

Short standfirst summary
Global sequencing and surveillance capacity for SARS-CoV-2 must be strengthened and combined with multidisciplinary studies of infectivity, virulence, and immune escape, in order to track the unpredictable evolution of the ongoing COVID-19 pandemic.
In June 2020, the World Health Organization (WHO) SARS-CoV-2 evolution working group was established to track SARS-CoV-2 variants and their specific genetic changes and to monitor virus characteristics and their impact on medical and non-medical countermeasures, including COVID-19 vaccines. In November 2021, this working group transitioned to a formal WHO Technical Advisory Group on Virus Evolution (TAG-VE), with the aim to develop and implement a global risk-monitoring framework for SARS-CoV-2 variants, based on a multidisciplinary approach that includes in silico, virological, clinical, and epidemiological data.
Tracking variants
The main role of the TAG-VE is to function as an integrative forum for exchange of information from global surveillance and research studies to monitor early warning signals, and perform timely assessment of the need for public health action in response to emerging variants1. It uses a Delphi consensus method to establish which emerging variants are considered variants of interest (VOI) or variants of concern (VOC; see Ref 2 for definitions)2. To avoid stigmatizing countries that first identify and report variants, a naming scheme that follows WHO guidelines3 and assigns Greek letters to VOIs and VOCs was adopted for global discourse in June 20214. As of 26 March 2022, eight VOIs and fiveVOCs have been designated, and are further described as previously or currently circulating variants to reflect changes in their epidemiology over time2,5.
The first available data on emerging variants that are assessed by TAG-VE are viral sequences and their associated metadata shared on publicly accessible genetic sequence databases (e.g. GISAID, Genbank, European Nucleotide Archive, DNA Database of Japan). Because all current VOIs and VOCs originate from ancestral variants, available sequences are still compared to the 2019 index virus (GISAID Accession ID: EPI_ISL_402124) with the primary focus on regions of the genome that are known to encode important viral, immunity- or infection-associated proteins. Mutations in the gene encoding the spike protein aregiven highest priority,, as this contains the receptor binding domain (RBD), essential for docking of the virus on host cells, as well as major determinants of both virus transmissibility (e.g. polybasic cleavage site) and antigenic make-up (e.g. N-terminal domain and RBD), and have therefore the highest prior probability of being clinically important. However, non-spike mutations have also been found to be important and should not be neglected. The constellation of mutations detected in any emerging variant is compared to a list of annotated mutations, which are suspected or known to play a role in one or more of the viral characteristics included in the VOI/VOC definitions: transmissibility, immune escape, disease severity, detectability and susceptibility to available treatments. Figure 1 depicts the evolution of SARS-CoV-2 and the variant-defining sets of spike protein amino acid substitutions seen in VOCs. To date, researchers have studied more than two dozen spike proteins, including those with phenotypes conferring immune escape (Figure 2). The list mutations and associated phenotypic impact, directly demonstrated in laboratory studies or inferred using in silico methods, is updated regularly, based on timely research presentations from the WHO partner network, as well as searches of preprints and peer-reviewed published literature.2 This annotated list of mutations has proven useful, as the same mutations have independently arisen in different lineages of SARS-CoV-2, strongly suggesting viral adaptation to the human host and selective pressure from population immunity.
Figure 1. Overview of VOCs and spike amino acid substitutions.
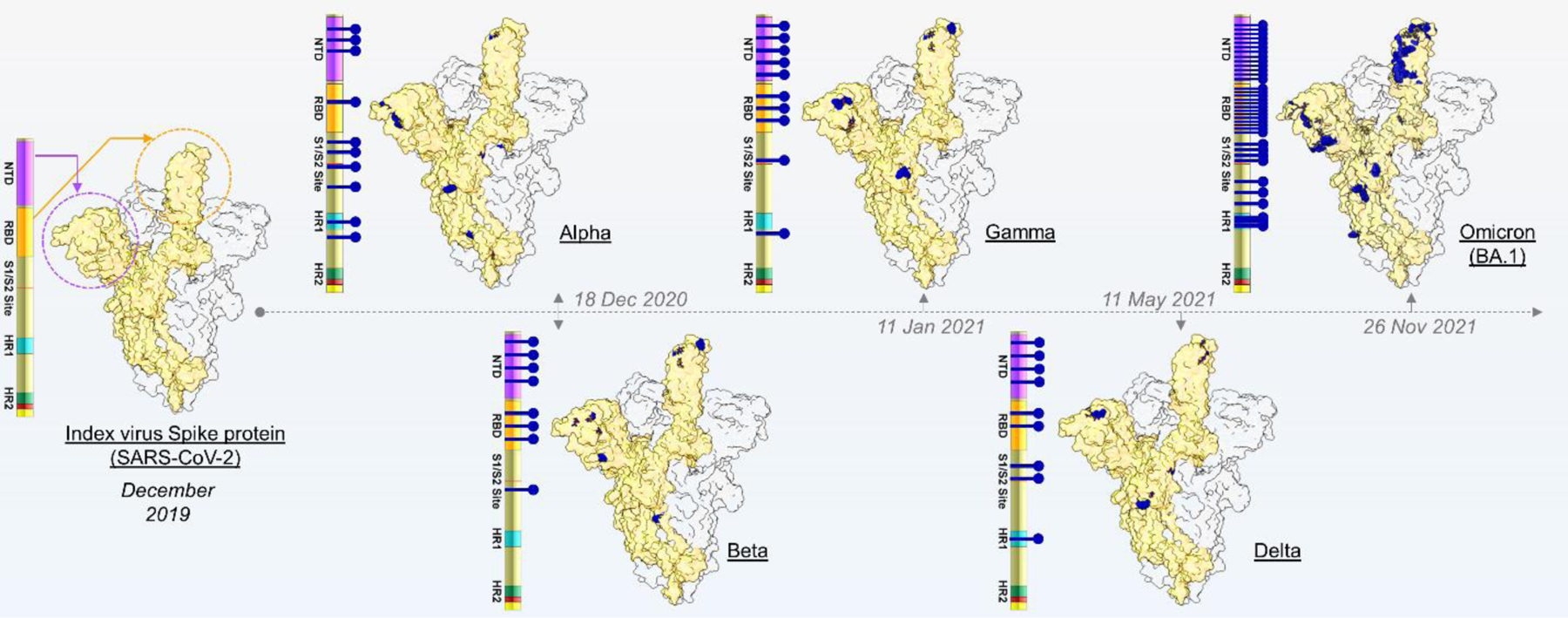
The spike structure of the index virus is shown, together with key domains, including N-terminal domain (NTD), Receptor Binding Domain (RBD), the S1/S2 junction (including the polybasic cleavage site), and the heptapeptide repeat domains (HR1, HR2) highlighted in different colours. The five VOCs and the characterising amino acids are marked with blue lines, both on the spike protein schematic and protein structure. VOCs are plotted chronologically according to date of identification.
Figure 2. Schematic of spike amino acid substitutions with known impact.
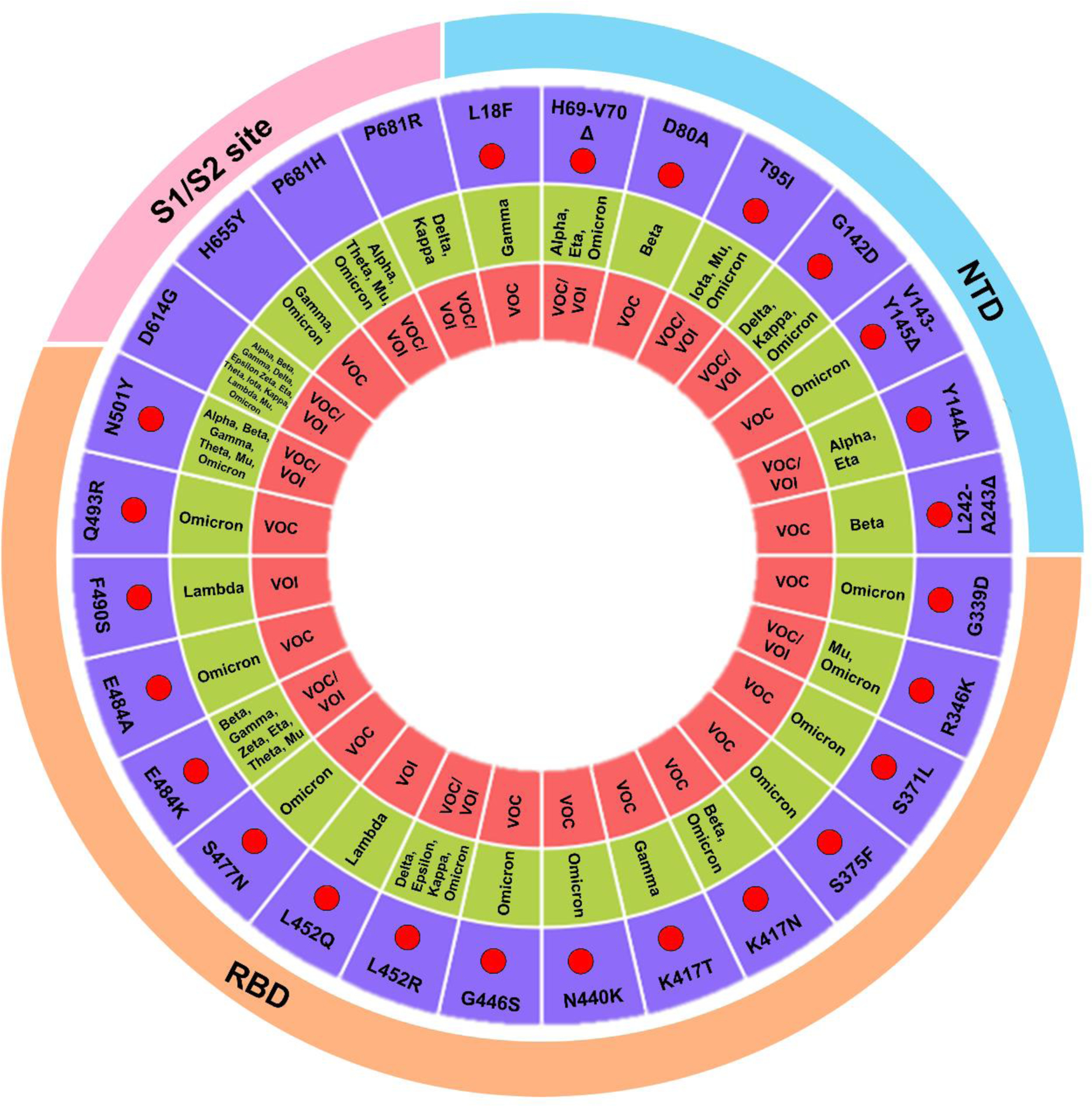
The circular plot shows key amino acid substitutions and their associated lineage, and presence in N-terminal domain, RBD, and S1/S2 polybasic cleavage site. The red are marked to emphasize mutations involved in immune evasion. The complete list of mutations is shown in Table 1.
Importantly, the detection of a new constellation of mutations in a variant does not necessarily translate into an increased public health threat, suggesting that some mutations, or combinations of mutations, may impart a fitness cost, rather than benefit. For example, the VOI Theta had a constellation of mutations that alerted scientists because it included top-ranked mutations E484K, N501Y, D614G and P681H in the spike protein (Figure 2), but there were other uncommon mutations in the spike and outside the spike that may have limited its potential to spread. To establish whether a new variant poses a serious threat, TAG-VE looks for early epidemiological signals of spread and clinical signals derived from surveillance data or specific studies. This includes assessing measures of how quickly cases are increasing, in what geographical areas and population subgroups a variant emerges and spreads, as well as changes to disease severity indicators, such as hospitalization. Careful consideration is also given to assessing evidence of relative transmissibility, compared to other circulating variants, including secondary attack rates observed in household transmission studies. Finally, the assessment of the threat posed by a variant also needs to consider vaccine- or infection-derived population immunity, which has progressively grown to high levels in many countries, as well as become more complex in terms of different permutations of hybrid immunity from vaccines, infection, or both)6. Therefore, studies that look at prevalence of re-infections or vaccine breakthroughs are also reviewed, where available.
Access to samples
The experience gained since the launch of the virus evolution working group, later becoming the TAG-VE, has shown that the speed of spread of new SARS-CoV-2 variants can outpace our current ability to assess their threat. Variant sequence determination has now become part of the global surveillance, and systematic phenotypic characterization of emerging variants is a critical component that needs to be added to the surveillance core toolbox. It is therefore important to define gaps that need to be addressed to enable better, and more timely, response to new variant threats.
While genomic indicators for transmissibility and immune escape of VOCs are conceptually relatively straightforward to assess, the wide diversity of assays, the lack of centralized biobanks of viruses and clinical specimens, the difficulties in international shipping of materials, and the globally fragmented funding landscape, makes the comparability of data amassed in real-time challenging. To improve the global capacity to rapidly perform phenotypic characterization, virus isolates of emerging variants should be promptly generated and shared to enable researchers in different laboratories to work with viruses that carry the same constellation of mutations. To address this challenge, the WHO is developing a bio hub system and recently established a Biohub Facility7. Its aim is to offer a reliable, safe, and transparent mechanism for WHO Member States to voluntarily share novel biological materials, without replacing or competing with existing systems such as EVAg and the NIH BEI repository.
Sequence data
Another important challenge concerns the availability, representativeness, and quality of genetic sequence data, which vary depending on sample quality, processing, experience, and the platforms, protocols, analytical tools used, and access to all of these. This challenge is exemplified in the large bias in the volume of sequences contributed by a small number of countries to global databases, as well as the release of sequences with low-confidence genomic regions and gaps.. For example, amplicon-based methods are sensitive to mutations, leading to amplification errors that can lead to sequence errors or poor-quality sequence in certain regions of the genome. The consequences of viral diversification call for sustained, if not enhanced, investment from governments in the capacity of reference laboratories to match the global public health demand for high quality sequences and virus characterisation for SARS-CoV-2, which can then be used for future public health threats posed by any infectious disease.
Verification of sequence quality through analysis of raw reads is a critical quality check, which becomes even more critical for the detection of recombination between SARS-CoV-2 genomes. Verified detection of SARS-CoV-2 circulating recombinant forms since Omicron emerged has increased, most likely because of increased availability of genomic surveillance, as well as natural factors. The higher number of lineage-defining mutations in Omicron makes the detection of recombinant forms easier, and re-infection with immune escape variants increases the chances of co-infection and therefore recombination, especially if two variants co-circulate in the community at high levels. While it is impossible to predict whether circulating recombinant forms with specific genomic breakpoints (i.e. where the two genomes recombine) may become more transmissible, for a new variant to spread widely, it must inherit traits from the parental viruses that provide a selective advantage.
Infectivity and virulence
Immune escape of VOCs can be shown through live virus neutralisation studies, with results from different laboratories showing similar results. In contrast, there are many methodological gaps in how to assess infectivity and virulence in vitro, ahead of clinical and epidemiological data. Some critical sites and amino acid substitutions have been identified, but this list is not comprehensive; these sites influence determinants of infectivity such as receptor binding, S1/S2 cleavage, and cellular entry (Table 1). In vivo animal studies can help understand specific features that may be difficult to estimate from epidemiologic data, such as assessing cross-neutralisation between variants, which is challenging in human subjects, as, unlike animal models, these may have unknown prior exposures. Animal models can also help assess virulence without the confounding effects of background immunity, but it remains to be seen how animal data correlate with disease severity in humans8–11.
Table 1:
Spike protein amino acid substitutions associated with laboratory evidence of phenotypic impact.
| Amino acid substitution(s) | Spike-domain | VOC/VOI (Pango lineages) | Transmissibility | Immune escape | Miscellaneous | References |
|---|---|---|---|---|---|---|
| L18F | NTD | VOC Gamma (P.1) | Increased | Reduced neutralization; abrogated binding of NTD specific mAb |
https://doi.org/10.3390/v13030392, https://doi.org/10.1016/j.cell.2021.03.028 https://doi.org/10.1038/s41586-021-03398-2 https://www.science.org/doi/10.1126/scitranslmed.abj6824 |
|
| H69-V70del | NTD | VOC Alpha (B.1.1.7), VOI Eta (B.1.525), VOC Omicron (BA.1, BA.3, BA.4, BA.5) | Increased replication in upper airway of hamsters and primary human airway cells | Escape from NTD-specific antibodies | Increased infectivity associated with enhanced incorporation of cleaved spike into virions | https://doi.org/10.1016/j.celrep.2021.109292, https://doi.org/10.1038/s41586-021-04245-0 + DOI: 10.1126/science.abf6950 |
| D80A | NTD | VOC Beta (B.1.351) | Loss of recognition by T-cells, but overall T-cell response preserved; abrogated binding to NTD specific mAb | https://www.science.org/doi/10.1126/scitranslmed.abj6824, | ||
| T95I | NTD | VOI Iota (B.1.526), VOI Mu (B.1.621), VOC Omicron (BA.1) | Very minor effect | https://doi.org/10.1016/j.ebiom.2021.103626 | ||
| G142D | NTD | VOC Delta (B.1.617.2), VOI Kappa (B.1.617.1), VOC Omicron (BA.1, BA.2, BA.3, BA.4, BA.5) |
Reduced neutralisation | https://www.science.org/doi/10.1126/science.abf6950, https://doi.org/10.1128/mBio.02473-21, https://dx.doi.org/10.1016%2Fj.cell.2021.03.028 | ||
| V143-Y145del | NTD | VOC Omicron (BA.1, BA.3) | Reduced neutralisation | https://www.science.org/doi/10.1126/science.abf6951, | ||
| Y144del | NTD | VOC Alpha (B.1.1.7), VOI Eta (B.1.525) | Reduced neutralisation | https://www.science.org/doi/10.1126/science.abf6952 | ||
| L242-A243del | NTD | VOC Beta (B.1.351) | Reduced neutralisation | https://doi.org/10.1038/s41591-021-01285-x | ||
| G339D | RBD | VOC Omicron (BA.1, BA.2, BA.3, BA.4, BA.5) | Escape from a subset of NAbs | Increased affinity for ACE-2 | https://doi.org/10.1038/s41586-021-04385-3,https://www.biorxiv.org/content/10.1101/2022.02.24.481899v1 | |
| R346K | RBD | VOI Mu (B.1.621), VOC Omicron (BA.1.1) | Reduced neutralisation | https://www.nature.com/articles/s41586-022-04594-4_reference.pdf | ||
| S371L | RBD | VOC Omicron (BA.1) | Escape from a subset of NAbs | https://doi.org/10.1038/s41586-021-04385-3 | ||
| S375F | RBD | VOC Omicron (BA.1, BA.2, BA.3, BA.4, BA.5) | Escape from a subset of NAbs | https://www.nature.com/articles/s41586-021-04385-3#citeas | ||
| K417N | RBD | VOC Beta (B.1.351), VOC Omicron (BA.1, BA.2, BA.3, BA.4, BA.5) | Reduced neutralisation | Decreased affinity for ACE-2 | https://doi.org/10.1038/s41586-021-04385-3, https://doi.org/10.1038/s41591-021-01294-w, https://doi.org/10.1101/2021.01.15.426911, | |
| K417T | RBD | VOC Gamma (P.1) | Reduced neutralisation | Decreased affinity for ACE-2 | https://doi.org/10.1038/s41586-021-04385-3 | |
| N440K | RBD | VOC Omicron (BA.1, BA.2, BA.3, BA.4, BA.5) | Escape from a subset of NAbs | Increased affinity for ACE-2 | https://doi.org/10.1038/s41586-021-04385-3, https://doi.org/10.1101/2020.11.30.405472, https://doi.org/10.7554/eLife.61312,https://doi.org/10.1126/science.abf4830,https://www.biorxiv.org/content/10.1101/2022.02.24.481899v1 | |
| G446S | RBD | VOC Omicron (BA.1) | Reduced neutralisation | https://doi.org/10.1038/s41586-021-04385-3 | ||
| L452R | RBD | VOC Delta (B.1.617.2), VOI Epsilon (B.1.427, B.1.429), VOI Kappa (B.1.617.1) VOC Omicron (BA.4, BA.5) |
Reduced neutralisation | Increased affinity for ACE-2; increased infectivity | https://doi.org/10.1016/j.cell.2021.04.025,https://doi.org/10.1038/s41579-021-00573-0,https://www.biorxiv.org/content/10.1101/2022.02.24.481899v1, https://doi.org/10.1016/j.chom.2021.06.006 | |
| L452Q | RBD | VOI Lambda (C.37) | Reduced neutralisation | Increased affinity for ACE-2; increased infectivity | https://doi.org/10.1080/22221751.2021.2008775,https://www.biorxiv.org/content/10.1101/2022.02.24.481899v1,https://doi.org/10.1101/2021.07.02.450959 | |
| S477N | RBD | VOC Omicron (BA.1, BA.2, BA.3, BA.4, BA.5) | Reduced neutralisation | Increased affinity for ACE-2 | https://www.biorxiv.org/content/10.1101/2022.02.24.481899v1,https://doi.org/10.1016/j.cell.2020.08.012, https://dx.doi.org/10.1016%2Fj.chom.2021.01.014 | |
| E484K | RBD | VOC Beta (B.1.351), VOC Gamma (P.1), VOI Zeta (P.2), VOI Eta (B.1.525), VOI Theta (P.3), VOI Mu (B.1.621) | Reduced neutralisation | Increased affinity for ACE-2 |
doi:10.1016/j.chom.2021.02.003 doi:10.1073/pnas.2103154118 doi:10.1016/j.chom.2021.01.014, https://doi.org/10.1038/s41586-021-03398-2 |
|
| E484A | RBD | VOC Omicron (BA.1, BA.2, BA.3) | Escape from a subset of Nabs; reduced neutralization by convalescent human sera and mAbs | doi:10.1016/j.chom.2021.01.014, https://doi.org/10.1038/s41586-021-04385-3, https://doi.org/10.1016/j.chom.2021.02.003 | ||
| F486V | RBD | VOC Omicron (BA.4, BA.5) | Escape from a subset of NAbs | Increased affinity for ACE-2 | https://doi.org/10.1038/s41467-021-24435-8, 10.1016/j.cell.2020.08.012 | |
| F490S | RBD | VOI Lambda (C.37) | Reduced neutralisation | https://doi.org/10.1016/j.chom.2021.01.014 + https://doi.org/10.1080/22221751.2021.2008775 | ||
| Q493R | RBD | VOC Omicron (BA.1, BA.2, BA.3) | Escape from a subset of NAbs | https://doi.org/10.1038/s41586-021-04385-3 | ||
| N501Y | RBD | VOC Alpha (B.1.1.7), VOC Beta (B.1.351), VOC Gamma (P.1), VOI Theta (P.3), VOI Mu (B.1.621), VOC Omicron (BA.1, BA.2, BA.3, BA.4, BA.5) | Increased replication in upper airway of hamsters and primary human airway cells | Reduced neutralisation | Increased affinity for ACE-2 | https://doi.org/10.1016/j.cell.2021.02.042, https://doi.org/10.1038/s41586-021-03412-7, https://www.biorxiv.org/content/10.1101/2022.02.24.481899v1 |
| D614G | SD2 | VOC Alpha (B.1.1.7), VOC Beta (B.1.351), VOC Gamma (P.1), VOC Delta (B.1.617.2), VOI Epsilon (B.1.427, B.1.429) VOI Zeta (P.2), VOI Eta (B.1.525), Theta (P.3), VOI Iota (B.1.526), VOI Kappa (B.1.617.1), VOI Lambda (C.37), VOI Mu (B.1.621), VOC Omicron (BA.1, BA.2, BA.3, BA.4, BA.5) | Increased infectivity in vitro; enhanced transmission in animal model; enhanced replication in upper respiratory tract | https://doi.org/10.1016/j.cell.2020.11.020, https://doi.org/10.1126/science.abe8499 | ||
| H655Y | Around S1/S2 site | VOC Gamma (P.1), VOC Omicron (BA.1, BA.2, BA.3, BA.4, BA.5) | Increased transmissibility in vivo | Escape from a subset of NAbs | https://doi.org/10.1016/j.chom.2022.01.006, https://www.science.org/doi/10.1126/science.abd0831 | |
| P681H | Around S1/S2 site | VOC Alpha (B.1.1.7), VOI Theta (P.3), VOI Mu (B.1.621), VOC Omicron (BA.1, BA.2, BA.3, BA.4, BA.5) | Enhanced cleavage; viral entry or cell-to-cell spread not significantly impacted | https://doi.org/10.1016/j.isci.2021.103589 | ||
| P681R | Around S1/S2 site | VOC Delta (B.1.617.2), VOI Kappa (B.1.617.1) | Enhanced cleavage, significant resistance to several mAbs and vaccine elicited NAb | https://dx.doi.org/10.1101%2F2021.08.12.456173, https://doi.org/10.1038/s41586-021-04266-9 |
Abbreviations: mAbs – monoclonal antibodies; NAbs – neutralising antibodies. NTD – N-terminal domain; RBD – Receptor Binding Domain; ACE2 – Angiotensin converting enzyme 2
Disease severity in the clinical setting can be especially difficult to determine rapidly and accurately because of challenges posed by ascertainment, inclusion, quality of care, confounding and colliding biases from empirical observational studies. A key role will be played by the interoperability of electronic health records, which should allow easy linkage of sequencing data with patient clinical records. Unbiased, systematic clinical and epidemiological data collection with comprehensive biological sampling and rigorous characterization of virulence and correlates of protection remain the gold standard for variant threat assessment. The generation of such data from well-designed studies across diverse healthcare settings and different geographies is expected to remain challenging, especially once the COVID-19 response is de-escalated in the post-acute phase of the pandemic. Integration of such studies into newly established routine surveillance system workplans may help ensure that clinical and epidemiological studies continue in the future.
Until November 2021, the emergence of VOCs such as Alpha and Delta was mainly associated with an increase in transmissibility, most likely driven by viral adaptation to the human host, with modest degrees of immune escape12. The emerging evidence on Omicron variants suggests that immune escape was a significant driver for its observed displacement over Delta, and its selective advantage was driven by increasing population immunity, in addition to the increased transmissibility13. While the lower virulence of Omicron was most likely a chance event, the preservation of cellular immunity that protects against severe infection is likely to be a recurring theme14,15. In addition, so-called hybrid immunity in individuals who have experienced a breakthrough infection appears to broaden immune responses, which suggests that population immunity may be able to tolerate significant continued evolution in SARS-CoV-2. 16,17 However, because transmission and virulence are uncoupled for SARS-CoV-2, it cannot be assumed that the next variant will be less virulent. Future variants may be of similar, higher or lower virulence than Omicron.
An early warning system
A recent retrospective analysis suggested that some of the key variant-defining mutations could potentially have been detected much earlier, showing the importance of early warning bioinformatics tools from globally shared data, an approach in its infancy.18 The identification of key mutations could then trigger a subsequent virus-, variant- or mutation- characterization pipeline. Machine learning algorithms to determine the potential impact of key mutations are being developed and validated, but a full assessment of variants still require epidemiological data and in vivo and /or in vitro experiments, as a VOC cannot be ascertained solely using genomic data.
The future of the pandemic is difficult to predict, for several reasons. Firstly, SARS-CoV-2 variants do not always emerge from the most recently dominant circulating virus, unlike other respiratory viruses such as human influenza viruses.19. Secondly, chronically infected patients can allow the acceleration of intra-host evolution 20. Thirdly, a wide number of susceptible mammals may act as secondary reservoirs, including cervids, mustelids and rodents, with potential for reverse zoonoses21. Fourthly, too much of the world’s population remains unvaccinated to date, and current vaccines are suboptimal in preventing transmission (future vaccines may remain so). Given these challenges in predicting the evolution of the virus, targeted surveillance of suspected high-risk populations, such as chronically infected patients, or improving detection systems such as wastewater and a One Health approach to animal surveillance are critical for the early detection of future variants.
The evolving virus and the uncertainty of predicting the trajectory of the pandemic calls for strengthened surveillance and continued monitoring of SARS-CoV-2. The TAG-VE will continue to critically appraise state-of-the-art methodologies towards predicting further evolution of SARS-CoV-2 and to rapidly determine the threat levels posed by new variants. The pandemic is not over and SARS-CoV-2 is spreading at a high level globally. Now is the time to enhance global sequencing capacities, focusing on widening coverage to include previous geographical and populations’ blind spots, and build a global consensus towards continued concerted multidisciplinary efforts, under the leadership of the WHO Research & Development Blueprint for action to prevent epidemics, to track and assess the threat posed by future SARS-CoV-2 variants.
Acknowledgments
We acknowledge scientists, public health professionals and Ministries of Health across the world for early generation and sharing of data on SARS-CoV-2 variants.
Footnotes
Competing interests
No financial and non-financial competing interests declared
References
- 1.World Health Organization. Terms of Reference for the Technical Advisory Group on SARS-CoV-2 Virus Evolution (TAG-VE).
- 2.World Health Organization. Tracking SARS-CoV-2 variants. https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/.
- 3.World Health Organization Best Practices for the Naming of New Human Infectious Diseases. https://apps.who.int/iris/bitstream/handle/10665/163636/WHO_HSE_FOS_15.1_eng.pdf.
- 4.Konings F et al. Nat Microbiol 6, 821–823 (2021). [DOI] [PubMed] [Google Scholar]
- 5.World Health Organization. Weekly Epidemiological Updates. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports. [Google Scholar]
- 6.Bergeri I et al. http://medrxiv.org/lookup/doi/10.1101/2021.12.14.21267791 (2021) doi: 10.1101/2021.12.14.21267791. [DOI]
- 7.World Health Organization. WHO Biohub System. https://www.who.int/initiatives/who-biohub.
- 8.CMMID COVID-19 Working Group et al. Nature 593, 270–274 (2021).33723411 [Google Scholar]
- 9.Twohig KA et al. The Lancet Infectious Diseases 22, 35–42 (2022).34461056 [Google Scholar]
- 10.Munster VJ et al. Sci. Adv 7, eabj3627 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muñoz-Fontela C et al. PLoS Pathog 18, e1010161 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oude Munnink BB et al. Nat Med 27, 1518–1524 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Viana R et al. Nature (2022) doi: 10.1038/s41586-022-04411-y. [DOI]
- 14.Gao Y et al. Nat Med (2022) doi: 10.1038/s41591-022-01700-x. [DOI]
- 15.Keeton R et al. Nature (2022) doi: 10.1038/s41586-022-04460-3. [DOI]
- 16.Kitchin D et al. Cell Reports Medicine 100535 (2022) doi: 10.1016/j.xcrm.2022.100535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He W et al. http://biorxiv.org/lookup/doi/10.1101/2021.09.08.459480 (2021) doi: 10.1101/2021.09.08.459480. [DOI]
- 18.Maher MC et al. Sci. Transl. Med 14, eabk3445 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones S et al. Sci Rep 9, 14690 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corey L et al. N Engl J Med 385, 562–566 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oude Munnink BB et al. Science 371, 172–177 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]