overall HDAC activities.
Figure 2.

HDAC3 variants near catalytic sites display deficient HDAC activity
(A) Western blot analyses were conducted on HEK293T cells transduced with either empty vector (EV), FLAG-tagged wild-type (WT) HDAC3, or selected HDAC3 variants (p.Ala110Thr, p.Gly267Ser, p.Leu266Ser, and p.Arg359Cys) using a lentiviral expression system. The detection of HDAC3; FLAG-HDAC3; acetylated histones H3K9, H3K27, and H3; total H3; NCoR1; NCoR2; and GAPDH (loading control) was visualized. Notably, increased acetylation levels of H3K9, H3K27, and H3 were observed in p.Gly267Ser and p.Leu266Ser variants (red dashed rectangle), indicating a defective deacetylation function.
(B) Quantitative analysis of acetylation levels at histone sites H3K9, H3K27, and H3 from (A). The p.Gly267Ser and p.Leu266Ser variants show increased acetylation levels compared to the WT at H3K9, H3K27, and H3, indicating impaired histone deacetylation function. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, n = 3/data point. Data are plotted as mean ± SD.
(C) HDAC assays were performed using H3K27ac-mononucleosomes or H3/H4Kac-mononucleosomes as substrates at various concentrations (0, 60, 120 nM) in conjunction with 100 nM of acetylated mononucleosomes. The deacetylation activities of complexes comprising either the NCoR1 DAD domain-HDAC3 WT or variant forms were measured for histone H3K27ac, H3ac, and H4ac in triplicate (n = 3/data point). The results are plotted as mean ± SD. The p.Gly267Ser and p.Leu266Ser variants do not result in histone acetylation levels, indicating defective HDAC activity. In contrast, the p.Ala110Thr and p.Arg359Cys variants retain deacetylation activities comparable to the WT form.
To further validate whether the HDAC3 variants directly influence HDAC activity, we purified WT and variant HDAC3 proteins in combination with the NCoR1 DAD domain, which is the minimal requirement for optimal HDAC activity, using a baculovirus expression system.12,13 Although the interaction with the full-length NCoR1 may have defects in the cell (see proteomic analysis using Co-IP), the HDAC3 variants did not exhibit any defects in their interaction with the NCoR1 DAD domain (Figure S2). Subsequently, we performed HDAC assays in triplicate using either mono-nucleosomes acetylated at H3K27 or mono-nucleosomes with acetylation on H3 and H4 (Figures 2C, S3, and S4). Consistent with the findings from the cellular experiments, the combination of the NCoR1 DAD domain with variants near catalytic sites (p.Asp93Asn, p.Pro201Ser, p.Leu266Ser, and p.Gly267Ser) displayed a notable impairment in HDAC activity. Similarly, when the NCoR1 DAD domain was paired with either p.Ala110Thr or p.Arg359Cys, no defects in HDAC activity were observed. Taken together, HDAC3 variants near catalytic sites (p.Asp93Asn, p.Pro201Ser, p.Leu266Ser, and p.Gly267Ser) exhibited significantly dampened HDAC activity, while p.Ala110Thr and p.Arg359Cys variants did not.
Proteomic analysis using Co-IP
To further delineate the underlying mechanism for the HDAC3 variants with retained HDAC activity, specifically for p.Ala110Thr and p.Arg359Cys, we explored potential changes in protein-protein interactions using Co-IP. Proteomic analysis was conducted on co-immunoprecipitated samples from HEK293T cells expressing HDAC3 WT and the four selected variants (p.Ala110Thr, p.Leu266Ser, p.Gly267Ser, and p.Arg359Cys). We normalized protein abundance to WT levels by using FC and iBAQ as a quantitative measurement (Table S4). Volcano plot analysis (Figure 3A) of HDAC3 variants revealed decreased protein interactions (highlighted in blue) with NCoR1/2, TBL1X/Y, GPS2, KDM1A, RCOR1/3, and GSE1, which are associated with the NCoR and CoREST complexes (Figure 3B).10 Conversely, proteins such as PSMA7/8, PSMC1, and PSMD14, involved in proteasome degradation pathways, exhibited increased interaction with HDAC3 variants (highlighted in red). Overall, the heatmap presentations of interactome changes show decreased protein interactions with molecules participating in the NCoR and CoREST complexes and decreased protein interactions with molecules involved in proteasome degradation pathways (Figure 3C). Notably, a universal disruption with KDM1A interaction was observed across all HDAC3 variants, implicating a critical role of the CoREST complex integrity in the disease mechanism. Furthermore, attenuation of the NCoR complex association was significant in the p.Ala110Thr, p.Leu266Ser, and p.Gly267Ser variants. In contrast, common upregulation of the proteasome pathways suggested that the HDAC3 variants may be more susceptible to proteasome-mediated degradation due to their impaired interactions with key multi-protein complexes. Remarkably, quantified protein levels in iBAQ intensity showed a substantial decrease in NCoR1/2 and KDM1A (Figure 3D), which further corroborated previous results. Moreover, pathway enrichment analysis focused on consistently reduced or increased protein interactions revealed several interesting Gene Ontology (GO) terms (Figure S5). Specifically, there was a significant reduction in interactions with proteins involved in histone deacetylation (GO: 0016575) and glial cell differentiation (GO: 0010001), while interactions with ATP-dependent protein folding chaperones were significantly increased (GO: 0140662).
Figure 3.

Proteomic analysis using co-immunoprecipitation reveals reduced interactions between HDAC3 variants and the subunits of NCoR and CoREST complexes
(A) Volcano plots of protein interaction profiles for four selected HDAC3 variants (p.Ala110Thr, p.Gly267Ser, p.Leu266Ser, and p.Arg359Cys) compared to HDAC3 wild-type (WT) in HEK293T cells. The plots highlight important proteins involved in the NCoR and CoREST complexes or the proteasome pathway, with the x axis representing the log2 fold change (FC) and the y axis showing the −log10p value (measured in triplicate for each data point).
(B) Schematic presentation of the NCoR and CoREST complexes, with the position of HDAC3 and its interacting subunits within each complex.
(C) A heatmap summarizes the log2 FC in protein interactions for HDAC3 variants relative to WT. Each column represents a distinct protein, as highlighted in (A), and each row corresponds to the four tested HDAC3 variants. Blue shades denote decreased, and red shades indicate increased interaction strength. The data show decreased interactions with subunits of the NCoR and CoREST complexes and contrasted with an increased interaction tendency with subunits of the proteasome pathway, illustrating variant-specific effects on HDAC3 function and complex integrity.
(D) Bar graphs display quantified protein levels using the intensity-based absolute quantification (iBAQ) method for three major proteins: NCoR1, NCoR2, and KDM1A. The data reveal a consistent reduction in association with the HDAC3 variants for these proteins. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001. Data are plotted as mean ± SD (n = 3/data point).
To validate these proteomic findings, we employed silver staining and western blot analyses. Immunoprecipitation assays in HEK293T cells expressing either HDAC3 WT or variants delineated distinct protein interaction profiles visualized by silver staining (Figures 4A and S4). In particular, the p.Ala110Thr variant displayed a diminished band correlating with the NCoR complex interaction, and a universally reduced interaction with KDM1A was evident across all variants. Western blot analysis (Figures 4B, 4C, and S4) further substantiated the significant disruption of the p.Ala110Thr variant’s interactions with full-length NCoR1 and NCoR2. In addition, the p.Leu266Ser variant presented more severe impairments than the p.Gly267Ser variant with these proteins, consistent with our proteomic analysis (Figure 3D). Furthermore, all HDAC3 variants showed diminished interactions with KDM1A. Collectively, our experiments demonstrate that HDAC3 variants perturb interactions with the CoREST and NCoR complexes, particularly involving KDM1A and NCoR1/2, which we posit as a central pathogenic mechanism underlying the clinical findings observed in the affected individuals.
Figure 4.

Impaired interactions of HDAC3 variants with NCoR1/2 and KDM1A
(A) Silver-stained SDS-PAGE analysis demonstrating the protein complexes co-immunoprecipitated (IP) with FLAG-tagged HDAC3 from HEK293T cells. Tested conditions include an empty vector (EV), wild type (WT), and HDAC3 variants (p.Ala110Thr, p.Gly267Ser, p.Leu266Ser, and p.Arg359Cys). The band intensities corresponding to the NCoR complex (indicated by red arrow) and KDM1A (indicated by blue arrow) are reduced in the variant forms. Specifically, the p.Ala110Thr shows a remarkable decrease in NCoR complex band intensity (red rectangle), and all variants exhibit diminished KDM1A bands (blue rectangle).
(B) Western blot analyses confirm the differential co-immunoprecipitation of NCoR1, NCoR2, and KDM1A with HDAC3 variants, using an anti-FLAG antibody for immunoprecipitation. FLAG-tagged HDAC3 and GAPDH serve as a reference for protein levels and loading control, respectively.
(C) Quantification of co-immunoprecipitated NCoR1, NCoR2, and KDM1A, normalized to the WT HDAC3 levels, based on the Western blot data in (B). This panel quantitatively depicts the significant interaction deficits in the p.Ala110Thr and p.Arg359Cys variants with NCoR1, NCoR2, and KDM1A, despite these variants retaining deacetylase activity comparable to WT. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001. Data are plotted as mean ± SD (n = 3/data point).
Diminished nuclear localization
Next, we investigated the localization of HDAC3 within HEK293T cells using fluorescence microscopy to assess its subcellular distribution (Figure 5A). The localization of WT or variant forms of HDAC3 protein was detected with an anti-FLAG antibody, while DAPI staining was employed to outline the nuclear boundary. HDAC3 was predominantly localized in the nucleus in cells expressing the WT, whereas cells expressing the variants displayed a discernible reduction in nuclear localization. Quantitative analysis of immunofluorescent intensities, represented as the nuclear-to-cytoplasmic (N/C) ratio, revealed a significant decrease in nuclear signal for the p.Ala110Thr, p.Leu266Ser, and p.Arg359Cys variants (median N/C ratios: 0.67, 0.92, and 1.55, respectively) compared to the WT cells, which had a median N/C ratio of 2.03 (Figure 5B). This reduction is consistent with our proteomic data (Figure 3), which showed diminished interactions with components of the NCoR complex, potentially leading to decreased nuclear stability and increased proteasome degradation, particularly for the p.Ala110Thr and p.Leu266Ser variants. Moreover, the p.Arg359Cys variant lies within the nuclear localization signal (NLS) sequence (amino acids 313–428 of HDAC3).48,49 The p.Arg359Cys variant could therefore undermine nuclear localization, resulting in a lower N/C ratio compared to the WT cells. Furthermore, the p.Gly267Ser variant, with a median N/C ratio of 1.95, exhibits a nuclear localization similar to the WT, although the broad distribution within the cell population may indicate small portions of mildly defective cells. These findings not only emphasize the potential pathogenicity of HDAC3 variants but also highlight protein mislocalization as a critical mechanism, elucidating the consequences of impaired protein interactions within cellular compartments.
Figure 5.

Differential nuclear localization of HDAC3 variants in HEK293T cells
(A) Immunofluorescence analysis of HEK293T cells transduced with wild-type (WT) or HDAC3 variants (p.Ala110Thr, p.Gly267Ser, p.Leu266Ser, and p.Arg359Cys). Cells were stained with an anti-FLAG antibody to visualize HDAC3 or its variants (green) and with DAPI to mark DNA (blue). HDAC3 WT prominently localizes in the nucleus, as shown by intense green fluorescence. In contrast, the p.Ala110Thr and p.Leu266Ser variants show notably weaker nuclear fluorescence and relatively stronger cytoplasmic fluorescence, implying compromised nuclear localization of HDAC3. In addition, the p.Gly267Ser variant exhibits relatively conserved nuclear localization similar to the WT, although the overall nuclear fluorescent intensity was weaker than the WT.
(B) Quantitative assessment of nuclear localization. This panel presents the quantification of nuclear versus cytoplasmic fluorescence intensity, expressed as the nuclear-to-cytoplasmic (N/C) ratio, in cells transduced with WT or variant forms of HDAC3. Individual data points correspond to measurements from single cells, with the median value of each variant depicted by a horizontal red line (cell counts of approximately 30–50 cells were used for each condition in the analysis). The N/C ratios for p.Ala110Thr, p.Leu266Ser, and p.Arg359Cys variants are reduced in comparison to the WT, indicating a deficiency in nuclear localization. Conversely, the p.Gly267Ser variant exhibits an N/C ratio similar to the WT, though the broad spread of data points indicates variable localization within the cell population. Statistical significance is indicated by asterisks (∗∗∗∗p < 0.0001).
Discussion
This study investigated the association between HDAC3 dysfunction and a wide spectrum of neurodevelopmental disorders in six individuals who displayed variable clinical presentations, such as intellectual disability, developmental delay, skeletal abnormalities, and epilepsy. We observed that all six variants were de novo missense variants located at evolutionarily conserved sites. Our functional analyses (Table S5) revealed that the p.Asp93Asn, p.Pro201Ser, p.Leu266Ser, and p.Gly267Ser variants, located near the catalytic pocket, have defects in HDAC activities. In contrast, the p.Ala110Thr and p.Arg359Cys variants, located away from the catalytic pocket, retained intact HDAC activities with the NCoR DAD domain. Further exploration using co-IP revealed a consistent disruption of interaction with KDM1A, a key molecule of the CoREST complex, across all variants. Moreover, disruption in interactions with the NCoR complex was also observed in the p.Asp93Asn, p.Ala110Thr, p.Pro201Ser, p.Leu266Ser, and p.Gly267Ser variants, but not in the p.Arg359Cys variant, indicating that variant position may have different effects on downstream pathways. Furthermore, immunofluorescence analyses also identified that the p.Ala110Thr, p.Leu266Ser, and p.Arg359Cys variants led to protein mislocalization, highlighting the importance of nuclear localization for the biological function of HDAC3. The p.Ala110Thr and p.Leu266Ser variants, which remarkably compromise complex integrity, may be more prone to ubiquitin degradation machinery, resulting in diminished levels of HDAC3 within the nucleus.
In our cohort, the clinical profiles and disease severity of each individual varied widely (Table 1). The predominant clinical findings in our cohort include intellectual disability (83.3%), musculoskeletal abnormalities (66.7%), and epilepsy (50.0%; Table 2). These symptoms are consistent with those evidenced in previous studies on mice with conditional Hdac3 knockout that displayed neurodevelopmental21,22,23 and musculoskeletal28,29 phenotypes. Individuals 1 and 2 presented with severe symptoms, including syndromic features such as facial dysmorphism, microcephaly, congenital heart defects, and hand and foot deformities similar to Kabuki syndrome (MIM:147920; Figure 1B). The essential role of HDAC3 in myocardial growth, as reported in a previous study, may explain the congenital heart disease observed in these two individuals.24 The p.Ala110Thr and p.Arg359Cys variants found in these individuals are located away from the catalytic pocket and exhibited no defects in HDAC activity when paired with the NCoR1 DAD domain. However, the Ala110 residue near the NCoR2 DAD domain interface (Figure 1C) made the p.Ala110Thr variant significantly defective in interacting with the NCoR complex, as shown by biochemical and proteomic experiments (Figure 3). Additionally, the interaction between the Arg359 and His234 residues is potentially crucial for alpha-helix interactions and the stability of the complex, as well as other protein interactions such as KDM1A. Further structural studies are warranted to elucidate the detailed mechanism.
In contrast, individuals 3 and 4 presented with gray matter heterotopia, indicating neuronal migration defects as observed in their brain MRI findings. This phenotypic association aligns with observations in Hdac3 conditional knockout mice, highlighting the role of HDAC3 in neuronal migration.23 Specifically, the p.Asp93Asn and p.Leu266Ser variants, which exhibited significant defects in HDAC activities, were positioned very close to the catalytic site (Figure 1C). Individuals 5 and 6 also had HDAC3 variants with defective HDAC activities but did not exhibit gray matter heterotopia. The p.Gly267Ser and p.Pro201Ser variants, though slightly distanced from the catalytic site compared to the previous ones, showed more severe defects in interactions with NCoR1/2 and KDM1A. Remarkably, individual 5 had a relatively severe clinical profile, including hearing impairment, and recently developed diabetes. In contrast, individual 6, with the p.Pro201Ser variant, exhibited the mildest phenotypes among the cohort. Hearing loss was observed in two individuals: individual 1 (sensorineural hearing loss with microtia) and individual 5 (mixed hearing loss with dilatated vestibular ducts revealed by brain MRI). These phenotypes may result from HDAC3 dysfunction, as knockdown of hdac3 in zebrafish showed reduced numbers of sensory hair cells and disrupted auditory organ development.50 Furthermore, diabetes in individual 5 may also be associated with HDAC3 dysfunction; a previous study demonstrated that β-cell-specific Hdac3 depletion in mice results in decreased pancreatic insulin content, disrupted insulin secretion, and increased susceptibility to streptozotocin-induced diabetes.51
We made efforts to find additional cases related to HDAC3 variants in publicly available databases. This search led us to identify two missense variants, c.893A>G (p.Tyr298Cys; SCV000742177.3) and c.902G>A (p.Arg301Gln; SCV000740901.3), classified as likely pathogenic in the ClinVar database.52 Intellectual disability and epilepsy were both observed with the p.Tyr298Cys and p.Arg301Gln variants. In addition, the p.Tyr298Cys variant was found in a female with gait disturbance and coordination abnormalities, while the p.Arg301Gln variant was identified in a male presenting with syndromic features, including microcephaly, synophrys, agenesis of the corpus callosum, white matter abnormalities, hemiparesis, generalized limb muscle weakness, hearing impairment, and otitis media. The Tyr298 residue is located close to the catalytic pocket, while the Arg301 residue is situated at the inositol phosphate 4 (IP4) binding sites. IP4 serves as an “intermolecular glue” in the formation of the HDAC3-NCoR complex, thereby stabilizing both HDAC3 and the DAD domain of NCoR.40 The pathogenicity of these variants is further supported by previous functional studies reporting defects with different amino acid substitutions at the same residues (p.Tyr298Phe, p.Tyr298His, and p.Arg301Ala).26,53 Collectively, these two variants and the observed phenotypic information further strengthen the diverse spectrum of phenotypic associations in HDAC3 dysfunction. Depending on variant position or genetic background, the penetrance or expressivity may vary in the HDAC3-related disorder, as observed in HDAC4.54 Future investigations involving larger cohorts that explore the association between variant position and disease severity may provide further insights into the genotype-phenotype relationship.
To gain further insights into the underlying mechanisms and genotype-phenotype correlations, we also analyzed copy-number variations (CNVs) involving HDAC3 using the DECIPHER database (Figure S6).55 The probability of haploinsufficiency (pHaplo) and triplosensitivity (pTriplo) scores for HDAC3 are 0.62 and 1.00,56 respectively, indicating a significant impact of gene dosage on the disorder. CNV analysis revealed 11 individuals (Table S6): five with large deletions and six with large duplications, most suspected to be de novo. While phenotypic information was lacking for three with large duplications, the other eight demonstrated intellectual disability (6/8, 75.0%), musculoskeletal abnormalities (4/8, 50.0%), and epilepsy (4/8, 50.0%). We acknowledge that these phenotypes may be non-specific and that the potential impact of gene dosage from other genes due to the large CNV sizes cannot be discounted. Nonetheless, these findings support the phenotypic associations observed with HDAC3 missense variants.
The proteomics study revealed the pivotal role of the disrupted interaction with KDM1A in the CoREST complex (Figure 3), a shared mechanism among all tested HDAC3 variants. Notably, three individuals with de novo variants in KDM1A (MIM: 609132) have been recently described with a Mendelian disorder characterized by “cleft palate, psychomotor retardation, and distinctive facial features” (MIM: 616728).57 The clinical findings in these individuals showed similarities to Kabuki syndrome, and functional assessment of the identified KDM1A variants (GenBank: NM_001009999.3; c.1207G>A [p.Glu403Lys], c.1739A>G [p.Asp580Gly], and c.2353T>C [p.Tyr785His]) demonstrated compromised catalytic activities and altered interactions with transcription factors.58 Considering the closed phenotypic overlap between individuals 1 and 2 in our cohort and the KDM1A-associated disorder, there may be a potential link to pathways involving the CoREST complex. Another notable observation was the universally diminished interaction with AMER1 among all variants, as depicted in Figure S5B. Given that pathogenic variations in AMER1 cause “osteopathia striata with cranial sclerosis” (MIM: 300373), the diminished interaction with AMER1 may be further linked to the facial dysmorphism and skeletal abnormalities, such as scoliosis and joint contractures, observed in some individuals. Similarly, diminished interactions were detected with GANAB, AKAP8L, and EIF3B across all examined variants. Prior reports have associated the gene dosage of AKAP8L (MIM: 609475) with the microcephaly phenotype in humans,59,60 suggesting that decreased interaction with AKAP8L may also contribute to microcephaly phenotypes in our subjects. Future investigations focusing on these proteins are expected to offer a more comprehensive understanding of genotype-phenotype correlations for the HDAC3-related disorder.
To explore the mechanism of variant effects, we reviewed the probability of loss-of-function (LOF) intolerance (pLI 1.00) and the LOF observed/expected upper bound fraction (LOEUF 0.46) scores of HDAC3 in the gnomAD v.4.1.0 database.61 These indices have been updated from a pLI of 0.57 and a LOEUF of 0.41 in the previous version of the gnomAD v.2.1.1 database. The significant change in the pLI score likely reflects the inclusion of a larger and more diverse dataset in the latest gnomAD version, as well as the transition from hg19 to the hg38 reference genome, which may have improved the overall statistical power for calculating the pLI score. These indices denote that HDAC3 is a highly constrained gene, suggesting that haploinsufficiency may be a likely underlying mechanism. Our investigation of 6,099 in-house exomes identified no alleles with predicted LOF (pLOF) variants in HDAC3,36 supporting the constraint on it. However, all individuals in our cohort harbored missense variants, and no individuals with pLOF variants were discovered, suggesting that a dominant-negative (DN) mechanism might also be at play. Systematic analysis of missense variants indicates that those with DN effects tend to cluster in three-dimensional space and are highly enriched at protein interfaces, whereas LOF variants are more dispersed.62 In addition, a recent study has shown that a DN effect results in “poisoning” protein complex assembly; therefore, complexes with known DN effects tend to expose their interfaces late during translation, lessening the likelihood of cotranslational assembly.63 Our experimental data imply that key mechanisms may involve not only changes in the catalytic function of HDAC3 but also altered interactions with multi-protein complexes. Hence, the mechanisms of HDAC3-related disorders may be multifaceted, potentially involving both LOF and DN effects that depend on the variant locations and the affected protein complexes. A recent study demonstrated that protein mislocalization, mainly attributed to effects on protein stability and membrane insertion, is a primary mechanism for the pathogenicity and different disease severity of many missense variants.64 Consequently, disrupted interactions with multi-protein complexes and the mislocalization of the HDAC3 complex may play a more critical role than the defects in the catalytic function itself.
In conclusion, our study highlights the role of HDAC3 dysfunction in a broad spectrum of neurodevelopmental disorders by providing evidence from six individuals with de novo heterozygous missense variants. The experimental data demonstrated that the HDAC3 variants not only impair deacetylase function but also disrupt interactions with the CoREST and NCoR complexes, leading to protein mislocalization in a variant-specific manner. These findings shed light on the integrative functions of HDAC3 as an epigenetic regulator in human physiology. Future research with an expanded cohort will be required to solidify the association between HDAC3 variants and the spectrum of clinical presentations and to advance our understanding of the genotype-phenotype relationships.
Data and code availability
The data supporting the findings of this study are available within the article and the supplemental information or can be made available upon reasonable request to the corresponding authors.
Acknowledgments
This study was supported by the Institute of Information & Communications Technology Planning & Evaluation (IITP) grant, funded by the Korean government (MSIT) (grant numbers 2022-0-00333 and RS-2023-00223069). Also, it was supported by the National Research Foundation of Korea (grant numbers NRF2021R1C1C1013220, NRF2022R1A5A102641311, and NRF2020R1A5A1019023) and the BK21 Four Biomedical Science Program. The SNUH Kun-hee Lee Child Cancer and Rare Disease Project Foundation, Republic of Korea (grant number 22B-001-0100), the Research Resettlement Fund for the new faculty of Seoul National University, the Creative-Pioneering Researchers Program through Seoul National University, grants from Seoul National University College of Medicine, and the AI-Bio Research Grant through Seoul National University also supported this study. Furthermore, it was supported by the Fellowship for Fundamental Academic Fields provided by Seoul National University. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009-003), a parallel funding partnership between Wellcome and the UK Department of Health, and the Wellcome Sanger Institute (grant number WT098051). The views expressed in this publication are those of the author(s) and not necessarily those of Wellcome or the Department of Health. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12, granted by the Republic of Ireland REC). The research team acknowledges the support of the National Institute for Health Research through the Comprehensive Clinical Research Network. The figures were created with BioRender.com.
Author contributions
Conceptualization: J.G.Y., S.-K.L., J.-H.C., and C.-H.L.; data curation: J.G.Y., S.L., J.C., S.Y.K., H.Y.K., A.H.P., V.R., P.V., M.J.G., J.M.K., and J.-H.C.; investigation: J.G.Y., S.-K.L., H.S., H.Y.K., D.H., and C.-H.L.; supervision: J.-H.C. and C.-H.L.; writing-original draft: J.G.Y., S.-K.L., and C.-H.L. All authors reviewed and approved the final version of the manuscript.
Declaration of interests
The authors declare no conflicts of interest.
Published: July 23, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2024.06.015.
Contributor Information
Jong-Hee Chae, Email: chaeped1@snu.ac.kr.
Chul-Hwan Lee, Email: chulhwan@snu.ac.kr.
Web resources
DECIPHER, https://www.deciphergenomics.org/
HPO, https://hpo.jax.org/
ImageJ, https://github.com/imagej/ImageJ
Korea Human Gene Bank, https://genbank.kribb.re.kr/
MaxQuant, https://www.maxquant.org/
OMIM, https://www.omim.org/
PyMOL, https://pymol.org/2/
UniProt, https://www.uniprot.org/
Supplemental information
References
- 1.Kuo M.H., Allis C.D. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays. 1998;20:615–626. doi: 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 2.Yang X.-J., Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26:5310–5318. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- 3.Gallinari P., Di Marco S., Jones P., Pallaoro M., Steinkühler C. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 2007;17:195–211. doi: 10.1038/sj.cr.7310149. [DOI] [PubMed] [Google Scholar]
- 4.Chen H.P., Zhao Y.T., Zhao T.C. Histone Deacetylases and Mechanisms of Regulation of Gene Expression. Crit. Rev. Oncog. 2015;20:35–47. doi: 10.1615/CritRevOncog.2015012997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee C.-H., Wu J., Li B. Chromatin Remodelers Fine-Tune H3K36me-Directed Deacetylation of Neighbor Nucleosomes by Rpd3S. Mol. Cell. 2013;52:255–263. doi: 10.1016/j.molcel.2013.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hyun K., Jeon J., Park K., Kim J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017;49:e324. doi: 10.1038/emm.2017.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu J.-R., Lee C.-H., Oksuz O., Stafford J.M., Reinberg D. PRC2 is high maintenance. Genes Dev. 2019;33:903–935. doi: 10.1101/gad.325050.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee S.H., Li Y., Kim H., Eum S., Park K., Lee C.-H. The role of EZH1 and EZH2 in development and cancer. BMB Rep. 2022;55:595–601. doi: 10.5483/BMBRep.2022.55.12.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park S.-Y., Kim J.-S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020;52:204–212. doi: 10.1038/s12276-020-0382-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bantscheff M., Hopf C., Savitski M.M., Dittmann A., Grandi P., Michon A.-M., Schlegl J., Abraham Y., Becher I., Bergamini G., et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011;29:255–265. doi: 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- 11.Mottis A., Mouchiroud L., Auwerx J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev. 2013;27:819–835. doi: 10.1101/gad.214023.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.You S.-H., Lim H.-W., Sun Z., Broache M., Won K.-J., Lazar M.A. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat. Struct. Mol. Biol. 2013;20:182–187. doi: 10.1038/nsmb.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J., Guo C., Rood C., Zhang J. A C terminus–dependent conformational change is required for HDAC3 activation by nuclear receptor corepressors. J. Biol. Chem. 2021;297 doi: 10.1016/j.jbc.2021.101192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee M.G., Wynder C., Cooch N., Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437:432–435. doi: 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- 15.Shi Y.-J., Matson C., Lan F., Iwase S., Baba T., Shi Y. Regulation of LSD1 Histone Demethylase Activity by Its Associated Factors. Mol. Cell. 2005;19:857–864. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 16.Song Y., Dagil L., Fairall L., Robertson N., Wu M., Ragan T.J., Savva C.G., Saleh A., Morone N., Kunze M.B.A., et al. Mechanism of Crosstalk between the LSD1 Demethylase and HDAC1 Deacetylase in the CoREST Complex. Cell Rep. 2020;30:2699–2711.e8. doi: 10.1016/j.celrep.2020.01.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xue Y., Wong J., Moreno G.T., Young M.K., Côté J., Wang W. NURD, a Novel Complex with Both ATP-Dependent Chromatin-Remodeling and Histone Deacetylase Activities. Mol. Cell. 1998;2:851–861. doi: 10.1016/S1097-2765(00)80299-3. [DOI] [PubMed] [Google Scholar]
- 18.Emmett M.J., Lazar M.A. Integrative regulation of physiology by histone deacetylase 3. Nat. Rev. Mol. Cell Biol. 2019;20:102–115. doi: 10.1038/s41580-018-0076-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moumné L., Campbell K., Howland D., Ouyang Y., Bates G.P. Genetic Knock-Down of Hdac3 Does Not Modify Disease-Related Phenotypes in a Mouse Model of Huntington’s Disease. PLoS One. 2012;7 doi: 10.1371/journal.pone.0031080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Montgomery R.L., Potthoff M.J., Haberland M., Qi X., Matsuzaki S., Humphries K.M., Richardson J.A., Bassel-Duby R., Olson E.N. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J. Clin. Invest. 2008;118:3588–3597. doi: 10.1172/JCI35847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Norwood J., Franklin J.M., Sharma D., D’Mello S.R. Histone Deacetylase 3 Is Necessary for Proper Brain Development. J. Biol. Chem. 2014;289:34569–34582. doi: 10.1074/jbc.M114.576397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nott A., Cheng J., Gao F., Lin Y.-T., Gjoneska E., Ko T., Minhas P., Zamudio A.V., Meng J., Zhang F., et al. Histone deacetylase 3 associates with MeCP2 to regulate FOXO and social behavior. Nat. Neurosci. 2016;19:1497–1505. doi: 10.1038/nn.4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L., Jin J., Yang X.-J. Histone Deacetylase 3 Governs Perinatal Cerebral Development via Neural Stem and Progenitor Cells. iScience. 2019;20:148–167. doi: 10.1016/j.isci.2019.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jang J., Song G., Pettit S.M., Li Q., Song X., Cai C.L., Kaushal S., Li D. Epicardial HDAC3 Promotes Myocardial Growth Through a Novel MicroRNA Pathway. Circ. Res. 2022;131:151–164. doi: 10.1161/CIRCRESAHA.122.320785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knutson S.K., Chyla B.J., Amann J.M., Bhaskara S., Huppert S.S., Hiebert S.W. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J. 2008;27:1017–1028. doi: 10.1038/emboj.2008.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun Z., Feng D., Fang B., Mullican S.E., You S.-H., Lim H.-W., Everett L.J., Nabel C.S., Li Y., Selvakumaran V., et al. Deacetylase-Independent Function of HDAC3 in Transcription and Metabolism Requires Nuclear Receptor Corepressor. Mol. Cell. 2013;52:769–782. doi: 10.1016/j.molcel.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y., Frank D.B., Morley M.P., Zhou S., Wang X., Lu M.M., Lazar M.A., Morrisey E.E. HDAC3-Dependent Epigenetic Pathway Controls Lung Alveolar Epithelial Cell Remodeling and Spreading via miR-17-92 and TGF-β Signaling Regulation. Dev. Cell. 2016;36:303–315. doi: 10.1016/j.devcel.2015.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molstad D.H.H., Mattson A.M., Begun D.L., Westendorf J.J., Bradley E.W. Hdac3 regulates bone modeling by suppressing osteoclast responsiveness to RANKL. J. Biol. Chem. 2020;295:17713–17723. doi: 10.1074/jbc.RA120.013573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song S., Wen Y., Tong H., Loro E., Gong Y., Liu J., Hong S., Li L., Khurana T.S., Chu M., Sun Z. The HDAC3 enzymatic activity regulates skeletal muscle fuel metabolism. J. Mol. Cell Biol. 2019;11:133–143. doi: 10.1093/jmcb/mjy066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emmett M.J., Lim H.-W., Jager J., Richter H.J., Adlanmerini M., Peed L.C., Briggs E.R., Steger D.J., Ma T., Sims C.A., et al. Histone deacetylase 3 prepares brown adipose tissue for acute thermogenic challenge. Nature. 2017;546:544–548. doi: 10.1038/nature22819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim T.H., Yoo J.-Y., Choi K.-C., Shin J.-H., Leach R.E., Fazleabas A.T., Young S.L., Lessey B.A., Yoon H.-G., Jeong J.-W. Loss of HDAC3 results in nonreceptive endometrium and female infertility. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aaf7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou W., He Y., Rehman A.U., Kong Y., Hong S., Ding G., Yalamanchili H.K., Wan Y.-W., Paul B., Wang C., et al. Loss of function of NCOR1 and NCOR2 impairs memory through a novel GABAergic hypothalamus–CA3 projection. Nat. Neurosci. 2019;22:205–217. doi: 10.1038/s41593-018-0311-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bjornsson H.T. The Mendelian disorders of the epigenetic machinery. Genome Res. 2015;25:1473–1481. doi: 10.1101/gr.190629.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wakeling E., McEntagart M., Bruccoleri M., Shaw-Smith C., Stals K.L., Wakeling M., Barnicoat A., Beesley C., DDD Study. Hanson-Kahn A.K., et al. Missense substitutions at a conserved 14-3-3 binding site in HDAC4 cause a novel intellectual disability syndrome. HGG Adv. 2021;2 doi: 10.1016/j.xhgg.2020.100015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deardorff M.A., Bando M., Nakato R., Watrin E., Itoh T., Minamino M., Saitoh K., Komata M., Katou Y., Clark D., et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature. 2012;489:313–317. doi: 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoon J.G., Lee S., Cho J., Kim N., Kim S., Kim M.J., Kim S.Y., Moon J., Chae J.-H. Diagnostic uplift through the implementation of short tandem repeat analysis using exome sequencing. Eur. J. Hum. Genet. 2024;32:584–587. doi: 10.1038/s41431-024-01542-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaplanis J., Samocha K.E., Wiel L., Zhang Z., Arvai K.J., Eberhardt R.Y., Gallone G., Lelieveld S.H., Martin H.C., McRae J.F., et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. 2020;586:757–762. doi: 10.1038/s41586-020-2832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koh H.Y., Smith L., Wiltrout K.N., Podury A., Chourasia N., D’Gama A.M., Park M., Knight D., Sexton E.L., Koh J.J., et al. Utility of Exome Sequencing for Diagnosis in Unexplained Pediatric-Onset Epilepsy. JAMA Netw. Open. 2023;6 doi: 10.1001/jamanetworkopen.2023.24380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park S., Jang S.S., Lee S., Kim M., Sim H., Jeon H., Hong S.E., Lee J., Lee J., Jeon E.Y., et al. Systematic analysis of inheritance pattern determination in genes that cause rare neurodevelopmental diseases. Front. Genet. 2022;13 doi: 10.3389/fgene.2022.990015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watson P.J., Fairall L., Santos G.M., Schwabe J.W.R. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature. 2012;481:335–340. doi: 10.1038/nature10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kong S.H., Lee J.-H., Bae J.M., Hong N., Kim H., Park S.Y., Choi Y.J., Lee S., Rhee Y., Kim S.W., et al. In-depth proteomic signature of parathyroid carcinoma. Eur. J. Endocrinol. 2023;188:385–394. doi: 10.1093/ejendo/lvad046. [DOI] [PubMed] [Google Scholar]
- 42.Kim S.I., Hwangbo S., Dan K., Kim H.S., Chung H.H., Kim J.-W., Park N.H., Song Y.-S., Han D., Lee M. Proteomic Discovery of Plasma Protein Biomarkers and Development of Models Predicting Prognosis of High-Grade Serous Ovarian Carcinoma. Mol. Cell. Proteomics. 2023;22 doi: 10.1016/j.mcpro.2023.100502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tyanova S., Temu T., Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016;11:2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 44.Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 45.Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 46.Kases K., Schubert E., Hajikhezri Z., Larsson M., Devi P., Darweesh M., Andersson L., Akusjärvi G., Punga T., Younis S. The RNA-binding protein ZC3H11A interacts with the nuclear poly(A)-binding protein PABPN1 and alters polyadenylation of viral transcripts. J. Biol. Chem. 2023;299 doi: 10.1016/j.jbc.2023.104959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang W.-M., Tsai S.-C., Wen Y.-D., Fejér G., Seto E. Functional Domains of Histone Deacetylase-3. J. Biol. Chem. 2002;277:9447–9454. doi: 10.1074/jbc.M105993200. [DOI] [PubMed] [Google Scholar]
- 49.Park H., Kim Y., Park D., Jeoung D. Nuclear localization signal domain of HDAC3 is necessary and sufficient for the expression regulation of MDR1. BMB Rep. 2014;47:342–347. doi: 10.5483/BMBRep.2014.47.6.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He Y., Wang Z., Sun S., Tang D., Li W., Chai R., Li H. HDAC3 Is Required for Posterior Lateral Line Development in Zebrafish. Mol. Neurobiol. 2016;53:5103–5117. doi: 10.1007/s12035-015-9433-6. [DOI] [PubMed] [Google Scholar]
- 51.Chen W.-B., Gao L., Wang J., Wang Y.-G., Dong Z., Zhao J., Mi Q.-S., Zhou L. Conditional ablation of HDAC3 in islet beta cells results in glucose intolerance and enhanced susceptibility to STZ-induced diabetes. Oncotarget. 2016;7:57485–57497. doi: 10.18632/oncotarget.11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Landrum M.J., Chitipiralla S., Brown G.R., Chen C., Gu B., Hart J., Hoffman D., Jang W., Kaur K., Liu C., et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 2020;48:D835–D844. doi: 10.1093/nar/gkz972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lahm A., Paolini C., Pallaoro M., Nardi M.C., Jones P., Neddermann P., Sambucini S., Bottomley M.J., Lo Surdo P., Carfí A., et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. USA. 2007;104:17335–17340. doi: 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Le T.N., Williams S.R., Alaimo J.T., Elsea S.H. Genotype and phenotype correlation in 103 individuals with 2q37 deletion syndrome reveals incomplete penetrance and supports HDAC4 as the primary genetic contributor. Am. J. Med. Genet. 2019;179:782–791. doi: 10.1002/ajmg.a.61089. [DOI] [PubMed] [Google Scholar]
- 55.Bragin E., Chatzimichali E.A., Wright C.F., Hurles M.E., Firth H.V., Bevan A.P., Swaminathan G.J. DECIPHER: database for the interpretation of phenotype-linked plausibly pathogenic sequence and copy-number variation. Nucleic Acids Res. 2014;42:D993–D1000. doi: 10.1093/nar/gkt937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Collins R.L., Glessner J.T., Porcu E., Lepamets M., Brandon R., Lauricella C., Han L., Morley T., Niestroj L.-M., Ulirsch J., et al. A cross-disorder dosage sensitivity map of the human genome. Cell. 2022;185:3041–3055.e25. doi: 10.1016/j.cell.2022.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chong J.X., Yu J.-H., Lorentzen P., Park K.M., Jamal S.M., Tabor H.K., Rauch A., Saenz M.S., Boltshauser E., Patterson K.E., et al. Gene discovery for Mendelian conditions via social networking: de novo variants in KDM1A cause developmental delay and distinctive facial features. Genet. Med. 2016;18:788–795. doi: 10.1038/gim.2015.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pilotto S., Speranzini V., Marabelli C., Rusconi F., Toffolo E., Grillo B., Battaglioli E., Mattevi A. LSD1/KDM1A mutations associated to a newly described form of intellectual disability impair demethylase activity and binding to transcription factors. Hum. Mol. Genet. 2016;25:2578–2587. doi: 10.1093/hmg/ddw120. [DOI] [PubMed] [Google Scholar]
- 59.Nebel R.A., Kirschen J., Cai J., Woo Y.J., Cherian K., Abrahams B.S. Reciprocal Relationship between Head Size, an Autism Endophenotype, and Gene Dosage at 19p13.12 Points to AKAP8 and AKAP8L. PLoS One. 2015;10 doi: 10.1371/journal.pone.0129270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Souza L.C., Sgardioli I.C., Gil-da-Silva-Lopes V.L., Vieira T.P. A recognizable phenotype related to 19p13.12 microdeletion. Am. J. Med. Genet. 2018;176:1753–1759. doi: 10.1002/ajmg.a.38842. [DOI] [PubMed] [Google Scholar]
- 61.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gerasimavicius L., Livesey B.J., Marsh J.A. Loss-of-function, gain-of-function and dominant-negative mutations have profoundly different effects on protein structure. Nat. Commun. 2022;13:3895. doi: 10.1038/s41467-022-31686-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Badonyi M., Marsh J.A. Buffering of genetic dominance by allele-specific protein complex assembly. Sci. Adv. 2023;9 doi: 10.1126/sciadv.adf9845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lacoste J., Haghighi M., Haider S., Lin Z.-Y., Segal D., Reno C., Qian W.W., Xiong X., Shafqat-Abbasi H., Ryder P.V., et al. Pervasive mislocalization of pathogenic coding variants underlying human disorders. bioRxiv. 2023 doi: 10.1101/2023.09.05.556368. Preprint at. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available within the article and the supplemental information or can be made available upon reasonable request to the corresponding authors.