

Abstract
Leishmania donovani adenosine kinase (LdAdK) plays a pivotal role in scavenging of purines from the host. Exploiting interspecies homology and structural co-ordinates of the enzyme from other sources, we generated a model of LdAdK that led us to target several amino acid residues (namely Gly-62, Arg-69, Arg-131 and Asp-299). Replacement of Gly-62 with aspartate caused a drastic reduction in catalytic activity, with decreased affinity for either substrate. Asp-299 was found to be catalytically indispensable. Mutation of either Arg-131 or Arg-69 caused a significant reduction in kcat. R69A (Arg-69→Ala) and R131A mutants exhibited unaltered Km for either substrate, whereas ATP Km for R69K increased 6-fold. Importance of both of the arginine residues was reaffirmed by the R69K/R131A double mutant, which exhibited approx. 0.5% residual activity with a large increase in ATP Km. Phenylglyoxal, which inhibits the wild-type enzyme, also inactivated the arginine mutants to different extents. Adenosine protected both of the Arg-69 mutants, but not the R131A variant, from inactivation. Binding experiments revealed that the AMP-binding property of R69K or R69A and D299A mutants remained largely unaltered, but R131A and R69K/R131A mutants lost their AMP binding ability significantly. The G62D mutant did not bind AMP at all. Free energy calculations indicated that Arg-69 and Arg-131 are functionally independent. Thus, apart from the mandatory requirement of flexibility around the diglycyl (Gly-61–Gly-62) motif, our results identified Asp-299 and Arg-131 as key catalytic residues, with the former functioning as the proton abstractor from the 5′-OH of adenosine, while the latter acts as a bidentate electrophile to stabilize the negative charge on the leaving group during the phosphate transfer. Moreover, the positive charge distribution of Arg-69 probably helps in maintaining the flexibility of the α-3 helix needed for proper domain movement. These findings provide the first comprehensive biochemical evidence implicating the mechanistic roles of the functionally important residues of this chemotherapeutically exploitable enzyme.
Keywords: active site, adenosine kinase, Leishmania donovani, purine salvage, ribokinase, site-directed mutagenesis
Abbreviations: AdK, adenosine kinase; Ado, adenosine; LdAdK, Leishmania donovani AdK; Ni-NTA, Ni2+-nitrilotriacetate; PfkB, phosphofructokinase B; PGO, phenylglyoxal
INTRODUCTION
AdK (adenosine kinase), an enzyme of the purine-salvage pathway, catalyses magnesium-dependent transfer of the terminal phosphate from ATP to Ado (adenosine). Besides phosphorylating Ado, the enzyme also recognizes a wide variety of Ado analogues as substrates, thus making it a prospective target for chemotherapy [1]. Although the enzyme was kinetically characterized from a number of sources, including higher eukaryotes [2–4], parasitic protozoa [5–8], plants [9] and bacteria [10], the mechanism of substrate recognition and phosphorylation of various nucleosides/analogues by this enzyme was the subject of substantial debate until recently. Using 6-methylmercaptopurine riboside as the substrate, Henderson et al. [11] claimed that the enzyme from Ehrlich ascites tumour cells carried out a reaction in which ATP was the first substrate to bind and 6-methylmercaptopurine riboside monophosphate was the last product to be released. However, subsequent results from most other laboratories were consistent with a reaction sequence in which Ado is the first substrate to bind and AMP is the last product to be released [2,4,12]. Mechanistic studies showing net stereochemical inversion of the γ-phosphate group suggested a direct in-line transfer of the phosphoryl group from ATP to Ado, as opposed to a doubledisplacement mechanism involving a phosphoenzyme intermediate [13]. This was established further by the inhibition studies with the bi-substrate analogues of AdK [14].
The translated amino acid sequence of the enzyme from different sources [15–17] showed that AdK, unlike other nucleoside and nucleotide kinases, is a member of the PfkB (phosphofructokinase B) family of carbohydrate kinases, namely ribokinase, inosine-guanosine kinase, fructokinase and 1-phosphofructokinase [18,19]. This conclusion is based on the presence of two common structural motifs characterized by short sequence stretches, one containing a highly conserved diglycine motif located near the N-terminal end and the other containing a conserved DTXGAGD motif, positioned near the C-terminus of all enzymes of this family [18,19].
Apart from these studies, the recently solved crystal structures of human and Toxoplasma gondii AdKs gave further insight into the structure–function relationship of AdK [20–22]. Their findings revealed that it consists of two unequal-sized domains. The large domain is a three-layered sandwich of α-helices and β-sheets over which the small domain forms a lid. The cleft formed between the two domains contains the catalytic Ado-binding site. Comparison of the structures of T. gondii AdK, in the presence and absence of the substrate, further revealed both global and local changes in the protein structure upon binding of Ado [22]. The most striking among them is the Ado-induced 30° hinge-bending motion leading to domain closure. This allows ATP binding, followed by subsequent conformational modulation of the protein, which ultimately results in the phosphotransfer reaction. Similar to the ribokinase family of proteins, a diglycyl motif (Gly-68–Gly-69), present in T. gondii AdK, has been proposed to be instrumental in this conformational switching. Another interesting observation was that upon Ado-induced domain closure, the small domain residue Arg-136 is translocated 13.7 Å (1 Å=0.1 nm) into the active site [22]. This positively charged residue has been postulated to interact with the γ-phosphate of ATP. Apart from these, several key residues were implicated to play a role in either substrate recognition or catalysis. Among them, the carboxy group of a highly conserved aspartate residue (Asp-318 for T. gondii AdK, and Asp-300 for human AdK) has been shown to form a hydrogen bond with the ribosyl O5′ hydroxy group of Ado and is predicted to have important role in catalysis [20,22]. Despite the predictions made from the structural analysis, apart from a preliminary report [23], actual biochemical evidence explaining the catalytic mechanism and substrate-binding characteristics still remains to be validated.
Leishmania donovani, a purine auxotrophic protozoan parasite that has a dimorphic life cycle, is the causative agent of visceral leishmaniasis in humans. This protozoan exists as a flagellated promastigote (extracellular form) in the sandfly vector and is transformed into amastigote (intracellular form) in the human macrophages. Since purines are essential components for survival, this group of organisms, in order to overcome their inability to synthesize purines de novo, have developed a unique series of purine-salvage enzymes by which purine bases could be scavenged from the host [24,25]. This made the purine-salvage enzymes an attractive target for designing anti-leishmanial drug.
LdAdk (L. donovani AdK), which is known to be a developmentally regulated stage-specific peak-function enzyme, is one of such candidates [24,26]. In contrast with its mammalian counterparts, LdAdK is not subject to inhibition by Ado and magnesium [5]. Moreover, unlike the results of studies by Maj et al. [27], our purified enzyme preparation failed to show dependency for quinquivalent and phosphate ions (R. Datta and I. Das, unpublished work). Until now, the only information on the active site architecture of LdAdK is based on our chemical modification studies [28–30]. With the use of group-specific chemical modifiers, it was shown that the L. donovani enzyme harbours at least one important arginine and two conformationally vicinal cysteine residues at or close to the Ado-binding site [29,30].
Previously, the enzyme has been cloned in our laboratory and hyperexpressed in Escherichia coli [17], thereby enabling us for the first time to undertake its structure–function analysis in a systematic manner. In the absence of structural information of LdAdK, a homology-based model of the protein utilizing available co-ordinates from the crystal structures of AdK from human and T. gondii has been constructed. Amino acid sequence alignment of LdAdK with its counterparts from different sources (Figure 1A) revealed that: (i) in LdAdK, Gly-62 appears to be a member of the diglycine (Gly-61–Gly-62) signature motif that is essential for maintaining the conformational flexibility of this family of proteins; (ii) among the seven arginine residues present in LdAdK, only Arg-131, corresponding to Arg-136 and Arg-132 of T. gondii and human AdK respectively, is absolutely conserved among AdKs from all species; (iii) interestingly, the arginine residue located at the 69th position of LdAdK seems to be conserved mostly among the lower eukaryotes [7,8,17]; contrastingly, in higher eukaryotes, this residue is replaced with lysine [15,16]; and (iv) Asp-299 of LdAdK, which aligns respectively with Asp-318 and Asp-300 of T. gondii and human AdK, is located on the second fingerprint motif (DTXGAGD) of the ribokinase family, and has been predicted to be an important catalytic residue [20,22].
Figure 1. Amino acid sequence alignment and homology modelling of LdAdK.

(A) Alignment of LdAdK amino acid sequence with that of T. gondii (AAF01261), Babesia canis (CAA11263), Saccharomyces cerevisiae (P47143), human (AAA97893) and Chinese hamster ovary (CHO; AAA91648) AdK (numbers in parentheses indicate GenBank® accession numbers for respective proteins). Boxes indicate amino acid signature motifs distinctive of this family of proteins. The residues selected for site-directed mutagenesis, i.e. Gly-62, Arg-69, Arg-131 and Asp-299, of LdAdK are indicated in bold. The boxes represent amino acid sequence motifs that are distinctive of the PfkB family of carbohydrate kinases. (B) Homology model showing the overall structure of LdAdK with α-helices drawn as ribbons and β-strands as arrows. Spatial position of Ado (magenta), ATP (yellow), Gly-62 (cyan), Arg-69 (red), Arg-131 (white) and Asp-299 (pink) are shown in space-fill mode. (C) Zoomed picture of the active-site residues, with inter-atomic distances shown by broken lines.
In the present study, we specifically altered the above amino acids either singly or in appropriate combination to investigate their biochemical role in substrate binding and catalysis. Our data show that alteration of the diglycine (Gly-61–Gly-62) motif resulted in almost complete loss of functional activity of the enzyme, thus supporting the results obtained from the structural studies, and suggested important mechanistic roles of Arg-131 and Asp-299 in the phosphotransfer reaction of the enzyme. A role for Arg-69 in the functional expression of the enzyme, which could not be predicted from the modelled structure of the enzyme, has also been highlighted. Based on these results, a concerted reaction mechanism for LdAdK is proposed.
EXPERIMENTAL
Materials
Unless otherwise mentioned, all reagents were purchased from Sigma Chemicals. Restriction enzymes, Taq DNA polymerase, T4 DNA ligase and oligonucleotides were obtained from Gibco BRL. Mutagenesis kits were purchased from either Amersham Biosciences or Stratagene. N-terminal His-tag expression vector (pQE30) and Ni-NTA (Ni2+-nitrilotriacetic acid)–agarose resin were the products of Qiagen.
Site-directed mutagenesis
Mutagenesis was performed using either the Sculptor™ in vitro mutagenesis system or Stratagene's QuikChange kit following the respective manufacturer's protocol. For the Sculptor™ kit, the open reading frame of the wild-type gene was cloned into the EcoRI/BamHI sites of the vector M13mp8.
For carrying out the desired mutations, the following oligonucleotides, with their substitution sites underlined, were used: R69K, 5′-GATCCACTGCGCAACCTTGGCGGTGTTGAGGCC-3′; R69A, 5′-TGCGCCACGGCGGCGGTGT-3′; and G62D, 5′-AGGCCAGAGTCGCCAGGGAC-3′.
However, to carry out mutation using the QuikChange kit, the N-terminal His-tagged expression plasmid (pQE30) harbouring the wild-type AdK gene was used directly as the template. The following sense primers (along with their antisense counterparts), with their substitution sites underlined, were used: R131A, 5′-CATCTCAGGCAAGGATGCCTCGCTGGTGGCGAAC-3′, and D299A, 5′-AACGGCGCCGGTGCCGCCTTCGTTGGC-3′.
To generate the double mutant R69K/R131A, the same primers used for generating the R131A mutant, were used on the mutant R69K template. Mutations were confirmed by automated DNA sequencing in a PerkinElmer ABI Prism™ DNA sequencer.
Expression and purification of the wild-type and mutant enzymes with His-tag
For the expression of mutant genes generated using the Sculptor™ kit, double-stranded RF (replicative form) DNA was isolated from mutant M13mp8 bacteriophages following the manufacturer's protocol and subjected to PCR amplification using the following primers: initiation primer, 5′-CTCGGGATCCGCGCTTCCGCAGCTCTAC-3′, and termination primer, 5′-CTCGGGTACCTCACGGAGAGATGGACGG-3′.
The PCR products obtained were digested with BamHI/KpnI and ligated into the BamHI/KpnI sites of the pQE30 vector. Since the QuikChange method permits direct use of the expression plasmid (pQE30) for carrying out the desired mutation, the mutant DNA generated by this method was used as such for expression. E. coli M15 [pREP4] cells harbouring the wild-type and mutant plasmids were used for induction of the protein. The IPTG (isopropyl β-D-thiogalactoside)-induced proteins were purified using a Ni-NTA–agarose column following the manufacturer's suggested protocol. In brief, the induced cells were harvested by centrifugation at 5000 g for 10 min at 4 °C and subjected to lysis using buffer containing 50 mM potassium phosphate, pH 7.5, 150 mM NaCl, 0.5% (v/v) Triton X-100, 1 mM PMSF, 10 mM 2-mercaptoethanol, 20 mM imidazole and 1 mg/ml lysozyme. The resultant suspension was sonicated three to four times for 30 s each at maximal output on a low setting in a Labsonic 2000 sonicator (B. Braun, Melsungen, Germany) and centrifuged at 27000 g for 30 min at 4 °C. The supernatant was directly loaded on to a Ni-NTA column previously equilibrated with Buffer A [50 mM potassium phosphate, pH 7.5, 300 mM NaCl, 0.5% Triton X-100, 1 mM PMSF, 10% (v/v) glycerol, 0.2 M urea, 0.25 M sorbitol and 10 mM 2-mercaptoethanol] containing 20 mM imidazole. The flowthrough was recycled three times. The column was then washed with the same buffer to remove non-specifically bound proteins. The bound proteins were then eluted with Buffer A containing 80 mM imidazole. The purity of the proteins was checked by SDS/PAGE (13% gel), followed by Coomassie Brilliant Blue staining.
AdK assay and initial velocity determinations
The AdK assay involved determination of AMP formation from [2-3H]Ado (50 μM) in presence of ATP (1 mM) and magnesium (1.5 mM). The assay was carried out at 30 °C [5]. For initial rate determination, the linearity of all plots was checked graphically for all substrate concentrations, and the initial velocities of reactions were fitted into the Michaelis–Menten equation, i.e. rectangular hyperbola and double-reciprocal plots were generated according to Cleland [31]. Secondary plots were used to determine kinetic constants.
CD spectroscopy
Spectral acquisition was performed on a Jasco-700 spectropolarimeter. Wild-type or mutant LdAdK (2.5 μM) in 20 mM phosphate buffer (pH 7.5) was placed in a cuvette with a 2 mm path length and a volume of 1.2 ml. Five scans were made at 50 nm/min between 195 and 260 nm in 1.00 nm increments at room temperature (25 °C). After subtraction of the buffer spectra, the data were converted into molecular ellipticity units.
AMP-affinity chromatography
AMP-affinity chromatography of the wild-type and mutant enzymes was carried out as described previously [5], with some minor modifications. Briefly, aliquots (500 μl) from the original stock (200 μg/ml) of each purified protein preparation (both wild-type and various mutants) were loaded on to several identically sized (500 μl bed volume) AMP–agarose columns previously equilibrated with Buffer B [100 mM potassium phosphate, pH 7.5, 1 mM dithiothreitol and 5% (v/v) glycerol]. In each case, the flowthrough was recycled three times to ensure proper binding. Columns were then washed extensively with Buffer B containing 300 mM KCl. Protein that remained bound to the column was then eluted with 3 ml of Buffer B containing 5 mM Ado. The entire 3 ml eluate was then freeze-dried to a final volume of 500 μl. For direct comparison of binding to the AMP column, an identical volume (15 μl) from the original stock (200 μg/ml) and the freeze-dried eluate of each protein were subjected to SDS/PAGE analysis.
Chemical modification studies
The modification reactions were carried out in Buffer C [20 mM Tris/HCl, pH 7.5, 20% (v/v) glycerol and 1 mM dithiothreitol]. PGO (phenylglyoxal), freshly dissolved in DMSO, was used. DMSO up to a concentration of 10–15% did not have any effect on the enzyme activity. Wild-type or mutant enzymes (200 μg/ml) were incubated with PGO (15 mM) in a final volume of 50 μl. Incubation of the enzyme with DMSO served as the control. After 30 min, aliquots were withdrawn and suitably diluted in Buffer C to stop further reaction. From the diluted mixture, enzyme (50 ng) was withdrawn and assayed for enzyme activity. For the substrate-protection experiment, Ado (300 μM) was added before PGO treatment. The extent of inactivation was monitored by measuring residual enzyme activity in the chemically modified enzyme with respect to unmodified enzyme as the control.
Modelling studies
As no experimentally determined three-dimensional structure of LdAdK is available as yet, we constructed a three-dimensional model by homology modelling. The published X-ray crystallographic structures of human and T. gondii AdK were used as reference templates [20,22]. Prediction of the three-dimensional structure of LdAdK was done by knowledge-based homology modelling using the InsightII 98.0 of MSI (Biosym Technologies, San Diego, CA, U.S.A.), ABGEN [32] and our in-house package of MODELYN and ANALYN [33] in UNIX as well as in the Microsoft Windows environment. Energy minimization and molecular dynamics were performed with the InsightII 98.0/Discover package using a CFF91 forcefield on a Silicon Graphics® OCTANE workstation. Energy minimization was carried out with a convergence criterion of 1 cal/mol (1 cal≈4.184 J/mol), using a combination of steepest descent and conjugate gradient methods (100 steps each). Molecular dynamics simulations were carried out using a time step of 1 fs for 1000 steps after 100 steps of equilibration. For regularization of the structures in which major alterations were carried out, alternative steps of minimizations and dynamics were repeated until satisfactory conformational parameters were obtained. The ribbon structure was determined using the program Ribbons [34]. Multiple sequence alignment was performed using CLUSTAL W [35].
RESULTS
Structural analysis of LdAdK
Similar to the AdK from various sources, LdAdK does not bear any sequence similarity with other nucleoside kinases [15–17]. Although the enzyme from other higher eukaryotes is more than 95% conserved among themselves, homology alignment studies revealed that LdAdK has only approx. 40% and 31% identity with the enzymes from human and T. gondii respectively (Figure 1A). Despite this limited identity, the primary sequence of LdAdK possesses all the characteristics typical of AdK from all known sources. First, similar to that of other AdKs, LdAdK lacks the consensus P-loop motif [17] and secondly, LdAdK harbours two amino acid sequence motifs (denoted by boxes in Figure 1A) that are distinctive of the PfkB family of carbohydrate kinases [18,19].
Based on the sequence alignment of LdAdK with that of human and T. gondii AdK (Figure 1A) and available co-ordinates of the human and T. gondii AdK crystal structures, we generated an energy-minimized structure of LdAdK using the InsightII molecular modelling software (Figure 1B). The ribbon diagram of the model indicated that, despite limited sequence identity with its human and T. gondii counterparts, LdAdK possesses a high level of overall structural and active site geometric symmetry. The protein appears to be organized into two domains: a large domain consisting of a three-layered sandwich of ten α-helices and nine β-strands which is connected by four peptide segments to the smaller domain (the lid), which consists of five β-strands running perpendicular to two α-helices. The distance between the terminal phosphate group of ATP and the 5′-OH group of the ribose moiety of Ado was found to be 1.68 Å, close enough for a direct in-line phosphate transfer. A careful examination of the structure showed that Gly-62 is located underneath the Ado-binding site of the peptide connecting β4 sheet of the small domain with α3 helix of the large domain. A closer view of the active site (Figure 1C) revealed that the peptide N of Gly-62 is 2.44 Å and 2.16 Å away from the O2′ and O3′ group of the adenosyl ribose respectively, suggesting its possible involvement in Ado binding. Furthermore, of the seven arginine residues present in LdAdK, only one arginine residue (Arg-131), located on the β8 sheet of the small domain, appeared to be spatially close to the active site. In fact, its NH1 and NH2 domains are at potentially H-bonding distance of 2.2 Å and 2.3 Å respectively from the O2G and O3G group of the terminal phosphate of ATP. This observation is clearly distinct from that of T. gondii AdK, where, instead of two terminal amino groups, NH2 and NE were predicted to interact with the terminal phosphate [22]. Spatial location of Arg-69, located on the α-3 helix of the large domain, was found to be quite far away from either of the substrates (>10.0 Å). The carboxy side chain of Asp-299 is proximally located to the ribosyl O5′ hydroxy group of Ado (2.5 Å), indicating that it might serve as the catalytic base.
Expression and purification of the wild-type and mutant enzymes
Based on the sequence analysis and structural studies, the following mutants of LdAdK were generated: G62D, R131A, R69K, R69A, R69K/R131A and D299A. It must be reiterated here that, although Arg-69 is quite far away from either of the substrates (>10.0 Å), its choice for mutation was based on the finding that the residue is strictly conserved among the lower eukaryotes, but is replaced with lysine in all of the higher eukaryotes so far reported. Moreover, LdAdK, as opposed to AdK from the higher eukaryotes, was inactivated with arginine-modifying agents [29]. The wild-type and mutant LdAdKs were expressed and purified as N-terminal His-tagged protein. The extent of purity of each protein was ascertained by separation on a SDS/13% polyacrylamide gel, followed by staining with Coomassie Brilliant Blue. All of the mutants, along with the wild-type, were found to be more than 95% pure and of similar molecular mass (results not shown).
CD spectra
Comparison of the CD spectra showed that all the mutants were similar to that of the wild-type enzyme. All of them exhibited minima at 209 nm and 221 nm (results not shown). These results indicated that none of the mutations caused appreciable change in the secondary structure of the enzyme.
Kinetic properties of different mutants
Kinetic parameters of the purified wild-type and the mutant enzymes were determined using Ado and ATP as substrates (Table 1). Replacement of Gly-62 with an aspartate residue resulted in a drastic loss in the catalytic efficiency (∼0.3% of wild-type) of the mutant enzyme. The affinity of the enzyme for Ado was reduced to such an extent that its Km for Ado could not be determined due to non-saturation kinetics. The apparent Km for ATP for the G62D mutant showed an approx. 3-fold higher value as compared with the control. Since CD studies confirmed almost no alteration in the overall structure of the mutant enzyme from its wild-type counterpart, the biochemical experiment provided additional support to the inference drawn from the structural studies [21,22] that rotation of the diglycyl (Gly-61–Gly-62) peptide bond, a prerequisite for proper binding of ATP and subsequent functional activity of the enzyme, was probably hindered due to replacement of the glycine with a bulky aspartate residue. That this was indeed the case was supported when Gly-62 was replaced with aspartate in the modelled structure (Figure 2). From the model of the wild-type enzyme, it is quite clear that the backbone amide nitrogen of Gly-62, located in the peptide connecting the two domains, is within the hydrogen-bonding distance from the O2′ and O3′ groups of the ribose moiety of Ado, and therefore has every possibility of forming bonds (Figure 2A). However, upon replacement of the glycine residue with a bulky aspartate, two additional hydrogen-bonding interactions developed with neighbouring Leu-65 and Asn-66 residues. Formation of these two additional hydrogen bonds probably resulted in a restricted rotation of Asp-62, thereby making the ‘hinge-bending’ of domain 2 over domain 1 difficult (Figure 2B).
Table 1. Comparison of kinetic data of wild-type and the mutant forms of LdAdK.
Km, Vmax and kcat values were determined from double-reciprocal plots against initial velocities. The assays were performed at 30 °C. Results are means±S.D. for three replicate experiments.
| Ado | ATP | |||||
|---|---|---|---|---|---|---|
| Enzyme | Km (μM) | kcat (min−1) | kcat/Km (μM−1·min−1) | Km (μM) | kcat (min−1) | kcat/Km (μM−1·min−1) |
| Wild-type | 20±5 | 86±4 | 4.3 | 42±4 | 72±6 | 1.71 |
| G62D | ND* | ND* | ND* | 139±3† | 0.20±0.008† | 0.001† |
| R131A | 16±1 | 3.9±0.5 | 0.24 | 71±2 | 3.1±0.3 | 0.043 |
| R69K | 17±2 | 2.6±0.1 | 0.153 | 278±10 | 1.94±0.02 | 0.007 |
| R69A | 15±3 | 2.3±0.2 | 0.153 | 68±2 | 1.75±0.01 | 0.025 |
| R69K/R131A | 16±2‡ | 0.37±0.04‡ | 0.023‡ | 1282±12 | 0.46±0.01 | 0.0004 |
| D299A | ND§ | ND§ | ND§ | ND§ | ND§ | ND§ |
* Values could not be determined due to non-saturation kinetics.
† When the Km value for Ado could not be determined owing to non-saturation kinetics, the Km and kcat values for ATP are apparent.
‡ These are apparent values owing to the fact that saturating concentration of ATP could not be used.
§ Kinetic parameters could not be determined because activity was too low.
Figure 2. Comparison of hydrogen bond network of the wild-type and the G62D mutant by molecular modelling.

Homology models showing the hydrogen bond network of (A) Gly-62 residue in wild-type LdAdK and (B) Asp-62 residue in the G62D mutant of LdAdK. The arrow indicates two extra hydrogen bonds observed in the G62D mutant.
In order to reassess the results of our earlier PGO-modification studies [29] and the predictions of the functional role of arginine residue deduced from the crystal structure of T. gondii AdK [22], we mutated two arginine residues (Arg-131 and Arg-69) based on the rationale described before. The R131A mutant showed a substantial loss of activity with more than 95% reduction in turnover with respect to the wild-type enzyme, despite limited alteration of Km for either Ado or ATP, implicating the involvement of this residue in catalysis rather than binding of the substrate. Interestingly, mutation of Arg-69, which appeared to be far away from the active site, to either lysine or alanine also showed a 30–40-fold reduction in the kcat. Although there was no significant change in the Ado Km for either the R69K or R69A mutants, a 6-fold increase in the Km for ATP of the mutant R69K was observed. Since the increase in ATP Km was not observed for the R69A mutant, it is likely that the positive charge of the side chain is not important for ATP binding. Because mutation of either Arg-69 or Arg-131 did not cause a total loss of catalytic activity of the enzyme, it was of interest to test the pattern of activity in the double mutant, R69K/R131A. The effect was so drastic that the kcat value decreased to ∼0.5% of the wild-type. While the Ado Km of the double mutant remained unchanged, the ATP Km increased almost 30-fold with a concomitant reduction in the kcat/Km value >4000-fold, indicating a significant alteration in the ATP-binding property. Kinetic results of the double mutant thus signified importance of both the arginine residues.
To have experimental evidence as to whether the hydrogen bonding between the carboxy group of Asp-299 and 5′-hydroxy group of adenosyl ribose, as proposed from the structural studies, plays any role either in substrate recognition or in the phosphate transfer step, we mutated this acidic residue to a neutral alanine. The catalytic activity of the D299A mutant was completely knocked out, indicating that this residue is absolutely indispensable for catalytic function of LdAdK. Our result reaffirmed the importance of this highly conserved residue that had been suggested by other investigators [20,22,23].
Comparison of AMP-binding property of the wild-type with that of the mutant enzymes
Observations from a number of laboratories have shown that AMP, one of the products of the reaction, acts as an inhibitor of AdK by competing for the Ado-binding site [12]. This property allows binding of the enzyme to the AMP–agarose affinity column, which subsequently can be eluted with Ado [5]. Given this, it would be reasonable to extrapolate that any change in the AMP-binding property of the enzyme should be reflected in its affinity for Ado. To explore whether the mutations created above caused any alterations in the Ado-binding behaviour, equal amount of the purified enzyme preparations from the wild-type and the various mutants were passed separately through identically sized AMP–agarose columns under conditions where most of the wild-type enzyme binds. Following extensive washing, the protein bound to the column was eluted with buffer containing Ado. Figure 3 shows a comparative account of the amount of enzymes (either wild-type or a mutant) loaded on to the column (lane 1) and the amount which remained bound to the column after extensive washing (lane 2). From the results, it is clear that there was no significant alteration in the AMP-binding property of the R69K, R69A or D299A mutant proteins, suggesting that these residues are not obligatory for binding of Ado. On the contrary, the G62D mutant did not bind to the column at all, implicating involvement of this residue in Ado binding. These findings are consistent with our structural and kinetic data. Surprisingly, the AMP-binding affinity of both R131A and R69K/R131A mutants was significantly affected, despite very little change in their Km for Ado, indicating that the positive charge of Arg-131 possibly exerts some additional influence while binding to the negatively charged AMP as opposed to neutral Ado. Support for this explanation comes from the fact that the arginine residue is well known for binding or stabilizing the binding of negatively charged groups of substrates or co-enzymes [36–38].
Figure 3. AMP-binding property of wild-type and mutant LdAdKs.

AMP-binding affinity of wild-type and mutant enzymes was compared by passing each of them through AMP–agarose affinity column. Lane 1 shows the amount of protein loaded on the column, and lane 2 shows the amount that remained bound to the column after extensive washing.
Chemical modification of the arginine mutants
Our earlier studies on chemical modification experiments had suggested that, in contrast with AdK from higher eukaryotes, LdAdK harbours at least one important arginine residue; modification of which led to inactivation of the enzyme. Based on Ado protection experiments, it was concluded that the residue probably was located at or close to the Ado-binding site [29]. The kinetic studies of the present study suggested that Arg-131 and Arg-69 are the two arginine residues that have important functional roles. Construction of mutant proteins with substitutions at these two residues therefore allowed us to reinvestigate the consequence of PGO modification of these mutant enzymes (Figure 4). Results showed that all the single arginine mutants (R131A and R69K or R69A) were inactivated to variable extents following modification with PGO, but the extent of inactivation of the R131A variant was somewhat reproducibly less than the R69K or R69A variants. Moreover, Ado afforded protection only to the R69K and R69A mutants, but not at all to the R131A mutant, despite the fact that its Ado Km remained unaltered. These results suggest that, of the seven arginine residues present in LdAdK, both Arg-131 and Arg-69 residues are the targets for PGO modification that leads to inactivation. Furthermore, our Ado protection experiment supported the modelling data that suggested closer proximity of the Arg-131 residue to the active site in comparison with Arg-69 and hence somewhat enhanced susceptibility to PGO inactivation.
Figure 4. Effect of PGO on arginine variants of LdAdK.

Unmodified activity of each enzyme was considered to be 100% (white bars). PGO (15 mM) for 30 min was used for modification (black bars) and 0.3 mM Ado was used for the substrate-protection experiment (grey bars). Actual specific activities of R131A, R69K and R69A are 5.3, 3.6 and 3.2% of wild-type respectively. Similar experiments could not be performed with the R69K/R131A double mutant because its activity is too low for this kind of inhibition studies.
DISCUSSION
To understand precisely (i) the mechanism by which the LdAdK carries out phosphotransfer reaction, (ii) the similarities and the distinguishing features of its active site from the counterpart enzyme from other sources, and (iii) the specific mechanistic role of different amino acid residues located in and around the interface of the two domains, we combined both molecular modelling and site-directed mutational analysis of its active site.
The kinetic parameters of the G62D variant, its elution behaviour from the AMP column and the modelling studies confirmed that, among the cascade of steps that ultimately result in the phosphate transfer reaction, the first step that involves Ado-induced domain rotation clearly did not occur due to the absence of necessary steric complementarity and lack of a requisite hydrogen-bonding network in the mutant enzyme. Obviously, replacement of glycine with the relatively bulky aspartate residue resulted in such distortion of the active site structure thereby affecting the binding of Ado. Loss of nucleoside-binding affinity of the enzyme observed due to replacement of O2′ hydroxy group of Ado by a methoxy group and reduction of affinity by substitution with a hydrogen indirectly support this proposal [22]. The reduced affinity of the mutant enzyme for ATP is consistent with the structural prediction that Ado-induced domain rotation helps in proper binding of ATP. The notion of a freely rotatable glycine residue as the underlying principle in the process of conformational switching is not unique to AdK, and has been well documented for other kinases [39,40], of which hexokinase is a classical example [41]. Moreover, binding of the lead substrate in an ordered enzymatic reaction has often been associated with such conformational alteration of the active site [40]. Our findings, along with other structural studies carried on AdKs [20,22], support the proposal that Ado is the first substrate to bind, followed by ATP.
The catalytic role of an aspartate residue acting as a general base is well established for a number of phosphoryl transfer enzymes [42,43]. A preliminary analysis of the hamster AdK by Maj et al. [23] provided more direct evidence for the involvement of an aspartate residue in proper functioning of the enzyme. Our model of LdAdK also showed that Asp-299 (corresponding to Asp-300 of human and Asp-318 of T. gondii AdK) is ideally positioned for interaction with the 5′ hydroxy group of ribose. Mutation of the Asp-299 to alanine led to a complete loss of activity, suggesting a crucial role of the residue. Unaltered CD spectra and AMP-binding affinity of the D299A mutant led us to conclude that the overall secondary structure and the Ado-binding pocket of the mutant are unaffected. Moreover, the role of this negatively charged aspartate residue in binding ATP is unlikely. Thus it appears that Asp-299 of LdAdK is one of the key catalytic residues which facilitate deprotonation of the sugar hydroxy group so as to activate it for a nucleophilic attack on the γ-phosphate of ATP. This explains our earlier observation, where it was shown that there was a drastic reduction in the activity of the enzyme at pH values around 5, which is close to the pKa value of aspartic acid [5]. The fact that a highly conserved aspartate residue is known to play similar role in the ribokinase family of proteins is consistent with our proposal [44].
In a number of cases, arginine residues have been implicated to serve as the cationic site for binding of anionic substrates or co-enzymes [36–38]. Several examples are also known where arginine residues are apparently involved in catalysis [45–47]. Prediction that an arginine might be a catalytically important residue for LdAdK was based on our original chemical modification studies [29], which, later on, drew support from the crystallographic analysis of T. gondii AdK [22]. Based on this notion, when the conserved Arg-131 was replaced with an alanine, the enzyme displayed significant loss in activity without much alteration in the Km for either of the substrates. The decreased activity of the R131A mutant might therefore be caused by either a direct inhibition of the phosphate transfer or decreased ability to release the reaction products, i.e. AMP or ADP. However, the decrease in affinity of the mutant enzyme for AMP and lack of any evidence for tighter ADP binding rules out the second alternative. On the other hand, since Arg-131 seemed to interact with the γ-phosphate of ATP, as depicted from the modelling studies, it appears that this residue, located on the β-sheet of the small domain, may stabilize the negative charge on the oxygen of the leaving group, thereby facilitating the phosphate transfer reaction. In addition to this, Arg-131 probably increases the electrophilicity of the γ-phosphate group by withdrawing negative charge from the oxygen atom. This kind of mechanistic role for arginine residues is observed in a number of enzymes, including alkaline phosphatase [48] and nucleoside monophosphate kinase [49]. The crystal structure of T. gondii AdK also predicted a similar mechanism for Arg-136 which is present on the lid domain and translocates into the active centre upon Ado binding [22]. Apart from establishing the role of Arg-131 in charge stabilization during the phosphate transfer, the binding parameters and the chemical modification studies of the R131A mutant provided additional insights. The reduced affinity of the mutant enzyme towards AMP indicated that the arginine residue, owing to its positive charge and close proximity to the active site, might be playing an important role in AMP binding. Moreover, on being subjected to PGO treatment, the mutant displayed significant inactivation, even in the presence of saturating concentrations of Ado. This finding is in sharp contrast with that observed with the wild-type enzyme which exhibited substantial protection by Ado against PGO-mediated inactivation. Given the fact that the Ado Km of the R131A mutant is almost unaffected, these data suggest that, besides Arg-131, at least one more arginine residue, distal from the active site, might be important for optimal activity of the enzyme. Interestingly, our results suggest that Arg-69 is indeed that residue. Although its distance is more than 10 Å away from the active site and seemed unlikely to have any direct role in either substrate binding or phosphate transfer, mutation of this residue to either lysine or neutral alanine resulted in appreciable reduction in the catalytic activity. Both the mutants (R69K and R69A) largely retained their AMP-binding property, and Ado afforded protection to each mutant from PGO-mediated inactivation. Collectively, these observations are consistent with the interpretation that, while both Arg-69 and Arg-131 are functionally important, it is the positive charge of the Arg-131 residue that influences AMP binding. Consequently, sequestration of this residue upon Ado-induced domain closure provided protection to the arginine residue from PGO-modification and hence prevented inactivation of the enzyme. It, however, remains to be determined as to why AdK from the higher eukaryotes, despite harbouring this critical residue, could not be inactivated by PGO treatment [29]. A possibility might lie in the restricted accessibility of the chemical modifier to the corresponding arginine residue of the eukaryotic enzyme. It is also worth mentioning here that the increased ATP Km observed with the R69K mutant tends to approach the high ATP Km values of AdKs from higher eukaryotes, which naturally harbour a lysine residue at the equivalent position [2,3,15,16,27]. Since Arg-69 is located on the α3-helix of the large domain that is connected to the small domain by a short peptide loop, the positive charge on the guanidinium group of Arg-69 by forming a salt-bridge with another putative amino acid residue might possibly maintain the required conformational flexibility of the helix. When replaced with a lysine residue, a different charge distribution probably restricts the movement of this helix, thereby affecting the domain movement and ultimately in efficient binding of ATP. Moreover, since Arg-69 is spatially close to Asp-299, an alternative possible role of this basic residue acting as a secondary proton acceptor to increase the nucleophilicity of the Asp-299 cannot be ruled out completely. A similar role of arginine residues has been observed in case of NADP-dependent isocitrate dehydrogenase, where positive charges of arginine residues were shown to lower the pKa value of the nearby catalytic base to facilitate its ionization [50]. But the question as to why the R69A mutant shows unaltered affinity for ATP, despite exhibiting a similar decrease in the catalytic activity of R69K mutant, should await an actually determined X-ray structure of LdAdK. Not-withstanding these limitations, our results established beyond doubt the importance of this hitherto unidentified residue, which reflects subtle differences between the parasite and host AdK.
The importance of both Arg-69 and Arg-131 was confirmed further by creating the R69K/R131A double mutant. The activity of the enzyme variant was drastically reduced, with a significant decrease in the affinity for ATP. Double-mutant cycles are widely used as a powerful tool for analysing structure–function relationships of proteins [51]. The free energy changes (ΔΔG) have been used to determine whether the two mutations have additive effects on catalysis. Large deviation from the simple additivity can occur when the sites of mutation interact strongly with one another (by making direct contact or indirectly through electrostatic interactions or large structural perturbations) and/or when both the sites function co-operatively [51]. If the free energy change due to the R131A mutation is denoted by ΔΔGR131A and that due to R69K mutation by ΔΔGR69K, the change in the free energy observed in the double mutant, ΔΔGR131A/R69K, can be expressed as
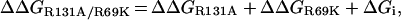 |
where ΔGi (also known as coupling energy) represents an interaction energy between the sites [52]. When ΔGi becomes near zero, the two sites behave independently, and the double mutant displays simple additivity. Changes in the transition state stabilization energy (ΔΔGT) caused by a mutation can be calculated from the following equation:
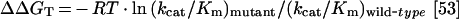 |
in which R is the gas constant, T is the absolute temperature, kcat is the catalytic centre activity and Km is the Michaelis constant for the wild-type and the mutant enzyme against a fixed substrate. Thus, at fixed ATP concentration, change in the transition state stabilization energy for the double mutant is 2.82 kcal/mol which is close to the sum of ΔΔGT (3.31 kcal/mol) for the single mutants R131A (1.52 kcal/mol) and R69K (1.79 kcal/mol). Similar additivity was observed when transition state stabilization energies were calculated at fixed Ado concentrations (results not shown). Thus it appears that Arg-131 and Arg-69 act independently during the catalytic function of LdAdK.
In the light of our results and the results obtained from other investigators, a schematic model showing a concerted mechanism for the phosphate transfer reaction is proposed in Figure 5. In the series of events, binding of Ado first induces a conformational change leading to a relatively closed positioning of the lid. The hinge located at the diglycine (Gly-61–Gly-62) motif and possibly Arg-69 on the α3 helix facilitates such domain movement. This favours ATP binding and subsequent translocation of Arg-131 into the active site [20,22]. The active-site Asp-299 functions as a general base that accepts a proton from the 5′ hydroxy group of the ribose of Ado. The negatively charged O5′ atom could then make a direct nucleophilic attack on the γ-phosphate group of ATP by an in-line SN2 mechanism. Arg-131 acts as a bidentate electrophile to stabilize the resulting quinquivalent transition state by interacting with two negatively charged oxygen groups of the terminal phosphate of ATP. Additionally, Arg-131 may contribute towards increasing the electrophilicity of the γ-phosphorous atom by withdrawing the negative charge via its interaction with the oxygen atoms. In the proposed mechanism, a magnesium ion, which, besides charge neutralization, probably creates the correct geometry for phosphoryl transfer, has been assumed to be present between α and β phosphates, as observed in T. gondii AdK, as opposed to the normal β and γ position in most kinases [22,54].
Figure 5. Schematic representation of the possible reaction mechanism of LdAdK.

Ado-induced domain rotation around the flexible diglycine motif (Gly-61–Gly-62) places the enzyme in pre-catalytic conformation. ATP binding causes further conformational changes, resulting in the initiation of a series of events in which Asp-299 first withdraws a proton from the 5′ hydroxy group of Ado (solid line) followed by a direct nucleophilic attack on the γ-phosphate of ATP (broken line). The resulting quinquivalent transition state is stabilized by Arg-131. Arg-131 also increases the electrophilicity (δ+) of the γ-phosphorus group.
In conclusion, the data presented here showed that the four amino acid residues (Gly-62, Arg-69, Arg-131 and Asp-299) are critical for the expression of optimal activity of LdAdK. In view of their absolute conservation, the functional roles of the residues Gly-62, Arg-131 and Asp-299 can probably be extended to the AdKs from all other sources, whereas the function of Arg-69 might be restricted to lower eukaryotes only. The homology model of LdAdK has allowed us to visualize the active site of the enzyme and analyse the results of the mutagenesis experiments. Although it cannot substitute for an actual crystal structure, this model can be useful for suggesting additional sites for mutagenesis and conceptualizing the results until the structure of LdAdK is determined experimentally.
Acknowledgments
This work was supported by Grants SP/SO/D-38/2000 from the Department of Science and Technology and CSIR Network Project SMM003, Government of India. R.D. and B.S. were each supported by individual fellowships from the Council of Scientific and Industrial Research, Government of India. We thank Mr Jaganmoy Guin of Bose Institute, Kolkata, for his technical support with CD experiments.
References
- 1.Miller R. L., Adamczyk D. L., Miller W. H., Koszalka G. W., Rideout J. L., Beacham L. M., 3rd, Chao E. Y., Haggerty J. J., Krenitsky T. A., Elion G. B. Adenosine kinase from rabbit liver: II. Substrate and inhibitor specificity. J. Biol. Chem. 1979;254:2346–2352. [PubMed] [Google Scholar]
- 2.Palella T. D., Andres C. M., Fox I. H. Human placental adenosine kinase: kinetic mechanism and inhibition. J. Biol. Chem. 1980;255:5264–5269. [PubMed] [Google Scholar]
- 3.Fisher M. N., Newsholme E. A. Properties of rat heart adenosine kinase. Biochem. J. 1984;221:521–528. doi: 10.1042/bj2210521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hawkins C. F., Bagnara A. S. Adenosine kinase from human erythrocytes: kinetic studies and characterization of adenosine binding sites. Biochemistry. 1987;26:1982–1987. doi: 10.1021/bi00381a030. [DOI] [PubMed] [Google Scholar]
- 5.Datta A. K., Bhaumik D., Chatterjee R. Isolation and characterization of adenosine kinase from Leishmania donovani. J. Biol. Chem. 1987;262:5515–5521. [PubMed] [Google Scholar]
- 6.Bhaumik D., Datta A. K. Immunochemical and catalytic characteristics of adenosine kinase from Leishmania donovani. J. Biol. Chem. 1989;264:4356–4361. [PubMed] [Google Scholar]
- 7.Darling J. A., Sullivan W. J., Jr, Carter D., Ullman B., Roos D. S. Recombinant expression, purification, and characterization of Toxoplasma gondii adenosine kinase. Mol. Biochem. Parasitol. 1999;103:15–23. doi: 10.1016/s0166-6851(99)00109-7. [DOI] [PubMed] [Google Scholar]
- 8.Carret C., Delbecq S., Labesse G., Carcy B., Precigout E., Moubri K., Schetters T. P., Gorenflot A. Characterization and molecular cloning of an adenosine kinase from Babesia canis rossi. Eur. J. Biochem. 1999;265:1015–1021. doi: 10.1046/j.1432-1327.1999.00806.x. [DOI] [PubMed] [Google Scholar]
- 9.Moffatt B. A., Wang L., Allen M. S., Stevens Y. Y., Qin W., Snider J., von Schwartzenberg K. Adenosine kinase of Arabidopsis: kinetic properties and gene expression. Plant Physiol. 2000;124:1775–1785. doi: 10.1104/pp.124.4.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Long M. C., Escuyer V., Parker W. B. Identification and characterization of a unique adenosine kinase from Mycobacterium tuberculosis. J. Bacteriol. 2003;185:6548–6555. doi: 10.1128/JB.185.22.6548-6555.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henderson J. F., Mikoshiba A., Chu S. Y., Caldwell I. C. Kinetic studies of adenosine kinase from Ehrlich ascites tumor cells. J. Biol. Chem. 1972;247:1972–1975. [PubMed] [Google Scholar]
- 12.Bhaumik D., Datta A. K. Reaction kinetics and inhibition of adenosine kinase from Leishmania donovani. Mol. Biochem. Parasitol. 1988;28:181–187. doi: 10.1016/0166-6851(88)90002-3. [DOI] [PubMed] [Google Scholar]
- 13.Richard J. P., Carr M. C., Ives D. H., Frey P. A. The stereochemical course of thiophosphoryl group transfer catalyzed by adenosine kinase. Biochem. Biophys. Res. Commun. 1980;94:1052–1056. doi: 10.1016/0006-291x(80)90525-2. [DOI] [PubMed] [Google Scholar]
- 14.Bone R., Cheng Y. C., Wolfenden R. Inhibition of adenosine and thymidylate kinases by bisubstrate analogs. J. Biol. Chem. 1986;261:6410–6413. [PubMed] [Google Scholar]
- 15.Spychala J., Datta N. S., Takabayashi K., Datta M., Fox I. H., Gribbin T., Mitchell B. S. Cloning of human adenosine kinase cDNA: sequence similarity to microbial ribokinases and fructokinases. Proc. Natl. Acad. Sci. U.S.A. 1996;93:1232–1237. doi: 10.1073/pnas.93.3.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh B., Hao W., Wu Z., Eigl B., Gupta R. S. Cloning and characterization of cDNA for adenosine kinase from mammalian (Chinese hamster, mouse, human and rat) species: high frequency mutants of Chinese hamster ovary cells involve structural alterations in the gene. Eur. J. Biochem. 1996;241:564–571. doi: 10.1111/j.1432-1033.1996.00564.x. [DOI] [PubMed] [Google Scholar]
- 17.Sinha K. M., Ghosh M., Das I., Datta A. K. Molecular cloning and expression of adenosine kinase from Leishmania donovani: identification of unconventional P-loop motif. Biochem. J. 1999;339:667–673. [PMC free article] [PubMed] [Google Scholar]
- 18.Wu L. F., Reizer A., Reizer J., Cai B., Tomich J. M., Saier M. H., Jr Nucleotide sequence of the Rhodobacter capsulatus fruK gene, which encodes fructose-1-phosphate kinase: evidence for a kinase superfamily including both phosphofructokinases of Escherichia coli. J. Bacteriol. 1991;173:3117–3127. doi: 10.1128/jb.173.10.3117-3127.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bork P., Sander C., Valencia A. Convergent evolution of similar enzymatic function on different protein folds: the hexokinase, ribokinase, and galactokinase families of sugar kinases. Protein Sci. 1993;2:31–40. doi: 10.1002/pro.5560020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathews I. I., Erion M. D., Ealick S. E. Structure of human adenosine kinase at 1.5 Å resolution. Biochemistry. 1998;37:15607–15620. doi: 10.1021/bi9815445. [DOI] [PubMed] [Google Scholar]
- 21.Cook W. J., DeLucas L. J., Chattopadhyay D. Crystal structure of adenosine kinase from Toxoplasma gondii at 1.8 Å resolution. Protein Sci. 2000;9:704–712. doi: 10.1110/ps.9.4.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schumacher M. A., Scott D. M., Mathews I. I., Ealick S. E., Roos D. S., Ullman B., Brennan R. G. Crystal structures of Toxoplasma gondii adenosine kinase reveal a novel catalytic mechanism and prodrug binding. J. Mol. Biol. 2000;298:875–893. doi: 10.1006/jmbi.2000.3753. [DOI] [PubMed] [Google Scholar]
- 23.Maj M. C., Singh B., Gupta R. S. Structure–activity studies on mammalian adenosine kinase. Biochem. Biophys. Res. Commun. 2000;275:386–393. doi: 10.1006/bbrc.2000.3307. [DOI] [PubMed] [Google Scholar]
- 24.Looker D. L., Berens R. L., Marr J. J. Purine metabolism in Leishmania donovani amastigotes and promastigotes. Mol. Biochem. Parasitol. 1983;9:15–28. doi: 10.1016/0166-6851(83)90053-1. [DOI] [PubMed] [Google Scholar]
- 25.Gottlieb M. The surface membrane 3′-nucleotidase/nuclease of trypanosomatid protozoa. Parasitol. Today. 1989;5:257–260. doi: 10.1016/0169-4758(89)90259-7. [DOI] [PubMed] [Google Scholar]
- 26.Konigk E., Putfarken B. Stage-specific differences of a perhaps signal-transferring system in Leishmania donovani. Tropenmed Parasitol. 1980;31:421–424. [PubMed] [Google Scholar]
- 27.Maj M., Singh B., Gupta R. S. The influence of inorganic phosphate on the activity of adenosine kinase. Biochim. Biophys. Acta. 2000;1476:33–42. doi: 10.1016/s0167-4838(99)00220-4. [DOI] [PubMed] [Google Scholar]
- 28.Bhaumik D., Datta A. K. Active site thiol(s) in Leishmania donovani adenosine kinase: comparison with hamster enzyme and evidence for the absence of regulatory adenosine binding site. Mol. Biochem. Parasitol. 1992;52:29–38. doi: 10.1016/0166-6851(92)90033-g. [DOI] [PubMed] [Google Scholar]
- 29.Ghosh M., Datta A. K. Probing the function(s) of active-site arginine residue in Leishmania donovani adenosine kinase. Biochem. J. 1994;298:295–301. doi: 10.1042/bj2980295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bagui T. K., Ghosh M., Datta A. K. Two conformationally vicinal thiols at the active site of Leishmania donovani adenosine kinase. Biochem. J. 1996;316:439–445. doi: 10.1042/bj3160439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cleland W. W. Steady state kinetics. In: Boyer P. D., editor. The Enzymes, vol. 2. 3rd edn. New York: Academic Press; 1970. pp. 1–65. [Google Scholar]
- 32.Mandal C., Kingery B. D., Anchin J. M., Subramaniam S., Linthicum D. S. ABGEN: a knowledge-based automated approach for antibody structure modeling. Nat. Biotechnol. 1996;14:323–328. doi: 10.1038/nbt0396-323. [DOI] [PubMed] [Google Scholar]
- 33.Mandal C. MODELYN: a molecular modeling program version PC-1.0. Indian Copyright No. 9/98. 1998.
- 34.Carson M. “Ribbons”. Methods Enzymol. 1997;277:493–505. [PubMed] [Google Scholar]
- 35.Thompson J. D., Higgins D. G., Gibson T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pal P. K., Colman R. F. The importance of arginine residues in the catalytic and regulatory functions of bovine-liver glutamate dehydrogenase. Eur. J. Biochem. 1976;68:437–443. doi: 10.1111/j.1432-1033.1976.tb10831.x. [DOI] [PubMed] [Google Scholar]
- 37.Riordan J. F., McElvany K. D., Borders C. L., Jr Arginyl residues: anion recognition sites in enzymes. Science. 1977;195:884–886. doi: 10.1126/science.190679. [DOI] [PubMed] [Google Scholar]
- 38.Riordan J. F. Arginyl residues and anion binding sites in proteins. Mol. Cell. Biochem. 1979;26:71–92. doi: 10.1007/BF00232886. [DOI] [PubMed] [Google Scholar]
- 39.Cox S., Radzio-Andzelm E., Taylor S. S. Domain movements in protein kinases. Curr. Opin. Struct. Biol. 1994;6:893–901. doi: 10.1016/0959-440x(94)90272-0. [DOI] [PubMed] [Google Scholar]
- 40.Sigrell J. A., Cameron A. D., Mowbray S. L. Induced fit on sugar binding activates ribokinase. J. Mol. Biol. 1999;290:1009–1018. doi: 10.1006/jmbi.1999.2938. [DOI] [PubMed] [Google Scholar]
- 41.Steitz T. A., Shoham M., Bennett W. S., Jr Structural dynamics of yeast hexokinase during catalysis. Philos. Trans. R. Soc. London B Biol. Sci. 1981;293:43–52. doi: 10.1098/rstb.1981.0058. [DOI] [PubMed] [Google Scholar]
- 42.Hellinga H. W., Evans P. R. Mutations in the active site of Escherichia coli phosphofructokinase. Nature (London) 1987;327:437–439. doi: 10.1038/327437a0. [DOI] [PubMed] [Google Scholar]
- 43.Pompliano D. L., Peyman A., Knowles J. R. Stabilization of a reaction intermediate as a catalytic device: definition of the functional role of the flexible loop in triosephosphate isomerase. Biochemistry. 1990;29:3186–3194. doi: 10.1021/bi00465a005. [DOI] [PubMed] [Google Scholar]
- 44.Sigrell J. A., Cameron A. D., Jones T. A., Mowbray S. L. Structure of Escherichia coli ribokinase in complex with ribose and dinucleotide determined to 1.8 Å resolution: insights into a new family of kinase structures. Structure. 1998;6:183–193. doi: 10.1016/s0969-2126(98)00020-3. [DOI] [PubMed] [Google Scholar]
- 45.Kremer A. B., Egan R. M., Sable H. Z. The active site of transketolase: two arginine residues are essential for activity. J. Biol. Chem. 1980;255:2405–2410. [PubMed] [Google Scholar]
- 46.Tripp B. C., Tu C., Ferry J. G. Role of arginine 59 in the γ-class carbonic anhydrases. Biochemistry. 2002;41:669–678. doi: 10.1021/bi010768b. [DOI] [PubMed] [Google Scholar]
- 47.Li Y., Wu Y., Blaszczyk J., Ji X., Yan H. Catalytic roles of arginine residues 82 and 92 of Escherichia coli 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase: site-directed mutagenesis and biochemical studies. Biochemistry. 2003;42:1581–1588. doi: 10.1021/bi026800z. [DOI] [PubMed] [Google Scholar]
- 48.Butler-Ransohoff J. E., Kendall D. A., Kaiser E. T. Use of site-directed mutagenesis to elucidate the role of arginine-166 in the catalytic mechanism of alkaline phosphatase. Proc. Natl. Acad. Sci. U.S.A. 1994;85:4276–4278. doi: 10.1073/pnas.85.12.4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schlichting I., Reinstein J. Structures of active conformations of UMP kinase from Dictyostelium discoideum suggest phosphoryl transfer is associative. Biochemistry. 1997;36:9290–9296. doi: 10.1021/bi970974c. [DOI] [PubMed] [Google Scholar]
- 50.Soundar S., Danek B. L., Colman R. F. Identification by mutagenesis of arginines in the substrate binding site of the porcine NADP-dependent isocitrate dehydrogenase. J. Biol. Chem. 2000;275:5606–5612. doi: 10.1074/jbc.275.8.5606. [DOI] [PubMed] [Google Scholar]
- 51.Wells J. A. Additivity of mutational effects in proteins. Biochemistry. 1990;29:8509–8517. doi: 10.1021/bi00489a001. [DOI] [PubMed] [Google Scholar]
- 52.Carter P. J., Winter G., Wilkinson A. J., Fersht A. R. The use of double mutants to detect structural changes in the active site of the tyrosyl-tRNA synthetase (Bacillus stearothermophilus) Cell. 1984;38:835–840. doi: 10.1016/0092-8674(84)90278-2. [DOI] [PubMed] [Google Scholar]
- 53.Wilkinson A. J., Fersht A. R., Blow D. M., Winter G. Site-directed mutagenesis as a probe of enzyme structure and catalysis: tyrosyl-tRNA synthetase cysteine-35 to glycine-35 mutation. Biochemistry. 1983;22:3581–3586. doi: 10.1021/bi00284a007. [DOI] [PubMed] [Google Scholar]
- 54.Knowles J. R. Enzyme-catalyzed phosphoryl transfer reactions. Annu. Rev. Biochem. 1980;49:877–919. doi: 10.1146/annurev.bi.49.070180.004305. [DOI] [PubMed] [Google Scholar]