

Abstract
Prolyl endopeptidases have potential for treating coeliac sprue, a disease of the intestine caused by proteolytically resistant peptides from proline-rich prolamins of wheat, barley and rye. We compared the properties of three similar bacterial prolyl endopeptidases, including the known enzymes from Flavobacterium meningosepticum (FM) and Sphingomonas capsulate (SC) and a novel enzyme from Myxococcus xanthus (MX). These enzymes were interrogated with reference chromogenic substrates, as well as two related gluten peptides (PQPQLPYPQPQLP and LQLQPFPQPQLPYPQPQLPYPQPQLPYPQPQPF), believed to play a key role in coeliac sprue pathogenesis. In vitro and in vivo studies were conducted to evaluate the activity, specificity and acid/protease stability of the enzymes. All peptidases were relatively resistant to acid, pancreatic proteases and membrane peptidases of the small intestinal mucosa. Although their activities against reference substrates were similar, the enzymes exhibited substantial differences with respect to chain length and subsite specificity. SC hydrolysed PQPQLPYPQPQLP well, but had negligible activity against LQLQPFPQPQLPYPQPQLPYPQPQLPYPQPQPF. In contrast, the FM and MX peptidases cleaved both substrates, although the FM enzyme acted more rapidly on LQLQPFPQPQLPYPQPQLPYPQPQLPYPQPQPF than MX. Whereas the FM enzyme showed a preference for Pro–Gln bonds, SC cleaved both Pro–Gln and Pro–Tyr bonds with comparable efficiency, and MX had a modest preference for Pro–(Tyr/Phe) sites over Pro–Gln sites. While a more comprehensive understanding of sequence and chain-length specificity may be needed to assess the relative utility of alternative prolyl endopeptidases for treating coeliac sprue, our present work has illustrated the diverse nature of this class of enzymes from the standpoint of proteolysing complex substrates such as gluten.
Keywords: coeliac sprue, chain-length specificity, gliadin peptide, prolyl endopeptidase, serine protease, subsite specificity
Abbreviations: BBM, brush border membrane; DPP IV, dipeptidyl peptidase IV; FM, Flavobacterium meningosepticum; MX, Myxococcus xanthus; Ni-NTA, Ni2+-nitrilotriacetate; pNA, p-nitroanilide; SC, Sphingomonas capsulata; Suc, succinyl; TFA, trifluoroacetic acid; Z, benzyloxycarbonyl
INTRODUCTION
Prolyl endopeptidases, or prolyl oligopeptidases, are a family of serine proteases with the unique ability to hydrolyse the peptide bond on the carboxyl side of a proline residue. These proline-specific enzymes are widely distributed in bacteria, fungi, animals and plants [1–5]. The isolation, cloning and heterologous expression of several prolyl endopeptidases have been reported, including those from Flavobacterium meningosepticum [3,5], Sphingomonas capsulate [1], Aeromonas hydrophila [6], porcine brain [2] and human T-lymphocytes [7]. These proteolytic enzymes are not only important in physiological processes, but also have considerable potential in biochemical and clinical applications [8]. Since most serine endoproteases are unable to hydrolyse proline residues, proline specific proteases may be therapeutic keys in digestive diseases. In the case of small intestinal disease, coeliac sprue, proline-rich gluten peptides from wheat, rye and barley are relatively resistant to gastrointestinal digestion, and therefore remain in the intestinal lumen to elicit immunopathology in genetically susceptible individuals [9]. Prolyl endopeptidase-catalysed endoproteolytic cleavage of these harmful peptides has been proposed to accelerate their clearance [10,11]. A key prerequisite of this therapeutically attractive concept is to obtain a fundamental understanding of the physical and chemical properties of this family of enzymes.
Crystal structures of the porcine prolyl endopeptidase and its mutated derivatives have revealed a unique two-domain architecture of the enzyme, comprising a catalytic domain and a propeller domain [12–15]. It has been suggested that the activity of prolyl endopeptidase is restricted to oligopeptides comprising no more than 30 amino acid residues [14], possibly by a gating filter mechanism [12]. However, most kinetic studies on prolyl endopeptidases have utilized short synthetic substrates, typically consisting of 2–4 residues [1,5,7,16]. A few studies have used longer peptide substrates, although these too have fewer than 10 residues [15]. Since many immunogenic peptides in gluten are longer than 10 residues, including a recently identified 33-amino-acid peptide [11], it is of interest to understand the activity of prolyl endopeptidases against substrates with various lengths.
To gain further insight into the similarities and differences between naturally occurring prolyl endopeptidases, we have systematically compared the properties of three similar enzymes from different bacterial sources. Our studies have utilized two known recombinant enzymes from Flavobacterium meningosepticum (FM) and Sphingomonas capsulata (SC), and a novel one from Myxococcus xanthus (MX) that we have expressed for the first time as a heterologous recombinant protein. Previous reports on the FM and SC prolyl endopeptidases had focused mainly on the cloning, sequencing and limited active site probing of the FM enzyme [17]. In the present study, the enzymic activities of these enzymes were quantitatively analysed using model substrates, as well as key gluten-derived peptides with important physiological relevance to coeliac sprue pathogenesis, PQPQLPYPQPQLP and LQLQPFPQPQLPYPQPQLPYPQPQLPYPQPQPF (33-mer). The peptide PQPQLPYPQPQLP contains an epitope found in α-gliadins that has been shown to play an immunodominant role, causing T-cell response to gluten in many coeliac sprue patients [11,18–21]. It cannot be cleaved by gastric or pancreatic proteases and is highly resistant to digestion by intestinal BBM (brush border membrane) peptidases, with only dipeptidyl carboxypeptidase I able to act upon it at a very limited rate [10]. Thus the efficiency of intestinal metabolism of this peptide can be expected to improve in the presence of an exogenous prolyl endopeptidase. The 33-mer peptide was selected as an essential probe for the present studies, because it is an unusually stable physiologically derived product of gastric and pancreatic digestion of α-gliadin, and strongly stimulates proliferation of T-cells from virtually all coeliac sprue patients tested to date [11]. The endoproteolytic breakdown of this 33-mer peptide represents an especially challenging goal for an exogenous prolyl endopeptidase in the detoxification of gluten in coeliac patients. We have probed the influence of substrate chain length, pH, pancreatic proteases and intestinal BBM peptidases on the activity of each enzyme. Both in vivo and ex vivo experiments were performed as part of these studies.
EXPERIMENTAL
Cloning of prolyl endopeptidase genes
Genes were amplified from the genomic DNA from the corresponding bacterial strains (F. meningosepticum: A.T.C.C. 13253; S. capsulata: A.T.C.C. 14666; M. xanthus: A.T.C.C. 25232). The sequence of the putative MX prolyl endopeptidase is available from the NCBI database (accession number AAD31004). Oligonucleotides used for PCR amplification included (with the restriction site underlined): (1) FM first half: 5′-AAC CAA TCA TAT GAA GTA CAA CAA ACT TTC TGT G (NdeI), 5′-GAT AAA AAC GGA AAG CTT GTA AGG GC (HindIII); FM second half: 5′-GCC CTT ACA AGC TTT CCG TTT TTA TC (HindIII) and 5′-CCC TTA ATT TTC AAA TTT TAG CTC GAG TTT ATG ATT TAT A (SacI); (2) SC first half: 5′-AGG ATA TCC ATA TGA AGA ACC GCT TGT GG (NdeI), 5′-GAC AAC CTC GAA TCC GTC GGC ATT G (HinfI); SC second half: 5′-CAA TGC CGA CGG ATT CGA GGT TGT C (HinfI), 5′-CGC GGG GAC CTC GAG TAG AAA CTG (SacI); (3) MX: 5′-CT CCC CAT ATG TCC TAC CCG GCG ACC (NdeI) and 5′-GTG GCG GCG CAG GGC CGC AAG CTT CCC AAG CG (HindIII). The amplified genes were cloned into a pET28b plasmid (Novagen).
Expression and purification of prolyl endopeptidases
Expression plasmids were introduced via transformation into Escherichia coli BL21(DE3) cells. Transformants were grown at 37 °C, and induced in the presence of 100 μM IPTG (isopropyl β-D-thiogalactoside) at 22 °C overnight. Low temperature induction was found to improve the yield of active enzyme.
All purification steps were performed at 4 °C unless noted otherwise. Since FM and SC enzymes naturally possess a signal sequence, they are secreted into the periplasmic space of E. coli. A modified osmotic shock protocol (EMD Biosciences, San Diego, CA, U.S.A.) was therefore used to obtain an enriched protein lysate containing either protein. Cell pellets (4 litres of culture) were resuspended in 30 ml of 30 mM Tris/HCl, pH 8, 20% sucrose and 1 mM EDTA, and stirred slowly at room temperature (25 °C) for 10 min. The suspension was centrifuged at 10000 g for 15 min, and the cell pellet was resuspended in ice-cold distilled water and stirred slowly on ice for 10 min. The shocked cells were then centrifuged again at 40000–50000 g for 30 min. The supernatant containing the periplasmic proteins was treated for 1–2 h with 1 M NaCl solution (to a final concentration of 300 mM NaCl), 1 M imidazole solution (to a final concentration 5 mM imidazole) and 1 ml of Ni-NTA (Ni2+-nitrilotriacetate) resin (Qiagen). The crude protein was then loaded on to a column containing additional 1 ml of Ni-NTA resin. After thorough wash steps using the wash buffer (50 mM phosphate buffer/300 mM NaCl, pH 7.0) with 0–10 mM imidazole, the protein was eluted with 150 mM imidazole, 50 mM phosphate and 300 mM NaCl, pH 8. The FM peptidase was further purified on a FPLC system (Amersham Pharmacia, Piscataway, NJ, U.S.A.) through a HiTrap-SP cation exchange column. Prior to application on the HiTrap-SP column, the protein was exchanged into 20 mM phosphate buffer (pH 7). Following injection, prolyl endopeptidase was eluted with a salt gradient from 20 mM phosphate, pH 7 (buffer A) to 20 mM phosphate/500 mM NaCl, pH 7 (buffer B) at a flow rate of 1 ml/min. MX prolyl endopeptidase, a cytosolic protein, was initially purified from a whole-cell lysate via Ni-NTA affinity chromatography (as above). The protein was further purified on a Superdex 200 gel filtration column (Amersham) with an isocratic gradient of 20 mM Hepes/2 mM dithiothreitol, pH 7.0, at 1 ml/min.
Activity assays
Post-proline cleavage activity was measured using Z-Gly-Pro-pNA and Suc-Ala-Pro-pNa (Z-, benzyloxycarbonyl-; -pNA, -p-nitroanilide; Suc-, succinyl-) (Bachem, Torrance, CA, U.S.A.). Z-Gly-Pro-pNA was dissolved in a PBS/water/dioxane (8:1.2:0.8, by vol.) assay mixture [22]. The concentration of Z-Gly-Pro-pNA was varied from 100–600 μM. Although the substrate Z-Gly-Pro-pNA was effective in detecting enzyme activity, its insolubility at higher concentrations precluded kinetic measurements under substrate-saturated conditions. In contrast, Suc-Ala-Pro-pNA [16], had the advantage of high water solubility at all pH values tested, and was therefore a preferred substrate for kinetic studies.
Hydrolysis of Suc-Ala-Pro-pNA by the FM, SC and MX peptidases was monitored in a reaction mixture (300 μl) consisting of 30 μl of 10×PBS buffer, a final concentration of 0.01–0.02 μM enzyme, and Suc-Ala-Pro-pNA (5 mM stock) at final concentrations between 100 μM to 4 mM. The release of the pNa was spectrophotometrically detected at a wavelength of 410 nm. The initial velocity of the reaction was determined by the increase in absorbance at 410 nm, which was used to calculate KM and kcat according to the Michaelis–Menten relationship.
For measurement of the influence of pH on the enzyme activity, a series of pH buffer solutions were prepared using 100 mM sodium phosphate for pH values from 6.0 to 8.0. Reaction mixtures (300 μl) consisted of 30 μl of 10×pH buffer, final concentration of 0.01 μM enzyme, and Suc-Ala-Pro-pNA to final concentrations between 100 μM to 4 mM.
pH stability
The ability to retain enzyme activity after exposure to acidic environments was determined. Hydrochloric acid solutions (10 μl) at pH values between 1.5 to 4.0 were mixed with 1 μl of enzyme for 10–20 min. The acidic mixtures were then neutralized with 40 μl of 10×PBS solution, 60 μl of 5 mM substrate to a final volume of 300 μl. The recovered enzyme activity was measured spectrophotometrically and compared with non-acid treated controls under identical conditions.
Gastric and pancreatic protease stability
In a 96-well U-bottomed plate, 5 μl of 2×reaction solution (40 mM Na2HPO4, pH 6.5 for pancreatic enzymes or 20 mM HCl for pepsin) was placed, and 1 μl of the degrading enzyme (either 1 mg/ml pepsin or a cocktail of 1 mg/ml trypsin, 1 mg/ml chymotrypsin, 0.2 mg/ml elastase and 0.2 mg/ml carboxypeptidase A) followed by 4 μl of prolyl endopeptidase (5–10 units/ml) were added. The plate was incubated at 37 °C for various times (e.g. 0, 5, 10, 20 and 30 min), with 190 μl of substrate solution [2 μl of Z-Gly-Pro-pNA (16.8 mg/ml in dioxane), 14 μl of dioxane, 24 μl of water, 150 μl of 10 mM PBS buffer, pH 7.5] added to each well. Absorption was measured at 410 nm for 1 to 2 min every 10 s to assay residual activity. Each buffer also contained 5 mg/ml gluten. Untreated gluten was used for pepsin, whereas gluten previously proteolysed with pepsin/0.01 M HCl, pH 2.0, (1:50, w/w), for 2 h at 37 °C was used for all other enzymes. Wells containing acid (pH 2.0) were neutralized by addition of 10 μl of 0.1 M NaOH before addition of the substrate. Enzyme activities are expressed as a percentage of the maximum activity, typically observed at the zero time point.
Substrate specificity
In addition to the reference substrates above, enzyme specificity was also evaluated using two immunogenic peptides derived from the sequence of α-gliadin proteins in gluten. Both peptides were synthesized using solid-phase peptide synthesis. The peptide PQPQLPYPQPQLP contains the immunodominant αII-epitope [20], and is resistant to proteolysis by pepsin or any pancreatic enzyme [9]. Prolyl endopeptidase specificity toward this substrate was assessed in a competitive assay in which 100 μM PQPQLPYPQPQLP and 100 μM Suc-Ala-Pro-pNA were mixed and allowed to react with 0.02 μM enzyme at 25 °C. The initial velocity of Suc-Ala-Pro-pNA cleavage was measured spectrophotometrically, whereas the initial velocity of PQPQLPYPQPQLP hydrolysis was determined via HPLC. The apparent specificity, kcat/KM, for the hydrolysis of PQPQLPYPQPQLP could be determined based on the known kcat/KM of the enzyme for Suc-Ala-Pro-pNA and the observed reaction rates of the two substrates, as follows:
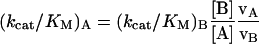 |
where A is PQPQLPYPQPQLP and B is Suc-Ala-Pro-pNA.
In addition to PQPQLPYPQPQLP, the specificity of prolyl endopeptidase for the more complex, but physiologically relevant, peptide LQLQPFPQPQLPYPQPQLPYPQPQLPYPQPQPF (33-mer [11]) was also assessed. Proteolysis reactions were performed at 37 °C in PBS buffer with 5–100 μM peptide and 0.1 μM enzyme for time periods of 1 min–4 h. The decrease in substrate concentration, as well as concomitant intermediate and product build-up, were monitored with HPLC analysis. Reverse-phase HPLC was performed on a system consisting of Beckman or Rainin Dynamax SD-200, a Varian 340 UV detector set at 215 nm and 280 nm. Solvent A was water with 0.1% TFA (trifluoroacetic acid) and solvent B was acetonitrile with 0.1% TFA. The gradient used was: 0–5% B in 0–15 min, 5–30% B in 15–30 min, 30–100% B in 30–35 min, 100% B for 5 min; flow rate, 1 ml/min. Separation was performed on a 4.6 mm×150 mm reverse-phase C-18 column (Vydac, Hesperia, CA, U.S.A.). Samples were centrifuged for 10 min at 13400 g, prior to injection of 10–100 μl of the supernatant.
Both PQPQLPYPQPQLP and the 33-mer peptide have multiple post-proline endoproteolytic sites. Thus multiple peptides accumulate during the course of the reaction, some of which are secondary substrates in themselves. Electrospray ionization trap MS/MS coupled with a UV HPLC (LCQ Classic/Surveyor; Thermo-Finnigan) was used to identify the preferred cleavage sites in PQPQLPYPQPQLP and the 33-mer.
For further evaluation of the proteolysis of the 33-mer and PQPQLPYPQPQLP in the appropriate physiological environment, gluten (30 g/l) was suspended in 0.01 M HCl (pH 2.0) and incubated in the presence of pepsin (600 mg/l) for 2 h at 37 °C. The resulting solution was neutralized using 10 M NaOH and diluted to 10 g/l in a phosphate buffer (40 mM, pH 6.5). An aliquot of this suspension (25 μl) was then supplemented with the 33-mer (0.1 mg/ml), PQPQLPYPQPQLP (0.08 mM), trypsin (0.1 mg/ml), chymotrypsin (0.1 mg/ml), elastase (0.02 mg/ml) and carboxypeptidase A (0.02 mg/ml). Prolyl endopeptidase (FM or MX; 1×, 500 m-unit/ml; 5×, 2.5 units/ml; 10×, 5 units/ml) and rat intestinal surface BBMs [1×, 40 m-unit/ml; 2×, 80 m-unit/ml, DPP IV (dipeptidyl peptidase IV) activity] were added to a total volume of 150 μl. The mixture was incubated at 37 °C and 25 μl aliquots were taken at 0, 5, 10, 30 and 60 min and immediately heat deactivated. To examine the chain-length specificity of individual prolyl endopeptidases, we performed competitive reactions containing both gluten-derived peptides, subjected the reaction mixture to reverse-phase HPLC, and monitored the disappearance of each substrate as a function of time. The peak areas of the 33-mer (32.5 min), PQPQLPYPQPQLP (27.5 min) were integrated.
In vivo endopeptidase activity
An adult (female or male) rat was anesthetized and maintained at 36–37 °C during the entire surgical procedure. The peritoneal cavity was opened, and a small incision was made at the beginning and the end of a 15–20 cm jejunum segment. Polyethylene catheters were inserted and secured at either end. The input catheter was connected to a pump-driven syringe filled with the experimental solution. The jejunum segment was perfused initially with PBS buffer to remove any residual debris at a flow rate of 0.4 ml/min. Purified peptide solutions (peptide concentration ranges from 25–100 μM) were then perfused at 0.4 ml/min through the jejunum segment with a 10–40 min residence time. In the case of a co-perfusion, the input catheter was connected to two syringes, one with a peptide solution and the other with the prolyl endopeptidase solution (concentration ranges from 50–500 μ-unit/μl), which were injected simultaneously. Fluid from the output catheter was collected into small centrifuge tubes in solid CO2 for subsequent analysis. The collected digestive products were analysed by HPLC on a C18 column.
RESULTS
Prolyl endopeptidase protein expression
FM and SC enzymes have their own signal sequences, and were therefore expressed as secreted soluble enzymes in the periplasmic space of E. coli. A simple freeze–thaw lysis procedure led to recovery of periplasmic protein without significant contamination by cytoplasmic proteins. In contrast, MX lacks a native signal sequence, and was therefore expressed as a cytoplasmic protein. Prolyl endopeptidase was purified from each lysate by Ni-NTA affinity purification, followed by a second chromatographic step. The yields of active FM, SC and MX prolyl endopeptidases were 1 mg/l, 60 mg/l and 30 mg/l respectively. The purity of the various enzymes was determined by SDS/PAGE to be >90% (Supplementary Figure 1, http://www.BiochemJ.org/bj/383/bj3830311add.htm).
Kinetic analysis with reference substrates
The activity of each prolyl endopeptidase was initially evaluated using the standard chromogenic substrate Suc-Ala-Pro-pNA. Release of the pNA was detected at A410, and kinetic data was fitted to the Michaelis–Menten equation (Supplementary Figure 2, http://www.BiochemJ.org/bj/383/bj3830311add.htm). Suc-Ala-Pro-pNA was selected as a reference substrate instead of the more commonly used Z-Gly-Pro-pNA due to the low solubility of the latter substrate, which necessitated use of co-solvents. The calculated kcat and KM values of FM, MX and SC prolyl endopeptidases for Suc-Ala-Pro-pNA are shown in Table 1. While these enzymes all exhibited a level of activity comparable with that of a serine protease, the MX enzyme had a higher specificity than FM, whereas SC had an intermediate level of specificity. The higher specificity of the MX peptidase could be attributed mainly to its higher affinity for the substrate, as reflected in the KM.
Table 1. Kinetic parameters for Suc-Ala-Pro-pNA hydrolysis by FM, MX and SC prolyl endopeptidases.
For details, see Supplementary Figure 2 (http://www.BiochemJ.org/bj/383/bj3830311add.htm) and the text.
| Enzyme | kcat (s−1) | KM (mM) | kcat/KM (mM−1/s−1) |
|---|---|---|---|
| FM | 39±6 | 1±0.02 | 44±7 |
| MX | 46±5 | 0.4±0.03 | 122±23 |
| SC | 149±5 | 2±0.01 | 70±2 |
Enzyme activity versus pH
The luminal environment of the duodenum is approximately at pH 6–7. Therefore, a therapeutically useful prolyl endopeptidase must retain high specific activity at that pH. The kcat and kcat/KM of each enzyme were titrated in the pH range 6–8 using Suc-Ala-Pro-pNA, (Table 2). In all cases the kcat/KM was optimal at or near pH 7.
Table 2. Effect of pH on the catalytic-centre activity (‘turnover number’) (kcat) and specificity (kcat/KM) of FM, MX and SC prolyl endopeptidases.
Phosphate buffer was used for all assays. n.d., not determined due to inability to saturate the enzyme at substrate concentrations up to 1 mM. Units: kcat, s−1; kcat/KM, mM−1·s−1.
| pH… | 6 | 7 | 8 | ||||
|---|---|---|---|---|---|---|---|
| Enzyme | kcat | kcat/KM | kcat | kcat/KM | kcat | kcat/KM | |
| FM | n.d. | 17±3 | 30±2 | 30±12 | 30±2 | 34±13 | |
| MX | n.d. | 49±6 | 48±6 | 97±26 | 61±4 | 63±21 | |
| SC | n.d. | 45±5 | 75±9 | 55±21 | n.d. | 24±2 | |
Prolyl endopeptidase stability
Although orally administered therapeutic proteins can be formulated to protect them from the acidic and proteolytic environment of the stomach [23], intrinsic acid stability of a prolyl endopeptidase is likely to be a desirable characteristic in its use as a therapeutic agent for coeliac sprue. We therefore evaluated the extent to which the activity of each enzyme remained intact after 10 min of incubation at selected pH values between 1.6 and 3.9. Within this pH range, FM retained 50–70% of its original activity; MX retained 70–90% activity, and SC retained 30–80% activity. Thus, although all prolyl endopeptidases appear to be moderately acid stable, the MX prolyl endopeptidase is most versatile.
Since therapeutic efficacy would require a prolyl endopeptidase to act upon gluten in conjunction with pancreatic proteases that are secreted into the duodenum, the resistance of the FM and MX peptidases toward both gastric and pancreatic enzymes was evaluated. For this we pre-incubated the enzymes with physiological quantities of either pepsin (at pH 2) or a cocktail comprising trypsin, chymotrypsin, elastase and carboxypeptidase A (at pH 6.5). As seen in Figure 1, both FM and MX were highly susceptible to pepsin-catalysed proteolysis, whereas they appeared to be remarkably stable to destruction in the presence of physiological quantities of the pancreatic enzymes.
Figure 1. Resistance of the FM and the MX prolyl endopeptidases to inactivation by gastric and pancreatic enzymes.

Pancreatic enzyme stability was evaluated by treating 5 units/ml of the FM and the MX enzymes with 1 mg/ml trypsin, 1 mg/ml chymotrypsin, 0.2 mg/ml elastase and 0.2 mg/ml carboxypeptidase A (40 mM phosphate, pH 6.5). Pepsin stability (gastric) was tested by treating FM and MX (5 units/ml) with 1 mg/ml pepsin (pH 2, 20 mM HCl).
Kinetic analysis using PQPQLPYPQPQLP as a substrate
The immunogenic peptide PQPQLPYPQPQLP is a recurring sequence in α-gliadins, and is resistant to proteolysis by gastric and pancreatic proteases. It is also highly resistant to digestion by intestinal brush border peptidases, with only DCP1 (dipeptidyl carboxypeptidase I) able to act upon it [10]. Treatment of this peptide with prolyl endopeptidase results in cleavage at internal proline residues, which in turn generates new recognition sites for brush border aminopeptidases. Thus, PQPQLPYPQPQLP represents a good test substrate to probe the specificity of prolyl endopeptidases.
The kcat/KM values of each enzyme were determined in an assay mixture containing PQPQLPYPQPQLP, as well as Suc-Ala-Pro-pNA as a competing substrate. The rates of disappearance of both substrates were determined by independent detection methods. The initial rate of disappearance of PQPQLPYPQPQLP was measured by HPLC, whereas the rate of consumption of Suc-Ala-Pro-pNA was measured spectrophotometrically. The kcat/KM values for the gluten peptide were found to be 228±17 (FM), 480±60 (MX) and 430±80 (SC). Both the FM and MX peptidases had a 5-fold higher specificity for the gluten peptide as compared with the chromogenic substrate, whereas the SC peptidase showed a 7-fold increase in specificity for the gluten peptide. This increase in specificity suggests that longer peptides may provide additional anchors at the catalytic site, a hypothesis that is consistent with the observation that Ala-Pro-pNA (which lacks an N-terminal Suc- or Z- group) did not react with any of the prolyl endopeptidases (L. Shan, unpublished work).
To analyse the regiospecificity of hydrolysis of PQPQLPYPQPQLP by individual prolyl endopeptidases, samples corresponding to early time points were further analysed by LC–MS/MS. The results, shown in Figure 2, revealed that each enzyme has unique subsite preferences. While the preferred site of cleavage by FM was at the PQPQLPPYP↓QPQLP position (cleavage site indicated by the arrow), MX preferentially cleaved the same peptide at the PQPQLP↓YPQPQLP position. SC had comparable preference for either site of cleavage. All enzymes preferentially cleaved the peptide at a proline located near the middle of the sequence, highlighting their functional difference from prolyl-specific exopeptidases such as DPP IV.
Figure 2. Site specificity of PQPQLPYPQPQLP hydrolysis by individual prolyl endopeptidasess.

UV HPLC (215 nm) traces are shown for each reaction mixture. Initial cleavage fragments (100 μM PQPQLPYPQPQLP, 0.1 μM enzyme, t=5 min) were identified by tandem MS. The starting material PQPQLPYPQPQLP and the cleavage fragments (A, PQPQLPYP; B, YPQPQLP; C, PQPQLP; D, QPQLP) are indicated in the traces. AU, arbitrary units of absorbance.
Chain-length tolerance and selectivity
It has been suggested that prolyl endopeptidases from the serine protease family are limited with regard to chain lengths of potential substrates [12]. To test this hypothesis in the context of the three bacterial prolyl endopeptidases studied here, we compared their hydrolytic activities with a physiologically relevant 33-mer peptide sequence from wheat gliadin [10,11], LQLQPFPQPQLPYPQPQLPYPQPQLPYPQPQPF (Figure 3A). The FM enzyme (0.1 μM) was able to hydrolyse 10 μM of the 33-mer in approx. 2–3 min, whereas the SC peptidase required >1 h to reach a comparable end point (results not shown). Based on initial rates, the FM enzyme was estimated to act 5-fold faster on the 33-mer than the MX enzyme, and >20-fold faster than the SC peptidase. Thus SC appears to have a severe chain-length restriction for long peptide substrates.
Figure 3. Hydrolysis of the 33-mer peptide by FM, MX and SC prolyl endopeptidases.

(A) Time dependence of hydrolysis in the presence of 10 μM substrate and 0.1 μM enzyme. The substrate appears as a doublet at a retention time of approx. 18 min, due to the presence of equal quantities of the 32-mer from which the N-terminal leucine residue is deleted; presence of this contaminant does not affect analysis. From the residual peak areas, the rates of substrate (33-mer+32-mer) disappearance were calculated as 2.3 μM/min (FM), 0.43 μM/min (MX) and 0.07 μM/min (SC). (B) Initial cleavage fragments observed due to hydrolysis by the FM enzyme (t=1 min) and the MX enzyme (t=5 min). (C) Summary of initial cleavage fragments from FM and MX prolyl endopeptidases catalysed hydrolysis of the 33-mer substrate. MW, molecular mass.
The intermediates and products from hydrolysis of the 33-mer by the FM and MX prolyl endopeptidases were analysed by LC–MS/MS (Figures 3B and 3C). Several features are noteworthy. First, even at relatively early time points, the digestion products of MX were predominantly small fragments, whereas FM digestion yielded a significant pool of long intermediates such as LQLQPFPQPQLPYPQPQLP, LQLQPFPQPQLPYP and LQLQPFPQPQLP. Thus, although both prolyl endopeptidases are able to effectively proteolyse the 33-mer, they have distinct hydrolytic patterns on this complex substrate. In particular, either the MX peptidase appears to be processive (i.e. for each 33-mer substrate molecule, it sequentially cleaves all the preferred sites in the chain prior to release), or alternatively the enzyme has a strong bias toward shorter chain substrates. It could also be noted that the C-terminal fragments generated by the two enzymes are different (QPQPF for FM, and YPQPQPF for MX). This finding is consistent with observed subsite preference in the case of PQPQLPYPQPQLP digestion.
To directly investigate chain-length selectivity of the three enzymes, we co-incubated PQPQLPYPQPQLP and the 33-mer peptide with each prolyl endopeptidase (Supplementary Figure 3, http://www.BiochemJ.org/bj/383/bj3830311add.htm), Both the SC and MX enzymes showed a clear preference for the 13-mer peptide, whereas FM showed comparable selectivity for both peptides.
To further evaluate the substrate preferences, PQPQPLPYPQPQLP and the 33-mer were mixed with pepsin-treated gluten, and allowed to react with pancreatic enzymes in the presence of BBM and either FM or MX enzymes. As seen in the HPLC traces (Figures 4A and 4B), the 33-mer had the longest retention time, whereas PQPQLPYPQPQLP and other medium-length gluten peptides eluted earlier. Here too FM proteolysed PQPQLPYPQPQLP, the 33-mer and other gluten peptides at comparable rates (Figure 4A). In the MX digestion, PQPQLPYPQPQLP and other smaller peptides were rapidly broken down (in 10 min), whereas hydrolysis of the 33-mer occurred at a slower rate (Figure 4B).
Figure 4. Competitive proteolysis of PQPQLPYPQPQLP (50 μM) and the 33-mer peptide (10 μM) in the presence of 30 mg/ml pepsin-treated gluten.

This complex mixture of substrates was treated under physiological conditions with a mixture of pancreatic enzymes (trypsin, chymotrypsin, carboxypeptidase and elastase), BBM enzymes (derived from rat small intestine) and either (A) FM or (B) MX. For details, see the text.
In vivo hydrolysis
To validate the implications of the above biochemical observations for peptide digestion in the intact small intestine, each prolyl endopeptidase was co-perfused in the rat jejunum with the 33-mer peptide substrate, and the effluent collected at a distance of 15–20 cm from the point of perfusion was analysed. In this live animal model, the impact of concerted action of the perfused (luminal) enzyme and the brush border (surface) peptidases is assessed. As shown by the in vitro results above, while the BBM enzymes were insufficient to process the 33-mer, FM promoted more complete breakdown of the 33-mer than both MX and SC (Figure 5). Within an enzyme dose range of 50–500 μ-unit/μl, the extent of 33-mer hydrolysis increased with increasing dose (results not shown), demonstrating that higher doses of prolyl endopeptidase could accelerate gluten breakdown in the mammalian gut.
Figure 5. Proteolysis of the 33-mer peptide (5 μM) co-perfused with individual prolyl endopeptidases (0.1 μM) in the small intestinal lumen of an anesthetized rat.

Each enzyme-substrate mixture was introduced via a catheter into a 15–20 cm segment of the upper jejunum. Samples were collected at the other end of the segment, and analysed by UV HPLC (at a wavelength of 215 nm). The control without any prolyl endopeptidases is shown in the top trace. AU, arbitrary units.
DISCUSSION
Coeliac sprue is a severe immune disease of the small intestine for which no therapeutic option to a strict life-long gluten-exclusion diet is currently available. Although gluten proteins from food grains such as wheat, rye and barley are important components of a nutritious diet, they can be highly toxic to patients suffering from coeliac sprue. In light of recent findings that related the strong antigenicity of gliadin peptides to their exceptional digestive resistance [10,11], prolyl endopeptidases, a class of enzymes not ordinarily involved in the intra-intestinal digestion of dietary proteins, were identified as a potentially interesting family of enzymes for oral coeliac sprue therapy. Understanding the properties of these enzymes is an essential prerequisite for such use. In our study, prolyl endopeptidases from three bacterial sources were selected and expressed in E. coli as recombinant proteins, and were subsequently purified and characterized. Two of these enzymes (from FM and SC) have been reported previously [1,8,22], whereas the third enzyme (from MX) represents a new member of the prolyl endopeptidase family.
In order to examine the endoproteolytic properties of these enzymes, it is important to utilize peptide substrates with internal cleavage sites. Although model substrates such as Z-Gly-Pro-pNA or Suc-Ala-Pro-pNA have been frequently used to identify and characterize prolyl endopeptidases [5,8,13,16,22], these substrates alone do not provide adequate insight to differentiate endopeptidases from each other or from proline-specific aminopeptidases (such as DPP IV). In the context of coeliac sprue, two peptides (PQPQLPYPQPQLP and LQLQPFPQPQLPYPQPQLPYPQPQLPYPQPQPF) have been recognized as useful probes for studying the fundamental properties of prolyl endopeptidases, as well as for their potential for detoxifying gluten.
Our investigations into the molecular recognition features of three bacterial prolyl endopeptidases for gluten peptides have revealed at least two interesting and potentially important characteristics of these enzymes. First, although all three enzymes tested here exhibited high specific activity against reference chromogenic substrates (Table 1 and Supplementary Figure 2, http://www.BiochemJ.org/bj/383/bj3830311add.htm), they showed remarkable differences in chain-length specificity (Figures 3A–3C). Whereas the SC and MX enzymes had higher specificity for PQPQLPYPQPQLP than FM, the reverse was true for the longer 33-mer gliadin peptide (Figure 3A), especially in the case of SC, which had extremely poor activity against the 33-mer. Structural and biochemical analysis led to the proposal that the activity of prolyl endopeptidases is limited to substrates containing fewer than 30 amino acid residues [14]. In that light, the activity of the MX enzyme and especially the FM enzyme against the 33-mer peptide is surprising. The broad chain length tolerance of the FM peptidase is vividly demonstrated in competitive in vitro and in vivo assays (Figures 4 and 5, and Supplementary Figure 3, http://www.BiochemJ.org/bj/383/bj3830311add.htm), where the FM peptidase was able to process longer and shorter substrates at comparable rates. Secondly sequence analysis of the major proteolytic products derived from both gliadin substrates demonstrated that the prolyl endopeptidase had distinct subsite specificity, as well as regiospecificity, in the context of the longer repetitive sequence. For example, the FM enzyme preferentially cleaved at PQPQLPYP↓QPQLP, whereas the MX enzyme preferred the PQPQLP↓YPQPQLP site, and SC had comparable activity toward either site (Figure 2). Similarly, sequence analysis of initial hydrolytic products of the 33-mer peptide underscored regiochemical differences between FM and MX peptidases (Figures 3A–3C). Whereas the MX enzyme treatment generated fragments mostly of 4–5 residues (presumably processed sequentially from both termini), FM yielded longer intermediates (presumably as a result of a preferential cleavage near the centre of the peptide). Thus the active sites of these enzymes are clearly different, which in turn has potential implications for the use of these enzymes in the detoxification of dietary gluten of a coeliac sprue patient. A more thorough mapping of sequence and chain-length specificity of these enzymes would facilitate identification of a therapeutic candidate that is most well suited for cleaving the major inflammatory epitopes in whole gluten.
In addition to analysing substrate specificity, we have also investigated other therapeutically relevant properties of the three prolyl endopeptidases. They include pH dependence of enzyme activity, acid tolerance of the protein and resistance towards inactivation by gastric, pancreatic and intestinal proteases/peptidases. All enzymes have a pH activity profile that is well matched to the mildly acidic environment of the upper small intestine (pH 6–7) (Table 2). They also appear to be moderately stable toward acid exposure as well as pancreatic protease (but not pepsin) action, with MX being the most stable (Figure 1). The enzymes also retain activity in the intact small intestinal lumen of a rat, indicative of their stability toward both intestinal secretions, as well as BBM peptidases (Figure 5). Together, these properties suggest that the prolyl endopeptidases evaluated in this study could be used as prophylactic agents for treating coeliac sprue, so long as they can be formulated to protect them from gastric inactivation by an enteric coating which rapidly hydrolyses when both enzyme and food are emptied into the weakly acidic environment of the upper small intestine. Finally, the expression levels of these enzymes vary significantly in recombinant E. coli. Specifically, in comparison to the FM enzyme, the expression levels of SC and MX prolyl endopeptidases were substantially superior.
Future studies on this family of proteases are likely to benefit from high resolution structural and mechanistic efforts, as have been recently pioneered by Polgar and co-workers in the context of the porcine brain prolyl endopeptidase [12–14,24,25]. The porcine brain enzyme has a di-domain architecture, including an unusual β-propeller domain that appears to regulate proteolysis. Pairwise sequence alignments between this structurally characterized enzyme and FM, MX and SC prolyl endopeptidases reveal 39% (49%), 36% (45%) and 40% (48%) identity (similarity) respectively. These alignments also suggest that the bacterial prolyl endopeptidases comprise a catalytic and a β-propeller domain. Since their active sites are predicted to lie near the interface between the two domains, mutagenesis at the inter-domain interface could alter protein dynamics and in turn affect substrate tolerance and specificity.
In summary, this report on the comparative biochemical analysis of three bacterial prolyl endopeptidases has shed light on several properties that are relevant for their potential use in coeliac sprue therapy. A comprehensive evaluation of such therapeutic use would require, among other aspects, a more extensive analysis of their specificity toward other known immunogenic gluten epitopes [21,26,27], as well as their stability in the presence of bile salts and buffer components ordinarily found in the gut. Nonetheless, our results provide a solid starting point for future protein engineering efforts aimed at enzyme optimization. We also note that this family of serine proteases includes numerous other putative homologues whose cDNAs have been sequenced, but whose gene products remain to be characterized. In light of the favourable properties of the MX prolyl endopeptidase, which was expressed and characterized for the first time as part of the present study, it would be useful to screen additional wild-type enzymes.
Online data
Acknowledgments
We thank Felix Hausch and Nilda Santiago for technical assistance, and Dale Kaiser for supplying Myxococcus xanthus genomic DNA. Allis Chien and Andrew Guzzetta (Stanford Vincent Coates Mass Spectrometry Facility) are acknowledged for their assistance with mass spectrometric analysis. This research is supported by a grant (DK063158) from the National Institutes if Health to C. K, G. M. G. and L. M. S. L. S. is a recipient of a Stanford Graduate Fellowship.
References
- 1.Kabashima T., Fujii M., Meng Y., Ito K., Yoshimoto T. Prolyl endopeptidase from Sphingomonas capsulata: isolation and characterization of the enzyme and nucleotide sequence of the gene. Arch. Biochem. Biophys. 1998;358:141–148. doi: 10.1006/abbi.1998.0836. [DOI] [PubMed] [Google Scholar]
- 2.Rennex D., Hemmings B. A., Hofsteenge J., Stone S. R. cDNA cloning of porcine brain prolyl endopeptidase and identification of the active-site seryl residue. Biochemistry. 1991;30:2195–2203. doi: 10.1021/bi00222a025. [DOI] [PubMed] [Google Scholar]
- 3.Yoshimoto T., Kanatani A., Shimoda T., Inaoka T., Kokubo T., Tsuru D. Prolyl endopeptidase from Flavobacterium meningosepticum: cloning and sequencing of the enzyme gene. J. Biochem. (Tokyo) 1991;110:873–878. doi: 10.1093/oxfordjournals.jbchem.a123682. [DOI] [PubMed] [Google Scholar]
- 4.Yoshimoto T., Miyazaki K., Haraguchi N., Kitazono A., Kabashima T., Ito K. Cloning and expression of the cDNA encoding prolyl oligopeptidase (prolyl endopeptidase) from bovine brain. Biol. Pharm. Bull. 1997;20:1047–1050. doi: 10.1248/bpb.20.1047. [DOI] [PubMed] [Google Scholar]
- 5.Yoshimoto T., Walter R., Tsuru D. Proline-specific endopeptidase from Flavobacterium. Purification and properties. J. Biol. Chem. 1980;255:4786–4792. [PubMed] [Google Scholar]
- 6.Kanatani A., Yoshimoto T., Kitazono A., Kokubo T., Tsuru D. Prolyl endopeptidase from Aeromonas hydrophila: cloning, sequencing, and expression of the enzyme gene, and characterization of the expressed enzyme. J. Biochem. (Tokyo) 1993;113:790–796. doi: 10.1093/oxfordjournals.jbchem.a124120. [DOI] [PubMed] [Google Scholar]
- 7.Vanhoof G., Goossens F., Hendriks L., De Meester I., Hendriks D., Vriend G., Van Broeckhoven C., Scharpe S. Cloning and sequence analysis of the gene encoding human lymphocyte prolyl endopeptidase. Gene. 1994;149:363–366. doi: 10.1016/0378-1119(94)90177-5. [DOI] [PubMed] [Google Scholar]
- 8.Diefenthal T., Dargatz H., Witte V., Reipen G., Svendsen I. Cloning of proline-specific endopeptidase gene from Flavobacterium meningosepticum: expression in Escherichia coli and purification of the heterologous protein. Appl. Microbiol. Biotechnol. 1993;40:90–97. doi: 10.1007/BF00170434. [DOI] [PubMed] [Google Scholar]
- 9.Sollid L. M. Coeliac disease: dissecting a complex inflammatory disorder. Nat. Rev. Immunol. 2002;2:647–655. doi: 10.1038/nri885. [DOI] [PubMed] [Google Scholar]
- 10.Hausch F., Shan L., Santiago N. A., Gray G. M., Khosla C. Intestinal digestive resistance of immunodominant gliadin peptides. Am. J. Physiol. Gastrointest. Liver. Physiol. 2002;283:G996–G1003. doi: 10.1152/ajpgi.00136.2002. [DOI] [PubMed] [Google Scholar]
- 11.Shan L., Molberg O., Parrot I., Hausch F., Filiz F., Gray G. M., Sollid L. M., Khosla C. Structural basis for gluten intolerance in celiac sprue. Science. 2002;297:2275–2279. doi: 10.1126/science.1074129. [DOI] [PubMed] [Google Scholar]
- 12.Fulop V., Szeltner Z., Polgar L. Catalysis of serine oligopeptidases is controlled by a gating filter mechanism. EMBO Rep. 2000;1:277–281. doi: 10.1093/embo-reports/kvd048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fulop V., Szeltner Z., Renner V., Polgar L. Structures of prolyl oligopeptidase substrate/inhibitor complexes. Use of inhibitor binding for titration of the catalytic histidine residue. J. Biol. Chem. 2001;276:1262–1266. doi: 10.1074/jbc.M007003200. [DOI] [PubMed] [Google Scholar]
- 14.Polgar L. The prolyl oligopeptidase family. Cell Mol. Life Sci. 2002;59:349–362. doi: 10.1007/s00018-002-8427-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szeltner Z., Renner V., Polgar L. The noncatalytic β-propeller domain of prolyl oligopeptidase enhances the catalytic capability of the peptidase domain. J. Biol. Chem. 2000;275:15000–15005. doi: 10.1074/jbc.M000942200. [DOI] [PubMed] [Google Scholar]
- 16.Heins J., Welker P., Schonlein C., Born I., Hartrodt B., Neubert K., Tsuru D., Barth A. Mechanism of proline-specific proteinases: (I) Substrate specificity of dipeptidyl peptidase IV from pig kidney and proline-specific endopeptidase from Flavobacterium meningosepticum. Biochim. Biophys. Acta. 1988;954:161–169. doi: 10.1016/0167-4838(88)90067-2. [DOI] [PubMed] [Google Scholar]
- 17.Bordusa F., Jakubke H. D. The specificity of prolyl endopeptidase from Flavobacterium meningoseptum: mapping the S′ subsites by positional scanning via acyl transfer. Bioorg. Med. Chem. 1998;6:1775–1780. doi: 10.1016/s0968-0896(98)00145-x. [DOI] [PubMed] [Google Scholar]
- 18.Anderson R. P., Degano P., Godkin A. J., Jewell D. P., Hill A. V. In vivo antigen challenge in celiac disease identifies a single transglutaminase-modified peptide as the dominant A-gliadin T-cell epitope. Nat. Med. 2000;6:337–342. doi: 10.1038/73200. [DOI] [PubMed] [Google Scholar]
- 19.Arentz-Hansen E. H., McAdam S. N., Molberg O., Kristiansen C., Sollid L. M. Production of a panel of recombinant gliadins for the characterisation of T cell reactivity in coeliac disease. Gut. 2000;46:46–51. doi: 10.1136/gut.46.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arentz-Hansen H., Korner R., Molberg O., Quarsten H., Vader W., Kooy Y. M., Lundin K. E., Koning F., Roepstorff P., Sollid L. M., McAdam S. N. The intestinal T cell response to α-gliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. J. Exp. Med. 2000;191:603–612. doi: 10.1084/jem.191.4.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arentz-Hansen H., McAdam S. N., Molberg O., Fleckenstein B., Lundin K. E., Jorgensen T. J., Jung G., Roepstorff P., Sollid L. M. Celiac lesion T cells recognize epitopes that cluster in regions of gliadins rich in proline residues. Gastroenterology. 2002;123:803–809. doi: 10.1053/gast.2002.35381. [DOI] [PubMed] [Google Scholar]
- 22.Chevallier S., Goeltz P., Thibault P., Banville D., Gagnon J. Characterization of a prolyl endopeptidase from Flavobacterium meningosepticum. Complete sequence and localization of the active-site serine. J. Biol. Chem. 1992;267:8192–8199. [PubMed] [Google Scholar]
- 23.Sinha V. R., Kumria R. Binders for colon specific drug delivery: an in vitro evaluation. Int. J. Pharm. 2002;249:23–31. doi: 10.1016/s0378-5173(02)00398-8. [DOI] [PubMed] [Google Scholar]
- 24.Fulop V., Bocskei Z., Polgar L. Prolyl oligopeptidase: an unusual β-propeller domain regulates proteolysis. Cell. 1998;94:161–170. doi: 10.1016/s0092-8674(00)81416-6. [DOI] [PubMed] [Google Scholar]
- 25.Polgar L., Patthy A. Cleavage of the Lys196–Ser197 bond of prolyl oligopeptidase: enhanced catalytic activity for one of the two active enzyme forms. Biochemistry. 1992;31:10769–10773. doi: 10.1021/bi00159a018. [DOI] [PubMed] [Google Scholar]
- 26.Vader L. W., de Ru A., van der Wal Y., Kooy Y. M., Benckhuijsen W., Mearin M. L., Drijfhout J. W., van Veelen P., Koning F. Specificity of tissue transglutaminase explains cereal toxicity in celiac disease. J. Exp. Med. 2002;195:643–649. doi: 10.1084/jem.20012028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vader W., Kooy Y., Van Veelen P., De Ru A., Harris D., Benckhuijsen W., Pena S., Mearin L., Drijfhout J. W., Koning F. The gluten response in children with celiac disease is directed toward multiple gliadin and glutenin peptides. Gastroenterology. 2002;122:1729–1737. doi: 10.1053/gast.2002.33606. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.