

Abstract
Background:
Atopic dermatitis (AD) is a common disease, with particularly high prevalence found in Africa. It is increasingly recognized that patients with AD of different ethnic backgrounds have unique molecular signatures in the skin, potentially accounting for treatment response variations. Nevertheless, the skin profile of patients with AD from Africa is unknown, hindering development of new treatments targeted to this patient population.
Objective:
To characterize the skin profile of patients with AD from Africa.
Methods:
Gene expression studies, including RNA sequencing (using threshold of fold change of >2 and false discovery rate of <0.05) and real-time polymerase chain reaction, were performed on skin biopsies of Tanzanian patients with moderate-to-severe AD and controls.
Results:
Tanzanian AD skin presented robust up-regulations of multiple key mediators of both T helper 2 (TH2) (interleukin 13 [IL-13], IL-10, IL-4R, CCL13,CCL17,CCL18,CCL26) and TH22 (IL22, S100As) pathways. Markers related to TH17 and IL-23 (IL-17A, IL-23A, IL-12, PI3, DEFB4B) and TH1 (interferon gamma, CXCL9,CXCL10, CXCL11) were also significantly overexpressed in AD tissues (FDR<.05), albeit to a lesser extent. IL-36 isoforms revealed substantial up-regulations in African skin. The barrier fingerprint of Tanzanian AD revealed no suppression of hallmark epidermal barrier differentiation genes, such as filaggrin, loricrin, and periplakin, with robust attenuation of lipid metabolism genes (ie, AWAT1).
Conclusion:
The skin phenotype of Tanzanian patients with AD is consistent with that of African Americans, exhibiting dominant TH2 and TH22 skewing, minimal dysregulation of terminal differentiation, and even broader attenuation of lipid metabolism-related products. These data highlight the unique characteristic of AD in Black individuals and the need to develop unique treatments targeting patients with AD from these underrepresented populations.
Introduction
Atopic dermatitis (AD) is characterized by eczematous patches and persistent pruritus.1 Although these clinical features are shared across different ethnic backgrounds and respective genetic variations, AD has a heterogeneous molecular signature, with distinct features in different populations.2,3 Furthermore, although a predominant TH2 and TH22 skewing is consistent across AD subtypes, varying degrees of TH1 and TH17 polarization are also present depending on the subtype. For example, TH1-related cytokines and chemokines are substantially up-regulated in adult vs pediatric AD,4 whereas Asian AD exhibits a stronger TH17 skewing in comparison with European American AD.4,5 The African American phenotype presents a relatively attenuated TH1 and TH17 polarization with a robust enrichment of the TH2 and TH22 pathways compared with European American patients, as recently reported.6 Nevertheless, African Americans, mainly descendants of West African populations, have been found to have different genetic and clinical profiles compared with the genetic and clinical profiles of East Africans.7,8 Moreover, African Americans also represent an admixture with other American populations.7,8 Despite the high prevalence of AD and atopy-related genetic variants, such as the interleukin (IL)-4R polymorphism in the African population,9–11 to the best of our knowledge, AD skin has not been comprehensively investigated in East Africans.12 This unmet need is made more important by the fact that African individuals are largely underinvestigated and underrepresented in clinical trials. As AD treatment is developing toward precision medicine approached in the globe, inclusion of African patients in molecular studies is imperative to promote targeted treatments for this population.
We thus performed a comprehensive molecular characterization of the immune and barrier abnormalities in skin of Tanzanian patients with AD vs ethnicity-matched healthy controls. We found that AD in these patients has a significant (FDR<.05) and robust TH2 and TH22 skewing with lesser TH1 and TH17 polarization and a unique barrier fingerprint, with sparse dysregulations of “classic” AD terminal differentiation genes (eg, filaggrin [FLG] and loricrin [LOR]), yet a marked lipid metabolism/synthesis disruption.
Methods
Clinical Characteristics
A total of 10 untreated patients with moderate-to-severe AD and 10 healthy controls were recruited, after signing an institutional review board–approved informed consent, at the outpatient clinic of the Regional Dermatology Training Center in Moshi, Tanzania. All participants were of Tanzanian origin (Fitzpatrick skin type 6). Clinical information on patient’s personal medical history (including allergic comorbidities), family history, and medication was documented using a questionnaire. Severity of AD was classified as mild, moderate, and severe using scoring of AD (SCORAD), investigator global assessment, and body surface area (BSA). Lesional and nonlesional skin samples of patients with AD and samples from controls were collected using 5-mm punch biopsies. Control biopsy samples were collected from volunteers’ back for consistency, whereas in patients with AD, skin biopsy samples were collected in the same body region in lesional and nonlesional skin. The biopsy samples were then immediately put into RNAlater (Qiagen, Hilden, Germany), incubated at 4°C overnight, and then stored at −80°C until further use.
RNA Sequencing
RNA was obtained from skin biopsy samples using Qiagen miRNeasy Mini kit, as previously described.13 Libraries were generated using TruSeq Stranded mRNA Library Prep kit (Illumina, San Diego, California). A cDNA library was then prepared by reverse transcription from extracted mRNA. Next-generation sequencing was performed with Illumina NovaSeq6000 (Illumina Inc) with single-ended read 100 cycles. Image analysis and base calling were performed in real time by the Illumina analysis pipeline.
Quantitative Real-Time Polymerase Chain Reaction
Reverse transcription to complementary DNA (cDNA) was performed using the High-Capacity cDNA reverse transcription (Thermo Fisher Scientific, Waltham, Massachusetts) after previous preamplification of all samples. TaqMan Low-Density Array (TLDA) cards (Thermo Fisher Scientific, Waltham, Massachusetts) were used for quantitative reverse-transcriptase–polymerase chain reaction (qRT-PCR). Primers are listed in eTable 1. A total of 100 ng total RNA was used for PreAMP pool and TLDA. Eukaryotic 18S recombinant RNA (rRNA) was used as an endogenous control. Expression values were normalized to Rplp0.
Statistical Analyses
Statistical analyses were performed using statistical language R (www.R-project.org). RNA sequencing data were preprocessed using standard pipeline incorporating quality control metrics, such as FastQC and MultiQC, sequence alignment based on STAR RNA-sequencing aligner, and sequencing read assignment to genomic features by featureCounts and voom-transformed. Mixed-effect model on R’s limma framework was used, and P values for the moderated t tests and paired t test were adjusted by Benjamini-Hochberg procedure. Differentially expressed genes were defined by fold-change (FCH) greater than 2.0 and false discovery rate (FDR) lesser than 0.05. qRT-PCR data were profiled using TLDA. Unsupervised clustering of the markers was performed using Euclidean distance and average agglomeration criteria. Threshold cycles [Ct] were normalized to Rplp0 by negatively transforming the Ct values to –dCt. The undetected expression values were estimated for each gene as 20% of the minimum unlogged expression across all samples. Log2-scale qRT-PCR expression data were modeled by a linear mixed-effect model with biopsy type as a fixed effect and a random intercept for each patient. Means of each group were estimated using lsmeans, and comparisons of interest were tested using contrast.
Results
Lesional and nonlesional skin samples from 10 adult Tanzanian patients with AD (3 female, 7 male, average age 43 ± 1 years old) with moderate-to-severe AD (mean SCORAD14 45 ± 14 and a mean body surface area [BSA] of 48% ± 23%) were compared with skin samples from 10 healthy Tanzanian controls with no personal or family history of atopy (5 female, 5 male, mean age 33 ± 1.3 years old). Of 10 patients with AD, 9 reported conomitant atopic diseases including allergic rhinoconjunctivitis and asthma. Patient characteristics are given in Table 1. Because healthy controls were younger than patients with AD, we provide, in eFigure 1, age-adjusted analysis that revealed comparable gene dysregulation as found in our unadjusted cohort (detailed below).
Table 1.
Patient Characteristics
| Characteristic | AD (n = 10) | Controls (n = 10) |
|---|---|---|
|
| ||
| Sex (n, male) | 7 | 5 |
| Age (mean years ± SD) | 43 ± 1.0 | 33 ± 1.2 |
| African race, N(%) | 10 (100) | 10 (100) |
| SCORAD (±SD) | 45 ± 14 | N/A |
| BSA(±SD) | 48 ± 23 | N/A |
| Total serum IgE levels (±SD) | 2607.9 (3522) | 504.3 (603.1) |
| Atopic conditions other than AD (n) | 10 | 0 |
| Family history of atopy (n) | Positive: 4 Negative: 6 |
Negative: 9 Unknown: 1 |
Abbreviations: AD, atopic dermatitis; BSA, body surface area; IgE, immunoglobulin E; SCORAD, scoring of AD.
Global Assessment of Immune Genes Reveals Robust TH2 and TH22/TH17 Up-Regulations
Global molecular profiling of the AD transcriptomes by RNA-seq identified 1946 differentially expressed genes (DEGs, 619 up-regulated, 1327 down-regulated) in lesional AD skin vs skin of healthy controls using threshold of FCH greater than 2 and false discovery rate (FDR) lesser than 0.05 (top DEGs in lesional AD vs normal skin are presented in eTables 2 and 3). Although nonlesional AD skin presented similar trends of gene modulations using P values (187 up-regulated, 497 down-regulated), there were only few gene dysregulations by FDR (6 up-regulated, 2 down-regulated).
Among genes significantly modulated in lesional vs normal skin were those related to cytokines and chemokines across various immune components, as depicted in a heatmap of a curated immune gene subset (FDR < .05; Fig 1).6,15 DEGs presenting the most robust up-regulations in lesional vs normal skin (FCH > 5, FDR < 0.05) included markers of cellular activation and recruitment of T-cells and dendritic cells (DCs) (CCR7, TNFRSF9, CD1B, CD80), general inflammation (MMP12), that presented exceedingly high up-regulation in nonlesional skin as well, and multiple genes related to TH2 (IL-13, IL-10, IL-4R, CCL18, CCL26, CCL13, CCL17, CCL7, CCL22, CCR4) and TH17 and TH22 (IL22, IL-26, CXCL1, S100A9, S100A8, S100A12, S100A7) pathways, all of which are implicated in AD pathogenesis across all subtypes.2,16 Unlike the robust and significant up-regulations found in TH2 and TH22 pathways, more modest up-regulations were found in some TH1- and TH17-related markers (FCH > 2, FDR < 0.05). Innate immune gene products, including IL-1β and IL-6, and the keratinocyte-derived cytokine IL-17C, were not up-regulated in lesional AD vs normal skin.
Figure 1.

Summary heatmap of selected immune genes by RNA-seq, representing multiple pathways in lesional and nonlesional AD vs normal Tanzanian skin by criteria of FCH greater than 2 and FDR lesser than 0.05. Red indicates up-regulated whereas blue indicates down-regulated mRNA expression values. Genes are sorted by hierarchical clustering. The triple asterisk indicates FDR < 0.001, the double asterisk indicates FDR < 0.01, the asterisk indicates FDR < 0.05, and +FDR < 0.1. AD, atopic dermatitis; NL, nonlesional; LS, lesional; FDR, false discovery rate; FCH, fold change.
Gene Set Variation Analysis of previously published AD-related genes by immune pathways, including the Meta-Analysis–Derived AD (MADAD) transcriptome16 and key immune pathways,17,18 revealed that the MADAD signature was significantly enriched in both lesional and nonlesional Tanzanian AD skin vs controls, as were the TH2 and TH22 pathways (P < .05; eFig 2).
Lipid Synthesis/Metabolism Genes Present the Most Significant Down-Regulations Among Epidermal Barrier-Related Genes
To evaluate the epidermal barrier, we analyzed a barrier gene subset6,19–21 including epidermal differentiation complex (EDC), tight junction and lipid genes, as illustrated as a heatmap in Figure 2. Particularly robust and significant down-regulations were found in genes related to lipid metabolism (eg, HAO2, ELOVL3, GAL, FAR2, AWAT1, DGAT2, FADS1 and 2, FABP7). Significant down-regulations were also noted in genes encoding for tight and adherent junctions, including cadherins (CDH12, CDH10, CDH19, CDH20) and claudins (CLDN8, CLDN10, CLDN1) in lesional skin vs controls and keratin genes (KRT79, KRT77, KRT33A) (FDR < 0.05). Nevertheless, key terminal differentiation genes that are characteristically down-regulated in European American AD (eg, FLG, LOR, sciellin, PSORS1C2, and late cornified envelope-LCE proteins)16,22 were not significantly down-regulated in lesional Tanzanian AD skin vs normal skin (FDR > 0.05). Furthermore, genes associated with FLG breakdown products were also found in comparable levels across AD and normal skin (ie, UCA1, UROC1, OPLAH, GGCT, ARG, CASP14, and TYR) (eTable 4). The intact keratinocyte terminal differentiation in AD lesional skin is supported by a Gene Set Variation Analysis for EDC-related genes, compared with lipid synthesis/metabolism genes that are significantly down-regulated (FDR < .05; eFig 2).
Figure 2.

Summary heatmap of selected epidermal barrier genes by RNA-seq in lesional and nonlesional AD vs normal Tanzanian skin by criteria of FCH greater than 2 and FDR lesser than 0.05. Red indicates up-regulated whereas blue indicates down-regulated mRNA expression values. Genes are sorted by hierarchical clustering. The triple asterisk indicates FDR < 0.001, the double asterisk indicates FDR < 0.01, the asterisk indicates FDR < 0.05, and +FDR < 0.1. AD, atopic dermatitis; NL, nonlesional; LS, lesional; FDR, false discovery rate; FCH, fold change.
To understand how these RNA-seq data in Tanzanian patients with AD reflect the previously published phenotype of African American patients with AD with moderate-to-severe disease,6 we performed a scatterplot comparison of relative differences vs normal skin (fold-changes) in either lesional or nonlesional AD, in the 2 patient populations (Fig 3). We found strong and significant correlations in lesional and nonlesional skin in the 2 phenotypes (r = 0.89 for lesional, r = 0.65 for nonlesional, P < .001). Nevertheless, we also found some differences, with a relative down-regulation of lipid synthesis and metabolism-related genes in Tanzanian skin compared with African American skin (ie, AWAT1, HAO2, FAR2). Further analysis using the same methods to compare Tanzanian and European American/White AD is presented in eFigure 3, which was consistent with overall agreement between the phenotypes (r = 0.85 for lesional, r = 0.49 for nonlesional, P < .001). Nevertheless, similar to the comparison of Tanzanian and African American AD, there was a relative down-regulation of lipid synthesis and metabolism-related genes in Tanzanian compared with European American lesional and or nonlesional skin (ie, FADS1, FADS2, FA2H). Moreover, terminal differentiation markers (eg, FLG, LOR, FLG2, LCE2B) presented greater down-regulation in European American AD (eTable 5).
Figure 3.
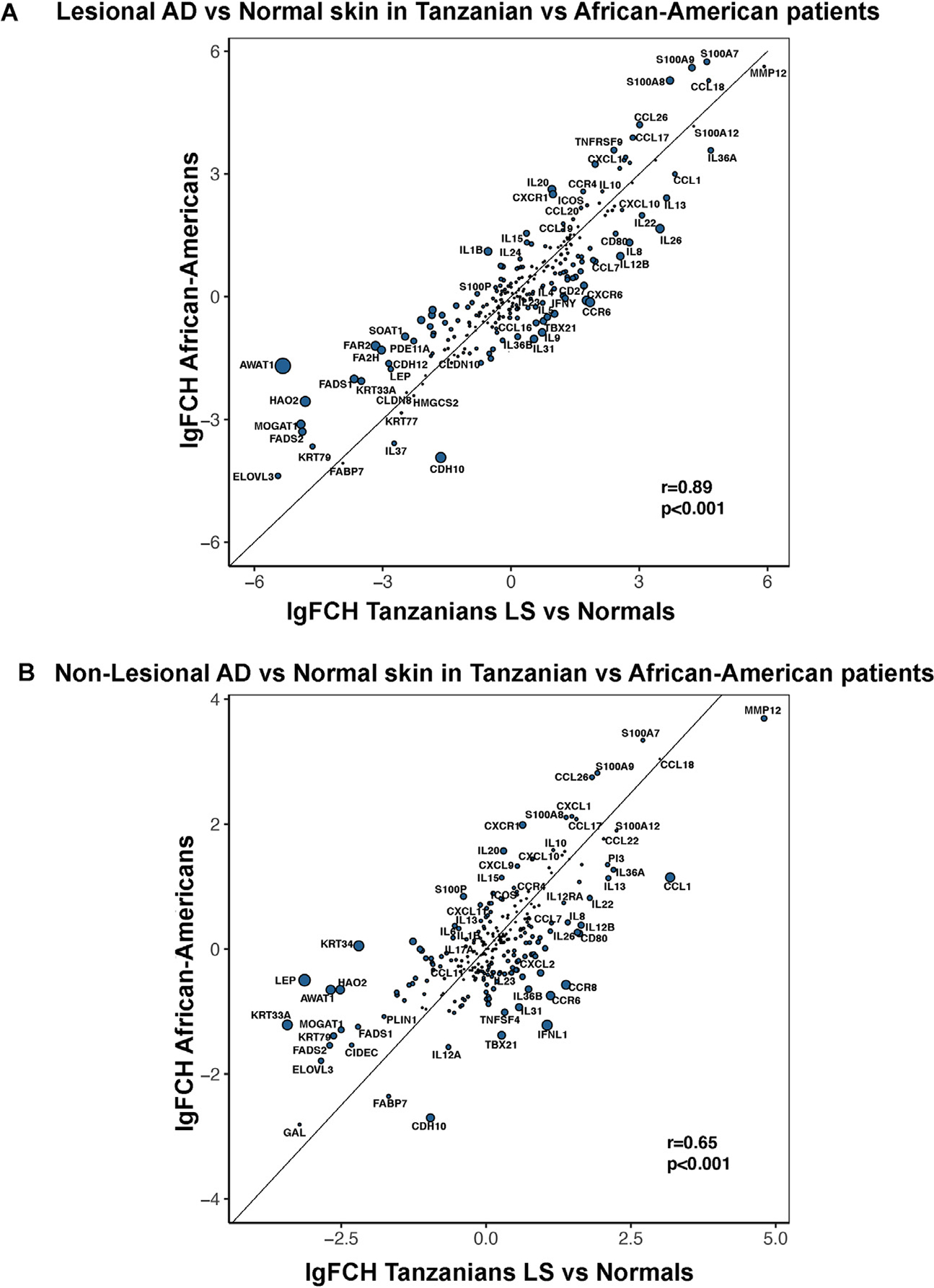
Scatterplots comparing relative FCH differences in Tanzanian vs African American AD samples (from a previously published cohort) in (A) lesional and (B) nonlesional skin. Circle size represents the magnitude of the differences in log2FCH between the populations. The location of the circles represents the respective expression in each population. AD, atopic dermatitis; FCH, fold change; LS, lesional.
RT-PCR Studies Further Refine TH2 and TH22 Up-Regulations and Barrier Impairment
To validate RNA-seq data and to evaluate key inflammatory and epidermal differentiation markers that are often below the detection threshold in RNA-seq, we also performed RT-PCR using a large panel of 87 AD-related genes (Fig 4, eFigs 4 and 5).
Figure 4.

qRT-PCR analysis of selected genes in AD vs normal Tanzanian skin. Values reveal log2 expression and are presented as means ± SEMs. Black symbols indicate significance of comparison to normal; red symbols indicate significance of comparison between lesional and nonlesional skin. The triple asterisk indicates P < .001, the double asterisk indicates P < .01, the asterisk indicates P < .05, and +P < .1. N, normal; NL, nonlesional; LS, lesional; qRT-PCR, quantitative reverse transcriptase–polymerase chain reaction; AD, atopic dermatitis.
Generally, PCR data were in line with our RNA-seq analysis. AD skin presented up-regulation of T-cell activation/migration markers, primarily in lesions compared with both nonlesional and control skin (ie, ICOS, CCR7, IL-2, IL-15, and CCL19), whereas MMP12 was up-regulated in both lesional and nonlesional tissues (P < .05) (Fig 4, eFig 4). Markers of innate immunity were not significantly up-regulated in Tanzanian AD tissues (IL-1β, IL-6, IL-17C;P<.05). NOS2 (inducible nitric oxidase synthase), a biomarker of disease activity in psoriasis, has been found to be up-regulated in psoriasis and down-regulated in AD in European Americans, helping to accurately classify these diseases.23–25 Indeed, in this study, we also found NOS2 to be significantly down-regulated in both lesional and nonlesional Tanzanian AD compared with controls. TH2 and TH22 dysregulations (compared with normal skin) were captured in lesional and/or nonlesional AD (ie, IL-13, IL-22, IL-5, CCL13, CCL18, CCL22, CCL26, S100A7, IL4, IL-4R, OX40; P < .05) (Fig 4, eFigs 4 and 5).
Similar to the AD skin phenotype in other ethnicities,5,6 several TH17 markers were increased in lesional skin (IL-17A, IL-21, IL-26, IL-23A, IL-12, PI3, CXCL1 and 2, DEFB4B) (P < .05 compared with nonlesional and controls). Key TH1-related genes were significantly up-regulated in Tanzanian AD lesions vs nonlesional and/or controls (ie, interferon gamma [IFN-γ], CXCL9,CXCL10,CXCL11; P < .05). The TH17-related IL-36 isoforms (IL-36α, IL-36γ, and IL-36RN) were up-regulated in both lesional and nonlesional skin in Tanzanian AD (P < .05). The negative regulators IL-34 and IL-37 displayed significant reductions only in lesional skin, whereas the regulatory markers IL-10, FOXP3, and CTLA4 were significantly modulated in both AD tissues (P < .05) (Fig 4, eFigs 4 and 5).
Overall, genes associated with lipid metabolism were significantly suppressed already in nonlesional AD (eg, ELOVL3/5, GAL, FA2H, FAR2, AWAT1/DGA2) (P < .05), with further decreases in lesional skin (P < .001). Significant up-regulation of epidermal hyperplasia markers (KRT16 and SERPINB3) and down-regulations of the terminal differentiation marker FLG2 and tight junction markers (CLDN1, CLDN8, CLDN23) were found in lesional AD vs nonlesional skin and controls, reflecting greater barrier impairment in lesional skin (P < .05) (Fig 4, eFig 5). Similar to RNA-seq data, FLG, LOR, and periplakin (PPL), hallmark markers of the epidermal differentiation complex,22,26 were not markedly down-regulated in AD tissues vs controls. A summary heatmap of expression profiles by PCR is in eFigure 6.
Disease Severity of Tanzanian Patients With AD Correlates With Polar Immune Pathways
Spearman correlation of immune and barrier markers measured by RT-PCR and disease severity (evaluated by SCORAD) revealed that clinical severity of Tanzanian AD strongly correlates with pivotal AD immune markers in both lesional and nonlesional skin and that correlations are not limited to a specific component of the immune response (r > 0.6, P < .05) (Table 2, eTable 6). In lesional skin, these included the T-cell activation/migration marker CCL19, the general inflammation marker MMP12, and the TH2 markers IL-5 and CCL13 (r > 0.6, P < .05). Moreover, key TH1-(CXCL9,CXCL10,CXCL11, IFN-γ) and TH17-related genes (IL-17A, IL21 and IL-8) were robustly and significantly correlated with SCORAD (r ≥ 0.7, P < .05). The TH9-cytokine IL-9 and the regulatory marker CD279/PD-1 also significantly correlated with SCORAD in lesions of AD (r > 0.6, P < .05). In nonlesional skin, robust correlations of SCORAD with TH2-(IL-5, CCL18) and TH1-(CXCL9,CXCL10,CXCL11) related markers were detected, in addition to CD279/PD-1 and the TH17-related marker IL-19 (r > 0.6, P < .05).
Table 2.
Spearman Correlations
| Lesional skin |
Nonlesional skin |
||||
|---|---|---|---|---|---|
| Marker | R | P value | Marker | r | P value |
|
| |||||
| MMP12 | 0.85 | <.001 | CXCL11 | 0.75 | .01 |
| CXCL10 | 0.84 | <.001 | CXCL9 | 0.74 | .01 |
| IL-17A | 0.81 | .01 | IL-5 | 0.70 | .02 |
| CXCL9 | 0.80 | .01 | CXCL10 | 0.70 | .02 |
| IFN-γ | 0.78 | .01 | PDCD1 | 0.69 | .03 |
| CXCL11 | 0.75 | .02 | CCL18 | 0.69 | .03 |
| CCL19 | 0.75 | .02 | IL-19 | 0.65 | .04 |
| IL-9 | 0.72 | .03 | CCL26 | 0.60 | .07 |
| IL-5 | 0.72 | .03 | CCL13 | 0.59 | .07 |
| IL-21 | 0.70 | .03 | CXCL2 | 0.59 | .07 |
| CCL13 | 0.69 | .04 | PI3 | 0.55 | .10 |
| CXCL8 | 0.68 | .04 | |||
| PDCD1 | 0.68 | .04 | |||
| CCL11 | 0.63 | .07 | |||
| CCL18 | 0.63 | .07 | |||
| CXCL2 | 0.62 | .08 | |||
| CCL7 | 0.61 | .08 | |||
| STAT1 | 0.59 | .10 | |||
| CLDN8 | −0.66 | .05 | |||
Abbreviations: IFN-g, interferon gamma; IL, interleukin.
Discussion
African and African American populations have perhaps the highest global rates of AD, affecting up to 18.9% of Tanzanians and 19.3% of African Americans, respectively.3,10,27,28 Populations of African ancestry also exhibit increased AD severity and higher rates of hospitalization29 and higher prevalence and/or morbidity of other atopic diseases, including asthma, allergic rhinitis, allergic rhinoconjunctivitis, and eosinophilic esophagitis.30–33 Furthermore, the clinical presentation of AD in patients of color differs from White patients, as they are more likely to have lichenified and postinflammatory manifestations.34,35 African populations and African Americans have overrepresentation of IL-4Rα polymorphisms, likely conferring a greater risk for type 2 diseases.9–11,36 In addition, different serologic immune signatures and sensitization patterns in sub-Saharan vs central European patients with AD.37 These unique characteristics of AD in patients of African ancestry may have an effect on therapeutic management of these patient populations. The need for development of treatments or dosing regimens that uniquely target African populations has been suggested by the reduced efficacy of the approved dose of the IL-4Rα antagonist, dupilumab, in African Americans compared with other races.38 Although African Americans share a genetic background with native Africans, they are also mixed with other ethnicities.7,8 As AD is a heterogenous disease with differential involvement of various immune axes in different disease phenotypes, a “one-size-fits-all” treatment may not be applicable to all patient populations, necessitating a precision medicine approach to improve clinical outcomes across the spectrum.2,39 Despite the high prevalence of AD in Africa and the reported differences in disease manifestations and genetics vs other races, advocating for investigation of the African AD subtype, the molecular phenotype in skin has never been characterized, inhibiting development of tailored treatment approaches for this population.12 We thus evaluated the skin phenotype of patients from Tanzania (East Africa) with moderate-to-severe AD using a global transcriptomic approach.
We found that the Tanzanian AD phenotype is highly concordant with the African American AD skin profile, with a dominant TH2 and TH22 skewing, which seems to be a common feature across all ethnic populations with AD.5,6,40,41 Significantly up-regulated markers in our cohort included pivotal type 2–related mediators (eg, IL-13, IL-10, IL-4R, CCL17, CCL18, CCL26 (FDR < .05) and key TH17- and TH22-related products (eg, IL-22, IL-26, CXCL1, S100As). Tanzanian AD was found to have some up-regulations of TH1- and TH17-related markers, but the key cytokines of these pathways (IFN-γ and IL-17A, respectively) were not captured by RNA sequencing. IL-36 isoforms were highly up-regulated in both lesional and nonlesional tissues, whereas IL-36 cytokines in European Americans were only considerably up-regulated in lesional AD skin.15 As IL-36 has been highly associated with psoriasis and psoriasiform dermatitis,42,43 this finding could potentially contribute to the more lichenified lesions characterizing AD in African individuals.34,35 In addition, the IL-21 cytokine, that is highly up-regulated in lesional Tanzanian AD skin, has been reported to induce both IL-17, IL-4 and IL-13, possibly linking the activation of IL-21 synergizes with IL-4 to increase IgE production to greater levels than each cytokine alone.44
Similar to other AD ethnic/race background, this AD cohort also presented significant up-regulations of markers associated with T-cell and DC activation and migration (eg, CCR7, ICOS, CD1B, CD80 in lesional skin; FDR < .05) and general inflammation (MMP12, in both lesional and nonlesional skin). We also detected down-regulation of NOS2, a marker that distinguishes AD from psoriasis across all phenotypes.23–25 Tanzanian AD skin largely lacked significant up-regulations of markers related to innate immunity, including IL-1b, IL-6, and IL-17C, a trend that was also observed in African American AD but not in European American patients.6
A hallmark of AD across all phenotypes is alterations in epidermal barrier, with up-regulation of TH2-/TH22-related markers (eg, IL-4, IL-13, IL-22) playing an important role in the suppression of epidermal barrier genes.45–47 Nevertheless, previous reports outlined some differences in barrier abnormalities among various ethnicities/races. For example, African Americans lack marker dysregulations of FLG, while having significant down-regulations in LOR and FLG2 (FDR < .05),6 and European American patients present extensive down-regulations of multiple terminal differentiation genes, including FLG, LOR, PPL, and FLG2, multiple LCEs, and PSORS1C2.15,45 Differential expression of FLG among different races can be explained in part by the variable rates of FLG null mutation and copy number variations (CNV), detected in up to 50%, 27%, and 12.2% of European, Asian, and African American patients with AD, respectively.48–53 Studies in African populations suggest present yet weaker association between FLG null mutations/CNV and the development of AD than those reported in European populations,54,55 and, as Zhu et al56 have recently found, FLG variation revealed in African American patients with AD differ from that of European patients. In our cohort, genes of terminal differentiation including FLG but also LOR, PPL, SCEL, multiple LCEs, and PSORS1C2 were not significantly down-regulated in lesional and nonlesional tissues. Although similar trends were detected in African American AD,6 some of these key barrier genes did attain significance in this population vs controls (including LOR and LCEs), highlighting the differences between African American and African AD. The lack of FLG null mutations suggests that other pathogenic molecules may play key roles in African patients with AD.3,57 These could include FLG2 (closely associated with FLG gene in both location and physiological activity, diminishing during acute spongiotic dermatitis)58 and CLDN1 (a tight junction gene) mutations, both associated with AD in subjects of African ancestry.59,60 Indeed, we found that these barrier markers were both significantly down-regulated in lesional AD in our cohort.
In addition, as the disrupted barrier of AD is also characterized by down-regulations of lipid synthesis/metabolism genes (eg, ELOVL5, FA2H, AWAT1),16,61–63 we also evaluated the expression of these lipid-related markers. Despite the observed differences in the expression of terminal differentiation genes, genes of lipid synthesis and metabolism were robustly down-regulated in Tanzanian AD, consistent with reports in other AD phenotypes and ethnicities.6,16 Moreover, on comparing the relative dysregulations of African American and Tanzanian AD skin, we found that lipid-related genes, including AWAT1, HAO2, and FAR2, were more suppressed vs controls in the latter. These results are also supported by previous reports on normal skin across different races, in which African American skin had the highest protein levels with significantly reduced lipid levels compared with that of both White and Asian subjects.64,65
This study has a few limitations. First, although our cohort was homogeneous and included only Tanzanian patients with AD and controls, our sample size was small. Second, although we captured up-regulation of TH2- and TH22-related cytokines but normal FLG expression, we did not have tissue left to conduct any protein analysis studies to evaluate FLG protein and breakdown products. We are thus not able to exclude that the transcriptomic data identified profilaggrin rather than end products of FLG. Third, DNA analyses were not performed, and we were therefore unable to evaluate gene polymorphism and mutation status. Finally, a direct comparison to patients with AD of other ethnicities was not feasible, as studies were not run simultaneously. Nevertheless, a comparison of the relative FCH of a previously published African American AD cohort6 with our Tanzanian AD cohort revealed a largely concordant immune and barrier dysregulation of patients of African heritage.
In conclusion, this study portrays the molecular characteristics of AD in African patients, an underinvestigated and underrepresented population, despite its high prevalence of AD and atopy. It suggests an overall high correlation between African American and Tanzanian AD phenotypes, although some expression differences were detected, mainly in lipid-related genes. Though a “magic bullet” treatment for AD across the various phenotypes is not available, our results may guide future drug developments for patients of African ancestry with AD and help reduce the health disparities associated with this population.
Supplementary Material
Funding:
Dr Lang reports being supported by a research grant from the Bruno Bloch Foundation, Zurich, to realize a fellowship abroad. Dr Renert-Yuval reports being supported, in part, by the National Center for Advancing Translational Sciences, National Institutes of Health, through The Rockefeller University, grant #UL1TR001866.
Disclosures:
Dr Guttman-Yassky is an employee of Mount Sinai; reports receiving research funds (grants paid to the institution) from AbbVie Inc, Celgene, Eli Lilly and Company, Janssen, MedImmune/AstraZeneca, Novartis, Pfizer, Regeneron, Vitae, Glenmark, Galderma, Asana, Innovaderm, Dermira, and UCB; and is a consultant for Sanofi Aventis, Regeneron, Stiefel/GlaxoSmithKline, MedImmune, Celgene, Anacor, AnaptysBio, Dermira, Galderma, Glenmark, Novartis, Pfizer, Vitae, Leo Pharma, AbbVie Inc, Eli Lilly and Company, Kyowa, Mitsubishi Tanabe, Asana Biosciences, and Promius. Dr Krueger reports receiving research support (grants paid to his institution) and/or personal fees from Pfizer, Amgen, Janssen, Lilly, Merck, Novartis, Kadmon, Dermira, Boehringer, Innovaderm, Kyowa, Bristol Myers Squibb, Serono, BiogenIdec, Delenex, AbbVie Inc, Sanofi, Baxter, Paraxel, Xenoport, and Kineta. Dr Schmid-Grendelmeier reports receiving research grants and/or personal fees from AbbVie Inc, Eli Lilly and Company, Leo Pharma, Novartis, Pfizer, and Sanofi Genzyme.
Footnotes
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.anai.2021.04.023.
References
- 1.Weidinger S, Novak N. Atopic dermatitis. Lancet. 2016;387(10023):1109–1122. [DOI] [PubMed] [Google Scholar]
- 2.Czarnowicki T, He H, Krueger JG, Guttman-Yassky E. Atopic dermatitis endotypes and implications for targeted therapeutics. J Allergy Clin Immunol. 2019;143(1):1–11. [DOI] [PubMed] [Google Scholar]
- 3.Brunner PM, Guttman-Yassky E. Racial differences in atopic dermatitis. Ann Allergy Asthma Immunol. 2019;122(5):449–455. [DOI] [PubMed] [Google Scholar]
- 4.Esaki H, Brunner PM, Renert-Yuval Y, et al. Early-onset pediatric atopic dermatitis is TH2 but also TH17 polarized in skin. J Allergy Clin Immunol. 2016;138(6): 1639–1651. [DOI] [PubMed] [Google Scholar]
- 5.Noda S, Suárez-Fariñas M, Ungar B, et al. The Asian atopic dermatitis phenotype combines features of atopic dermatitis and psoriasis with increased TH17 polarization. J Allergy Clin Immunol. 2015;136(5):1254–1264. [DOI] [PubMed] [Google Scholar]
- 6.Sanyal RD, Pavel AB, Glickman J, et al. Atopic dermatitis in African American patients is TH2/TH22-skewed with TH1/TH17 attenuation. Ann Allergy Asthma Immunol. 2019;122(1):99–110. e6. [DOI] [PubMed] [Google Scholar]
- 7.Bryc K, Auton A, Nelson MR, et al. Genome-wide patterns of population structure and admixture in West Africans and African Americans. Proc Natl Acad Sci U S A. 2010;107(2):786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tishkoff SA, Reed FA, Friedlaender FR, et al. The genetic structure and history of Africans and African Americans. Science. 2009;324(5930):1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caggana M, Walker K, Reilly AA, Conroy JM, Duva S, Walsh AC. Population-based studies reveal differences in the allelic frequencies of two functionally significant human interleukin-4 receptor polymorphisms in several ethnic groups. Genet Med. 1999;1(6):267–271. [DOI] [PubMed] [Google Scholar]
- 10.Deckers IA, McLean S, Linssen S, Mommers M, van Schayck CP, Sheikh A. Investigating international time trends in the incidence and prevalence of atopic eczema 1990–2010: a systematic review of epidemiological studies. PLoS One. 2012;7(7):e39803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ait-Khaled N, Odhiambo J, Pearce N, et al. Prevalence of symptoms of asthma, rhinitis and eczema in 13- to 14-year-old children in Africa: the International Study of Asthma and Allergies in Childhood Phase III. Allergy. 2007;62(3):247–258. [DOI] [PubMed] [Google Scholar]
- 12.Schmid-Grendelmeier P, Takaoka R, Ahogo KC, et al. Position Statement on Atopic Dermatitis in Sub-Saharan Africa: current status and roadmap. J Eur Acad Dermatol Venereol. 2019;33(11):2019–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suarez-Farinas M, Ungar B, Noda S, et al. Alopecia areata profiling shows TH1, TH2, and IL-23 cytokine activation without parallel TH17/TH22 skewing. J Allergy Clin Immunol. 2015;136(5):1277–1287. [DOI] [PubMed] [Google Scholar]
- 14.Hurault G, Schram ME, Roekevisch E, Spuls PI, Tanaka RJ. Relationship and probabilistic stratification of EASI and oSCORAD severity scores for atopic dermatitis. Br J Dermatol. 2018;179(4):1003–1005. [DOI] [PubMed] [Google Scholar]
- 15.Suarez-Farinas M, Ungar B, Correa da Rosa J, et al. RNA sequencing atopic dermatitis transcriptome profiling provides insights into novel disease mechanisms with potential therapeutic implications. J Allergy Clin Immunol. 2015;135(5):1218–1227. [DOI] [PubMed] [Google Scholar]
- 16.Ewald DA, Malajian D, Krueger JG, et al. Meta-analysis derived atopic dermatitis (MADAD) transcriptome defines a robust AD signature highlighting the involvement of atherosclerosis and lipid metabolism pathways. BMC Med Genomics. 2015;8:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dhingra N, Shemer A, Correa da Rosa J, et al. Molecular profiling of contact dermatitis skin identifies allergen-dependent differences in immune response. J Allergy Clin Immunol. 2014;134(2):362–372. [DOI] [PubMed] [Google Scholar]
- 18.Guttman-Yassky E, Ungar B, Noda S, et al. Extensive alopecia areata is reversed by IL-12/IL-23p40 cytokine antagonism. J Allergy Clin Immunol. 2016;137(1):301–304. [DOI] [PubMed] [Google Scholar]
- 19.Guttman-Yassky E, Bissonnette R, Ungar B, et al. Dupilumab progressively improves systemic and cutaneous abnormalities in atopic dermatitis patients. J Allergy Clin Immunol. 2019;143(1):155–172. [DOI] [PubMed] [Google Scholar]
- 20.Guttman-Yassky E, Diaz A, Pavel AB, et al. Use of tape strips to detect immune and barrier abnormalities in the skin of children with early-onset atopic dermatitis. JAMA Dermatol. 2019;155(12):1358–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suárez-Fariñas M, Tintle SJ, Shemer A, et al. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J Allergy Clin Immunol. 2011;127(4). 954–964:e1-e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim BE, Leung DY, Boguniewicz M, Howell MD. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin Immunol. 2008;126(3):332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garzorz N, Eyerich K. NOS2 and CCL27: clinical implications for psoriasis and eczema diagnosis and management. Expert Rev Clin Immunol. 2015;11(2):167–169. [DOI] [PubMed] [Google Scholar]
- 24.Quaranta M, Knapp B, Garzorz N, et al. Intraindividual genome expression analysis reveals a specific molecular signature of psoriasis and eczema. Sci Transl Med. 2014;6(244):244ra290. [DOI] [PubMed] [Google Scholar]
- 25.He H, Bissonnette R, Wu J, et al. Tape strips detect distinct immune and barrier profiles in atopic dermatitis and psoriasis. J Allergy Clin Immunol. 2021;147(1):199–212. [DOI] [PubMed] [Google Scholar]
- 26.Howell MD, Kim BE, Gao P, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol. 2009;124(3 suppl 2):R7–R12. [DOI] [PubMed] [Google Scholar]
- 27.Odhiambo JA, Williams HC, Clayton TO, Robertson CF, Asher MI, Group IPTS. Global variations in prevalence of eczema symptoms in children from ISAAC Phase Three. J Allergy Clin Immunol. 2009;124(6):1251–1258. e23. [DOI] [PubMed] [Google Scholar]
- 28.Wander K, Shell-Duncan B, Brindle E, O’Connor K. Hay fever, asthma, and eczema and early infectious diseases among children in Kilimanjaro, Tanzania. Am J Hum Biol. 2017;29(3). [DOI] [PubMed] [Google Scholar]
- 29.Daya M, Barnes KC. African American ancestry contribution to asthma and atopic dermatitis. Ann Allergy Asthma Immunol. 2019;122(5):456–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Almoguera B, Vazquez L, Mentch F, et al. Identification of four novel loci in asthma in European American and African American populations. Am J Respir Crit Care Med. 2017;195(4):456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffmans R, Wagemakers A, van Drunen C, Hellings P, Fokkens W. Acute and chronic rhinosinusitis and allergic rhinitis in relation to comorbidity, ethnicity and environment. PLoS One. 2018;13:(2) e0192330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katelaris CH, Lee BW, Potter PC, et al. Prevalence and diversity of allergic rhinitis in regions of the world beyond Europe and North America. Clin Exp Allergy. 2012;42(2):186–207. [DOI] [PubMed] [Google Scholar]
- 33.Weiler T, Mikhail I, Singal A, Sharma H. Racial differences in the clinical presentation of pediatric eosinophilic esophagitis. J Allergy Clin Immunol Pract. 2014;2(3):320–325. [DOI] [PubMed] [Google Scholar]
- 34.Vachiramon V, Tey HL, Thompson AE, Yosipovitch G. Atopic dermatitis in African American children: addressing unmet needs of a common disease. Pediatr Dermatol. 2012;29(4):395–402. [DOI] [PubMed] [Google Scholar]
- 35.Mei-Yen Yong A, Tay YK. Atopic dermatitis: racial and ethnic differences. Dermatol Clin. 2017;35(3):395–402. [DOI] [PubMed] [Google Scholar]
- 36.Tachdjian R, Mathias C, Al Khatib S, et al. Pathogenicity of a disease-associated human IL-4 receptor allele in experimental asthma. J Exp Med. 2009;206(10):2191–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lang C, Masenga J, Semango G, et al. Evidence for different immune signatures and sensitization patterns in sub-Saharan versus central European atopic dermatitis patients. J Eur Acad Dermatol Venereol. 2021;35(2):e140–e142. [DOI] [PubMed] [Google Scholar]
- 38.Alexis AF, Rendon M, Silverberg JI, et al. Efficacy of dupilumab in different racial subgroups of adults with moderate-to-severe atopic dermatitis in three randomized, placebo-controlled phase 3 trials. J Drugs Dermatol. 2019;18(8):804–813. [PubMed] [Google Scholar]
- 39.Renert-Yuval Y, Guttman-Yassky E. What’s new in atopic dermatitis. Dermatol Clin. 2019;37(2):205–213. [DOI] [PubMed] [Google Scholar]
- 40.Chan TC, Sanyal RD, Pavel AB, et al. Atopic dermatitis in Chinese patients shows TH2/TH17 skewing with psoriasiform features. J Allergy Clin Immunol. 2018;142(3):1013–1017. [DOI] [PubMed] [Google Scholar]
- 41.Wen HC, Czarnowicki T, Noda S, et al. Serum from Asian patients with atopic dermatitis is characterized by TH2/TH22 activation, which is highly correlated with nonlesional skin measures. J Allergy Clin Immunol. 2018;142(1):324–328. e11. [DOI] [PubMed] [Google Scholar]
- 42.D’Erme AM, Wilsmann-Theis D, Wagenpfeil J, et al. IL-36gamma (IL-1F9) is a biomarker for psoriasis skin lesions. J Invest Dermatol. 2015;135(4):1025–1032. [DOI] [PubMed] [Google Scholar]
- 43.Tortola L, Rosenwald E, Abel B, et al. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J Clin Invest. 2012;122(11):3965–3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Avery DT, Ma CS, Bryant VL, et al. STAT3 is required for IL-21-induced secretion of IgE from human naive B cells. Blood. 2008;112(5):1784–1793. [DOI] [PubMed] [Google Scholar]
- 45.Kim BE, Leung DYM. Significance of skin barrier dysfunction in atopic dermatitis. Allergy Asthma Immunol Res. 2018;10(3):207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Howell MD, Kim BE, Gao P, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol. 2007;120(1):150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Furue M Regulation of filaggrin, loricrin, and involucrin by IL-4, IL-13, IL-17A, IL-22, AHR, and NRF2: pathogenic implications in atopic dermatitis. Int J Mol Sci. 2020;21(15):5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Polcari I, Becker L, Stein SL, Smith MS, Paller AS. Filaggrin gene mutations in African Americans with both ichthyosis vulgaris and atopic dermatitis. Pediatr Dermatol. 2014;31(4):489–492. [DOI] [PubMed] [Google Scholar]
- 49.Margolis DJ, Apter AJ, Gupta J, et al. The persistence of atopic dermatitis and filaggrin (FLG) mutations in a US longitudinal cohort. J Allergy Clin Immunol. 2012;130(4):912–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown SJ, McLean WH. One remarkable molecule: filaggrin. J Invest Dermatol. 2012;132(3 Pt 2):751–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown SJ, McLean WH. Eczema genetics: current state of knowledge and future goals. J Invest Dermatol. 2009;129(3):543–552. [DOI] [PubMed] [Google Scholar]
- 52.Park J, Jekarl DW, Kim Y, Kim J, Kim M, Park YM. Novel FLG null mutations in Korean patients with atopic dermatitis and comparison of the mutational spectra in Asian populations. J Dermatol. 2015;42(9):867–873. [DOI] [PubMed] [Google Scholar]
- 53.Margolis DJ, Mitra N, Gochnauer H, et al. Uncommon filaggrin variants are associated with persistent atopic dermatitis in African Americans. J Invest Dermatol. 2018;138(7):1501–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thawer-Esmail F, Jakasa I, Todd G, et al. South African amaXhosa patients with atopic dermatitis have decreased levels of filaggrin breakdown products but no loss-of-function mutations in filaggrin. J Allergy Clin Immunol. 2014;133(280–282):e1–e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Winge MC, Bilcha KD, Lieden A, et al. Novel filaggrin mutation but no other loss-of-function variants found in Ethiopian patients with atopic dermatitis. Br J Dermatol. 2011;165(5):1074–1080. [DOI] [PubMed] [Google Scholar]
- 56.Zhu Y, Mitra N, Feng Y, Tishkoff S, Hoffstad O, Margolis D. FLG variation differs between European Americans and African Americans [e-pub ahead of print]. J Invest Dermatol. doi: 10.1016/j.jid.2020.12.022, accessed March 5, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fernandez K, Asad S, Taylan F, et al. Intragenic copy number variation in the filaggrin gene in Ethiopian patients with atopic dermatitis. Pediatr Dermatol. 2017;34(3):e140–e141. [DOI] [PubMed] [Google Scholar]
- 58.Seykora J, Dentchev T, Margolis DJ. Filaggrin-2 barrier protein inversely varies with skin inflammation. Exp Dermatol. 2015;24(9):720–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Margolis DJ, Gupta J, Apter AJ, et al. Filaggrin-2 variation is associated with more persistent atopic dermatitis in African American subjects. J Allergy Clin Immunol. 2014;133(3):784–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Asad S, Winge MC, Wahlgren CF, et al. The tight junction gene Claudin-1 is associated with atopic dermatitis among Ethiopians. J Eur Acad Dermatol Venereol. 2016;30(11):1939–1941. [DOI] [PubMed] [Google Scholar]
- 61.De Benedetto A, Rafaels NM, McGirt LY, et al. Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol. 2011;127(3). 773–786:e1-e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leung DY. New insights into atopic dermatitis: role of skin barrier and immune dysregulation. Allergol Int. 2013;62(2):151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brunner PM, Leung DYM, Guttman-Yassky E. Immunologic, microbial, and epithelial interactions in atopic dermatitis. Ann Allergy Asthma Immunol. 2018;120(1):34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muizzuddin N, Hellemans L, Van Overloop L, Corstjens H, Declercq L, Maes D. Structural and functional differences in barrier properties of African American, Caucasian and East Asian skin. J Dermatol Sci. 2010;59(2):123–128. [DOI] [PubMed] [Google Scholar]
- 65.McColl M, Boozalis E, Aguh C, Eseonu AC, Okoye GA, Kwatra SG. Pruritus in black skin: unique molecular characteristics and clinical features. J Natl Med Assoc. 2021;113(1):30–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.