

Abstract
Spinobulbar muscular atrophy (SBMA) is an X-linked neuromuscular disease caused by polyglutamine (polyQ) expansion in the androgen receptor (AR) gene. SBMA belongs to the family of polyQ diseases, which are fatal neurodegenerative disorders mainly caused by protein-mediated toxic gain-of-function mechanisms and characterized by deposition of misfolded proteins in the form of aggregates. The neurotoxicity of the polyQ proteins can be modified by phosphorylation at specific sites, thereby providing the rationale for the development of disease-specific treatments. We sought to identify signaling pathways that modulate polyQ-AR phosphorylation for therapy development. We report that cyclin-dependent kinase 2 (CDK2) phosphorylates polyQ-AR specifically at Ser96. Phosphorylation of polyQ-AR by CDK2 increased protein stabilization and toxicity and is negatively regulated by the adenylyl cyclase (AC)/protein kinase A (PKA) signaling pathway. To translate these findings into therapy, we developed an analog of pituitary adenylyl cyclase activating polypeptide (PACAP), a potent activator of the AC/PKA pathway. Chronic intranasal administration of the PACAP analog to knock-in SBMA mice reduced Ser96 phosphorylation, promoted polyQ-AR degradation, and ameliorated disease outcome. These results provide proof of principle that noninvasive therapy based on the use of PACAP analogs is a therapeutic option for SBMA.
INTRODUCTION
The X-linked neuromuscular disease spinobulbar muscular atrophy (SBMA) is caused by polyglutamine (polyQ) expansion in the androgen receptor (AR) gene. Expansions over 38 glutamine-encoding CAG trinucleotide tandem repeats in AR give rise to the polyQ-AR protein (1). SBMA is characterized by the selective degeneration of lower motor neurons and skeletal muscle atrophy (2) and belongs to the polyQ subfamily of neurodegenerative diseases, which also includes Huntington’s disease (HD), dentatorubral-pallidoluysian atrophy, and six types of spinocerebellar ataxia (SCA) (3). PolyQ diseases are characterized by progressive dysfunction and death of specific neuronal populations in the central nervous system (CNS) and accumulation of toxic aggregation-prone proteins in the form of inclusions and microaggregates. PolyQ diseases are inherited in an autosomal dominant fashion, and neurodegeneration occurs mainly through toxic gain-of-function mechanisms. Several strategies have been developed to decrease the accumulation and toxicity of polyQ proteins, from inhibiting mutant gene transcription (4–6) to targeting the disease protein for degradation (7, 8). Nonetheless, at the present time, there is no effective therapy to arrest or delay the onset and progression of these fatal diseases.
SBMA is the only polyQ disease with X-linked transmission and sex specificity. SBMA primarily manifests in males because they have high serum levels of androgens. Although therapeutic approaches to reduce androgen levels in the serum have been pursued (9, 10), several undesired side effects limit the viability of this approach. The binding of polyQ-AR to its natural ligands—testosterone and its more potent derivative dihydrotestosterone (DHT)—is necessary and sufficient to convert the mutant protein into a neurotoxic species (11). Posttranslational modifications such as phosphorylation regulate important aspects of AR biology and modify the toxicity of polyQ-AR (7, 8, 12), thereby offering opportunities for therapy development. AR is a highly phosphorylated protein (13–15), and AR phosphorylation sites are mostly serine/threonine residues followed by one proline residue at position +1 [(S/T)P], all of which are consensus sites for phosphorylation by cyclin-dependent kinases (CDKs), a family of cell cycle checkpoint enzymes (16). Here, we developed a strategy to reduce polyQ-AR accumulation and toxicity in degenerating cells by modulating the AR phosphorylation state.
RESULTS
Accumulation of phosphorylated AR in neuronal cells is decreased by the AC/PKA pathway
In proliferating cells, such as the human embryonic kidney (HEK) 293T cell line, both nonexpanded AR and polyQ-AR undergo extensive phosphorylation, resulting in the generation of a doublet by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. 1A) (13, 14). The upper isoform of both nonexpanded AR and polyQ-AR represented about 55 to 60% of total AR, indicating that polyQ expansion does not modify the accumulation of this isoform. Binding of DHT stabilizes the AR (7). DHT treatment increased by about 1.2-fold accumulation of the upper AR isoform, although this increase was not statistically significant. In vitro dephosphorylation by incubation of protein lysates with λ phosphatase resulted in complete loss of the upper isoform, indicating that it is composed of phosphorylated AR (Fig. 1B). The AR phosphorylation state results from androgen signaling acting in concert with several cellular pathways, such as the adenylyl cyclase (AC)/protein kinase A (PKA) signaling pathway (17), which was previously reported to reduce non-expanded AR phosphorylation (18). Overexpression of either wild-type PKA (wtPKA) or constitutively active PKA (caPKA), but not dominant-negative PKA (dnPKA), as well as treatment of the cells with forskolin to activate endogenous AC decreased the accumulation of the upper isoform of polyQ-AR (Fig. 1, C and D, and fig. S1). Upper-isoform accumulation was increased to more than 70% of the total polyQ-AR in primary rat cortical neurons and mouse motor neuron–derived MN-1 cells compared to nonneuronal cells and was decreased by forskolin treatment (Fig. 1, E and F) (19). Motivated by these observations, we sought to elucidate the underlying molecular mechanisms and the biological relevance of this phosphorylation event in SBMA pathogenesis.
Fig. 1. Accumulation of a phosphorylated isoform of AR is increased in neuronal cells and modified by the AC/PKA pathway.
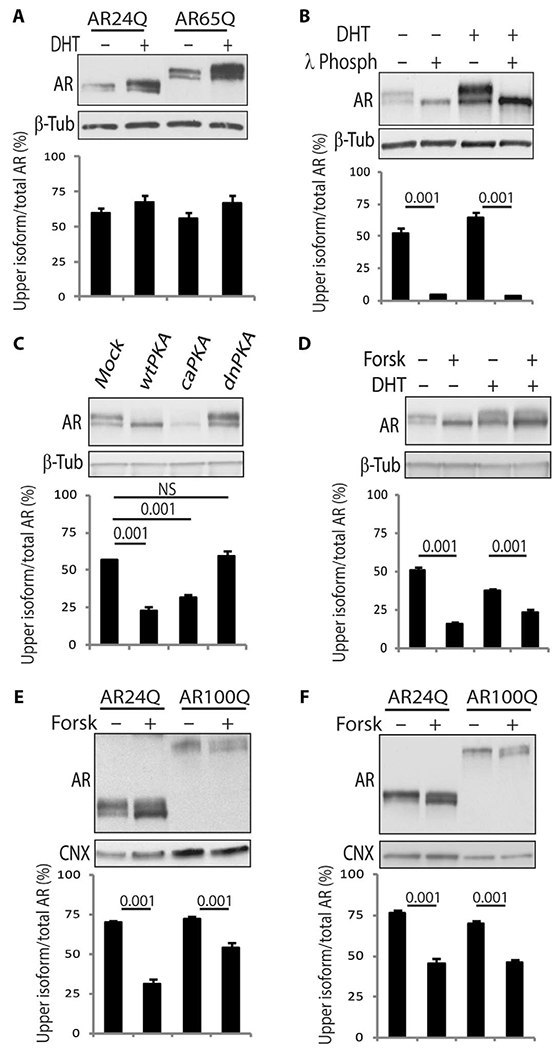
(A and B) Western blotting analysis of nonexpanded AR and polyQ-AR in HEK293T cells treated with vehicle and DHT [10 nM; 24 hours (A) and 5 hours (B)]. In (B), cell extracts were incubated with λ phosphatase (λ Phosph). n = 3 to 5 (A) and 2 to 3 (B) independent experiments. (C and D) Western blotting analysis in HEK293T cells expressing AR55Q and transfected with empty vector (mock), or vectors expressing wtPKA, caPKA, and dnPKA, or treated with 10 nM DHT and 10 μM forskolin (Forsk) for 5 hours. n = 3 independent experiments. NS, not significant. (E and F) Western blotting analysis in MN-1 cells (E) and rat primary cortical neurons (F) expressing AR24Q and AR100Q and treated with vehicle and 10 μM forskolin (5 hours). n = 3 (E) and 4 (F) independent experiments. AR was detected with a specific antibody, and β-tubulin (β-Tub) and calnexin (CNX) were used as loading controls. Graphs, means ± SEM; two-way analysis of variance (ANOVA) (A, B, E, and F) and one-way ANOVA (C and D) were used.
Reduction of AR phosphorylation by the PACAP/AC/PKA pathway is protective in SBMA
Next, we sought to determine whether activation of the AC/PKA pathway modifies polyQ-AR toxicity. Forskolin treatment and overexpression of wtPKA were associated with a significant decrease in activity of the apoptosis mediator caspase 3 in SBMA MN-1 cells (Fig. 2, A to C). Conversely, the inhibition of endogenous PKA by the PKA inhibitor (PKI) fragment (6–22) amide had the opposite effect (Fig. 2B). Pituitary adenylyl cyclase activating polypeptide (PACAP) is a potent activator of the AC/PKA pathway with strong neuroprotective properties, whereas vasoactive intestinal peptide (VIP) belongs to the same family of neuropeptides but has a reduced ability to activate AC (20, 21). Using a bioluminescence resonance energy transfer (BRET) cyclic adenosine monophosphate (cAMP) biosensor (22), we found that PACAP, but not VIP, induced cAMP release in SBMA MN-1 cells similarly to forskolin, and the effect was dose-dependent (Fig. 2, D and E, and fig. S2A and B). Furthermore, PACAP, and not VIP, reduced the accumulation of the upper isoform of polyQ-AR by 35% (Fig. 2F and fig. S3) and significantly reduced caspase 3 activity and cell death through activation of PKA in SBMA MN-1 cells (Fig. 2, G to I, and fig. S4). These data show that the PACAP/AC/PKA pathway reduces polyQ-AR phosphorylation and toxicity and imply that PACAP is a good candidate for SBMA treatment.
Fig. 2. PACAP/AC/PKA signaling protects SBMA cells from the toxicity of polyQ-AR.
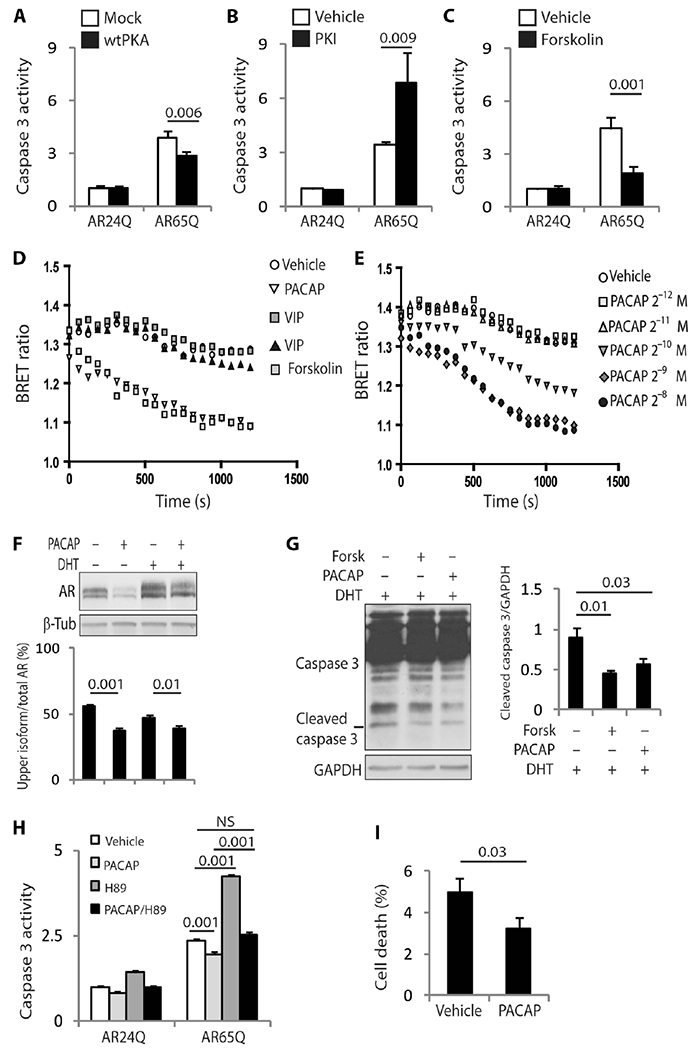
(A to C) Caspase 3 assays in MN-1 cells stably expressing AR24Q and AR65Q and transfected with either empty vector or vector expressing wtPKA, or treated with 10 μM PKI and 10 μM forskolin for 48 hours. n = 6 to 8 (A), 8 (B), and 9 (C) independent experiments. (D and E) BRET assays in MN-1 cells expressing AR65Q treated with 2.5 × 10−6 M forskolin, 2 × 10−8MVIP(gray), 2 × 10−7 MVIP (black), and 2 × 10−9 MPACAP, or different concentrations of PACAP for 20 min. n = 4 independent experiments. (F) Western blotting analysis of AR55Q in HEK293T cells expressing AR55Q treated with 10 nM DHT and 100 nM PACAP for 5 hours. n = 9 independent experiments. (G) Western blotting analysis of caspase 3 cleavage in MN-1 cells stably expressing AR100Q treated with 10 nM DHT, 10 μM forskolin, and 100 nM PACAP for 72 hours. n = 3 independent experiments. (H) Caspase 3 assay in MN-1 cells expressing AR24Q and AR65Q treated with 10 μM H89, and 100 nM PACAP for 48 hours. n = 3 independent experiments. (I) Trypan blue assay in MN-1 cells expressing AR100Q treated with either vehicle or 100 nM PACAP for 96 hours. n = 8 independent experiments. AR and caspase 3 were detected with specific antibodies, and β-tubulin and GAPDH were used as loading controls. Graphs, means ± SEM; two-way ANOVA (A to C, and H), one-way ANOVA (F and G), and Student’s t test (I) were used.
CDK2 phosphorylates AR at Ser96
To gain insights into the mechanism that regulates AR phosphorylation, we initially undertook an unbiased approach and treated HEK293T cells expressing AR55Q with inhibitors that target several protein phosphatases and kinases (fig. S5). Roscovitine, which mainly inhibits CDK1, CDK2, CDK5, and CDK7, specifically decreased the accumulation of the upper isoform of polyQ-AR. Next, we treated the cells with inhibitors that target both CDK1 and CDK5, which had been previously shown to phosphorylate nonexpanded AR (23, 24), and CDK2, which had not been previously linked to AR biology. Manipulation of CDK1 and CDK5 activity had no effect on the accumulation of AR isoforms (fig. S6, A to E). A CDK1/CDK2 inhibitor and overexpression of dnCDK2 reduced the accumulation of the upper AR isoform by 38 and 24%, respectively, whereas the overexpression of wtCDK2 increased it by 1.32-fold and blocked the effect of forskolin and PACAP (Fig. 3, A to C). By immunoprecipitation assay, polyQ-AR was shown to form a complex with endogenous CDK2 (Fig. 3D). CDK2 works in complex with either cyclin A or cyclin E (16). In an in vitro phosphorylation assay performed in HEK293 cells expressing AR55Q, we established that the CDK2/cyclin E complex phosphorylates polyQ-AR (Fig. 3E). CDK2 was expressed and colocalized with nonexpanded AR and polyQ-AR in SBMA MN-1 and doxycycline-inducible rat PC12 cells expressing AR112Q, brainstem and spinal cord motor neurons and skeletal muscle fibers from wild-type and knock-in SBMA mice in which exon 1 of the endogenous AR gene was replaced with human AR exon 1 with 113 glutamine residues (AR113Q), SBMA patient-derived induced pluripotent stem cells differentiated to motor neurons and neural progenitor cells (NPCs), and the spinal cord motor neurons of an SBMA patient (Fig. 3F and fig. S7 to S12) (19). Notably, the overexpression of CDK2 significantly exacerbated DHT-induced cell death in SBMA PC12 cells (Fig. 3G). Okadaic acid (OA; a well-characterized inhibitor of serine/theorine protein phosphatases) as well as PACAP and forskolin induced the expression of the CDK2/cyclin E complex inhibitor p21Cip1 (fig. S13, A to D), increased the binding of CDK2 to p21Cip1 in SBMA cells (fig. S13E), and decreased accumulation of the upper AR isoform, and their effect was blocked by p21Cip1 knockdown (Fig. 3, H to J, and fig. S13, B and C) (25). These results indicate that CDK2 phosphorylates polyQ-AR and suggest that p21Cip1-mediated inhibition of CDK2 is protective in SBMA.
Fig. 3. PolyQ-AR is a substrate of CDK2.
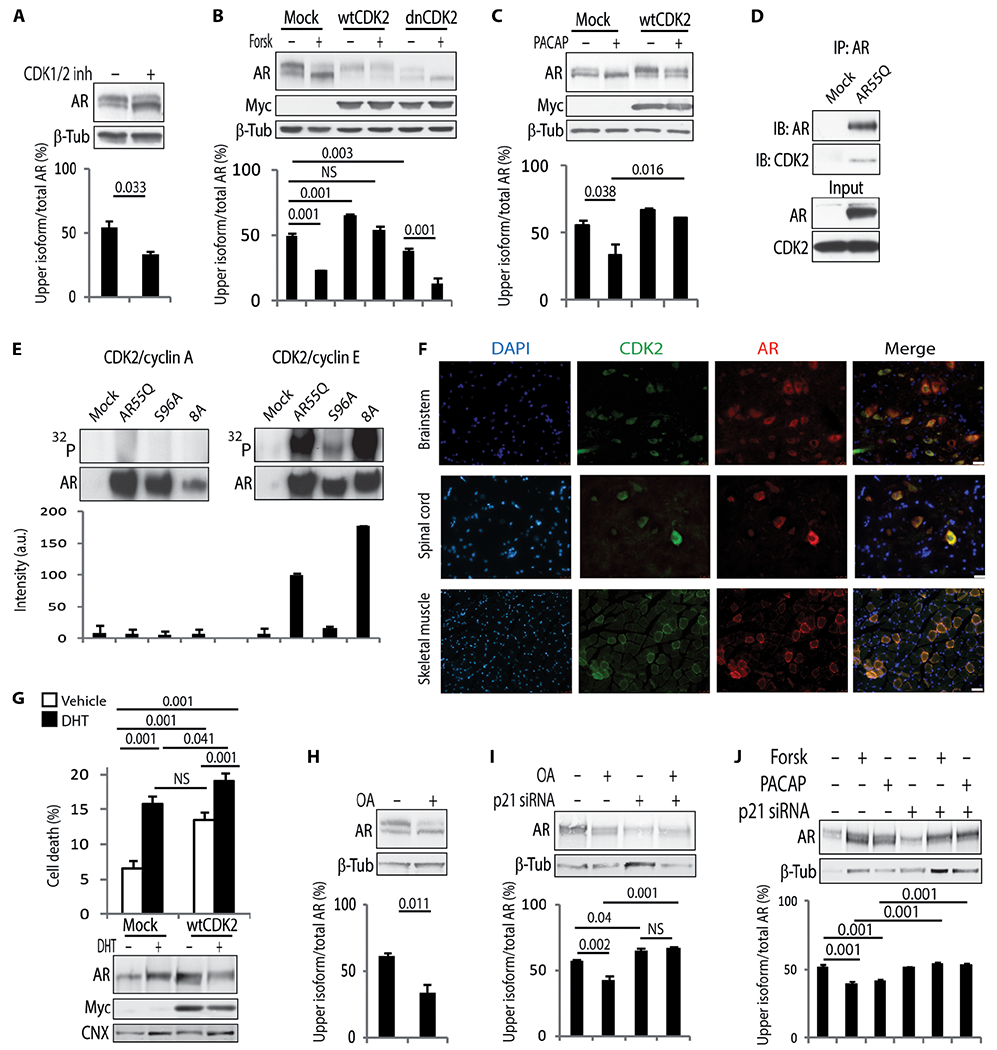
(A to C) Western blotting analysis of AR55Q in HEK293T cells either (A) treated with 30 μM CDK1/2 inhibitor (CDK1/2 inh) or (B and C) transfected with empty vector or vectors expressing Myc-tagged wtCDK2 and dnCDK2 and treated with 10 μM forskolin and 100 nM PACAP for 5 hours. n = 3 independent experiments. (D) Immunoprecipitation (IP) analysis in HEK293T cells transfected with empty vector or vector expressing AR55Q and immunoblotting (IB) analysis of AR and endogenous CDK2. Input, 10% of total protein extract. n = 3 independent experiments. (E) In vitro phosphorylation assay. AR55Q, AR55Q-S96A, and AR55Q-8A with all the (S/T)P sites substituted with alanine except for Ser96 (fig. S15A) were immunopurified from HEK293T cells and incubated with recombinant CDK2 and either cyclin A or cyclin E. Top: Autoradiography. Bottom: Western blotting analysis of AR levels. Graph, means ± SD. n = 2 independent experiments. a.u., arbitrary units. (F) Immunofluorescence analysis of polyQ-AR and CDK2 in tissues from 180-day-old AR113Q mice. Nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI). Scale bars, 25 μm (brainstem and spinal cord) and 100 μm (muscle). (G) Trypan blue assay in doxycycline-inducible PC12 cells stably expressing AR112Q, transfected with empty vector and vector expressing wtCDK2, and treated with doxycycline (10 μg/ml) and 50 μM DHT for 72 hours. Bottom: Western blotting analysis of AR and Myc-tagged wtCDK2 expression levels. n = 4 independent experiments. (H to J) Western blotting analysis of AR55Q in HEK293T cells treated with (H) 100 nM OA or (I and J) transfected with three small interfering RNAs (siRNAs) against p21Cip1 and treated with 5 nM OA, 10 μM forskolin, and 100 nM PACAP for 24 hours. n = 3 independent experiments. AR, Myc-tagged CDK2, and CDK2 were detected with specific antibodies, and β-tubulin and calnexin were used as loading control. Graphs, means ± SEM; Student’s t test (A and H), one-way ANOVA (B to C, and I and J), and two-way ANOVA (G) were used.
Next, we sought to identify the site(s) of AR that are modified by CDK2. AR has nine (S/T)P site motifs (Fig. 4A). By replacing these serine and threonine residues with phosphoresistant alanine, we found that Ser96-to-alanine (S96A) substitution was the only phosphorylation-resistant AR variant that abolished formation of the upper isoform of AR (Fig. 4B). Ser96 is highly conserved throughout evolution (fig. S14). Substrate phosphorylation by CDKs can occur at multiple sites in a coordinated and processive manner (26). Alanine substitution of at least two and up to all of the (S/T)P sites except Ser96 (mutant 8A) and of other sites relevant to SBMA did not alter the accumulation of the upper AR isoform (fig. S15, A and B) (7, 8, 12, 27). PACAP and forskolin modified the mobility shift of all the phosphoresistant AR variants at (S/T) P sites, except S96A (Fig. 4B and fig. S15A and B). In an in vitro phosphorylation assay, S96A, but not mutant 8A, prevented phosphorylation by CDK2 (Fig. 3E). To determine whether CDK2 phosphorylates Ser96 in vivo, we generated an antibody that recognizes phosphorylated Ser96, which, as expected, did not recognize polyQ-AR-S96A (Fig. 4, C and D). The overexpression of dnCDK2 and treatment with PACAP and forskolin significantly reduced the amount of polyQ-AR phosphorylated at Ser96. Together, these observations demonstrate that CDK2 specifically phosphorylates polyQ-AR at Ser96.
Fig. 4. CDK2 phosphorylates nonexpanded AR and polyQ-AR at Ser96.
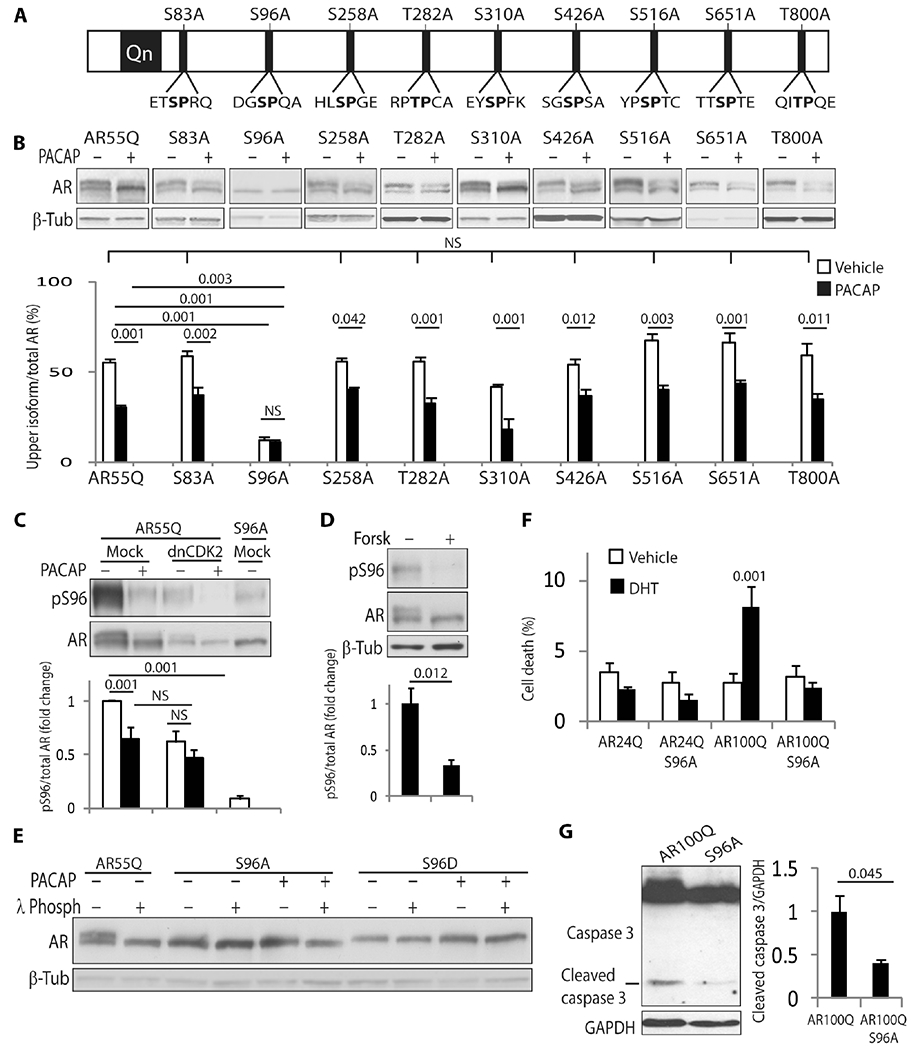
(A) Schematic representation of the (S/T)P sites in human AR (NM_000044). (B) Western blotting analysis of the indicated AR55Q phosphodefective alanine variants in HEK293T cells treated with 100 nM PACAP (5 hours). n = 3 independent experiments. (C and D) Western blotting analysis of Ser96-phosphorylated and total AR55Q and AR55Q-S96A in HEK293T cells treated with 10 μM forskolin and 100 nM PACAP for 5 hours. n = 9 to 10 (C) and 5 (D) independent experiments. (E) Western blotting analysis of AR55Q, AR55Q-S96A, and AR55Q-S96D in HEK293T cells treated with vehicle and 100 nM PACAP (5 hours) and incubated with λ phosphatase (λ Phosph), as indicated. n = 4 independent experiments. (F) Trypan blue assay in MN-1 cells stably expressing the indicated AR variants and treated with vehicle and 10 μM DHT (96 hours). Graph, means ± SEM. n = 4 to 8 independent experiments. (G) Western blotting analysis of caspase 3 cleavage in MN-1 cells stably expressing AR100Q and AR100Q-S96A. n = 3 independent experiments. Ser96-phosporylated and total AR were detected with specific antibodies, and β-tubulin and GAPDH were used as loading controls. Graphs, means ± SEM; one-way ANOVA (B), two-way ANOVA (C and F), and Student’s t test (D and G) were used.
To further elucidate the biological relevance of phosphorylation of polyQ-AR at Ser96, we generated a phosphomimetic Ser96-to-aspartate (S96D) AR variant. By SDS-PAGE, the S96A and S96D phosphomutants migrated as a single band that corresponded to the lower and upper AR isoforms, respectively, and their mobility was not affected by λ phosphatase, wtCDK2, dnCDK2, PACAP, or forskolin (Fig. 4E and fig. S16, A to C). Neither the gain nor the loss of Ser96 phosphorylation altered phosphorylation at sites close to the pathogenic polyQ tract (fig. S16D) or affected AR nuclear translocation induced by androgens (fig. S17, A and B). Rather, S96A substitution reduced cell death and caspase 3 cleavage (Fig. 4, F and G), suggesting that the inhibition of Ser96 phosphorylation is protective in SBMA.
Phosphorylation affects aggregation and turnover of polyQ-AR
A hallmark of SBMA and other neurodegenerative diseases is the deposition of microaggregates, which can be detected as high–molecular weight species that accumulate in the stacking gel by SDS-PAGE (8). PACAP and forskolin reduced polyQ-AR aggregation in HEK293T and PC12 cells (Fig. 5A and fig. S18, A and B). By atomic force microscopy, polyQ expansion shifts the deposition of AR from annular oligomers to fibrillar oligomers (28), an effect reversed by forskolin and S96A substitution (Fig. 5B). Reduced polyQ-AR aggregation might result from increased degradation (7, 8). Consistent with this hypothesis, both forskolin and overexpression of caPKA reduced the accumulation of polyQ-AR by 80%, whereas the overexpression of dnPKA increased it by twofold (Fig. 5C and fig. S19, A and B), without altering AR transcript levels (fig. S19C). The effect of PKA and OA was abolished by treatment of the cells with the ubiquitin-proteasome system (UPS) inhibitor, MG132, indicating that AC/PKA signaling promotes AR degradation via the UPS (Fig. 5D and fig. S20). We then asked whether phosphorylation of polyQ-AR affects protein stability. Eight hours after inhibition of protein synthesis, accumulation of the upper and lower isoforms was decreased by 52 and 71%, respectively, indicating that the lower isoform has a faster turnover than the upper isoform (Fig. 5C). Forskolin decreased the accumulation of the upper and lower isoforms by 85 and 75%, respectively. Similar results were obtained upon overexpression of caPKA and S96A substitution, whereas overexpression of dnPKA and S96D substitution increased the accumulation of both isoforms by twofold (fig. S19, B and D). These results indicate that Ser96 phosphorylation increases AR stability and activation of the AC/PKA pathway promotes the turnover of phosphorylated and total polyQ-AR.
Fig. 5. AC/PKA signaling decreases aggregation and promotes turnover of polyQ-AR.
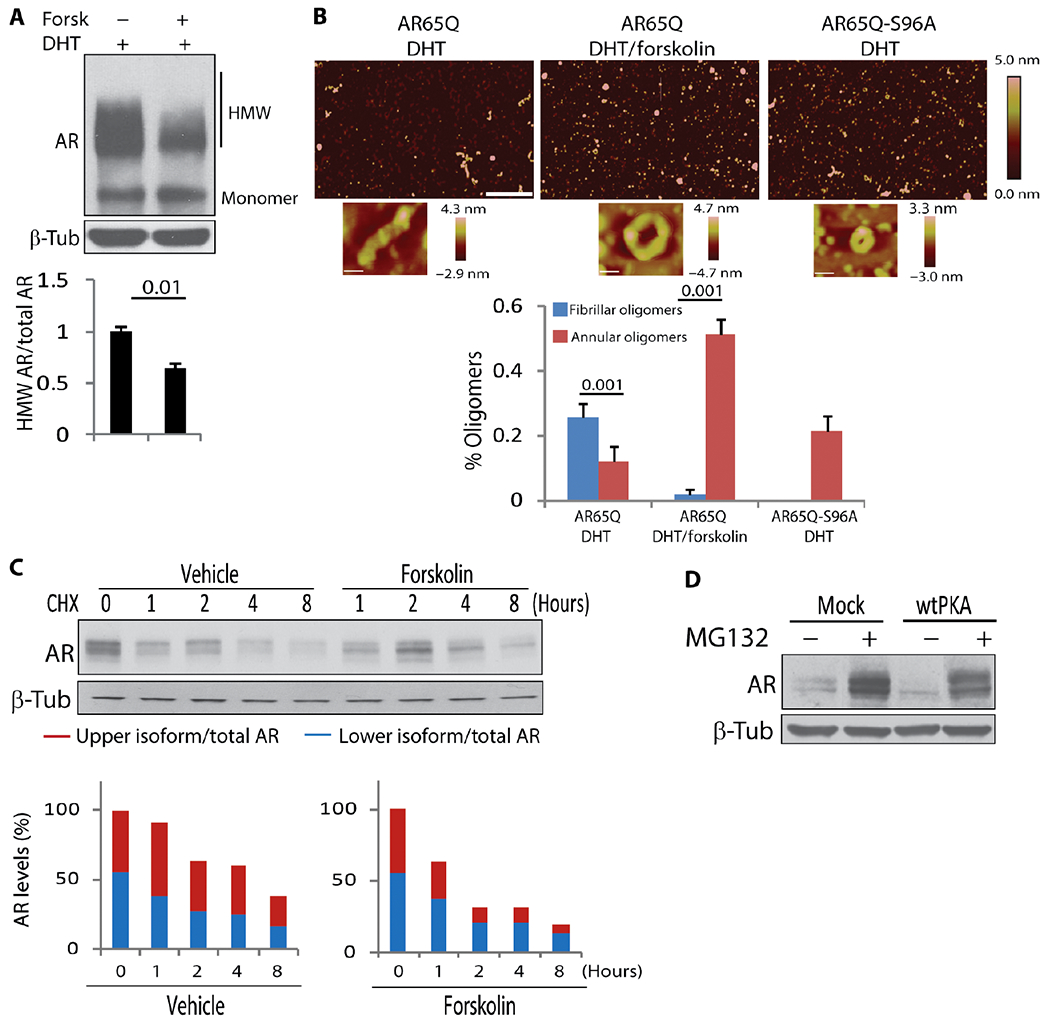
(A) Western blotting analysis of AR112Q aggregation in PC12 cells treated with doxycycline (10 μg/ml), 10 nM DHT, and 10 μM forskolin for 24 hours. n = 3 independent experiments. HMW, high–molecular weight species. (B) Atomic force microscopy analysis of polyQ-AR aggregation in Sf9 insect cells. Representative images of cells treated with 10 nM DHT and 10 μM forskolin for 24 hours. Scale bars, 600 nm. Magnification of oligomers is shown at the bottom. n = 15 to 25. (C) Western blotting analysis of AR55Q turnover in HEK293T cells treated with cycloheximide (CHX; 10 μg/ml) and 10 μM forskolin. n = 5 independent experiments. (D) Western blotting analysis of AR55Q in HEK293T cells transfected with empty vector and vector expressing wtPKA and treated with 10 μM MG132 (5 hours). n = 3 independent experiments. AR was detected with a specific antibody, and β-tubulin was used as loading control. Graphs, means ± SEM; Student’s t test (A and B) and one-way ANOVA (C) were used.
Chronic intranasal administration of a PACAP analog ameliorates the phenotype of knock-in SBMA mice and patient-derived cells
Next, we sought to translate this information into a therapy for SBMA. The clinical application of PACAP is limited by its poor metabolic stability and scarce penetration into target tissues (20, 21). These unfavorable pharmacokinetic properties led to the development of PACAP analogs with increased stability, enhanced receptor selectivity, and proven efficacy in several models of neurodegenerative diseases, six of which (hereafter referred to as peptides 1 to 6) were tested here (table S1) (20, 21, 29–31). In addition, we developed a new peptide (peptide 7) that is similar to peptide 6 but lacks the propilamide modification at the C terminus. Peptides 2, 4, and 7 stimulated cAMP production to levels similar to PACAP in MN-1 cells (table S1) and decreased the accumulation of Ser96-phosphorylated polyQ-AR in a dose-dependent manner, with significant effects at a concentration of 10 nM for peptides 2 and 4, and 1 nM for peptide 7 (Fig. 6A). Treatment with these three peptides increased the viability of SBMA MN-1 cells (Fig. 6B). Among these peptides, peptide 7 was the most effective in modifying polyQ-AR phosphorylation, and it enhanced polyQ-AR turnover in PC12 cells (fig. S19A).
Fig. 6. The PACAP analog, peptide 7, decreases CDK2-mediated polyQ-AR phosphorylation in vitro and induces cAMP production in vivo.
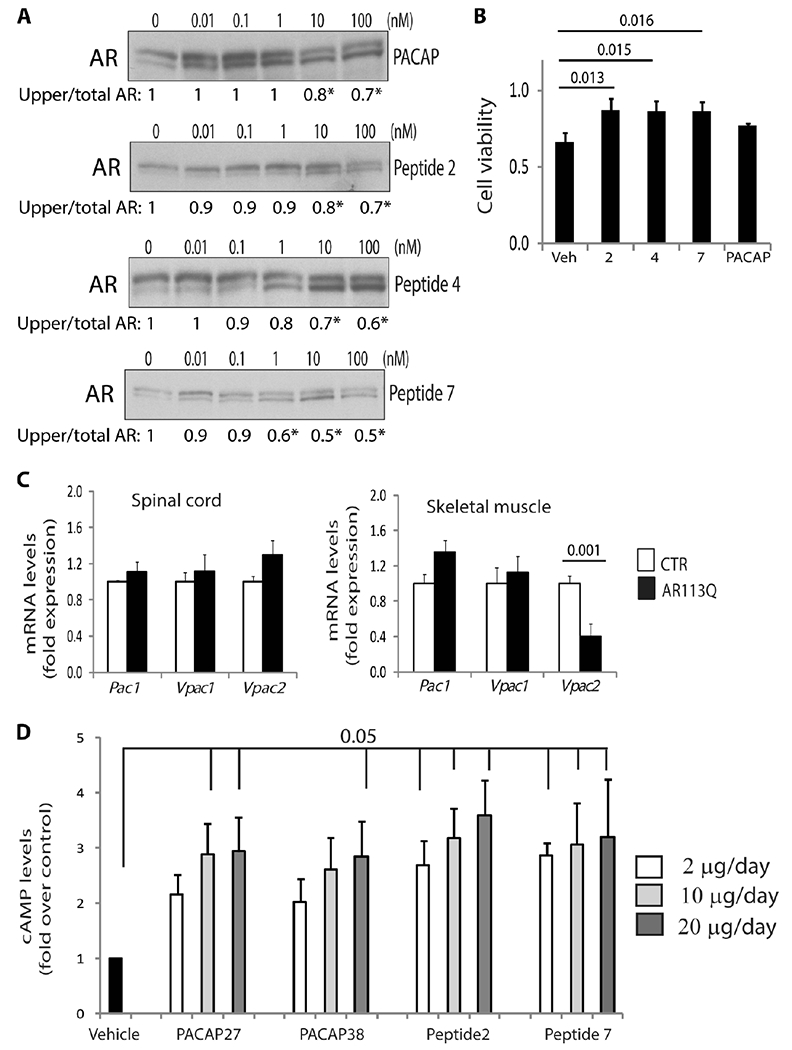
(A) Western blotting analysis of AR55Q in HEK293T cells treated with PACAP and peptides 2, 4, and 7. Quantification is shown at the bottom. n = 3 to 5 independent experiments; *P < 0.05. (B) XTT [2,3-bis (2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino) carbonyl]-2H-tetrazolium hydroxide] assay in MN-1 cells expressing AR65Q treated with peptides 2, 4, and 7 and 100 nM PACAP for 48 hours. n = 3 independent experiments. Veh, vehicle. (C) Real-time polymerase chain reaction (PCR) analysis of the transcript levels of Pac1, Vpac1, and Vpac2 receptors in the spinal cord and skeletal muscle of 180-day-old control (CTR; wild type) and AR113Q mice and normalized to hypoxanthine phosphoribosyltransferase 1 (Hprt1) and Pgk1 in spinal cord and to Hprt1 and β-glucuronidase (Gusβ) in skeletal muscle. n = 6 mice for each group. (D) cAMP level analysis in the spinal cord of control mice treated with PACAP27, PACAP38, peptide 2, and peptide 7. n = 3 mice for each group. Graphs, means ± SEM; one-way ANOVA was used.
On the basis of these results, the efficacy of peptide 7 was further tested in SBMA knock-in mice (32). These mice reproduce important features of the disease, such as sex-specific and age-dependent body weight loss and motor dysfunction. SBMA knock-in mice have an average median survival of 24 weeks of age, and ~50% of these mice die prematurely as a result of urinary tract obstructions caused by altered muscle membrane excitability. First, we analyzed expression of the PACAP-specific receptor, PAC1, and VPAC1 and VPAC2, which bind both PACAP and VIP (20, 21). Pac1 and Vpac1 were expressed to a similar extent in the spinal cord and muscle of SBMA and control mice, whereas the Vpac2 receptor was down-regulated specifically in muscle (Fig. 6C). To target the CNS using a noninvasive strategy, peptide 7 was administered intranasally at increasing concentrations into control mice. Administration of peptide 7 at concentrations of 2, 10, and 20 μg/day stimulated cAMP production in the brain, spinal cord, and muscle to levels similar to those obtained with PACAP (Fig. 6D and fig. S21). Of these concentrations, we used 2 μg/day (hereafter referred to as “low”) and 10 μg/day (“high”) for our preclinical study. To mirror the clinical course of the disease, treatment was initiated at 3 months of age, which corresponds to disease onset (32, 33). Mice were treated for 3 months, 5 days/week, instead of 7 days/week to avoid PACAP receptor desensitization. During the last week of treatment, the dose was gradually reduced to prevent withdrawal effects and to allow adaptation at the receptor level. Peptide 7 affected neither body weight nor food consumption in control and SBMA mice (fig. S22, A and B).
Treatment of AR113Q mice with low and high doses of peptide 7 increased the median survival of the mice by 10 weeks (χ2 log rank = 3.293, P =0.069) and 16 weeks (χ2 log rank = 5.417, P = 0.019), respectively (Fig. 7A and fig. S22C). Treatment of the AR113Q mice with high-dose peptide 7 significantly increased muscle force and improved motor coordination (Fig. 7, B and C). It is note-worthy that motor coordination of the vehicle-treated AR113Q mice was significantly different from that of control mice at 5 months of age, and this difference was abrogated by peptide 7 treatment. Stimulation with peptide 7 increased expression of the neuromuscular junction (NMJ) marker, muscle-specific kinase (Musk), and decreased expression of the denervation marker, runt-related transcription factor 1 (Runx1) (Fig. 7D), which are up-regulated in the quadriceps muscles of AR113Q mice (33). NR4A1 is an orphan nuclear receptor that induces p21Cip1 expression (34), is regulated by the AC/PKA signaling (35), and is decreased in SBMA muscle (36). Notably, peptide 7 increased expression of Nr4a1 in the muscle of AR113Q mice (Fig. 7D). Skeletal muscles composed of glycolytic and oxidative fibers, such as quadriceps, undergo a glycolytic-to-oxidative fiber-type switch in SBMA mice, with reduced expression of key glycolytic genes, including glyceraldehyde-3-phosphate dehydrogenase (Gapdh) and phosphoglycerate kinase 1 (Pgk1), and glycolytic fibers more severely affected than oxidative fibers (33). By nicotinamide adenine dinucleotide (NADH) staining, peptide 7 significantly (P = 0.001) decreased the number of oxidative fibers by 31%, increased the cross-sectional area (CSA) of glycolytic fibers by 11%, and restored the expression of Gapdh and Pgk1 in AR113Q mice (Fig. 7E and fig. S23, A to C). Next, we assessed the efficacy of peptide 7 in SBMA NPCs, in which expression of endogenous polyQ-AR causes mitochondrial membrane depolarization (37). Peptide 7 significantly attenuated mitochondrial membrane depolarization in SBMA NPCs (Fig. 7F). Mechanistically, intranasal administration of peptide 7 significantly decreased Ser96-phosphorylated and total AR by 15 to 30% in the brain, brainstem, spinal cord, and skeletal muscle (Fig. 8, A and B, and fig. S24, A and B). Peptide 7 did not alter the expression of CDK2 (fig. S24C). Rather, peptide 7 induced the expression of p21Cip1 (Fig. 8C), but not of the other CDK2/cyclin E inhibitors, p27Kip1 and p57Kip2 (fig. S24D). Together, these results show that stimulation of AC/PKA signaling in vivo reduces the accumulation of Ser96-phosphorylated and total polyQ-AR and ameliorates the phenotype of SBMA mice and patient-derived NPCs, further supporting our conclusion that activation of the AC/PKA signaling by PACAP is beneficial in SBMA.
Fig. 7. Intranasal administration of peptide 7 ameliorates the phenotype of knock-in SBMA mice.
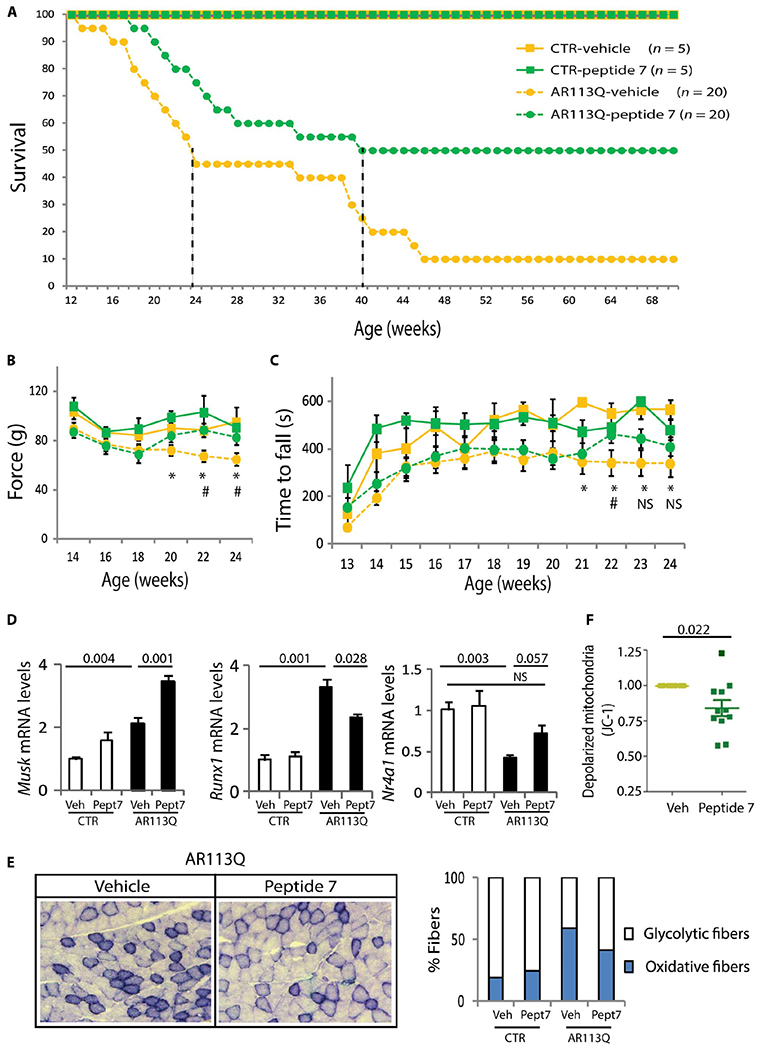
(A) Kaplan-Meier analysis of survival of control (CTR; squares) and AR113Q (circles) mice treated with either vehicle (yellow) or peptide 7 (10 μg/day; green). Median survival is indicated by the dashed vertical lines. (B) Grip-strength analysis of muscle force in CTR and AR113Q mice. n = as in (A). Week 20, *P = 0.045 CTR-vehicle versus AR113Q-vehicle; week 22, *P = 0.018 CTR-vehicle versus AR113Q-vehicle and #P = 0.002 AR113Q-vehicle versus AR AR113Q–peptide 7; week 24, *P = 0.0001 CTR-vehicle versus AR113Q-vehicle and #P = 0.002 AR113Q-vehicle versus AR113Q–peptide 7. (C) Rotarod analysis of motor coordination in CTR and AR113Q mice. n = as in (A). Week 21, *P = 0.004 CTR-vehicle versus AR113Q-vehicle; week 22, *P = 0.02 CTR-vehicle versus AR113Q-vehicle and #P = 0.05 AR113Q-vehicle versus AR113Q–peptide 7; week 23 *P = 0.0024 CTR-vehicle versus AR113Q-vehicle and C TR-vehicle versus AR113Q–peptide 7; week 23 *P = 0.006 CTR-vehicle versus AR113Q-vehicle and CTR-vehicle versus AR113Q–peptide 7. (D) Real-time PCR analysis in the quadriceps muscle of 180-day-old CTR and AR113Q mice treated with vehicle and peptide 7 (Pept7) and normalized to β-actin. n = 3 to 6 mice. (E) NADH analysis of the number of oxidative (blue) and glycolytic (white) fibers in the quadriceps muscle of 180-day-old CTR and AR113Q mice treated with vehicle and peptide 7. Number of fibers: n = 793 (CTR-vehicle), n = 384 (CTR-peptide 7), n = 1635 (AR113Q-vehicle), and n = 1145 (AR113Q–peptide 7). Left: Representative images of AR113Q mice. (F) Mitochondrial membrane depolarization in NPCs derived from three SBMA patients treated with vehicle or 100 nM peptide 7. n = 11 independent experiments. Graphs, means ± SEM; two-way ANOVA (B to E) and Student’s t test (F) were used.
Fig. 8. Peptide 7 decreases the accumulation of phosphorylated and total AR in vivo.
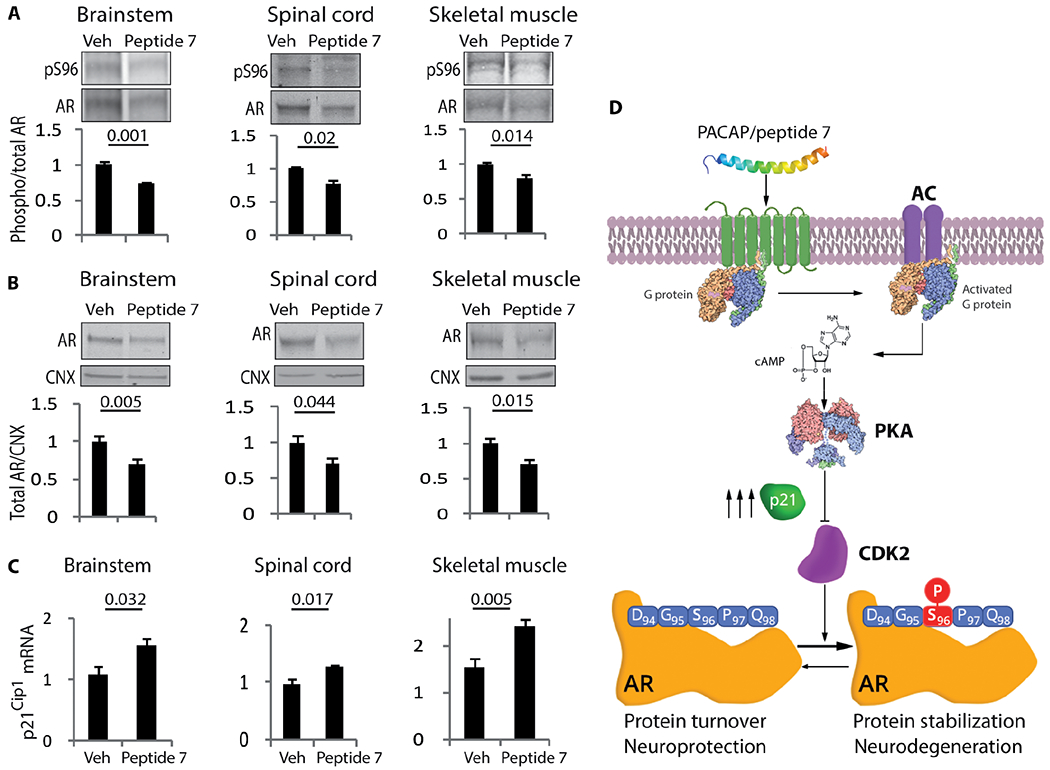
(A and B) Western blotting analysis of S96-phosphorylated (A) and total (B) AR in 180-day-old AR113Q mice treated with either vehicle or peptide 7 (10 μg/day). n = 5 to 6 mice. (C) Real-time PCR analysis of p21Cip1 transcript levels in 180-day-old AR113Q mice treated with vehicle or peptide 7 (10 μg/day) and normalized to β-actin. n = 4 to 6 mice. (D) Working model. Phosphorylated and total AR were detected with specific antibodies, and calnexin was used as loading control. Graphs, means ± SEM; Student’s t test was used.
DISCUSSION
Here, we show that modulation of polyQ protein phosphorylation has a key effect on neurotoxicity (Fig. 8D). AR is a highly phosphorylated protein, and most of the phosphorylation sites are consensus sites for the CDKs. Phosphorylation regulates two major aspects of AR biology: protein stabilization and function. Ser83, Ser96, Ser310, and Ser651 residues are the most highly phosphorylated AR sites in proliferating cells (13, 14, 38, 39). Ser83 and Ser310 phosphorylation by CDK1, CDK5, and CDK11 regulates AR stability (23, 40), whereas Ser83, Ser310, and Ser651 phosphorylation by CDK9, CDK11, and stress kinases regulates AR native functions (41, 42). Ser96 phosphorylation is independent of androgens, predominantly occurs in the cytosol (13, 15, 43), and, as shown here, is mediated by the CDK2/cyclin E complex and regulates AR stability. Ser96 phosphorylation is more abundant in neuronal than in nonneuronal cells, and we speculate that cell-specific mechanisms operating in postmitotic cells are responsible for Ser96 phosphorylation, leading to increased stability of the disease protein, thereby contributing to enhance neuronal vulnerability in SBMA.
CDK2 hyperphosphorylates the microtubule-associated protein tau (44), whereas the inhibition of CDK2 reduces the accumulation of RNA regulatory element (TAR) DNA binding protein 43 (TDP-43) into stress granules (45). This evidence suggests a role for CDK2 in neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS). Although the role of CDK2 in cell cycle progression is well established, its physiological function in neurons remains to be clarified. Our findings suggest a functional interaction between CDK2 and AR in postmitotic cells in a physiological context, which can become detrimental in pathological conditions, such as SBMA. The CDK2/cyclin E complex is inhibited by p21Cip1, which is emerging as a key factor in neurodegenerative diseases. p21Cip1 has antiapoptotic functions (46), p21Cip1 allele variants with reduced cell cycle inhibitory activity are associated with an increased risk for AD and PD (47), and p21Cip1 levels of expression are increased in transgenic mice expressing mutant tau (48). Our findings highlight a role for the CDK2/cyclin E complex and p21Cip1 in SBMA, supporting the concept that components on the cell cycle machinery are involved in the pathogenesis of neurodegenerative diseases. Mechanistically, our data suggest that PACAP/AC/PKA signaling requires p21Cip1 to inhibit CDK2/cyclin E. In addition, the PACAP/AC/PKA pathway might also activate a cellular phosphatase, which can have either a direct effect on polyQ-AR or an indirect effect through CDK2, ultimately leading to dephosphorylation of polyQ-AR at Ser96. Moreover, PACAP signaling might have additional effects that are independent of CDK2 and p21Cip1 and work additively or synergistically to reduce polyQ-AR toxicity. Further analysis is required to identify the cellular phosphatases and kinases that regulate AR Ser96 phosphorylation in neurons upon activation of pathways that modulate cAMP levels.
Modulation of cellular pathways involved in polyQ protein phosphorylation might have diverse outcomes, depending on the impact of a specific phosphorylation site on neurotoxicity. The phosphatidylinositide 3-kinase/Akt pathway reduces toxicity in both HD (49) and SBMA (7, 8) through direct modification of the disease proteins. On the other hand, activation of PACAP/AC/PKA signaling is protective in SBMA, yet activation of PKA is deleterious in SCA1 (50). This concept implies that disease-specific therapy can be designed on the basis of biochemical evidence of the effect of polyQ protein phosphorylation on neurotoxicity. Our findings have clinical relevance for SBMA therapy development. Here, we provide preclinical evidence that intranasal administration of PACAP analogs reduces disease manifestations in SBMA mice. PACAP belongs to the secretin/glucagon/VIP superfamily, is broadly expressed in the central and peripheral nervous systems, and is well conserved throughout evolution. Both PACAP38 and PACAP27, the active forms of PACAP, stimulate cAMP production. PACAP38 is the main peptide in mammals with strong neuroprotective effects in animal models of AD (51), PD (52), and HD (53). Mechanistically, PACAP neuroprotective effects involve inhibition of apoptosis and reduction of inflammation and oxidative stress. In addition to general CNS neuroprotective effects, our findings show that the PACAP/AC/PKA axis modulates specifically CDK2 activity to modify polyQ-AR phosphorylation and toxicity. Genetic variations in an estrogen-responsive element in the promoter of the gene that encodes the PAC1 receptor mediate response to stress in patients suffering from posttraumatic stress disorder, indicating a genetic link between steroid hormone receptors and PACAP signaling (54). Because Vpac2 is down-regulated in SBMA muscle, further analysis may clarify whether PACAP signaling is influenced by androgen regulation in patients suffering from neurodegenerative diseases.
Delivery of drugs and peptides to the CNS is hampered by the blood-brain barrier. This limitation can be circumvented by intranasal delivery, a route of administration that reduces side effects associated with systemic delivery, prevents exposure of the peptides to the circulation (PACAP is rapidly degraded in the blood), and is noninvasive, thus allowing long-term treatments that are necessary for management of chronic neurodegenerative diseases (55). Intranasal administration of PACAP38 in healthy male subjects was well tolerated (56). Notably, in pharmacological rodent models of PD and HD, PACAP treatment preceded brain lesion development (52, 53). To provide evidence for the efficacy of peptide 7 in the perspective of future clinical trials, we started treatment of SBMA mice at disease onset. Three-month treatment of SBMA mice with a newly developed PACAP analog ameliorated phenotype and extended life span, which suggests this treatment as a valid strategy for SBMA. One of the most striking effects of intranasal administration of PACAP was observed in skeletal muscle, a primary target tissue of polyQ-AR toxicity (57). Peptide 7 decreased the expression of the denervation marker Runx1 and enhanced expression of the NMJ marker MuSK. MuSK is a synaptic receptor tyrosine kinase involved in muscle–to–motor neuron retrograde signaling, and a threefold increase in MuSK is sufficient to delay denervation in mutant superoxide dismutase 1–linked ALS mice (58). Therefore, the effect of peptide 7 on the phenotype of SBMA mice might result from decreased denervation and increased retrograde signaling at the NMJ. Peptide 7 reduced the glycolytic-to-oxidative metabolic switch that occurs in SBMA muscles, restored the expression of key glycolytic genes, and ameliorated motor dysfunction. Thus, we provide proof of principle that chronic intranasal administration of a PACAP analog has a major effect on a peripheral tissue, skeletal muscle, which controls body metabolism and is a primary target of polyQ-AR toxicity. Long-lasting treatment with this newly developed PACAP analog has therapeutic potential for SBMA and possibly for other neurodegenerative diseases.
MATERIALS AND METHODS
Study design
The objective of this study was to identify phosphorylation sites on AR that modify neurotoxicity in SBMA and that can be modulated by pharmacologic intervention for therapy development. To achieve this objective, we undertook a molecular and biochemistry approach to analyze AR phosphorylation in nonneuronal and neuronal cells, knock-in SBMA mice, and SBMA patient-derived cells. We used activators and inhibitors of cellular kinases and phosphatases, activators of the AC/PKA pathway, and analogs of the neuropeptide, PACAP. We assessed AR phosphorylation in cultured cells and in vivo. We treated the knock-in mice with a PACAP analog, and we assessed the effect of treatment with measures of survival, motor function, histopathology, and biochemistry. Knock-in SBMA and control mice were randomized and assigned to the vehicle- or peptide-treated groups, and the effect on phenotype was assessed by an investigator who was blind for genotype and treatment. We performed at least three independent experiments for each data shown in the manuscript, unless differently indicated in the figure legends. Raw data for experiments with samples sizes n < 20 are presented in fig. S25.
Statistical analysis
Descriptive statistics are means ± SEM. For normal data, two-sample Student’s t tests and one-way ANOVAs with group as between-subject variable were used to assess differences for two groups and more than two groups, respectively. In case of two independent predictors for normal data, we used two-way ANOVAs. Fisher’s least significant difference and Dunnett’s tests were used for post hoc comparisons. Log-rank tests were used to compare Kaplan-Meier survival curves of treatment and vehicle. P values less than 0.05 were considered significant. All tests were two-tailed.
Expression vectors
Vectors for expression of PKA variants were obtained from G. Stanley McKnight (University of Washington, USA). Human CDK1 and CDK2 expression vectors were generated as follows: Total RNA was extracted from HEK293T cells, and full-length CDK1 (NM_001786.4) and CDK2 (NM_001798.3) complementary DNAs (cDNAs) were obtained by PCR using the following forward (F) and reverse (R) primers: CDK1-F, 5′-AAAAAAGGCCATGGAGGCCATGGAAGATTATACCAAAAT-3′; CDK1-R, 5′-AAAAAACTCGAGCTACATCTTCAATCTGATTGTCCA-3′; and CDK2-F, 5′-AAAAAAGGCCATGGAGGCCATGGAGAACTTCCAAAAGGT-3′; CDK2-R, 5′-AAAAAACTCGAGTCAGAGTCGAAGATGGGGTAC-3′.
Dominant-negative Asp145-to-asparagine (D145N) mutants were obtained by site-directed mutagenesis using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent) with template cDNAs obtained from Addgene (1885 and 1889). The siRNA duplexes to target human CDK1 and p21Cip1 were purchased from OriGene Technologies (siRNA CDK1 TL320288 and siRNA p21Cip1 TL305469). The transfection of the siRNA into HEK293T cells was optimized according to the instructions provided by OriGene Technologies. Briefly, HEK293T cells were transfected with 50 nM siRNA targeting CDK1 and 10 nM siRNA targeting p21Cip1, together with 0.5 μg of pEF-AR55Q, using Lipofectamine 2000 (Invitrogen).
The human AR (NM_000044) phosphorylation mutants were obtained by site-directed mutagenesis (QuikChange Lightning Site-Directed Mutagenesis Kit, Agilent) using the following primers: S83A-F, 5′-GCAGCAGCAAGAGACTGCCCCCAGGCAGCAGCAGC-3′; S83A-R, 5′-GCTGCTGCTGCCTGGGGGCAGTCTCTTGCTGCTGC-3′; S96A-F, 5′-GCAGGGTGAGGATGGTGCTCCCCAAGCCCATCGTAGAGG-3′; S96A-R, 5′-CCTCTACGATGGGCTTGGGGAGCACCATCCTCACCCTGC-3′; S96D-F, 5′-GCAGGGTGAGGATGGTGATCCCCAAGCCCATCGTAGAGG-3′; S96D-R, 5′-CCTCTACGATGGGCTTGGGGATCACCATCCTCACCCTGC-3′; S258A-F, 5′-GGCGTTGGAGCATCTGGCTCCAGGGGAACAGCT-3′; S258A-R, 5′-AGCTGTTCCCCTGGAGCCAGATGCTCCAACGCC-3′; S310A-F, 5′-GATACTGCTGAGTATGCCCCTTTCAAGGGAGG-3′; S310A-R, 5′-CCTCCCTTGAAAGGGGCATACTCAGCAGTATC-3′; S426A-F, 5′-GGGACCCGGTTCTGGG GCACCCTCAGCCGCCGCTTCC-3′; S426A-R, 5′-GGAAGCGGCGGCTGAGGGTGCCCCAGAACCGGGTCCC-3′; S282A-F, 5′-CCCGCTGTGCGTCCCGCTCCTTGTGCCCCATTGGC-3′; S282A-R, 5′-GCCAATGGGGCACAAGGAGCGGGACGCACAGCGGG-3′; and S800A-F, 5′-GGATGGCTCCAAATCGCCCCCCAGGAATTCC-3′; S800A-R, 5′-GGAATTCCTGGGGGGCGATTTGGAGCCATCC-3′.
Cell cultures, transfections, and treatments
All the cell types used in this study were previously validated, as recently described (19). HEK293T cells (American Type Culture Collection, CRL-1573) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) of heat-inactivated fetal bovine serum (FBS), penicillin/streptomycin (100 U/ml), and 2 mM l-glutamine at 37°C in a humidified atmosphere at 95% air and 5% CO2, as previously described (7). The cells were plated in standard culture medium.
The day after, the cells were transfected with polyethylenimine (408727, Sigma-Aldrich) following standard procedures. Transfection efficiency was 70%.
The inducible PC12 cells were cultured in DMEM supplemented with 10% (v/v) of heat-inactivated horse serum, 5% FBS, penicillin/streptomycin (100 U/ml), and 2 mM l-glutamine, as previously described (12, 59). The cells were transfected with Lipofectamine 2000, according to the manufacturer’s protocol (Invitrogen). Transfection efficiency was 80%. The cells were treated with doxycycline (10 μg/ml; D9891, Sigma-Aldrich) to induce AR expression.
MN-1 cells were cultured in DMEM supplemented with 10% (v/v) FBS, 2 mM l-glutamine, and gentamicin (0.05 mg/ml) at 37°C in a humidified atmosphere at 95% air and 5% CO2, as previously described (7). MN-1 cells were transfected with Lipofectamine 2000, according to the manufacturer’s protocol (Invitrogen). Transfection efficiency was 68%. Generation of stable MN-1 cell clones was carried out by lentiviral transduction. Full-length AR24Q and AR100Q cDNAs were cloned into the lentiviral vector #945.PCCL.sin.cPPT.SV40ployA.eGFP.minCMV.hPGK.deltaLNGFR.Wpre, in which the transgene expression is driven by the human PGK promoter, and the enhanced green fluorescent protein (EGFP) expression is driven by the cytomegalovirus promoter. The production of vesicular stomatitis virus–pseudotyped third-generation lentiviruses was performed as previously described (60). The infected cell lines were cultured and expanded in DMEM with 10% heat-inactivated FBS, penicillin/streptomycin (100 U/ml), and 2 mM l-glutamine at 37°C in a humidified atmosphere containing 5% CO2. After 6 weeks postinfection, the cells were sorted by flow cytometry with the BD FACSAria II (Becton Dickinson) cell sorter, on the basis of EGFP expression.
Primary cortical neurons were obtained from Sprague-Dawley rats (Harlan) at embryonic day 18. Cortices were dissected out of the embryos, and neurons were prepared with a papain digestion method [papain (20 U/ml), deoxyribonuclease (125 μg/ml), and cysteine (1.1 mg/ml) in Neurobasal medium], as previously reported (61). Cells were plated at 40,000 cells/cm2 on poly-l-lysine (0.1 mg/ml)–coated plates and maintained in Neurobasal medium supplemented with 2% B-27 supplement, 1% GlutaMAX, and penicillin/streptomycin (50 μg/ml) (Invitrogen). After 6 to 7 days in vitro (DIV), neurons were transduced with 10 multiplicity of infection of lentiviral particles expressing AR24Q or AR100Q and EGFP, as previously described (62). Neurons were used for Western blot analysis at 14 to 15 DIV.
For induced pluripotent stem cell (iPSC) derivation, primary fibroblast cells were obtained under the National Institutes of Health protocol 00-N-0043 with Institutional Review Board (IRB) approval from forearm skin biopsies of adult control and SBMA patients. These cells were cultured in DMEM supplemented with 10% FBS, penicillin/streptomycin (100 U/ml), and 2 mM l-glutamine. For NPC generation, fibroblasts were collected from dermal biopsies from three European Caucasian male SBMA patients aged between 55 and 65 years upon informed consent in compliance with an IRB protocol (#130337ZF) approved by the University of California, San Diego. Fibroblasts were cultured in 1× DMEM with 10% (v/v) FBS and 5% (v/v) antibiotics [penicillin/streptomycin (10,000 U/ml)]. Low-passage fibroblasts were reprogrammed as previously described (37, 63). Briefly, Oct4, c-Myc, Klf4, and Sox2 human cDNAs were obtained from Addgene and used in a retroviral system to infect. Two days after infection, fibroblasts were transferred onto irradiated mouse embryonic fibroblasts (Chemicon) with human embryonic stem cell (hESC) medium. After 10 to 15 days, hESC-like cells were manually selected and moved to feeder-free conditions on Matrigel-coated dishes (BD Biosciences) with mTeSR1 (STEMCELL Technologies). Standard G-banding chromosome analysis of putative iPSC clones was performed at Children’s Hospital Los Angeles (Los Angeles, CA). NPCs were also obtained as previously described (37). Briefly, iPSC medium was removed and switched to DMEM/F12 (Invitrogen) with 1×N2 and 1 μM dorsomorphin (Calbiochem). After 2 days, the colonies were mechanically dissociated and kept under rotation for 5 to 7 days at 37°C to form embryoid bodies (EBs). The EBs were plated onto dishes coated with poly-l-ornithine/laminin (Sigma-Aldrich), where the rosettes would arise on the plates after 7 days. Manually collected neural rosettes were enzymatically dissociated and plated again to be expanded onto coated dishes with DMEM/F12 medium, enriched with 0.5× N2/0.5× B-27/FGF2.
HEK293T, MN-1, and PC12 cell treatments were carried out in 10% charcoal/dextran-stripped FBS. The cells were treated with VIP (05-23-2101, Calbiochem; resuspended in 5% acetic acid), PACAP27 (05-23-2151, Calbiochem; resuspended in 5% acetic acid), forskolin [344273, Calbiochem; resuspended in dimethyl sulfoxide (DMSO)], H-89 (371962, Calbiochem), PKI (476485, Calbiochem; resuspended in H2O), PD98059 (513001, Calbiochem), LY294002 (440204, Calbiochem), rapamycin (553211, Calbiochem), MG132 (474790, Calbiochem), cycloheximide (239765, Calbiochem), DHT [10300-5G-F, Sigma-Aldrich; resuspended in ethanol (EtOH)], OA (495609, Calbiochem), CDK1/2 inhibitor (217713, Calbiochem; resuspended in DMSO), CDK1 inhibitor (CGP74514A, Calbiochem), CDK1/5 inhibitor (217720, Calbiochem), GSK-3b inhibitor VIII (361557, Calbiochem), roscovitine (557364, Calbiochem), fostriecin (344280, Calbiochem; resuspended in EtOH), tautomycin (580551, Calbiochem; resuspended in EtOH), cyclosporin A (C1832, Sigma-Aldrich; resuspended in DMSO), and FK-506 monohydrate (F4679, Sigma-Aldrich) for 5 hours unless differently indicated.
Seven peptides were designed as described in the legend to table S1. Synthesis was carried out by Hy-Labs. Peptide 1 was derived from the C-terminal portion of PACAP (also common to VIP), has neurotrophic properties similar to full-length VIP, and was effective in animal models of AD (64, 65). Peptides 2, 3, and 4 correspond to PACAP1–27, VIP, and PACAP1–38, respectively, in which the methionine at position 17 that is susceptible to oxidation was replaced by norleucine (Nle), a stable unnatural amino acid (66). Peptide 5 is acetyl–PACAP1–38[Nle17] and was designed to stabilize the C terminus of the peptide. Peptide 6 is acetyl–[Ala15, Ala20]PACAP1–38–propylamide, a superagonist of the PAC1 receptor (30). Peptide 7 is peptide 6 without the propylamide group.
Cell toxicity assays
For caspase 3 assays, the cells were cultured for 48 hours in charcoal/dextran-stripped medium and processed as previously described (7). For trypan blue assay, the SBMA PC12 and MN-1 cells were treated with DHT. At least 200 cells per sample were counted, and the percentage of trypan blue-positive cells was calculated. To measure cell viability by XTT assay, the cells were serum-starved and treated with aphidicolin (400 ng/ml; 178273, Calbiochem) for 48 hours using the Cell Proliferation Kit II (Roche Diagnostics), according to the manufacturer’s instructions.
BRET assay
All cell culture reagents and buffers were from Invitrogen and Sigma-Aldrich. Coelenterazine h was purchased from Promega. MN-1 cells were transfected with vector expressing the EPAC (exchange protein activated by cAMP) cAMP biosensor (67). This biosensor consists of a modified form of EPAC tagged with Renilla luciferase and a yellow fluorescent protein at the N and C termini of EPAC, respectively. Binding to cAMP results in a decrease in signal emitted because of a conformational change that increases the distance of the two tags. Transient transfections were performed 24 hours after cell seeding using Lipofectamine 2000, according to the manufacturer’s protocol (Invitrogen). Twenty-four hours after transfection, the cells were plated in poly-d-lysine–coated 96-well microplates (well assay plate with clear bottom; Fisher Scientific) at a density of 80,000 cells per well in phenol red–free minimum essential medium containing 2% of FBS, 10 mM Hepes, and 2 mM l-glutamine. The cells were then cultured for an additional 24 hours in the presence or absence of 10 nM DHT. The phenol red–free medium was removed from the cells and replaced by phosphate-buffered saline (PBS) containing calcium and magnesium and 0.003% (w/v) ascorbic acid. Ten microliters of the cell-permeant substrate specific for Renilla luciferase, coelenterazine h, was added to the well to yield a final concentration of 5 μM. For time course experiments, the plate was read immediately after the addition of the agonist for about 20 min. All the compounds were tested at the initial concentration of 10 μM. Then, for active compounds, a dose response was performed to calculate the EC50 (median effective concentration) values. All the experiments were conducted in the presence of the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (Sigma-Aldrich) at the final concentration of 200 μM. Readings were collected using a Tecan Infinite instrument that allows the sequential integration of the signals detected in the 465- to 505-nm and 515- to 555-nm windows using filters with the appropriate band pass using iControl software. The acceptor/donor ratio was calculated as previously described (68). Curve was fitted using a non-linear regression and one site–specific binding with GraphPad Prism 5.
Quantitative real-time PCR analysis
Total RNA was extracted with TRIzol (Invitrogen). RNA was retrotranscribed using the SuperScript III First-Strand Synthesis System Kit (Invitrogen) or iScript Reverse Transcription Supermix (Bio-Rad) following the manufacturer’s instructions. For Pac1, Vpac1, and Vpac2, gene expression was measured by real-time quantitative PCR (RT-qPCR) with specific TaqMan assays [Adcyap1r1 Mm00431680_m1, Mm00431683_m1; Adcyap1 Mm00437433_m1, Hs00153869_m1; Vip Mm00660234_m1; Vipr1 Mm00449214_m1, Hs00270351_m1; Vipr2 Mm00437316_m1, Hs00173643_m1 (Applied Biosystems)] on a 7900 HT Fast Real-Time PCR System (Applied Biosystems). Other gene expression analyses were made by RT-qPCR with SYBR Green method on the C1000 Touch Thermal Cycler–CFX96 Real-Time System (Bio-Rad). Gene expression was measured by RT-qPCR using the 7900 HT Fast Real-Time PCR System (Applied Biosystems) and the C1000 Touch Thermal Cycler–CFX96 Real-Time System (Bio-Rad). Specific primers (Invitrogen) were as follows: Hprt1-F, 5’-TTGCTGACCTGCTGGATTAC-3’; Hprt1-R, 5’-CCCGTTGACTGATCATTACAGTAG-3’; β-actin-F, 5’-GACAGGATGCAGAAGGAGATTACTG-3’; β-actin–R, 5’-CTCAGGAGGAGCAATGATCTTGAT-3’; Musk-F, 5’-ATCACCACGCCTCTTGAAAC-3’; Musk-R, 5’-TGTCTTCCACGCTCAGAATG-3’; Runx1-F, 5’-GAAGAACCAGGTAGCGAGATTC-3’; Runx1-R, 5’-GTAAAGACGGTGATGGTCAGAG-3’; NR4A1-F, 5’-GGCATGGTGAAGGAAGTTGTA-3’; NR4A1-R, 5’-CATCTGGAGGCTGCTTGG-3’; Cdk2-F, 5’-GCAGACTTTGGACTAGCAAGA-3’; Cdk2-R, 5’-AAGAATTTCAGGTGCTCGGT-3’; p21Cip1 (mouse)–F, 5’-CTGAGCGGCCTGAAGATT-3’; p21Cip1 (mouse)–R, 5’-ATCTGCGCTTGGAGTGATAG-3’; p21Cip1 (human)–F, 5’-GGAGACTCTCAGGGTCGAAA-3’; p21Cip1 (human)–R, 5’-GCTTCCTCTTGGAGAAGATCAG-3’; p27Kip1-F, 5’-CCTGACTCGTCAGACAATCC-3’; p27Kip1-R, 5’-TCTGTTCTGTTGGCCCTTT-3’; p57Kip2-F, 5’-AGGACGAGAATCAAGAGCAG-3’; p57Kip2-R, 5’-GAAGAAGTCGTTCGCATTGG-3’; Pgk1-F, 5’-ACCATTGTGCATTGTAGA-3’; Pgk1-R, 5’-AGTCAAGAACAGTGAGTG-3’; and Gapdh-F, 5’-TAGACAAAATGGTGAAGGT-3’; Gapdh-R, 5’-AGTTGAGGTCAATGAAGG-3’.
Biochemistry analyses
For Western blotting analysis, cells were processed as previously described (7, 8, 12). For biochemical analysis in vivo, brain, brainstem, spinal cord, and quadriceps were pulverized using pestle and mortar. For Western blotting analysis, proteins were extracted from pulverized tissues using a lysis buffer containing 2% SDS, 150 mM NaCl, 2 mM EDTA, and 10 mM Hepes (pH 7.4). Lysates from brain, brainstem, and spinal cord were sonicated and centrifuged at 15,000 rpm for 15 min at room temperature. Lysates from quadriceps were homogenized (homogenizer RZR 2052 control, Heidolph) at 600 rpm (20 times); passed through syringes of 18-, 22-, and 25-gauge needles; and centrifuged at 15,000 rpm for 15 min at room temperature. Protein concentration was measured using the bicinchoninic acid assay method. Equal amounts of protein were subjected to 7% tris-HCl SDS-PAGE. Gels were blotted overnight onto 0.45-μm nitrocellulose membranes (Bio-Rad, 162-0115). Western blotting signals were detected using the enhanced chemiluminescence reagent (GE Healthcare Life Sciences) or the Li-Cor Odyssey Infrared Imaging System. Quantifications were performed using ImageJ 1.45 software. Antibodies used were AR (sc-7305, sc-816, and sc-13062), CDK2 (sc-6248), and p21Cip1 (C-19, sc-397) from Santa Cruz Biotechnology; tubulin (T7816) and myc (C3956) from Sigma-Aldrich; calnexin (ADI-SPA-860) from Enzo Life Sciences; caspase 3 (9662) and CDK5 (2506) from Cell Signaling Technology; HDAC1 (MA1-20354) from Thermo Scientific; and CDK1 (ab38384) and CDK2 (ab7954) from Abcam. The polyclonal anti-AR (phospho S96) antibody used was generated and affinity-purified by Biomatik. For nuclear/cytosolic fractionation, we used the NE-PER Nuclear and Cytoplasmic Extraction (Pierce), according to the manufacturer’s instructions. For the in vitro dephosphorylation assay, cells were lysed in lysis buffer [50 mM tris (pH 7.5), 1% Triton X-100, and protease inhibitors], sonicated, centrifuged at 4°C at 13,000 rpm, and incubated with 1500 U of λ phosphatase (New England Biolabs) for 6 hours at 30°C in 1× protein metallophosphatase buffer (New England Biolabs) supplemented with 1 mM MnCl2. For the immunoprecipitation assay, HEK293T cells were washed with cold 1× PBS and then lysed with lysis buffer [50 mM tris-HCl (pH 8), 150 mM NaCl, 0.1% SDS, 1% NP-40, and protease inhibitor cocktail]. Before the start of the immunoprecipitation assay, 1 mg of total cell lysate was precleared by 1-hour incubation with the protein G dynabeads (10003D, Thermo Fisher) on a wheel at 4°C. Then, the lysate was incubated overnight at 4°C with 3 μg of specific antibody (anti-AR N20; sc-816, Santa Cruz). The day after, the antibody-protein complexes were bound to the dynabeads, washed three times with the lysis buffer, and processed for Western blotting analysis.
In vitro kinase assay
AR was immunoprecipitated using monoclonal anti-AR antibody (clone 441, Santa Cruz Biotechnology) and incubated with CDK2/cyclin A or CDK2/cyclin E (Millipore). HEK293 cells were transfected with AR55Q or AR55Q-S96A and AR55Q-S8A mutants, and after 48 hours, immune complexes obtained from 1 mg of each extract were incubated in phosphorylation buffer [20 mM NaF, 20 mM Mops (pH 7.2), 25 mM β-glycerophosphate, 10 mM MgCl2, 25 μM adenosine triphosphate (ATP), 1 mM Na3VO4, 5 mM EGTA, and 1 mM dithiothreitol], followed by the addition of 15 ng of recombinant CDK2/cyclin A or CDK2/cyclin E and 5.0 μCi [γ-32P]ATP (3000 Ci/mmol; PerkinElmer). After 30 min of incubation at 30°C, samples were subjected to 7.5% SDS-PAGE and transferred on a polyvinylidene difluoride membrane. Signals were detected by autoradiography.
Luciferase assay
Luciferase assays were performed using the Dual-Luciferase Reporter Assay System, as we have previously described (7).
Atomic force microscopy
PolyQ-extended AR (ARQ65) and the S96A mutant (ARQ65S96A) were expressed in Sf9 insect cells and processed as previously described in (28, 69).
Immunofluorescence analyses
Cells were fixed in 4% paraformaldehyde for 20 min, washed with PBS, and permeabilized in 0.1% Triton X-100 in PBS for 10 min. Cells were incubated with blocking buffer containing 10% normal goat serum (NGS) and 5% milk powder diluted in tris-buffered saline (TBS) for 45 min. Cells were then incubated with the following primary antibodies: AR (sc-13062, Santa Cruz Biotechnology) and CDK2 (sc-6248, Santa Cruz Biotechnology) overnight at 4°C. Cells were then washed with TBS and incubated for 1 hour with secondary antibodies [Alexa Fluor 546 goat anti-rabbit immunoglobulin G (IgG) and Alexa Fluor 488 goat anti-mouse IgG; Invitrogen], followed by washes in TBS and 5-min incubation with DAPI (Invitrogen). Slices were then mounted with Aqua-Poly/Mount (Polyscience Inc.). Mouse tissues were collected, snap-frozen in isopentane precooled in liquid nitrogen and stored at −80°C. Sections of brainstem, spinal cord, and quadriceps were taken using Leica CM1850 UV cryostat. The sections were subjected to antigen retrieval for 30 min at 95°C in a solution of 10 mM sodium citrate (pH 6), followed by blocking in a solution containing 10% NGS, 5% bovine serum albumin, and 0.5% fish gelatin in TBS–Tween 0.01%, overnight at 4°C. The incubation with the primary antibodies for detection of AR and CDK2 was performed overnight (AR, sc-13062; CDK2, sc-6248) after 1-hour incubation with secondary antibodies (Alexa Fluor 546 goat anti-rabbit IgG and Alexa Fluor 488 goat anti-mouse IgG, Invitrogen) and mounted in fluoroshield mounting medium with DAPI (ab104139, Abcam). For Nissl staining of motor neurons in brainstem and spinal cord, tissues were cut and mounted in slices. The samples were gradually dehydrated (70, 95, and 100% EtOH), stained with a solution containing 0.1% cresyl violet for 5 min, washed in water, gradually dehydrated (70, 95, and 100% EtOH), cleared in xylene, and mounted in Eukitt (Bio Optica). All the images were acquired with the inverted Zeiss Observer Z1 microscope. NADH staining was performed as previously described (33).
Maintenance and treatment of AR113Q knock-in mice
Animal care and experimental procedures were conducted in accordance with the Italian Institute of Technology and the University of Trento (Italy) ethics committees and were approved by the Italian Ministry of Health. Generation and genotyping of knock-in AR113Q mice were previously described (70, 71). Here, only male mice were used. The mice were housed in filtered cages in a temperature-controlled room with a 12-hour light/dark cycle with ad libitum access to water and food. Peptide 7 was dissolved in intranasal solution containing 7.5 mg of NaCl, 1.7 mg of citric acid monohydrate, 3 mg of disodium phosphate dihydrate, and 0.2 mg of benzalkonium chloride solution (50%) per milliliter (65, 72). Mice were treated with 5 μl (2.5 μl per nostril) of intranasal solution containing 10 or 2 μg of peptide 7 or vehicle. Treatment was carried out 5 days per week to avoid desensitization of PACAP receptors. Animals were treated with fixed doses per day for 12 weeks. During the 13th week, the dose of peptide 7 was reduced day by day gradually to prevent a possible withdrawal response. For grip strength, a grip-strength meter (Ugo Basile instruments) was used to measure forelimb grip strength. The grip-strength meter was positioned horizontally, and the mice were held by the tail and lowered toward the apparatus. Mice were allowed to grasp the smooth metal triangular pull bar with their forelimbs only and then were pulled backward. The force applied to the bar at the moment the grasp was released was recorded as the peak tension. The test was repeated three consecutive times within the same session, and the highest value from the three trials was used to evaluate muscle strength. The test was performed every other week. Motor coordination was measured using the rotarod task (Ugo Basile instruments). Mice were trained the week before starting the test. Mice performed the rotarod test weekly, and the time performed in three consecutive trials at 21 rpm for 600 s was recorded. The results of the test were shown as the average time of three trials. For food intake analysis, mice were housed individually in cages with a fixed and an excess amount of food. Every day for 5 days, the amount of food left in the cage was weighted. For tissue analysis, 6 wild-type mice and 11 AR113Q mice per treatment were sacrificed after 12 weeks of treatment, 1 hour after the last administration. The mice were sacrificed by cervical dislocation to avoid the use of anesthetic drugs that can modify the levels of cAMP. Tissues were collected, snap-frozen in isopentane precooled in liquid nitrogen, and stored at −80°C until further processing.
In vivo analysis of cAMP levels
Mice were treated with vehicle [intranasal solution containing 7.5 mg of NaCl, 1.7 mg of citric acid monohydrate, 3 mg of disodium phosphate dihydrate, and 0.2 mg of benzalkonium chloride solution (50%) per milliliter; PACAP38 (137061-48-4, Calbiochem); or peptide 7 (2, 10, and 20 μg) dissolved in vehicle (51)]. Mice were treated daily for 5 days, 5 μl/day, and 2.5 μl per nostril. One hour after the last administration, mice were sacrificed, and tissues were snap-frozen in liquid nitrogen. Frozen tissues were weighted and pulverized. Five volumes (milliliters per gram of tissues) of 5% trichloroacetic acid (TCA) in water were added. The samples were homogenized on ice (4°C) and centrifuged at 1500g for 10 min. TCA was extracted from samples using water-saturated ether. cAMP levels were measured using enzyme-linked immunosorbent assay cAMP kit (Cayman, 581001) according to the manufacturer’s instructions.
Mitochondrial membrane potential assay
The mitochondrial membrane potential was analyzed using the NC-3000 image system (ChemoMetec) as previously described (37). Briefly, cells were stained with JC-1 and DAPI. Cellular JC-1 monomers and aggregates are detected as green and red fluorescence, respectively, and the amount of blue, green, and red fluorescence of individual cells was quantified. Mitochondrial depolarization was revealed as a decrease in red/green fluorescence intensity ratio.
Supplementary Material
Table S1. List of PACAP analogs and cAMP release in MN-1 cells treated with either PACAP or the indicated PACAP analogs.
Fig. S1. Overexpression of PKA reduces the accumulation of the upper isoform of polyQ-AR.
Fig. S2. PACAP stimulates the release of cAMP in MN-1 cells expressing AR24Q and AR65Q.
Fig. S3. VIP does not modify the accumulation of the upper isoform of polyQ-AR.
Fig. S4. Expression of polyQ-AR in MN-1 cells results in caspase 3 activation.
Fig. S5. Analysis of polyQ-AR phosphorylation upon inhibition of specific cellular phosphatases and kinases.
Fig. S6. Modulation of CDK1 and CDK5 activity does not affect the accumulation of the upper isoform of polyQ-AR.
Fig. S7. Nonexpanded AR and polyQ-AR colocalize with endogenous CDK2 in the absence and presence of androgens in MN-1 and PC12 cells.
Fig. S8. Nonexpanded AR and polyQ-AR colocalize with endogenous CDK2 in the brainstem motor neurons of control and knock-in SBMA mice.
Fig. S9. Nonexpanded AR and polyQ-AR colocalize with endogenous CDK2 in the motor neurons of the lumbar spinal cord of control and knock-in SBMA mice.
Fig. S10. Nonexpanded AR colocalizes with endogenous CDK2 in the quadriceps of control mice.
Fig. S11. Endogenous nonexpanded AR and polyQ-AR colocalize with endogenous CDK2 in control and SBMA patient-derived motor neurons and NPCs.
Fig. S12. Endogenous polyQ-AR colocalizes with endogenous CDK2 in the spinal cord of an SBMA patient.
Fig. S13. Forskolin, PACAP, and the pan-phosphatase inhibitor OA stimulate p21Cip1 expression.
Fig. S14. Ser96 of AR is conserved throughout evolution.
Fig. S15. Analysis of phosphoresistant polyQ-AR variants.
Fig. S16. Ser96 phosphorylation is responsible for the formation of the upper isoform of polyQ-AR.
Fig. S17. Phosphodefective and phosphomimetic substitution of Ser96 does not affect polyQ-AR subcellular localization.
Fig. S18. Forskolin and PACAP, and not VIP, reduce polyQ-AR aggregation.
Fig. S19. Activation of the AC/PKA signaling increases the turnover of polyQ-AR.
Fig. S20. OA reduces the accumulation of phosphorylated polyQ-AR by inducing degradation through the UPS.
Fig. S21. PACAP induces cAMP production in vivo.
Fig. S22. Effect of intranasal administration of peptide 7 on body weight, food intake, and survival of AR113Q mice.
Fig. S23. Peptide 7 increases the CSA of glycolytic fibers in the skeletal muscle of AR113Q mice and restores the expression of glycolytic genes to normal levels.
Fig. S24. Intranasal administration of peptide 7 decreases polyQ-AR phosphorylation and accumulation in SBMA mice without altering CDK2, p27Kip1, and p57Kip2 expression levels.
Fig. S25. Data presented for experiments with sample sizes of less than 20.
Acknowledgments:
We thank S. Robbiati and M. Tarter (University of Trento, Italy) for technical assistance, G. Stanley McKnight (University of Washington) for providing us with PKA expression vectors, and D. E. Merry (Thomas Jefferson University) for giving us SBMA PC12 cells. We thank L. Tosatto (University of Trento; supported by Akira Arimura Foundation) for comments on the manuscript.
Funding:
This work was supported by Kennedy’s Disease Association (to M.P., S.P., and H.C.M.), the Muscular Dystrophy Association (196646 to M.P. and 277469 to A.R.L.S.), Telethon-Italy and Provincia Autonoma di Trento–Italy (TCP12013 to M.P.), Bando Progetti Strategici di Ateneo–University of Trento (to M.P.), Marie Curie Reintegration Grants (FP7-256448 to M.P. and FP7-276981 to F.S.), the Italian Ministry of Health (RF-2011-02350097 to M.P.), Association Française contre les Myopathies (18722 to M.P.), the Russian Science Foundation (14-50-00069 to R.R.G.), NIH (R01 NS055746 to A.P.L. and R01 NS041648 to A.R.L.S.), Fondazione Umberto Veronesi Fellowship (to M.C.), and Marie Curie International Outgoing Fellowships (PIOF-GA-2011-300723 to S.P.).
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/8/370/370ra181/DC1
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH, Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352, 77–79 (1991). [DOI] [PubMed] [Google Scholar]
- 2.Kennedy WR, Alter M, Sung JH, Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology 18, 671–680 (1968). [DOI] [PubMed] [Google Scholar]
- 3.Orr HT, Zoghbi HY, Trinucleotide repeat disorders. Annu. Rev. Neurosci 30, 575–621 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, Artates JW, Weiss A, Cheng SH, Shihabuddin LS, Hung G, Bennett CF, Cleveland DW, Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 74, 1031–1044 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pourshafie N, Lee PR, Chen K-I, Harmison GG, Bott LC, Katsuno M, Sobue G, Burnett BG, Fischbeck KH, Rinaldi C, MiR-298 counteracts mutant androgen receptor toxicity in spinal and bulbar muscular atrophy. Mol. Ther 24, 937–945 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lieberman AP, Yu Z, Murray S, Peralta R, Low A, Guo S, Yu XX, Cortes CJ, Bennett BF, Monia BP, La Spada AR, Hung G, Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep. 7, 774–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palazzolo I, Burnett BG, Young JE, Brenne PL, La Spada AR, Fischbeck KH, Howell BW, Pennuto M, Akt blocks ligand binding and protects against expanded polyglutamine androgen receptor toxicity. Hum. Mol. Genet 16, 1593–1603 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Palazzolo I, Stack C, Kong L, Musaro A, Adachi H, Katsuno M, Sobue G, Taylor JP, Sumner CJ, Fischbeck KH, Pennuto M, Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron 63, 316–328 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernández-Rhodes LE, Kokkinis AD, White MJ, Watts CA, Auh S, Jeffries NO, Shrader JA, Lehky TJ, Li L, Ryder JE, Levy EW, Solomon BI, Harris-Love MO, La Pean A, Schindler AB, Chen C, Di Prospero NA, Fischbeck KH, Efficacy and safety of dutasteride in patients with spinal and bulbar muscular atrophy: A randomised placebo-controlled trial. Lancet Neurol. 10, 140–147 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katsuno M, Banno H, Suzuki K, Takeuchi Y, Kawashima M, Yabe I, Sasaki H, Aoki M, Morita M, Nakano I, Kanai K, Ito S, Ishikawa K, Mizusawa H, Yamamoto T, Tsuji S, Hasegawa K, Shimohata T, Nishizawa M, Miyajima H, Kanda F, Watanabe Y, Nakashima K, Tsujino A, Yamashita T, Uchino M, Fujimoto Y, Tanaka F, Sobue G; Japan SBMA Interventional Trial for TAP-144-SR (JASMITT) study group, Efficacy and safety of leuprorelin in patients with spinal and bulbar muscular atrophy (JASMITT study): A multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 9, 875–884 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Sang C, Kobayashi Y, Doyu M, Sobue G, Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 35, 843–854 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Scaramuzzino C, Casci I, Parodi S, Lievens PMJ, Polanco MJ, Milioto C, Chivet M, Monaghan J, Mishra A, Badders N, Aggarwal T, Grunseich C, Sambataro F, Basso M, Fackelmayer FO, Taylor JP, Pandey UB, Pennuto M, Protein arginine methyltransferase 6 enhances polyglutamine-expanded androgen receptor function and toxicity in spinal and bulbar muscular atrophy. Neuron 85, 88–100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gioeli D, Ficarro SB, Kwiek JJ, Aaronson D, Hancock M, Catling AD, White FM, Christian RE, Settlage RE, Shabanowitz J, Hunt DF, Weber MJ, Androgen receptor phosphorylation. Regulation and identification of the phosphorylation sites. J. Biol. Chem 277, 29304–29314 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Jenster G, de Ruiter PE, van der Korput HA, Kuiper GG, Trapman J, Brinkmann AO, Changes in the abundance of androgen receptor isotypes: Effects of ligand treatment, glutamine-stretch variation, and mutation of putative phosphorylation sites. Biochemistry 33, 14064–14072 (1994). [DOI] [PubMed] [Google Scholar]
- 15.Wong HY, Burghoorn JA, van Leeuwen M, de Ruiter PE, Schippers E, Blok LJ, Li KW, Dekker HL, de Jong L, Trapman J, Grootegoed JA, Brinkmann AO, Phosphorylation of androgen receptor isoforms. Biochem. J 383, 267–276 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Errico A, Deshmukh K,Tanaka Y, Pozniakovsky A, Hunt T, Identification of substrates for cyclin dependent kinases. Adv. Enzyme Regul 50, 375–399 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Koryakina Y, Ta HQ, Gioeli D, Androgen receptor phosphorylation: Biological context and functional consequences. Endocr. Relat. Cancer 21, T131–T145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blok LJ, de Ruiter PE, Brinkmann AO, Forskolin-induced dephosphorylation of the androgen receptor impairs ligand binding. Biochemistry 37, 3850–3857 (1998). [DOI] [PubMed] [Google Scholar]
- 19.Pennuto M, Basso M, In vitro and in vivo modeling of spinal and bulbar muscular atrophy. J. Mol. Neurosci 58, 365–373 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Reglodi D, Kiss P, Lubics A, Tamas A, Review on the protective effects of PACAP in models of neurodegenerative diseases in vitro and in vivo. Curr. Pharm. Des 17, 962–972 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Vaudry D, Falluel-Morel A, Bourgault S, Basille M, Burel D, Wurtz O, Fournier A, Chow BKC, Hashimoto H, Galas L, Vaudry H, Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol. Rev 61, 283–357 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Barak LS, Salahpour A, Zhang X, Masri B, Sotnikova TD, Ramsey AJ, Violin JD, Lefkowitz RJ, Caron MG, Gainetdinov RR, Pharmacological characterization of membrane-expressed human trace amine-associated receptor 1 (TAAR1) by a bioluminescence resonance energy transfer cAMP biosensor. Mol. Pharmacol 74, 585–594 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen S, Xu Y, Yuan X, Bubley GJ, Balk SP, Androgen receptor phosphorylation and stabilization in prostate cancer by cyclin-dependent kinase 1. Proc. Natl. Acad. Sci. U.S.A 103, 15969–15974 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsu F-N, Chen M-C, Chiang M-C, Lin E, Lee Y-T, Huang P-H, Lee G-S, Lin H, Regulation of androgen receptor and prostate cancer growth by cyclin-dependent kinase 5. J. Biol. Chem 286, 33141–33149 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Messner DJ, Ao P, Jagdale AB, Boynton AL, Abbreviated cell cycle progression induced by the serine/threonine protein phosphatase inhibitor okadaic acid at concentrations that promote neoplastic transformation. Carcinogenesis 22, 1163–1172 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Kõivomägi M, Valk E, Venta R, Iofik A, Lepiku M, Balog ER, Rubin SM, Morgan DO, Loog M, Cascades of multisite phosphorylation control Sic1 destruction at the onset of S phase. Nature 480, 128–131 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zboray L, Pluciennik A, Curtis D, Liu Y, Berman-Booty LD, Orr C, Kesler CT, Berger T, Gioeli B, Paschal BM, Merry DE, Preventing the androgen receptor N/C interaction delays disease onset in a mouse model of SBMA. Cell Rep. 13, 2312–2323 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jochum T, Ritz ME, Schuster C, Funderburk SF, Jehle K, Schmitz K, Brinkmann F, Hirtz M, Moss D, Cato ACB, Toxic and non-toxic aggregates from the SBMA and normal forms of androgen receptor have distinct oligomeric structures. Biochim. Biophys. Acta 1822, 1070–1078 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Doan N-D, Bourgault S, Dejda A, Létourneau M, Detheux M, Vaudry D, Vaudry H, Chatenet B, Fournier A, Design and in vitro characterization of PAC1/VPAC1-selective agonists with potent neuroprotective effects. Biochem. Pharmacol 81, 552–561 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Bourgault S, Vaudry D, Botia B, Couvineau A, Laburthe M, Vaudry H, Fournier A, Novel stable PACAP analogs with potent activity towards the PAC1 receptor. Peptides 29, 919–932 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Lee EH, Seo SR, Neuroprotective roles of pituitary adenylate cyclase-activating polypeptide in neurodegenerative diseases. BMB Rep. 47, 369–375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu Z, Dadgar N, Albertelli M, Scheller A, Albin RL, Robins DM, Lieberman AP, Abnormalities of germ cell maturation and Sertoli cell cytoskeleton in androgen receptor 113 CAG knock-in mice reveal toxic effects of the mutant protein. Am. J. Pathol 168, 195–204 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rocchi A, Milioto C, Parodi S, Armirotti A, Borgia D, Pellegrini M, Urciuolo A, Molon S, Morbidoni V, Marabita M, Romanello V, Gatto P, Blaauw B, Bonaldo P, Sambataro F, Robins DM, Lieberman AP, Soraru G, Vergani L, Sandri M, Pennuto M, Glycolytic-to-oxidative fiber-type switch and mTOR signaling activation are early-onset features of SBMA muscle modified by high-fat diet. Acta Neuropathol. 132, 127–144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee S-O, Chintharlapalli S, Liu S, Papineni S, Cho SD, Yoon K, Safe S, p21 expression is induced by activation of nuclear nerve growth factor–induced Bα (Nur77) in pancreatic cancer cells. Mol. Cancer Res 7, 1169–1178 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lam BY, Zhang W, Ng DC-H, Maruthappu M, Roderick HL, Chawla S, CREB-dependent Nur77 induction following depolarization in PC12 cells and neurons is modulated by MEF2 transcription factors. J. Neurochem 112, 1065–1073 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Giorgetti E, Yu Z, Chua JP, Shimamura R, Zhao L, Zhu F, Venneti S, Pennuto M, Guan Y, Hung G, Lieberman AP, Rescue of metabolic alterations in AR113Q skeletal muscle by peripheral androgen receptor gene silencing. Cell Rep. 17, 125–136 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cortes CJ, Miranda HC, Frankowski H, Batlevi Y, Young JE, Le A, Ivanov N, Sopher BL, Carromeu C, Muotri AR, Garden GA, La Spada AR, Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat. Neurosci 17, 1180–1189 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou ZX, Kemppainen JA, Wilson EM, Identification of three proline-directed phosphorylation sites in the human androgen receptor. Mol. Endocrinol 9, 605–615 (1995). [DOI] [PubMed] [Google Scholar]
- 39.Zhu Z, Becklin RR, Desiderio DM, Dalton JT, Identification of a novel phosphorylation site in human androgen receptor by mass spectrometry. Biochem. Biophys. Res. Commun 284, 836–844 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Lindqvist J, Imanishi SY, Torvaldson E, Malinen M, Remes M, Örn F, Palvimo JJ, Eriksson JE, Cyclin-dependent kinase 5 acts as a critical determinant of AKT-dependent proliferation and regulates differential gene expression by the androgen receptor in prostate cancer cells. Mol. Biol. Cell 26, 1971–1984 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gioeli D, Black BE, Gordon V, Spencer A, Kesler CT, Eblen ST, Paschal BM, Weber MJ, Stress kinase signaling regulates androgen receptor phosphorylation, transcription, and localization. Mol. Endocrinol 20, 503–515 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Mellinghoff IK, Vivanco I, Kwon A, Tran C, Wongvipat J, Sawyers CL, HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell 6, 517–527 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Kesler CT, Gioeli D, Conaway MR, Weber MJ, Paschal BM, Subcellular localization modulates activation function 1 domain phosphorylation in the androgen receptor. Mol. Endocrinol 21, 2071–2084 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Baumann K, Mandelkow E-M, Biernat J, Piwnica-Worms H, Mandelkow E, Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett. 336, 417–424 (1993). [DOI] [PubMed] [Google Scholar]
- 45.Moujalled D, James JL, Yang S, Zhang K, Duncan C, Moujalled DM, Parker SJ, Caragounis A, Lidgerwood G, Turner BJ, Atkin JD, Grubman A, Liddell JR, Proepper A, Boeckers TM, Kanninen KM, Blair I, Crouch PJ, White AR, Phosphorylation of hnRNP K by cyclin-dependent kinase 2 controls cytosolic accumulation of TDP-43. Hum. Mol. Genet 24, 1655–1669 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Asada M, Yamada T, Ichijo H, Delia D, Miyazono K, Fukumuro K, Mizutani S, Apoptosis inhibitory activity of cytoplasmic p21Cip1/WAF1 in monocytic differentiation. EMBO J. 18, 1223–1234 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagy Z, The dysregulation of the cell cycle and the diagnosis of Alzheimer’s disease. Biochim. Biophys. Acta 1772, 402–408 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Delobel P, Lavenir I, Ghetti B, Holzer M, Goedert M, Cell-cycle markers in a transgenic mouse model of human tauopathy: Increased levels of cyclin-dependent kinase inhibitors p21Cip1 and p27Kip1. Am. J. Pathol 168, 878–887 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Humbert S, Bryson EA, Cordelières FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F, The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves Huntingtin phosphorylation by Akt. Dev. Cell 2, 831–837 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Jorgensen ND, Andresen JM, Lagalwar S, Armstrong B, Stevens S, Byam CE, Duvick LA, Lai S, Jafar-Nejad P, Zoghbi HY, Clark HB, Orr HT, Phosphorylation of ATXN1 at Ser776 in the cerebellum. J. Neurochem 110, 675–686 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rat D, Schmitt U, Tippmann F, Dewachter I, Theunis C, Wieczerzak E, Postina R, van Leuven F, Fahrenholz F, Kojro E, Neuropeptide pituitary adenylate cyclaseactivating polypeptide (PACAP) slows down Alzheimer’s disease-like pathology in amyloid precursor protein-transgenic mice. FASEB J. 25, 3208–3218 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reglodi D, Lubics A, Tamás A, Szalontay L, Lengvári I, Pituitary adenylate cyclase activating polypeptide protects dopaminergic neurons and improves behavioral deficits in a rat model of Parkinson’s disease. Behav. Brain Res 151, 303–312 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Tamás A, Lubics A, Lengvári I, Reglódi D, Protective effects of PACAP in excitotoxic striatal lesion. Ann. N. Y. Acad. Sci 1070, 570–574 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Ressler KJ, Mercer KB, Bradley B, Jovanovic T, Mahan A, Kerley K, Norrholm SD, Kilaru V, Smith AK, Myers AJ, Ramirez M, Engel A, Hammack SE, Toufexis D, Braas KM, Binder EB, May V, Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature 470, 492–497 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Born J, Lange T, Kern W, McGregor GP, Bickel U, Fehm HL, Sniffing neuropeptides: A transnasal approach to the human brain. Nat. Neurosci 5, 514–516 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Doberer D, Gschwandtner M, Mosgoeller W, Bieglmayer C, Heinzl H, Petkov V, Pulmonary and systemic effects of inhaled PACAP38 in healthy male subjects. Eur. J. Clin. Invest 37, 665–672 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Sambataro F, Pennuto M, Cell-autonomous and non-cell-autonomous toxicity in polyglutamine diseases. Prog. Neurobiol 97, 152–172 (2012). [DOI] [PubMed] [Google Scholar]
- 58.Pérez-García MJ, Burden SJ, Increasing MuSK activity delays denervation and improves motor function in ALS mice. Cell Rep. 2, 497–502 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walcott JL, Merry DE, Ligand promotes intranuclear inclusions in a novel cell model of spinal and bulbar muscular atrophy. J. Biol. Chem 277, 50855–50859 (2002). [DOI] [PubMed] [Google Scholar]
- 60.De Palma M, Naldini L, Transduction of a gene expression cassette using advanced generation lentiviral vectors. Methods Enzymol. 346, 514–529 (2002). [DOI] [PubMed] [Google Scholar]
- 61.Bony G, Szczurkowska J, Tamagno I, Shelly M, Contestabile A, Cancedda L, Non-hyperpolarizing GABAB receptor activation regulates neuronal migration and neurite growth and specification by cAMP/LKB1. Nat. Commun 4, 1800 (2013). [DOI] [PubMed] [Google Scholar]
- 62.Pozzi D, Lignani G, Ferrea E, Contestabile A, Paonessa F, D’Alessandro R, Lippiello P, Boido A, Fassio A, Meldolesi J, Valtorta F, Benfenati F, Baldelli P, REST/NRSF-mediated intrinsic homeostasis protects neuronal networks from hyperexitability. EMBO J. 32, 2994–3007 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grunseich C, Zukosky K, Kats IR, Ghosh L, Harmison GG, Bott LC, Rinaldi C, Chen K.-l., Chen G, Boehm M, Fischbeck KH, Stem cell-derived motor neurons from spinal and bulbar muscular atrophy patients. Neurobiol. Dis 70, 12–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gozes I, Perl O, Giladi E, Davidson A, Ashur-Fabian O, Rubinraut S, Fridkin M, Mapping the active site in vasoactive intestinal peptide to a core of four amino acids: Neuroprotective drug design. Proc. Natl. Acad. Sci. U.S.A 96, 4143–4148 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gozes I, Bardea A, Reshef A, Zamostiano R, Zhukovsky S, Rubinraut S, Fridkin M, Brenneman DE, Neuroprotective strategy for Alzheimer disease: Intranasal administration of a fatty neuropeptide. Proc. Natl. Acad. Sci. U.S.A 93, 427–432 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gozes I, Lilling G, Glazer R, Ticher A, Ashkenazi IE, Davidson A, Rubinraut S, Fridkin M, Brenneman DE, Superactive lipophilic peptides discriminate multiple vasoactive intestinal peptide receptors. J. Pharmacol. Exp. Ther 273, 161–167 (1995). [PubMed] [Google Scholar]
- 67.Espinoza S, Salahpour A, Masri B, Sotnikova TD, Messa M, Barak LS, Caron MG, Gainetdinov RR, Functional interaction between trace amine-associated receptor 1 and dopamine D2 receptor. Mol. Pharmacol 80, 416–425 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Salahpour A, Espinoza S, Masri B, Lam V, Barak LS, Gainetdinov RR, BRET biosensors to study GPCR biology, pharmacology, and signal transduction. Front. Endocrinol. (Lausanne) 3, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jochum T, Cato AC, Analysis of the conformation of the androgen receptor in spinal bulbar muscular atrophy by atomic force microscopy. Methods Mol. Biol 1204, 197–204 (2014). [DOI] [PubMed] [Google Scholar]
- 70.Yu Z, Dadgar N, Albertelli M, Gruis K, Jordan C, Robins DM, Lieberman AP, Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J. Clin. Invest 116, 2663–2672 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Albertelli MA, Scheller A, Brogley M, Robins DM, Replacing the mouse androgen receptor with human alleles demonstrates glutamine tract length-dependent effects on physiology and tumorigenesis in mice. Mol. Endocrinol 20, 1248–1260 (2006). [DOI] [PubMed] [Google Scholar]
- 72.Alcalay RN, Giladi E, Pick CG, Gozes I, Intranasal administration of NAP, a neuroprotective peptide, decreases anxiety-like behavior in aging mice in the elevated plus maze. Neurosci. Lett 361, 128–131 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of PACAP analogs and cAMP release in MN-1 cells treated with either PACAP or the indicated PACAP analogs.
Fig. S1. Overexpression of PKA reduces the accumulation of the upper isoform of polyQ-AR.
Fig. S2. PACAP stimulates the release of cAMP in MN-1 cells expressing AR24Q and AR65Q.
Fig. S3. VIP does not modify the accumulation of the upper isoform of polyQ-AR.
Fig. S4. Expression of polyQ-AR in MN-1 cells results in caspase 3 activation.
Fig. S5. Analysis of polyQ-AR phosphorylation upon inhibition of specific cellular phosphatases and kinases.
Fig. S6. Modulation of CDK1 and CDK5 activity does not affect the accumulation of the upper isoform of polyQ-AR.
Fig. S7. Nonexpanded AR and polyQ-AR colocalize with endogenous CDK2 in the absence and presence of androgens in MN-1 and PC12 cells.
Fig. S8. Nonexpanded AR and polyQ-AR colocalize with endogenous CDK2 in the brainstem motor neurons of control and knock-in SBMA mice.
Fig. S9. Nonexpanded AR and polyQ-AR colocalize with endogenous CDK2 in the motor neurons of the lumbar spinal cord of control and knock-in SBMA mice.
Fig. S10. Nonexpanded AR colocalizes with endogenous CDK2 in the quadriceps of control mice.
Fig. S11. Endogenous nonexpanded AR and polyQ-AR colocalize with endogenous CDK2 in control and SBMA patient-derived motor neurons and NPCs.
Fig. S12. Endogenous polyQ-AR colocalizes with endogenous CDK2 in the spinal cord of an SBMA patient.
Fig. S13. Forskolin, PACAP, and the pan-phosphatase inhibitor OA stimulate p21Cip1 expression.
Fig. S14. Ser96 of AR is conserved throughout evolution.
Fig. S15. Analysis of phosphoresistant polyQ-AR variants.
Fig. S16. Ser96 phosphorylation is responsible for the formation of the upper isoform of polyQ-AR.
Fig. S17. Phosphodefective and phosphomimetic substitution of Ser96 does not affect polyQ-AR subcellular localization.
Fig. S18. Forskolin and PACAP, and not VIP, reduce polyQ-AR aggregation.
Fig. S19. Activation of the AC/PKA signaling increases the turnover of polyQ-AR.
Fig. S20. OA reduces the accumulation of phosphorylated polyQ-AR by inducing degradation through the UPS.
Fig. S21. PACAP induces cAMP production in vivo.
Fig. S22. Effect of intranasal administration of peptide 7 on body weight, food intake, and survival of AR113Q mice.
Fig. S23. Peptide 7 increases the CSA of glycolytic fibers in the skeletal muscle of AR113Q mice and restores the expression of glycolytic genes to normal levels.
Fig. S24. Intranasal administration of peptide 7 decreases polyQ-AR phosphorylation and accumulation in SBMA mice without altering CDK2, p27Kip1, and p57Kip2 expression levels.
Fig. S25. Data presented for experiments with sample sizes of less than 20.