

Abstract
RNA-capping enzymes are involved in the synthesis of the cap structure found at the 5′-end of eukaryotic mRNAs. The present study reports a detailed study on the thermodynamic parameters involved in the interaction of an RNA-capping enzyme with its ligands. Analysis of the interaction of the Saccharomyces cerevisiae RNA-capping enzyme (Ceg1) with GTP, RNA and manganese ions revealed significant differences between the binding forces that drive the interaction of the enzyme with its RNA and GTP substrates. Our thermodynamic analyses indicate that the initial association of GTP with the Ceg1 protein is driven by a favourable enthalpy change (ΔH=−80.9 kJ/mol), but is also clearly associated with an unfavourable entropy change (TΔS=−62.9 kJ/mol). However, the interaction between Ceg1 and RNA revealed a completely different mode of binding, where binding to RNA is clearly dominated by a favourable entropic effect (TΔS=20.5 kJ/mol), with a minor contribution from a favourable enthalpy change (ΔH=−5.3 kJ/mol). Fluorescence spectroscopy also allowed us to evaluate the initial binding of GTP to such an enzyme, thereby separating the GTP binding step from the concomitant metal-dependent hydrolysis of GTP that results in the formation of a covalent GMP–protein intermediate. In addition to the determination of the energetics of ligand binding, our study leads to a better understanding of the molecular basis of substrate recognition by RNA-capping enzymes.
Keywords: enzymology, fluorescence spectroscopy, mRNA-capping, thermodynamics, yeast
Abbreviations: GdmCl, guanidinium chloride
INTRODUCTION
The 5′-end of eukaryotic mRNAs harbours an m7G411N cap structure that plays a critical role in the translation, stability, splicing and transport of the mRNAs from the nucleus to the cytoplasm [1]. Addition of the cap structure is the first modification to occur on nascent pre-mRNAs. The cap structure consists of a 7-methylguanosine residue linked by a 5′–5′-triphosphate bridge to the RNA transcript and is synthesized by a series of three sequential enzymic reactions. The first step involves the hydrolysis of the RNA 5′-triphosphate end of the nascent RNA by an RNA triphosphatase to form a diphosphate end. The addition of GMP to the diphosphate end is then mediated by an RNA guanylyltransferase or RNA-capping enzyme. Finally, the G411N cap is methylated by an RNA (guanine-N7) methyltransferase [2]. The importance of the cap structure for RNA metabolism is highlighted by genetic analyses in Saccharomyces cerevisiae that showed that the triphosphatase, guanylyltransferase and methyltransferase components of the capping apparatus are essential for cell growth [3–8].
Eukaryotic RNA-capping enzymes catalyse a two-stage Ping-Pong reaction in which GMP is transferred to the 5′-diphosphate terminus of RNA. In the first step of the reaction, the enzyme uses GTP as a substrate, and the reaction proceeds with the formation of a covalent GMP–enzyme intermediate and concomitant release of pyrophosphate [9,10]. The second step of the reaction entails the catalytic transfer of the GMP moiety to the diphosphate end of the RNA [9,10]. Both steps require a bivalent metal ion cofactor [9,10]. The reaction is mechanistically related to the reactions catalysed by ATP-dependent DNA ligases, RNA ligases and tRNA ligases, in which a covalent protein–ATP intermediate is formed [11]. All these enzymes share many conserved motifs and are part of a conserved family of covalent nucleotidyl transferases [11,12].
The crystal structures of five different members of the covalent nucleotidyl transferase superfamily have been determined [13,14] and they provide insightful information on the reaction chemistry [15]. Members of the family are characterized by a common tertiary structure that consists of an N-terminus, which encompasses the nucleotide-binding pocket, and a C-terminal oligonucleotide binding-fold domain. Examination of the Chlorella virus RNA guanylyltransferase crystals suggested that a large conformational change occurs on GTP binding, shifting the structure from an open to a closed state [13,14]. On the basis of these crystallographic studies, a model has been suggested, in which the conformational change encountered on GTP binding would promote metal ion binding and guanylylation [13,14].
Crystallography has provided important information regarding the specific residues that participate in ligand binding and also regarding the conformational changes that occur in the RNA guanylyltransferase reaction. However, proteins are not static, and some conformational species may not be represented in the different crystalline forms. A detailed thermodynamic description is highly desirable to complement the structural data. In this paper, we describe a detailed thermodynamic study of ligand binding to the S. cerevisiae RNA guanylyltransferase (Ceg1 protein) using fluorescence spectroscopy. We focused on the interaction of the enzyme with GTP, manganese and RNA, to (i) evaluate the relative contributions of both the enthalpy (ΔH) and entropy (ΔS) changes to the binding of the respective ligands, (ii) determine the roles of ionic equilibria and electrostatic interactions in ligand recognition and (iii) monitor potential conformational changes during reaction chemistry. Significant differences were found between the respective forces that drive the interaction of the enzyme with its ligands. These differences are discussed in terms of the relative contributions of both the enthalpy and entropy changes, thereby providing a thermodynamic basis for the forces driving complex formation. In addition to determination of the energetics of ligand binding, our study leads to a better understanding of the molecular basis of the RNA-capping reaction.
EXPERIMENTAL
Expression and purification of the Ceg1 protein
A plasmid for the expression of the Ceg1 protein was generated by inserting the CEG1 gene between the NheI and HindIII sites of the pET28a expression plasmid (Novagen). In this context, the Ceg1 protein is fused in-frame with an N-terminal peptide containing six tandem histidine residues, and expression of the His-tagged protein is driven by a T7 RNA polymerase promoter. The resulting recombinant plasmid, pET-Ceg1, was transformed into Escherichia coli BL21(DE3). A 100 ml culture of E. coli BL21(DE3)/pET-Ceg1 was grown at 37 °C in Luria–Bertani medium containing 30 μg/ml kanamycin until the absorbance A600 reached 0.5. The culture was adjusted to 0.4 mM isopropyl β-D-thiogalactoside, and the incubation was continued at 18 °C for 20 h. The cells were then harvested by centrifugation, and the pellet was stored at −80 °C. All subsequent procedures were performed at 4 °C. The thawed bacterial pellets were resuspended in 5 ml of lysis buffer A [50 mM Tris/HCl, pH 7.5, 150 mM NaCl and 10% (w/v) sucrose] and cell lysis was achieved by the addition of lysozyme and Triton X-100 to final concentrations of 50 μg/ml and 0.1% respectively. The lysates were sonicated to reduce the viscosity, and the insoluble material was removed by centrifugation at 10000 g for 45 min. The soluble extract was applied to a 2 ml column of Ni2+-nitrilotriacetic acid–agarose (Qiagen) that had been equilibrated with buffer A containing 0.1% Triton X-100. The column was washed with the same buffer and then eluted stepwise with buffer B [50 mM Tris/HCl, pH 8.0, 0.1 M NaCl and 10% (v/v) glycerol] containing 50, 100, 200, 500 and 1000 mM imidazole. The polypeptide composition of the column fractions was monitored by SDS/PAGE. The recombinant Ceg1 protein was retained on the column and recovered in the 200 mM imidazole eluate. This fraction was applied to a 2 ml column of phosphocellulose that had been equilibrated with buffer C (50 mM Tris/HCl, pH 8.0, 50 mM NaCl and 10% glycerol). The column was washed with the same buffer and then eluted stepwise with buffer C containing 0.1, 0.2, 0.3, 0.4, 0.5 and 1.0 M NaCl. The recombinant protein was retained on the column and recovered predominantly in the 0.3 M NaCl fraction. The fraction was then dialysed against buffer C that was supplemented with potassium pyrophosphate (5 mM) to ensure a homogeneous non-guanylylated enzyme. The phosphocellulose preparation was stored at −80 °C. The protein concentration was determined by the Bio-Rad dye binding method using BSA as the standard.
Fluorescence measurements
Fluorescence was measured using a Hitachi F-2500 fluorescence spectrophotometer. Background emission was eliminated by subtracting the signal from either buffer alone or buffer containing an appropriate quantity of the substrate.
The extent to which ligands bind to the Ceg1 protein was determined by monitoring the fluorescence emission of a fixed concentration of proteins and titrating with a given ligand. The binding can be described by
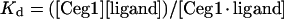 |
(1) |
where Kd is the apparent dissociation constant, [Ceg1] the concentration of the protein, [Ceg1·ligand] the concentration of the complexed protein and [ligand] the concentration of the unbound ligand.
The proportion of ligand-bound protein as described by eqn (1) is related to the measured fluorescence emission intensity by
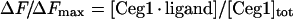 |
(2) |
where ΔF is the magnitude of the difference between the observed fluorescence intensity at a given concentration of ligand and the fluorescence intensity in the absence of ligand, ΔFmax the difference at infinite [ligand] and [Ceg1]tot the total protein concentration.
If the total ligand concentration, [ligand]tot, is in large molar excess relative to [Ceg1]tot, then it can be assumed that [ligand] is approximately equal to [ligand]tot. Equations (1) and (2) can then be combined to give
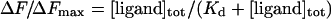 |
(3) |
The Kd values were determined from a non-linear least-squares regression analysis of the titration data by using eqn (3). The stoichiometry of binding was established from a linear version of the Hill equation:
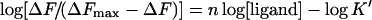 |
(4) |
where n is the order of the binding reaction with respect to ligand concentration and K′ the concentration of ion that yields 50% of ΔFmax.
The thermodynamic parameters ΔG (Gibbs free energy of binding), ΔH and ΔS were determined using the equation
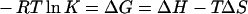 |
(5) |
where R is the gas constant and T is the absolute temperature.
RNA substrate
An RNA substrate of 84 nucleotides was synthesized by in vitro transcription using the T7 RNA polymerase (New England Bio-labs). The RNA transcript was synthesized from the pBS-KSII+plasmid (Stratagene), which had been linearized with HindIII. The RNA substrate was purified on a denaturing 20% (w/v) polyacrylamide gel and visualized by UV shadowing. The corresponding band was excised and then eluted from the gel by an overnight incubation in 0.1% SDS/0.5 M ammonium acetate. The RNA was then precipitated with ethanol and quantified by spectrophotometry. Alternatively, an RNA substrate harbouring a diphosphorylated 5′-end was synthesized by incubating the RNA with 2 μg of purified S. cerevisiae RNA triphosphatase, as described previously [8]. An aliquot of the reaction was analysed by TLC on a polyethyleneimine–cellulose plate and visualized under UV light.
Thermodynamics of binding
The temperature dependence of the association constant Kas for ligand binding was analysed according to the van't Hoff isobaric equation, assuming that the entropy change (ΔSo) and the enthalpy change (ΔHo) remained constant over the whole range of temperatures:
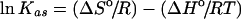 |
(6) |
CD spectroscopy measurements
CD measurements were performed with a Jasco J-810 spectropolarimeter. The samples were analysed in quartz cells with path lengths of 1 mm. Far- and near-UV wavelength scans were recorded from 200 to 250 nm and from 250 to 340 nm respectively. All dichroic spectra were corrected by subtraction of the background for the spectrum obtained with either buffer alone or buffer containing GTP. The average of six wavelength scans is presented. Ellipticity results were expressed in terms of the mean residue ellipticity, [θ], in degrees·cm2·dmol−1.
Equilibrium unfolding experiments
A 100 nM solution of Ceg1 was adjusted to the desired final concentration of GdmCl (guanidinium chloride) and incubated for 60 min at 22 °C. The parameters ΔGou (free energy of unfolding in the absence of denaturant), m (co-operativity of unfolding) and Cm (the midpoint concentration of denaturant required to unfold half of the total amount of protein) were obtained as outlined previously using the following equations:
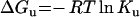 |
(7) |
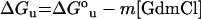 |
(8) |
RESULTS
Expression, purification and intrinsic fluorescence properties of the Ceg1 protein
The S. cerevisiae RNA-capping enzyme (Ceg1 protein) contains motifs shared by various nucleotidyl transferases, and its RNA guanylyltransferase activity has been shown to be dependent on the presence of manganese ions [4]. To characterize the ligand-binding activity of the enzyme, the Ceg1 protein was expressed in E. coli as described under the Experimental section. SDS/PAGE analysis showed that the 55 kDa Ceg1 protein was the predominant polypeptide in the purified fraction (Figure 1A). The amount of Ceg1 protein in this fraction corresponded to 150 μg/ml. Note that potassium pyrophosphate was added during the purification steps to ensure a homogeneous non-guanylylated enzyme. The ability of the Ceg1 protein to form a covalent GMP–enzyme intermediate was detected by label transfer from [α-32P]GTP to the enzyme. A single SDS-stable GMP–enzyme complex that migrated as a 55 kDa species was detected after SDS/PAGE (Figure 1B). Note that labelling of the enzyme was not detected in the absence of bivalent cations (results not shown). The yield of protein–GMP formation by the Ceg1 protein increased with GTP concentrations up to 300 μM and leveled off thereafter (Figure 1C). Note that the amount of protein–GMP complex formed during a 10 min incubation at 37 °C in the presence of 1 mM [α-32P]GTP was proportional to the amount of Ceg1 protein added, and formation of the enzyme–GMP complex was dependent on the presence of a bivalent ion (results not shown).
Figure 1. Expression, purification and fluorescent properties of Ceg1.

(A) An aliquot (3 μg) of the purified preparation of Ceg1 (P) was analysed by electrophoresis through a 12.5% polyacrylamide gel containing 0.1% SDS and visualized by staining with Coomassie Blue dye. The positions and masses (in kDa) of the molecular-mass standards are indicated on the left (M). (B) RNA guanylyltransferase activity of the Ceg1 protein. The enzyme (3 μg) was incubated for 5 min at 37 °C with 10 μM [α-32P]GTP in a buffer containing 50 mM Tris/HCl (pH 8.0), 5 mM dithiothreitol and 5 mM MgCl2. The reaction was stopped by the addition of EDTA and SDS to final concentrations of 10 mM and 1%. The reaction was analysed by electrophoresis through a 12.5% polyacrylamide gel containing 0.1% SDS. An autoradiogram of the gel is shown. The positions and masses (in kDa) of the molecular-mass standards are indicated on the left. (C) The extent of GMP–enzyme formation is plotted as a function of the substrate concentration. (D) Background-corrected fluorescence emission spectra of Ceg1. Curve 1, purified protein in 50 mM Tris/HCl (pH 7.5) and 50 mM KOAc; curve 2, purified protein after a 2 h exposure to an 8 M solution of urea at 25 °C. Fluorescence spectra were recorded at an excitation wavelength of 290 nm. A graph of the molar fluorescence of Ceg1 is shown in the inset. Various concentrations of the purified Ceg1 protein were assayed in 50 mM Tris/HCl (pH 7.5) and 50 mM KOAc. Emission was monitored at 339 nm and excitation was performed at 290 nm.
The fluorescence emission spectrum of purified Ceg1 in standard buffer at 22 °C is shown in Figure 1(D). To obtain the maximal emission peak at low concentrations of the protein, which is required to determine accurately the Kd values, excitation was performed at 290 nm. Both tyrosine and tryptophan absorb at this wavelength [16]. However, varying the excitation wavelength from 254 nm, where the contribution of tyrosine fluorescence to the emission spectrum would be the greatest, to 295 nm, where the emission spectrum would arise almost exclusively from tryptophan, produced no change in either the emission maximum (λmax=339 nm) or the spectral bandwidth (75 nm at half-height) (results not shown). Therefore, despite the fact that Ceg1 contains 15 tyrosine residues in addition to the seven tryptophan residues, the emission spectrum is dominated by the indole fluorophores. This dominance is due, in part, to the higher molar absorption coefficient of tryptophan and to resonance energy transfer from tyrosine to tryptophan.
The λmax of the enzyme (339 nm) is blue-shifted relative to that of free L-tryptophan, which under the same conditions is observed to be at 350 nm. The λmax of tryptophan is highly sensitive to the polarity of the microenvironment in which its indole side chain is localized. Blue-shifts of protein emission spectra have been ascribed to shielding of the tryptophan residues from the aqueous phase [17]. This shielding is a result of the three-dimensional structure of the protein. Accordingly, denaturation of Ceg1 with 8 M urea results in a red-shift of λmax towards 350 nm (Figure 1D).
The molar intensity of the fluorescence emission spectrum of Ceg1 was also determined. This spectrum was obtained to determine whether significant protein aggregation or loss of protein from the solution through adhesion, could influence the data. As shown in Figure 1(D), a decrease in fluorescence is observed with increase in the concentration of Ceg1. A linear change of 0.11 fluorescence intensity units/1 nM protein was observed over the range examined. This relatively small change can be attributed to the minor aggregation occurring at higher Ceg1 concentrations. All subsequent binding experiments were therefore performed at a protein concentration of 100 nM, with the assumption that the binding equilibrium was not complicated by the presence of an aggregation equilibrium.
Binding of GTP to the Ceg1 protein
The traditional guanylyltransferase assay relies on the formation of a covalent GMP–protein complex after label transfer from [α-32P]GTP to the enzyme [9,10]. Such an assay does not permit the detection of the GTP binding activity itself, but permits only the detection of the metal-dependent hydrolysis of GTP and the concomitant formation of a covalent guanylylated intermediate. One of our goals in the present study was to characterize the initial GTP binding step of the RNA-capping reaction. Fluorescence spectroscopy has been used in recent years to monitor the binding of nucleotides to various enzymes [18–21]. Fluorescence spectroscopy assays were performed in the absence of metal ions to monitor the initial GTP binding step, thereby separating the binding activity from catalysis. We observed that the binding of GTP to the enzyme resulted in a significant modification in the intensity of intrinsic fluorescence of the protein. The addition of increasing amounts of GTP produced a decrease in the fluorescence intensity, and the λmax shifted from 339 nm for the free protein to 336 nm in the presence of higher concentrations of GTP, indicating movement of the tryptophan residues to a more hydrophobic environment (results not shown). The binding of GTP could not be detected when the enzyme was preincubated at 60 °C in the presence of 0.1% (results not shown). Approx. 73% of the intrinsic protein fluorescence was accessible to the quencher GTP (Table 1). As a consequence, we were able to evaluate the Gibbs free energy of binding ΔG, as well as both the enthalpy (ΔH) and entropy (ΔS) change associated with the binding of GTP to the enzyme. Evaluation of these thermodynamic parameters yields significant insight into the nature of the GTP binding reaction. Our binding studies indicate that the ΔG for the interaction of GTP with the enzyme was −18.0 kJ/mol. Although the free energy of binding provides the overall description of the system, defining the entropic and enthalpic contributions to the free energy provides a more complete understanding of the forces that drive the protein–GTP association. The enthalpic and entropic contributions to the free energy of binding were determined by measuring the initial binding of GTP to the enzyme as a function of temperature. The GTP binding reaction was shown to be exothermic at 25 °C with a high enthalpy of association, ΔH=−80.9 kJ/mol. Analysis of a van't Hoff plot for the interaction between GTP and Ceg1 (Figure 2A) revealed that the TΔS value for the binding reaction was −62.9 kJ/mol, clearly indicating that the initial GTP binding step is primarily driven by enthalpy, with an unfavourable entropic contribution.
Table 1. Maximal decreases in fluorescence (ΔF/Fo)max and thermo-dynamic parameters for the interaction of Ceg1 with various ligands.
n.d., not determined.
| Ligand | (ΔF/Fo)max | ΔG (kJ/mol) | ΔH (kJ/mol) | ΔS (kJ/mol) |
|---|---|---|---|---|
| GTP | 0.73 | −18.0 | −80.9 | −62.9 |
| RNA | 0.48 | −25.8 | −5.3 | 20.5 |
| Mn2+ | −0.17 | −14.2 | n.d. | n.d. |
Figure 2. Binding of GTP to the Ceg1 protein.

(A) A van't Hoff plot for the interaction between GTP and Ceg1 is shown. Increasing amounts of GTP were added to a 100 nM solution of the enzyme in binding buffer (50 mM Tris/HCl, pH 7.5, and 50 mM KOAc) and the emission spectrum was scanned from 310 to 440 nm. The effect of temperature on the association constant was evaluated at pH 7.0. (B) The effect of increasing ionic strength on the association constant of Ceg1 for GTP was investigated. Increasing concentrations of KCl were added to the reactions to generate the desired ionic strengths. (C) Kinetic analysis of real-time binding of GTP to the Ceg1 protein. A 100 nM solution of the enzyme was incubated with 5 mM GTP. Emission was monitored under constant agitation for 60 s at 339 nm and excitation was performed at 290 nm. (D) Binding of GTP to the Ceg1 protein as a function of pH.
An important component of the energetics of protein–ligand binding is the interaction of protein residues with the electrostatic field of the ligand. The salt dependence of the Ceg1–GTP interaction provides information about the contribution of electrostatic interactions. The experimentally observed equilibrium constant was clearly dependent on the ionic strength of the solution. Analysis of the data revealed that the interaction is strongly attenuated at increased KCl concentrations (Figure 2B). Evaluation of the Gibbs energy due to electrostatic interactions (ΔΔGES) by extrapolation of the binding reaction to an ionic strength of 1 M, the standard state where electrostatic interactions are effectively eliminated, revealed that 31% of the binding energy is derived from electrostatic interactions.
The use of fluorescence spectroscopy also allowed us to monitor the kinetics of the initial GTP binding to the Ceg1 protein (Figure 2C). The progress of the binding reaction was followed for 60 s on addition of saturating amounts of GTP. The results show that there is a rapid exponential decrease in fluorescence intensity after the addition of GTP. An apparent association rate of 2.7 μM−1·s−1 was estimated from the data. Half-maximal quenching was observed at approx. 1.5 s, whereas maximal quenching was achieved after 5 s of incubation with GTP and remained constant thereafter. The exponential decrease in fluorescence observed after the addition of GTP was not due to photobleaching, since similar results were obtained when the Ceg1 protein was incubated away from the light source. Finally, the pH dependence of GTP binding was analysed (Figure 2D). Binding of GTP covered a wide pH range and the optimum pH for the binding was found to be 7.03.
Binding of RNA to the Ceg1 protein
Fluorescence spectroscopy was also used to monitor the RNA binding activity of Ceg1 and revealed that the binding of RNA to the Ceg1 protein results in a significant decrease in emission fluorescence intensities (Table 1). The binding of Ceg1 to a 5′-diphosphate-terminated RNA transcript of 84 nucleotides was evaluated in the absence of metal ions. An apparent Kd of 30 μM could be estimated for the RNA substrate (Figure 3A). Similar values were also drawn from electrophoretic mobility-shift assays (results not shown).
Figure 3. Characterization of the interaction between RNA and Ceg1.

(A) Increasing amounts of RNA were added to a 100 nM solution of the enzyme in binding buffer (50 mM Tris/HCl, pH 7.5, and 50 mM KOAc) and the emission spectrum was scanned from 310 to 440 nm. (B) A van't Hoff plot for the interaction between RNA and Ceg1 is shown. The effect of temperature on the association constant was evaluated at pH 7.0. (C) The effect of increase in ionic strength on the apparent Kd of Ceg1 for RNA was investigated. Increasing concentrations of KCl were added to the reactions to generate the desired ionic strengths.
To determine the origins of the specificity of interaction between the Ceg1 protein and RNA, we sought to investigate the thermodynamics of the binding reaction. Again, we examined the energetics of RNA binding to the enzyme using fluorescence spectroscopy. Measurement of the RNA binding activity as a function of temperature allowed us to dissect the standard Gibbs free energy for the binding process into its enthalpic and entropic components (Figure 3B). These standard thermodynamic functions provide information about the change in the intrinsic properties of the protein on Ceg1–RNA complex formation. The RNA binding reaction was characterized by a ΔG=−25.8 kJ/mol. In contrast with the GTP binding activity, the association process between the enzyme and RNA appeared to be entropically favoured (TΔS=20.5 kJ/mol), with a minor contribution coming from a favourable enthalpy change (ΔH=−5.3 kJ/mol).
The contribution of specific electrostatic interactions to the binding of RNA to the Ceg1 protein was investigated by monitoring the salt dependence of the binding process. In solution, nucleic acids are associated with a layer of positive counterions, which neutralizes the high negative charge of the phosphodiester backbone [22]. These counterions can be displaced by electrostatic interactions with positively charged groups of the protein, leading to a dependence of the binding reaction on salt concentration. As shown in Figure 3(C), the RNA–enzyme interaction was attenuated at increased KCl concentrations. The equilibrium binding experiments showed that the apparent Kd at 500 mM KCl was 140 μM, almost five times higher than that at 10 mM KCl. This change in binding affinity corresponds to a ΔΔG=3.1 kJ/mol. These findings indicate that hydrogen-bonding or electrostatic interactions with the phosphodiester backbone are not a major force that can contribute to formation of the RNA–Ceg1 complex. In fact, evaluation of the ΔΔGES reveals that 12% of the binding energy is derived from electrostatic interactions.
Binding of metal ions to the Ceg1 protein
The Ceg1 protein requires manganese ions to mediate its RNA guanylyltransferase reaction [4]. We intended to use fluorescence spectroscopy to monitor the binding of manganese to Ceg1. Increasing amounts of manganese ions were added to the purified Ceg1 protein, and the fluorescence intensity was monitored. However, the interaction of the free protein with metal ions could not be efficiently and repeatedly detected using fluorescence spectroscopy. No modification of the fluorescence intensity could be observed, even at high metal ion concentrations (up to 100 mM), suggesting that the Ceg1 protein does not bind to manganese ions (results not shown). However, as suggested by previous crystallographic studies [13,14], binding of metal ions was observed when the enzyme was preincubated with the GTP substrate. Consequently, increasing concentrations of Mn2+ ions were added to the Ceg1 protein that had been preincubated with saturating amounts of GTP. Significant modifications of the fluorescence intensities could then be detected (Figure 4A). The addition of increasing concentrations of manganese ions resulted in an increase in the fluorescence intensity. Furthermore, the λmax value was blue-shifted by 5 nm, indicating movement of the tryptophan residues into a relatively more hydrophobic region. The fluorescence spectroscopy assays allowed us to determine the Kd value for the Mn2+ ions by titrating the binding of increasing amounts of metal ion to a fixed concentration of the Ceg1 protein. Typical emission spectra obtained from the titration of MnCl2 are shown in Figure 4(A). The corresponding saturation isotherm generated by plotting the change in fluorescence intensity as a function of the amount of MnCl2 added is shown in Figure 4(B). Quenching saturated at millimolar Mn2+ concentrations, and a Kd value of 3.1 mM could be estimated for Mn2+ from a fit of eqn (3) to the generated saturation isotherm. The addition of EDTA to the reaction reversed the effects on fluorescence and showed that the change in fluorescence observed on the addition of the metal ion is not solely due to a change in the ionic strength of the solution (results not shown). Binding of manganese ions to the Ceg1 protein was characterized by a ΔG of −14.2 kJ/mol. It should be noted that we were not able to measure accurately and repeatedly the change in enthalpy and/or entropy that occurs on metal ion binding.
Figure 4. Titration of Ceg1 with manganese ions.

(A) Increasing amounts of MnCl2 were added to a 100 nM solution of the enzyme in a binding buffer (50 mM Tris/HCl and 50 mM KOAc, pH 7.5) and the emission spectrum was scanned from 310 to 440 nm. (B) A saturation isotherm can be generated from these data by plotting the change in fluorescence intensity at 339 nm as a function of the amount of MnCl2 added. A double-reciprocal plot is shown in the inset.
Thermodynamic stability
Thermodynamic stability of the Ceg1 protein bound to its ligands was then investigated by GdmCl denaturation assays performed at 30 °C. The change in the microenvironment of the tryptophan residues as a function of the GdmCl concentration was investigated by integration of the fluorescence intensity. With an increase in the GdmCl concentration, the λmax of the unliganded Ceg1 protein shifted to 350 nm (results not shown), reflecting the transfer of tryptophan residues to a more polar environment. The protein structure reacts very sensitively to the slightest changes in concentration over the lower concentration range of 0.5–2.0 M, where the strongest effects on emission changes are observed (Figure 5). No changes could be visualized at GdmCl concentrations higher than 3 M.
Figure 5. GdmCl-induced unfolding equilibrium of Ceg1.

Transition curves for GdmCl-induced unfolding of the free Ceg1 protein (○) or the protein bound to either GTP (□) or GMP (●) were determined. Formation of the enzyme–GTP and enzyme–GMP complexes was induced by incubating the Ceg1 protein (100 nM) with either 1 mM GTP or 1 mM GTP+50 mM MnCl2 respectively. Equilibrium unfolding transitions were monitored by integration of the fluorescence intensity.
The change in the integrated fluorescence intensity as a function of the GdmCl concentration for the Ceg1 protein incubated with various ligands is shown in Figure 5. All the samples display smooth transition curves, indicating a co-operative unfolding event. The results suggest that the binding of GTP or RNA to the Ceg1 protein does not significantly stabilize the protein structure (Table 2). However, formation of the covalent protein–GMP intermediate, through the addition of 50 mM MnCl2 and 1 mM GTP to the Ceg1 protein, resulted in a significant modification of the protein stability. Higher concentrations of GdmCl were required to unfold the protein with bound GMP. The transition extends from approx. 1.0 to 3.0 M. Analysis of the thermodynamic parameters reveals that formation of the protein–GMP intermediate is thermodynamically more stable than the unliganded Ceg1 protein or the Ceg1 protein with bound GTP or RNA (Table 2). Therefore these results indicate that formation of the Ceg1–GMP covalent intermediate stabilizes the protein structure, rendering it less susceptible to denaturation.
Table 2. Thermodynamic unfolding parameters measured by equilibrium GdmCl denaturation.
The parameters ΔGou, m and Cm were determined by GdmCl denaturation and from the integration of the fluorescence intensity. The differences in Cm and ΔGou values in comparison with the free Ceg1 protein are also shown (ΔCm and ΔΔGou respectively).
| Protein | Cm (M) | ΔCm (M) | m (kJ·mol−1·M−1) | ΔGou (kJ/mol) | ΔΔGou (kJ/mol) |
|---|---|---|---|---|---|
| Ceg1 | 1.01 | 0.00 | 1.62 | 3.21 | 0.00 |
| Ceg1·GTP | 1.03 | 0.02 | 1.36 | 2.96 | −0.25 |
| Ceg1·GMP | 2.02 | 1.02 | 3.23 | 7.98 | 4.77 |
| Ceg1·RNA | 1.08 | 0.08 | 1.24 | 3.26 | 0.05 |
CD analysis
CD studies were then performed to monitor the conformational changes that occur on ligand binding. Far-UV CD spectra can provide useful information on the secondary-structure features of a protein, whereas the CD spectra in the near-UV region reflect the environments of the aromatic amino acid side chains, thereby yielding information on the tertiary structure of a protein. To determine whether the binding of GTP results in a modification of the Ceg1 structure, far- and near-UV CD spectra were recorded both in the absence and presence of GTP. Analysis of the near-UV CD spectra of the Ceg1 protein in both the absence and presence of GTP was performed over the range 250–340 nm. As shown in Figure 6(A), a marked negative ellipticity when compared with that of the protein alone was observed over the 280–300 nm region when the protein is incubated with GTP. These significant changes in the near-UV CD spectra clearly indicate that a significant conformational change occurs in the tertiary structure of Ceg1 on GTP binding.
Figure 6. Structural consequences of substrate binding to the Ceg1 protein.

(A) Far-UV CD spectra were recorded for the free Ceg1 protein (curve 1) or the protein bound to either GTP (curve 2) or GMP (curve 3). Formation of the enzyme–GTP and enzyme–GMP complexes was induced by incubating the Ceg1 protein (100 nM) with either 1 mM GTP or 1 mM GTP+50 mM MnCl2 respectively. The spectra were recorded from 200 to 250 nm, and the average of six wavelength scans is presented. (B) Near-UV CD spectra were recorded for the free Ceg1 protein (curve 1) or the protein bound to either GTP (curve 2) or GMP (curve 3). The spectra were recorded from 250 to 340 nm, and the average of six wavelength scans is presented.
Figure 6(B) shows the far-UV CD spectrum of Ceg1 both in the absence and presence of GTP. Analysis of the far-UV spectrum of Ceg1 suggests that the secondary structure of the protein contains 15% α-helix and 36% β-sheet [23]. Examination of the far-UV CD spectra (Figure 6B) of the Ceg1–GTP complex revealed that the binding of GTP to the Ceg1 protein does not induce a significant modification of the secondary structure of the protein (13% α-helix and 39% β-sheet). Thus, despite the major conformational changes, the overall secondary-structure content of the protein does not appear to be strongly altered on GTP binding.
CD studies were also performed with the Ceg1 protein covalently bound to GMP (Figure 6). The Ceg1–GMP covalent intermediate was formed by the addition of 1 mM GTP and 50 mM MnCl2 to the enzyme. Analysis of both the near- and far-UV spectra reveals that the protein adopts a conformation that is similar to that of the free Ceg1 protein. No significant differences could be observed at the secondary- and tertiary-structure levels (Figure 6). Finally, CD analyses performed on the Ceg1–RNA complex revealed no significant differences for the unliganded enzyme, indicating that the binding of RNA is not accompanied by a significant modification of the structure of the protein.
DISCUSSION
In addition to the data obtained from crystallographic analyses, binding studies performed with proteins in solution are of equal importance for a full comprehension of enzymic reactions in a quantitative way. Furthermore, analysis of protein–ligand interactions is a necessary counterpart to high-resolution structural studies in the design of potential antifungal/antiviral agents. In the present study, the use of fluorescence spectroscopy allowed, for the first time, a precise quantification of the thermodynamic parameters associated with the binding of substrates to an mRNA-capping enzyme. Although the thermodynamic parameters of binding do not provide a complete picture of the binding activity, they can suggest which features would be important for the interaction and provide a framework to construct an accurate model of the complex. Fluorescence spectroscopy also allowed us to evaluate, for the first time, the initial binding of GTP to such an enzyme, thereby separating the GTP binding step from the concomitant metal-dependent hydrolysis of GTP that results in the formation of the covalent GMP–protein intermediate.
In evaluating the energetics of ligand binding, our study leads to a better understanding of the molecular basis of substrate recognition by Ceg1. Changes in both the magnitude and the sign of ΔH and ΔS, in conjunction with structural data generated by crystallography, can provide crucial information about the structural alterations that accompany ligand binding in terms of (i) changes in salvation state, (ii) interactions between ligand and protein, such as electrostatic and hydrophobic interactions and (iii) changes in conformation/dynamics induced by ligand binding. Our thermodynamic analyses indicate that the initial association of GTP with the Ceg1 protein is driven by a favourable enthalpy change, but is also clearly associated with an unfavourable entropy change. Favourable negative enthalpy changes are generally associated with contributions from hydrogen bonds, van der Waal's interactions or ionic interactions [24], whereas unfavourable negative entropy changes are associated with the exposure of hydrophobic surfaces to the surface of the protein and/or to a decrease in conformational flexibility [25]. Although our salt dependence assays clearly established the importance of electrostatic interactions for the binding of GTP to the Ceg1 protein, both our fluorescence spectroscopy and CD studies indicate that aromatic residues move to a more hydrophobic environment after binding, suggesting that a decrease in conformational flexibility is probably encountered on GTP binding. On the basis of sequence alignment, the structure of the S. cerevisiae RNA-capping enzyme will probably resemble that of the Chlorella virus enzyme [15]. Various crystal structures of the Chlorella virus RNA-capping enzyme have now been determined. Analysis of these structures revealed the importance of hydrogen bonds, van der Waal's interactions and hydrophobic interactions for the binding of GTP. For instance, the 6-oxo group of the guanine ring of GTP is involved both in hydrogen bonds with a lysine residue (Lys-188) and in van der Waal's interactions with the side chain of a tryptophan residue (Trp-190). The 2-amino group of GTP is also involved in a hydrogen bond with a proline residue (Pro-59). Furthermore, the ribose hydroxyls and triphosphate moieties of GTP make additional hydrogen bonds and van der Waal's interactions with numerous residues of the enzyme active site [13,14]. It should also be noted that the guanosine base is buried in a hydrophobic pocket in a parallel stacking interaction between a phenylalanine (Phe-146) and an isoleucine (Ile-216). Interestingly, analysis of the Ceg1 structure using computer algorithms reveals that both the Chlorella virus and S. cerevisiae RNA-capping enzymes adopt very similar conformations (Figure 7A). Remarkably, the Ceg1–GTP interaction involves many similar contacts more than those reported for the interaction among GTP and the Chlorella RNA-capping enzyme (Figure 7B). Key interactions between the ribose hydroxyls, triphosphate chain and the guanine ring of GTP all appeared to be conserved in the predicted structural model for the Ceg1–GTP interaction.
Figure 7. Three-dimensional model of the S. cerevisiae RNA-capping enzyme active site.

(A) Ribbon representations of the active sites of both the Chlorella virus (blue) and S. cerevisiae (white) RNA-capping enzymes. The predicted three-dimensional structure of the S. cerevisiae RNA-capping enzyme active site was generated with the Deep View program [28]. (B) Predicted interactions between GTP and key residues of the S. cerevisiae RNA-capping enzymes. Hydrogen bonds are depicted in white.
In comparison with the GTP binding analyses, our thermodynamic studies of the interaction between Ceg1 and RNA revealed a completely different mode of binding. Binding to RNA was dominated by a favourable entropic effect (TΔS=20.5 kJ/mol). Such entropic changes can result from conformational changes and/or from the release of water molecules to the bulk solvent on RNA binding [24]. However, the use of CD did not allow the detection of any significant structural change in the Ceg1 protein on RNA binding, suggesting that release of water molecules occurs on RNA binding. Hydrophobic interactions are also associated with a relatively small ΔH (compared with ΔG) and a positive ΔS. Since the parameters for the Ceg1–RNA interaction fit these characteristics, it is tempting to speculate that hydrophobic interactions are a major determinant in the thermostability of the complex. In accordance with this hypothesis, examination of the ionic strength dependence for the interaction between RNA and Ceg1 reveals that the affinity of the enzyme for RNA is only slightly affected by an increased ionic strength. Taken together with the thermodynamic parameters, these results suggest that hydrophobic and stacking interactions probably play a more significant role in the formation of the Ceg1–RNA complex when compared with ionic contacts, which are expected to be completely disrupted after an increase in the ionic strength of the solvent. In comparison with protein–DNA interactions, very few studies have dealt with the thermodynamics of protein–RNA interactions. Nonetheless, most of the previously characterized protein–RNA interactions appear to be characterized by a small enthalpy change and are generally driven by a favourable entropy change [26], as is the case with the Ceg1–RNA interactions. These characteristics are, most frequently, supposed to result from the displacement of bound cations from the negatively charged phosphodiester backbone [26].
Conformational changes in proteins are often critical for their function and/or regulation. The presence of various protein conformers in the crystal structures of both the Chlorella virus and Candida albicans RNA-capping enzymes suggested that conformational changes occur during the RNA-capping reaction [13–15]. In the present study, the use of fluorescence spectroscopy, which relies on the endogenous fluorescence of the enzyme, coupled with the use of CD, which gives information on the secondary and tertiary structures of proteins, provided additional insights into the conformational changes associated with ligand binding and reaction chemistry. Both spectroscopic approaches clearly demonstrated the existence of conformational changes in Ceg1 following the binding of ligands. The results indicate that the Ceg1 protein undergoes conformational changes on GTP binding, and again on the formation of the covalent GMP–protein intermediate. Furthermore, the CD results suggest that the Ceg1–GMP and Ceg1–RNA complexes adopt an overall conformation that is similar to the unliganded protein. The presence of two distinct conformations is reminiscent of the two conformational states that have been observed in crystals of the Chlorella virus RNA-capping enzyme crystals [13,14]. Analysis of the crystal structure of the PBCV-1 RNA guanylyltransferase provided evidence of important conformational changes that occur during both substrate binding and reaction chemistry, most notably the opening and closing of the gap between the N- and C-terminal domains [13,14]. In the open configuration, the C-terminal domain moves away from the N-terminal domain to widen the cleft, whereas in the closed configuration, the gap has been narrowed after movement of the C-terminal domain towards the N-terminal domain [13,14]. The closed conformation is required for the catalytic formation of the enzyme–GMP intermediate, since only in the closed conformation is the enzyme able to undergo catalysis and form the GMP–enzyme intermediate [13,14]. By analogy, it is tempting to speculate that the binding of GTP to the Ceg1 protein results in a modification of the protein architecture that could be similar to that observed in the Chlorella virus enzyme. Our CD data indicated that the formation of the GMP–enzyme intermediate is accompanied by a conformational change that results in the formation of a tridimensional structure similar to that of the free protein. However, this intermediate appears to be significantly more stable when compared with either the unliganded Ceg1 protein or the protein bound to GTP, as illustrated by the denaturation studies. It is interesting to note that such an open form with bound GMP has recently been identified from crystallographic analysis of the C. albicans RNA-capping enzyme [15]. An open RNA-capping configuration has been suggested to be required at three distinct phases of the guanylyltransferase reaction: (i) for binding of the GTP substrate, (ii) for the binding of the ppRNA acceptor molecule and (iii) for the release of the capped RNA product [15,27]. As shown by our CD analysis, the presence of similar structures for the unliganded Ceg1 protein, which binds to GTP, and for the Ceg1–GMP intermediate, which ultimately binds to the ppRNA (5′ diphosphate RNA) molecules, is in agreement with such a model.
The fluorescence results reported here provide a surprisingly simple picture of ligand binding to the S. cerevisiae RNA-capping enzyme. All the kinetic processes measured in the present study were adequately analysed using single exponential rate equations. In no case was there any evidence for another significant kinetic barrier during binding that would require the use of a second exponential equation to fit the data satisfactorily. Furthermore, our binding studies clearly demonstrated that the metal ion cofactor-binding site of the Ceg1 protein can be formed only after the binding of GTP. Our fluorescence studies demonstrated that the binding of the metal ion cofactor does not occur on the free protein. This confirms the model that has been suggested previously from the crystallographic analysis of the Chlorella virus RNA-capping enzyme, in which only the closed form of the enzyme is capable of binding metal ions [13,14].
The present study is the first detailed study on the thermodynamics involved in the interaction of an RNA-capping enzyme with its ligands. Surprisingly, significant differences were found between the binding forces that drive the interaction of the enzyme with its RNA and GTP substrates. Most of the free energy of binding for the Ceg1–GTP interaction is due to a favourable ΔH and is entropy-opposed, whereas formation of the Ceg1–RNA complex is an entropy-driven reaction. Although knowledge of the mechanisms underlying RNA-capping activity is still incomplete, characterization of the individual biochemical steps involved in catalysis should provide the basis for further studies in this direction. Analysis of enzymic reactions requires reliable measurements of the protein–ligand interactions as a necessary counterpart to high-resolution structural studies. Physical measurements in solution should enable us to correlate the conformation and kinetic parameters of ligand recognition, and add another dimension to investigate the individual biochemical steps involved in the RNA-capping reaction. Given the recent interest in the development of selective inhibitors that can be used as antimicrobial and/or antiviral agents, understanding the thermodynamics of ligand interaction for proteins involved in RNA-capping chemistry is of particular importance for the future design of selective drugs.
Acknowledgments
We thank Dr S. Labbé for helpful comments on this paper and Dr P. Lavigne for his generosity and expert assistance with CD spectroscopy measurements. This work was supported by grants from the CIHR (Canadian Institutes for Health Research) and Fonds de la Recherche en Santé du Québec. M.B. is supported by a New Investigator Scholarship from the CIHR.
References
- 1.Furuichi Y., Shatkin S. Viral and cellular mRNA-capping: past and prospects. Adv. Virol. Res. 2000;55:135–184. doi: 10.1016/S0065-3527(00)55003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shuman S. Structure, mechanism, and evolution of the mRNA-capping apparatus. Prog. Nucleic Acids Res. Mol. Biol. 2000;66:1–40. doi: 10.1016/s0079-6603(00)66025-7. [DOI] [PubMed] [Google Scholar]
- 3.Shibagaki Y., Itoh N., Yamada H., Nagata S., Mizumoto K. mRNA-capping enzyme. Isolation and characterization of the gene encoding mRNA guanylyltransferase subunit from Saccharomyces cerevisiae. J. Biol. Chem. 1992;267:9521–9528. [PubMed] [Google Scholar]
- 4.Schwer B., Shuman S. Mutational analysis of yeast mRNA-capping enzyme. Proc. Natl. Acad. Sci. U.S.A. 1994;91:4328–4332. doi: 10.1073/pnas.91.10.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mao X., Schwer B., Shuman S. Mutational analysis of the Saccharomyces cerevisiae ABD1 gene: cap methyltransferase activity is essential for cell growth. Mol. Cell. Biol. 1996;16:475–480. doi: 10.1128/mcb.16.2.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang S. P., Shuman S. Structure-function analysis of the mRNA cap methyltransferase of Saccharomyces cerevisiae. J. Biol. Chem. 1997;272:14683–14689. doi: 10.1074/jbc.272.23.14683. [DOI] [PubMed] [Google Scholar]
- 7.Tsukamoto T., Shibagaki Y., Imajoh-Ohmi S., Murakoshi T., Suzuki M., Nakamura A., Gotoh H., Mizumoto K. Isolation and characterization of the yeast mRNA-capping enzyme beta subunit gene encoding RNA 5′-triphosphatase, which is essential for cell viability. Biochem. Biophys. Res. Commun. 1997;239:116–122. doi: 10.1006/bbrc.1997.7439. [DOI] [PubMed] [Google Scholar]
- 8.Ho C. K., Pei Y., Shuman S. Yeast and viral RNA 5′ triphosphatases comprise a new nucleoside triphosphatase family. J. Biol. Chem. 1998;273:34151–34156. doi: 10.1074/jbc.273.51.34151. [DOI] [PubMed] [Google Scholar]
- 9.Cong P., Shuman S. Covalent catalysis in nucleotidyl transfer. A KTDG motif essential for enzyme-GMP complex formation by mRNA-capping enzyme is conserved at the active sites of RNA and DNA ligases. J. Biol. Chem. 1993;268:7256–7260. [PubMed] [Google Scholar]
- 10.Niles E. G., Christen L. Identification of the vaccinia virus mRNA guanyltransferase active site lysine. J. Biol. Chem. 1993;268:24986–24989. [PubMed] [Google Scholar]
- 11.Shuman S., Liu Y., Schwer B. Covalent catalysis in nucleotidyl transfer reactions: essential motifs in Saccharomyces cerevisiae RNA-capping enzyme are conserved in Schizosaccharomyces pombe and viral capping enzymes and among polynucleotide ligases. Proc. Natl. Acad. Sci. U.S.A. 1994;91:12046–12050. doi: 10.1073/pnas.91.25.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Subramanya H. S., Doherty A. J., Ashford S. R., Wigley D. B. Crystal structure of an ATP-dependent DNA ligase from bacteriophage T7. Cell (Cambridge, Mass.) 1996;85:607–615. doi: 10.1016/s0092-8674(00)81260-x. [DOI] [PubMed] [Google Scholar]
- 13.Hakansson K., Doherty A. J., Shuman S., Wigley D. B. X-ray crystallography reveals a large conformational change during guanyl transfer by mRNA-capping enzymes. Cell (Cambridge, Mass.) 1997;89:545–553. doi: 10.1016/s0092-8674(00)80236-6. [DOI] [PubMed] [Google Scholar]
- 14.Hakansson K., Wigley D. B. Structure of a complex between a cap analogue and mRNA guanylyl transferase demonstrates the structural chemistry of RNA-capping. Proc. Natl. Acad. Sci. U.S.A. 1998;95:1505–1510. doi: 10.1073/pnas.95.4.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fabrega C., Shen V., Shuman S., Lima C. D. Structure of an mRNA-capping enzyme bound to the phosphorylated carboxy-terminal domain of RNA polymerase II. Mol. Cell. 2003;11:1549–1561. doi: 10.1016/s1097-2765(03)00187-4. [DOI] [PubMed] [Google Scholar]
- 16.Painter G. R., Wright L. L., Hopkins S., Furman P. A. Initial binding of 2′-deoxynucleoside 5′-triphosphates to human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem. 1991;266:19362–19368. [PubMed] [Google Scholar]
- 17.Eftink M. R., Ghiron C. A. Exposure of tryptophanyl residues in proteins. Quantitative determination by fluorescence quenching studies. Biochemistry. 1976;15:672–680. doi: 10.1021/bi00648a035. [DOI] [PubMed] [Google Scholar]
- 18.Flowers S., Biswas E. E., Biswas S. B. Conformational dynamics of DnaB helicase upon DNA and nucleotide binding: analysis by intrinsic tryptophan fluorescence quenching. Biochemistry. 2003;42:1910–1921. doi: 10.1021/bi025992v. [DOI] [PubMed] [Google Scholar]
- 19.Pan J. Y., Sanford J. C., Wessling-Resnick M. Effect of guanine nucleotide binding on the intrinsic tryptophan fluorescence properties of Rab5. J. Biol. Chem. 1995;270:24204–24208. doi: 10.1074/jbc.270.41.24204. [DOI] [PubMed] [Google Scholar]
- 20.Zhou T., Rosen B. P. Tryptophan fluorescence reports nucleotide-induced conformational changes in a domain of the ArsA ATPase. J. Biol. Chem. 1997;272:19731–19737. doi: 10.1074/jbc.272.32.19731. [DOI] [PubMed] [Google Scholar]
- 21.Henn A., Shi S. P., Zarivach R., Ben-Zeev E., Sagi I. The RNA helicase DbpA exhibits a markedly different conformation in the ADP-bound state when compared with the ATP- or RNA-bound states. J. Biol. Chem. 2002;277:46559–46565. doi: 10.1074/jbc.M207438200. [DOI] [PubMed] [Google Scholar]
- 22.Record M. T., Lohman M. L., de Haseth P. L. Ion effects on ligand-nucleic acid interactions. J. Mol. Biol. 1976;107:145–158. doi: 10.1016/s0022-2836(76)80023-x. [DOI] [PubMed] [Google Scholar]
- 23.Andrade M. A., Chacon P., Merelo J. J., Moran F. Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 1993;6:383–390. doi: 10.1093/protein/6.4.383. [DOI] [PubMed] [Google Scholar]
- 24.Beaudette N. V., Langerman N. The thermodynamics of nucleotide binding to proteins. Crit. Rev. Biochem. 1980;9:145–169. doi: 10.3109/10409238009105433. [DOI] [PubMed] [Google Scholar]
- 25.Tame J. R. H., O'Brien R., Ladbury J. E. Isothermal titration calorimetry of biomolecules. In: Ladbury J. E., Chowdhry B. Z., editors. Biocalorimetry: Applications of Calorimetry in the Biological Sciences. London: John Wiley and Sons; 1998. pp. 27–38. [Google Scholar]
- 26.Hall K. B., Kranz J. K. Thermodynamics and mutations in RNA-protein interactions. Methods Enzymol. 1995;259:261–281. doi: 10.1016/0076-6879(95)59048-x. [DOI] [PubMed] [Google Scholar]
- 27.Sawaya R., Shuman S. Mutational analysis of the guanylyltransferase component of mammalian mRNA-capping enzyme. Biochemistry. 2003;42:8240–8249. doi: 10.1021/bi034396d. [DOI] [PubMed] [Google Scholar]
- 28.Schwede T., Kopp J., Guex N., Peitsch M. C. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]