

SUMMARY
Long bones are generated by mesoderm-derived skeletal progenitor/stem cells (SSCs) through endochondral ossification, a process of sequential chondrogenic and osteogenic differentiation tightly controlled by the synergy between intrinsic and microenvironment cues. Here, we report that loss of TRIM28, a transcriptional corepressor, in mesoderm-derived cells expands the SSC pool, weakens SSC osteochondrogenic potential, and endows SSCs with properties of ectoderm-derived neural crest cells (NCCs), leading to severe defects of skeletogenesis. TRIM28 preferentially enhances H3K9 trimethylation and DNA methylation on chromatin regions more accessible in NCCs; loss of this silencing upregulates neural gene expression and enhances neurogenic potential. Moreover, TRIM28 loss causes hyperexpression of GREM1, which is an extracellular signaling factor promoting SSC self-renewal and SSC neurogenic potential by activating AKT/mTORC1 signaling. Our results suggest that TRIM28-mediated chromatin silencing establishes a barrier for maintaining the SSC lineage trajectory and preventing a transition to ectodermal fate by regulating both intrinsic and microenvironment cues.
In brief
Liu et al. show that loss of TRIM28 in developing mesoderm causes severe skeletal defects, expands the skeletal stem cell pool, and switches them to an ectodermal neural crest-like state. Thus, TRIM28-mediated chromatin silencing generates a barrier between ectoderm and mesoderm, thereby securing skeletal stem cell fate to safeguard skeletogenesis.
Graphical Abstract
INTRODUCTION
The epigenetic landscape of a cell, meaning its pattern of DNA methylation and histone modification, reflects its history of lineage progression and predetermines its future identity.1–3 Recent studies revealed that increased global DNA methylation occurs at the exit of pluripotency, followed by a gradual and asymmetric demethylation and loss of chromatin accessibility on germ layer-specific enhancers during gastrulation.4 In addition, H3K9 trimethylation (me3) is more dynamically altered than H3K27me3 marks on protein-coding genes during gastrulation.5 H3K9me3 generally increases as germ layers develop from the epiblast and then decreases as precursor cells are specified, leading to the derepression of cell-type-specific genes.5 This suggests that H3K9me3 and DNA methylation marks generate epigenetic barriers in defining germs layers during gastrulation.
In the mammalian skeletal system, appendicular bones and most axial bones develop from and are maintained by mesoderm-derived skeletal precursors/stem cells (later unified as “skeletal stem cells” [SSCs]6), while the majority of craniofacial bones arise from ectoderm-derived neural crest cells (NCCs).7 SSCs have a default osteogenic or chondrogenic fate,6 while NCCs have a default neural/glial and epidermal fate but acquire an osteochondrogenic potential through transient reactivation of pluripotency during craniofacial bone formation.8
As with other stem cells, lineage-specific transcription factors9 and the microenvironment cooperatively regulate SSC maintenance and differentiation,9–11 but their effectiveness depends on the permissiveness of the epigenetic landscape.12 Accumulated work has shown that DNA methylation and H3K9me3 regulate SSC homeostasis necessary for skeletal health,12–14 but there is still a lack of understanding as to whether and how these two essential chromatin silencing marks contribute to establish SSC identity and guide SSC lineage progression for skeletogenesis.
TRIM28 is a unique chromatin silencing protein suitable to address this question as a key scaffolding protein that recruits histone H3K9 methyltransferases (SETDB1 and SUV39H1) and DNA methyltransferases (DNMTs) to promote both H3K9me3 and DNA methylation, resulting in the formation of heterochromatin and the silencing of genes as well as transposable elements (TEs).15–18 Loss of TRIM28 in oocytes causes a large variability in the size of embryos.19 Trim28-null embryos are arrested at embryonic day 5.5 (E5.5),20 while global Trim28 haplo-insufficiency causes bistable epigenetic obesity.21 In contrast, TRIM28 loss at postnatal stages does not cause distinctive phenotypes.21,22 These results suggest that TRIM28 establishes an essential epigenetic landscape in early embryogenesis but becomes dispensable postnatally. Here, we demonstrate that TRIM28 loss disrupts SSC identity and lineage fidelity, leading to severe defects in skeletogenesis.
RESULTS
TRIM28 loss in mesodermal cells disrupts skeletal development
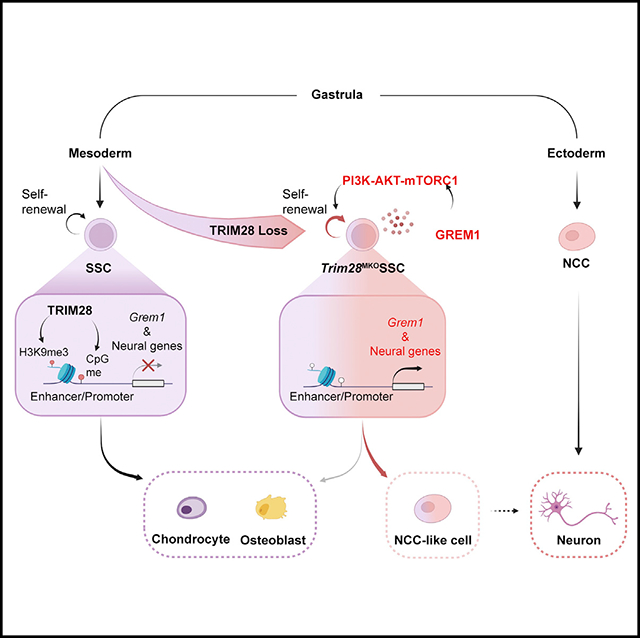
We generated Dermo1-Cre; Trim28f/f mice to knock out Trim28 specifically in mesodermal cells (hereafter Trim28MKO) from E9.5, the stage when cells migrate from the lateral plate mesoderm to the limb field for limb bud formation.23 The Trim28MKO mice died perinatally with small skeletal frames, narrow rib cages, short and thick bones and cartilage, and under-mineralized sternums and vertebrae (Figures 1A, S1A, and S1B). Trim28MKO mice also had high penetrance of cleft palate (45/86) (Figure S1C). Developmental defects were observed in other organs where Dermo1-Cre is expressed, including skin and lungs, the latter of which is a possible cause of perinatal death (Figure S1D).
Figure 1. TRIM28 loss in mesodermal cells disrupts skeletal development.
(A) Skeletal phenotypes of WT and Trim28MKO mice at E19.5 visualized with Alcian blue and Alizarin red staining; arrowheads indicate thicker and shorter ribs and over-expanded hindlimb cartilage. Scale bar, 0.5 cm.
(B) E17.5 mouse proximal tibial sections stained with Alcian blue Hematoxylin/Orange G. Gross image (left) and magnified views of each zone (right) are shown(RZ, resting zone; PZ, proliferating zone; HZ, hypertrophic zone). Scale bar, 200 μm.
(C) Increased cell proliferation in the Trim28MKO proximal tibial sections (E17.5) as indicated by Ki67 immunohistochemistry (IHC). Bottom panel: area boxed in red is the magnified perichondrium (PC), while the yellow boxed area is the magnified periarticular resting zone (PRZ). Scale bars, 200 μm.
(D) Type II collagen IHC of proximal tibial sections (E17.5). Area boxed in red is magnified in the bottom panels showing high levels of type II collagen retention inthe Trim28MKO HZ. Scale bars, 200 μm.
(E) Type X collagen IHC staining of distal femoral sections (E17.5). The red box area is magnified in the bottom panel, and red dotted lines define type X collagen-expressing areas. Scale bars, 200 μm.
See also Figure S1.
Histologically, the Trim28MKO mice had wider and severely disorganized growth plates (GPs) (Figure 1B). Normally, GPs contain chondrocytes at sequential stages of differentiation in three consecutive zones24 and are an important reservoir of SSCs.11,25,26 The resting zone, which usually consists of naive round upper zone chondrocytes, is overexpanded in Trim28MKO mice (Figure 1B). The proliferating zone, comprised of flat proliferating column-forming chondrocytes, is diminished and replaced with a mixture of flat and round chondrocytes Trim28MKO (Figure 1B). And the hypertrophic zone, which typically contains prehypertrophic column-forming chondrocytes and terminally differentiated hypertrophic chondrocytes, was shortened (Figure 1B). In addition, we detected elevated cell proliferation (Figure 1C), without a change in apoptosis (Figure S1E), in the periarticular resting zone and perichondrium, the connective tissue that surrounds the GP cartilage. A delay in chondrocyte maturation was noted in the mutant hypertrophic zone, as assessed by the abnormal retention of type II collagen and a reduction in type X collagen and Indian hedgehog (IHH) (boxed area, Figures 1D, 1E, and S1F). In contrast, deleting Trim28 at E12.5 in differentiated chondrocytes and a small portion of chondroprogenitors using Col2a1-Cre27 produced a less severe skeletal phenotype (Figure S1G). These results collectively demonstrate that TRIM28 is crucial for the function of nascent mesodermal cells more so than in committed progenitors or chondrocytes.
Loss of TRIM28 generates a new cell cluster abundant in SSCs in the GP
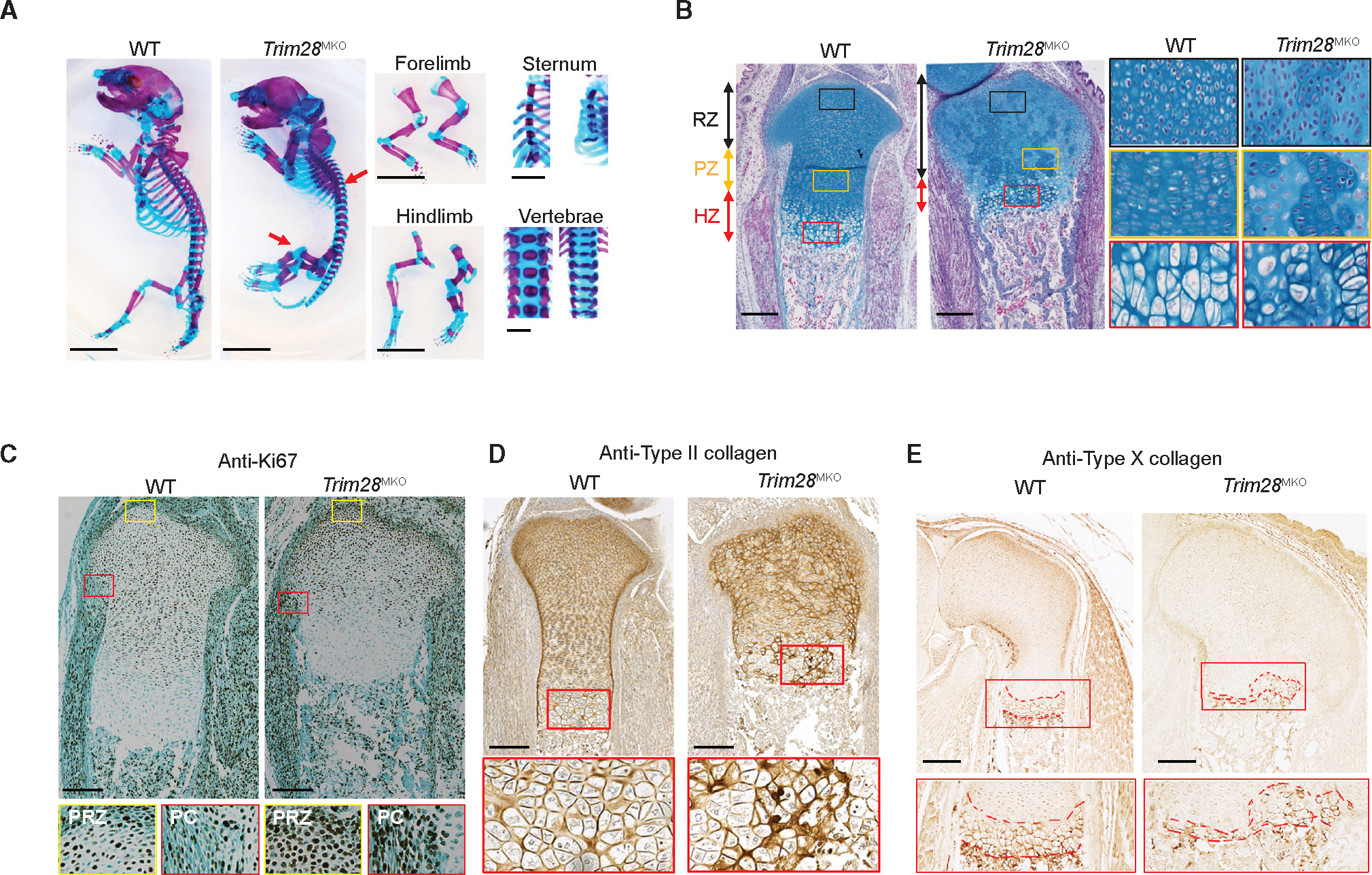
The striking disorganization of the Trim28MKO GP motivated us to evaluate how TRIM28 loss affected the cellular composition and lineage hierarchy within GPs. We utilized single-cell RNA sequencing (RNA-seq) analysis to characterize cells isolated from wild-type (WT) and Trim28MKO hindlimb GPs at the same stage as our phenotypic studies (E17.5; Figure S2A). By unsupervised clustering, cells were categorized into nine clusters (Figure 2A) and annotated based on their transcriptomic profiles (Figure S2B; Table S1). The Trim28MKO GPs contained fewer upper zone (cluster 0) and column-forming (cluster 2) chondrocytes but more borderline chondrocytes (a chondrocyte type located at the periphery of the GP immediately adjacent to the perichondrium that behaves as transient precursor cells,28 cluster 4) compared with WT GPs (Figures 2B and 2C). Notably, a large unique cell cluster (cluster 1, 42.3% of total Trim28MKO GP cells, hereafter called the Trim28MKO-U cluster, where “U” stands for “unique”) was identified in Trim28MKO GPs (Figures 2B and 2C). This cell type is virtually absent in the counterpart E17.5 WT GP (0.14%) and is likewise not enriched in limbs of younger WT embryos (E10.5: 0.43%; E11: 0.06%; E12: 0.48%),29 suggesting that this cluster is unique to Trim28MKO mice. Compared with the combined GP cells from WT and Trim28MKO, the Trim28MKO-U cluster was enriched in genes involved in endochondral ossification, PI3K-AKT and mTOR signaling, neural crest differentiation, adipogenesis, axon guidance, and synaptic vesicle cycle (Figure 2D). Notably, key SSC markers,6 including Grem1,30 Cd200,11 Prrx1,31 Nes,32 and Ctsk,33 as well as genes that belong to embryonic stem cell (ESC) pluripotency and PluriNetWork pathways were enriched in the Trim28MKO-U cluster (Figures 2E and 2D).
Figure 2. Loss of TRIM28 generates a new cell cluster abundant in SSCs in the growth plates.
(A) Distribution of 13,683 qualified single cells from WT (6,235) and Trim28MKO (7,448) hindlimb GPs in 8 clusters were visualized by uniform manifold approximation and projection (UMAP).
(B) UMAP in (A) is divided into two genotype-specific UMAPs.
(C) The proportion of each cell cluster in total WT or Trim28MKO hindlimb GPs.
(D) Enriched Wikipathways and KEGG pathways in Trim28MKO-U cluster versus all cells, which includes total WT and Trim28MKO cells.
(E) Violin plots of the expression of previously reported mesenchymal progenitor/SSC marker genes in each of the eight cell clusters identified in (A).
(F) Distribution of SSCs in scRNA-seq UMAPs (the same as B). The criteria for defining SSCs here are cells positive for Itgav and Cd200 and negative for Ptprc, Tek, Thy1, Enpep, and Eng in mRNA expression. Dashed black line indicates the boundary of the Trim28MKO-U cluster.
(G) Colony-formation assay initiated by seeding 1,000 (top panels) or 100 (bottom panels) rib GP cells. Clones containing at least 50 cells from the 100-cell seeding group are quantified in the bottom (n = 6). p value = 0.0081. Scale bar, 1 cm. Comparisons are conducted by Student’s t test, two tailed. Data are presented as mean ± SEM.
(H) Representative flow cytometric analysis of the proportion of SSCs (CD45− Ter119− TIE2 CD51+ 6C3− THY1.2− CD105− CD200+) and pre-BCSPs (CD45− Ter119− TIE2− CD51+ 6C3 THY1.2− CD105− CD200−) in hindlimb GPs (n = 3 for each genotype). The representative flow image in the bottom shows the Trim28MKO sample has a population shift from pre-BCSP to SSC when gated by CD200 expression. p values are shown in the figure. All comparisons are conducted by Student’s t test, two tailed. Data are presented as mean ± SEM.
We thus hypothesized that the Trim28MKO GPs, especially the Trim28MKO-U cluster, harbor more SSCs than WT GPs. By using a well-established panel of SSC markers,11 we found that Trim28MKO GPs contained proportionally more SSCs (cells positive for Itgav and Cd200 and negative for Ptrprc, Tek, Thy1, Enpep, and Eng11), most of which were in the Trim28MKO-U cluster (55.6%; Figure 2F). Furthermore, Trim28MKO rib GP cells have stronger colony-forming activity ex vivo (Figure 2G). In addition, we found a 4- to 5-fold increase in SSCs and a decrease in SSC progeny cells—the bone, cartilage, and stromal progenitors (pre-BCSPs)—from Trim28MKO GPs (Figure 2H). The overexpanded SSC pool and the emergence of a new cell cluster (Trim28MKO-U), combined with the diminished pre-BCSPs and two chondrocyte types (upper zone and column-forming chondrocytes) in Trim28MKO GPs, indicates that loss of TRIM28 impairs SSC lineage fidelity during skeletogenesis.
Loss of TRIM28 endows SSCs with NCC-like properties while impairing osteochondrogenesis
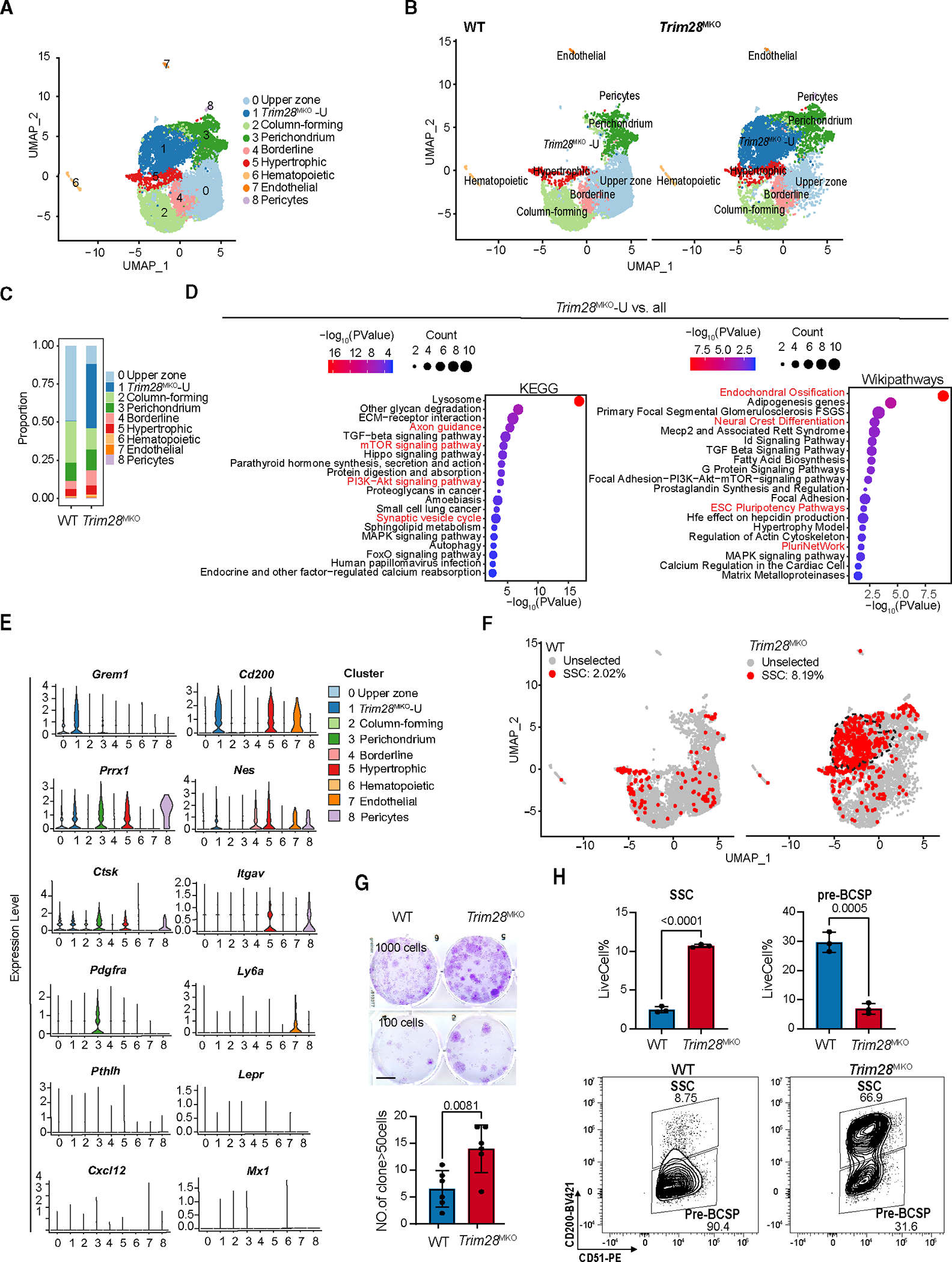
To further decipher how SSC lineage specification is affected by TRIM28 loss, we investigated the transcriptomic difference between WT and Trim28MKO SSCs. We found that Trim28MKO SSCs had a global reduction in osteochondrogenic markers34 (Figure 3A). Ex vivo cultured fluorescence-activated cell sorting (FACS)-isolated GP SSCs (CD45− Ter119− TIE2− CD51+ 6C3− THY1.2− CD105− CD200+)11 or total rib GP cells from Trim28MKO mice had impaired osteogenesis and chondrogenesis (Figures 3B, S3A, and S3B). These data demonstrate that loss of Trim28 restricts the osteochondrogenic potential of SSCs.
Figure 3. Loss of TRIM28 endows SSCs with neural crest cell-like prosperities while impairing osteochondrogenesis.
(A) Dot plots comparing the expression of selected lineage/niche markers in total WT SSCs versus Trim28MKO SSCs.
(B) Ex vivo chondrogenesis (top panels) and osteogenesis (bottom panels) of FACS WT or Trim28MKO GP SSCs. Representative staining is shown (n = 3 per genotype). Scale bar, 400 μm.
(C) UMAP of 5 subclusters of SSCs from combined WT and Trim28MKO hindlimb GP cells (top panel). Genotype-specific UMAPs of SSCs are shown in the bottom panels.
(D) RNA velocity trajectory inference analysis of gross SSCs (left). The separated WT (top panel) and Trim28MKO (bottom panel) velocity trajectories are shown on the right.
(E) Proportion of each SSC subtype in WT or Trim28MKO GP SSCs.
(F) Enriched WiKiPathways and MSigDB_Hallmarks in Trim28MKO-NSSCs versus all SSCs (WT plus Trim28MKO).
(G) Dot plots comparing the expression of osteochondrogenic marker genes in WT and Trim28MKO root-SSCs, Trim28MKO-NSSCs, WT chondro- and osteo-SSCs.
(H) Transcriptomic profiles of Trim28MKO SSCs (MKO), but not WT SSCs (WT), show a strong similarity with neural crest cells (NCCs) according to Spearman correlation analysis.
(I) Pseudo-bulk-level transcriptomic similarity analysis of NCC and SSC subtypes. The size of dots represents the cell number of each subcluster.
(J) FACS Trim28MKO GP SSCs exhibit more potent neurogenic differentiation than WT SSCs. Representative bright-field images are shown in the left panels, and percentages of neuron-like cell/total cell and neurite length/neuron-like cell are quantified in the right panel (n = 4 per genotype). Scale bar, 100 μm. p < 0.0001. Data comparisons are conducted by Student’s t test, two-tailed. Data are presented as mean ± SEM.
(K) Trim28MKO SSCs express higher levels of neurogenic markers than WT SSCs after 5 days of neurogenic induction (n = 3 per genotype). p values are shown in the figure. Data comparisons are conducted by Student’s t test, two-tailed. Data are presented as mean ± SEM. See also Figures S3 and S4 and Table S2.
SSC is a heterogeneous population with cells at varying priming stages (chondro-SSC, osteo-SSC, stromal-SSC etc.), which can be discerned by their subtle transcriptomic differences.35 To evaluate whether TRIM28 regulates SSC identity and priming, we applied unsupervised subclustering and cell trajectory analyses (Figures 3C and 3D) and identified the “root SSC” (cluster 1, Figure 3D); chondro-SSC (cluster 2); osteo-SSC (cluster 3); and “perichondrial-SSC” (cluster 4) subclusters35,36 (Figures S4A and S4B; Table S2). We found that in Trim28MKO SSCs, the proportion of chondro- and osteo-SSCs was reduced 3–4 times (Figure 3E). Interestingly, 49.2% of all Trim28MKO SSCs belong to a new SSC subcluster (cluster 0, hereafter called Trim28MKO-NSSCs) (Figures 3C–3E). 90.1% of this SSC subcluster belongs to the Trim28MKO -U cluster (Figure S4C). Moreover, this new SSC subcluster had high expression of Grem1 (Figure S4B) and was enriched with neural crest differentiation, ESC pluripotency, spinal cord injury, and mTORC1 signaling genes (Figure 3F). At the same time, the expression of osteochondrogenic markers was decreased in both the Trim28MKO root-SSCs (versus WT root-SSCs) and Trim28MKO-NSSCs (versus WT chondro-SSCs or WT osteo-SSCs) (Figure 3G).
The above data suggest that Trim28 loss impairs the osteochondrogenic priming of root-SSCs and directs them toward a new fate, that of Trim28MKO-NSSCs, with an altered SSC identity. Upregulation of pluripotency genes along with the activation of neural crest differentiation pathway and neural genes (Figure 3F) suggests that SSCs adopt a NCC-like fate following Trim28 deletion. NCCs are derived from the dorsal neural tube of vertebrate embryos and undergo an epithelial-to-mesenchymal transition prior to differentiating into either ectoderm lineage cells, including neurons and glia cells, or mesoderm lineage cells, such as melanocytes, osteoblasts, and chondrocytes.8 Notably, mesoderm-derived SSCs can also be reprogrammed to a neural crest-like state to direct regeneration of the adult mandible.37 To test whether TRIM28 establishes a barrier to restrict SSCs from reprogramming toward the neural crest-like state, we compared the single-cell RNA-seq (scRNA-seq) data from our WT and Trim28MKO GPs with a dataset published from mouse embryonic NCCs.29 We found that NCCs have a much higher degree of similarity with Trim28MKO SSCs (correlation coefficient = 0.93) than WT SSCs (correlation coefficient = 0.53) (Figure 3H). By conducting a pseudo-bulk-level transcriptomic similarity analysis, we found that the root-, osteo-, and chondro-subclusters of Trim28MKO SSCs were all shifted toward NCCs and away from their WT counterparts (Figure 3I). Moreover, the largest descendant subcluster of the Trim28MKO root-SSCs, the Trim28MKO-NSSCs (Figure 3D), are more preferentially primed toward the NCC state (Figure 3I).
Next, we investigated whether Trim28MKO SSCs are functionally similar to NCCs by evaluating their potential to differentiate into neuron-like cells ex vivo. We conducted neural differentiation experiments on FACS SSCs from the GPs of WT and Trim28MKO mice. Classical neuronal appearance and expression of neurogenic markers (Mapt2, Tubb3, and Nes) was enhanced in Trim28MKO SSCs after induction in mesenchymal stem cell (MSC) neurogenic media compared with WT SSCs (Figures 3J and 3K). Similar preferential neurogenic tendency was observed in the Trim28MKO GP cells (Figures S3C and S3D). Collectively, these results suggest that loss of Trim28 leads to overactivated self-renewal, loss of identity, impaired osteochondrogenic priming, and adoption of NCC-like features of SSCs.
TRIM28 loss changes the epigenetic landscape and derepresses neural genes
To investigate how the transcriptomic changes we observed in scRNA-seq correlate with potential TRIM28-mediated epigenetic silencing, we performed multi-omics (epigenomic [H3K9me3, H3K27ac, and DNA methylation] and transcriptomic [bulk RNA]) profiling of WT and Trim28MKO rib GPs. Bulk RNA-seq (Figure 4A; Table S3) identified PI3K-AKT-mTORC1 signaling as the most upregulated signaling pathway in Trim28MKO GP cells (Figure 4B). Also, other major changes include a global activation of TEs (mainly long terminal repeat [LTR] and long interspersed nuclear element [LINE] species) (Table S4).
Figure 4. TRIM28 loss changes the epigenetic landscape and derepresses neural genes.
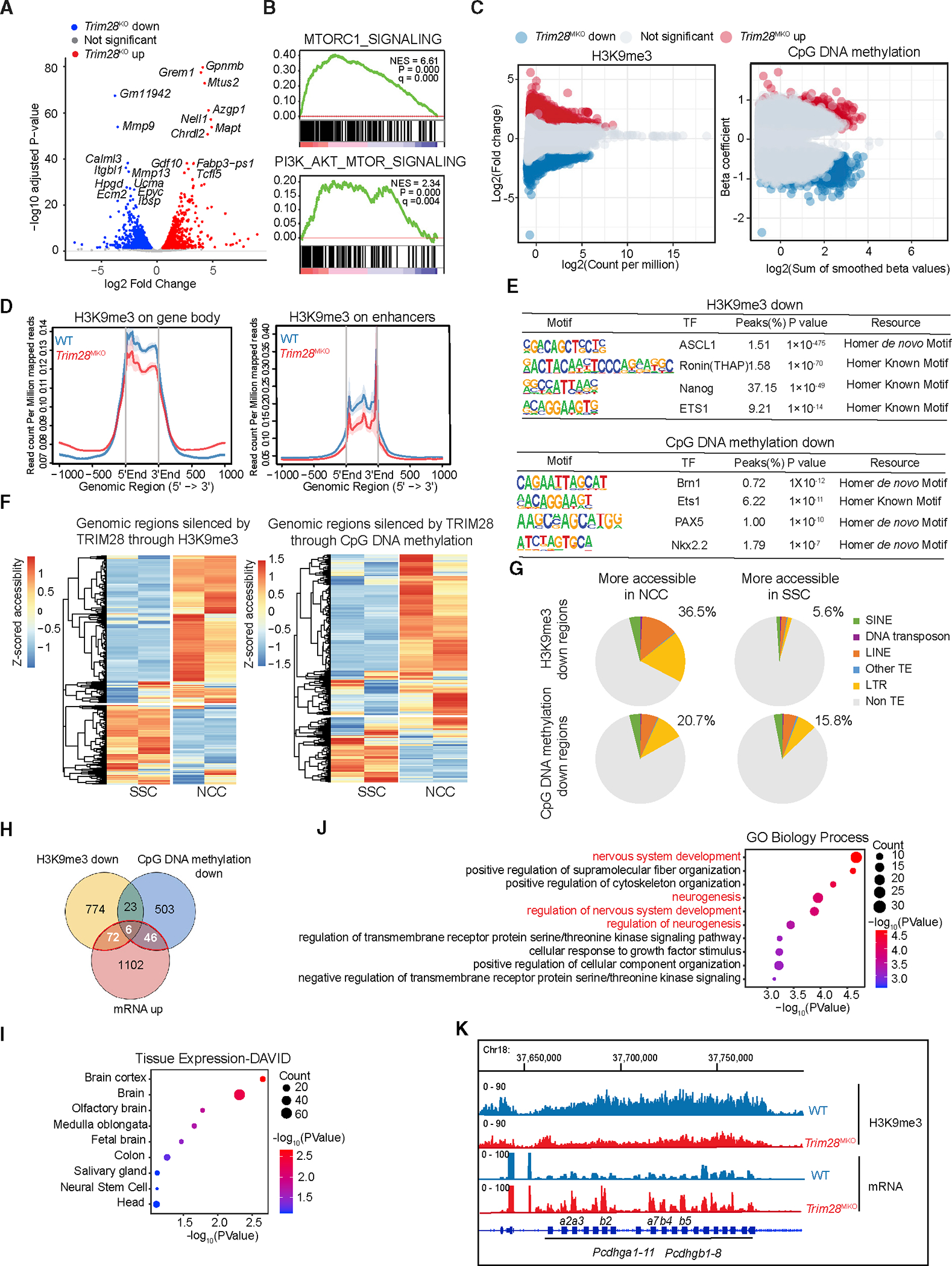
(A) Volcano plot of RNA-seq data demonstrating significant changes in gene expression between WT and Trim28MKO rib GP cells. Top 10 up- and down-regulated genes are shown.
(B) mTORC1 and PI3K-AKT pathways are enriched in Trim28MKO samples by gene set enrichment analysis (GSEA).
(C) Global changes of H3K9me3 (left panel) and DNA methylation (right panel) caused by Trim28 deletion.
(D) Trim28MKO GP cells have decreased global H3K9me3 modifications at gene bodies (left panel) and enhancers (right panel).
(E) Regions with down-regulated H3K9me3 (upper panel) and CpG DNA methylation (lower panel) show enriched binding motifs of TFs related to pluripotency and neurogenesis.
(F) Chromatin accessibility status of SSCs and NCCs at the genomic regions that are silenced by TRIM28 through H3K9me3 (left panel) and CpG DNA methylation (right panel).
(G) Proportion of TRIM28-silenced regions that contain TEs. The chromatin accessibility information is from (F).
(H) Venn diagram of genes with upregulated mRNA expression (mRNA up), genes silenced by TRIM28 through H3K9me3 (H3K9me3 down), and genes silenced by TRIM28 through CpG DNA methylation (CpG DNA methylation down) identified 124 TRIM28-silenced genes.
(I) DAVID tissue expression analysis of the 124 TRIM28-silenced genes.
(J) DAVID GO biology process analysis of the 124 TRIM28-silenced genes.
(K) TRIM28 silences the gamma-protocadherin gene cluster through H3K9me3.
The epigenetic profiling analysis showed that among 86,587 total H3K9me3 peaks, Trim28MKO rib GPs had 11,105 loci significantly decreased and10,528 loci significantly increased (false discovery rate [FDR] % 0.05, abs(log2FC) ≥ 1) compared with WT cells (Figure 4C; Table S5). In addition, we observed a global decrease in H3K9me3 modifications in the gene body and enhancer regions of the Trim28MKO GP cells (Figure 4D). Using whole-genome bisulfite sequencing (WGBS), we identified 2,472 decreased and 2,143 increased differentially methylated regions (DMRs) (p % 0.01) in Trim28MKO versus WT cells (Figure 4C; Table S6). By motif analysis using HOMER (v.4.11.1), we found that the regions silenced by TRIM28 through H3K9me3 marks were enriched with the transcription factor (TF) binding motifs of Nanog and Ronin (TFs essential for maintaining pluripotency), EST1 (neural crest regulator), and ASCL1 (a master regulator of neurogenesis) (Figure 4E). Similarly, regions silenced by TRIM28 through CpG DNA methylation were also enriched with binding motifs of neurogenesis regulators, such as Brn1, Ets1, PAX5, and Nkx2.2 (Figure 4E). These findings demonstrate that TRIM28 loss markedly changes the chromatin silencing landscape to a more pluripotent and neurogenic state.
A recent study reported that mechanical stress promotes the transition of SSC to NCC with a distinctive chromatin accessibility rearrangement on genes and TEs,37 reminiscent of our observation in Trim28MKO GP cells. We speculated that TRIM28 loss caused a similar shift in the epigenetic landscape to that of SSCs under mechanical stress. To determine if TRIM28 silenced regions (the 11,105 H3K9me3 loci and the 2,472 DMRs that were decreased in Trim28MKO GP) are pivotal for the maintenance of SSC identity, we compared the chromatin states of SSC and NCC at those TRIM28-silenced regions by analyzing the published assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) data.37,38 We found that most TRIM28-silenced regions (65.1% H3K9me3 silenced regions, 71.3% of DNA methylation silenced regions) are more accessible in NCCs than in SSCs (Figure 4F). In addition, these highly accessible regions in NCCs are abundant in TEs (36.5% of H3K9me3 silenced regions, 20.7% of CpG methylation silenced regions), among which LTR and LINE are the most enriched types (Figure 4G). This is in line with the previous findings that LINE is the largest TE class that contains active enhancers in the NCCs.39
To explore the contribution of TRIM28-repressed genes to the NCC-like phenotype in Trim28MKO cells, we conducted an integrated analysis of the RNA-seq and epigenetic profiling data. We identified 124 candidate genes that were derepressed in Trim28MKO rib GPs with reduced H3K9me3 or CpG DNA methylation at their promoter or enhancer regions (Figure 4H; Table S7). According to DAVID40 tissue expression and Gene Ontology (GO) analysis, 61% of the 124 genes were enriched in brain and neural tissues (Figure 4I), with a large portion belonging to neurogenesis and nervous system development GO terms (Figure 4J; Table S7). These genes include glial-specific genes (S100b and Ndrg4) and neuronal genes (Slc6a17, Kif1a, Cspg4/Ng2, Prph, Ntn4, and Impact) (Table S7). For example, the large gamma-protocadherin gene (Pcdhg) cluster, which encodes a family of cell surface homophilic-binding proteins important in axon guidance, synapse formation, and neuronal survival,41 was repressed by TRIM28 through H3K9me3 (Figure 4K). Moreover, we found that among the 124 genes, 39 are silenced through TE overlapped enhancers or promoters, and 15 of these are neural genes (Table S7). This data suggests that the NCC-like phenotype caused by Trim28 deletion is due to the derepression of neural genes, a large portion of which are TE associated. Thus, TRIM28 forms an epigenetic barrier that restricts neural gene expression to fortify SSC identity.
TRIM28 epigenetically silences Grem1 expression to suppress PI3K-AKT-mTORC1 signaling
The epigenetic landscape of cells can modulate the tissue microenvironment by controlling the expression of signaling regulators.42 Also, SSC fate is profoundly impacted by extracellular signaling.11 Thus, we next explored the signaling pathways altered by TRIM28 loss. The PI3K-AKT-mTORC1 pathway was the most upregulated signaling pathway in Trim28MKO GP (Figure 4B) and is known to control SSC self-renewal in the GP.25 It is significantly elevated in both the Trim28MKO-U cell cluster and the Trim28MKO-NSSC subcluster (Figures 2D and 3F). We confirmed the activation of PI3K-AKT-mTORC1 signaling in the GPs by immunoblot and immunohistochemistry (Figures 5A and 5B). Our observation that mTOR is activated, as a consequence of TRIM28 loss, is in seeming opposition to previous findings, which reported that TRIM28 activates mTOR signaling via formation of ubiquitin E3 ligase complexes with MAGE3/6.43 However, this mechanism is not expected to be present in our system because MAGE3/6 are oncoproteins found only in cancer or germ cells43,44 and are not expressed in GPs (RPKM = 0 in GP bulk RNA-seq; Table S3). Notably, Grem1, which encodes the established SSC marker30 and extracellular signaling molecule required for skeletal development and homeostasis,45 GREM1, is one of the most upregulated genes in the Trim28KO GP (Figure 4A) and is highly enriched in both the Trim28KO-U cell cluster and in Trim28KO-NSSCs (Figures 2E and S4B). GREM1 is known as an antagonist of BMP signaling and was recently found to activate PI3K-AKT signaling in endothelial cells.46 In addition, GREM1+ mesenchymal cells have higher PI3K-AKT signaling than their GREM1− counterparts.30 Therefore, we hypothesized that upregulation of GREM1 is responsible for the activation of PI3K-AKT-mTORC1 signaling in Trim28MKO cells. In support of this hypothesis, we found that Trim28KO GPs have higher expression of GREM1 protein than WT GPs (Figure 5C) and that stimulation with recombinant GREM1 protein induced PI3K-AKT-mTORC1 signaling in immortalized chondrocyte-like cells (ADTC5) as indicated by a rapid increase in phosphorylated AKT (pAKT; an mTOR upstream activator) and delayed increase in p4E-BP1 and p-P70S6K (mTORC1 downstream effectors) (Figure 5D). Moreover, knocking down Grem1 with short hairpin RNA (shRNA) in Trim28MKO rib GP cells normalized pAKT and pS6 levels (Figure 5E). We observed no difference in the major downstream effectors of BMPs pSMAD1,5,8 in Trim28MKO GPs (Figure 5A). This is consistent with a previous study reporting that BMP signaling is higher in Grem1-expressing than in Grem1-negative mesenchymal cells.30 These results together suggest that in the context of the developing GP, where BMP levels are high, GREM1 is less effective as a BMP antagonist but acts as an activator of the PI3K-AKT-mTORC1 pathway.
Figure 5. TRIM28 epigenetically silences Grem1 expression to suppress PI3K-AKT-mTORC1 signaling.
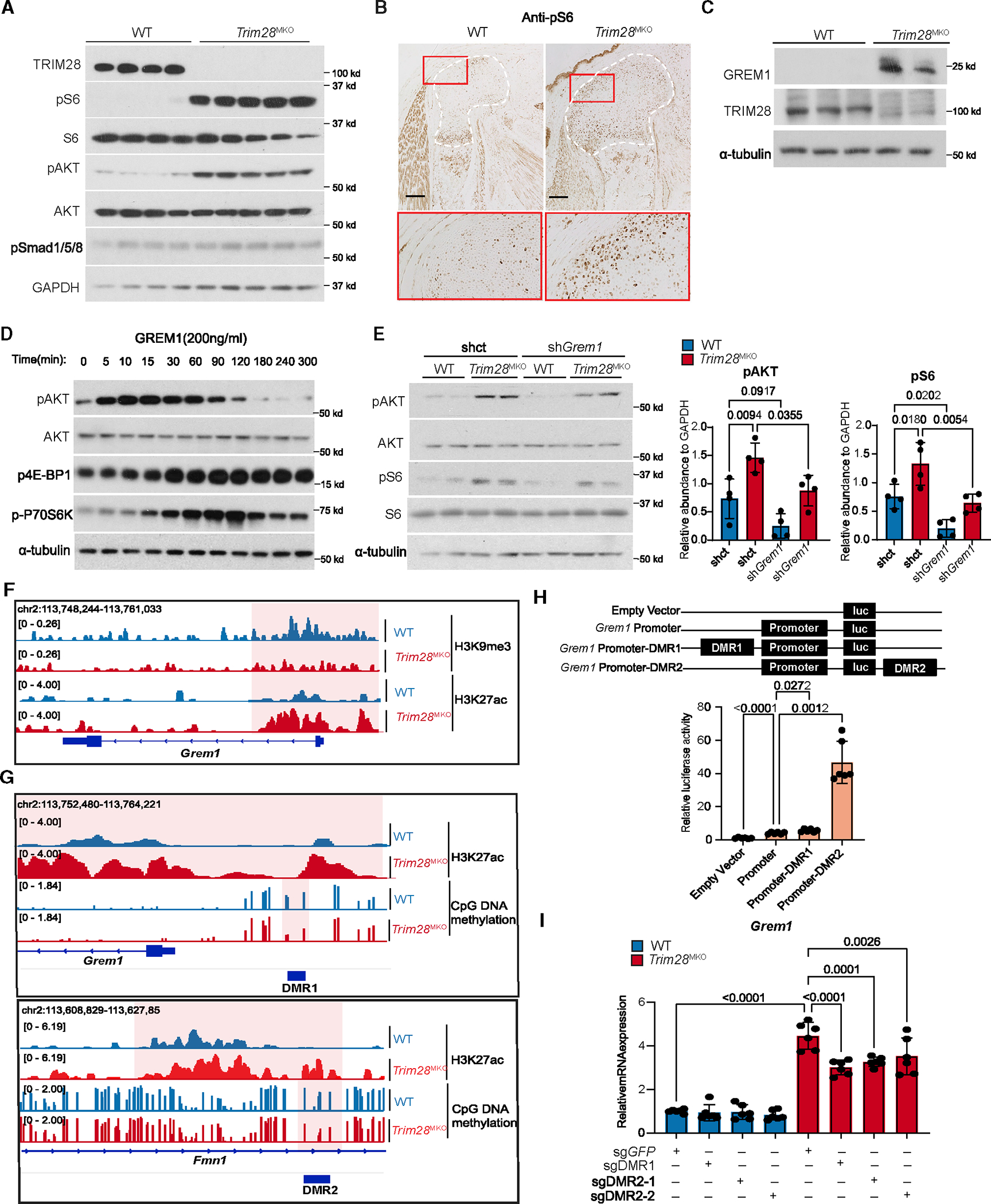
(A) pS6 (an mTORC1 downstream effector) and pAKT (an mTORC1 upstream activator) levels are increased in E17.5 Trim28MKO rib GPs (WT, n = 4; Trim28MKO, n = 5).
(B) pS6 level is increased in E17.5 Trim28MKO mouse distal femur. GP areas are demarcated by white dashed lines. Red boxes are magnified in the bottom panel. Scale bars, 200 μm.
(C) GREM1 and TRIM28 protein expressions are inversely related in the rib GP cells (WT, n = 3; Trim28MKO, n = 2).
(D) mTORC1 and AKT pathways are activated by recombinant GREM1 in ATDC5 cells in a time-dependent manner.
(E) Levels of pAKT and pS6 were reduced by Grem1 gene knockdown with shRNA targeting Grem1 (shGrem1) in cultured Trim28MKO rib GP cells compared with control shRNA (shct). Densitometry quantification (n = 4) is shown on the right. p values are shown in the figure. Comparisons are conducted using one-way ANOVA analyses with Tukey’s post-hoc test. Data are presented as mean ± SEM.
(F) H3K27ac and H3K9me3 marks at the promoter region of the Grem1 gene.
(G) The occupancy status of CpG DNA methylation and H3K27ac at DMR1 and DMR2. The most distinctive changes are highlighted, and DMR regions are indicated with dark blue bars.
(H) Luciferase reporter assays indicating the transcriptional activity of the Grem1 enhancers (DMR1 or DMR2) and promoter in ATDC5 cells. Schematics of the reporter constructs are in the top panel. Empty pGL3-basic vector was used as a negative control. Representative quantified results are shown in the bottom panel (n = 6). p values are shown in the figure. Comparisons are achieved using Student’s t test, two-tailed. Data are presented as mean ± SEM.
(I) Grem1 mRNA levels in control (sgGFP) and CRISPR-Cas9-mediated DMR1 (sgDMR1) or DMR2 (sgDMR2–1, sgDMR2–2) deleted WT and Trim28MKO rib GP cells (n = 6). p values are shown in the figure. Comparisons are conducted using one-way ANOVA analyses with Tukey’s post-hoc test. Data are presented as mean ± SEM.
See also Figure S5.
To investigate how TRIM28 regulates Grem1 expression, we analyzed epigenetic changes at the Grem1 locus in WT versus Trim28MKO GP cells. TRIM28 is known as a crucial silencing factor of TEs, and its loss leads to activation of TEs and their nearby genes in ESCs and neural progenitor cells.47–50 However, the two TEs adjacent to the Grem1 loci with reduced H3K9me3 marks did not gain H3K27ac modification in Trim28MKO GP cells (Figure S5A). Likewise, their deletion did not affect Grem1 expression (data not shown), thus they are unlikely to be responsible for the upregulation of Grem1. We did observe, however, a loss of H3K9me3 and a gain of H3K27ac modification in Trim28MKO rib GP cells at the Grem1 promoter (Figure 5F). We additionally identified two DMRs (DMR1 and DMR2, respectively; Figure 5G) as hypothetical Grem1 enhancers. DMR1 is located approximately 1,000 bp upstream of the Grem1 transcription start site (TSS) and DMR2 is located between exons 18 and 19 of the neighboring Fmn1 gene (Figure S5A). DMR1 is moderately conserved and located within a proximal enhancer as annotated by ENCODE51 (Figure S5A), and DMR2 contains a 603 bp highly conserved region with 82.4% identity to the human counterpart (Figures S5C and S5D). The DMR2 homologous human genomic region (hDMR2) contains a cis-regulatory element annotated by ENCODE (Figure S5B). We found that DMR1 and DMR2 and their surrounding regions had more H3K27ac modifications in Trim28MKO cells than in WT cells (Figure 5G). Moreover, DMR2 and hDMR2 are located within the Grem1/GREM1 topologically-associated domain (TAD) in mouse myoblasts52 and H1-ESCs (H1 human ESC line),53 respectively (Figures S5E and S5F). To determine whether these two DMRs affect transcription from the Grem1 promoter, we cloned them and conducted luciferase reporter assays. We found that both DMRs significantly increased the transcriptional activity of the Grem1 promoter, with DMR2 having higher activity (Figure 5H). Notably, deletion of either of the two DMRs using single-guide RNA (sgRNA)/Cas9 in Trim28MKO primary GP cells resulted in a significant decrease in Grem1 expression (Figure 5I). These data suggest that TRIM28 silences Grem1 expression by establishing H3K9me3 marks at the Grem1 promoter and establishing DNA methylation at the two Grem1 enhancers.
Hyperactivated GREM1-PI3K-AKT-mTORC1 signaling contributes to the skeletal defects of Trim28MKO mice
To test whether GREM1-PI3K-AKT-mTORC1 signaling accounts for the skeletal defects observed in Trim28MKO mice, we injected pregnant mice intraperitoneally with an AKT inhibitor (LY294002, 20 mg/kg/dose) or an mTORC1 inhibitor (rapamycin 3 mg/kg/dose) on days E12.5 and E14.5 and analyzed fetuses at E17.5. Trim28MKO skeletal phenotypes were partially, but distinctively, rescued in both the LY294002- and rapamycin-injected Trim28MKO mice as indicated by the expanded rib cage, increased bone length, and thinning of the femoral and tibial GPs (Figure 6A). Histologically, GP zonal organization, chondrocyte size, and increased proliferation were all rescued (Figures 6B and S6A).
Figure 6. Hyperactivated GREM1-PI3K-AKT-mTORC1 signaling contributes to the skeletal defects of Trim28MKO mice.
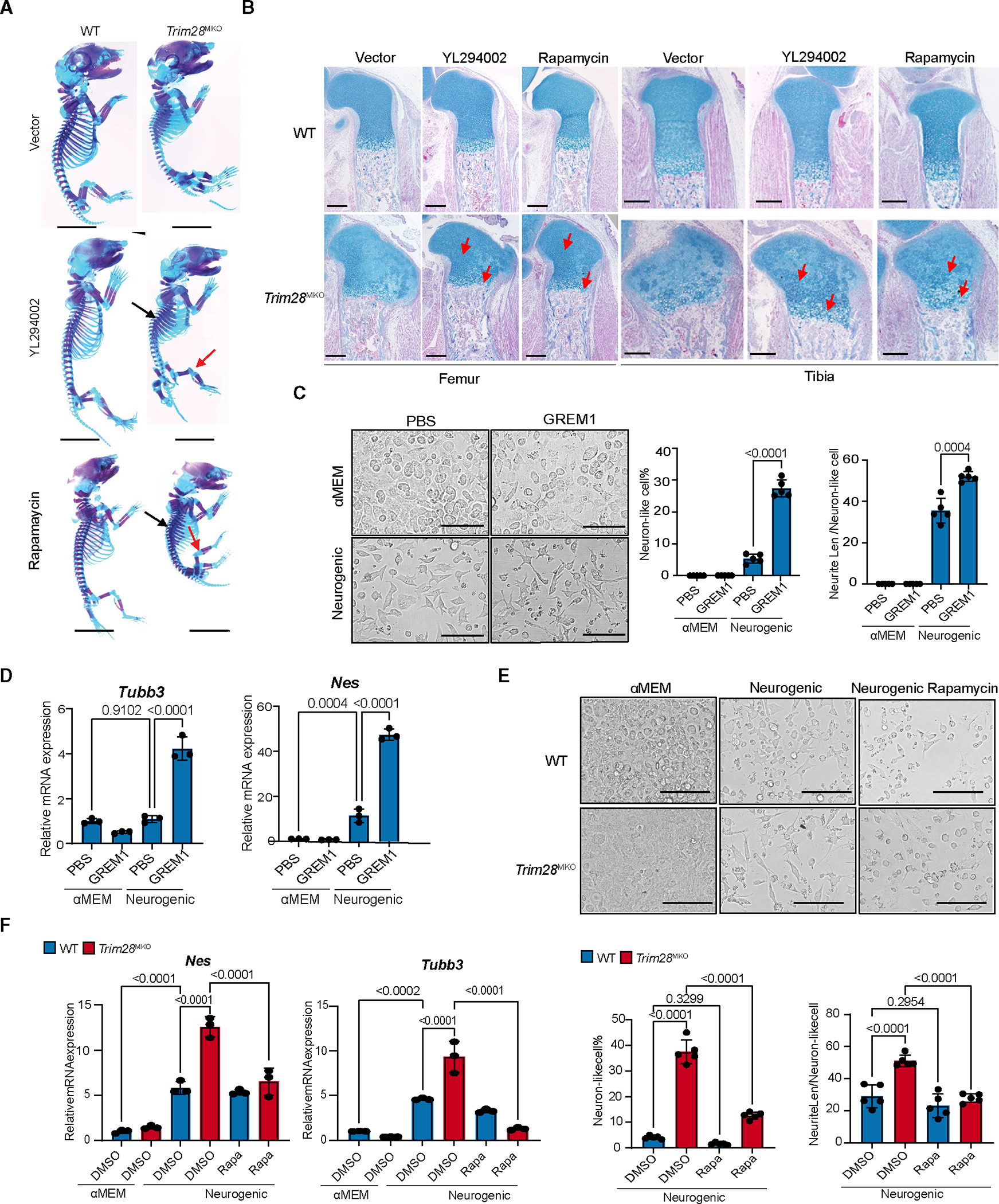
(A) Skeletal phenotypes of the E17.5 vector-, YL294002-, or rapamycin-treated mice visualized with Alcian blue and Alizarin red staining. Red arrows indicate reduced knee cartilage and black arrows indicate expanded rib cages in YL294002-and rapamycin-treated mice. Scale bars, 0.5 cm.
(B) Representative Alcian blue Hematoxylin/Orange G stain of proximal tibial (right) and distal femoral (left) sections from mice treated with vector, YL294002, and rapamycin. Red arrows indicate rescued proliferating and hypertrophic zones. Scale bars, 200 μm.
(C) Ex vivo neurogenesis of GP cells isolated from WT hindlimb treated with either GREM1 (300 ng/mL) or PBS in either neurogenic media or αMEM (n = 5 per group). p values are shown in the figure. Comparisons are achieved by one-way ANOVA analyses with Tukey’s post-hoc test. Data are presented as mean ± SEM. Scale bars, 100 μm.
(D) Expression of key neurogenic markers (n = 3 per group) in cells cultured as described in (C). p values are shown in the figure. Comparisons are achieved by one-way ANOVA analyses with Tukey’s post-hoc test. Data are mean ± SEM.
(E) Rapamycin (100 nM) treatment partially rescues the enhanced neurogenesis of hindlimb GP cells caused by Trim28 loss. DMSO was used as a negative control (n = 5 per group). p values are shown in the figure. Comparisons are achieved by one-way ANOVA analyses with Tukey’s post-hoc test. Data are presented as mean ± SEM. Scale bars, 100 μm.
(F) mRNA expression of key neurogenic markers from cells treated as described in (E) (n = 3 per group). p values are shown in the figure. Comparisons are achieved by one-way ANOVA analyses with Tukey’s post-hoc test. Data are presented as mean ± SEM.
Because previous studies found that GREM154 and PI3K-AKT-mTORC1 signaling promote self-renewal and neurogenesis of neural stem cells,55,56 we evaluated whether the GREM1-PI3K-ATK-mTORC1 axis contributed to the NCC-like phenotype of Trim28MKO cells. We found that recombinant GREM1 protein promoted neurogenic differentiation of WT GP cells (Figures 6C and 6D) and that this enhanced neurogenesis can be reversed by inhibiting mTORC1 with rapamycin treatment (Figure S6B). Moreover, rapamycin treatment inhibited the neurogenic differentiation of Trim28MKO GP cells (Figures 6E and 6F). These data demonstrate that the activation of GREM1-PI3K-AKT-mTORC1 signaling promotes the neurogenic tendency in Trim28MKO cells. Taken together, our data suggest that TRIM28 restrains GREM1-PI3K-AKT-mTORC1 signaling to safeguard skeletal development.
DISCUSSION
Our work shows the pivotal role of TRIM28-governed chromatin silencing during organogenesis of the skeletal system. We find that in GP SSCs, TRIM28 mediates H3K9me3 and DNA methylation preferentially at the enhancers and promoters (with an ample overlap with TEs) of neural genes and that the loss of this gene silencing redirects SSCs into other fates of ectoderm-derived cells (including NCCs and neurons). It was found that during gastrulation, pluripotent epiblast cells are epigenetically primed for a default ectodermal fate, and their subsequent diversion toward endodermal or mesodermal paths requires a comprehensive and coordinated epigenetic rearrangement, especially through H3K9me3 and DNA methylation.4,5 Our findings extend this model by showing that later, during organogenesis, an epigenetic barrier established and maintained by TRIM28 remains crucial in defining the mesodermal lineage trajectory and in preventing its regression to the default ectodermal fate.
Previous studies reported that enhanced H3K9me3 or DNA methylation by loss of H3K9me3 demethylase KDM4B, or DNA demethylase TET1 and TET2, in SSCs does not significantly impact embryonic skeletogenesis13,14 but instead impairs SSC renewal and promotes SSC senescence at postnatal stages, resulting in stem cell exhaustion and low bone mass. Another study showed that targeted knock in of a catalytically inactive SuV39H1, a H3K9 methyltransferase, accelerates cellular senescence of MSCs. One possibility for these observed differences is that chromatin-modifying enzymes target heterochromatin at different genomic regions. We postulate, based on these studies and our data, that strengthening chromatin silencing by H3K9me3 and DNA methylation is crucial for securing skeletal organogenesis, but abnormally heightened or reduced chromatin silencing at postnatal stages can undermine SSC homeostasis and skeletal health.
Importantly, our findings reveal that TRIM28 silences Grem1 expression by augmenting H3K9me3 and DNA methylation at evolutionally conserved cis-regulatory elements. Hyperexpressed GREM1 in Trim28MKO cells activates the PI3K/AKT/mTORC1 pathway, which enhances self-renewal, partially promotes the neurogenic tendency of SSCs, and impairs skeletogenesis of the Trim28MKO mice. This highlights a model in which TRIM28 safeguards SSC identity by integrating the repression of non-skeletal genes with the establishment of a proper tissue microenvironment. Given that TRIM28 is widely expressed in other organ systems and is frequently altered in cancers and other diseases, whether and how TRIM28 safeguards stem cell function and impacts the tissue microenvironment in those developmental and pathological processes deserves further investigation.
Limitations of the study
Due to the SSC numbers in fetal GP being insufficient for generating high-quality epigenetic profiling data, we opted to use total fetal GP cells to perform epigenetic profiling studies and relied on scRNA-seq data to correlate the epigenetic changes with SSC activities. We expect that this may have decreased our ability to detect more subtle epigenetic changes, because of the heterogeneity of cell populations in the GP. In addition, we identified that TRIM28 regulates SSC renewal by silencing two Grem1 enhancers that are evolutionally conserved between mouse and human. However, because TRIM28 can regulate gene expression through silencing TEs, which are highly variable among species,18,47,48 it is unclear to what extent TRIM28 silences the NCC-state-related genes that contain TEs in their cis-regulatory elements in an evolutionally conserved manner. Interestingly, a recent study reported that species-specific TEs, acting as cis-regulatory elements, yield evolutionary conserved gene isoforms essential for development.57 It is possible that TRIM28 also uses similar evolutionally conserved mechanisms among species to suppress those genes controlled by TE cis-regulatory elements. To demonstrate this, further investigations should be done in human samples, and the data need to be thoroughly compared with the mouse data to understand their similarities and divergences.
Another consideration that should be made is that in this study we used Dermo1-Cre, instead of Prx1-Cre to delete TRIM28. Prx1-Cre is relatively more specific to the skeletal cells and was first attempted to utilize in this study. However, we failed to yield any Prx1-Cre; Trim28f/f deletion after trying multiple mating schemes; this is likely because Prx1-Cre is inserted in the vicinity of the Trim28 locus on chromosome 7, preventing our ability to obtain the desired genotypes (Prx1-Cre+; Trim28f/f).
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the lead contact, Tao Yang (Tao.Yang@vai.org).
Materials availability
Materials generated in this study are available upon request. All requests need to execute a suitable materials transfer agreement.
Data and code availability
All sequencing data generated in this study have been submitted to and deposited at NCBI GEO: GSE202060. Data are publicly available as of the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Trim28 f/f, Dermo1-Cre and Col2a1-Cre mice were purchased from Jackson lab (Cat#:018552, Cat#:008712, Cat#: 003554). Mice were housed in pathogen-free conditions under 12-h light and 12-h dark cycle with constant temperature(19–23°C) and humidity (55% ± 10%). Mice of both sexes were used in all experiments. Mice at development stages E17.5-E19.5 were used in this study. The exact age for each experiment is indicated in figure legend or in the methods. All animal experiments were approved by the Institutional Animal care and Use Committee (IACUC) at the Van Andel Institute and were conducted based on the guidelines from American Association for Accreditation of Laboratory Animal Care (AALAC).
METHOD DETAILS
Skeletal staining
Skeletal staining was performed as described previously.60 Briefly, skinned and eviscerated embryonic mice were fixed in 95% ethanol overnight, washed in acetone for 24 h and then placed in Alcian blue solution (0.015% Alcian blue 8GX in 20% acetic acid and 76% ethanol) overnight to stain cartilage. After washing with 95% ethanol for 3 h, mice were transferred to 2% KOH solution for 24–48 h, then stained with Alizarin Red S solution (0.005% Alizarin red S in 1% KOH) overnight to stain bone. Skeletons were cleared by incubation in 1% KOH with 20% glycerol for 2–3 days and stored in a mixture of ethanol and glycerol (1:1).
Microcomputed tomography (μ-CT)
μ-CT was performed by following the JBMR-recommended μ-CT guidelines using the SkyScan 1172 microCT imaging system (Bruker microCT, Kontich, Belgium). Scanning was carried out at 6.92-μm voxel size, 59 kV, 167 μA, 0.5-mm aluminum filter, with 0.4° rotation steps, exposure time of 380 ms and six-frame averaging. Reconstruction was carried out using the RNecon software (SkyScan). CTan and CTVol software were used for creating 3D images.
Histology
Skeletal tissues were fixed overnight at 4°C in 4% paraformaldehyde. After three washes in double distilled water, fixed tissues were dehydrated, paraffin-embedded and sectioned at a thickness of 5 μm. Alcian blue Hematoxylin/Orange G stain was performed using standard procedures.
Immunohistochemistry (IHC) and antibodies
Skeletal tissues sections were obtained as described above and incubated at 60°C for 3 h, dewaxed in xylene, rehydrated in gradient ethanol and antigen-unmasked in citrate buffer. Tissue sections were then incubated with 5% H2O2 for 10 min, permeabilized with 0.1% Triton X-100 in PBS for 5 min, blocked with normal serum blocker (NSB) buffer (5% normal serum, 1% BSA in PBS) for 1 h, then reacted with primary antibodies (in 1% BSA) overnight at 4°C. The next day, sections were washed three times with PBS, incubated with biotinylated secondary antibody for 1 h, and further processed with Vectastain® abc kit (Vector Laboratories) and DAB substrate kit (Vector Laboratories) for peroxidase reactions. Sections were counterstained with fast green or nuclear fast red. For type X collagen antibody IHC, tissues were digested with hyaluronidase (250U/mL, Sigma) at 37°C for 1 h, before H2O2 treatment. Antibodies used for IHC are anti-pS6 (Cell Signaling Technology; 4858S), anti-Ki67 (Thermo Fisher; MA5–14520), anti-Collagen II (Thermo Fisher; MA5–13026), anti-Collagen X (DSHB; X-AC9), anti-BrdU (Thermo Fisher; MA5–11285), anti-IHH (Millipore; MABF23), and anti-p4E-BP1 (Cell Signaling Technology; 2855T).
TUNEL assays
Immunofluorescent staining was performed on paraffin-embedded distal femoral GP sections from E17.5 WT and Trim28MKO mouse, using an in-situ Cell Death Detection Kit (Roche Diagnostics) according to the manufacturer’s instructions.
Rib and hindlimb GP cell isolation
Rib cage or hindlimb joint cartilage from E17.5 mouse embryos was dissected, and the surrounding muscles and other soft tissues were removed as much as possible. After two PBS washes, the rib cages or the joint cartilage was digested with Pronase (Roche life science), and Collagenase Type II (Gibco) as described.61 After digestion, the cells were washed twice with PBS, filtered through a 40 μm cell strainer and subjected to downstream experiments and analyses.
Cell lines and primary chondrocyte culture
Human embryonic kidney cell derivative 293T and human chondrogenic cell derived from teratocarcinoma AT805 ATDC5 cells were purchased from Millipore Sigma and cultured in DMEM medium supplied with 10% FBS and 1% penicillin-streptomycin (PS). Primary mouse rib and hindlimb GP cells were isolated as described above and cultured in α-MEM medium supplied with 10% FBS and 1% PS. For recombinant Gremlin1 protein stimulation, ATDC5 cells and primary rib GP cells were serum-starved in serum-free αMEM medium overnight, treated with 200 ng/mL recombinant Human Gremlin1 protein (PEPRPTECH, 120–42) in serum free αMEM medium for the indicated time prior to lysis and immunoblotting. For shRNA experiments, Trim28MKO rib GP cells were transfected with control shRNA or shRNA targeting the Grem1 gene. 2 days after transfection, cells were serum-starved in serum free α-MEM medium overnight prior to lysis. Colony-formation assays were conducted using either 100 or 1000 rib GP cells per well. Cells were grown in αMEM medium supplied with 10% FBS and 1% PS for 14 days, prior to fixation and staining with 2.5% Crystal violet solution. Clones with over 50 cells from the 100-cell seeding group were counted.
Immunoblotting and antibodies
Total protein from mouse tissue or cultured cells was extracted using SDS cell lysis buffer (150 mM NaCl, 2% SDS,10% glycerol, 50 mM Tris-HCl pH 7.5) supplied with1 × protease inhibitor cocktail (Sigma, P8340),1 mM PMSF, 2 mM NaVO3 and 5 mM NaF. 20 μg of protein from each sample was separated by SDS-PAGE gel electrophoresis, transferred to nitrocellulose, blocked with 5% non-fat milk in PBS with 0.5% Tween 20, followed by incubation with primary antibodies overnight at 4°C. Primary antibodies were recognized with horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary antibodies for 1 h, prior to chemiluminescent visualization. α-tubulin or GAPDH antibodies were used as loading controls. Primary antibodies used in this paper are anti-TRIM28 (Cell Signaling Technology; 4123S), anti-phospho-S6 (Cell Signaling Technology; 4858S), anti-S6 (Cell Signaling Technology; 2217S), anti-phospho-Akt (Cell Signaling Technology, 4060S), anti-Akt (Cell Signaling Technology; 4685), anti-GAPDH (Proteintech; 10494-I-AP), anti-GREM1 (ABGENT; AP12247B), anti-α-Tubulin (Thermo Fisher; T8203), anti-phospho-4E-BP1(Cell Signaling Technology; 2855T), and anti-phospho-p70 S6 Kinase (Cell Signaling Technology; 9234T), anti-phospho-pSmad1/5/8 (Cell Signaling Technology; 9511s).
Quantitative RT-PCR
Total RNA from cultured cells or mouse tissue was extracted with TRIZol reagent (Invitrogen), and further purified with a RNeasy kit (Qiagen) following the manufacturer’s protocol. First-strand cDNA was synthesized with a VILO cDNA synthesis kit (Invitrogen). qRT-PCR was performed using SYBR Green QPCR Master Mix (Invitrogen). The Gapdh (neurogenesis assay) or Actb (all other qRT-PCR analysis) gene was used as the internal control for normalization. The mRNA levels of genes were normalized to the average levels of Actb or Gapdh, relative expression was calculated as the ratio to control (defined as 1).
Bulk RNA-seq and data processing
Total RNA from WT and Trim28MKO mouse rib GP was extracted as described above. RNA was sequenced by Novogene RNA-seq service (Hongkong, China). The raw reads were mapped to the Mus musculus genome (mm10), and the read counts on transcripts/gene exons were estimated by Subjunc v1.6.462 and featureCounts v1.6.4.63 The estimated read counts were used as the input for differential gene expression analysis using edgeR v3.32.1.64 The Benjamini-Hochberg adjusted p values (i.e., false discovery rate, FDR) were taken to determine differentially expressed genes (DEGs). In total, 2,238 genes were defined as DEGs between Trim28MKO and WT samples (FDR ≤0.05). To visualize the RNA-seq data in the IGV genome browser, bigwig files from the mapped reads were created. Briefly, the mapped reads were converted into bedGraph format to compute the coverage with bedtools v2.29.2 (genomecov -ibam -bg -split -scale 1). bedGraph files were sorted (LC_COLLATE = C sort -k1,1 -k2,2n) before converting to bigwig format using bedGraphToBigWig (ucsc-2020.06.11). The chromatin size was downloaded from http://hgdownload.cse.ucsc.edu/goldenPath/mm10/bigZips/mm10.chrom.sizes. We used RepEnrich265 to estimate transposable element expression in WT and Trim28MKO. Briefly, the transposable element counts were generated by using Repenrich2 algorithm with mm10 genome, and edgeR v3.32.164 was used to identify differential expressions.
Whole genome bisulfite sequencing (WGBS)
Genomic DNA was isolated from rib GP cells of E17.5 mouse embryos, using Quick-DNA Miniprep Plus kit (Zymo Research; Irvine, CA) according to the manufacturer’s protocol. 5 μg of DNA from each sample was sent to Fulgent Genetics (Temple City, CA). Fulgent performed the library prep and sequencing. The library was sequenced as 150 bp paired end reads on an Illumina HiSeq2500 platform to obtain 30×coverage. Adaptor sequences and low-quality reads were trimmed from WGBS sequences using Trim Galore (v0.6). Trimmed reads were aligned to the reference index that was generated using BISulfite-seq CUI Toolkit, BISCUIT, (0.3.16; github) based on mm10 reference genome (“index” and ‘align” modules in BISCUIT). SAMBLASTER v0.1.2466 was used to mark duplicates, and Samtools v1.9 was used to sort and index resulting bam files. SAMBLASTER v0.1.2466 was used to mark duplicates, and Samtools v1.9 was used to sort and index resulting bam files. Next, “pileup” and “vcf2bed” modules from BISCUIT were used to create VCF and bed files which contained methylation and SNP information. R package dmrseq v1.10.1 was used to identify differentially methylated regions (DMR) with default setting. CpGs with zero reads in every sample were filtered out prior to DMR analysis. Transcription factor binding motif enrichment analysis for hypomethylated DMRs was performed using HOMER v4.11.1 with parameters ‘findMotifsGenome.pl mm10 -size 200 -mask’. The enriched de novo and known motifs, as well as their matching transcription factors, were searched and scanned.
H3K27ac chromatin immunoprecipitation sequencing
1×106 freshly prepared rib GP cells were fixed using cross-linking buffer (50 mM HEPES-KOH pH 7.5, 100 mM NaCl, 1 mM EDTA pH 8.0, 0.5 mM EGTA pH 8.0, and 1% formaldehyde) for 8 min with rotation, followed by 5 min incubation in 125 mM glycine. Cells were washed twice in PBS; nuclei were then isolated. The nuclei were re-suspended in shearing buffer (0.1% SDS, 1 mM EDTA pH 8.0, and 10mM Tris pH 8.1) and sonicated with Covaris E220 sonicator (Covaris; Woburn, MA) at 5% duty cycle, 4 intensity, 200 cycles per burst at 4°C for 12.5 min. After adding 150 mM NaCl and 1% Triton X-100 to the sonicated samples, the solution was cleared by spinning at 4°C for 10 min at 13,000 rpm. 25 μL of cleared chromatin was saved as an input sample. The chromatin sample was incubated with anti-H3K27ac antibody (Active Motif; Carlsbad, CA) at 4°C overnight with rotation. 20 μL of Dynabeads Protein G (Thermo Fisher Scientific) were added to both the input and samples and immunoprecipitated by incubation at 4°C for 2 h with rotation. The samples were washed twice with low salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM HEPES-KOH pH 7.9, and 150 mM NaCl) and twice with high salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM HEPES-KOH pH 7.9, and 500 mM NaCl), followed by washing with LiCl wash buffer (100 mM Tris-HCl pH 7.5, 0.5 M LiCl 1% NP-40, and 1% Sodium Deoxycholate). The ChIP DNA was reverse X-linked with 1 μL (20 mg/mL) of Proteinase K by incubating at 65°C overnight. The libraries for ChIP DNA sequencing were prepared using TruSeq ChIP Library Preparation kit (Illumina; San Diego, CA) according to the manufacturer’s instructions. The amplified libraries were quantitated using a Qubit 3.0 fluorometer and ds DNA HS Assay kit, and the size distribution was determined using a Bioanalyzer and sequenced as 150 bp paired-end sequencing run on an Illumina Hiseq2500 platform to obtain around 40 million reads.
H3K9me3 CUT&RUN sequencing
Nuclei were isolated from 1×106 freshly prepared cells and incubated with 25 μL of Concanavalin A-coated magnetic beads (Bangs Laboratories, #BP531) per sample for 10 min with rotation. The bead-bound nuclei were then incubated with anti-H3K9me3 antibody (Abcam, #8898) in 200 μL antibody buffer including 2 mM EDTA and 0.05% digitonin buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM spermidine, and 0.05% digitonin) at 4°C overnight with rotation. The nuclei were washed twice with digitonin and incubated with in-house purified protein A-micrococcal nuclease (pA-MN) (700 ng/mL)67 in digitonin at 4°C for 1 h. The samples were washed twice with digitonin buffer and once with low salt buffer (20 mM HEPES pH 7.5, 0.5 mM spermidine, and 0.05% digitonin), then incubated in incubation buffer (3.5 mM HEPES pH 7.5, 10 mM CaCl2, and 0.05% digitonin) at 0°C for 5 min. To chelate Ca2+ and allow diffusion out of chromatin DNA, nuclei were incubated in 200 μL of stop buffer (170 mM NaCl, 20 mM EGTA, 50 μg/mL RNAse A, 25 μg/mL glycogen, and 0.05% digitonin) at 37°C for 30 min. DNA fragments were purified using Quick-DNA Miniprep Plus kit (Zymo Research).
To construct the sequencing library for CUT&RUN, we used NEBNext Ultra II DNA Library Prep Kit (NEB; Ipswich, MA) according to the manufacturer’s protocol. To amplify the library by PCR, the eluted library was mixed with 12.5 μL of NEBNext Ultra II Q5 Master mix (2x), 1 μL of 10 μM NEBNext i5 and 1 μL of 10 μM NEBNext i7 primers (NEB) in a final volume of 25 μL. The amplification reaction mixture was incubated at 98°C for 30 s, followed by 13 cycles of PCR at 98°C for 10 s and 65°C for 75 s, followed by a final extension step at 65°C for 5 min. The amplified library was purified with 25 μL (1x) of Ampure XP beads (Beckman Coulter; Indianapolis, IN), and was quantitated using a Qubit 3.0 fluorometer and ds DNA HS Assay kit (Thermo Fisher Scientific). The size distribution of the sequencing library was determined using Bioanalyzer (Agilent; Santa Clara, CA) and sequenced as 150 bp paired-end sequencing runs on an Illumina Hiseq2500 platform to obtain around 40 million reads.
Analysis of H3K9me3 CUT&RUN and H3K27ac ChIP-seq
Reads were mapped to the mm10 reference genome by bowtie2 v2.3.5 and samtools v1.2 using default parameters. Peaks were called using MASC2 peak caller with the following commands: ‘macs2 callpeak -t -g mm –broad –broad-cutoff 0.01 -p 0.001’ (H3K9me3) and ‘macs2 callpeak -t -c -g mm –broad –broad-cutoff 0.01 -p 0.01’ (H3K27ac). Raw peak regions were merged using bedtools v2.29.2 (mergeBed -d 300).68 Reads falling into raw peak regions were counted for individual samples using bedtools. Read counts were normalized as count-per-million (CPM) based on relative log expression normalization (RLE) methods implemented in edgeR v3.18.1. p values were adjusted for comparison using the false discovery rate (FDR). The criteria for differential H3K9me3 and H3K27ac modified region/peak was FDR ≤0.05 and abs(log2FC) ≥ 1; p value %0.05. 21,633 out of 86,587 peaks were defined as differential H3K9me3 peaks; 4,287 out of 368,121 peaks were defined as differential H3K27ac peaks between Trim28MKO and WT samples. Transcription factor binding motif enrichment analysis for downregulated H3K9me3 loci was conducted as detailed above for WGBS.
ATAC-seq data analysis
Chromatin accessibility analysis of TRIM28-silenced regions in SSCs and NCCs was achieved by reanalyzing published ATAC-seq data downloaded from the Gene Expression Omnibus (accession number: GSE89436 for NCC, GSE104473 for SSC). Briefly, reads were mapped to the genome (mm10) using Bowtie2 v2.3.5. Peaks were called using MASC2 peak caller with the following commands: ‘macs2 callpeak -t -g mm –broad –broad-cutoff 0.01 -p 0.001’. Read counts on TRIM28-silenced regions were estimated by ‘bedtools multicov’. The CPM normalized readcounts were used to generate the heatmap by using R package pheatmap v1.0.12. To analyze the proportion of accessible loci that were TEs in NCCs or SSCs, we intersected TRIM28-silenced regions against all TE insertions downloaded from the RepeatMasker database (http://www.repeatmasker.org/).
Definition of TRIM28-silenced genes
Genes were categorized by the following conditions.
the gene’s mRNA expression is increased in the MKO sample;
its enhancer (based on Enhancer Atlas V2.0 database69) has increased H3K27ac signals;
its enhancer has decreased H3K9me3 signals;
its enhancer has decreased DMRs;
it has down-regulated H3K9me3 signals within ±1.5kb adjacent to the gene’s TSS;
it has hypomethylation DMRs within ±1.5kb adjacent to the TSS;
The TRIM28-silenced genes were defined by the Formula.
Targeted DNA deletion
Oligonucleotides containing gRNAs targeting the 3′ side of the DMRs were cloned into LentiCRISPRv2 puro vector58 (Addgene), and oligonucleotides containing gRNAs targeting the 5′ side of the DMRs were cloned into Lenti-CRISPRv2 blast vector. Lentivirus was produced in HEK293T cells by transfecting 4 μg lentiviral plasmid, 2 μg pMD2.G (Addgene) and 4 μg psPAX2 (Addgene) using PEI reagent. gRNAs targeting GFP were used as a negative control. Primary rib GP cells were seeded into 12-well plates and transfected with lentiviral supernatant containing gRNAs targeting both the 3′ end and 5′ end of the target sequence. 24 h after transfection, positively transfected cells were selected with blasticidin (5 μg/mL) and puromycin (1 μmg/mL) for 1 week. Total RNA was extracted for qRT-PCR assay.
shRNA-mediated gene silencing
Non-targeting or Grem1 shRNA oligos were cloned into PLKO.1 puro plasmid59(Addgene). Lentivirus was produced in HEK293T cells as described above. Primary rib GP cells were seeded into 12-well plates and transfected with lentiviral supernatant. 24 h after transfection, positively transfected cells were selected with puromycin (1 μg/mL) for 1 week. Total protein was extracted for Western blot.
Enhancer luciferase assay
ATDC5 cells in 12-well plates were transfected with pGL3-Basic empty vector, pGL3-Basic vector inserted with 600bp Grem1 promoter, pGL3-Basic vector inserted with 600bp Grem1 promoter plus Grem1 putative enhancers (1 μg/well). Each transfection included pRL-Renilla (100 ng, as transfection control). Cells were harvested and subjected to luciferase assays using the Dual-Luciferase Reporter assay kit (Promega).
Flow cytometry and cell sorting
Dissociated hindlimb GP cells were stained by standard protocols with fluorochrome-conjugated antibodies against the following surface markers: CD45 (Tonbo bioscience; 35–0451), TER119 (Tonbo bioscience; 20–5921), TIE2 (Biolegend; 124010), CD51 (eBio-science; 12–0512), THY1.2 (Thermo Fisher; 25–0902-81), 6C3 (Biolegend; 46–5891-82), CD105 (BD Bioscience; 562762) and CD200 (BD Biosciences; 565547). Viable cells were stained with Fixable Viability Dye eFluoraâ780 (eBioscience; 65–0865) for flow cytometry and with DAPI for cell sorting. SSC was defined by CD45−Ter119−TIE2−CD51+6C3−THY1.2−CD105−CD200+, pre-BCSP was defined by CD45−Ter119−TIE2−CD51+6C3−THY1.2−CD105−CD200 − as previously reported.11 Flow cytometry was performed on the Cytek Aurora cytometer while SSC population was sorted on BD FACSymphonyä S6 in the Van Andel Institute Flow Cytometry Core Facilities. Acquired raw data were further analyzed on FlowJo software and sorted cells were cultured for further experiments.
Single-cell RNA sequencing and data processing
Hindlimb GP cells from WT and Trim28MKO mice were isolated using the method described above. Libraries were generated and sequenced by the Van Andel Institute Genomics Core. Briefly, cells were processed with 10× Chromium Next GEM Single Cell 3ʹ GEM kit (103Genomics, v.3.1) according to the manufacturer’s instruction to target 6,000 cells per group. Quality and quantity of the finished libraries were assessed using a combination of Agilent DNA High Sensitivity chip (Agilent Technologies, Inc.) and QuantiFluor® dsDNA System (Promega Corp., Madison, WI, USA). 28 × 98, paired end sequencing was performed on an Illumina NovaSeq 6000 sequencer using an SP reagent kit (v1.5) (Illumina Inc., San Diego, CA, USA). Base calling was done by Illumina RTA3, and output was demultiplexed and converted to FASTQ format with Cell Ranger (10×Genomics, v3.1.0). FASTQ files were aligned to mm10 reference genome using cell ranger count pipeline v4.0.0 with the default settings. The unique molecular identified (UMI) count matrixes were analyzed with Seurat R package v4.0.3,70 cells with <200 or >6,000 genes and cells with >5% mitochondrial gene mapped reads were filtered, and the ribosomal genes were removed from the downstream analysis. In total, 7,448 cells from Trim28MKO and 6,235 cells from WT were selected. After merging the Seurat objects of two samples, SCTransform function was used for normalization and cell cycle score regression. Cell clusters were annotated based on the expression of cell type specific markers. SSC were sorted using the gene expression signatures: Itgav+, Cd200+, Ptprc−, Tek−, Thy1− Enpep−, Eng−. ScVelo v0.2.571 was used to estimate RNA velocity and cellular dynamics.
Gene set enrichment analysis
R package enrichR v3.072 or DAVID73 were used to identify enriched gene sets. The selected gene lists were input to query enrichment for gene ontology (Biological Process 2021), WikiPathways (mouse 2019), KEGG pathways (mouse 2019), and tissue expression. The top terms ranked by −log10(p value) were visualized by R package ggplot2 v3.3.5. Gene set enrichment analysis (GSEA) v4.0.1 was used for all ranked gene lists (−log10(FDR) * sign(log2FC)) from bulk RNA-seq to identify enriched pathways that differed between WT and Trim28MKO72 or DAVID73 were used to identify enriched gene sets. The selected gene lists were input to query enrichment for gene ontology (Biological Process 2021), WikiPathways (mouse 2019), KEGG pathways (mouse 2019), and tissue expression. The top terms ranked by −log10(p value) were visualized by R package ggplot2 v3.3.5. Gene set enrichment analysis (GSEA) v4.0.1 was used for all ranked gene lists (−log10(FDR) * sign(log2FC)) from bulk RNA-seq to identify enriched pathways that differed between WT and Trim28MKO.
Integration analysis of SSC and NCC
The NCC cluster from an E15.0 mouse limb 10×Genenomics scRNA-seq dataset29 was integrated with our WT and Trim28MKO SSCs scRNA-seq data. Canonical correlation analysis (CCA) from Seurat v4.0.3 was performed on the top 20 PCs (principal components) between the NCCs and SSCs for batch correction. A pseudo bulk RNA UMAP was generated with the average embedding scores of each cell type and the dot size represents the number of cells.
Differentiation assays
For osteogenic differentiation, rib GP cells were seeded into 12-well plates at 1 × 105 cells/well or sorted SSCs into 96-well plates at 1×104 cells/well and induced with osteogenic medium (α-MEM containing 10% FBS, 100 nM dexamethasone, 10 mM β-glycerophosphate, and 0.05 mM L-ascorbic acid-2-phosphate) at 80% confluence. After 4 days of differentiation, cells were fixed and subjected to alkaline phosphatase (ALP) staining using standard protocols. Alizarin Red S staining was performed 14 days after induction.
For chondrogenic differentiation of GP cells, 10 μL GP cell suspensions containing 1×105 cells in α-MEM were seeded into each well of a 48-well plate to form high cell density Micromass and allowed to attach for 1 h. 300 μL chondrogenic medium (DMEM containing 1mM Pyruvate, 10 μg/mL insulin, 0.1 μM dexamethasone, 0.05 mM L-ascorbic acid, 10 ng/mL TGFβ1) was added to induce differentiation. Medium was changed every 2 days. 5 days after induction, cells were fixed and stained with 1% Alcian blue at 37°C overnight. For SSC chondrogenesis, 1×104 cells were seeded into a 96-well plate, the same chondrogenic process was used to induce differentiation after they had reached 100% confluence.
For neurogenic differentiation, 8×104 cells were seeded into a 24-well plate. After the cells reached 80% confluence, 400 μL Mesenchymal Stem Cell Neurogenic Differentiation Medium (PromoCell, C-28015) was added to each well. 4 (SSC) or 8 (hindlimb GP cell) days after induction, brightfield images were taken, and cells were harvested for qPCR assay. Neurite outgrowth was quantified by using NeuronJ (a plugin in the ImageJ package).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was determined using unpaired Student’s t test to compare two groups, and one-way ANOVA with Tukey’s multiple-comparison test when more than two groups were compared, unless otherwise specified in the figure legend. Data were considered statistically significant when p value ≤0.05. All data points refer to biological replicates, unless otherwise indicated in the figure legend, and are presented as mean ± standard error of the mean (SEM). Statistical analyses were performed using GraphPad Prism 9.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-TRIM28 | Cell Signaling Technology | Cat #4123; RRID:AB_2256670 |
| Anti-Ki67 | Thermo Fisher Scientific | Clone SP6, Cat # MA5–14520; RRID:AB_10979488 |
| Anti-Collagen II | Thermo Fisher Scientific | Clone 6B3, Cat # MA5–13026; RRID: AB_10979392 |
| Anti-Collagen X | DSHB | Clone X-AC9, Cat # X-AC9; RRID: AB_528175 |
| Anti-IHH | Millipore | Cat # MABF23; RRID: AB_10807445 |
| Anti-Phospho-S6 | Cell Signaling Technology | Cat # 4858; RRID: AB_916156 |
| Anti-Phospho-Akt | Cell Signaling Technology | Clone D9E, Cat # 4060; RRID: AB_2315049 |
| Anti-S6 | Cell Signaling Technology | Clone 5G10, Cat # 2217; RRID: AB_331355 |
| Anti-Akt | Cell Signaling Technology | Cat # 4685; RRID: AB_2225340 |
| Anti-GAPDH | Proteintech | Cat # 10494–1-AP; RRID: AB_2263076 |
| Anti-GREM1 | Abgent | Cat # AP12247b; RRID: AB_10819593 |
| Anti-α-Tubulin | Sigma-Aldrich | Clone AA13, Cat # T8203; RRID: AB_1841230 |
| Anti-phospho-4E-BP1 | Cell Signaling Technology | Clone 236B4, Cat # 2855; RRID: AB_560835 |
| Anti-phospho-p70 S6 Kinase | Cell Signaling Technology | Clone 108D2, Cat # 9234; RRID: AB_2269803 |
| Anti-phospho-smad1/5/8 | Cell Signaling Technology | Cat # 9 511; RRID: AB_331671 |
| Anti-H3K9me3 | Abcam | Cat # ab8898; RRID: AB_306848 |
| Anti-H3K27ac | Active Motif | Cat # 39133; RRID: AB_2561016 |
| Anti-CD45 | Tonbo Biosciences | Clone 30-F11 Cat # 35–0451 RRID: AB_2621689 |
| Anti-TER-119 | Tonbo Bioscience | Clone TER-119 Cat # 20–5921 RRID: AB_2621609 |
| Anti-Tie-2 | BioLegend | Clone TEK4 Cat # 124010 RRID: AB_10897106 |
| Anti-CD51 | Thermo Fisher Scientific | Clone RMV-7 Cat # 12–0512-81 RRID: AB_465703 |
| Anti-Thy-1.2 | Thermo Fisher Scientific | Clone 53–2.1 Cat # 25–0902–81 RRID: AB_469641 |
| Anti-6C3 | Thermo Fisher Scientific | Clone 6C3 Cat # 46–5891-82 RRID: AB_2573797 |
| Anti-CD105 | BD Biosciences | Clone MJ7/18 Cat # 562762 RRID: AB_2737776 |
| Anti-CD200 | BD Biosciences | Clone OX-90 Cat # 565547 RRID: AB_2739289 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| One Shot™ Stbl3™ chemically competent E. coli | Thermo Fisher Scientific | Cat #C737303 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Recombinant Human Gremlin1 protein | PEPROTECH | Cat # 120–42 |
| YL294002 | Millipore Sigma | Cat # 440202 |
| Rapamycin | Selleckchem | Cat #S1039 |
|
| ||
| Critical commercial assays | ||
|
| ||
| VECTASTAIN® Elite® ABC-HRP Kit | Vector Laboratories | Cat # PK-6102 |
| ImmPACT® DAB EqV Substrate Kit, Peroxidase (HRP) | Vector Laboratories | Cat # SK-4103 |
| in-situ Cell Death Detection Kit | Millipore Sigma | Cat # 11684795910 |
| RNeasy kit | Qiagen | Cat # 74004 |
| SYBR Green QPCR Master Mix | Applied biosystems™ | Cat # 4309155 |
| VILO cDNA synthesis kit | Invitrogen | Cat # 11754050 |
| Quick-DNA Miniprep Plus kit | Zymo Research | Cat #D4068 |
| TruSeq ChIP Library Preparation kit | Illumina | Cat # IP-202–1012 Cat # IP-202–1024 |
| NEBNext Ultra II DNA Library Prep Kit | NEB; Ipswich, MA | Cat #E7645 |
| ds DNA HS Assay kit | Thermo Fisher Scientific | Cat #Q32851 |
| AMPure XP beads | Beckman Coulter | Cat # A63880 |
| Dual-Luciferase Reporter assay kit | Promega | Cat #E1910 |
| 10x Chromium Next GEM Single Cell 3' GEM kit, v.3.1 | 10xGenomics | Cat # PN-1000121 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and analyzed data | This paper | GEO: GSE202060 |
|
| ||
| Experimental models: Cell lines | ||
| Human: 293T | ATCC | Cat # CRL-3216™ |
| Mouse: ATDC5 | Millipore Sigma | Cat # 99072806 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: Trim28f/f | Jackson Laboratories | Cat # 018552 |
| Mouse: Dermo1-Cre | Jackson Laboratories | Cat # 008712 |
| Mouse: Col2a1-Cre | Jackson Laboratories | Cat # 003554 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Actb qPCR primers | Integrated DNA Technologies | See Table S8 for sequences |
| Trim28 qPCR primers | Integrated DNA Technologies | See Table S8 for sequences |
| Grem1 qPCR primers | Integrated DNA Technologies | See Table S8 for sequences |
| Tubb3 qPCR primers | Integrated DNA Technologies | See Table S8 for sequences |
| Nes qPCR primers | Integrated DNA Technologies | See Table S8 for sequences |
| Mapt qPCR primers | Integrated DNA Technologies | See Table S8 for sequences |
| Gapdh qPCR primers | Integrated DNA Technologies | See Table S8 for sequences |
| Trim28-flox genotyping primer | Integrated DNA Technologies | See Table S8 for sequences |
| Cre genotyping primer | Integrated DNA Technologies | See Table S8 for sequences |
|
| ||
| Recombinant DNA | ||
|
| ||
| Lenti-CRISPRv2-puro | Sanjana et al.58 | Cat# 52961 RRID: Addgene_52961 |
| Lenti-CRISPRv2-blast | This paper | N/A |
| Lenti-CRISPRv2_sgGFP | This paper | N/A |
| Lenti-CRISPRv2_sgDMR1 | This paper | N/A |
| Lenti-CRISPRv2_sgDMR2–1 | This paper | N/A |
| Lenti-CRISPRv2_sgDMR2–2 | This paper | N/A |
| pGL3-Basic vector | Promega | Catalog #E1751 |
| pGL3_Grem1_Promter | This paper | N/A |
| pGL3_Grem1_Promter_DMR1 | This paper | N/A |
| pGL3_Grem1_Promter_DMR2 | This paper | N/A |
| pMD2.G | pMD2.G was a gift from Didier Trono (http://n2t.net/addgene:12259) | Cat# 12259 RRID: Addgene_12259 |
| psPAX2 | psPAX2 was a gift from Didier Trono (Addgene plasmid # 12260; http://n2t.net/addgene:12260) | Cat# 12260 RRID: Addgene_12260 |
| PLKO.1 puro | Stewart et al.59 | Cat# 8453 RRID: Addgene_8453 |
| PLKO.1 puro_shct | This paper | N/A |
| PLKO.1 puro_shGrem1 | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ software | GraphPad | https://www.graphpad.com/ |
| NeuronJ | Biomedical Imaging Group Rotterdam of Erasmus University Medical Center | https://imagescience.org/meijering/software/neuronj/ |
| FlowJo™ v10 | BD Biosciences | https://www.bdbiosciences.com/en-us/products/software/flowjo-v10-software |
| R version 4.1.0 | R Foundation for Statistical Computing | https://www.R-project.org |
Highlights.
Loss of TRIM28 in developing mesoderm causes severe skeletal development defects
Loss of TRIM28 endows SSCs with neural crest cell-like properties
TRIM28 loss expands the SSC pool by activating GREM1-AKT-mTORC1 signaling
TRIM28 silences neural genes and strengthens the barrier between mesoderm and ectoderm
ACKNOWLEDGMENTS
We thank Drs. Peter A. Jones, J. Andrew Pospisilik, Bart O. Williams, Scott Rothbart, Piroska Szabo, Tim Triche, Patrik Brundin, Hongyuan Chu, Stephanie Grainger, Josh Jang, Leslie Yang, and Hyo Sik Jang for their invaluable expertise and suggestions regarding epigenetics, bioinformatics analyses, signaling, and neurogenesis. We also thank the VAI Vivarium and Transgenics Core, Flow Cytometry Core, Genomics Core, Bioinformatics & Biostatistics Core, and Pathology Core for their excellent technical services. This work was supported by NIH funds (R01AG061086 to T.Y.; R01AR079174 to G.P.P.; R01CA204020 to X.S.), VARI startup funds (T.Y.), and the Arthritis National Research Foundation (30–40379) (T.Y.)
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests. Received: July 15, 2022
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112012.
REFERENCES
- 1.Allaway KC, Gabitto MI, Wapinski O, Saldi G, Wang CY, Bandler RC, Wu SJ, Bonneau R, and Fishell G (2021). Genetic and epigenetic coordination of cortical interneuron development. Nature 597, 693–697. 10.1038/s41586-021-03933-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yadav T, Quivy JP, and Almouzni G (2018). Chromatin plasticity: a versatile landscape that underlies cell fate and identity. Science 361, 1332–1336. 10.1126/science.aat8950. [DOI] [PubMed] [Google Scholar]
- 3.Atlasi Y, and Stunnenberg HG (2017). The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet. 18, 643–658. 10.1038/nrg.2017.57. [DOI] [PubMed] [Google Scholar]
- 4.Argelaguet R, Clark SJ, Mohammed H, Stapel LC, Krueger C, Kapourani CA, Imaz-Rosshandler I, Lohoff T, Xiang Y, Hanna CW, et al. (2019). Multi-omics profiling of mouse gastrulation at single-cell resolution. Nature 576, 487–491. 10.1038/s41586-019-1825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicetto D, Donahue G, Jain T, Peng T, Sidoli S, Sheng L, Montavon T, Becker JS, Grindheim JM, Blahnik K, et al. (2019). H3K9me3-heterochromatin loss at protein-coding genes enables developmental lineage specification. Science 363, 294–297. 10.1126/science.aau0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ambrosi TH, Longaker MT, and Chan CKF (2019). A revised perspective of skeletal stem cell biology. Front. Cell Dev. Biol. 7, 189. 10.3389/fcell.2019.00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cordero DR, Brugmann S, Chu Y, Bajpai R, Jame M, and Helms JA (2011). Cranial neural crest cells on the move: their roles in craniofacial development. Am. J. Med. Genet. 155A, 270–279. 10.1002/ajmg.a.33702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zalc A, Sinha R, Gulati GS, Wesche DJ, Daszczuk P, Swigut T, Weissman IL, and Wysocka J (2021). Reactivation of the pluripotency program precedes formation of the cranial neural crest. Science 371, eabb4776. 10.1126/science.abb4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan WCW, Tan Z, To MKT, and Chan D (2021). Regulation and role of transcription factors in osteogenesis. Int. J. Mol. Sci. 22, 5445. 10.3390/ijms22115445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kurenkova AD, Medvedeva EV, Newton PT, and Chagin AS (2020). Niches for skeletal stem cells of mesenchymal origin. Front. Cell Dev. Biol. 8, 592. 10.3389/fcell.2020.00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan CKF, Seo EY, Chen JY, Lo D, McArdle A, Sinha R, Tevlin R, Seita J, Vincent-Tompkins J, Wearda T, et al. (2015). Identification and specification of the mouse skeletal stem cell. Cell 160, 285–298. 10.1016/j.cell.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sui BD, Zheng CX, Li M, Jin Y, and Hu CH (2020). Epigenetic regulation of mesenchymal stem cell homeostasis. Trends Cell Biol. 30, 97–116. 10.1016/j.tcb.2019.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Deng P, Yuan Q, Cheng Y, Li J, Liu Z, Liu Y, Li Y, Su T, Wang J, Salvo ME, et al. (2021). Loss of KDM4B exacerbates bone-fat imbalance and mesenchymal stromal cell exhaustion in skeletal aging. Cell Stem Cell 28, 1057–1073.e7. 10.1016/j.stem.2021.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang R, Yu T, Kou X, Gao X, Chen C, Liu D, Zhou Y, and Shi S (2018). Tet1 and Tet2 maintain mesenchymal stem cell homeostasis via demethylation of the P2rX7 promoter. Nat. Commun. 9, 2143. 10.1038/s41467-018-04464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quenneville S, Turelli P, Bojkowska K, Raclot C, Offner S, Kapopoulou A, and Trono D (2012). The KRAB-ZFP/KAP1 system contributes to the early embryonic establishment of site-specific DNA methylation patterns maintained during development. Cell Rep. 2, 766–773. 10.1016/j.celrep.2012.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iyengar S, and Farnham PJ (2011). KAP1 protein: an enigmatic master regulator of the genome. J. Biol. Chem. 286, 26267–26276. 10.1074/jbc.R111.252569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang P, Wang Y, and Macfarlan TS (2017). The role of KRAB-ZFPs in transposable element repression and mammalian evolution. Trends Genet. 33, 871–881. 10.1016/j.tig.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, Maillard PV, Layard-Liesching H, Verp S, Marquis J, et al. (2010). KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463, 237–240. 10.1038/nature08674. [DOI] [PubMed] [Google Scholar]
- 19.Messerschmidt DM, de Vries W, Ito M, Solter D, Ferguson-Smith A, and Knowles BB (2012). Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science 335, 1499–1502. 10.1126/science.1216154. [DOI] [PubMed] [Google Scholar]
- 20.Cammas F, Mark M, Dollé P, Dierich A, Chambon P, and Losson R (2000). Mice lacking the transcriptional corepressor TIF1beta are defective in early postimplantation development. Development 127, 2955–2963. 10.1242/dev.127.13.2955. [DOI] [PubMed] [Google Scholar]
- 21.Dalgaard K, Landgraf K, Heyne S, Lempradl A, Longinotto J, Gossens K, Ruf M, Orthofer M, Strogantsev R, Selvaraj M, et al. (2016). Trim28 haploinsufficiency triggers Bi-stable epigenetic obesity. Cell 164, 353–364. 10.1016/j.cell.2015.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rousseaux MW, Revelli JP, Vázquez-Vélez GE, Kim JY, Craigen E, Gonzales K, Beckinghausen J, and Zoghbi HY (2018). Depleting Trim28 in adult mice is well tolerated and reduces levels of alpha-synuclein and tau. Elife 7, e36768. 10.7554/eLife.36768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, and Ornitz DM (2003). Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 130, 3063–3074. 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- 24.Long F, and Ornitz DM (2013). Development of the endochondral skeleton. Cold Spring Harb. Perspect. Biol. 5, a008334. 10.1101/cshperspect.a008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newton PT, Li L, Zhou B, Schweingruber C, Hovorakova M, Xie M, Sun X, Sandhow L, Artemov AV, Ivashkin E, et al. (2019). A radical switch in clonality reveals a stem cell niche in the epiphyseal growth plate. Nature 567, 234–238. 10.1038/s41586-019-0989-6. [DOI] [PubMed] [Google Scholar]
- 26.Mizuhashi K, Ono W, Matsushita Y, Sakagami N, Takahashi A, Saunders TL, Nagasawa T, Kronenberg HM, and Ono N (2018). Resting zone of the growth plate houses a unique class of skeletal stem cells. Nature 563, 254–258. 10.1038/s41586-018-0662-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ovchinnikov DA, Deng JM, Ogunrinu G, and Behringer RR (2000). Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis 26, 145–146. [PubMed] [Google Scholar]
- 28.Mizuhashi K, Nagata M, Matsushita Y, Ono W, and Ono N (2019). Growth Plate borderline chondrocytes behave as transient mesenchymal precursor cells. J. Bone Miner. Res. 34, 1387–1392. 10.1002/jbmr.3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He P, Williams BA, Trout D, Marinov GK, Amrhein H, Berghella L, Goh ST, Plajzer-Frick I, Afzal V, Pennacchio LA, et al. (2020). The changing mouse embryo transcriptome at whole tissue and single-cell resolution. Nature 583, 760–767. 10.1038/s41586-020-2536-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Worthley DL, Churchill M, Compton JT, Tailor Y, Rao M, Si Y, Levin D, Schwartz MG, Uygur A, Hayakawa Y, et al. (2015). Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 160, 269–284. 10.1016/j.cell.2014.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilk K, Yeh SCA, Mortensen LJ, Ghaffarigarakani S, Lombardo CM, Bassir SH, Aldawood ZA, Lin CP, and Intini G (2017). Postnatal calvarial skeletal stem cells expressing PRX1 reside exclusively in the calvarial sutures and are required for bone regeneration. Stem Cell Rep. 8, 933–946. 10.1016/j.stemcr.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Méndez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma’ayan A, Enikolopov GN, and Frenette PS (2010). Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466, 829–834. 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang W, Wang J, Moore DC, Liang H, Dooner M, Wu Q, Terek R, Chen Q, Ehrlich MG, Quesenberry PJ, and Neel BG (2013). Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature 499, 491–495. 10.1038/nature12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ambrosi TH, Sinha R, Steininger HM, Hoover MY, Murphy MP, Koepke LS, Wang Y, Lu WJ, Morri M, Neff NF, et al. (2021). Distinct skeletal stem cell types orchestrate long bone skeletogenesis. Elife 10, e66063. 10.7554/eLife.66063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ambrosi TH, Marecic O, McArdle A, Sinha R, Gulati GS, Tong X, Wang Y, Steininger HM, Hoover MY, Koepke LS, et al. (2021). Aged skeletal stem cells generate an inflammatory degenerative niche. Nature 597, 256–262. 10.1038/s41586-021-03795-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Späth SS, Andrade AC, Chau M, Baroncelli M, and Nilsson O (2018). Evidence that rat chondrocytes can differentiate into perichondrial cells. JBMR Plus 2, 351–361. 10.1002/jbm4.10056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ransom RC, Carter AC, Salhotra A, Leavitt T, Marecic O, Murphy MP, Lopez ML, Wei Y, Marshall CD, Shen EZ, et al. (2018). Mechanoresponsive stem cells acquire neural crest fate in jaw regeneration. Nature 563, 514–521. 10.1038/s41586-018-0650-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Minoux M, Holwerda S, Vitobello A, Kitazawa T, Kohler H, Stadler MB, and Rijli FM (2017). Gene bivalency at Polycomb domains regulates cranial neural crest positional identity. Science 355, eaal2913. 10.1126/science.aal2913. [DOI] [PubMed] [Google Scholar]
- 39.Prescott SL, Srinivasan R, Marchetto MC, Grishina I, Narvaiza I, Selleri L, Gage FH, Swigut T, and Wysocka J (2015). Enhancer divergence and cis-regulatory evolution in the human and chimp neural crest. Cell 163, 68–83. 10.1016/j.cell.2015.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sherman BT, Hao M, Qiu J, Jiao X, Baseler MW, Lane HC, Imamichi T, and Chang W (2022). DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221. 10.1093/nar/gkac194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pancho A, Aerts T, Mitsogiannis MD, and Seuntjens E (2020). Protocadherins at the crossroad of signaling pathways. Front. Mol. Neurosci. 13, 117. 10.3389/fnmol.2020.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dai HQ, Wang BA, Yang L, Chen JJ, Zhu GC, Sun ML, Ge H, Wang R, Chapman DL, Tang F, et al. (2016). TET-mediated DNA demethylation controls gastrulation by regulating Lefty-Nodal signalling. Nature 538, 528–532. 10.1038/nature20095. [DOI] [PubMed] [Google Scholar]
- 43.Pineda CT, Ramanathan S, Fon Tacer K, Weon JL, Potts MB, Ou YH, White MA, and Potts PR (2015). Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell 160, 715–728. 10.1016/j.cell.2015.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Plaen E, Arden K, Traversari C, Gaforio JJ, Szikora JP, De Smet C, Brasseur F, van der Bruggen P, Lethé B, Lurquin C, et al. (1994). Structure, chromosomal localization, and expression of 12 genes of the MAGE family. Immunogenetics 40, 360–369. 10.1007/BF01246677. [DOI] [PubMed] [Google Scholar]
- 45.Canalis E, Parker K, and Zanotti S (2012). Gremlin1 is required for skeletal development and postnatal skeletal homeostasis. J. Cell. Physiol. 227, 269–277. 10.1002/jcp.22730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiang Q, Hong D, Liao Y, Cao Y, Liu M, Pang J, Zhou J, Wang G, Yang R, Wang M, and Xiang AP (2017). Overexpression of Gremlin1 in mesenchymal stem cells improves hindlimb ischemia in mice by enhancing cell survival. J. Cell. Physiol. 232, 996–1007. 10.1002/jcp.25578. [DOI] [PubMed] [Google Scholar]
- 47.Rowe HM, Kapopoulou A, Corsinotti A, Fasching L, Macfarlan TS, Tarabay Y, Viville S, Jakobsson J, Pfaff SL, and Trono D (2013). TRIM28 repression of retrotransposon-based enhancers is necessary to preserve transcriptional dynamics in embryonic stem cells. Genome Res. 23, 452–461. 10.1101/gr.147678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fasching L, Kapopoulou A, Sachdeva R, Petri R, Jönsson ME, Männe C, Turelli P, Jern P, Cammas F, Trono D, and Jakobsson J (2015). TRIM28 represses transcription of endogenous retroviruses in neural progenitor cells. Cell Rep. 10, 20–28. 10.1016/j.celrep.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grassi DA, Jönsson ME, Brattås PL, and Jakobsson J (2019). TRIM28 and the control of transposable elements in the brain. Brain Res. 1705, 43–47. 10.1016/j.brainres.2018.02.043. [DOI] [PubMed] [Google Scholar]
- 50.Trono D (2015). Transposable elements, polydactyl proteins, and the genesis of human-specific transcription networks. Cold Spring Harb. Symp. Quant. Biol. 80, 281–288. 10.1101/sqb.2015.80.027573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.ENCODE Project Consortium; Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, Kawli T, Davis CA, Dobin A, Kaul R, et al. (2020). Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710. 10.1038/s41586-020-2493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doynova MD, Markworth JF, Cameron-Smith D, Vickers MH, and O’Sullivan JM (2017). Linkages between changes in the 3D organization of the genome and transcription during myotube differentiation in vitro. Skelet. Muscle 7, 5. 10.1186/s13395-017-0122-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. (2015). Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336. 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ichinose M, Suzuki N, Wang T, Kobayashi H, Vrbanac L, Ng JQ, Wright JA, Lannagan TRM, Gieniec KA, Lewis M, et al. (2021). The BMP antagonist gremlin 1 contributes to the development of cortical excitatory neurons, motor balance and fear responses. Development 148, dev195883. 10.1242/dev.195883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Han J, Wang B, Xiao Z, Gao Y, Zhao Y, Zhang J, Chen B, Wang X, and Dai J (2008). Mammalian target of rapamycin (mTOR) is involved in the neuronal differentiation of neural progenitors induced by insulin. Mol. Cell. Neurosci. 39, 118–124. 10.1016/j.mcn.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 56.Hartman NW, Lin TV, Zhang L, Paquelet GE, Feliciano DM, and Bordey A (2013). mTORC1 targets the translational repressor 4E-BP2, but not S6 kinase 1/2, to regulate neural stem cell self-renewal in vivo. Cell Rep. 5, 433–444. 10.1016/j.celrep.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 57.Modzelewski AJ, Shao W, Chen J, Lee A, Qi X, Noon M, Tjokro K, Sales G, Biton A, Anand A, et al. (2021). A mouse-specific retrotransposon drives a conserved Cdk2ap1 isoform essential for development. Cell 184, 5541–5558.e22. 10.1016/j.cell.2021.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen ISY, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li J, Lu D, Dou H, Liu H, Weaver K, Wang W, Li J, Yeh ETH, Williams BO, Zheng L, and Yang T (2018). Desumoylase SENP6 maintains osteochondroprogenitor homeostasis by suppressing the p53 pathway. Nat. Commun. 9, 143. 10.1038/s41467-017-02413-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu H, Li J, Lu D, Li J, Liu M, He Y, Williams BO, Li J, and Yang T (2018). Ginkgolic acid, a sumoylation inhibitor, promotes adipocyte commitment but suppresses adipocyte terminal differentiation of mouse bone marrow stromal cells. Sci. Rep. 8, 2545. 10.1038/s41598-018-20244-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liao Y, Smyth GK, and Shi W (2013). The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108. 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 64.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Criscione SW, Zhang Y, Thompson W, Sedivy JM, and Neretti N (2014). Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics 15, 583. 10.1186/1471-2164-15-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Faust GG, and Hall IM (2014). SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505. 10.1093/bioinformatics/btu314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Skene PJ, and Henikoff S (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6, e21856. 10.7554/eLife.21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gao T, and Qian J (2020). EnhancerAtlas 2.0: an updated resource with enhancer annotation in 586 tissue/cell types across nine species. Nucleic Acids Res. 48, D58–D64. 10.1093/nar/gkz980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bergen V, Lange M, Peidli S, Wolf FA, and Theis FJ (2020). Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414. 10.1038/s41587-020-0591-3. [DOI] [PubMed] [Google Scholar]
- 72.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dennis G Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, and Lempicki RA (2003). DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 4, P3. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data generated in this study have been submitted to and deposited at NCBI GEO: GSE202060. Data are publicly available as of the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.