

Abstract
High-resolution crystallographic studies of a number of inhibited forms of bovine F1-ATPase have identified four independent types of inhibitory site: the catalytic site, the aurovertin B-binding site, the efrapeptin-binding site and the site to which the natural inhibitor protein IF1 binds. Hitherto, the binding sites for other inhibitors, such as polyphenolic phytochemicals, non-peptidyl lipophilic cations and amphiphilic peptides, have remained undefined. By employing multiple inhibition analysis, we have identified the binding sites for these compounds. Several of them bind to the known inhibitory sites. The amphiphilic peptides melittin and synthetic analogues of the mitochondrial import pre-sequence of yeast cytochrome oxidase subunit IV appear to mimic the natural inhibitor protein, and the polyphenolic phytochemical inhibitors resveratrol and piceatannol compete for the aurovertin B-binding site (or sites). The non-peptidyl lipophilic cation rhodamine 6G acts at a separate unidentified site, indicating that there are at least five inhibitory sites in the F1-ATPase. Each of the above inhibitors has significantly different activity against the bacterial Bacillus PS3 α3β3γ subcomplex compared with that observed with bovine F1-ATPase. IF1 does not inhibit the bacterial enzyme, even in the absence of the ε-subunit. An understanding of these inhibitors may enable rational development of therapeutic agents to act as novel antibiotics against bacterial ATP synthases or for the treatment of several disorders linked to the regulation of the ATP synthase, including ischaemia–reperfusion injury and some cancers.
Keywords: amphiphilic peptide, inhibitory site, mitochondrial F1-ATPase, non-peptidyl lipophilic cation, polyphenolic phytochemical
Abbreviations: IF1; natural inhibitor protein of the mitochondrial F1F0-ATPase; PMF, proton-motive force
INTRODUCTION
The mitochondrial ATP synthase (F1Fo-ATPase) is a multi-subunit, membrane-bound assembly central to biological energy conversion. The enzyme comprises a globular F1 catalytic domain (subunit composition α3β3γ1δ1ε1) and a membrane-bound Fo proton translocating domain, linked together by central and peripheral stalks [1]. ATP synthase couples the transmembrane PMF (proton-motive force) to the synthesis of ATP from ADP and Pi. Several covalent and non-covalent inhibitors of mitochondrial F1-ATPase have been identified. The covalent inhibitors include NBD-Cl (4-chloro-7-nitrobenzofurazan), DCCD (N,N′-dicyclohexylcarbodi-imide), 8-azido-ATP, 2-azido-ATP, 5′-p-fluorosulphonylbenzoyladenosine and 5′-p-fluorosulphonylbenzoylinosine [2,3]. The non-covalent inhibitors include non-hydrolysable substrate analogues [4,5], the natural inhibitor protein IF1 [6], the efrapeptins [7], the aurovertins [8,9], polyphenolic phytochemicals [10,11], non-peptidyl lipophilic cations and amphiphilic peptides [12].
Four inhibitory sites have been identified from high-resolution crystallographic studies of inhibited forms of F1-ATPase from bovine heart mitochondria. The non-hydrolysable NTP analogues adenylylimidodiphosphate [13], ADP aluminium fluoride [14] and ADP beryllium fluoride [15] bind to the catalytic site. The covalent inhibitors act by modification of residues near the catalytic sites [16,17]. The antibiotics aurovertin and efrapeptin bind at two independent inhibitory sites in the catalytic β-subunits: efrapeptin to a site located in the central cavity of the enzyme, making contacts with the central γ-subunit, with subunit βE and with two adjacent α-subunits [18], and aurovertin to two equivalent sites situated between the nucleotide-binding and C-terminal domains of the βE- and βTP-subunits [19]. The natural inhibitor protein, IF1, binds to a fourth site at the catalytic interface between the C-terminal domains of the αDP- and βDP-subunits, and is in contact with the γ-subunit [20].
The polyphenolic phytochemical inhibitors resveratrol and piceatannol inhibit the ATP synthase by targeting the F1 domain. They exhibit non-competitive and mixed inhibition of F1-ATPase activity respectively [10,11]. A number of amphiphilic peptide inhibitors of F1-ATPase have also been identified. They include the bee venom peptide melittin and synthetic analogues (SynA2 and SynC) of the mitochondrial import pre-sequence of yeast cytochrome oxidase subunit IV [12]. All adopt cationic amphiphilic α-helical structures [21–23]. Melittin and SynA2 exhibit non-competitive inhibition of F1-ATPase, whereas SynC shows mixed inhibition. The non-peptidyl lipophilic cation rhodamine 6G also acts as a mixed inhibitor of F1-ATPase [12]. It has been suggested previously that an interfacial region near the C-terminus of α- and β-subunits might form part of the binding site for the amphiphilic peptides and non-peptidyl lipophilic cations [24].
The modes of inhibition (mixed or non-competitive) of polyphenolic phytochemicals, non-peptidyl lipophilic cations and amphiphilic peptides indicate they do not bind to the catalytic site. However, their binding sites and inhibitory mechanisms have remained unclear. In particular, it is not known if they bind to unidentified sites, or whether they interact with previously characterized sites. In the present study, multiple inhibition analysis has been used to determine the sites to which the amphiphilic peptides, non-peptidyl lipophilic cations and polyphenolic inhibitors bind to the F1-ATPase. Several of these inhibitors bind to previously characterized inhibitory sites, but we show that at least one additional distinct inhibitory site is present. In addition, we investigated the effects of several of these inhibitors on the bovine mitochondrial F1-ATPase compared with their effects on the bacterial F1-ATPase subcomplex from thermophilic Bacillus PS3. Each inhibitor showed significantly different activity against the bacterial enzyme compared with the bovine F1-ATPase.
EXPERIMENTAL
Materials
Melittin (GIGAVLKVLTTGLPALISWIKRKRQQ-NH2), rhodamine 6G, dequalinium, efrapeptin (F and G subtypes), aurovertin B, resveratrol and piceatannol (>95% purity) were all obtained from Sigma (St. Louis, MO, U.S.A.). The synthetic analogues of the mitochondrial import pre-sequence of yeast cytochrome oxidase subunit IV, SynA2 (MLSRLSLRLLSRLSLRLLSRYLL-NH2) and SynC (MLSSLLRLRSLSLLRLRLSRYLL-NH2) (>95% purity) [12] were synthesized by Jerini Peptide Technologies (Berlin, Germany). The purified α3β3γ subcomplex of F1-ATPase from thermophilic Bacillus PS3 was a gift from Professor William Allison (Department of Chemistry and Biochemistry, University of California at San Diego, La Jolla, CA, U.S.A.).
Purification of F1-ATPase and IF1
The purification of bovine F1-ATPase [16] and recombinant bovine IF1 [25] were carried out as described previously.
Inhibition assay
The activity of F1-ATPase was measured in the presence of various concentrations of inhibitors using an ATP-regenerating system. ATPase activity was estimated by addition of either 1.5 μg of purified bovine F1-ATPase or 4.0 μg of the purified Bacillus PS3 α3β3γ subcomplex to 1 ml of assay mixture at 37 °C and following the decrease in absorbance of NADH at 340 nm. Initial rates (v) over a period of 60 s were recorded. The ATPase assay mixture contained 50 mM Pipes/NaOH, pH 6.6, 50 mM KCl, 2 mM MgCl2, 12–15 units/ml pyruvate kinase, 12–15 units/ml lactate dehydrogenase (Sigma), 0.2 mM NADH and 1 mM phosphoenolpyruvate. For multiple inhibition experiments and determination of the IC50, 2 mM MgATP was used as substrate. IC50 values were calculated by unweighted non-linear regression fits to the median effect equation [26]. To determine Ki values for single inhibitor steady-state kinetics, initial rates were recorded using MgATP concentrations ranging from 0.2 to 2 mM. Characterization of inhibition was carried out using the complementary methods of Dixon [27] and Cornish-Bowden [28]. Separate measurements with MgADP ensured that the inhibitors had no effect on the coupled assay system.
Kinetics of inhibition of F1-ATPase activity by multiple inhibitors
A semi-generalized formulation of single-enzyme multiple inhibition by two reversible inhibitors (I1, I2) has been reported previously (Scheme 1) [29]. It is assumed that I1·I2, I1·S, I2·S and I1·I2·S are not formed and that no other type of interaction, except those shown, takes place. For the binary complexes, inhibitor dissociation constants are designated as K with the complex name in subscript. Dissociation constants of ternary complexes are represented similarly with the ligand that dissociates from the complex written as the last subscript term. For example, KEI1I2 refers to the dissociation of I2 from the E·I1·I2 complex.
Scheme 1. Equilibria among enzyme species in the presence of substrate and two inhibitors [29].

Factors α and β represent the change in substrate affinity induced by I1 and I2 respectively, or, alternatively, the alteration in the affinity for the inhibitors due to the bound substrate. Factor γ represents the mutual influence of the two inhibitors on the binding of each other. Inhibitor binding is independent when γ=1, whereas values of γ lower or greater than unity denote mutual facilitation or hindrance respectively. The rate equation for Scheme 1 can be written as:
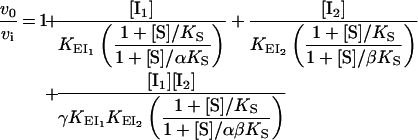 |
where vi and v0 are the velocities of the inhibited and uninhibited reactions respectively [29].
The Yonetani–Theorell plot [30] was used to evaluate the interaction between two inhibitors. From eqn (1) it follows that, for situations where the binding of two inhibitors to the enzyme is mutually exclusive (i.e. γ=∞), a plot of v0/vi against [I1] at a range of fixed [I2] would give a series of parallel lines. On the other hand, if they can bind simultaneously to the enzyme (i.e. ∞>γ>0), the slope will depend on [I2] and the series of lines will intersect. A value for the factor γ can be calculated from fits of the kinetic data to eqn (1) for several pairs of mutually non-exclusive inhibitors. Since factor γ provides information on the mutual influence of any two inhibitors on each other, this serves as a useful parameter for comparison. The strength of mutual hindrance or facilitation can be easily assessed by the size of the numerical value for γ as described above.
As a test of this approach, the Yonetani–Theorell analysis [30] was conducted on the combination of either efrapeptin or aurovertin B with IF1. Both plots indicated mutual non-exclusive binding, i.e. separate binding sites (results not shown). This is in agreement with the high-resolution crystal structures of the F1-ATPase inhibited by efrapeptin [18], aurovertin B [19] and IF1 [20], which indicate that neither efrapeptin nor aurovertin B bind to the same site as IF1. This confirms that our approach is a valid way of evaluating inhibitor mutual exclusivity on the F1-ATPase.
RESULTS
IF1 and the cationic amphiphilic peptides bind to the same site in F1-ATPase
Three amphiphilic peptides were studied: melittin, SynA2 and SynC. The combinations of each peptide and IF1 were analysed by means of the Yonetani–Theorell plot [30] (Figure 1). For each peptide, the plots of v0/vi against peptide (I1) concentration at various concentrations of IF1 (I2) gave a series of parallel straight lines, indicating mutually exclusive binding to F1-ATPase (γ=∞).
Figure 1. Effect of the combination of IF1 and the amphiphilic α-helical peptides melittin (A), SynA2 (B) and SynC (C) on bovine F1-ATPase.

The series of parallel straight lines evident in the Yonetani–Theorell plots [30] indicates mutually exclusive binding. The concentrations of I1 (melittin, SynA2 or SynC) are shown on the abscissa, and those of I2 (IF1) are: (●) 0 μM, (○) 0.1 μM, (■) 0.2 μM, (□) 0.5 μM, (▲) 1.0 μM. Linear regression lines are drawn through the data points. Each point on the graphs is independent of all others. The use of at least 20 points is sufficient to determine the nature of the interaction between multiple inhibitors. The slight deviation from absolute parallel lines at higher inhibitor concentrations is insignificant, the data are consistent with mutual exclusivity.
Resveratrol and piceatannol bind to the same site as aurovertin B
Each combination of efrapeptin, aurovertin B and IF1 with resveratrol was analysed as above to determine whether polyphenolic phytochemical inhibitors compete with other inhibitors for the known inhibitory sites or whether they act at a separate site. The Yonetani–Theorell plot [30] obtained for aurovertin B and resveratrol gave a series of parallel lines (Figure 2A). All other combinations gave intersecting lines (results not shown). Parallel lines were also observed in a similar plot for piceatannol with aurovertin B (Figure 2B).
Figure 2. Effect of the combination of aurovertin B and the polyphenolic phytochemicals resveratrol (A) and piceatannol (B) on bovine F1-ATPase.

The series of parallel straight lines evident in the Yonetani–Theorell plots [30] indicates mutually exclusive binding. The concentrations of I1 (piceatannol or resveratrol) are shown on the abscissa, and those of I2 (aurovertin B) are: (●) 0 μM, (○) 0.05 μM, (■) 0.125 μM, (□) 0.25 μM, (▲) 0.625 μM (data using 0.625 μM aurovertin B in combination with resveratrol were not determined). Linear regression lines are drawn through the data points; for further details, see the legend for Figure 1.
Rhodamine 6G binds to a distinct site in F1-ATPase
Each combination of efrapeptin, aurovertin B and IF1 with rhodamine 6G was analysed as above to determine whether non-peptidyl lipophilic cations compete with other inhibitors for the known inhibitory sites or whether they act at a separate site. All combinations gave a series of intersecting lines (∞>γ>0), indicating simultaneous binding to the enzyme (mutually non-exclusive binding). A representative plot is shown (Figure 3). Consequently, rhodamine 6G and other structurally related non-peptidyl lipophilic cations appear to bind to a site distinct from the four inhibitory sites characterized by X-ray crystallography.
Figure 3. Effect of the combination of efrapeptin and rhodamine 6G on bovine F1-ATPase.

The series of intersecting straight lines evident in the Yonetani–Theorell plot [30] indicates mutually non-exclusive binding. The concentrations of I1 (efrapeptin) are shown on the abscissa, and those of I2 (rhodamine 6G) are: (●) 0 μM, (○) 10.4 μM, (■) 26 μM, (□) 52 μM, (▲) 78 μM. Linear regression lines are drawn through the data points; for further details, see the legend for Figure 1.
Data for the rhodamine 6G double inhibition experiments were fitted to eqn (1) by unweighted non-linear regression. For each system, the set of data points provides sufficient information to enable determination of four of the six unknown parameters: α, β, γ, KS, KEI1 (efrapeptin, aurovertin B or IF1) and KEI2 (rhodamine 6G). Steady-state single-inhibitor kinetic parameters for IF1, rhodamine 6G, efrapeptin and aurovertin B were determined independently and their values kept fixed during fitting to eqn (1). All of the inhibitors exhibited mixed inhibition of F1-ATPase activity, except aurovertin B, which exhibited uncompetitive inhibition. Values for factor γ (representing the mutual influence of the two inhibitors on the binding of each other) were determined (Table 1).
Table 1. Mutual influence of IF1, efrapeptin, aurovertin B (I1) and rhodamine 6G (I2) on the binding of each other.
Values for the mutual influence factor, γ, were calculated by unweighted non-linear regression fits to eqn (1). Kinetic parameters derived from single inhibitor steady state kinetic experiments were assigned for the fits to eqn (1): IF1 (KEI=0.25 μM, α=0.72), efrapeptin (KEI=0.12 μM, α=1.4), aurovertin B (α=0.05) and rhodamine 6G (KEI=91 μM, α=0.29). KEI represents the inhibition constant for the competitive component. Factor α represents the change in substrate affinity induced by inhibitor binding, or, alternatively, the alteration in the affinity for the inhibitor due to the bound substrate. The inhibition constant for the uncompetitive component KESI=αKEI. A mean value of 370 μM was obtained for KS. The KEI value for aurovertin B could not be determined accurately, since the inhibition was predominantly uncompetitive and hence it was not included as a fixed parameter in the double inhibition non-linear regression fits to eqn (1). The data are expressed as the means±S.D.
| Variable inhibitor | γ | Fold change in inhibition 1/γ |
|---|---|---|
| IF1 | 2.8±0.3 | 0.36 |
| Efrapeptin | 0.38±0.02 | 2.6 |
| Aurovertin B | 1.3±0.3 | 0.77 |
These values indicate that IF1 and rhodamine 6G act antagonistically, hindering the binding of one another, whereas efrapeptin and rhodamine 6G act synergistically. Aurovertin B and rhodamine 6G appear to bind independently. The interactions between I1 and I2 in the E·I1·I2 complex could arise from any number of effects, including ion-dipole, interionic, interdipole, hydrophobic and hydrophilic interactions, as well as simple steric hindrance and protein conformational changes. A γ value greater than unity could be interpreted as two binding sites being in close proximity and, as such, the binding of one inhibitor, while not causing mutual exclusivity, may hinder the approach and interaction of a second inhibitor through binding site overlap or protein conformational changes. Independent inhibitor binding would suggest that the sites do not interact at all. Synergism is more difficult to explain, and again could be a consequence of global conformational changes in the enzyme. Any of the inhibitors described here may lock the F1-ATPase in a conformation that hinders or facilitates the binding of subsequent inhibitor molecules, even if they operate at distinct sites.
Comparison of inhibitory activities against bovine F1-ATPase and the α3β3γ subcomplex of Bacillus PS3
IC50 values were determined for each of the amphiphilic peptides, IF1, rhodamine 6G, dequalinium, piceatannol and resveratrol against both the bovine F1-ATPase and the bacterial α3β3γ subcomplex (Table 2). The order of effectiveness of the inhibitors on bovine F1-ATPase as assessed by IC50 values was: SynC (most effective), IF1, SynA2, piceatannol, resveratrol, melittin, rhodamine 6G and dequalinium (least effective). Each of the above inhibitors has significantly different activity against the bacterial α3β3γ subcomplex compared with that observed with bovine F1-ATPase. IF1, resveratrol and piceatannol have no inhibitory activity on the bacterial enzyme subcomplex. However, melittin and rhodamine 6G stimulate the ATPase activity of the bacterial subcomplex. The stimulatory effect seen for melittin is much weaker than that observed for rhodamine 6G, which appears to stimulate ATPase activity by more than 3-fold at low concentrations. As the concentrations are increased, the stimulatory effect of both melittin and rhodamine 6G decreases until, at sufficiently high concentrations, the compounds act as inhibitors. SynA2 and SynC have much weaker inhibitory activity on the bacterial enzyme when compared with the bovine enzyme. The IC50 values determined for SynA2 and SynC on the bacterial α3β3γ subcomplex are 6- and 10-fold higher respectively than those observed on the bovine enzyme. Dequalinium, on the other hand, shows approx. 2-fold greater inhibitory activity on the bacterial enzyme compared with the bovine enzyme.
Table 2. Comparison of inhibitory activities on bovine F1-ATPase and the α3β3γ subcomplex of Bacillus PS3.
The data are expressed as the means±S.D.
| IC50 (μM) | ||
|---|---|---|
| Inhibitor | Bovine F1-ATPase | Bacillus PS3 α3β3γ |
| IF1 | 0.25±0.01 | No inhibition |
| Melittin | 12±0.1 | Stimulatory |
| SynA2 | 0.29±0.02 | 1.7±0.3 |
| SynC | 0.16±0.01 | 1.6±0.4 |
| Resveratrol | 6.4±0.1 | No inhibition |
| Piceatannol | 6.1±0.1 | No inhibition |
| Rhodamine 6G | 27±1 | Stimulatory |
| Dequalinium | 46±1 | 19±1 |
DISCUSSION
Bovine IF1 is an 84-residue protein, which inhibits ATP hydrolysis by forming a 1:1 complex with ATP synthase. Binding of the inhibitor protein requires hydrolysis of MgATP in the absence of the PMF and is optimal below pH 7.0 [31,32]. In vitro, the active form of bovine IF1 is a dimer [25] associated by an antiparallel α-helical coiled-coil between the C-terminal regions of monomers [33,34]. This arrangement places the N-terminal minimal inhibitory sequences (residues 14–47) [35] in opposition, allowing the dimeric IF1 to bind two F1 domains simultaneously [20,36]. Each monomer folds into a single cationic amphiphilic α-helix approx. 95 Å in length. Melittin, SynA2 and SynC also adopt cationic amphiphilic α-helical structures [21–23].
In light of the structural similarity between the amphiphilic cationic peptides and IF1, (Figure 4) it is reasonable to conclude that their mutual exclusivity of binding is a consequence of competition for the same inhibitory site in F1. Furthermore, high-affinity binding to the IF1 site must be largely dependent on the adoption of a cationic, amphiphilic helical structure, rather than sequence similarity to the inhibitor protein. This suggestion is consistent with the amphiphilic nature of the F1-ATPase–IF1 binding surface [20]. Previously, it had been proposed that the binding site for amphiphilic peptides and non-peptidyl lipophilic cations involves an interfacial region near the C-terminus of the α- and β-subunits [24]. The crystal structure of the F1-ATPase–IF1 complex [20] has now identified this region as a critical part of the IF1-binding site. These observations are in agreement with our kinetic analysis.
Figure 4. High-resolution crystal structures of IF1 (A) and melittin (B).

Ribbon and electron density mesh (contoured at 1σ) representations of the structures of the N-terminal segment (residues 4–40) of bovine IF1 taken from the F1-ATPase–IF1 crystal structure [20] (Protein DataBank accession code 1OHH) and full-length bee venom melittin (residues 1–26) [21] (Protein DataBank accession code 2MLT). The N- and C-termini are indicated. Both molecules adopt a cationic amphiphilic helix–turn–helix motif. Images were produced with the program PyMOL (DeLano Scientific, San Carlos, CA, U.S.A.).
Inhibitor proteins from yeast and bovine mitochondria are able to inhibit the F1-ATPase from each other [37]. Of the 32 bovine F1-ATPase residues involved in binding to IF1, 25 are identical in yeast F1-ATPase. A total of 21 of the 32 IF1-binding residues are also identical in the Escherichia coli enzyme [20], and in Bacillus PS3 19 of them are identical. Since the IF1 interface is well conserved in both Bacillus PS3 and E. coli, it has remained unclear why the inhibitor protein has no effect on bacterial F1-ATPases. One explanation has been that the ε-subunit, which has been shown to inhibit ATP hydrolysis in vitro, may impede the inhibitory effect of IF1 on bacterial enzymes [38–40].
The α3β3γ subcomplex of Bacillus PS3 F1-ATPase used in our present study is not inhibited by IF1. Thus the ε-subunit is not responsible for the lack of inhibition by IF1. Furthermore, it might be expected that amphiphilic peptides acting at the same site as IF1 on the bovine enzyme would exhibit significantly reduced affinity for the bacterial enzyme. Indeed, melittin, SynA2 and SynC all show poor inhibitory activity against the bacterial α3β3γ subcomplex compared with bovine F1-ATPase, supporting the conclusion that they compete for the same site as IF1. However, in light of the sequence and structural similarity between Bacillus PS3 F1-ATPase and bovine F1-ATPase [41], it remains unclear as to why the inhibitor protein has no effect on the bacterial enzyme, even in the absence of the ε-subunit, and yet is a potent regulator of the mitochondrial enzyme.
Also, melittin has been shown to bind to calmodulin, inhibiting its activation and interaction with target enzymes [42,43]. IF1 also interacts specifically with calmodulin [44]. These observations are consistent with our proposal of amphiphilic α-helical peptides mimicking the action of the inhibitor protein.
The mutually exclusive binding of the polyphenolic phytochemical inhibitors and aurovertin B to F1-ATPase indicates that resveratrol, piceatannol and probably other related polyphenolic phytochemicals bind to the aurovertin B site (or sites). Aurovertin B binds to bovine F1 at two equivalent sites on βTP and βE, in a cleft between the nucleotide-binding and C-terminal domains [19]. It appears to inhibit the F1-ATPase by preventing closure of the catalytic interfaces necessary for cyclic inter-conversion of catalytic sites. Our kinetic data and the modest structural similarity (Figure 5) between these two classes of molecules suggest that polyphenolic inhibitors may act on the F1-ATPase by a similar mechanism.
Figure 5. Chemical structures of aurovertin B (A) and resveratrol/piceatannol (B).

Boxed areas indicate the structurally similar features of the two molecules. Aurovertin B comprises a (left-hand side) substituted pyrone ring linked by a rigid spacer containing conjugated double bonds to a (right-hand side) substituted dioxadicyclo[3.2.1]octane (or aglycone) ring. Resveratrol and piceatannol consist of two phenolic rings linked by a rigid spacer containing a double bond.
The Bacillus PS3 α3β3γ subcomplex remains uninhibited in the presence of the polyphenolic phytochemicals resveratrol and piceatannol. Previously, it has been shown that Bacillus PS3 is naturally insensitive to aurovertin and does not bind the antibiotic [45,46]. In the structure of bovine F1-ATPase complexed with aurovertin B [19], β-subunit(Arg-412) makes an important hydrogen bonding interaction with the carbonyl group on the substituted pyrone ring of aurovertin B. In addition, β-subunit(Tyr-458) forms a crucial staggered stacking interaction with the pyrone ring. A similar structural element to the aurovertin B pyrone ring is also present in the polyphenolic phytochemical inhibitors. However, in Bacillus PS3, β-subunit(Arg-412) is replaced by a phenylalanine residue and β-subunit(Tyr-458) is also changed to an arginine residue. Therefore, in Bacillus PS3, this hydrogen bonding interaction and stacking interaction with the pyrone of aurovertin B or the phenolic ring of the phytochemical inhibitors cannot form. Thus the insensitivity of Bacillus PS3 to polyphenolic phytochemicals observed in the present study is entirely consistent with them acting at the aurovertin B-binding site (or sites). Previously, on the basis of photochemical labelling studies, it has been argued that dequalinium might mimic aurovertin B when it inhibits the mitochondrial F1-ATPase [47].
Rhodamine 6G appears not to share a binding site with any of the other inhibitors. We suggest, on the basis of the mutual hindrance observed between IF1 and rhodamine 6G, that the binding site for the non-peptidyl lipophilic cations is located at the C-terminal domain of the α-subunit close to the IF1-binding site. In agreement with this proposal is the previous observation that rhodamine 6G protects against photo-inactivation of F1-ATPase by dequalinium, which binds to the C-terminal α-helical bundle domains of both α- and β-subunits [24,47]. The C-terminal region of the βDP-subunit forms the major part of the IF1-binding site. However, IF1 also forms limited interactions with the adjacent C-terminal domain of the αDP-subunit [20] and in particular with Phe-403, which is derivatized during photo-inactivation by dequalinium [24]. Rhodamine 6G, being much smaller than the inhibitor protein, may consequently bind to this region of the α-subunit, and its binding would not exclude the binding of IF1 (and vice versa). Phe-406 is also derivatized by dequalinium [24]. The aromatic rings of these phenylalanine residues are close enough to allow an aromatic ring of rhodamine 6G and related cationic dyes to stack between them. The mutual hindrance observed for IF1 and rhodamine 6G binding can then be explained by the limited interactions the inhibitor protein forms with this region of the α-subunit. Previously, it had been suggested that there may be more than one binding site for rhodamine 6G [12]. Therefore, it is possible that it binds to the C-terminal region in more than one of the α-subunits.
Medical implications
It is becoming increasingly evident that the ATP synthase plays a crucial role in the pathophysiology of several human disorders, including ischaemia–reperfusion injury, tumour growth and progression and cholesterol homoeostasis. Moreover, inhibitors of mitochondrial and ectopic ATP synthase could form the basis of effective therapeutic strategies.
Numerous studies have been conducted into the pathophysiological significance of IF1 and the ATP synthase, primarily in the context of myocardial ischaemia and tumour growth [32]. When a cell is deprived of oxygen, for example under conditions of ischaemia, the PMF across the inner mitochondrial membrane collapses, and cellular ATP is provided by glycolysis, leading to a drop in the pH of both the cytosol and the mitochondrial matrix. The reduction in pH activates IF1, which binds to the catalytic domain of ATP synthase and prevents futile hydrolysis of ATP [31]. Ischaemic conditions may also be found in tumour cells, where oxygen levels vary from the perivascular regions to the anoxic necrotic centres. It has been suggested that ATPase activity of the ATP synthase might be more tightly regulated by IF1 in tumour cells [32].
The ectopic localization of catalytically active ATP synthase has been reported on human endothelial cells [48] and hepatocytes [49], where ATP metabolism might be linked to the promotion of angiogenesis and cholesterol homoeostasis respectively. This immediately suggests a novel therapeutic use for inhibitors of the ATP synthase for the control of cholesterol levels and the inhibition of tumour growth – fundamental issues in cardiovascular and cancer research respectively.
Dietary polyphenolic phytochemicals interact with numerous biological processes and have been shown to exhibit a range of medically beneficial effects. For example, the major biological activities of resveratrol and piceatannol include antioxidant and free-radical scavenging properties, cardiovascular protective activity, anti-osteoporotic and anti-inflammatory activities, anti-cancer activity, anti-bacterial and fungicidal activities and oestrogenic activity [50–53]. These and other polyphenolic phytochemicals may exert their many effects through inhibition of ATP synthase, both on the plasma membrane, acting as anti-angiogenic factors, and in the mitochondrion, disrupting energy production [10,11]. Disruption of mitochondrial function is implicated in many medically important cellular processes [54].
Of particular interest are the differential effects of the inhibitors studied here on the mitochondrial and bacterial F1-ATPases. These differential effects suggest the binding sites might be quite different in bacterial enzymes. Dequalinium, and possibly other non-peptidyl lipophilic cations, are thought to interact at different sites on the bovine and Bacillus F1-ATPases [55]. Through systematic investigation we have been able to show that selective inhibition of the ATP synthase from one species over another is possible. The identification of inhibitors capable of acting selectively on bacterial ATPases immediately suggests a novel avenue for the development of antibiotics. Also, dequalinium appears to exhibit preference for the Bacillus PS3 subcomplex over the bovine F1-ATPase. Dequalinium is a well-known antimicrobial and has been used for over 40 years in over-the-counter mouthwashes, lozenges, ointments and creams. In addition, dequalinium has been reported to mimic the anti-carcinoma activity of rhodamine 123 [56]. Rhodamine 6G and other non-peptidyl lipophilic cations protect the F1-ATPase activity against photo-inactivation by dequalinium and many are structurally similar to rhodamine 123 [12]. Therefore, it is likely that rhodamine 6G and other non-peptidyl lipophilic cations may also exhibit anticarcinoma activity.
Conclusions
We have identified the binding sites for a number of inhibitory molecules that were previously uncharacterized. To date, the number and nature of inhibitory sites present in the mitochondrial F1-ATPase have not been systematically studied. The amphiphilic α-helical peptides described here represent novel inhibitors that mimic the natural inhibitor protein. In addition, we have shown that polyphenolic phytochemical inhibitors act at the binding site (or sites) for the antibiotic aurovertin B. Non-peptidyl lipophilic cations, on the other hand, act at a distinct site yet to be characterized. Therefore, there are at least five distinct sites for inhibition. Each of the above inhibitors also exhibited significantly different activity against the bacterial Bacillus PS3 α3β3γ subcomplex compared with that observed with bovine F1-ATPase. IF1 does not inhibit the bacterial enzyme, even in the absence of the ε-subunit. Thus bacterial enzymes are unable to bind IF1, despite high sequence and structural similarity. It is becoming increasingly apparent that ATP synthase inhibitors may be of therapeutic value.
References
- 1.Walker J. E. ATP synthesis by rotary catalysis (Nobel Lecture) Angew. Chem. Int. Edn. Engl. 1998;37:2309–2319. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2308::AID-ANIE2308>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 2.Walker J. E., Cozens A. L., Dyer M. R., Fearnley I. M., Powell S. J., Runswick M. J. Studies of the genes for ATP synthases in eubacteria, chloroplasts and mitochondria – implications for structure and function of the enzyme. Chem. Scripta. 1987;27B:97–105. [Google Scholar]
- 3.Walker J. E., Fearnley I. M., Lutter R., Todd R. J., Runswick M. J. Structural aspects of proton-pumping ATPases. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1990;326:367–378. doi: 10.1098/rstb.1990.0018. [DOI] [PubMed] [Google Scholar]
- 4.Boyer P. D. The binding change mechanism for ATP synthase – some probabilities and possibilities. Biochim. Biophys. Acta. 1993;1140:215–250. doi: 10.1016/0005-2728(93)90063-l. [DOI] [PubMed] [Google Scholar]
- 5.Boyer P. D. The ATP synthase – a splendid molecular machine. Annu. Rev. Biochem. 1997;66:717–749. doi: 10.1146/annurev.biochem.66.1.717. [DOI] [PubMed] [Google Scholar]
- 6.Pullman M. E., Monroy G. C. A naturally occurring inhibitor of mitochondrial adenosine triphosphatase. J. Biol. Chem. 1963;238:3762–3769. [PubMed] [Google Scholar]
- 7.Lardy H., Reed P., Lin C. H. Antibiotic inhibitors of mitochondrial ATP synthesis. Fed. Proc. 1975;34:1707–1710. [PubMed] [Google Scholar]
- 8.Connelly J. L., Lardy H. A. Antibiotics as tools for metabolic studies. III. Effects of oligomycin and aurovertin on the swelling and contraction processes of mitochondria. Biochemistry. 1964;19:1969–1973. doi: 10.1021/bi00900a031. [DOI] [PubMed] [Google Scholar]
- 9.Lardy H. A., Connelly J. L., Johnson D. Antibiotic studies. II. Inhibition of phosphoryl transfer in mitochondria by oligomycin and aurovertin. Biochemistry. 1964;19:1961–1968. doi: 10.1021/bi00900a030. [DOI] [PubMed] [Google Scholar]
- 10.Zheng J., Ramirez V. D. Inhibition of mitochondrial proton F0F1-ATPase/ATP synthase by polyphenolic phytochemicals. Br. J. Pharmacol. 2000;130:1115–1123. doi: 10.1038/sj.bjp.0703397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng J., Ramirez V. D. Piceatannol, a stilbene phytochemical, inhibits mitochondrial F0F1-ATPase activity by targeting the F1 complex. Biochem. Biophys. Res. Commun. 1999;261:499–503. doi: 10.1006/bbrc.1999.1063. [DOI] [PubMed] [Google Scholar]
- 12.Bullough D. A., Ceccarelli E. A., Roise D., Allison W. S. Inhibition of the bovine-heart mitochondrial F1-ATPase by cationic dyes and amphipathic peptides. Biochim. Biophys. Acta. 1989;975:377–383. doi: 10.1016/s0005-2728(89)80346-9. [DOI] [PubMed] [Google Scholar]
- 13.Abrahams J. P., Leslie A. G., Lutter R., Walker J. E. Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature (London) 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 14.Menz R. I., Walker J. E., Leslie A. G. Structure of bovine mitochondrial F1-ATPase with nucleotide bound to all three catalytic sites: implications for the mechanism of rotary catalysis. Cell. 2001;106:331–341. doi: 10.1016/s0092-8674(01)00452-4. [DOI] [PubMed] [Google Scholar]
- 15.Kagawa R., Montgomery M. G., Braig K., Leslie A. G., Walker J. E. The structure of bovine F1-ATPase inhibited by ADP and beryllium fluoride. EMBO J. 2004;23:2734–2744. doi: 10.1038/sj.emboj.7600293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orriss G. L., Leslie A. G., Braig K., Walker J. E. Bovine F1-ATPase covalently inhibited with 4-chloro-7-nitrobenzofurazan: the structure provides further support for a rotary catalytic mechanism. Structure. 1998;6:831–837. doi: 10.1016/s0969-2126(98)00085-9. [DOI] [PubMed] [Google Scholar]
- 17.Gibbons C., Montgomery M. G., Leslie A. G., Walker J. E. The structure of the central stalk in bovine F1-ATPase at 2.4 Å resolution. Nat. Struct. Biol. 2000;7:1055–1061. doi: 10.1038/80981. [DOI] [PubMed] [Google Scholar]
- 18.Abrahams J. P., Buchanan S. K., van Raaij M. J., Fearnley I. M., Leslie A. G., Walker J. E. The structure of bovine F1-ATPase complexed with the peptide antibiotic efrapeptin. Proc. Natl. Acad. Sci. U.S.A. 1996;93:9420–9424. doi: 10.1073/pnas.93.18.9420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Raaij M. J., Abrahams J. P., Leslie A. G., Walker J. E. The structure of bovine F1-ATPase complexed with the antibiotic inhibitor aurovertin B. Proc. Natl. Acad. Sci. U.S.A. 1996;93:6913–6917. doi: 10.1073/pnas.93.14.6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cabezon E., Montgomery M. G., Leslie A. G., Walker J. E. The structure of bovine F1-ATPase in complex with its regulatory protein IF1. Nat. Struct. Biol. 2003;10:744–750. doi: 10.1038/nsb966. [DOI] [PubMed] [Google Scholar]
- 21.Terwilliger T. C., Eisenberg D. The structure of melittin. II. Interpretation of the structure. J. Biol. Chem. 1982;257:6016–6022. [PubMed] [Google Scholar]
- 22.Roise D., Horvath S. J., Tomich J. M., Richards J. H., Schatz G. A chemically synthesized pre-sequence of an imported mitochondrial protein can form an amphiphilic helix and perturb natural and artificial phospholipid bilayers. EMBO J. 1986;5:1327–1334. doi: 10.1002/j.1460-2075.1986.tb04363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roise D., Theiler F., Horvath S. J., Tomich J. M., Richards J. H., Allison D. S., Schatz G. Amphiphilicity is essential for mitochondrial presequence function. EMBO J. 1988;7:649–653. doi: 10.1002/j.1460-2075.1988.tb02859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allison W. S., Jault J. M., Zhuo S., Paik S. R. Functional sites in F1-ATPases: location and interactions. J. Bioenerg. Biomembr. 1992;24:469–477. doi: 10.1007/BF00762364. [DOI] [PubMed] [Google Scholar]
- 25.Cabezon E., Butler P. J., Runswick M. J., Walker J. E. Modulation of the oligomerization state of the bovine F1-ATPase inhibitor protein, IF1, by pH. J. Biol. Chem. 2000;275:25460–25464. doi: 10.1074/jbc.M003859200. [DOI] [PubMed] [Google Scholar]
- 26.Martinez-Irujo J. J., Villahermosa M. L., Alberdi E., Santiago E. A checkerboard method to evaluate interactions between drugs. Biochem. Pharmacol. 1996;51:635–644. doi: 10.1016/s0006-2952(95)02230-9. [DOI] [PubMed] [Google Scholar]
- 27.Dixon M. The determination of enzyme inhibitor constants. Biochem. J. 1953;55:170–171. doi: 10.1042/bj0550170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cornish-Bowden A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors. Biochem. J. 1974;137:143–144. doi: 10.1042/bj1370143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez-Irujo J. J., Villahermosa M. L., Mercapide J., Cabodevilla J. F., Santiago E. Analysis of the combined effect of two linear inhibitors on a single enzyme. Biochem. J. 1998;329:689–698. doi: 10.1042/bj3290689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yonetani T., Theorell H. Studies on liver alcohol hydrogenase complexes. III. Multiple inhibition kinetics in the presence of two competitive inhibitors. Arch. Biochem. Biophys. 1964;106:243–251. doi: 10.1016/0003-9861(64)90184-5. [DOI] [PubMed] [Google Scholar]
- 31.Walker J. E. The regulation of catalysis in ATP synthase. Curr. Opin. Struct. Biol. 1994;4:912–918. doi: 10.1016/0959-440x(94)90274-7. [DOI] [PubMed] [Google Scholar]
- 32.Green D. W., Grover G. J. The IF1 inhibitor protein of the mitochondrial F1F0-ATPase. Biochim. Biophys. Acta. 2000;1458:343–355. doi: 10.1016/s0005-2728(00)00085-2. [DOI] [PubMed] [Google Scholar]
- 33.Gordon-Smith D. J., Carbajo R. J., Yang J. C., Videler H., Runswick M. J., Walker J. E., Neuhaus D. Solution structure of a C-terminal coiled-coil domain from bovine IF1: the inhibitor protein of F1 ATPase. J. Mol. Biol. 2001;308:325–339. doi: 10.1006/jmbi.2001.4570. [DOI] [PubMed] [Google Scholar]
- 34.Cabezon E., Runswick M. J., Leslie A. G., Walker J. E. The structure of bovine IF1, the regulatory subunit of mitochondrial F-ATPase. EMBO J. 2001;20:6990–6996. doi: 10.1093/emboj/20.24.6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Raaij M. J., Orriss G. L., Montgomery M. G., Runswick M. J., Fearnley I. M., Skehel J. M., Walker J. E. The ATPase inhibitor protein from bovine heart mitochondria: the minimal inhibitory sequence. Biochemistry. 1996;35:15618–15625. doi: 10.1021/bi960628f. [DOI] [PubMed] [Google Scholar]
- 36.Cabezon E., Arechaga I., Jonathan P., Butler G., Walker J. E. Dimerization of bovine F1-ATPase by binding the inhibitor protein, IF1. J. Biol. Chem. 2000;275:28353–28355. doi: 10.1074/jbc.C000427200. [DOI] [PubMed] [Google Scholar]
- 37.Cabezon E., Butler P. J., Runswick M. J., Carbajo R. J., Walker J. E. Homologous and heterologous inhibitory effects of ATPase inhibitor proteins on F-ATPases. J. Biol. Chem. 2002;277:41334–41341. doi: 10.1074/jbc.M207169200. [DOI] [PubMed] [Google Scholar]
- 38.Klionsky D. J., Brusilow W. S., Simoni R. D. In vivo evidence for the role of the ε-subunit as an inhibitor of the proton-translocating ATPase of Escherichia coli. J. Bacteriol. 1984;160:1055–1060. doi: 10.1128/jb.160.3.1055-1060.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsunoda S. P., Rodgers A. J., Aggeler R., Wilce M. C., Yoshida M., Capaldi R. A. Large conformational changes of the ε-subunit in the bacterial F1F0 ATP synthase provide a ratchet action to regulate this rotary motor enzyme. Proc. Natl. Acad. Sci. U.S.A. 2001;98:6560–6564. doi: 10.1073/pnas.111128098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suzuki T., Murakami T., Iino R., Suzuki J., Ono S., Shirakihara Y., Yoshida M. F0F1-ATPase/synthase is geared to the synthesis mode by conformational rearrangement of ε-subunit in response to proton motive force and ADP/ATP balance. J. Biol. Chem. 2003;278:46840–46846. doi: 10.1074/jbc.M307165200. [DOI] [PubMed] [Google Scholar]
- 41.Shirakihara Y., Leslie A. G., Abrahams J. P., Walker J. E., Ueda T., Sekimoto Y., Kambara M., Saika K., Kagawa Y., Yoshida M. The crystal structure of the nucleotide-free α3β3 subcomplex of F1-ATPase from the thermophilic Bacillus PS3 is a symmetric trimer. Structure. 1997;5:825–836. doi: 10.1016/s0969-2126(97)00236-0. [DOI] [PubMed] [Google Scholar]
- 42.Maulet Y., Cox J. A. Structural changes in melittin and calmodulin upon complex formation and their modulation by calcium. Biochemistry. 1983;22:5680–5686. doi: 10.1021/bi00293a035. [DOI] [PubMed] [Google Scholar]
- 43.Malencik D. A., Anderson S. R. Effects of calmodulin and related proteins on the hemolytic activity of melittin. Biochem. Biophys. Res. Commun. 1985;130:22–29. doi: 10.1016/0006-291x(85)90376-6. [DOI] [PubMed] [Google Scholar]
- 44.Pedersen P. L., Hullihen J. Inhibitor peptide of mitochondrial proton adenosine triphosphatase. Neutralization of its inhibitory action by calmodulin. J. Biol. Chem. 1984;259:15148–15153. [PubMed] [Google Scholar]
- 45.Saishu T., Kagawa Y., Shimizu R. Resistance of thermophilic ATPase (TF1) to specific F1-ATPase inhibitors including local anesthetics. Biochem. Biophys. Res. Commun. 1983;112:822–826. doi: 10.1016/0006-291x(83)91691-1. [DOI] [PubMed] [Google Scholar]
- 46.Douglas M. G., Koh Y., Dockter M. E., Schatz G. Aurovertin binds to the β-subunit of yeast mitochondrial ATPase. J. Biol. Chem. 1977;252:8333–8335. [PubMed] [Google Scholar]
- 47.Grodsky N. B., Allison W. S. The adenine pocket of a single catalytic site is derivatized when the bovine heart mitochondrial F1-ATPase is photoinactivated with 4-amino-1-octylquinaldinium. Cell Biochem. Biophys. 1999;31:285–294. doi: 10.1007/BF02738243. [DOI] [PubMed] [Google Scholar]
- 48.Moser T. L., Kenan D. J., Ashley T. A., Roy J. A., Goodman M. D., Misra U. K., Cheek D. J., Pizzo S. V. Endothelial cell surface F1–F0 ATP synthase is active in ATP synthesis and is inhibited by angiostatin. Proc. Natl. Acad. Sci. U.S.A. 2001;98:6656–6661. doi: 10.1073/pnas.131067798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martinez L. O., Jacquet S., Esteve J. P., Rolland C., Cabezon E., Champagne E., Pineau T., Georgeaud V., Walker J. E., Terce F., et al. Ectopic β-chain of ATP synthase is an apolipoprotein A-I receptor in hepatic HDL endocytosis. Nature (London) 2003;421:75–79. doi: 10.1038/nature01250. [DOI] [PubMed] [Google Scholar]
- 50.Setchell K. D. Phytoestrogens: the biochemistry, physiology, and implications for human health of soy isoflavones. Am. J. Clin. Nutr. 1998;68:1333S–1346S. doi: 10.1093/ajcn/68.6.1333S. [DOI] [PubMed] [Google Scholar]
- 51.Tham D. M., Gardner C. D., Haskell W. L. Clinical review 97: Potential health benefits of dietary phytoestrogens: a review of the clinical, epidemiological, and mechanistic evidence. J. Clin. Endocrinol. Metab. 1998;83:2223–2235. doi: 10.1210/jcem.83.7.4752. [DOI] [PubMed] [Google Scholar]
- 52.Fremont L. Biological effects of resveratrol. Life Sci. 2000;66:663–673. doi: 10.1016/s0024-3205(99)00410-5. [DOI] [PubMed] [Google Scholar]
- 53.Surh Y. J. Cancer chemoprevention with dietary phytochemicals. Nat. Rev. Cancer. 2003;3:768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- 54.Wallace D. C. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 55.Paik S. R., Jault J. M., Allison W. S. Inhibition and inactivation of the F1 adenosinetriphosphatase from Bacillus PS3 by dequalinium and activation of the enzyme by lauryl dimethylamine oxide. Biochemistry. 1994;33:126–133. doi: 10.1021/bi00167a016. [DOI] [PubMed] [Google Scholar]
- 56.Weiss M. J., Wong J. R., Ha C. S., Bleday R., Salem R. R., Steele G. D., Jr, Chen L. B. Dequalinium, a topical antimicrobial agent, displays anticarcinoma activity based on selective mitochondrial accumulation. Proc. Natl. Acad. Sci. U.S.A. 1987;84:5444–5448. doi: 10.1073/pnas.84.15.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]