

Abstract
Magnesium protoporphyrin IX methyltransferase (ChlM), an enzyme in the chlorophyll biosynthetic pathway, catalyses the transfer of a methyl group to magnesium protoporphyrin IX (MgP) to form magnesium protoporphyrin IX monomethyl ester (MgPME). S-Adenosyl-L-methionine is the other substrate, from which a methyl group is transferred to the propionate group on ring C of the porphyrin macrocycle. Stopped-flow techniques were used to characterize the binding of porphyrin substrate to ChlM from Synechocystis PCC6803 by monitoring tryptophan fluorescence quenching on a millisecond timescale. We concluded that a rapid binding step is preceded by a slower isomerization of the enzyme. Quenched-flow techniques have been employed to characterize subsequent partial reactions in the catalytic mechanism. A lag phase has been identified that has been attributed to the formation of an intermediate. Our results provide a greater understanding of this catalytic process which controls the relative concentrations of MgP and MgPME, both of which are implicated in signalling between the plastid and nucleus in plants.
Keywords: chlorophyll biosynthesis, magnesium protoporphyrin IX methyltransferase, methylation, porphyrin
Abbreviations: ChlM, magnesium protoporphyrin IX methyltransferase; MgD, magnesium deuteroporphyrin IX; MgDME, magnesium deuteroporphyrin IX monomethyl ester; MgP, magnesium protoporphyrin IX; MgPME, magnesium protoporphyrin IX monomethyl ester; SAH, S-adenosyl-L-homocysteine; SAM, S-adenosyl-L-methionine
INTRODUCTION
Chlorophyll biosynthesis is essential for the assembly of photosynthetic complexes, which harvest sunlight and convert it into chemical energy. The analysis of this biosynthetic pathway has been facilitated by the overexpression of genes encoding these enzymes, often from bacterial sources, which has resulted in the production of the large amounts of protein needed for detailed functional studies [1–9]. Magnesium protoporphyrin IX methyltransferase (ChlM; EC 2.1.1.11) catalyses the second committed step in chlorophyll biosynthesis, where magnesium protoporphyrin IX (MgP) is converted into magnesium protoporphyrin IX monomethyl ester (MgPME). These tetrapyrrole intermediates have been implicated in the plastid-to-nucleus signalling pathway. For example, feeding and inhibitor experiments have identified MgPME as the plastid-to-nucleus signal that leads to repression of the lhcb genes in Chlamydomonas reinhardtii and cress seedlings [10]. It has been shown that MgPME feeding of Chlamydomonas activates nuclear heat-shock genes through a light-responsive promoter element in dark-grown cultures [11,12]. Furthermore, several Arabidopsis thaliana gun mutants, previously shown to be uncoupled in this signalling pathway [13], are proposed to affect the same plastid-to-nucleus signalling by perturbing levels of protoporphyrin and MgP [11,14–16].
In order to understand the availability of these chlorophyll precursors for signalling as well as biosynthesis, it is important to understand the kinetics of MgP methylation, and, eventually, how this process is coupled to the preceding step, that of magnesium chelation. Despite the successful overexpression of bchM, the gene encoding the methyltransferase from Rhodobacter capsulatus and R. sphaeroides [17,18], little kinetic work has been undertaken. This is possibly because this enzyme does not lend itself to continuous assays by spectroscopic methods. Kinetic studies have been performed on MgP methyltransferase from Euglena gracilis [19], which imply the existence of a ternary complex. Subsequent inhibition studies demonstrated that ChlM from E. gracilis operates via a random ternary mechanism [20]. Steady-state kinetic studies, using purified ChlM, have demonstrated that the reaction proceeds via a random ternary mechanism, where the porphyrin substrate and S-adenosyl-L-methionine (SAM) may bind in either order [21].
It has been suggested that all methyltransferases operate via a direct displacement SN2 mechanism [22]. However, it cannot be assumed that nucleophilic displacement can fully describe the reaction mechanism; it should be noted that enzyme-bound intermediates have been identified in methyltransferases involved in cobalamin biosynthesis [23], the methyl cycle [24] and DNA methylation [25–29].
In the present paper we describe the use of stopped-flow techniques to demonstrate that porphyrin rapidly binds to ChlM, following a slower isomerization of the enzyme. Pre-steady-state catalysis was monitored using quenched-flow and HPLC techniques, which highlighted a lag phase prior to product evolution. This has been attributed to the formation of a stable intermediate, as the evolution and depletion of a third peak in the HPLC traces is characterized by a double exponential, a trademark of such a species. In light of these results, a putative reaction mechanism is discussed.
MATERIALS AND METHODS
Materials
All pigments were purchased from Porphyrin Products (Logan, UT, U.S.A.). The remaining chemicals were purchased from Sigma–Aldrich unless otherwise specified.
Protein expression and purification
The plasmid pET9a-His6-ChlM [30] was transformed into Escherichia coli BL21 (DE3) cells and the chlM gene was induced for 15 h at 20 °C using 0.4 mM IPTG (isopropyl β-D-thiogalactoside). The cells were harvested by centrifugation (3000 g, 20 min, 4 °C) and cells from a 2-litre culture were resuspended in 20 ml of chilled binding buffer [20 mM citrate/KOH (pH 5.8), 500 mM NaCl, 500 mM glycerol and 5 mM imidazole]. The cells were disrupted by sonication for 6×30 s on ice, and the cell debris was removed by centrifugation (39000 g, 30 min, 4 °C) at 4 °C. The supernatant was loaded at 2 ml/min on to a 2.0 cm×5.0 cm column packed with Chelating Sepharose Fast-Flow resin (Pharmacia) charged with 50 mM NiSO4 and pre-equilibrated with 3 column vol. of binding buffer. The column was washed with 10 column vol. of binding buffer and 6 column vol. of binding buffer containing 60 mM imidazole (wash buffer) to remove any loosely bound contaminants. The His-tagged ChlM was eluted with binding buffer containing 250 mM imidazole (eluent buffer). A 50 ml column of P-6 desalting gel (Bio-Rad) was equilibrated with chilled buffer [50 mM citrate/KOH (pH 5.8), 300 mM glycerol and 200 mM NaCl] and was used to remove imidazole from the protein sample. A typical yield was 15 mg of protein from a 2-litre culture of E. coli.
Substrates and products
Porphyrin solutions were prepared weekly by dissolving a small amount of porphyrin in buffer. Porphyrin concentrations were determined in 0.1 M HCl, using the ε398 of 433000 M−1·cm−1 [30], after Mg2+ had been removed from the porphyrin by a 5 min incubation in 1 M acetic acid. SAM and S-adenosyl-L-homocysteine (SAH) stock solutions were prepared daily in 0.1 M HCl and 0.1 M NaOH respectively. Their concentrations were determined using the ε256 of 15200 M−1·cm−1 in 1 M HCl for SAM and the ε260 of 16000 M−1·cm−1 at pH 7 for SAH [32].
ChlM assays
Reactions were carried out at 30 °C in 100 mM Tris (pH 7.5)/0.5 μM ChlM/500 μM SAM/100 μM magnesium deuteroporphyrin IX (MgD). The assay mixtures were incubated at 30 °C in the absence of SAM for 5 min to allow for thermal equilibration. SAM was added, 20 μl aliquots were taken every 2 min over a period of 8 min and then quenched in 400 μl of stop solution (acetone/water/33% ammonia solution, 80:20:1, by vol.). These aliquots were centrifuged at 20000 g for 5 min to pellet any aggregated protein. Pigments were separated using HPLC reversed-phase chromatography as described by Shepherd et al. [21]. A 20 μl portion of the soluble phase was loaded on to a Beckman ODS Ultrasphere column (150 mm×4.6 mm). The pigments were separated by a 7 min linear gradient from 0 to 67% solvent B at 2 ml/min, and then the gradient was paused for a further 5 min for the porphyrins to be eluted.
The peaks were integrated using Waters Millennium software. Known amounts of magnesium deuteroporphyrin IX monomethyl ester (MgDME) were analysed in the same way to produce a standard curve. The maximum rate during an assay was taken as the steady-state rate, which occurred at the beginning of the reaction.
Stopped-flow measurements
ChlM–porphyrin binding was followed using an Applied Photophysics SF18 MV stopped-flow spectrofluorimeter. The reaction cell was maintained at a constant temperature of 30 °C by circulation of water from a thermostatically controlled water bath [Grant Instruments (Cambridge) Ltd]. Tryptophan fluorescence was measured by excitation at 280 nm through a 9-nm band-pass filter. Emission was observed through a Schott WG305 high-pass filter and an Oriel 51664 low-pass 450 nm filter, positioned in front of the observation photomultiplier. Values of the observed rate constant (kobs) were obtained by analysis of the reaction trace as a single exponential [Ft=F1exp (−kobs·t)+c], using the on-line data analysis package. The kobs-versus-S data were fitted to a single hyperbolic decay.
Quenched-flow measurements
Pre-steady-state time samples from ChlM-catalysed methyltranferase reactions were obtained using a Hi-Tech rapid quenched-flow system. The reaction cell was maintained at a constant temperature of 30 °C by circulation of water from a thermostatically controlled water bath (Grant Instruments). Reactions were quenched in stop solution (acetone/water/33% ammonia solution, 80:20:1, by vol.) and the concentrations of MgDME and intermediate were determined using HPLC reversed-phase chromatography, in the same way that steady-state assays were monitored. The data obtained were analysed using non-linear regression (Sigmaplot), and apparent rate constants for the evolution/decay of intermediate were obtained by modelling the data using a double exponential [y=a(1−e−bx)+c(1−e−dx)]. The data points for the evolution of product could not easily be fitted to exponential curves, so these data were modelled to a double exponential with a linear phase [y=a(1−e−bx)+c(1−e−dx)+mx] using the rate constants for the formation/decay of intermediate. The kobs value for the evolution of intermediate corresponds to the lag phase in MgDME evolution, and the kobs value for the decay of intermediate corresponds to the burst of MgDME formation.
RESULTS
Stopped-flow substrate-binding kinetics
When the binding of MgD to ChlM was performed, the observed association rate constant appeared to decrease with increasing substrate concentration (Figure 1). This is characteristic of a reaction scheme whereby two consecutive reversible reactions exist, the second of which is faster than the first. This is summarized in Scheme 1, where the transition from E to E′ represents a conformational change. The data are fitted to eqn (1), which describes a single hyperbolic decay. When the [MgD] is very high, the first equilibrium is essentially irreversible as E′ is transformed completely to E′·MgD, and kobs tends towards k1. When [MgD] is very low, there is very little E′·MgD, and kobs tends towards k1+k−1. Hence, the observed rate constant appears to decrease as more MgD is added. The values of k1 and K2 were 3.09 s−1 and 3.36 μM respectively. The value of k−1 could not be accurately determined, as this requires the extrapolation of the data in Figure 1 to the y-axis. However, this value could be approximated as 600 s−1.
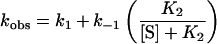 |
(1) |
Figure 1. Plots of observed rates of ChlM tryptophan fluorescence decay against MgD concentration.

The data are fitted to a single hyperbolic decay, and the fitted parameters correspond to events in Scheme 1. The curve levels off at kobs=3.09 s−1, which corresponds to k1. The fitted equilibrium constant is 3.36 μM, which corresponds to K2. The y-axis intercept, corresponding to k1+k−1·k−1, is approx. 600 s−1.
Scheme 1. Proposed isomerization step that precedes MgP binding.

Quenched-flow analysis of pre-steady state methyltransferase turnover
In order to establish the conditions for a pre-steady-state analysis, a steady-state stopped assay was performed over an 8-min duration. The individual time points were analysed by HPLC, which shows the evolution of the MgDME product peak, as well as a peak of constant amplitude in the steady-state. Figure 2 shows the chromatograms obtained in a steady-state assay, which shows the evolution of MgDME at 12.3 min and the presence of an extra peak at 12.7 min, later to be attributed to an intermediate species. When pre-steady state kinetics were performed using quenched flow, the evolution of MgDME and evolution/depletion of a putative intermediate could be monitored on the millisecond timescale. Figure 3(A) shows selected HPLC chromatograms to illustrate the relative concentrations of these two species at different time-points in the reaction. The t=4.3 ms chromatogram in Figure 3(A) demonstrates that a small amount of intermediate has been formed, within the dead time of the quenched-flow apparatus. The next time point illustrates that after 100 ms more intermediate has been formed, yet very little MgDME has evolved. These demonstrate that the intermediate evolves before MgDME is produced. The absorbance and fluorescence spectra of the intermediate are identical with that of MgDME. Figure 3(B) shows the evolution/depletion of intermediate, represented by the peaks at 12.7 min in the HPLC chromatograms. These data are fitted to a double exponential, the first phase of which has a rate constant of 11.9±0.5 s−1 and the decay has a rate constant of 11.8±0.5 s−1. This behaviour is characteristic of a reaction intermediate. Figure 3(C) shows the evolution of MgDME, represented by the peaks at 12.3 min in the HPLC chromatograms. These data could not be accurately fitted to a double exponential so the rate constants for the evolution/decay of intermediate were used as constraints to fit a double exponential with a linear phase. The linear phase corresponds to a steady-state rate of 0.41±0.04 μM·min−1. The decay of the intermediate species coincides with the evolution of MgDME, which implies that MgDME is formed via the decay of this species.
Figure 2. Steady-state methyltransferase assay using 0.5 μM ChlM, 500 μM SAM and 100 μM MgD.

The four chromatograms correspond to quenched aliquots taken at 2, 4, 6 and 8 min. The pigments were bound to a C-18 reversed-phase column with a 2 ml bed volume. A gradient of acetonitrile from 0–100% over 15 ml was used to elute substrate and product at different points. The elution of porphyrins was detected using a fluorescence detector with excitation and emission wavelengths of 400 nm and 580 nm respectively. Peaks at 10 min and 12.3 min correspond to MgD and MgDME respectively. The peaks at 12.7 min correspond to a putative intermediate.
Figure 3. Demonstration of an intermediate in the reaction mechanism using a combination of quenched-flow techniques and HPLC reversed-phase chromatography.

(A) HPLC chromatograms demonstrating the pre-steady-state turnover of 0.5 μM ChlM with 1 mM SAM and 30 μM MgD. The elution of porphyrins was detected using a fluorescence detector with excitation and emission wavelengths of 400 nm and 580 nm respectively. This sample of chromatograms illustrates the evolution/depletion of intermediate and evolution of MgDME. (B) Evolution and depletion of the intermediate that is eluted at 12.7 min during HPLC analysis. Fluorescence excitation and emission wavelengths were 400 nm and 580 nm respectively. These data have been fitted to a double exponential. k1=11.9±0.5 s−1, k2=11.8±0.5 s−1. (C) Evolution of MgDME calculated from the peaks at 12.3 min on the HPLC chromatograms. MgDME concentrations have been fitted to a double exponential with a linear phase, using the rate constants from (B). The steady-state rate is 0.41±0.04 μM·min−1.
DISCUSSION
MgD has previously been shown to bind to free ChlM [21]. Figure 1 depicts the rate of quenching of ChlM tryptophan fluorescence upon binding to MgD. The observed rate constants, which describe the rate of E′ accumulation (Scheme 1), decrease with increasing porphyrin concentration, which is characteristic of a slower step that precedes rapid substrate binding [33]. The first step is likely to be a domain reorganization that alters the conformation of the MgD-binding site. This has been observed in the case of M.HhaI [27], another member of the methyltransferase family of enzymes; in this case it has been shown that SAM may bind to two conformationally distinct enzyme species. Similarly, SAM may bind to ChlM before or after MgD binding [21], suggesting that SAM may bind to both E and E′ (Schemes 1 and 2).
Scheme 2. Proposed reaction mechanism of ChlM.

The scheme depicts a random ternary mechanism, whereby both substrates may bind in either order. KmSAM=38 μM, KdMgD=2.37 μM, kcat/KmSAM=1500 M−1·s−1, Ki≈Ki′ [21]. k1=3.09 s−1, k−1≈600 s−1, K2=3.36 μM. E, enzyme; E′, enzyme with altered conformation; Int, putative intermediate.
The evolution of MgDME was followed on the millisecond timescale using quenched flow combined with HPLC reversed-phase chromatography (Figures 3A and 3C). The steady-state rate was 0.41±0.04 μM·min−1. This is consistent with previous kinetic analysis of ChlM from Synechocystis [21]. The burst of MgDME evolution corresponds to the time at which the depletion of porphyrin intermediate commences (Figure 3B). This suggests that MgDME is formed directly from this modified porphyrin.
The transition between E and E′ at 30 μM MgD (Figure 1) has an observed rate constant of approx. 70 s−1. This is faster than the evolution of intermediate in Figure 3(B) (kobs=11.9±0.5 s−1). This is consistent with Scheme 1, as the isomerization step must occur before MgD binds and the intermediate is formed. Scheme 1 assumes that MgD binding is very rapid (>600 s−1) and, at present, further resolution of this rate constant is outside the capabilities of the rapid-mixing techniques used in this study. Therefore, the difference in rates between enzyme isomerization and intermediate formation may be attributable to a change in the conformation of bound SAM or MgD substrates and/or the breaking/formation of chemical bonds.
The t=4.3 ms time point chromatogram in Figure 3(A) demonstrates that a small amount of intermediate has been formed. The next time point illustrates that after 100 ms more intermediate has been formed, yet very little MgDME has evolved. The last time point shown is the 800 ms sample, which clearly demonstrates the concomitant depletion of intermediate and evolution of MgDME. The fluorescence yields of MgD and MgDME are very similar when measured in the HPLC solvents used. Also, the absorption/fluorescence characteristics of the intermediate are the same as those of MgD and MgDME. Given that the porphyrin macrocycle chemistry should remain largely unchanged during the methyltransferase reaction, it seems likely that the intermediate will have a similar fluorescence yield. If this is the case, the concentration of intermediate at 150 ms is approx. 0.5 μM. This is equal to the concentration of enzyme used and suggests that the majority of ChlM is active.
These results, along with recent steady-state analysis [21], are consistent with the reaction mechanism in Scheme 2. This depicts a random ternary mechanism, established as a result of the steady-state kinetic studies described previously [21]. The Kd for the binding of MgD to E′ (3.36 μM) is in close agreement with previous data, where tryptophan fluorescence quenching was used to measure the binding of MgD to ChlM at equilibrium (Kd=2.37 μM) [21]. MgDME and SAH formation is likely to proceed via the production of an intermediate, as suggested in the present study. Since the rates of formation and decay of the intermediate are much faster than kcat (0.057 s−1) [21], the rate-limiting reaction is likely to involve the product-release steps. The most simple methyltransferase reaction is a direct displacement involving nucleophilic attack on the methyl carbon atom. However, there are precedents for the formation of intermediates in methyltransferases [23–29], but this is the first time that rapid-mixing techniques have been used to characterize such a species in the field of chlorophyll biosynthesis. Further characterization of this intermediate has proved difficult, as the isolation of acceptable yields will require large amounts of protein and a preparative chromatography protocol.
The natural substrate and product for the ChlM catalysed reaction have been implicated in plastid-to-nucleus signalling [10–14]. A recent study has highlighted the role of MgP in repressing the expression of nuclear-encoded photosynthesis genes [15]. This further emphasizes the importance of quantitative studies, not only of the methyltransferase and magnesium chelatase, but also of the interaction between these enzymes. Such interactions could be crucial in determining the availability of MgP for both signalling and biosynthetic roles in the chloroplast.
Acknowledgments
This research was funded by the Biotechnology and Biological Sciences Research Council, U.K.
References
- 1.Gibson L. C. D., Jensen P. E., Hunter C. N. Magnesium chelatase from Rhodobacter sphaeroides: initial characterization of the enzyme using purified subunits and evidence for a BchI–BchD complex. Biochem. J. 1999;337:243–251. [PMC free article] [PubMed] [Google Scholar]
- 2.Jensen P. E., Gibson L. C. D., Hunter C. N. Determinants of catalytic activity with the use of purified I, D and H subunits of the magnesium protoporphyrin IX chelatase from Synechocystis PCC6803. Biochem. J. 1998;334:335–344. doi: 10.1042/bj3340335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heyes D. J., Martin G. E., Reid R. J., Hunter C. N., Wilks H. M. NADPH:protochlorophyllide oxidoreductase from Synechocystis: overexpression, purification and preliminary characterisation. FEBS Lett. 2000;483:47–51. doi: 10.1016/s0014-5793(00)02081-0. [DOI] [PubMed] [Google Scholar]
- 4.Oster U., Bauer C. E., Rüdiger W. Characterization of chlorophyll a and bacteriochlorophyll a synthases by heterologous expression in Escherichia coli. J. Biol. Chem. 1997;272:9671–9676. doi: 10.1074/jbc.272.15.9671. [DOI] [PubMed] [Google Scholar]
- 5.Keller Y., Bouvier F., d'Harlingue A., Camara B. Metabolic compartmentation of plastid prenyllipid biosynthesis – evidence for the involvement of a multifunctional geranylgeranyl reductase. Eur. J. Biochem. 1998;251:413–417. doi: 10.1046/j.1432-1327.1998.2510413.x. [DOI] [PubMed] [Google Scholar]
- 6.Addlesee H. A., Hunter C. N. Physical mapping and functional assignment of the geranylgeranyl-bacteriochlorophyll reductase gene, bchP, of Rhodobacter sphaeroides. J. Bacteriol. 1999;181:7248–7255. doi: 10.1128/jb.181.23.7248-7255.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hansson A., Willows R. D., Roberts T. H., Hansson M. Three semidominant barley mutants with single amino acid substitutions in the smallest magnesium chelatase subunit form defective AAA+ hexamers. Proc. Natl. Acad. Sci. U.S.A. 2002;99:13944–13949. doi: 10.1073/pnas.212504499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willows R. D., Gibson L. C. D., Kanangara C. G., Hunter C. N., von Wettstein D. Three separate proteins constitute the magnesium chelatase of Rhodobacter sphaeroides. Eur. J. Biochem. 1996;235:438–443. doi: 10.1111/j.1432-1033.1996.00438.x. [DOI] [PubMed] [Google Scholar]
- 9.Willows R. D., Beale S. I. Heterologous expression of the Rhodobacter capsulatus BchI, -D, and -H genes that encode magnesium chelatase subunits and characterization of the reconstituted enzyme. J. Biol. Chem. 1998;273:34206–34213. doi: 10.1074/jbc.273.51.34206. [DOI] [PubMed] [Google Scholar]
- 10.Johanningmeier U. Possible control of transcript levels by chlorophyll precursors in Chlamydomonas. Eur. J. Biochem. 1988;177:417–424. doi: 10.1111/j.1432-1033.1988.tb14391.x. [DOI] [PubMed] [Google Scholar]
- 11.Kropat J., Oster U., Rudiger W., Beck C. F. Chlorophyll precursors are signals of chloroplast origin involved in light induction of nuclear heat-shock genes. Proc. Natl. Acad. Sci. U.S.A. 1997;94:14168–14172. doi: 10.1073/pnas.94.25.14168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kropat J., Oster U., Rudiger W., Beck C. F. Chloroplast signalling in the light induction of nuclear HSP70 genes requires the accumulation of chlorophyll precursors and their accessibility to cytoplasm/nucleus. Plant J. 2000;24:523–531. doi: 10.1046/j.1365-313x.2000.00898.x. [DOI] [PubMed] [Google Scholar]
- 13.Susek R. E., Ausubel F. M., Chory J. Signal-transduction mutants of Arabidopsis uncouple nuclear cab and rbcs gene expression from chloroplast development. Cell. 1993;74:787–799. doi: 10.1016/0092-8674(93)90459-4. [DOI] [PubMed] [Google Scholar]
- 14.Mochizuki N., Brusslan J. A., Larkin R., Nagatani A., Chory J. Arabidopsis genomes uncoupled 5 (GUN5) mutant reveals the involvement of Mg-chelatase H subunit in plastid-to-nucleus signal transduction. Proc. Natl. Acad. Sci. U.S.A. 2001;98:2053–2058. doi: 10.1073/pnas.98.4.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strand A., Asami T., Alonso J., Ecker J. R., Chory J. Chloroplast to nucleus communication triggered by accumulation of Mg-protoporphyrinIX. Nature (London) 2003;421:79–83. doi: 10.1038/nature01204. [DOI] [PubMed] [Google Scholar]
- 16.Larkin R. M., Alonso J. M., Ecker J. R., Chory J. GUN4, a regulator of chlorophyll synthesis and intracellular signalling. Science. 2003;299:902–906. doi: 10.1126/science.1079978. [DOI] [PubMed] [Google Scholar]
- 17.Gibson L. C. D., Hunter C. N. The bacteriochlorophyll biosynthesis gene, bchm, of Rhodobacter sphaeroides encodes S-adenosyl-L-methionine:Mg protoporphyrin IX methyltransferase. FEBS Lett. 1994;352:127–130. doi: 10.1016/0014-5793(94)00934-1. [DOI] [PubMed] [Google Scholar]
- 18.Bollivar D. W., Jiang Z. Y., Bauer C. E., Beale S. I. Heterologous expression of the BchM gene product from Rhodobacter capsulatus and demonstration that it encodes S-adenosyl-L-methionine:Mg-protoporphyrin-IX-methyltransferase. J. Bacteriol. 1994;176:5290–5296. doi: 10.1128/jb.176.17.5290-5296.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hinchigeri S. B., Chan J. C. S., Richards W. R. Purification of S-adenosyl-L-methionine: magnesium protoporphyrin methyltransferase by affinity chromatography. Photosynthetica. 1981;15:351–359. [Google Scholar]
- 20.Hinchigeri S. B., Richards W. R. The reaction mechanism of S-adenosyl-L-methionine: magnesium protoporphyrin methyltransferase from Euglena gracilis. Photosynthetica. 1982;16:554–560. [Google Scholar]
- 21.Shepherd M., Reid J. D., Hunter C. N. Purification and kinetic characterization of the magnesium protoporphyrin IX methyltransferase from Synechocystis PCC6803. Biochem. J. 2003;371:351–360. doi: 10.1042/BJ20021394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takusagawa F., Fujioka M., Spies A., Schowen R. L. S-Adenosylmethionine (AdoMet)-dependent methyltransferases. In: Sinnott M., editor. Comprehensive Biological Catalysis. San Diego: Academic Press; 1998. pp. 1–30. [Google Scholar]
- 23.Woodcock S. C., Warren M. J. Evidence for a covalent intermediate in the S-adenosyl-L-methionine-dependent transmethylation reaction catalysed by sirohaem synthase. Biochem. J. 1996;313:415–421. doi: 10.1042/bj3130415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jarrett J. T., Hoover D. M., Ludwig M. L., Matthews R. G. The mechanism of adenosylmethionine-dependent activation of methionine synthase: A rapid kinetic analysis of intermediates in reductive methylation of cob(II)alamin enzyme. Biochemistry. 1998;37:12649–12658. doi: 10.1021/bi9808565. [DOI] [PubMed] [Google Scholar]
- 25.Wu J. C., Santi D. V. Kinetic and catalytic mechanism of HhaI methyltransferase. J. Biol. Chem. 1987;262:4778–4786. [PubMed] [Google Scholar]
- 26.Chen L., MacMillan A. M., Chang W., Ezaz-Nikpay K., Lane W. S., Verdine G. L. Direct identification of the active-site nucleophile in a DNA (cytosine-C5) methyltransferase. Biochemistry. 1991;30:11018–11025. doi: 10.1021/bi00110a002. [DOI] [PubMed] [Google Scholar]
- 27.Kumar S., Cheng X. D., Pflugrath J. W., Roberts R. J. Purification, crystallization and preliminary X-ray diffraction analysis of an M.HhaI–AdoMet complex. Biochemistry. 1992;31:8648–8653. doi: 10.1021/bi00151a035. [DOI] [PubMed] [Google Scholar]
- 28.O'Gara M., Zhang X., Roberts R. J., Cheng X. D. Structure of a binary complex of HhaI methyltransferase with S-adenosyl-L-methionine formed in the presence of a short non-specific DNA oligonucleotide. J. Mol. Biol. 1999;287:201–209. doi: 10.1006/jmbi.1999.2608. [DOI] [PubMed] [Google Scholar]
- 29.Lindstrom W. M., Flynn J., Reich N. O. Reconciling structure and function in HhaI DNA cytosine-C-5 methyltransferase. J. Biol. Chem. 2000;275:4912–4919. doi: 10.1074/jbc.275.7.4912. [DOI] [PubMed] [Google Scholar]
- 30.Falk J. E. Porphyrins and Metalloporphyrins: their General, Physical and Coordination Chemistry, and Laboratory Methods. Amsterdam: Elsevier; 1964. Appendix 1; p. 236. [Google Scholar]
- 31.Jensen P. E., Gibson L. C. D., Shephard F., Smith V., Hunter C. N. Introduction of a new branchpoint in tetrapyrrole biosynthesis in Escherichia coli by co-expression of genes encoding the chlorophyll-specific enzymes magnesium chelatase and magnesium protoporphyrin methyltransferase. FEBS Lett. 1999;455:349–354. doi: 10.1016/s0014-5793(99)00909-6. [DOI] [PubMed] [Google Scholar]
- 32.Dawson R. M. C., Elliot D. C., Elliot W. H., Jones K. M. Data for Biochemical Research. New York: Oxford University; 1986. pp. 2–3. [Google Scholar]
- 33.Fersht A. Enzyme Structure and Mechanism. New York: W. H. Freeman and Company; 1985. Measurement and magnitude of enzymatic rate constants; pp. 121–154. [Google Scholar]